

Abstract
Lysine acetylation is a widespread posttranslational modification affecting many biological pathways. Recent studies indicate that acetylated lysine residues mainly exhibit low acetylation occupancy, but challenges in sample preparation and analysis make it difficult to confidently assign these numbers, limiting understanding of their biological significance. Here, we tested three common sample preparation methods to determine their suitability for assessing acetylation stoichiometry in three human cell lines, identifying the acetylation occupancy in more than 1,300 proteins from each cell line. The stoichiometric analysis in combination with quantitative proteomics also enabled us to explore their functional roles. We found that higher abundance of the deacetylase sirtuin 1 (SIRT1) correlated with lower acetylation occupancy and lower levels of ribosomal proteins, including those involved in ribosome biogenesis and rRNA processing. Treatment with the SIRT1 inhibitor EX-527 confirmed SIRT1's role in the regulation of pre-rRNA synthesis and processing. Specifically, proteins involved in pre-rRNA transcription, including subunits of the polymerase I and SL1 complexes and the RNA polymerase I-specific transcription initiation factor RRN3, were up-regulated after SIRT1 inhibition. Moreover, many protein effectors and regulators of pre-rRNA processing needed for rRNA maturation were also up-regulated after EX-527 treatment with the outcome that pre-rRNA and 28S rRNA levels also increased. More generally, we found that SIRT1 inhibition down-regulates metabolic pathways, including glycolysis and pyruvate metabolism. Together, these results provide the largest data set thus far of lysine acetylation stoichiometry (available via ProteomeXchange with identifier PXD005903) and set the stage for further biological investigations of this central posttranslational modification.
Keywords: acetylation, cancer, histone deacetylase inhibitor (HDAC inhibitor) (HDI), sirtuin 1 (SIRT1), stoichiometry
Introduction
Cells of all kingdoms use the posttranslational modifications (PTMs)3 of proteins to regulate biological pathways and processes and to increase the complexity and variety of functions of their protein targets without increasing the number of primary protein sequences. Many PTMs have been described, and the implications for their targets and pathways are the main goal of an increasing number of scientific reports. Reversible protein phosphorylation is the most widely studied PTM so far; it targets proteins that are involved in the vast majority of biological processes (1, 2). Lysine acetylation is also a reversible and widespread PTM that was discovered in histones more than 5 decades ago (3). It is currently known to be involved in the regulation of a large number of biological pathways with targets in all cellular compartments (4–7). Among the most representative pathways and groups of proteins that have been identified with this PTM, beyond the group of histones, are chromatin regulators and modifying proteins, DNA repair, DNA replication, spliceosome, ribosome biogenesis in the nucleus, most of the metabolic enzymes, ribosomal and cytoskeleton proteins in the cytoplasm, and the oxidative phosphorylation and the tricarboxylic acid cycle in the mitochondrion. In addition, many proteins involved in the posttranslational modification of proteins such as kinases, lysine acetyltransferases, deacetylases, and ubiquitin conjugation proteins are also targeted by lysine acetylation.
Lysine acetylation is a highly dynamic PTM that is controlled by two groups of enzymes: lysine acetyltransferases and lysine deacetylases (KDACs). However, in the mitochondria, where no lysine acetyltransferase has yet been described, strong evidence indicates that lysine acetylation occurs non-enzymatically, favored by the mitochondrial matrix conditions of a high concentration of acetyl-CoA (0.1–1.5 mm) and a pH of 7.9–8.0 (8). KDACs are further divided into two major groups: the Zn2+-dependent members of KDAC classes I, II, and IV and KDAC class III or sirtuins, which are NAD+-dependent and exhibit a mono-ADP-ribosyltransferase activity, in addition to deacetylase (9–11). Particularly, sirtuins are involved in the regulation of several important processes and therefore have been linked to physiologic and pathologic states, including aging and cancer (12). Of the seven sirtuins reported in humans, SIRT1 is the most abundant and closely related to its yeast homolog, Sir2 (13). This protein is mostly nuclear and cytoplasmic and has been found to regulate many pathways in response to several physiological stresses or cell cycle phases (14). In cancer, SIRT1 has been linked to both tumor progression and tumor repression, and its functions are largely tumor-specific (15).
Lysine acetylation is largely a low-stoichiometric PTM, and for its large-scale identification using mass spectrometry-based proteomics, specific target approaches based on anti-acetyl-lysine antibodies are required (7). With this strategy, the number of identified acetylation sites in human cells rapidly increased to several thousand in the last decade. However, important limitations are associated with this strategy, including lack of information related to site occupancy; a bias in site identification, which is linked to the use of antibodies; and the amount of starting sample needed for the enrichment step (11). Recently, a method based on the chemical acetylation of untargeted lysine residues, carrying heavy stable isotopes to estimate the relative occupancy of endogenous acetylation, was reported and applied to the Escherichia coli proteome (16). The method uses the MS precursor intensities to estimate the relative lysine acetylation occupancy versus the untargeted residues that were chemically acetylated with heavy isotopes prior to the generation of peptides by trypsin digestion. The same strategy was applied to the stoichiometric analysis of mammalian cells to analyze the dynamics of acetylation stoichiometries after treatment with a deacetylase inhibitor (17).
Here, we combined three of the most widely used methods for sample preparation in proteomics with large-scale lysine acetylation stoichiometry determination based on the chemical acetylation of proteins with stable heavy isotopes in human cells. Our strategy incorporated an efficient acetylation reaction with N-acetoxysuccinimide-d3 (NAS-d3) and protein digestion with trypsin in the presence of sodium deoxycholate (SDC) to avoid degradation and losses due to protein precipitation. The integration of lysine acetylation stoichiometric analysis with quantitative proteomics allowed us to establish a functional link between lysine acetylation occupancy and the abundance of targeted proteins, pathway partners, and deacetylases in three human cell lines. In addition, we studied the dynamics of acetylation stoichiometry and the consequences of the inhibition of the deacetylase SIRT1 at the proteome level.
Results
Method development
For lysine acetylation stoichiometry analysis at the proteome level, several steps are mandatory. In the first step, the proteins subjected to the study must be obtained. Next, all lysine residues of proteins will be chemically acetylated with heavy isotopes to differentiate them from endogenous acetylation (natural composition of isotopes). Once the lysine residues of all proteins are fully acetylated, the proteins are digested with trypsin. Because trypsin cannot hydrolyze the peptide bond when acetyl-lysine is present, the generated peptides will be delimited by arginine residues. Finally, in the MS analysis, those peptides with some degree of endogenous acetylation occupancy at lysine residues will be seen as complex isotopic distributions. If the three hydrogens of the chemically incorporated acetyl group are replaced with deuterium, then in the MS spectrum the isotopic distribution of chemically acetylated lysine-containing peptides will be shifted three daltons relative to the endogenously acetylated peptide. Therefore, by analyzing the isotopic distribution, it is possible to determine the lysine acetylation stoichiometry compared with the endogenous untargeted lysine residues (chemically acetylated with heavy isotopes).
We combined an optimized method for lysine acetylation stoichiometry analysis with three of the most widely used procedures for sample preparation in proteomics: filter-aided sample preparation (FASP), gel sample preparation (GSP), and solution sample preparation (SSP). Briefly, we used a simple and highly efficient total protein extraction protocol, described under “Experimental procedures,” that relies on the incorporation of high concentrations of sodium dodecyl sulfate (SDS) (4%) and DTT (0.1 m). Equal amounts of proteins were submitted to the selected sample preparation procedure (Fig. 1). For the chemical derivatization of lysine residues, we used NAS-d3 instead of deuterated acetic anhydride because NAS-d3 is a significantly more efficient alkylating reagent, and a gentler buffer can be utilized during the acetylation reaction. Although the acetylation reaction with NAS-d3 leads to fewer collateral reactions in residues such as tyrosine, threonine, and serine, we included a hydroxylamine treatment to revert O-acetylation modifications completely. To avoid the precipitation of proteins after chemical acetylation, we introduced two detergents, SDS and SDC, during the reaction step and SDC during trypsin digestion. It is well known that trypsin works satisfactorily in high concentrations of SDC (18). SDC was easily removed by ethyl acetate extraction under acidic conditions. Finally, samples were submitted to a high-resolution LC-MS/MS system. Protein identification and quantification were performed with MaxQuant software, and the lysine acetylation stoichiometric analysis was achieved with Pview software.
Figure 1.

Schematic representation of the workflow to perform lysine acetylation stoichiometric analysis following the three sample preparation procedures, FASP, GSP, and SSP. In all strategies, untargeted lysine residues are chemically acetylated with the deuterated derivate of NHS (NAS-d3), and side reactions are reverted by treating proteins with hydroxylamine. To avoid unwanted protein precipitation in FASP and SSP, trypsin digestion was performed in the presence of SDC. Peptide mixtures were analyzed in a high-resolution LC-MS/MS system. Data analysis was performed by means of MaxQuant and Pview software.
Comparison of sample preparation methods
Three different cell lines were used to evaluate the three sample preparation procedures: HaCaT (non-cancerous immortalized human keratinocyte), SiHa (HPV-16-positive cervical cancer cell line), and CaLo (HPV-18-positive cervical cancer cell line). Three independent biological replicates were performed. Methods were evaluated according to the number of peptides and proteins identified as well as their mass and hydrophobicity distributions. To determine the potential advantages or potentialities of analyzed sample preparation methods, we performed a molecular mass and hydrophobicity distribution of identified peptides (Fig. 2, A and B) and proteins (Fig. 2, C and D). Despite the distributions of molecular masses and hydrophobicity having highly similar patterns at the protein level without any important bias, we found that at peptide level we missed certain kinds of peptides using the FASP method. The molecular mass distribution of peptides discarded the idea that large peptides generated from the trypsin digestion of proteins that were fully acetylated at their lysine residues would be lost. The distribution of identified peptides with a molecular mass above 2 kDa was highly similar for the three methods. In contrast, the distribution of identified peptides according to their grand average of hydropathy index indicated a bias for hydrophobic peptides in the FASP method. Although this bias did not have a substantial effect on the distribution at the protein level, probably because even the most hydrophobic proteins can generate hydrophilic peptides after trypsin digestion, for our purpose of lysine acetylation stoichiometry analysis it represents lower identification and quantification of their occupancy by acetylation of lysine-containing peptides. The number of peptides and proteins, their corresponding lysine content, and the number of identified acetylation sites are summarized in Table 1.
