

Abstract
The human transporter ATP7B delivers copper to the biosynthetic pathways and maintains copper homeostasis in the liver. Mutations in ATP7B cause the potentially fatal hepatoneurological disorder Wilson disease. The activity and intracellular localization of ATP7B are regulated by copper, but the molecular mechanism of this regulation is largely unknown. We show that the copper chaperone Atox1, which delivers copper to ATP7B, and the group of the first three metal-binding domains (MBD1–3) are central to the activity regulation of ATP7B. Atox1–Cu binding to ATP7B changes domain dynamics and interactions within the MBD1–3 group and activates ATP hydrolysis. To understand the mechanism linking Atox1–MBD interactions and enzyme activity, we have determined the MBD1–3 conformational space using small angle X-ray scattering and identified changes in MBD dynamics caused by apo–Atox1 and Atox1–Cu by solution NMR. The results show that copper transfer from Atox1 decreases domain interactions within the MBD1–3 group and increases the mobility of the individual domains. The N-terminal segment of MBD1–3 was found to interact with the nucleotide-binding domain of ATP7B, thus physically coupling the domains involved in copper binding and those involved in ATP hydrolysis. Taken together, the data suggest a regulatory mechanism in which Atox1-mediated copper transfer activates ATP7B by releasing inhibitory constraints through increased freedom of MBD1–3 motions.
Keywords: ATPase, copper transport, membrane protein, membrane transport, nuclear magnetic resonance (NMR), protein dynamic, protein NMR
Introduction
Copper is an essential but potentially toxic element (1), and its levels and distribution are tightly regulated in human cells. In the cytosol, copper is always bound to metal chaperones, such as Atox1, which delivers copper to ATP7B, a membrane transporter, which distributes copper to biosynthetic pathways and regulates copper levels in the liver and in the brain, among other tissues (2). Genetic inactivation of ATP7B leads to Wilson disease, a potentially fatal hepatoneurologic disorder. Copper homeostasis is also intertwined with cell resistance to platinum-based anticancer drugs. ATP7B expression level affects differentiation of tumor cells and correlates with the outcome of platinum-based chemotherapy in some cancers (3–7). Atox1 has been implicated in cancer sensitivity to platinum drugs as well (8–10). Given its multifaceted role in cell physiology and human health, the current mechanistic understanding of the Atox1-ATP7B pathway function and regulation is clearly insufficient.
ATP7B is a P-type ATPase (2, 11, 12), which uses the energy of ATP hydrolysis to translocate copper across cellular membranes. Copper binding to ATP7B is required for the formation of the phosphorylated enzyme intermediate, the key step in the P-type ATPase catalytic cycle, and therefore ATPase activity is copper-dependent. Copper also modulates intracellular localization of ATP7B and its modification by kinases (13–15). Copper binding to ATP7B appears to alter protein conformation (16–18), but the molecular mechanism of copper-dependent regulation of ATP7B remains largely unknown.
ATP7B is a multidomain membrane protein (see Fig. 1). The eight transmembrane helices of ATP7B, the nucleotide-binding (N),3 the phosphorylation (P), and the actuator (A) domains form the protein core, which couples ATP hydrolysis to copper translocation across the membrane and is highly conserved from bacteria to humans. In contrast, the N-terminal region, which is essential for regulation, has variable composition. In humans, the N-terminal region is 630 amino acids long. It includes a short segment, which is required for trafficking of ATP7B to the apical membrane of hepatocytes (19), followed by a chain of six metal-binding domains (MBDs). Each of the MBDs has an invariant CXXC motif responsible for binding copper (I) ions (20, 21). The MBD5–6 pair operates as a single unit (22), and MBD1–3 interact with each other, forming a dynamically correlated domain group (16). MBD4 does not significantly interact with either MBD1–3 or MBD5–6 and serves as a link between these two domain groups (16). This organization parallels a proposed functional distinction between the MBD1–4 group, which has a regulatory function, and the MBD5–6 pair, which is required for ATP7B transport activity (23, 24). MBD5–6 may be functionally related to the one or two metal-binding domains found in bacterial and archaeal copper ATPases (25, 26), but the regulatory domain group MBD1–4 is a distinct feature of the human copper transporters ATP7B and ATP7A and their homologs in higher vertebrates.
Figure 1.
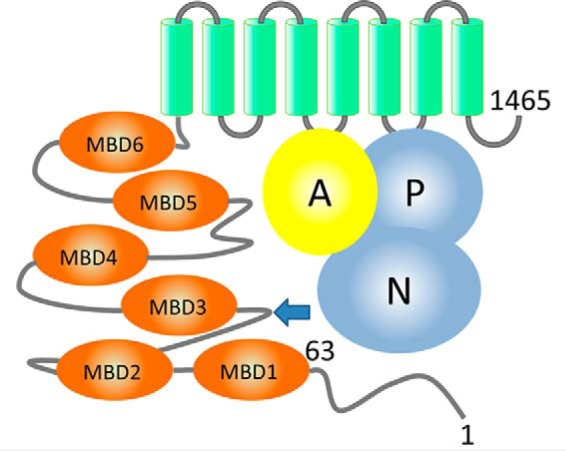
Domain organization of ATP7B. ATP7B includes the N-terminal peptide (ATP7B1–63, residues labeled), six metal-binding domains (MBD1–MBD6, orange), and eight transmembrane helices (green). The N and P domains (blue) together hydrolyze ATP, with participation of the A domain (yellow). The arrow indicates the site of the spontaneous ATP7B proteolysis in the cell.
