

Abstract
Previous studies on changes in murine brain gene expression associated with the selection for ethanol preference have used F2 intercross or heterogeneous stock (HS) founders, derived from standard laboratory strains. However, these populations represent only a small proportion of the genetic variance available in Mus musculus. To investigate a wider range of genetic diversity, we selected mice for ethanol preference using an HS derived from the eight strains of the collaborative cross. These HS mice were selectively bred (four generations) for high and low ethanol preference. The nucleus accumbens shell of naive S4 mice was interrogated using RNA sequencing (RNA-Seq). Gene networks were constructed using the weighted gene coexpression network analysis assessing both coexpression and cosplicing. Selection targeted one of the network coexpression modules (greenyellow) that was significantly enriched in genes associated with receptor signaling activity including Chrna7, Grin2a, Htr2a and Oprd1. Connectivity in the module as measured by changes in the hub nodes was significantly reduced in the low preference line. Of particular interest was the observation that selection had marked effects on a large number of cell adhesion molecules, including cadherins and protocadherins. In addition, the coexpression data showed that selection had marked effects on long non-coding RNA hub nodes. Analysis of the cosplicing network data showed a significant effect of selection on a large cluster of Ras GTPase-binding genes including Cdkl5, Cyfip1, Ndrg1, Sod1 and Stxbp5. These data in part support the earlier observation that preference is linked to Ras/Mapk pathways.
Keywords: Addiction, ethanol, HS-CC, mouse, network analysis, nucleus accumbens shell, RNA-Seq, selection, transcriptome
Selective breeding of mice for ethanol preference (and/or consumption) has been used to map quantitative trait loci (QTL) and to detect selection-related effects on brain gene expression (Bice et al. 2006; Bubier et al. 2014; Hitzemann et al. 2009; Hoffman et al. 2014; Metten et al. 1998, 2014; Mulligan et al. 2006). Some consistent themes have emerged. For example, Mulligan et al. (2006) analyzed brain microarray data obtained in three selected lines (and six inbred mouse strains) and detected several gene clusters associated with preference; annotation of these clusters included enrichment in mitogen-activated protein kinase (MAPK) signaling and transcription regulation pathways. Metten et al. (2014) used a dual selection paradigm for preference and acute withdrawal to examine selection effects on gene expression. Similar to Mulligan et al. (2006), the analysis of the expression data showed selection effects on MAPK-related genes. More generally, Bubier et al. (2014) reviewed 86 preference gene data sets and found that MAPK-related genes have been detected in multiple studies; genes frequently detected have included Map4k3, Map4k5 and Mapk8.
The founder populations in the selection studies noted above were either F2 intercross or heterogeneous stock (HS) mice formed from crossing four or eight standard inbred laboratory strains. The HS were seen as an advantage over the F2 intercross animals because of the greater genetic diversity. However, it is now clear that with those founder populations, only a fraction of the genetic diversity available in Mus musculus was actually screened for preference-related genes and gene networks (see Keane et al. 2011; Roberts et al. 2007). Recently, solutions to this potential genetic bottleneck have become available in the form of two outbred populations related to the collaborative cross (CC) [the HS-CC and the diversity outbred (DO)], and these are now available for selective breeding (Iancu et al. 2010, 2013b; Svenson et al. 2012). The CC itself is a large panel of recombinant inbred (RI) strains where the eight founder strains included three wild-derived strains; it is estimated that the eight founders encompass >90% of M. musculus genetic diversity (Churchill et al. 2004). The HS-CC and DO were created as complementary to the CC because large populations can be obtained relatively easily, can enable the precision mapping of complex traits and are ideal populations for selective breeding (Chesler 2014; Hitzemann et al. 2014).
In the current study, HS-CC founders were selectively bred for high and low ethanol preference using a standard 24/7, two-bottle choice paradigm (10% ethanol vs. water). The breeding scheme used was short term (four generations) in order to minimize the random fixation of genes unrelated to selection. Gene expression data (RNA-Seq) were collected from the nucleus accumbens (NAc) shell, a key component of the addiction circuit (Koob & Volkow 2010). Previously, we have shown the advantages of network-based approaches when examining gene expression data in animals selectively bred for high drinking in the dark (Iancu et al. 2013b), ethanol preference, and ethanol withdrawal (Metten et al. 2014). We observed that network data provide additional and largely discrete information from differential expression. We continue this approach in the current study and also include a novel approach for analyzing cosplicing networks (Iancu et al. 2015). Key coexpression and cosplicing hub nodes were identified.
Materials and methods
Husbandry
All mice used for the short-term selection (STS) were obtained from the colony at the Portland VA Medical Center, an Association for Assessment and Accreditation of Laboratory Animal Care-approved facility. All procedures were in accordance with the VA Institutional Animal Care and Use Committee and were performed according to NIH Guidelines for the Care and Use of Laboratory Animals. Mice were maintained at 21 ± 1°C in plastic cages (19 × 31 × 13 cm3) on Eco-Fresh bedding (Absorption Corp., Ferndale, WA, USA) with tap water and Purina 5001 chow (PMI Nutrition International, Brentwood, MO, USA). Pups were weaned and housed with same-sex littermates at postnatal day 21.
Animals
The HS-CC mice were the selection founders. The HS-CC was formed by interbreeding of the eight different inbred strains used to form the CC: 129Sv/Im, A/J, C57BL/6, CAST/Ei, NOD/Lt, NZO/HILt, PWK/Ph and WSB/EiJ. The intercrossing strategy was pseudo-random (Iancu et al. 2010). The HS-CC mice are maintained as 48 families using a rotational breeding scheme. The founder animals used for selection were from G23.
Behavior analysis: two-bottle choice ethanol preference
Ethanol self-administration was monitored in adult (8 weeks) HS-CC mice using the two-bottle choice preference test (10% ethanol vs. water) (see Metten et al. 2014). Mice were individually housed and given 1 week of habituation, with continuous access to food and tap water, accessible from two 25-ml graduated glass cylinders fitted with rubber stoppers and stainless steel sipper tubes. Mice were acclimated to the taste and effects of ethanol by progressively increasing the concentration of ethanol (0%, 5% and 10% v/v) every 4 days. Every other day, the position of the ethanol bottle was reversed to control for side preferences.
Selection
After testing, S0 mice were selected for breeding based on their preference for 10% ethanol. The 20 males and 20 females with the highest preference values were paired, with brother–sister matings avoided, to create a ‘high’ preference line; similarly, the 40 mice with the lowest preference scores were paired to create a ‘low’ preference line. Approximately 200 pups from each generation were weaned and tested at adulthood as described above for three subsequent generations; active selection concluded at S3. S4 alcohol-naive pups were used for genetic analyses.
Genotyping the high and low ethanol preferring lines and quantification of genetic variability
Eighty-eight S4 animals (22 males and 22 females from each selected line) were genotyped using the MegaMUGA mouse genotyping array (Geneseek, Lincoln, NE, USA). The array provides robust calls for >77 000 single nucleotide polymorphisms (SNPs). For this analysis, genetic distances were calculated for each pair of samples, based on the number of identical alleles at each marker and then summed over all markers. Next, the between-group and within-group distances were calculated following the analysis of molecular variance approach (Excoffier et al. 1992).