Figure 2.

Distribution of identified peptides and proteins for each sample preparation method according to molecular mass and hydrophobicity. A and B, peptide distribution according to molecular mass (A) and hydrophobicity (B). C and D, protein distribution according to molecular mass (C) and hydrophobicity (D). E, relative error (RE) distributions for reported acetylation stoichiometry results of acetylated peptides identified using the three sample preparation methods. GRAVY, grand average of hydropathy.
Table 1.
Identified proteins, peptides, lysine-containing peptides, acetylated peptides, and proteins by cells using different sample preparation methods
| HaCaT | CaLo | SiHa | All cells | |
|---|---|---|---|---|
| Identified proteins | ||||
| FASP | 4,896 | 4,537 | 3,910 | 5,800 |
| GSP | 4,788 | 4,894 | 5,164 | 6,105 |
| SSP | 4,821 | 4,522 | 4,548 | 5,825 |
| All methods | 6,180 | 6,116 | 6,071 | 7,196 |
| Identified Peptides | ||||
| FASP | 18,387 | 15,509 | 11,622 | 23,730 |
| GSP | 18,807 | 21,582 | 22,486 | 30,815 |
| SSP | 22,011 | 18,926 | 19,023 | 28,616 |
| All methods | 33,893 | 32,658 | 31,512 | 45,823 |
| Lysine-containing peptides | ||||
| FASP | 12,066 | 10,194 | 7,425 | 15,406 |
| GSP | 11,233 | 12,872 | 13,492 | 18,306 |
| SSP | 12,968 | 11,346 | 11,440 | 17,024 |
| All methods | 20,291 | 19,660 | 18,803 | 27,527 |
| Acetylated peptides | ||||
| FASP | 898 | 732 | 437 | 1,608 |
| GSP | 939 | 1,059 | 1,171 | 2,359 |
| SSP | 1,026 | 905 | 849 | 1,909 |
| All methods | 2,378 | 2,304 | 2,110 | 4,557 |
| Acetylated proteins | ||||
| FASP | 685 | 581 | 351 | 1,063 |
| GSP | 718 | 803 | 865 | 1,478 |
| SSP | 748 | 689 | 655 | 1,223 |
| All methods | 1,433 | 1,399 | 1,325 | 2,217 |
Although equal amounts of proteins were used to evaluate the three methods, to analyze similar quantities of peptides in the LC-MS/MS system, the amount of injected material was different for each method. In the case of the FASP method, an amount equivalent to 4 μg of the starting material was used for each LC-MS/MS run. In the GSP method, an amount equivalent to 5 μg was used. The best overall recoveries were achieved in SSP where we used an amount equivalent to 2 μg of the starting sample. As no important differences were observed in the distribution of proteins based on their molecular mass or their hydrophobicity, we believe that the final peptide recovery steps in the FASP and GSP methods are the most critical for the overall recovery of the samples. In the case of the FASP method, peptides with higher hydrophobicity are more difficult to recover. However, in the GSP method, no bias was observed for any particular group of peptides as all peptides were affected similarly. For the SSP method, there is no need to perform a peptide recovery step as the final step to eliminate the detergent (Fig. 1, right) is carried out by ethyl acetate extraction under acidic conditions, and the peptides remain in the aqueous solution.
Although some methods have disadvantages, we identified complementary sets of peptide and proteins. For example, the average number of peptides identified by individual methods for each cell was 18,700, and the average number of peptides identified for each cell line combining the three methods was 32,600. These results show a complementarity between methods as one single method can identify about 60% of the peptides that can be identified using all three methods. However, if a peptide prefractionation step is included before LC-MS/MS analysis, more overlap between methods could be observed. Conversely, we analyzed the consistency of the reported stoichiometric results between different methods. For those acetylated peptides identified in the three sample preparation methods, we report the coefficient of variation and the relative errors between methods (supplemental Table S1). The relative error distributions for each method showed high similarity (Fig. 2E), indicating that the sample preparation method is not associated with the labeling efficiency and that similar acetylation stoichiometry results can be obtained with different methods.
Lysine acetylation analysis
For the lysine acetylation stoichiometric analysis, we used the Pview software package that was recently validated and used to analyze the lysine acetylation stoichiometry in bacterial proteins (16). We combined the results achieved with the three sample preparation methods to report the acetylation stoichiometry of peptides for each cell line (supplemental Table S1). Additionally, we only report the acetylation occupancy for those peptides that were identified with an FDR lower than 1% by two independent search engines (MaxQuant and Pview), an isotopic distribution tolerance of less than 5 ppm, and at least three signals observed for both isotopic distributions endogenously (light) and chemically (heavy) acetylated. Approximately 25% of all identified proteins in each cell line had some degree of acetylation on lysine residues (1,433 in HaCaT, 1,399 in CaLo, and 1,325 in SiHa). The distribution of acetylated proteins according to their cellular compartment showed that two cellular structures were particularly enriched in proteins affected by this PTM, the ribosome and the nucleolus (Fig. 3A). We also noticed that SiHa cells are particularly less enriched in acetylated proteins for these two cellular structures. In addition, ∼1,200 membrane proteins were identified in each cell line, and it was found that this group of proteins is among the most enriched groups in acetylated proteins.
Figure 3.

A, proteins identified for each cell line, distributed according to their cellular compartment (bottom) and the percentage of these proteins that were found acetylated (top). B, acetylated proteins distributed according to their biological pathway. Proteins are represented as the percentage of all proteins identified for the pathway in each cell line. C–H, the stacked column charts show the number of peptides grouped in the six quantiles. The columns go from clear to darker indicating less to more occupation of the acetylation sites. The color of the columns represents the cell line: HaCaT, CaLo, and SiHa cells are represented in green, red, and blue, respectively. The six quantiles corresponding to the acetylation occupancy of peptides were: peptides with less than 1% of acetylation occupancy (<1%); peptides with acetylation stoichiometry between 1 and 5% (1–5%), 5 and 10% (5–10%), 10 and 25% (10–25%), and 25 and 50% (25–50%); and peptides with more than 50% of site occupation (>50%). C, the chart represents all acetylated peptides in the three cell lines. D–H, the charts correspond to the number of peptides from proteins involved in the enriched biological pathways using the online version of DAVID bioinformatics software. TCA, tricarboxylic acid.
As reported previously, lysine acetylation is a widespread PTM, targeting proteins involved in a large set of biological processes and pathways from all cellular compartments (4). The functional enrichment analysis revealed that acetylation is more frequently observed in proteins that are involved in carbon and fatty acid metabolism as well as in amino acid biosynthesis, targeting more than 40% of the proteins identified for these pathways (Fig. 3B and supplemental Table S2). Unexpectedly, we found that more proteins involved in these pathways are acetylated in SiHa cells. In addition, large proportions of acetylated proteins were also found in proteins involved in transcription and translation pathways of the three cell lines analyzed.
Our stoichiometric analysis confirmed that lysine acetylation is a low-stoichiometry PTM as reported previously for other cells (17, 19). The global distributions of peptides according to their acetylation occupancy in the three cell lines showed high similarity, revealing that half of the acetylated peptides displayed stoichiometries lower than 5% (Fig. 3C). However, the acetylation stoichiometry distributions for certain groups of proteins, involved in different pathways, show different stoichiometry patterns. Ribosomal proteins are among the groups of proteins with more acetylated elements identified (between 50–60% of identified proteins). Particularly, most of the acetylation sites of this group of proteins show lower occupancy than the average. More than 40% of ribosomal protein acetylation sites are less than 1% occupied by this PTM (Fig. 3G). Conversely, acetylation sites of proteins involved in fatty acid metabolism, degradation, and biosynthesis show greater acetylation stoichiometry. For these proteins, more than 85% of their identified acetylated peptides show more than 1% occupation (Fig. 3F). Besides, we observed differences in the stoichiometry distribution of peptides when analyzing proteins from different cellular compartments. Acetylated peptides from Golgi apparatus proteins show higher acetylation occupancy than endoplasmic reticulum, cytoplasmic, and nuclear proteins. Moreover, acetylation sites of membrane proteins are among the less occupied by this PTM. The distributions of acetylation occupancy of several groups of proteins are presented in supplemental Fig. S1.
The acetylation stoichiometry varies for different sites in the same proteins. Histones form a well known group of proteins targeted by acetylation. However, we found large differences in the stoichiometry of acetylation for the reported acetylation sites. In fact, the N-terminal tails of H2A, H3, and H4 contain lysine residues highly susceptible to acetylation with stoichiometries ranging from 5 to 30%. However, in their globular central domains, very few residues were acetylated, and in all cases, the acetylation occupancy was below 1%.