Unlike Ca2+- or Na+,K+-ATPase, which bind metal ions directly from the bulk phase, ATP7B in the cell receives copper from Atox1. Atox1 transfers copper to the individual MBDs by forming a transient complex, which is stabilized largely by the copper-coordinating cysteines. Presumably, copper is then transferred to the copper-binding site in the transmembrane domain, and, finally, to an acceptor on the other side of the membrane. However, the exact path of copper transfer is unknown. Direct copper transfer from the chaperone to the transmembrane site has been demonstrated for the archaeal transporter CopA, and such a bypass has been proposed as a general mechanism for copper ATPases (27).
The high-resolution structures of all the MBDs (8, 22, 28, 29) have been solved by NMR. However, how the domains work together remains unknown. Here we show that Atox1–Cu, but not free copper, stimulates ATPase activity of ATP7B. Investigating the mechanism of ATP7B regulation by Atox1, we found that Atox1 changes the dynamics of MBD1–3 group, which is directly linked to the nucleotide-binding domain of ATP7B through the interactions with the N terminus of the protein.
Results
Proteolysis within the N-terminal region of ATP7B stimulates ATP7B activity
To investigate the effect of Atox1 on ATPase activity, we expressed the full-length ATP7B in the insect and mammalian cells using baculovirus- and adenovirus-mediated infection, respectively. In addition to the full-length ATP7B (165 kDa), Western blots of cell membranes showed a prominent band at 130 kDa (supplemental Fig. S1A). The intensity of this band increased with time, concomitant with the decrease in the intensity of the full-length protein (supplemental Fig. S1B), indicating that the 130-kDa protein is a product of ATP7B proteolysis. To identify the exact cleavage site, we performed N-terminal sequencing of the purified proteolyzed recombinant ATP7B from the Sf9 cells. Two sequences were identified, matching 229STNPKRPLS237 and 238SANQNFNNS246 of ATP7B and indicating two closely spaced cleavage sites located between MBD2 and MBD3 (Fig. 1A and supplemental Fig. S2B). Unexpectedly, the truncated ATP7B showed higher ATPase activity than the full-length protein (supplemental Fig. S1C). Thus, site-specific proteolysis appears to activate ATP hydrolysis by releasing inhibitory interactions in the MBD1–3 region. To understand the nature of these interactions, we studied the fold and dynamics of MBD1–3 domain group by NMR and SAXS.
Interactions between MBD1 and MBD3 define conformation and dynamics of the MBD1–3 group
Increase in the ATPase activity following proteolytic cleavage between MBD2 and MBD3 suggested that changes in the folding of the MBD1–3 domain group may modulate ATP7B activity. Our recent analysis of the dynamics of MBD1–6 chain showed that, in the absence of copper, MBDs 1–3 interact in a transient fashion, without forming a stable compact structure (16). This interaction can be seen as an equilibrium between the closed conformation, where MBDs 1–3 associate with each other, and a continuum of open conformations, restricted only by the flexible interdomain linkers (16). Here, we have mapped the contact surfaces between the MBDs by chemical shift perturbation analysis. We produced MBD1, MBD2, and MBD3 individually and analyzed chemical shift changes in the 1H,15N-HSQC spectrum of each domain, caused by each of the other two MBDs (supplemental Fig. S3). MBD1 and MBD3 showed well-clustered reciprocal chemical shift changes consistent with domain–domain interactions. These secondary shifts likely arise from direct domain–domain contacts and not from long-range conformational changes, because MBD structures do not show any significant changes caused by either copper binding (30–33) or interdomain interactions (22). Docking MBD1 and MBD3 by HADDOCK (34) based on chemical shift perturbation data produced a top-scoring cluster of 73 structures of 200 calculated with a backbone RMSD of 2.02 ± 0.62 Å overall and 1.18 Å for the 10 top-scoring structures (Fig. 2, A and B).
Figure 2.