Dissection of tissue and extraction of RNA
Naive S4 mice (N = 12/line/sex) were euthanized, brains removed and immediately frozen on dry ice. Frozen brains were sliced in 55-μ coronal sections on a freezing microtome at −13°C and slices containing the NAc were mounted on polyethylene naphthalate-covered slides. Mounted slices were lightly thionin stained under RNAse-free conditions and dehydrated in increasing concentrations of ethanol diluted in RNAse-free water (50%, 70%, 95% and 100%) for 30 seconds each and then air-dried. The NAc shell was dissected bilaterally on a Leica LMD-6000 (Leica Microsystems Inc., Buffalo Grove, IL) using known anatomical landmarks (Paxinos & Franklin 2007). Dissected tissue was processed with the Arcturus Picopure Kit (ThermoFisher Scientific, Waltham, MA) yielding on average 200 ng of total RNA. RNA quality was assessed using the Caliper Labchip GX (PerkinElmer, Waltham, MA) and RNA Quality Scores (RQS).
RNA-Seq
Library formation (polyA+, stranded) and sequencing were all performed according to Illumina’s (San Diego, CA) specifications at the OHSU Massively Parallel Sequencing Shared Resource. Libraries were multiplexed 6 per lane, yielding approximately 25–30 million total reads per sample. FastQC was used for quality checks on the raw sequence data. Sequence data were then aligned using STAR [Spliced Transcripts Alignment to a Reference, 2.3.0e (Dobin et al. 2013)] allowing for a maximum of three mismatches per 100 bp read. For all samples, >85% of the reads were uniquely aligned. Using the Bedtools suite (2.26.0), reads were aligned to known genomic features to generate counts at the gene and exon level. Gene and exon expression data were imported into the R application environment; upper-quartile normalization was performed using the edgeR Bioconductor package (Robinson et al. 2010). The read density threshold for inclusion in the network analyses for genes and exons was 30 and 5, respectively. Network connectivity for both coexpression and cosplicing were calculated as described elsewhere (Iancu et al. 2015). Genes in the top 50% for both coexpression and cosplicing connectivity (see below) were used for further analysis (7545 genes). The expression data are available via NCBI Gene Expression Omnibus (accession number GSE65950, ID: 200065950).
DE and DV analyses
The DE (differentially expressed) was determined using edgeR, with the option of ‘tagwise’ dispersion; the threshold for significance was set at a false discovery rate (FDR) < 0.05. For gene DV (differentially variable), we utilized the ‘var.test’ procedure in the R ‘stats’ package. For exon DV, we computed pairwise distances between all samples (Canberra metric) and then utilized the ‘mrpp’ function in the ‘vegan’ R package. The mrpp function is sensitive to differences in spread/dispersion of pairwise distances, as well as within/between group distance differences.
Coexpression and Cosplicing network construction and effects of selection
The coexpression network was constructed by means of the Weighted Gene Coexpression Network Analysis (WGCNA) (Iancu et al. 2012; Langfelder & Horvath 2008) using a consensus module approach followed by assessment of selection effects on network structure (Ando et al. 2015; Gill et al. 2010; Iancu et al. 2013a; Ideker & Krogan 2012). Cosplicing networks were constructed using a procedure we have termed CoSplicEx (Iancu et al. 2015). The procedure for CoSplicEx network construction was identical to that for the coexpression networks, except that the Pearson correlations were replaced with Mantel correlations (Iancu et al. 2015). In the coexpression network, the difference in correlation strength was evaluated utilizing the ‘var.test’ R function. To mitigate the computational load, we restricted the search to Pearson correlations between individual genes that differed by ≥0.5 before power transformation. For CoSplicEx edges, we implemented a permutation procedure that compared differences in correlation strengths between the selected groups with differences in correlation strengths between two randomly assigned groups. This general procedure has been used to quantify network rewiring in both genomic (Gill et al. 2010) and neural imaging studies (Hosseini et al. 2012). Using this procedure, we identified the number of changed edges for each gene and then inquired as to whether some genes had a disproportionately high number of changing edges. For the latter, the binomial test was used with the following parameters. The average incidence of changing edges (the rate of the binomial test) was computed by dividing the number of changing edges (P < 0.01) by the total number of network edges. The number of trials (for each gene) was equal to the number of edges. The number of ‘successes’ was equal to the number of significantly changing edges (P < 0.01). Genes enriched in changing edges are denoted as differentially wired (DW).
Module characterization
Functional significance of all modules was evaluated using gene ontology (GO) enrichment analysis using the GO-stats R package (Ashburner et al. 2000; Falcon & Gentleman 2007). Because of the nested structure of the GO terms, a graph decorrelation procedure was used (Alexa et al. 2006). To implement a ranking procedure, we integrated differential network results at the module and gene summarization levels into a comprehensive gene screening procedure. Modules enriched in gene or edge changes were the primary focus of further annotations. At the individual gene level, we focused on module hubs with normalized intramodular connectivity above 0.8. The GOrilla algorithm (Eden et al. 2009) was used to provide a visual representation of GO annotation enrichment and to examine annotation enrichment of selected groups of genes against a background set of all network genes.
Results
Selection for ethanol preference
After three generations of bidirectional selection for ethanol preference, there was a marked difference between the high and low lines (Fig. 1). At S3, the difference in preference ratio was 0.49 vs. 0.15 (F1,209 = 103, P < 0.001). In both groups, females showed a preference than males (F1,209 = 20, P < 0.001). Notably, 65% of females and 37% of males of high preference lines achieved a preference ratio of >0.5 compared with 6.5% of females and 2.3% of males of low preference lines.
Figure 1. Differences in ethanol preference and consumption as a result of bidirectional selection.
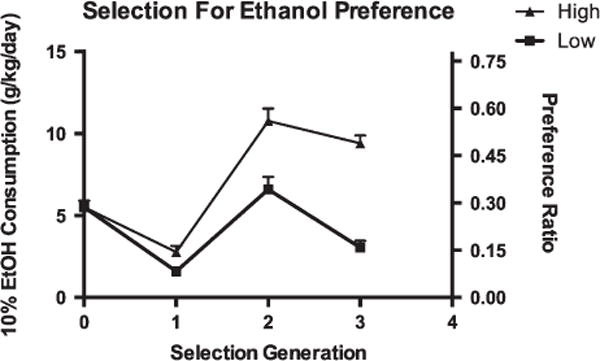
Left y-axis: consumption of 10% ethanol averaged from experimental days 10 and 12 expressed as g/kg ethanol consumed per day. Right y-axis: ethanol preference ratio (ratio of milliliters of 10% ethanol consumed to total milliliters of fluid consumed).
Genetic variability analysis of high and low preference selected lines
Only those SNPs on the genotyping array with an alternative allele frequency of >5% (N = 61 731) were included in the analysis. Total genetic distance among samples was calculated and plotted (Fig. 2). Genetic distances in the selected lines were relatively constrained when compared with the HS-CC founder strains. Strong divergence between the high and low preferring lines was observed. From a genetic distance perspective, the High line was closest to the WSB/EiJ inbred strain. Note that considerable genetic diversity was maintained in the selected lines.
Figure 2. Multidimensional scaling (MDS) plots of genome-wide differences between high (magenta) and low (turquoise) ethanol-preferring selected lines compared with the founding strains (B6 = red, AJ = brown, 129 = purple, NOD = black, NZO = orange, CAST = dark green, PWK = green, WSB = blue) of the HS-CC founder stock.