Acetylation stoichiometry dynamics
Acetylation cross-talks with other PTMs in residues near the acetylation site. We found evidence that methylation and phosphorylation can alter the acetylation status in at least nearby lysine residues. With our experiment design, it was confirmed that monomethylated lysine residues can be fully acetylated with the chemical reaction performed. However, we found no evidence of the coexistence of these PTMs endogenously. The peptide 10KSTGGKAPR18 from histone H3 illustrates the complexity of the interactions between different PTMs. The Lys10 residue was identified in the three cell lines under study as unmodified, mono-, di-, trimethylated, and acetylated; the Ser11 residue was found unmodified and phosphorylated; and the Lys15 residue was detected unmodified and acetylated (Fig. 4). The lysine acetylation stoichiometric analysis revealed that the Lys15 residue is largely more susceptible to acetylation than the Lys10 residue, which is a target for methylation. In the fraction corresponding to the non-methylated Lys10 residue, only a tiny fraction was detected with both lysine residues acetylated. The MS/MS spectrum of the signal 494.786 Th confirms that Lys15 is the preferred acetylation site even in the absence of methylation in the Lys10 residue (Fig. 4, G and H). For this peptide, we noticed that mono- and dimethylation in Lys10 did not affect the acetylation occupancy in Lys15 compared with the non-methylated Lys10. However, the presence of trimethylated Lys10 resulted in a decrease of 7–10% in the acetylation occupancy of the Lys15 residue (Fig. 4F). Conversely, phosphorylation in Ser11 favors the occurrence of acetylation in Lys15, increasing its occupancy 7–25% relative to non-phosphorylated peptide (Fig. 4, I and J). Also, we were unable to detect the co-occurrence of methylation or acetylation in Lys10 and phosphorylated Ser11. These findings clearly illustrate the complexity of the regulation of protein functions by means of PTMs and their cross-talk.
Figure 4.

MS/MS and MS spectra of the peptide 10KSTGGKAPR18 from histone H3 identified in the three cell lines under study with several PTMs in three of the residues. Lys10 was mono-, di-, and trimethylated and acetylated; Ser11 was phosphorylated; and Lys15 was acetylated. Representative MS/MS spectra of the peptide with mono-, di-, and trimethylated Lys10 are illustrated in A, C, and E, respectively. B, D, and F correspond to survey scans showing the isotopic distribution of the peptide mono-, di-, and trimethylated in Lys10 in the three cell lines. The reported values correspond to the degree of endogenous acetylation in the Lys15 residue, confirmed by MS/MS of the signals 501.794, 486.287, and 493.294 Th, respectively. G, representative MS/MS spectrum of the double-charge signal 494.785 Th corresponding to the peptide with Lys15 residue endogenously acetylated. H, survey scans showing the isotopic distribution of the peptide fully acetylated in the three cell lines. The reported values correspond to the degree of endogenous acetylation in one and both lysine residues. I, representative MS/MS spectrum of the double-charge signal 534.769 Th corresponding to the peptide with the Ser11 residue phosphorylated and the Lys15 residue endogenously acetylated. J, survey scans showing the isotopic distribution of the acetylated peptide with phosphorylated Ser11 in the three cell lines. The reported values correspond to the degree of endogenous acetylation in the Lys15 residue.
Not only do PTMs surrounding the acetylation site alter its occupancy, but small differences in the amino acid composition can also lead to variations in the stoichiometry of acetylation sites. Two variants of histone H3, H3.1 and H3.3, only differ in five residues, most of which are located in the globular region with relevance for their specific deposition in the genome (20). The N-terminal tails of these variants differ by a single amino acid at position 32. Ala32 from the canonical histone H3.1/H3.2 is substituted by Ser32 in histone H3.3. The peptides 28KSAPATGGVKKPHR41 from histone H3.1/H3.2 and 28KSAPSTGGVKKPHR41 from histone H3.3 were both identified in the three cell lines. According to our stoichiometric analysis, only one of the three lysine residues in both peptides was the target for acetylation, and its degree of occupancy in all cells and variants of peptides was below 5%. Besides, Lys28 and Lys37 residues were methylated in both histone variants. For the peptide corresponding to histones H3.1/H3.2, the acetylation dynamics differ from the peptide in histone H3.3. In the non-methylated peptides, the acetylation occupancy of the peptide from histone H3.3 was at least 2.5-fold greater than that from histone H3.1/H3.2. In both peptides, when the Lys28 residue was methylated, no acetylation was detected, indicating that both PTMs are probably competing for the same residue. We were unable to confirm by MS/MS the exact acetylation site due to the low stoichiometry of this PTM in these peptides. Unexpectedly, the presence of methylation in the Lys37 residue results in different outcomes in both peptides. For histone H3.1/H3.2, the acetylation occupancy of the peptide increases at least 2.5-fold compared with the non-methylated peptide, whereas for histone H3.3 the stoichiometry decreases at least 1.5-fold (supplemental Fig. S2). Histone H3.3 replaces the canonical H3.1/H3.2 in several regions of the genome, generally in the start sites and the transcribing regions of active genes, in the telomeres and other regulatory regions (21). According to our results, it is possible that the substitution of residue Ala32 for Ser32 in histone H3.3 could be responsible for the increase in the stoichiometry of acetylation of residue Lys28, and therefore this substitution may favor the active transcription of genes. In addition, this substitution could also be involved in the cross-talk between both acetylation and methylation in these peptides.
For histone H2A variants, several amino acid substitutions are located in the N-terminal tails (from Ac-Ser2 to Arg18) relative to the canonical variant. We identified and determined the acetylation stoichiometry of the two generated peptides and their variants after trypsin digestion (as described under “Experimental Procedures”). The determination of acetylation stoichiometry of the peptide 5GKQGGKAR12 from the canonical H2A and two other variants (5GKTGGKAR12 from H2AX and 5GKQGGKVR12 from H2AJ) uncovered slight but measurable differences. The peptide from histone H2AJ had higher acetylation occupancy than the canonical and H2AX variants. The role of histone H2AJ, as well as the conditions in which it normally replaces the canonical variant from nucleosomes, is unknown. In contrast, histone H2AX has been more extensively studied and found to have an important role in the cellular response to stress and DNA breakage. The two variants of the second peptide from the N-terminal tail of H2A histones (13AKAKSR18 from H2AC, H2AJ, and H2AX and 13AKAKTR18 from H2AB) displayed very low stoichiometry, below 0.20% for both peptide variants, in the three cell types.
Quantitative proteomics and acetylomics
To globally analyze our data, we performed a quantitative label-free proteomics analysis comparing the three cell types. For this analysis, we used the experimental data from the GSP and SSP methods, and we compared the protein profiles of the three cell types under study (supplemental Table S3). To connect quantitative proteomics and lysine acetylation stoichiometry, we first analyzed the relative abundance of the deacetylases identified by two or more unique peptides in the three cell types. According to our results, SIRT1 was the deacetylase with the greatest variation between cell types. SiHa contains ∼2-fold the amount of SIRT1 compared with HaCaT and CaLo cells. These results were confirmed by Western blot analysis (Fig. 5). Taking into account these findings, we selected the proteins that contain all the peptides for which we determined their acetylation stoichiometry in the three cell types and that showed less acetylation occupancy in SiHa cells compared with HaCaT and CaLo. We searched these proteins for significant enrichments in biological pathways and processes. In addition, we used the quantitative proteomics data to determine the protein profiles of the enriched groups of proteins.
Figure 5.
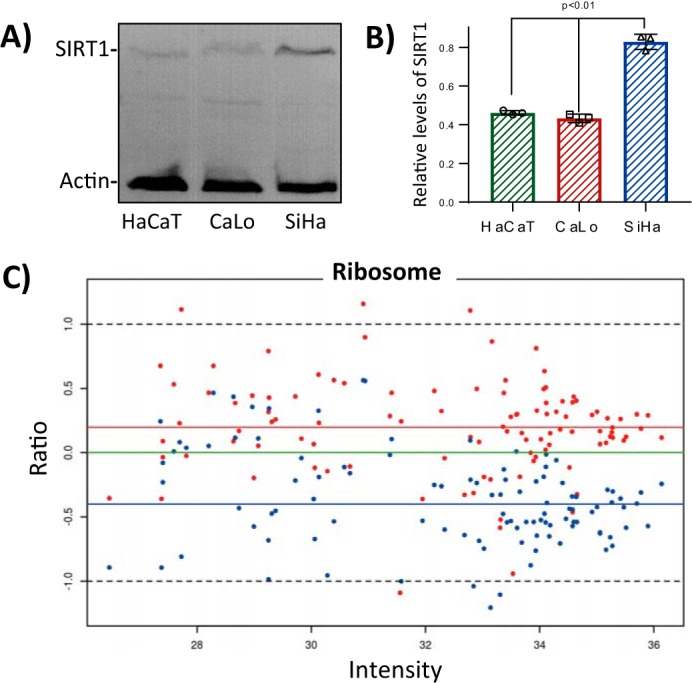
SIRT1 levels negatively correlate with the abundance of ribosomal proteins. Immunoblotting analysis confirmed that SIRT1 is significantly more abundant in SiHa cells than in CaLo and HaCaT cells. A, representative image of immunoblotting analysis of total protein extraction, from the three cell lines under study, using SIRT1 and actin antibodies. 10 g of total protein extract for each cell line were separated by 12% SDS-PAGE and transferred to a PVDF membrane. SIRT1 antibody was used at 1:1,000 dilution, and actin was used at 1:5,000 dilution. B, the levels of SIRT1 were calculated relative to actin with ImageJ software using three independent replicates. Error bars represent S.D. C, ratio-intensity plot to analyze the relative abundance of proteins from ribosomes of CaLo and SiHa relative to HaCaT cells from the label-free quantitative proteomics analysis. Red and blue dots represent CaLo and SiHa proteins, respectively, characterized by the ratio of intensities, log2(CaLo or SiHa/HaCaT), and their intensities in HaCaT cells, log10(HaCaT). Colored horizontal lines represent the medians for each cell line data set relative to HaCaT (green, HaCaT; red, CaLo; and blue, SiHa).