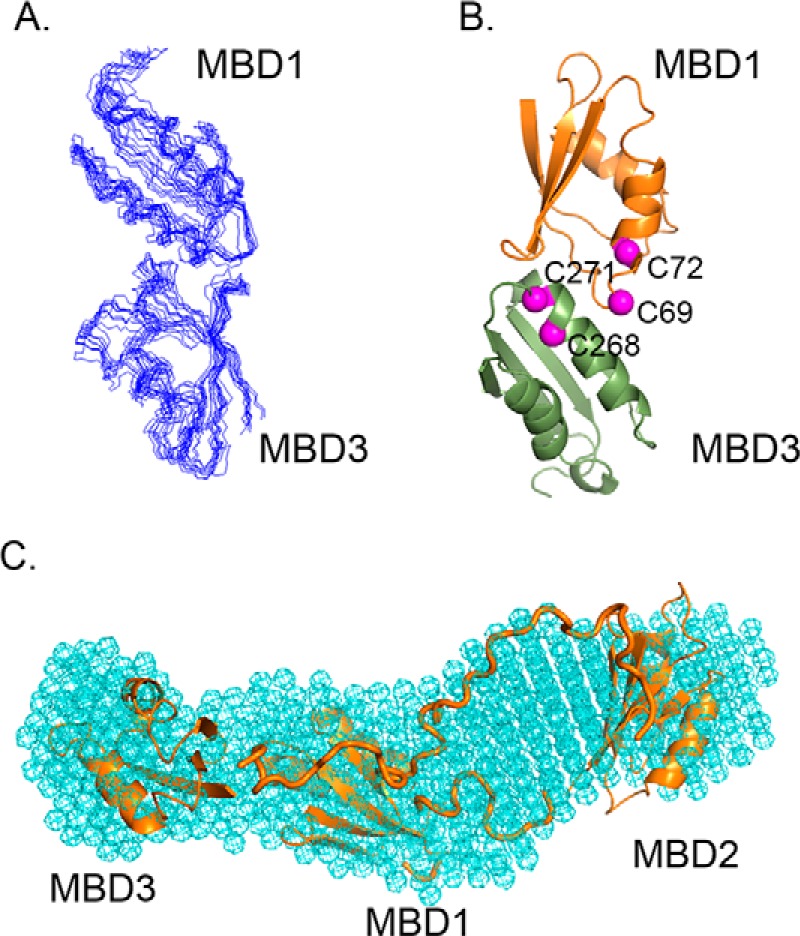
Conformational space of the MBD1–3 group revealed by molecular docking and SAXS. A, ensemble of the 10 top-scoring MBD1-MBD3 models produced by HADDOCK. B, ribbon diagram of MBD1–MBD3 complex with the α-carbons of the cysteine residues in the invariant CXXC motifs shown in magenta. C, overall shape of the MBD1–3 group determined by SAXS (cyan mesh) with MBDs 1–3 model including connecting loops (orange) fitted into the SAXS shape using SUPCOMB (54).
In the MBD1–MBD2 and MBD2–MBD3 pairs, significant chemical shift changes were observed only in MBD2, but not in MBD1 or MBD3. The weak interactions between MBD1–MBD2 and MBD2–MBD3 may not significantly affect the backbone amide chemical shifts of MBD1 and MBD3 and primarily involve side chains in these domains that are not observed in the 1H,15N-HSQC experiment. MBD2 may also show weak dimerization, and the observed chemical shift changes may reflect a shift in the MBD2 monomer-dimer equilibrium caused by MBD2 interaction with the other MBD, whereas MBD1–MBD2 and MBD2–MBD3 interactions themselves are not detected by the backbone amide chemical shifts. Consequently, attempts to dock these domains did not produce well-converged structure clusters.
To determine the overall conformational space of the MBD1–3 domain group, we combined the high resolution structures of MBDs 1, 2, and 3, domain–domain interaction data, and SAXS analysis of the MBD1–3 fragment of ATP7B. The calculated MBD1–3 model (Fig. 2C, supplemental Fig. S4) shows close contacts between MBD1 and MBD3 and a larger distance between MBD2 and MBD3, consistent with the chemical shift perturbation analysis and easy accessibility of the MBD2-MBD3 loop to proteolysis. The long connecting loop between MBD2 and MBD3 (44 residues) and the shorter one between MBD1 and MBD2 (14 residues) are easily accommodated within this fold and fit well in the SAXS envelope.
Atox1–Cu stimulates ATPase activity by docking to MBD2 and changing MBD1–3 conformation
Atox1 was previously shown to transfer copper to the isolated MBDs of ATP7B (35, 36), but the effect of Atox1 on ATP7B activity has not been investigated. As in the other copper ATPases, copper is required for the activity of ATP7B, and basal ATPase activity, stimulated by the trace copper, is inhibited by the copper chelator bathocuproinedisulfonate. We found that this basal ATPase activity of the membrane-bound ATP7B expressed in Sf9 cells (Fig. 3, A and B) is significantly stimulated by the Atox1–Cu complex (Fig. 3C). Neither apo–Atox1 nor copper added as CuCl2, Cu-TCEP, Cu-DTT, Cu-cysteine, or Cu-GSH activated ATP7B. This result implies that Atox1 has a more complex role than mere copper transfer to the MBDs. To better understand this role, we monitored interactions of Atox1 with MBD1–6 by measuring NMR transverse relaxation rates (R2) of the latter.
Figure 3.

ATPase activity of ATP7B is stimulated by Atox1–Cu, but not free copper. A, expression of ATP7B in baculovirus-infected Sf9 cells detected by Western blot with antibodies against the ATP7B C terminus (left panel) and against the Strep tag (right panel). B, ATPase activity of microsomes prepared from noninfected Sf9 cells (1 μg of total protein/data point), cells infected with the virus encoding no exogenous protein (mock), D1027A-ATP7B, or wild-type ATP7B (n = 3). a.u., absorption at 600 nm. D1027A is a catalytically inactive variant of ATP7B used as a control. C, effect of free copper with various reducing agents (2 mm TCEP, 2 mm DTT, 10 mm cysteine, or 2 mm GSH) and of Atox1–Cu on the ATPase activity of Sf9 microsomes expressing wild-type ATP7B. Atox1 was added at 1:1 molar ratio with copper (Cu–Atox1). Equal amount of apo–Atox1 was used as a control for each experimental point (apo–Atox1).