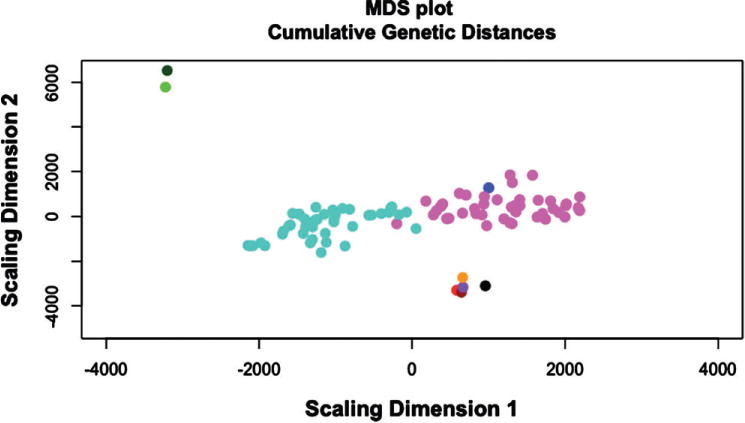
This figure illustrates strong genetic divergence between high and low preferring lines, in part because of the incorporation of wild-derived alleles in the low line. Additionally, it is notable the WSB/EiJ strain appears most genetically similar to the high preferring line.
Coexpression and cosplicing network construction
Consensus coexpression and cosplicing networks were constructed. A total of 7545 genes met the criteria for inclusion in the analyses. The adjacency matrices were clustered into 19 modules for coexpression and 23 modules for cosplicing; each module was randomly assigned a nominal color. The genes associated with each coexpression and cosplicing module are found in Tables S1 and S2 (Supporting information), respectively. The clustering results were validated using a Z-score quantification of module quality. All modules had at least a moderate (Z > 4) quality score in one (and most in more than one) measure of network quality [density, separability or connectivity; see Langfelder et al. (2011)]. The GO annotations for the coexpression and cosplicing modules are found in Tables S3 and S4, respectively.
Selection effects on gene-level expression
Selection resulted in 7 DE genes, 223 DV genes and 1974 DW genes (FDR < 0.05). The identity of these genes, their raw and FDR adjusted P-values, module assignment and intramodular connectivity values are found in Table S1. Genes previously reported as significantly associated with preference consumption and DE by Mulligan et al. (2006) were flagged. Given the small number of DE genes, these were not considered further.
For the DV genes (Table S5), higher variability was observed in the high line (208 genes with higher variability in the high line vs. 15 genes with higher variability in the low line). Annotation of the DV genes (Eden et al. 2009) showed significant enrichment in genes associated with cell–cell signaling and included the GO categories ‘Response to Endogenous Stimulus’ and ‘Signaling Receptor Activity’ (Table S5). The 19 genes in the latter category included Adra1a, Chrna7, Fzd3, Gabrb2, Grin2a, Grin2b, Htr2a, Kit, Oprd1 and Sort1. Fdz3, Kit and Sort1 were detected as DE by Mulligan et al. (2006). Coexpression network information was used to further annotate the DV genes. The DV genes exhibited significant enrichment in two coexpression modules: greenyellow (P < 10−42) and pink (P < 7 × 10−3) (Table S5). The GO annotation of these modules is found in Table S3. The greenyellow module was notably enriched in annotations associated with neurons, e.g. neuronal cell body (P < 1 × 10−5) and signaling receptor activity (P < 5 × 10−6). The pink module was enriched in annotation associated with glutamate secretion (P < 5 × 10−4).
Annotation of the DW genes showed significant enrichment in genes associated with cell–cell adhesion (Table S6) and neuron part. Genes in the former category included 18 protocadherin related genes; only one of these, Pdch15, was detected as DE between preferring and non-preferring animals by Mulligan et al. (2006). Genes associated with neuron part included a number of glutamate receptor subunits (Gria3, Grid2, Grik2, Grin2a, Grm1, Grm3, Grm4 and Grm7), voltage-gated potassium channels (Kcna4, Kcnab1, Kcnb1, Kcnb2 and Kcnd2) and kinesin-related genes (Kif1a, Kif1b, Kif5a, Kif5b and Kif5c). Of the genes associated with neuron part, 27 were also detected as DE by Mulligan et al. (2006); these included Grid2, Mapk1, Mapk8ip3, Pde4b, Psen1, Shank3 and Snap25. The DW genes were significantly enriched in five coexpression modules: brown (P < 3 × 10−14), greenyellow (P < 1 × 10−18), lightcyan (P < 3 × 10−10), pink (P < 3 × 10−4) and yellow (P < 2 × 10−7). Annotations for the greenyellow and pink module are discussed above. The brown module (Table S3) was enriched in annotations associated with synaptic function, including synaptic transmission (P < 2 × 10−5); the lightcyan module was enriched in annotation for the synapse (P < 1 × 10−5) and the yellow module was enriched in genes associated with the nuclear part (P < 9 × 10−7) and RNA splicing (P < 1 × 10−5).
The DW and coexpression network data were integrated to focus on those nodes that are hubs in only one of the selected lines and show a significant change in affected edges. Two hundred and fifty-three genes met these criteria (Table S7). Two modules, greenyellow (P < 10−7) and lightcyan (P < 3 × 10−3), were significantly enriched in affected genes; the brown module showed a trend (P > 0.08) toward significant enrichment. Of the affected hub nodes in the greenyellow and lightcyan modules (N = 65), only two genes had a higher intramodular connectivity in the low line (Gm12356 and Dpp6), and both were found in the lightcyan module. The effects of selection specifically on the greenyellow hub node Oprd1 and more generally on intramodular connectivity are illustrated in Fig. 3. Connectivity in the low line is reduced; for Oprd1, the decrease in connectivity was marked (0.88 to 0.46; see Table S7).
Figure 3. Effects of selection on connectivity in Oprd1, a greenyellow module hub with significant enrichment in affected edges.
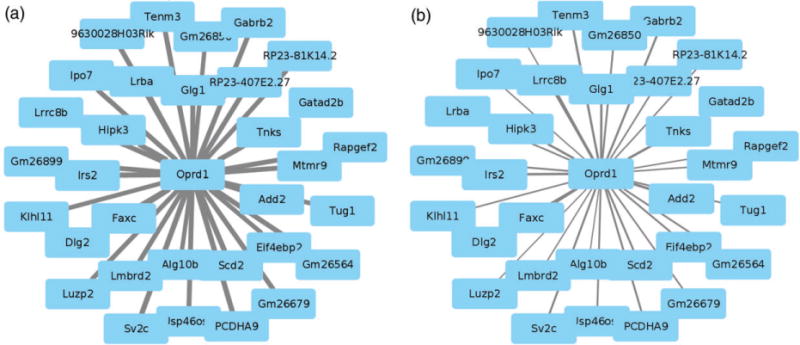
(a) Connectivity patterns of Oprd1 in the high line network. (b) Connectivity pattern of Oprd1 in the low line network. Edge thickness and transparency are proportional with connection strength.