A total of 215 peptides corresponding to 196 proteins had less acetylation occupancy in SiHa than in the other two cell lines (supplemental Table S4). Among the most enriched biological pathways and processes, we found proteins from the spliceosome, ribosome, glycolysis pathway, rRNA processing, ribosome biogenesis, RNA transport, DNA repair, gene expression, cell–cell adhesion, and protein processing in the endoplasmic reticulum among others. To establish a link between acetylation occupancy and the regulation of the biological pathways, we analyzed the protein abundance profiles of identified proteins in each enriched biological pathway, including those identified proteins where no acetylation was detected.
For this analysis, we used the protein levels from the non-cancerous HaCaT cells as reference to relatively measure the abundance of the proteins in CaLo and SiHa cells. One of the groups subjected to this study was the ribosome. For this group of proteins, the ratio-intensity plot clearly showed that these proteins are underrepresented in SiHa cells compared with CaLo and HaCaT cells. This observation was more evident for the most abundant proteins for which the reported quantification values are more trustworthy (Fig. 5C). Statistical analysis showed that the medians for this group of proteins are significantly different between the three cell lines (supplemental Fig. S3). Proteins involved in ribosome biogenesis and rRNA processing were also underrepresented in SiHa cells, suggesting that SIRT1 might play a negative role in these processes.
Acetylomics and proteomics analyses in cells treated with a SIRT1 inhibitor
EX-527 is a potent and selective inhibitor of SIRT1 with an IC50 of 38 nm. EX-527 is between 200- and 500-fold more selective for SIRT1 than for SIRT2 and SIRT3, the more closely related sirtuins (22). This inhibitor does not inhibit members of KDAC classes I, II, and IV (Zn2+-dependent histone deacetylases 1–11). We treated the three cell types (HaCaT, CaLo, and SiHa) with vehicle (control) or 1 μm EX-527 for 24 h. Inhibition of SIRT1 followed by integral analysis of lysine acetylation stoichiometry and quantitative proteomics will help to clarify the role of SIRT1 in controlling the acetylation of the previously identified biological pathways and processes. For this analysis, samples were prepared according to the SSP method. To increase the confidence and coverage of the study, we performed two independent replicates of the experiment, and a peptide prefractionation step, based on reverse-phase chromatography at high pH, was incorporated before LC-MS/MS.
The number of identified proteins in each cell type significantly increased with the incorporation of the peptide fractionation step from 6,120 identified proteins per cell type without peptide fractionation (Table 1) to 9,150 identified proteins after peptide fractionation. The number of identified acetylation sites was increased ∼60% compared with the previous results using SSP without peptide fractionation (supplemental Table S5). As we noticed previously, the complementarity between sample preparation methods is high, and we did not identify more acetylation sites in the current analysis than we did with the combined results of the three methods.
The inhibition of SIRT1 with EX-527 did not affect normal cell growth during the 24 h of treatment compared with untreated cells in any of the three cell lines. After acetylation stoichiometric analysis, we noticed that the number of identified acetylation sites in the non-cancerous HaCaT cells increased from 1,500 peptides in the untreated cells to 1,911 in EX-527-treated cells. However, the distribution in quantiles did not show a global increase in acetylation occupancy (supplemental Fig. S4). In cancer cells treated with the SIRT1 inhibitor, we identified fewer acetylated peptides from 1,696 and 1,639 peptides in untreated CaLo and SiHa to 1,588 and 1,510, respectively, after SIRT1 inhibition. However, in both cancerous cell lines, a slight increase in global acetylation occupancy was observed in the acetylation stoichiometry distribution after treatment (supplemental Fig. S4). Surprisingly, we found several peptides that indeed showed decreased acetylation occupancy when SIRT1 was inhibited. This observation was also noticed previously in a large-scale lysine acetylation analysis when 19 different deacetylase inhibitors were used to inhibit all KDAC classes (23).
We identified 57 proteins with sites that consistently showed increased lysine acetylation occupancy in the three cell lines after treatment with the SIRT1 inhibitor (supplemental Table S6 and Figs. S5–S12). Functional enrichment analysis confirmed that most of these proteins are involved in rRNA processing, mRNA splicing and spliceosome, translational initiation and ribosomal proteins, cell–cell adhesion, and signal recognition particle-dependent cotranslational protein targeting to the membrane. All these pathways or processes were previously found to be enriched in the group of proteins with lower acetylation occupancy in untreated SiHa compared with CaLo and HaCaT cells, confirming that SIRT1 regulates these processes through its deacetylase activity.
To gain a better understanding of the role of SIRT1, we performed a functional enrichment analysis with the proteins from the three cell lines that showed increased acetylation occupancy by more than 5% after treatment with the SIRT1 inhibitor EX-527 (supplemental Table S7). Although more than 30% of proteins involved in metabolic pathways were acetylated, very few of them showed at least a 5% increase in acetylation after SIRT1 inhibition (Fig. 6A). The largest number of proteins that showed increased acetylation after EX-527 treatment is involved in transcription and translation pathways. We also observed differences between the cell lines in response to the treatment. In SiHa cells, more proteins involved in metabolism had increased acetylation stoichiometry compared with the other cell lines. In CaLo cells treated with EX-527, more proteins from the pathways of genetic information processing showed increased acetylation occupancy compared with other cell lines. Finally, in the HaCaT cell line, the treatment increased the acetylation occupancy by at least 5% in more proteins involved in cellular processes such as endocytosis, regulation of the actin cytoskeleton, and phagosome compared with SiHa and CaLo cell lines (Fig. 6A).
Figure 6.

A, number of proteins with sites that showed an increase in acetylation occupancy greater than 5% after EX-527 treatment in each cell line, distributed according to their biological pathway. B, biological pathways and processes enriched in proteins that showed increased abundance after SIRT1 inhibition in the three cell types analyzed and therefore are negatively regulated by SIRT1. C, biological pathways and processes enriched in proteins that showed decreased abundance after SIRT1 inhibition in the three cell types analyzed and therefore are positively regulated by SIRT1. All enrichment analyses were performed with the online version of the DAVID bioinformatics software. TCA, tricarboxylic acid; ER, endoplasmic reticulum.
In addition, we used the quantitative proteomics data to determine to what extent the inhibition of the deacetylase activity of SIRT1 influences the regulation of the protein profile of its targets and their pathways (supplemental Table S8). For this analysis, we selected two groups of proteins, those that decreased in abundance after SIRT1 inhibition in the three cell types and those that were up-regulated in the three cell types during SIRT1 inhibition. For both groups, we searched for significant protein enrichments in cellular pathways and processes. Our results revealed that SIRT1 targets and negatively regulates pathways and processes, including rRNA processing, ribosome biogenesis, the spliceosome, the ribosome, DNA replication and repair, and cell division among others (Fig. 6B). In contrast, we discovered a positive regulation of SIRT1 in several metabolic pathways, in protein processing in the endoplasmic reticulum, in cytoskeleton organization, and in cellular processes such as endocytosis and lysosome (Fig. 6C).
SIRT1 inhibition promotes the synthesis and processing of the pre-rRNA
The quantitative proteomics and lysine acetylation stoichiometry analyses in our three cell lines provided evidence supporting a repressive effect of SIRT1 over the biogenesis of ribosomes, starting from the synthesis of pre-rRNA. To confirm these findings, we treated our cells with vehicle (control), 1 μm EX-527, or 5 μm EX-527. We measured by real-time quantitative PCR (RT-qPCR) the levels of pre-rRNA and the mature 28S rRNA. Our results reveal that the levels of pre-rRNA significantly increase in the three cell lines after SIRT1 inhibition in a dose-dependent manner (Fig. 7A). The measurement of the mature 28S rRNA also shows an increase in its levels after EX-527 treatment that was significant in HaCaT and SiHa cells (Fig. 7B). These results can be linked to the levels of SIRT1 in each cell line. SIRT1 levels in CaLo cells were slightly lower than in HaCaT cells, and the increase in pre-rRNA levels at the highest dose was 3-fold for CaLo cells and 5-fold for HaCaT cells. For SiHa cells, which significantly express higher levels of SIRT1 than HaCaT and CaLo cells, the increase in pre-rRNA levels was more than 20-fold. The increases in the mature 28S rRNA were lower than the increases in its precursor, indicating that SIRT1 activity is mostly linked to the initial steps of ribosomal biogenesis. The quantitative proteomics experiments comparing 1 μm EX-527-treated with untreated cells are in full agreement with these results. In Fig. 7C, the distributions of ratios of intensities of ribosomal and all identified proteins in treated versus control cells are represented. The means of ribosomal protein ratios were shifted toward cells where SIRT1 was inhibited compared with the means of all protein ratios. The CaLo cell line, which exhibited the lowest increase in 28S rRNA, was also found to have the lowest increase in the abundance of ribosomal proteins.
Figure 7.