Our previous detailed analysis of domain dynamics in MBD1–6 established R2 rates as reliable indicators of domain mobility changes caused by protein–protein interactions (16). At a 6:1 apo–Atox1/MBD1–6 molar ratio (1:1 Atox1/MBD ratio), 15N-relaxation rates of the amino acid residues in MBD2 and, to a lesser extent, in MBD1 showed major increases, indicating slower tumbling of these domains. The other MBDs showed a much smaller uniform increase in relaxation rates (Fig. 4A). This pattern is consistent with the preferential interaction of Atox1 with MBD2, whereas the increase in the relaxation rates of the other MBDs is likely caused by the propagation of the slower tumbling rate of MBD2 through the domain chain and possibly weak interactions of Atox1 with the other domains.
Figure 4.

Copper transfer from Atox1 increases mobility of MBDs 1–3. Difference in the transverse relaxation rate values (R2) between free MBD1–6 and MBD1–6 with either apo–Atox1 (A) or Atox1–Cu (B) is plotted as a function of the amino acid residue number. Negative difference corresponds to lower domain mobility in the presence of apo–Atox1 or Atox1–Cu, positive difference (e.g. for MBD1–3, B) to higher mobility.
The addition of Atox1–Cu to MBD1–6 (Fig. 4B) had a profoundly different effect on the relaxation rates of MBD1–6 than that of apo–Atox1. The relaxation rates of MBD1–MBD3 decreased, indicating higher tumbling rates of these domains, whereas MBD4–MBD6 were essentially unaffected. This result indicates that copper transfer from Atox1 breaks up the interactions within the MBD1–3 domain group, increasing the mobility of the individual domains. Taken together, the changes in the R2 relaxation rates indicate that both apo–Atox1 and Atox1–Cu interact with the MBDs, but upon transferring copper to the MBDs, Atox1 rapidly dissociates from MBD1–6, whereas in the absence of copper, apo–Atox1–MBD2 complex is more stable.
Interaction between the N-terminal peptide of ATP7B and the N domain
Changes in MBD1–6 conformation and dynamics caused by copper transfer from Atox1 constitute the first step in the regulation of ATP7B by copper, presumably followed by the changes in the interactions between the MBDs and the other domains of ATP7B (37). However, we were unable to detect interactions between any of the six MBDs, either individually or in the context of the complete MBD1–6 chain, and any other cytosolic domains of ATP7B. Consequently, we turned our attention to the N-terminal peptide (residues 1–63) preceding MBD1 in the primary sequence of ATP7B (Fig. 1A). This peptide is required for the copper-dependent trafficking of ATP7B to apical membrane (38), and contains the targeting signal within residues 37–45 (19). Taking these results into account, we analyzed interactions between the synthetic peptide comprising residues 33–63 of ATP7B (ATP7B33–63) and the N domain of ATP7B by NMR. The ATP7B33–63 peptide caused chemical shift changes in several residues clustered in the β-sheet and an adjacent α-helix in the structure of the N domain (39) (Fig. 5). Docking ATP7B33–63 to the N domain based on chemical shift perturbation data, using HADDOCK, placed the peptide at the interface between the A and N domains in the full-length protein (Fig. 5B). At this location, the ATP7B33–63 peptide would affect the movements of these two domains (40) and thus couple ATP hydrolysis to the copper-dependent changes in MBD1–6 conformation and dynamics.
Figure 5.