It was of interest to note that among the affected hub nodes in the greenyellow and lightcyan modules, 13 of the nodes were non-coding RNAs, largely antisense to protein-coding genes. One of these non-codes (Gm26672) is antisense to a gene family of the protocadherins described above and includes Pcdhga2, a5, a6, a7b2, b5 and b7. The EnrichR tool (Chen et al. 2013; Kuleshov et al. 2016) was used to determine if the greenyellow and lightcyan hub nodes were significantly enriched in transcription factor (TF) and/or micro-RNA (mi-RNA)-binding sites. No significant enrichment was detected for the greenyellow hubs; however, for the lightcyan module hubs there was a significant enrichment (corrected P < 0.01) in TCFAP2A, ZBT7A and NFKB1 TF-binding sites.
Selection effects on gene cosplicing
The cosplicing network was formed as described elsewhere (Iancu et al. 2015). Twenty-nine genes displayed alternative exon usage (AEU, identified as splicing significance in Table S2), no genes displayed DV in splicing patterns and 1633 genes exhibited DW (FDR < 0.05; Table S2). Only the DW genes were analyzed further. The DW genes were enriched in annotations associated with Golgi vesicle transport, the nuclear pore and Ras GTPase binding (Table S8). Genes in the latter category included a cluster of Rab-, Rap- and syntaxin-binding protein-associated genes. The DW genes were significantly enriched in three cosplicing modules [brown (P < 4 × 10−27), green (P < 1 × 10−21) and yellow (P < 7 × 10−8), Table S8]. The brown module was significantly enriched in annotations associated with GTPase binding, as was the green module. The green module also showed enrichment in neuronal membrane annotations, e.g. neuron part (P < 10−7) and post-synaptic density (P < 9 × 10−7). Genes in the latter category included Gria1, Gria3, Htr2a and Htr5a.
The DW and splicing network data were integrated to focus on those nodes that are hubs in only one of the selected lines and show a significant change in affected edges. Thirty-four genes met these criteria (Table S9). When compared with all DW genes, there are no significant functional, molecular or component GO annotation enrichments for this group of genes, nor was there a significant enrichment in one or more of the cosplicing modules. The number of significantly changing hubs in the high and low lines was equal. Several of the hub nodes have been directly associated with alcohol-related phenotypes: Tnik, Pde10a, Ndrg1, Sos1, Tmem208, Nploc4, Slc6a15 and Pcsk1 (Bell et al. 2009; Costin et al. 2013; Logrip & Zorrilla 2014; Parsons et al. 2012; Rodd et al. 2009). An additional 18 of the splicing hubs are associated with mouse preference QTL intervals (GeneWeaver.org) (Table S9).
Discussion
Brain gene expression data are available for mice selectively bred for ethanol preference from F2 intercross, HS4, HS/NPT and HS/Ibg founders and for inbred laboratory strains and RI strains that differ in ethanol preference (or consumption) (Bubier et al. 2014; Hoffman et al. 2014; Metten et al. 2014; Mulligan et al. 2006; Williams & Mulligan 2012). However, as sequence data became available from multiple inbred mouse strains (Keane et al. 2011; Roberts et al. 2007), it has become clear that the preference/consumption studies have engaged only a fraction of the genetic diversity available in M. musculus. By increasing the genetic diversity, one has the potential to detect new pathways regulating ethanol preference and consequently to detect new targets for manipulation. With this perspective in mind, the current study was undertaken using HS-CC animals as the founders for short-term selective breeding. Three generations of breeding were sufficient for significant segregation of the high and low preference lines; preference in the high line was approximately 10 times greater than in the low line. Genotyping the lines not only confirmed the segregation of the lines but also illustrated that considerable genetic diversity was retained (Fig. 2). The genotype data also showed that from a genetic perspective, the high line was closest to the WSB founder strain. These data were of interest in that it has been reported that in both the HS-CC and the related DO mice there is a WSB-dominant meiotic drive locus on chromosome 2 (Chesler et al. 2016); numerous preference QTL have been detected on chromosome 2, albeit in less genetically diverse populations (Belknap & Atkins 2001). Using a selective breeding protocol, the meiotic drive locus and the associated WSB alleles have been eliminated from the DO population (Chesler et al. 2016). We have compared these DO mice with the HS-CC; no difference was detected in either acute or chronic preference consumption (data not shown). For both the HS-CC and the ‘new’ DO, approximately 25% of the animals consume daily >10 g/kg of ethanol.
As described by Grahame et al. (1999) and Oberlin et al. (2011), replicate lines of high alcohol preferring and low alcohol preferring mice have been selected from HS/Ibg founders. These were long-term selections, continued for >20 generations and achieved considerable segregation of the high and low preferring lines. The characteristics of these mice are described in numerous publications (Oberlin et al. 2011). The HS/Ibg (named for the place of their derivation, the Institute of Behavioral Genetics, Boulder, CO) founders are genetically similar to the HS/NPT (derived at the Northport VAMC, North-port, NY) and have a relatively low genetic diversity when compared with the HS-CC. The STS protocol used in the current study and implanted to minimize the random fixation of genes unrelated to preference is similar to the design found in the studies of Metten et al. (1998, 2014). These studies focused on the effects of selection and the apparent inverse genetic relationship between preference and acute ethanol withdrawal. The current high and low preference lines were tested for differences in acute withdrawal following a 4 g/kg alcohol challenge; none were detected (data not shown). These studies also showed that there was no difference between the selected lines in the loss of and recovery of the righting reflex. The selected lines were not tested for differences in preference for other tastants. However, among the HS-CC founders, there is only a weak correlation (r = 0.21; N = 96) between preference for ethanol and preference for 0.2% saccharin.
The RNA-Seq data were analyzed at both the gene and exon level. Both analysis strategies confirmed earlier findings that the number of genes associated with ethanol preference is large and that these genes involve multiple functional pathways. However, the data differ from previous results (Mulligan et al. 2006) in that only a small number of genes (N = 7) were significantly DE (FDR < 0.05). We recognize that the sample sizes used in the current study were moderate (N = 24/line) and thus the power to detect significant changes was limited. Nonetheless, the data suggest that at least from the perspective of the NAc shell, numerous and large changes in gene expression are not necessary for selection.
The DV gene expression statistic is one approach for assessing whether there are differences in the regulation of gene expression between groups. Mar et al. (2011) have suggested that the variance in gene expression may be far more important in understanding disease etiology than previously recognized. Further, these authors noted that genes with high expression variability tend to function as cell surface receptors. In the current data set, we observed that 223 genes showed significant DV between the high and low preference lines, and consistent with Mar et al. (2011), this group of genes was enriched for cell–cell signaling, synaptic signaling and signaling receptor activity (Table S5). Importantly, for this group of genes, higher variability was significantly skewed to the high preference line (high vs. low: 208 vs. 15). Numerous members of the signaling receptor activity genes, including Chrna7, Gabrb2, Grin2a, Grin2b, Fzd3, Htr2a, Igf1r, Kit, Oprd1 and Sort1, have been associated with one or more alcohol phenotypes, including preference (Bowers et al. 2005; Bubier et al. 2014; Cao et al. 2014; de la Monte et al. 2012; Dick et al. 2006; Enoch 2013; Lo et al. 2016; Mulligan et al. 2006; Zhang et al. 2008).