The chemical inhibition of SIRT1 by treating cells with EX-527 increases the levels of pre-rRNA and the mature 28S rRNA in a dose-dependent manner. HaCaT, CaLo, and SiHa cells were treated with vehicle, 1 μm EX-527, and 5 μm EX-527 for 24 h, and the level of pre-rRNA (A) and 28S rRNA (B) were estimated by RT-qPCR relative to actin mRNA levels. Error bars represent S.D. C, the levels of the proteic components of the ribosomes were analyzed as the ratio of the protein intensities between cells treated with 1 μm EX-527 and control. The ratio of protein intensities was distributed on a log2 scale. Ribosomal proteins are represented in continuous lines, and all quantified proteins are shown in discontinuous lines. The means of all ratios are presented as continuous vertical lines for ribosomal proteins and discontinuous lines for all proteins. D, schematic representation of the synthesis of pre-rRNA and proteins involved in this process. SIRT1 inhibition increases the levels of proteins involved in the synthesis of pre-rRNA. Elements of SL1 complex required for the formation of the preinitiation complex were up-regulated in CaLo and SiHa cells. Most of the components specific for the Pol I complex as well as the protein RRN3 required for the activation of Pol I and for the interaction with SL1 complex at the transcription start site were up-regulated in the three cells lines analyzed. UBTF, nucleolar transcription factor 1; TTF-1, transcription terminator factor 1; PTRF, polymerase I and transcript release factor; TBP, TATA-binding protein; TWISTNB, TWIST neighbor.
Our results directly link SIRT1 to the regulation of the synthesis of pre-rRNA and its processing. In the schematic representation of the pre-rRNA synthesis process in Fig. 7D, the protein elements that were discovered to be up-regulated during SIRT1 inhibition are highlighted. SIRT1 does not affect the expression of nucleolar transcription factor 1 (UBTF) that recognizes the rRNA gene promoter, and in our experimental conditions, we were unable to detect any variation in the acetylation levels of their lysine residues. Similarly, we did not detect changes in the transcription terminator factor 1 (TTF-1) and polymerase I and transcript release factor (PTRF) responsible for releasing polymerase I (Pol I) and the pre-rRNA. Conversely, elements of the SL1 complex responsible for the recruitment of Pol I to the promoters were up-regulated after SIRT1 inhibition. In addition, most of the protein components specific for Pol I complex, including the catalytic subunit POLR1A, were up-regulated in the three cell lines. The RNA polymerase I-specific transcription initiation factor RRN3, which is required to activate Pol I and for the interaction with the SL1 complex at transcription initiation sites, was also overexpressed in the three cell lines treated with EX-527. As a consequence of the up-regulation of the Pol I components and its activator, pre-rRNA was overexpressed.
Several proteins involved in pre-rRNA processing to yield the mature rRNA molecules were up-regulated and/or showed increased acetylation site occupancy. Table 2 summarizes the ratios of abundance levels of several proteins involved in the regulation of pre-rRNA processing. The quantitative proteomics analysis revealed that the pre-rRNA processing pathway in CaLo cells is less affected by SIRT1 inhibition than that in HaCaT and SiHa cells. These results were confirmed by the determination of mature 28S rRNA and ribosomal protein levels (Fig. 7, B and C). We found proteins involved in different steps of rRNA processing that were consistently up-regulated in the three cell lines. In addition, two proteins showed increased acetylation occupancy after SIRT1 inhibition in all cells.
Table 2.
Relative levels of proteins involved in the regulation of rRNA processing in cells treated with the SIRT1 inhibitor EX-527 relative to control
The reported values correspond to the ratio of intensities for each protein between treated and control cells (EX-527/CTRL) for the three cell lines under study.
| Accession number | Gene | Protein names | Unique peptides | Molecular mass (kDa) | Q-value | Score | EX-527/CTRL |
||
|---|---|---|---|---|---|---|---|---|---|
| HaCaT | CaLo | SiHa | |||||||
| 47S pre-rRNA processing to yield 45S pre-rRNA | |||||||||
| Q9H8H0 | NOL11 | Nucleolar protein 11 | 2 | 81.123 | 0 | 96.59 | 2.11 | 1.19 | 2.74 |
| Q9Y5J1 | UTP18 | U3 small nucleolar RNA-associated protein 18 homolog | 9 | 62.003 | 0 | 75.95 | 0.91 | 1.14 | 5.03 |
| 45S pre-rRNA processing to yield 30S and 32S pre-RNAs | |||||||||
| Q9NW13 | RBM28 | RNA-binding protein 28 | 15 | 85.737 | 0 | 212.16 | 1.16 | 0.95 | 1.25 |
| Q01780 | EXOSC10 | Exosome component 10 | 21 | 100.83 | 0 | 180.36 | 1.85 | 1.36 | 1.41 |
| Q9UGY1 | NOL12 | Nucleolar protein 12 | 14 | 24.663 | 0 | 40.817 | 1.52 | 1.28 | 1.47 |
| P56182 | RRP1 | Ribosomal RNA processing protein 1 homolog A | 22 | 52.839 | 0 | 223.67 | 1.26 | 1.16 | 2.17 |
| Q14684 | RRP1B | Ribosomal RNA processing protein 1 homolog B | 18 | 82.175 | 0 | 157.89 | 1.51 | 1.57 | 2.08 |
| Q14137 | BOP1 | Ribosome biogenesis protein BOP1 | 22 | 83.629 | 0 | 116.56 | 1.68 | 1.47 | 1.70 |
| Q9H0D6 | XRN2 | 5′–3′ Exoribonuclease 2 | 28 | 108.58 | 0 | 288.2 | 1.31 | 0.64 | 1.11 |
| Q9Y2R4 | DDX52 | Probable ATP-dependent RNA helicase DDX52 | 11 | 67.497 | 0 | 30.176 | 1.85 | 1.55 | 2.10 |
| 30S pre-rRNA processing to yield 21S pre-rRNA | |||||||||
| P42677 | RPS27 | 40S ribosomal protein S27 | 3 | 9.461 | 0 | 130.44 | 0.66 | 1.27 | 1.03 |
| P15880 | RPS2 | 40S ribosomal protein S2 | 8 | 31.324 | 0 | 157.79 | 9.53 | 1.01 | 1.25 |
| P62847 | RPS24 | 40S ribosomal protein S24 | 17 | 15.423 | 0 | 126.66 | 0.69 | 1.34 | 1.16 |
| Q9H8H0 | NOL11 | Nucleolar protein 11 | 2 | 81.123 | 0 | 96.59 | 2.11 | 1.19 | 2.74 |
| P62081 | RPS7 | 40S ribosomal protein S7 | 11 | 21.312 | 0 | 117.54 | 1.51 | 1.56 | 4.02 |
| Q9Y324 | FCF1 | rRNA-processing protein FCF1 homolog | 13 | 23.369 | 0 | 35.869 | 1.86 | 3.88 | 5.03 |
| P62249 | RPS16 | 40S ribosomal protein S16 | 14 | 16.445 | 0 | 323.31 | 1.18 | 1.51 | 1.56 |
| P62277 | RPS13 | 40S ribosomal protein S13 | 10 | 17.222 | 0 | 46.946 | 3.30 | 2.36 | 3.09 |
| P61247 | RPS3A | 40S ribosomal protein S3a | 11 | 23.747 | 0 | 323.31 | 0.94 | 1.56 | 1.14 |
| P46782 | RPS5 | 40S ribosomal protein S5 | 8 | 25.333 | 0 | 118.21 | 1.77 | 2.45 | 2.34 |
| P62857 | RPS28 | 40S ribosomal protein S28 | 5 | 7.8409 | 0 | 193.73 | 0.77 | 0.84 | 1.08 |
| P62244 | RPS15A | 40S ribosomal protein S15a | 8 | 14.839 | 0 | 309.07 | 1.45 | 1.58 | 0.70 |
| P62241 | RPS8 | 40S ribosomal protein S8 | 19 | 24.205 | 0 | 323.31 | 0.67 | 1.35 | 1.03 |
| P62280 | RPS11 | 40S ribosomal protein S11 | 11 | 18.431 | 0 | 121.11 | 2.67 | 0.88 | 2.12 |
| Q8IY81 | FTSJ3 | Pre-rRNA processing protein FTSJ3 | 30 | 96.557 | 0 | 196.05 | 1.80 | 1.19 | 1.57 |
| P62753 | RPS6 | 40S ribosomal protein S6 | 16 | 28.68 | 0 | 323.31 | 1.11 | 0.76 | 1.02 |
| 21S pre-rRNA processing to yield 18S pre-rRNA | |||||||||
| Q9NQT4 | EXOSC5 | Exosome complex component RRP46 | 5 | 25.249 | 0 | 13.818 | 2.27 | 2.89 | 1.07 |
| P42285 | SKIV2L2 | Superkiller viralicidic activity 2-like 2 | 19 | 117.8 | 0 | 230.06 | 0.96 | 1.06 | 1.15 |
| Q01780 | EXOSC10 | Exosome component 10 | 21 | 100.83 | 0 | 180.36 | 1.85 | 1.36 | 1.41 |
| 18S pre-rRNA processing in pre-40S particles | |||||||||
| O43709 | WBSCR22 | Probable 18S rRNA (guanine-N7)-methyltransferase | 5 | 31.88 | 0 | 30.62 | 2.20 | 0.99 | 2.36 |
| 32S pre-rRNA processing to yield 12S pre-rRNA and 28S rRNA | |||||||||
| Q9BVP2 | GNL3 | Guanine nucleotide-binding protein-like 3 | 16 | 61.992 | 0 | 165 | 1.83 | 0.84 | 1.63 |
| Q9NVN8 | GNL3L | Guanine nucleotide-binding protein-like 3-like protein | 10 | 65.572 | 0 | 20.152 | 4.54 | 0.76 | 12.52 |
| Q99848 | EBNA1BP2 | Probable rRNA-processing protein EBP2 | 23 | 40.684 | 0 | 279.31 | 1.04 | 1.08 | 1.46 |
| Q9NR30 | DDX21 | Nucleolar RNA helicase 2 | 17 | 87.343 | 0 | 323.31 | 1.84 | 0.68 | 3.49 |
| O00541 | PES1 | Pescadillo homolog | 31 | 66.077 | 0 | 284.25 | 2.68 | 0.91 | 2.50 |
| Q9Y4W2 | LAS1L | Ribosomal biogenesis protein LAS1L | 18 | 81.242 | 0 | 57.623 | 2.03 | 1.02 | 1.28 |
| Q5SY16 | NOL9 | Polynucleotide 5-hydroxyl kinase NOL9 | 12 | 79.322 | 0 | 110.49 | 2.11 | 1.47 | 0.75 |
| 12S pre-rRNA processing to yield 5.8S rRNA | |||||||||
| P42285 | SKIV2L2 | Superkiller viralicidic activity 2-like 2 | 19 | 117.8 | 0 | 230.06 | 0.96 | 1.06 | 1.15 |
| Q9Y2L1 | DIS3 | Exosome complex exonuclease RRP44 | 20 | 109 | 0 | 115.25 | 0.63 | 0.73 | 0.51 |
| Q99547 | MPHOSPH6 | M-phase phosphoprotein 6 | 5 | 19.024 | 0.0004 | 3.9044 | 1.29 | 2.56 | 1.55 |
| Q01780 | EXOSC10 | Exosome component 10 | 21 | 100.83 | 0 | 180.36 | 1.85 | 1.36 | 1.41 |
The peptide 512GSPTGGAQLLKR523 from ribosomal RNA-processing protein 1 homolog B (RRP1B) was acetylated in the Lys522 residue in the three cell lines treated with EX-527 (HaCaT, 36.3%; CaLo, 38.4%; and SiHa, 22.4% site occupation), whereas in control cells no endogenous acetylation was detected (supplemental Fig. S9). This residue can be a potential SIRT1 target. Besides, the RRP1B protein levels were up-regulated in the three cell lines treated with the SIRT1 inhibitor (Table 2). Interestingly, the peptide 16LASSEKGIR24 from the N terminus of RRP1B was acetylated in the Lys21 residue in the three untreated cell lines (HaCaT, 2.8%; CaLo, 3.8%; and SiHa, 1.8% site occupation). However, in cells treated with the SIRT1 inhibitor, the Lys21 residue showed no traces of endogenous acetylation in any of the analyzed cells. This finding points to a new level of regulation of rRNA processing that involves SIRT1 regulation of the activity of lysine acetyltransferases and/or other lysine deacetylases that needs further attention. Another potential target of SIRT1 deacetylase activity is the probable ATP-dependent RNA helicase DDX52 required to process the 45S pre-rRNA molecule (Table 2). Treatment with EX-527 resulted in the up-regulation of this protein in the three cell lines. We identified the peptide 124ESKLTSGKLENLR136 with two potential acetylation sites that were both acetylated in treated cells, whereas in control cells no acetylation was detected in those sites (supplemental Fig. S11).