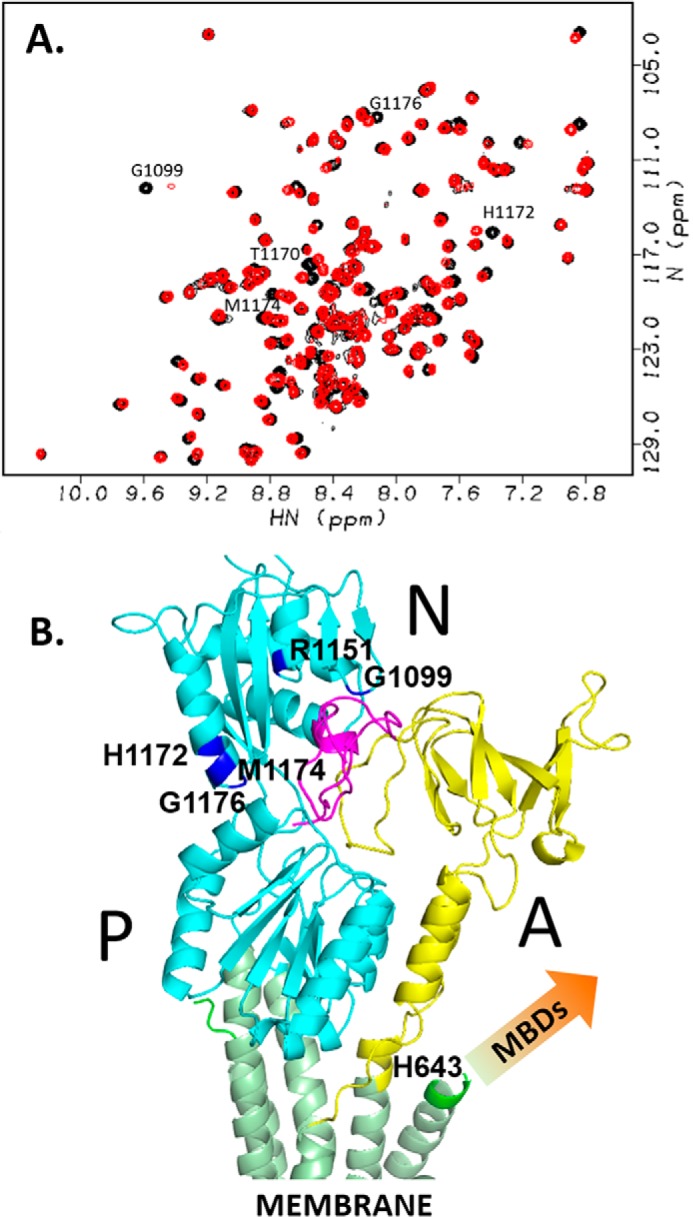
ATP7B33–63 peptide interacts with the N domain. A, 1H,15N-HSQC spectra of 0.5 mm N domain with (red) and without (black) 2.8 mm ATP7B33–63. B, the model structure of ATP7B33–63 peptide bound to the N domain of ATP7B (Protein Data Bank code 2ARF) calculated from the chemical shift perturbation data (A) and aligned to the homology model of ATP7B (12) based on the X-ray structure of copper ATPase CopA from Legionella pneumophila (58), by minimizing RMSD for the N domain. The N and P domains are in cyan, the A domain is in yellow, transmembrane helices are in green, and the ATP7B33–63 peptide is in magenta. Amino acid residues in the N domain with a combined chemical shift change Δδ > 0.03 (cf. A) are shown in blue and labeled. Metal-binding domains, which are absent from the homology model, are not shown (arrow).
Discussion
The N-terminal module of ATP7B comprising the first 63 amino acid residues, and the following chain of six metal-binding domains regulates the activity and intracellular localization of the protein, but the molecular basis of this regulation has not been understood. Previous studies ascribed the regulatory role to the MBD1–4 region (23). We have shown that MBDs 1–3 interact with each other, forming a dynamically correlated domain group, whereas MBD4 did not show any interactions with the other MBDs (16). These results suggest that MBD4 plays a mostly structural role, linking the MBD1–3 and MBD5–6 domain groups. This conclusion is supported by the absence of the CXXC motif in MBD4 in rat and in mouse ATP7B, which makes it unable to bind copper (37). Therefore MBD1–3 appear to be central to ATP7B regulation, and our studies focused on this region of the protein.
Although previous NMR studies show that the individual MBDs are highly mobile (35, 41), biochemical data indicate that the MBDs interact with each other. Copper binding to MBD2 changed proteolysis pattern in the region between MBD3 and MBD4 (42, 43). Accessibility of individual MBDs to the metal chaperone Atox1 appears to vary depending on the experimental conditions (22, 35, 36, 43). Finally, mutations in the copper-binding sites of MBD2 or MBD3 caused changes in copper binding to the other MBDs, suggesting communication between the individual domains (44). Taken together, these results suggest that dynamic interactions between the MBDs may form the basis of ATP7B regulation by copper.
In the present work, we mapped the interactions between MBDs 1–3 and determined the overall space envelope of the MBD1–3 domain group by SAXS. The interaction between MBD1 and MBD3 appears to be stronger than the interactions between MBD2 and the other two MBDs, which may facilitate preferential binding of Atox1 to MBD2. Based on the chemical shift changes (supplemental Fig. S3), the residues in the CXXC motifs of all three MBDs appear to be involved in domain–domain interactions, either through the transient disulfide bridge formation or through hydrogen bonding. The latter would explain previously reported functional differences between the AXXA and SXXS mutants of the copper-binding CXXC motifs (44).
ATP7B receives copper from the cytosolic protein Atox1, which is structurally similar to the MBDs (45). Although all six MBDs in ATP7B can accept copper from Atox1 in vitro (35, 36), MBD2 was found to be the preferred copper acceptor in the context of MBD1–6 (43). This observation is consistent with our finding that apo–Atox1 specifically binds to MBD2. MBDs 1, 2, and 4, but not the others, formed copper-bridged adducts with Atox1 (35). Although the Atox1–Cu–MBD intermediate is stabilized by copper bridging the CXXC motifs of the two proteins, differences in Atox1–MBD binding kinetics suggest significant contribution of other interactions, unique to the specific MBDs. Such interactions likely account for the binding of metal-free Atox1 to MBD2 observed in our experiments.