The DV genes were highly enriched (P < 10−42) in a single coexpression module (greenyellow). The module was of moderate size (227 members) and, not surprisingly, was significantly enriched in annotations associated with receptor signaling and neuronal membrane-related genes. Using our consensus module approach for selection line data (Iancu et al. 2013a, 2013b), it was observed that essentially all of the hub nodes in the greenyellow module were affected by selection. Using the EnrichR tool (Chen et al. 2013; Kuleshov et al. 2016), these hub nodes were not detected as significantly enriched in TF- or mi-RNA-binding sites. However, the protein–protein interaction hub protein tool showed that a significant number of the greenyellow hub nodes were functionally downstream from Fyn kinase (corrected P < 7 × 10−7). This cluster of hub nodes included Cdkl5, Dcc, Pde4d, Gpr63, Cbl, Dock1 and Add2. Fyn has been implicated in the regulation of a variety of ethanol phenotypes; Fyn knockout (KO) mice have been reported to show decreased or no difference in preference consumption and mice overexpressing Fyn have been reported to have decreased preference consumption (Boehm et al. 2003; Chen & Charness 2008; Farris & Miles 2013; Lee & Messing 2008; Yaka et al. 2003). Fyn is a member of the greenyellow module but was not significantly affected by selection. It also should be noted that among the Fyn-related cluster of hub nodes is Cdkl5, which has been associated with alcoholism (Liu et al. 2006; Sokolov et al. 2003).
The DW gene expression statistic assesses the change in connectivity of a single gene to all other genes in the network (N = 7545); the statistic counts the number of pairwise gene correlations that show a statistically significant change between the two selected groups, followed by identification of genes enriched in changing correlations/network edges. The data again point to the broad effect selection had on the shell transcriptome. Twenty-six percent of the genes included in the network analysis showed a significant change (FDR < 0.05), indicating moderate effects on transcription that are dispersed among many genes. Not surprisingly many of the DW genes have been associated with a variety of alcohol phenotypes, including preference for Grid2, Mapk1, Mapk8ip3, Pde4b, Psen1, Shank3 and Snap25 (Mulligan et al. 2006). Of some note, the DW genes were significantly enriched in cell–cell adhesion genes, including 4 cadherin and 18 protocadherin genes. Previous studies have implicated Cdh11, Cdh12, Cdh13, Cdh19 Pchha9 and Pcdh15 in alcoholism or preference (Edenberg et al. 2010; Liu et al. 2009; Lydall et al. 2011; Mulligan et al. 2006; Sokolov et al. 2003; Treutlein et al. 2009). However, the data presented here suggest very broad effects on the protocadherins. It is interesting to speculate that this broad effect in part is the result of selection effects on the non-coding RNA, Gm26672, a hub in the lightcyan module and antisense to a large cluster of the affected protocadherins. More generally, the data presented here are the first to show that preference selection has significant effects on several non-coding RNA hub nodes.
Preference selection had a marked effect on alternative splicing. The mammalian genome contains over 20 000 protein-coding genes and alternative splicing produces approximately 100 000 intermediate to highly expressed transcripts with the greatest diversity found in brain (Calarco et al. 2011; Li et al. 2007; Pan et al. 2008). There is evidence that drugs of abuse including alcohol affect alternative splicing and/or that the responses of drugs of abuse are transcript dependent (Acosta et al. 2011; Bulwa et al. 2011; Farris & Mayfield 2014; Glatt et al. 2011; Jin & Woodward 2006; Lee et al. 2014; Maiya et al. 2012; Raeder et al. 2008; Rothenfluh et al. 2006; Wernicke et al. 2010). We and others (see Iancu et al. 2015 and references therein) have observed that coordinated splicing has network properties amenable to analysis using tools such as WGCNA. While exon usage patterns can be represented in several ways, we have chosen a method that utilizes Mantel correlations of Canberra distance matrices (Iancu et al. 2015). This method detects exon inclusion rates but does not provide information on isoform identity, which requires greater read depth than the current study (Lee et al. 2014).
The Mantel correlations, similar to the Pearson correlations for coexpression, were used to assess DW. The annotations for the DW genes differed significantly from the coexpression annotations (see above). Enrichment was detected for Golgi vesicle-mediated transport, the nuclear pore and Ras GTPase binding. The latter annotation is consistent with several reports, although not from the perspective of splicing, that preference consumption is associated generally with intracellular signaling and specifically with Ras/MAPK pathways (Metten et al. 2014; Mulligan et al. 2006). The number of Ras GTPase affected genes was large (N = 65) and included several different gene families, e.g. Rab, Rap and Srga. From the network perspective, most of these Ras GTPase genes were ‘leaf’ nodes, i.e. showing moderate to low intramodular connectivity. However, there were five hub nodes, all of which were significantly affected by selection: superoxide dismutase (Sod1), TBC1 domain family member 9 (Tbc1d9), nucleoporin 50 [Nup50, cytoplasmic FMR1-interacting protein 1 (Cyfip1) and N-Myc-downstream regulated 1 (Ndrg1)]. The latter four are hub nodes in the low line and Sod1 is a hub in the high line. To our knowledge, none of these genes have been directly linked to ethanol preference. However, Ndrg1 has been shown to be affected by acute ethanol administration (Costin et al. 2013).
Thirty-six splicing hub nodes were significantly (FDR < 0.05) affected by selection. In contrast, 253 coexpression hub nodes were affected by selection (FDR < 0.05). The difference may reflect a greater stability of the splicing nodes and/or insufficient read depth to detect changes in the splicing network. Eighteen of these splicing hub nodes lie with known preference QTL intervals. However, the relevance of this observation will depend on which genes exhibit cis-regulated AEU. Of the remaining affected hub nodes, there is some gene expression evidence directly or indirectly linking eight of the hubs to ethanol phenotypes (Table S9); however, only one of these studies focused on preference risk. Carr et al. (2007) used inbred preferring and non-preferring rats to form congenic strains to determine what genes are DE in a Chr 4 QTL. Son of sevenless 1 (Sos1), a GTPase-activating protein, was found to show modestly higher expression in the preferring animals. However, this was not confirmed with quantitative reverse transcriptase-polymerase chain reaction.
Beginning with the selection of the long-sleep and short-sleep mice (see references in McClearn & Kakihana 1981), there is a long record of using outbred animals as the founders for ethanol-related selective breeding (Crabbe et al. 2016). The current study continues this strategy using a founder population (the HS-CC) that is three to four times more genetically diverse than other HS populations such as the HS/NPT or HS/Ibg (Hitzemann et al. 1994; Roberts et al. 2007). As noted above, there was a prediction that by using a substantially more diverse founder population, new pathways/mechanisms associated with preference consumption would be detected and these in turn could lead to novel therapeutic strategies. However, much of the data suggest that rather than detecting novel pathways, a more genetically diverse population engages familiar pathways but from novel perspectives. This conclusion is consistent with our observation that when comparing striatal gene regulation among F2, HS4 and HS-CC mice (Iancu et al. 2010) module annotation is more consistent than the alignment of genes within specific modules. For example, Ras/MAPK pathways have been repeatedly associated with the risk for excessive preference consumption (see above) but generally from the perspective of differential gene expression. In the current study, these pathways appeared prominent in the cosplicing data, suggesting a more subtle form of transcriptome regulation. However, we also note that selection from the HS-CC detected very broad effects on cell adhesion molecules and showed that long non-coding RNAs may have a prominent role in preference genetics.
Supplementary Material
Table S1: Genes associated with coexpression module.
Table S2: Genes associated with cosplicing module.