Discussion
For large-scale lysine acetylation stoichiometry analyses, we visualized two major challenges during sample processing. The first is related to the acetylation reaction, which is necessary for the labeling with heavy isotopes of untargeted lysine residues. The second involves the solubilization of fully acetylated proteins, which are known to show an increase in hydrophobicity after modification. For the acetylation reaction, we replaced the conventional acetic anhydride with the N-hydroxysuccinimide (NHS) derivate NAS-d3, which is a more stable and gentler reagent. In previous works, we optimized the reactions for several alkylating reagents in solution and in gel to completely modify lysine residues at the peptide and protein levels and applied them to proteomics studies (24–26).4 As we synthesized the NAS-d3 reagent starting from acetic anhydride-d6 and NHS, we used cytochrome c to verify the quality of the reagent and the efficiency of labeling (supplemental Fig. S13). The reaction with acetic anhydride generates acetic acid that lowers the pH. As a consequence, stronger basic buffers that can affect the stability of proteins are needed to maintain basic pH during the reaction. In addition, due to the high reactivity, acetic anhydride treatment can generate side reactions in residues such as tyrosine, threonine, and serine. The acetylation reaction with NAS-d3 generates NHS, which does not lower the pH, allowing the use of gentle buffers such as TEAB at a lower concentration. This reagent has been successfully used for quantitative proteomics and for lysine acetylation stoichiometric analysis (17, 27). As the acetylation reaction occurs at the protein level, which can significantly increase the hydrophobicity of proteins, we explored the use of different methods for sample preparation. We included detergents during the acetylation reaction and during trypsin digestion to avoid unwanted protein precipitation. We compared three of the most widely used methods for sample preparation in proteomics. FASP, GSP, and SSP were evaluated by comparing the peptide and protein distributions based on their hydrophobicity and molecular mass. The FASP method displayed an important bias in the analysis of hydrophobic peptides in comparison with the other two methods. Although the SSP method exhibited the best overall recoveries, we obtained complementary sets of identified acetyl-lysine peptides with the three sample preparation methods.
Our LC-MS/MS data set was also useful for quantitative label-free proteomics analysis between cell types and for comparing untreated versus SIRT1 inhibitor-treated cells. All samples from cell lines or experimental conditions were subjected to the same procedure of chemical acetylation with NAS-d3 and trypsin digestion. In addition, about half of the identified peptides do not contain lysine residues, and only 10% of the lysine-containing peptides had some degree of endogenous normal acetylation occupancy. However, half of the peptides determined to be acetylated presented a stoichiometry of less than 5%. According to our data, more than 95% of the identified peptides did not show variation in their isotopic distributions, behaving as non-labeled, and therefore can be useful for quantitative label-free analysis.
Our stoichiometric analysis revealed that lysine acetylation is a highly dynamic PTM and that it largely depends on the amino acid composition surrounding the acetylation site. Changes in one residue can alter the stoichiometry of acetylation; this is particularly true for histone variants where single-residue exchange led to variations in the acetylation occupancy of their N-terminal tails. In addition, methylation not only competes with acetylation for the same residues but also can alter the acetylation occupancy in nearby lysine residues. Phosphorylation in residues near the acetylation site can also alter its occupancy, not only for acetylation but also for methylation. Our results show the high degree of complexity of cross-talk between PTMs in a specific protein target. Similar observations were reported previously when Chen et al. (14) performed a quantitative acetylome analysis in mouse cells.
We integrated the quantitative proteomics and acetylation stoichiometry analyses in three cell types. We found that, among the group of deacetylase enzymes, SIRT1 was up-regulated more than 2-fold in SiHa cells compared with HaCaT and CaLo cells. We expected that at least some of the proteins that show low acetylation stoichiometry in SiHa cells could be targets of SIRT1, and therefore their pathways could be regulated by this enzyme. The functional analysis of the group of proteins with less acetylation occupancy in SiHa cells revealed a significant enrichment in ribosomal proteins, ribosome biogenesis, rRNA processing, and glycolysis among others. The proteomics analysis confirmed that these groups of proteins indeed showed differences between cell types. These results indicate that acetylation regulates these pathways and that SIRT1 is probably involved in controlling the acetylation status of these proteins and therefore involved in the regulation of their pathways.
To clarify the role of SIRT1, we treated cells with the SIRT1 inhibitor EX-527. With this analysis, we confirmed that SIRT1 regulates the acetylation status of several proteins involved in ribosome biogenesis and rRNA processing and ribosomal proteins. In fact, SIRT1 acts as a negative regulator of these pathways because in all cases both the direct identified targets and most of the members of these pathways had increased protein levels after SIRT1 inhibition. Previous reports also provided evidence indicating that SIRT1 plays a role in the repression of ribosomal biogenesis and rRNA processing (28, 29). In addition, our data provide evidence in three different human cell lines that SIRT1 is involved in the negative regulation of several pathways, most of which occur in the nuclear compartment, specifically in the nucleolus. From the proteomics experiments, we assumed that SIRT1 acts as a repressor of the synthesis of pre-rRNA and its processing. To confirm this finding, we measured the levels of pre-rRNA and 28S rRNA in untreated cells and after treatment with a SIRT1 inhibitor. As expected, the levels of pre-rRNA were significantly increased after SIRT1 inhibition, confirming the role of this deacetylase in the process. To a lesser extent, the role of SIRT1 in the processing of pre-rRNA to yield the mature rRNA molecules was also validated by the level of 28S rRNA. Several proteins in different steps of rRNA processing were up-regulated in the three cell lines. Two of them, RRP1B and DDX52, both involved in 45S pre-rRNA processing to yield 30S and 32S pre-RNAs, were also potential direct targets of SIRT1. In the three cell lines, both proteins had increased acetylation occupancy in at least one site.
In contrast, we found that SIRT1 positively regulates several metabolic pathways, cytoskeleton organization, and protein processing in the endoplasmic reticulum, which are mostly non-nuclear pathways. These results correspond to the functions of SIRT1 that were consistently conserved in the three cell lines analyzed. However, as we observed, SIRT1 is differentially expressed in these cell lines, and particular functions for each cell type can be described. For example, in SiHa cells, SIRT1 seems to have a more profound effect on metabolic proteins. In the cases where we found differences between cell lines, further validation is required to obtain more confident evidence about the role of SIRT1 in specific cell types.
With the integration of quantitative proteomics and large-scale lysine acetylation stoichiometry, we explored lysine acetylation sites, their stoichiometry dynamics, and the variations at the proteome level in several experimental conditions without the bias of specific target approaches. In fact, the same data set was useful for both quantitative label-free proteomics and lysine acetylation stoichiometry analyses. With this strategy, we compared three cell lines and analyzed their response to inhibition of the deacetylase SIRT1. More than 1,500 acetylated peptides were identified, and their acetylation occupancy was determined in each cell line and experimental condition. Altogether, we report the acetylation occupancy of more than 5,000 acetylated peptides in proteins from human cells. This study represents the largest data set published so far for a stoichiometric analysis at the proteome level. Finally, the procedures presented here can be applied to study integrally the role of acetylation and its enzymatic control in any biological system. In addition, several sample preparation methods can be used or explored, depending on the conditions and experiences of each laboratory.