Copper transfer from Atox1–Cu disrupts MBD1–3 interactions producing an open conformation with largely unrestricted domain motions. We speculated that dissociation of the MBD1–3 group may release inhibitory interactions between MBDs and the other domains of ATP7B, increasing its turnover rate and triggering trafficking or ATP7B to the membrane vesicles and plasma membrane. Previous work suggested that either the N domain (46) or the A domain (47, 48) may interact with the MBDs. However, neither we nor others (17) were able to detect any interaction between the MBDs and either the N or A domain by NMR.
These results prompted us to search for an alternative regulatory mechanism that does not involve direct MBD interactions with the ATP-hydrolase module of ATP7B. The N-terminal peptide comprising residues 1–63 of ATP7B (ATP7B1–63) is required for proper copper-dependent trafficking of ATP7B in the cell (38), and the targeting signal was localized to residues 37–45 (19). Following up on this work, we have found that ATP7B33–63 peptide binds to the N domain, and in the full-length ATP7B, the peptide-binding site would be located at the interface of the N domain and the A domain (Fig. 5B). Peptide binding at this site would affect large scale movements of the A and N domains, known to occur during the catalytic cycle of the P-type ATPases (40) and thus modulate enzymatic activity. Although binding of the isolated ATP7B33–63 to the N domain is copper-independent, copper-dependent changes in the dynamics and conformation of the metal-binding domain chain could strongly affect binding of the ATP7B1–63 peptide to the N domain in the full-length protein.
Taken together, our studies of domain interactions led us to the following model of ATP7B regulation by copper (Fig. 6). At low copper, MBD1–3 is predominantly in the closed conformation, and ATP7B1–63 peptide is bound to the N domain inhibiting ATP hydrolysis and copper transport. When copper level in the cell and, consequently, the Atox1–Cu/apo–Atox1 ratio increase, copper is transferred from Atox1 to MBD2 (Fig. 6, step 1), breaking up MBD1–3 association. The open conformation of MBD1–3 is further stabilized by copper transfer to MBDs 1 and 3 (Fig. 6, step 2).
Figure 6.

Proposed mechanism of ATP7B regulation by Atox1–Cu. At low copper, MBD1–3 associate with each other, and the N-terminal peptide is bound at the A-N domain interface, preventing ATP hydrolysis. (1) Copper transfer from Atox1 to MBD2 breaks up interactions between MBDs 1–3, and then apo–Atox1 dissociates. (2) Copper transfer from Atox1–Cu to MBD1 and MBD3 stabilizes the open conformation of MBD1–3. (3) Transition of MBD1–3 to the open conformation dislodges the N-terminal peptide (ATP7B1–63) from its binding site at the interface of the N and A domains, activating the enzyme and, possibly, inducing trafficking by making ATP7B1–63 accessible for interaction with a hypothetical partner protein X (right panel). (4) At low Atox1–Cu/apo–Atox1 ratios, apo–Atox1 removes copper from the MBDs, including, finally, MBD2 (5). (6) MBD1–3 reassociate, and ATP7B1–63 peptide binds at the A–N interface inhibiting ATP7B. For clarity, only regulatory copper transfer events are shown.
The Brownian pull of the open MBD chain dislodges the N-terminal peptide from its binding site, allowing unimpeded movement of the A and N domains and activating the enzyme (Fig. 6, step 3). Concomitantly, the now-accessible ATP7B1–63 peptide can interact with a hypothetical partner protein (19), triggering trafficking of ATP7B to the vesicles. Decreasing the Atox1–Cu/apo–Atox1 ratio at low copper levels would favor the reverse copper transfer from MBDs to Atox1 (Fig. 6, steps 4 and 5), reassociation of MBDs 1–3, repositioning of the N-terminal peptide to the A/N interface, and enzyme inhibition and reverse trafficking (Fig. 6, step 6).
Alternatively, the physical link between MBD1–3 and the N domain, formed by the ATP7B1–63 peptide, could directly couple mobility of MBD1–3 and of the A and N domains. In this case, dissociation of the MBD1–3 domain group following copper transfer from Atox1 would allow unimpeded movement of the A and N domains in the course of the catalytic cycle, with the ATP7B1–63 peptide still bound to the N domain. Further investigation is needed to distinguish between these two possibilities.
The proposed mechanism of ATP7B regulation is supported by the selective activation of ATP7B ATPase activity by Atox1–Cu observed in our experiments. Lack of activation by free copper confirms the essential role of Atox1–Cu in regulation suggested by its effect on the dynamics of MBD1–3. Activation of ATP7B by selective proteolytic cleavage between MBD2 and MBD3 is consistent with the notion that dissociation of the MBD1–3 domain group triggers enzyme activation.
Cooperativity of copper binding to MBD1–3 predicted by our model, combined with the dynamic equilibria based on reversible copper transfer and relatively weak domain interactions, provides for a finely tuned regulation of ATP7B in the cell. Copper-binding affinities of Atox1 and most MBDs are sufficiently close (36, 49) to suggest that copper transfer between Atox1 and MBDs can be reversible under the physiological conditions (49). In fact, transfer of copper from MBD4-Cu and MBD2-Cu of ATP7B back to Atox1, as well as copper transfer between various MBDs of ATP7B (22, 50) was shown experimentally. Copper also exchanges and equilibrates rapidly between the bacterial copper chaperone CopZ and cognate Cu-ATPase metal-binding domain (25, 51). These results support the notion that apo–Atox1/Atox1–Cu ratio in the cell regulates activity and trafficking of copper ATPases, which is central to our model.