Table S3: GO annotations for the coexpression module.
Table S4: GO annotations for the cosplicing module.
Table S5: Annotation of the DV genes.
Table S6: Annotation of the DW genes.
Table S7: DW and coexpression network data.
Table S8: DW genes enriched in three cosplicing modules.
Table S9: DW and splicing network data.
Acknowledgments
A.M.C. passed away on 5 August 2015. The data presented here are part of his thesis. A.M.C. was awarded a PhD posthumously on 10 June 2016. The thesis work was supported in part by (AA10034, AA13484 and AA10760).
Footnotes
The authors declare that they have no conflict of interest in presenting the data herein.
Supporting Information
Additional supporting information may be found in the online version of this article at the publisher’s web-site:
References
- Acosta G, Freidman DP, Grant KA, Hemby SE. Alternative splicing of AMPA subunits in prefrontal cortical fields of cynomolgus monkeys following chronic ethanol self-administration. Front Psychiatry. 2011;2:72. doi: 10.3389/fpsyt.2011.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexa A, Rahnenführer J, Lengauer T. Improved scoring of functional groups from gene expression data by decor-relating GO graph structure. Bioinformatics. 2006;22:1600–1607. doi: 10.1093/bioinformatics/btl140. [DOI] [PubMed] [Google Scholar]
- Ando T, Kato R, Honda H. Differential variability and correlation of gene expression identifies key genes involved in neuronal differentiation. BMC Syst Biol. 2015;9:82. doi: 10.1186/s12918-015-0231-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belknap JK, Atkins AL. The replicability of QTLs for murine alcohol preference drinking behavior across eight independent studies. Mamm Genome. 2001;12:893–899. doi: 10.1007/s00335-001-2074-2. [DOI] [PubMed] [Google Scholar]
- Bell RL, Kimpel MW, McClintick JN, Strother WN, Carr LG, Liang T, Rodd ZA, Mayfield RD, Edenberg HJ, McBride WJ. Gene expression changes in the nucleus accumbens of alcohol-preferring rats following chronic ethanol consumption. Pharmacol Biochem Behav. 2009;94:131–147. doi: 10.1016/j.pbb.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bice PJ, Foroud T, Carr LG, Zhang L, Liu L, Grahame NJ, Lumeng L, Li TK, Belknap JK. Identification of QTLs influencing alcohol preference in the High Alcohol Preferring (HAP) and Low Alcohol Preferring (LAP) mouse lines. Behav Genet. 2006;36:248–260. doi: 10.1007/s10519-005-9019-6. [DOI] [PubMed] [Google Scholar]
- Boehm SL, Peden L, Chang R, Harris RA, Blednov YA. Deletion of the fyn-kinase gene alters behavioral sensitivity to ethanol. Alcohol Clin Exp Res. 2003;27:1033–1040. doi: 10.1097/01.ALC.0000075822.80583.71. [DOI] [PubMed] [Google Scholar]
- Bowers BJ, McClure-Begley TD, Keller JJ, Paylor R, Collins AC, Wehner JM. Deletion of the alpha7 nicotinic receptor subunit gene results in increased sensitivity to several behavioral effects produced by alcohol. Alcohol Clin Exp Res. 2005;29:295–302. doi: 10.1097/01.alc.0000156116.40817.a2. [DOI] [PubMed] [Google Scholar]
- Bubier JA, Jay JJ, Baker CL, Bergeson SE, Ohno H, Metten P, Crabbe JC, Chesler EJ. Identification of a QTL in Mus musculus for alcohol preference, withdrawal, and Ap3m2 expression using integrative functional genomics and precision genetics. Genetics. 2014;197:1377–1393. doi: 10.1534/genetics.114.166165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulwa ZB, Sharlin JA, Clark PJ, Bhattacharya TK, Kilby CN, Wang Y, Rhodes JS. Increased consumption of ethanol and sugar water in mice lacking the dopamine D2 long receptor. Alcohol. 2011;45:631–639. doi: 10.1016/j.alcohol.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calarco JA, Zhen M, Blencowe BJ. Networking in a global world: establishing functional connections between neural splicing regulators and their target transcripts. RNA. 2011;17:775–791. doi: 10.1261/rna.2603911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Liu X, Han S, Zhang CK, Liu Z, Li D. Association of the HTR2A gene with alcohol and heroin abuse. Hum Genet. 2014;133:357–365. doi: 10.1007/s00439-013-1388-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr LG, Kimpel MW, Liang T, McClintick JN, McCall K, Morse M, Edenberg HJ. Identification of candidate genes for alcohol preference by expression profiling of congenic rat strains. Alcohol Clin Exp Res. 2007;31:1089–1098. doi: 10.1111/j.1530-0277.2007.00397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Charness ME. Ethanol inhibits neuronal differentiation by disrupting activity-dependent neuroprotective protein signaling. Proc Natl Acad Sci U S A. 2008;105:19962–19967. doi: 10.1073/pnas.0807758105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma’ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesler EJ. Out of the bottleneck: the diversity outcross and collaborative cross mouse populations in behavioral genetics research. Mamm Genome. 2014;25:3–11. doi: 10.1007/s00335-013-9492-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesler EJ, Gatti DM, Morgan AP, Strobel M, Trepanier L, Oberbeck D, McWeeney S, Hitzemann R, Ferris M, McMul-lan R, Clayshultle A, Bell TA, Manuel de Villena FP, Churchill GA. Diversity outbred mice at 21: maintaining allelic variation in the face of selection. G3 (Bethesda) 2016;6:3893–3902. doi: 10.1534/g3.116.035527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill GA, Airey DC, Allayee H, et al. The collaborative cross, a community resource for the genetic analysis of complex traits. Nat Genet. 2004;36:1133–1137. doi: 10.1038/ng1104-1133. [DOI] [PubMed] [Google Scholar]
- Costin BN, Wolen AR, Fitting S, Shelton KL, Miles MF. Role of adrenal glucocorticoid signaling in prefrontal cortex gene expression and acute behavioral responses to ethanol. Alcohol Clin Exp Res. 2013;37:57–66. doi: 10.1111/j.1530-0277.2012.01841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabbe JC, Schlumbohm JP, Hack W, Barkley-Levenson AM, Metten P, Lattal KM. Fear conditioning in mouse lines genetically selected for binge-like ethanol drinking. Alcohol. 2016;52:25–32. doi: 10.1016/j.alcohol.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick DM, Bierut L, Hinrichs A, Fox L, Bucholz KK, Kramer J, Kuperman S, Hesselbrock V, Schuckit M, Almasy L, Tischfield J, Porjesz B, Begleiter H, Nurnberger J, Xuei X, Edenberg HJ, Foroud T. The role of GABRA2 in risk for conduct disorder and alcohol and drug dependence across developmental stages. Behav Genet. 2006;36:577–590. doi: 10.1007/s10519-005-9041-8. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edenberg HJ, Koller DL, Xuei X, et al. Genome-wide association study of alcohol dependence implicates a region on chromosome 11. Alcohol Clin Exp Res. 2010;34:840–852. doi: 10.1111/j.1530-0277.2010.01156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enoch MA. Genetic influences on the development of alcoholism. Curr Psychiatry Rep. 2013;15:412. doi: 10.1007/s11920-013-0412-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier L, Smouse PE, Quattro JM. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics. 1992;131:479–491. doi: 10.1093/genetics/131.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcon S, Gentleman R. Using GOstats to test gene lists for GO term association. Bioinformatics. 2007;23:257–258. doi: 10.1093/bioinformatics/btl567. [DOI] [PubMed] [Google Scholar]