Experimental procedures
Cell culture and protein extraction
Non-cancerous HaCaT and cervical cancer cell lines SiHa (HPV-16-positive) and CaLo (HPV-18-positive) were cultured in RPMI 1640 medium (Gibco, Invitrogen) supplemented with 10% fetal bovine serum, penicillin, and streptomycin and maintained in a saturated-humidity atmosphere with 5% CO2 at 37 °C. At 70–90% confluence, fetal bovine serum was removed from the medium, and cells were harvested 24 h later. For cells treated with EX-527 (6-chloro-2,3,4,9-tetrahydro-1H-carbazole-1-carboxamide) and their corresponding untreated controls, 70–90% confluent cells, grown in the same conditions described above, were washed two times with PBS. Cells were incubated with fresh medium lacking fetal bovine serum and supplemented with 1 μm EX-527, 5 μm EX-527, or vehicle for 24 h.
For cell harvest, the culture medium was removed, and cells were washed two times with PBS and incubated 5 min with Versene solution at 37 °C. Collected cells were washed with cold PBS. Cells were lysed in 4% SDS, 0.1 m DTT, 0.1 m Tris, pH 8.6. Cells were incubated for 1 min in lysis buffer and briefly sonicated on ice (20 pulses, 1 s each). Samples were incubated for 30 min at 40 °C to reduce disulfide bridges completely. Free cysteine residues were modified with iodoacetamide for 30 min at room temperature in darkness. Protein content was estimated based on SDS-PAGE and blue silver staining of samples, aided by ImageJ software.
RNA extraction and RT-qPCR
Total RNA from HaCaT, CaLo, and SiHa cells was extracted using TRI Reagent from Zymo Research and treated with DNase I (RNase-free; Thermo Fisher Scientific). To generate cDNA, we used the RevertAid First Strand cDNA Synthesis kit from Thermo Fisher Scientific using random hexamer primers. For RT-qPCR, SYBR Green chemistry (Maxima SYBR Green/ROX qPCR Master Mix (2×) from Thermo Fisher Scientific) was used. Transcripts levels were analyzed in an ABI 7300 Real Time PCR System and were normalized using actin mRNA levels as a control. The primer sequences used in this project were previously reported to determine the levels of pre-rRNA and 28S rRNA (30).
Synthesis of NAS-d3
The synthesis of NAS-d3 reagent was performed as described previously (27, 31). Briefly, NHS was incubated with acetic anhydride-d6 for 16 h under continuous stirring at room temperature. The white residue (NAS-d3) was washed with hexane to eliminate excess reactants and vacuum-dried. NAS-d3 can also be acquired from Sigma-Aldrich (633259).
In-solution sample preparation
Proteins were precipitated with 9 volumes of cold ethanol overnight at −20 °C. The pellet was washed three times with 90% ethanol solution. The precipitate was solubilized with 0.5% SDS, 0.5% SDC, 0.1 m TEAB, pH 8.0. The chemical acetylation reaction of the unmodified lysine residues was performed in two consecutive additions of a 100-fold molar excess of NAS-d3 in DMSO and incubated at room temperature for 1 h, respectively. After the reaction was completed, O-acetylation was reverted by incubating the sample with 5% hydroxylamine for 20 min. An ethanol precipitation step, as described previously, was performed, and the sample was solubilized in 50 mm ABC, 0.5% SDC. Trypsin was added to a ratio of 1:50 (enzyme:substrate), and the sample was incubated for 16 h at 37 °C. SDC was removed by ethyl acetate extraction under acidic conditions (1 volume of ethyl acetate was added to the sample and acidified with 0.5% TFA). After vigorous vortexing and centrifugation, the organic phase was discarded. An additional step of ethyl acetate extraction without TFA was performed. Finally, the mixture of peptides was desalted with reverse-phase chromatography, dried, and stored at −80 °C until LC-MS/MS analysis.
In-gel sample preparation
Proteins were loaded on an SDS-polyacrylamide gel. When proteins entered the concentrator gel, the run was stopped, and the slice was cut. The gel was washed with water, dehydrated with acetonitrile, rehydrated with a solution of 0.1 m TEAB containing a 100-fold molar excess of NAS-d3, and incubated for 1 h at room temperature. This step was repeated once, and after washing the gel the O-acetylation was reverted by incubating the gel with 5% hydroxylamine for 20 min. After washing and dehydrating, the gel slice was rehydrated with 50 mm ABC and trypsin (1:50 enzyme:substrate) and then incubated at 37 °C for 16 h. The peptide mixture was extracted in the presence of water/acetonitrile/formic acid. Finally, peptides were desalted, dried, and stored at −80 °C until LC-MS/MS analysis.
In-filter-aided sample preparation
The protein sample was loaded onto a 10-kDa-cutoff filter; subjected to buffer exchange with 0.1 m TEAB, 0.5% SDS, 0.5% SDC; and incubated with a 100-fold molar excess of the lysine acetylation chemical reagent NAS-d3 for 1 h. A second addition of the acetylating reagent was included, and then the sample was incubated for another hour. O-Acetylation was reverted by exchanging the buffer with a solution containing 5% hydroxylamine and incubating the sample for 20 min. For protein digestion, the buffer was exchanged with 50 mm ABC, 0.5% SDC, and trypsin was added to a ratio of 1:50 (enzyme:substrate). The reaction was incubated for 16 h at 37 °C, and the mixture of peptides was collected by centrifugation. SDC elimination and the preparation of the sample for LC-MS/MS analysis were performed as described previously for the solution sample preparation method.
Peptide prefractionation
The mixtures of peptides were subjected to reverse-phase chromatography at high pH on a reverse-phase column (4.9 × 150 mm) from Waters by means of an UltiMate 3000 UPLC system from Dionex. The buffer system was composed of buffer A (50 mm ammonium formate in water, pH 10) and buffer B (50 mm ammonium formate in acetonitrile/water (60:40), pH 10). Samples were loaded onto the column at a flow rate of 0.8 ml/min in buffer A and desalted for 10 min. The elution gradient was from 0 to 80% buffer B in 50 min, from 80 to 100% in 5 min, and kept at 100% of buffer B for another 5 min. 30 sample fractions were collected every 2 min. Finally, fractions were pooled together in five final fractions, each of them composed of fractions separated by 5; for example, the final fraction 1 was composed by fractions 1, 6, 11, 16, 21, and 26. Each final fraction was dried using a SpeedVac, desalted, dried, and stored at −80 °C until LC-MS/MS analysis.
LC-MS/MS and data analysis
LC-MS/MS analysis was performed at the Proteomics Core Facility, Ecole Polytechnique Fédérale de Lausanne in Switzerland. Peptides were resuspended in initial chromatographic conditions and separated on a Dionex Ultimate 3000 RSLCnano UPLC system in line-coupled to a Q-Exactive high-resolution mass spectrometer (Thermo Fischer Scientific). Samples were first trapped on a home-made precolumn (Magic AQ C18; 3 μm, 200 Å, 2 cm × 100-μm inner diameter) and then separated following a 250-min elution gradient using a capillary column at 250 nl/min (Nikkyo Technos Co.; Magic AQ C18; 3 μm, 100 Å, 15 cm × 75-μm inner diameter). The mobile phases were as follows: A, 2% acetonitrile, 0.1% formic acid in water and B, 90:10 (v/v) acetonitrile:water, 0.1% formic acid. The mass spectrometer was operated in positive data-dependent acquisition mode, and the full MS range was from 300 to 2,000 m/z. The 10 most intense ions were isolated in the quadrupole and fragmented under high-energy collisional dissociation with a normalized collision energy of 27%. Precursor ions were measured at a resolution of 70,000 (at 200 m/z), and the fragments were measured at 17,500. Only ions with charge states of 2 and higher were fragmented with an isolation window of 2 Th. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (32) partner repository with the data set identifier PXD005903.
Protein identification and label-free quantification were performed using MaxQuant v1.5.3.30 (33) setting Arg-C as the digestion enzyme and carbamidomethylcysteine as a fixed modification. Lysine and N-terminal protein acetylation (normal (d0) and heavy (d3)) were included as variable modifications as well as phosphorylation (Ser/Thr/Tyr) and oxidation (Met). Proteins were identified with an FDR of 1% based on the target-decoy strategy provided by MaxQuant. The database used for protein identification was the human reference proteome UP000005640 from the UniProt repository downloaded on April 30, 2016. For label-free quantification, we considered proteins with at least two razor-unique peptides identified by MS/MS. For lysine acetylation stoichiometric analysis, we used Pview software (16), which calculates the stoichiometry of lysine acetylation based on the isotopic distribution of identified peptides in the MS spectrum. For the stoichiometry calculation, we allowed a 5-ppm tolerance for peaks in the isotopic distribution, and for MS/MS identification the tolerance was fixed to 15 ppm with an FDR of 1%. The grand average of hydropathy index of identified peptides and proteins was calculated online (http://www.gravy-calculator.de/)5 by the program developed by Dr. Stephan Fuchs from the University of Greifswald. The functional enrichments of the identified proteins were performed online on the site of DAVID Bioinformatics Resources v6.8 (34).
Author contributions
J. G. designed and performed experiments, analyzed data, and wrote the manuscript. A. R.-T., J. L.-P., B. A.-E., and S. C. performed experiments. D. C. performed mass spectrometry analysis. F. C. F.-R. performed bioinformatics analysis. S. E.-G. analyzed results, designed experiments, and conceived the study. All authors approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Dr. Marcela Lizano and Dr. Alejandro García Carrancá for helpful discussion during project development. We also thank Dr. Zia Khan from the Center for Bioinformatics and Computational Biology of the University of Maryland, for kindly providing the Pview software, the parameter file, and example data and for the stoichiometric analysis.