In summary, our work offers new insight into the complex interactions between the cytosolic and transmembrane copper transporters at the molecular level and establishes a novel regulatory role for copper chaperone Atox1. We show that Atox1 stimulates catalytic activity by increasing freedom of motion of the regulatory domains in ATP7B. This unexpected mechanism demonstrates how interactions with a metal chaperone, or another accessory protein, can modulate activity of the complex membrane transporters through changes in domain dynamics.
Experimental procedures
Protein expression and purification
Atox1, individual MBDs 1, 2, and 3, full-length MBD1–6 (residues 1–633 of ATP7B), MBD1–3 fragment (residues 56–329), and the N domain were expressed as fusions with the chitin-binding domain and intein using vector pTYB12 (New England BioLabs). The proteins were isotopically labeled with 15N and purified by chitin affinity chromatography combined with intein self-cleavage, essentially as described previously (16, 39). Prior to NMR and SAXS experiments, all proteins were dialyzed against 50 mm HEPES, pH 7.4, 50 mm NaCl, 5 mm TCEP. The ATP7B33–63 peptide corresponding to residues 33–63 of ATP7B was prepared by chemical synthesis (NEO group, Inc.).
ATP7B with the C-terminal Strep-tag was expressed in Sf9 insect cells and extracted from the cell membranes with 1% n-dodecyl-β-d-maltoside (DDM). The soluble fraction was incubated with Strep-Tactin-Sepharose (IBA) pre-equilibrated with buffer A containing 100 mm NaCl, 50 mm Tris-HCl, pH 8.0, 10% glycerol, 2 mm TCEP, 1 mm 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride, and 1% DDM for 2 h on ice. The ATP7B-bound Strep-Tactin resin was washed with buffer A, followed by buffer A supplemented with 0.1% DDM. The protein was eluted with buffer A supplemented with 0.1% DDM and 2.5 mm desthiobiotin.
NMR experiments
Most NMR experiments were conducted on a 600-MHz Bruker NMR spectrometer equipped with a Cryoprobe. NMR relaxation measurements with MBD1–6 were conducted on a 900-MHz Varian NMR spectrometer equipped with a cold probe as described previously (16). The data were processed using NMRPipe (52) and analyzed using NMRView (53).
SAXS data collection and processing
Prior to SAXS data collection, MBD1–3 was additionally purified by size-exclusion chromatography on Superdex 75 to remove protein aggregates. X-ray scattering data were collected on a Bruker Nanostar instrument. The scattering signal from the used dialysis buffer (see above) was subtracted from the protein scattering signal. The data were processed using ATSAS software suite (54). DAMMIF was used to generate 10 independent ab initio dummy atom models from the SAXS data, which were then averaged with DAMAVER (55).
Calculation of the MBD1–3 fold
Contact interfaces of MBD1–3 were mapped by chemical shift perturbation analysis of the 1H,15N-HSQC spectra using all possible pairwise combinations of MBD1, MBD2, and MBD3, where only one protein was 15N-labeled. Chemical shift perturbation data were used as input for domain docking using HADDOCK (34). Interresidue distance restraints derived from the 10 top-scoring MBD1–MBD3 HADDOCK structures were used as input for fitting MBD1, MBD2, and MBD3 structures into the MBD1–3 shape envelope determined by SAXS, using BUNCH (56).
ATPase activity assay
ATPase activity was measured by the release of inorganic phosphate using malachite green dye (57). ATPase reaction was performed in 20 mm Bis-Tris propane, pH 6.0, 5 mm MgCl2, 200 mm KCl, and 1 mm ATP for 30 min at 25 °C in 50 μl of volume, typically containing 1 μg of total membrane protein.
N-terminal protein sequencing
Approximately 30 μg of purified ATP7B was subjected to Laemmli SDS-PAGE on a 7.5% gel and then transferred to PVDF membrane by electroblotting. The protein bands, identified by Coomassie staining, were excised and destained prior to the N-terminal sequencing by Edman degradation.
Author contributions
C. H. Y., N. Y., J. B., M. T., S. N., and N. V. D. performed the experiments and analyzed the data. L. B. contributed to the study concept and experimental design. S. L. and O. Y. D. designed experiments, analyzed the data, and wrote the manuscript.
Supplementary Material
This work was supported by a Natural Sciences and Engineering Research Council of Canada Discovery Grant (to O. Y. D.) and National Institutes of Health Grant R01 DK071865 (to S. L.). NMR data were collected at the Saskatchewan Structural Sciences Center and at the National Magnetic Resonance Facility at Madison (NMRFAM), which is supported by National Institutes of Health Grant P41 GM103399 and by the University of Wisconsin-Madison. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. S1–S4.
- N domain
- nucleotide-binding domain
- P domain
- phosphorylation domain
- A domain
- actuator domain
- MBD
- metal-binding domain
- SAXS
- small angle X-ray scattering
- HSQC
- heteronuclear single quantum coherence
- TCEP
- tris-(2-carboxyethyl) phosphine
- DDM
- n-dodecyl-β-d-maltoside.