- Farris SP, Mayfield RD. RNA-Seq reveals novel transcriptional reorganization in human alcoholic brain. Int Rev Neurobiol. 2014;116:275–300. doi: 10.1016/B978-0-12-801105-8.00011-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farris SP, Miles MF. Fyn-dependent gene networks in acute ethanol sensitivity. PLoS One. 2013;8:e82435. doi: 10.1371/journal.pone.0082435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill R, Datta S, Datta S. A statistical framework for differential network analysis from microarray data. BMC Bioinformatics. 2010;11:95. doi: 10.1186/1471-2105-11-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatt SJ, Cohen OS, Faraone SV, Tsuang MT. Dysfunctional gene splicing as a potential contributor to neuropsychiatric disorders. Am J Med Genet B Neuropsychiatr Genet. 2011;156B:382–392. doi: 10.1002/ajmg.b.31181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grahame NJ, Li TK, Lumeng L. Limited access alcohol drinking in high- and low-alcohol preferring selected lines of mice. Alcohol Clin Exp Res. 1999;23:1015–1022. [PubMed] [Google Scholar]
- Hitzemann R, Hitzemann B, Rivera S, Gatley J, Thanos P, Shou LL, Williams RW. Dopamine D2 receptor binding, Drd2 expression and the number of dopamine neurons in the BXD recombinant inbred series: genetic relationships to alcohol and other drug associated phenotypes. Alcohol Clin Exp Res. 1994;27:1–11. doi: 10.1097/01.ALC.0000047862.40562.27. [DOI] [PubMed] [Google Scholar]
- Hitzemann R, Edmunds S, Wu W, Malmanger B, Walter N, Belknap J, Darakjian P, McWeeney S. Detection of reciprocal quantitative trait loci for acute ethanol withdrawal and ethanol consumption in heterogeneous stock mice. Psychopharmacology (Berl) 2009;203:713–722. doi: 10.1007/s00213-008-1418-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitzemann R, Bottomly D, Iancu O, Buck K, Wilmot B, Mooney M, Searles R, Zheng C, Belknap J, Crabbe J, McWeeney S. The genetics of gene expression in complex mouse crosses as a tool to study the molecular underpinnings of behavior traits. Mamm Genome. 2014;25:12–22. doi: 10.1007/s00335-013-9495-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman PL, Saba LM, Flink S, Grahame NJ, Kechris K, Tabakoff B. Genetics of gene expression characterizes response to selective breeding for alcohol preference. Genes Brain Behav. 2014;13:743–757. doi: 10.1111/gbb.12175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosseini SM, Hoeft F, Kesler SR. GAT: a graph-theoretical analysis toolbox for analyzing between-group differences in large-scale structural and functional brain networks. PLoS One. 2012;7:e40709. doi: 10.1371/journal.pone.0040709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iancu OD, Darakjian P, Walter NAR, Malmanger B, Oberbeck D, Belknap J, McWeeney S, Hitzemann R. Genetic diversity and striatal gene networks: focus on the heterogeneous stock-collaborative cross (HS-CC) mouse. BMC Genomics. 2010;11:585. doi: 10.1186/1471-2164-11-585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iancu OD, Kawane S, Bottomly D, Searles R, Hitzemann R, McWeeney S. Utilizing RNA-Seq data for de novo coexpression network inference. Bioinformatics. 2012;28:1592–1597. doi: 10.1093/bioinformatics/bts245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iancu OD, Oberbeck D, Darakjian P, Kawane S, Erk J, McWeeney S, Hitzemann R. Differential network analysis reveals genetic effects on catalepsy modules. PLoS One. 2013a;8:e58951. doi: 10.1371/journal.pone.0058951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iancu OD, Oberbeck D, Darakjian P, Metten P, McWeeney S, Crabbe JC, Hitzemann R. Selection for drinking in the dark alters brain gene coexpression networks. Alcohol Clin Exp Res. 2013b;37:1295–1303. doi: 10.1111/acer.12100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iancu OD, Colville A, Oberbeck D, Darakjian P, McWeeney SK, Hitzemann R. Cosplicing network analysis of mammalian brain RNA-Seq data utilizing WGCNA and Mantel correlations. Front Genet. 2015;6:174. doi: 10.3389/fgene.2015.00174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ideker T, Krogan NJ. Differential network biology. Mol Syst Biol. 2012;8:565. doi: 10.1038/msb.2011.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin C, Woodward JJ. Effects of 8 different NR1 splice variants on the ethanol inhibition of recombinant NMDA receptors. Alcohol Clin Exp Res. 2006;30:673–679. doi: 10.1111/j.1530-0277.2006.00079.x. [DOI] [PubMed] [Google Scholar]
- Keane TM, Goodstadt L, Danecek P, et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 2011;477:289–294. doi: 10.1038/nature10413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology. 2010;35:217–238. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, McDermott MG, Monteiro CD, Gundersen GW, Ma’ayan A. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–W97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Luo R, Oldham MC, Horvath S. Is my network module preserved and reproducible? PLoS Comput Biol. 2011;7:e1001057. doi: 10.1371/journal.pcbi.1001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AM, Messing RO. Protein kinases and addiction. Ann N Y Acad Sci. 2008;1141:22–57. doi: 10.1196/annals.1441.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Mayfield RD, Harris RA. Altered gamma-aminobutyric acid type B receptor subunit 1 splicing in alcoholics. Biol Psychiatry. 2014;75:765–773. doi: 10.1016/j.biopsych.2013.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Lee JA, Black DL. Neuronal regulation of alternative pre-mRNA splicing. Nat Rev Neurosci. 2007;8:819–831. doi: 10.1038/nrn2237. [DOI] [PubMed] [Google Scholar]
- Liu W, Thielen RJ, Rodd ZA, McBride WJ. Activation of serotonin-3 receptors increases dopamine release within the ventral tegmental area of Wistar and alcohol-preferring (P) rats. Alcohol. 2006;40:167–176. doi: 10.1016/j.alcohol.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Chen G, Ma C, Bower KA, Xu M, Fan Z, Shi X, Ke ZJ, Luo J. Overexpression of glycogen synthase kinase 3beta sensitizes neuronal cells to ethanol toxicity. J Neurosci Res. 2009;87:2793–2802. doi: 10.1002/jnr.22098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo CL, Lossie AC, Liang T, Liu Y, Xuei X, Lumeng L, Zhou FC, Muir WM. High resolution genomic scans reveal genetic architecture controlling alcohol preference in bidirectionally selected rat model. PLoS Genet. 2016;12:e1006178. doi: 10.1371/journal.pgen.1006178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logrip ML, Zorrilla EP. Differential changes in amygdala and frontal cortex Pde10a expression during acute and protracted withdrawal. Front Integr Neurosci. 2014;8:30. doi: 10.3389/fnint.2014.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lydall GJ, Saini J, Ruparelia K, Montagnese S, McQuillin A, Guerrini I, Rao H, Reynolds G, Ball D, Smith I, Thomson AD, Morgan MY, Gurling HMD. Genetic association study of GABRA2 single nucleotide polymorphisms and electroencephalography in alcohol dependence. Neurosci Lett. 2011;500:162–166. doi: 10.1016/j.neulet.2011.05.240. [DOI] [PubMed] [Google Scholar]