This work was supported in part by Consejo Nacional de Ciencia y Tecnología (CONACyT) Grant 220790 and Dirección General de Asuntos del Personal Académico-Programa de Apoyo a Proyectos de Investigación e Inovación Tecnológica Grants IN-206113 and IN-213216. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. S1–S13 and Tables S1–S8.
The mass spectrometric raw data and spectral libraries associated with this manuscript are available from ProteomeXchange with the accession number PXD005903.
Y. Ramos, Y. Cruz-García, A. Sanchez, J. Gil, L. J. Gonzalez, and V. Besada, unpublished results.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party-hosted site.
- PTM
- posttranslational modification
- SIRT1
- sirtuin 1
- KDAC
- lysine deacetylase
- NAS
- N-acetoxysuccinimide
- SDC
- sodium deoxycholate
- FASP
- filter-aided sample preparation
- GSP
- gel sample preparation
- SSP
- solution sample preparation
- FDR
- false discovery rate
- RT-qPCR
- real-time quantitative PCR
- Pol
- polymerase
- NHS
- N-hydroxysuccinimide
- TEAB
- triethylammonium bicarbonate
- HPV
- human papillomavirus
- ABC
- ammonium bicarbonate
- UPLC
- ultraperformance LC
- Th
- thomsons.
References
- 1. Olsen J. V., Blagoev B., Gnad F., Macek B., Kumar C., Mortensen P., and Mann M. (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648 [DOI] [PubMed] [Google Scholar]
- 2. Linding R., Jensen L. J., Ostheimer G. J., van Vugt M. A., Jørgensen C., Miron I. M., Diella F., Colwill K., Taylor L., Elder K., Metalnikov P., Nguyen V., Pasculescu A., Jin J., Park J. G., et al. (2007) Systematic discovery of in vivo phosphorylation networks. Cell 129, 1415–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Allfrey V. G., Faulkner R., and Mirsky A. E. (1964) Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl. Acad. Sci. U.S.A. 51, 786–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Norris K. L., Lee J.-Y., and Yao T.-P. (2009) Acetylation goes global: the emergence of acetylation biology. Sci. Signal. 2, pe76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Choudhary C., Kumar C., Gnad F., Nielsen M. L., Rehman M., Walther T. C., Olsen J. V., and Mann M. (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 [DOI] [PubMed] [Google Scholar]
- 6. Close P., Creppe C., Gillard M., Ladang A., Chapelle J. P., Nguyen L., and Chariot A. (2010) The emerging role of lysine acetylation of non-nuclear proteins. Cell. Mol. Life Sci. 67, 1255–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim S. C., Sprung R., Chen Y., Xu Y., Ball H., Pei J., Cheng T., Kho Y., Xiao H., Xiao L., Grishin N. V., White M., Yang X. J., and Zhao Y. (2006) Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell 23, 607–618 [DOI] [PubMed] [Google Scholar]
- 8. Wagner G. R., and Payne R. M. (2013) Widespread and enzyme-independent Nϵ-acetylation and Nϵ-succinylation in the chemical conditions of the mitochondrial matrix. J. Biol. Chem. 288, 29036–29045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. de Ruijter A. J., van Gennip A. H., Caron H. N., Kemp S., and van Kuilenburg A. B. (2003) Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J. 370, 737–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Seto E., and Yoshida M. (2014) Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 6, a018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gil J., Ramírez-Torres A., and Encarnación-Guevara S. (2017) Lysine acetylation and cancer: a proteomics perspective. J. Proteomics 150, 297–309 [DOI] [PubMed] [Google Scholar]
- 12. Bosch-Presegué L., and Vaquero A. (2011) The dual role of sirtuins in cancer. Genes Cancer 2, 648–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Simmons G. E. Jr., Pruitt W. M., and Pruitt K. (2015) Diverse roles of SIRT1 in cancer biology and lipid metabolism. Int. J. Mol. Sci. 16, 950–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen Y., Zhao W., Yang J. S., Cheng Z., Luo H., Lu Z., Tan M., Gu W., and Zhao Y. (2012) Quantitative acetylome analysis reveals the roles of SIRT1 in regulating diverse substrates and cellular pathways. Mol. Cell. Proteomics 11, 1048–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lin Z., and Fang D. (2013) The roles of SIRT1 in cancer. Genes Cancer 4, 97–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baeza J., Dowell J. A., Smallegan M. J., Fan J., Amador-Noguez D., Khan Z., and Denu J. M. (2014) Stoichiometry of site-specific lysine acetylation in an entire proteome. J. Biol. Chem. 289, 21326–21338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhou T., Chung Y. H., Chen J., and Chen Y. (2016) Site-specific identification of lysine acetylation stoichiometries in mammalian cells. J. Proteome Res. 15, 1103–1113 [DOI] [PubMed] [Google Scholar]
- 18. Lin Y., Zhou J., Bi D., Chen P., Wang X., and Liang S. (2008) Sodium-deoxycholate-assisted tryptic digestion and identification of proteolytically resistant proteins. Anal. Biochem. 377, 259–266 [DOI] [PubMed] [Google Scholar]
- 19. Weinert B. T., Moustafa T., Iesmantavicius V., Zechner R., and Choudhary C. (2015) Analysis of acetylation stoichiometry suggests that SIRT3 repairs nonenzymatic acetylation lesions. EMBO J. 34, 2620–2632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tagami H., Ray-Gallet D., Almouzni G., and Nakatani Y. (2004) Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell 116, 51–61 [DOI] [PubMed] [Google Scholar]
- 21. Zink L. M., and Hake S. B. (2016) Histone variants: nuclear function and disease. Curr. Opin. Genet. Dev. 37, 82–89 [DOI] [PubMed] [Google Scholar]
- 22. Gertz M., Fischer F., Nguyen G. T., Lakshminarasimhan M., Schutkowski M., Weyand M., and Steegborn C. (2013) Ex-527 inhibits sirtuins by exploiting their unique NAD+-dependent deacetylation mechanism. Proc. Natl. Acad. Sci. U.S.A. 110, E2772–E2781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schölz C., Weinert B. T., Wagner S. A., Beli P., Miyake Y., Qi J., Jensen L. J., Streicher W., McCarthy A. R., Westwood N. J., Lain S., Cox J., Matthias P., Mann M., Bradner J. E., et al. (2015) Acetylation site specificities of lysine deacetylase inhibitors in human cells. Nat. Biotechnol. 33, 415–423 [DOI] [PubMed] [Google Scholar]
- 24. Betancourt L., Gil J., Besada V., González L. J., Fernández-de-Cossio J., García L., Pajón R., Sanchez A., Alvarez F., and Padrón G. (2005) SCAPE: a new tool for the selective CApture of PEptides in protein identification. J. Proteome Res. 4, 491–496 [DOI] [PubMed] [Google Scholar]
- 25. Gil J., Betancourt L. Z., Sardiñas G., Yero D., Niebla O., Delgado M., García D., Pajón R., Sánchez A., González L. J., Padrón G., Campa C., Sotolongo F., Barberó R., Guillén G., et al. (2009) Proteomic study via a non-gel based approach of meningococcal outer membrane vesicle vaccine obtained from strain CU385: a road map for discovering new antigens. Hum. Vaccin. 5, 347–356 [DOI] [PubMed] [Google Scholar]
- 26. Sanchez A., Ramos Y., Solano Y., Gonzalez L. J., Besada V., Betancourt L., Gil J., Alvarez F., Rodriguez M., Perez L., Pujol M., and Padron G. (2007) Double acylation for identification of amino-terminal peptides of proteins isolated by polyacrylamide gel electrophoresis. Rapid Commun. Mass Spectrom. 21, 2237–2244 [DOI] [PubMed] [Google Scholar]
- 27. Chakraborty A., and Regnier F. E. (2002) Global internal standard technology for comparative proteomics. J. Chromatogr. A 949, 173–184 [DOI] [PubMed] [Google Scholar]
- 28. Voit R., Seiler J., and Grummt I. (2015) Cooperative action of Cdk1/cyclin B and SIRT1 is required for mitotic repression of rRNA synthesis. PLoS Genet. 11, e1005246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Murayama A., Ohmori K., Fujimura A., Minami H., Yasuzawa-Tanaka K., Kuroda T., Oie S., Daitoku H., Okuwaki M., Nagata K., Fukamizu A., Kimura K., Shimizu T., and Yanagisawa J. (2008) Epigenetic control of rDNA loci in response to intracellular energy status. Cell 133, 627–639 [DOI] [PubMed] [Google Scholar]
- 30. Ali M., Yan K., Lalonde M. E., Degerny C., Rothbart S. B., Strahl B. D., Côté J., Yang X. J., and Kutateladze T. G. (2012) Tandem PHD fingers of MORF/MOZ acetyltransferases display selectivity for acetylated histone H3 and are required for the association with chromatin. J. Mol. Biol. 424, 328–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ji J., Chakraborty A., Geng M., Zhang X., Amini A., Bina M., and Regnier F. (2000) Strategy for qualitative and quantitative analysis in proteomics based on signature peptides. J. Chromatogr. B Biomed. Sci. Appl. 745, 197–210 [DOI] [PubMed] [Google Scholar]
- 32. Vizcaíno J. A., Csordas A., del-Toro N., Dianes J. A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T., Xu Q. W., Wang R., and Hermjakob H. (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–D456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tyanova S., Temu T., and Cox J. (2016) The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 11, 2301–2319 [DOI] [PubMed] [Google Scholar]
- 34. Huang da W., Sherman B. T., and Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.