- Maiya R, Lee S, Berger KH, Kong EC, Slawson JB, Griffith LC, Takamiya K, Huganir RL, Margolis B, Heberlein U. DlgS97/SAP97, a neuronal isoform of discs large, regulates ethanol tolerance. PLoS One. 2012;7:e48967. doi: 10.1371/journal.pone.0048967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mar JC, Matigian NA, Mackay-Sim A, Mellick GD, Sue CM, Silburn PA, McGrath JJ, Quackenbush J, Wells CA. Variance of gene expression identifies altered network constraints in neurological disease. PLoS Genet. 2011;7:e1002207. doi: 10.1371/journal.pgen.1002207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClearn GE, Kakihana R. Selective breeding for ethanol sensitivity: short-sleep and long -sleep mice. In: McClearn GE, Dietrich RA, Erwin VG, editors. Development of Animal Models as Pharmacogenetic Tools. US Government Printing Office; Washington, DC: 1981. pp. 81–113. [Google Scholar]
- Metten P, Phillips TJ, Crabbe JC, Tarantino LM, McClearn GE, Plomin R, Erwin VG, Belknap JK. High genetic susceptibility to ethanol withdrawal predicts low ethanol consumption. Mamm Genome. 1998;9:983–990. doi: 10.1007/s003359900911. [DOI] [PubMed] [Google Scholar]
- Metten P, Iancu OD, Spence SE, Walter NAR, Oberbeck D, Harrington CA, Colville A, McWeeney S, Phillips TJ, Buck KJ, Crabbe JC, Belknap JK, Hitzemann RJ. Dual-trait selection for ethanol consumption and withdrawal: genetic and transcriptional network effects. Alcohol Clin Exp Res. 2014;38:2915–2924. doi: 10.1111/acer.12574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Monte S, Derdak Z, Wands JR. Alcohol, insulin resistance and the liver-brain axis. J Gastroenterol Hepatol. 2012;27:33–41. doi: 10.1111/j.1440-1746.2011.07023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan MK, Ponomarev I, Hitzemann RJ, Belknap JK, Tabakoff B, Harris RA, Crabbe JC, Blednov YA, Grahame NJ, Phillips TJ, Finn DA, Hoffman PL, Iyer VR, Koob GF, Bergeson SE. Toward understanding the genetics of alcohol drinking through transcriptome meta-analysis. Proc Natl Acad Sci U S A. 2006;103:6368–6373. doi: 10.1073/pnas.0510188103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberlin B, Best C, Matson L, Henderson A, Grahame N. Derivation and characterization of replicate high- and low-alcohol preferring lines of mice and a high-drinking crossed HAP line. Behav Genet. 2011;41:288–302. doi: 10.1007/s10519-010-9394-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- Parsons MJ, Grimm C, Paya-Cano JL, Fernandes C, Liu L, Philip VM, Chesler EJ, Nietfeld W, Lehrach H, Schalkwyk LC. Genetic variation in hippocampal microRNA expression differences in C57BL/6J X DBA/2J (BXD) recombinant inbred mouse strains. BMC Genomics. 2012;13:476. doi: 10.1186/1471-2164-13-476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin K. The Mouse Brain in Sterotaxic Coordinates. 4th. Academic Press; Cambridge, MA: 2007. [Google Scholar]
- Raeder H, Holter SM, Hartmann AM, Spanagel R, Moller HJ, Rujescu D. Expression of N-methyl-d-aspartate (NMDA) receptor subunits and splice variants in an animal model of long-term voluntary alcohol self-administration. Drug Alcohol Depend. 2008;96:16–21. doi: 10.1016/j.drugalcdep.2007.12.013. [DOI] [PubMed] [Google Scholar]
- Roberts A, Pardo-Manuel de Villena F, Wang W, McMillan L, Threadgill DW. The polymorphism architecture of mouse genetic resources elucidated using genome-wide resequencing data: implications for QTL discovery and systems genetics. Mamm Genome. 2007;18:473–481. doi: 10.1007/s00335-007-9045-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodd ZA, Bell RL, Kuc KA, Murphy JM, Lumeng L, McBride WJ. Effects of concurrent access to multiple ethanol concentrations and repeated deprivations on alcohol intake of high-alcohol-drinking (HAD) rats. Addict Biol. 2009;14:152–164. doi: 10.1111/j.1369-1600.2008.00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenfluh A, Threlkeld RJ, Bainton RJ, Tsai LTY, Lasek AW, Heberlein U. Distinct behavioral responses to ethanol are regulated by alternate RhoGAP18B isoforms. Cell. 2006;127:199–211. doi: 10.1016/j.cell.2006.09.010. [DOI] [PubMed] [Google Scholar]
- Sokolov BP, Jiang L, Trivedi NS, Aston C. Transcription profiling reveals mitochondrial, ubiquitin and signaling systems abnormalities in postmortem brains from subjects with a history of alcohol abuse or dependence. J Neurosci Res. 2003;72:756–767. doi: 10.1002/jnr.10631. [DOI] [PubMed] [Google Scholar]
- Svenson KL, Gatti DM, Valdar W, Welsh CE, Cheng R, Chesler EJ, Palmer AA, McMillan L, Churchill GA. High-resolution genetic mapping using the mouse diversity outbred population. Genetics. 2012;190:437–447. doi: 10.1534/genetics.111.132597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treutlein J, Cichon S, Ridinger M, et al. Genome-wide association study of alcohol dependence. Arch Gen Psychiatry. 2009;66:773–784. doi: 10.1001/archgenpsychiatry.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernicke C, Hellmann J, Finckh U, Rommelspacher H. Chronic ethanol exposure changes dopamine D2 receptor splicing during retinoic acid-induced differentiation of human SH-SY5Y cells. Pharmacol Rep. 2010;62:649–663. doi: 10.1016/s1734-1140(10)70322-4. [DOI] [PubMed] [Google Scholar]
- Williams RW, Mulligan MK. Genetic and molecular network analysis of behavior. Int Rev Neurobiol. 2012;104:135–157. doi: 10.1016/B978-0-12-398323-7.00006-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaka R, Tang KC, Camarini R, Janak PH, Ron D. Fyn kinase and NR2B-containing NMDA receptors regulate acute ethanol sensitivity but not ethanol intake or conditioned reward. Alcohol Clin Exp Res. 2003;27:1736–1742. doi: 10.1097/01.ALC.0000095924.87729.D8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Kranzler HR, Yang BZ, Luo X, Gelernter J. The OPRD1 and OPRK1 loci in alcohol or drug dependence: OPRD1 variation modulates substance dependence risk. Mol Psychiatry. 2008;13:531–543. doi: 10.1038/sj.mp.4002035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Genes associated with coexpression module.
Table S2: Genes associated with cosplicing module.
Table S3: GO annotations for the coexpression module.
Table S4: GO annotations for the cosplicing module.
Table S5: Annotation of the DV genes.
Table S6: Annotation of the DW genes.
Table S7: DW and coexpression network data.
Table S8: DW genes enriched in three cosplicing modules.
Table S9: DW and splicing network data.