References
- 1. Festa R. A., and Thiele D. J. (2011) Copper: an essential metal in biology. Curr. Biol. 21, R877–R883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lutsenko S., Barnes N. L., Bartee M. Y., and Dmitriev O. Y. (2007) Function and regulation of human copper-transporting ATPases. Physiol. Rev. 87, 1011–1046 [DOI] [PubMed] [Google Scholar]
- 3. Dmitriev O. Y. (2011) Mechanism of tumor resistance to cisplatin mediated by the copper transporter ATP7B. Biochem. Cell Biol. 89, 138–147 [DOI] [PubMed] [Google Scholar]
- 4. Kuo M. T., Chen H. H., Song I. S., Savaraj N., and Ishikawa T. (2007) The roles of copper transporters in cisplatin resistance. Cancer Metastasis Rev. 26, 71–83 [DOI] [PubMed] [Google Scholar]
- 5. Li X. P., Yin J. Y., Wang Y., He H., Li X., Gong W. J., Chen J., Qian C. Y., Zheng Y., Li F., Yin T., Gong Z. C., Zhou B. T., Zhang Y., Xiao L., et al. (2014) The ATP7B genetic polymorphisms predict clinical outcome to platinum-based chemotherapy in lung cancer patients. Tumour Biol. 35, 8259–8265 [DOI] [PubMed] [Google Scholar]
- 6. Martinez-Balibrea E., Martínez-Cardús A., Musulén E., Ginés A., Manzano J. L., Aranda E., Plasencia C., Neamati N., and Abad A. (2009) Increased levels of copper efflux transporter ATP7B are associated with poor outcome in colorectal cancer patients receiving oxaliplatin-based chemotherapy. Int. J. Cancer 124, 2905–2910 [DOI] [PubMed] [Google Scholar]
- 7. Yang T., Chen M., Chen T., and Thakur A. (2015) Expression of the copper transporters hCtr1, ATP7A and ATP7B is associated with the response to chemotherapy and survival time in patients with resected non-small cell lung cancer. Oncol. Lett. 10, 2584–2590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dolgova N. V., Nokhrin S., Yu C. H., George G. N., and Dmitriev O. Y. (2013) Copper chaperone Atox1 interacts with the metal-binding domain of Wilson's disease protein in cisplatin detoxification. Biochem. J. 454, 147–156 [DOI] [PubMed] [Google Scholar]
- 9. Hua H., Günther V., Georgiev O., and Schaffner W. (2011) Distorted copper homeostasis with decreased sensitivity to cisplatin upon chaperone Atox1 deletion in Drosophila. Biometals 24, 445–453 [DOI] [PubMed] [Google Scholar]
- 10. Safaei R., Maktabi M. H., Blair B. G., Larson C. A., and Howell S. B. (2009) Effects of the loss of Atox1 on the cellular pharmacology of cisplatin. J. Inorg. Biochem. 103, 333–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Petrukhin K., Lutsenko S., Chernov I., Ross B. M., Kaplan J. H., and Gilliam T. C. (1994) Characterization of the Wilson disease gene encoding a P-type copper transporting ATPase: genomic organization, alternative splicing, and structure/function predictions. Hum. Mol. Genet. 3, 1647–1656 [DOI] [PubMed] [Google Scholar]
- 12. Schushan M., Bhattacharjee A., Ben-Tal N., and Lutsenko S. (2012) A structural model of the copper ATPase ATP7B to facilitate analysis of Wilson disease-causing mutations and studies of the transport mechanism. Metallomics 4, 669–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hasan N. M., Gupta A., Polishchuk E., Yu C. H., Polishchuk R., Dmitriev O. Y., and Lutsenko S. (2012) Molecular events initiating exit of a copper-transporting ATPase ATP7B from the trans-Golgi network. J. Biol. Chem. 287, 36041–36050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pilankatta R., Lewis D., and Inesi G. (2011) Involvement of protein kinase D in expression and trafficking of ATP7B (copper ATPase). J. Biol. Chem. 286, 7389–7396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Roelofsen H., Wolters H., Van Luyn M. J., Miura N., Kuipers F., and Vonk R. J. (2000) Copper-induced apical trafficking of ATP7B in polarized hepatoma cells provides a mechanism for biliary copper excretion. Gastroenterology 119, 782–793 [DOI] [PubMed] [Google Scholar]
- 16. Huang Y., Nokhrin S., Hassanzadeh-Ghassabeh G., Yu C. H., Yang H., Barry A. N., Tonelli M., Markley J. L., Muyldermans S., Dmitriev O. Y., and Lutsenko S. (2014) Interactions between metal-binding domains modulate intracellular targeting of Cu(I)-ATPase ATP7B, as revealed by nanobody binding. J. Biol. Chem. 289, 32682–32693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mondol T., Åden J., and Wittung-Stafshede P. (2016) Copper binding triggers compaction in N-terminal tail of human copper pump ATP7B. Biochem. Biophys. Res. Commun. 470, 663–669 [DOI] [PubMed] [Google Scholar]
- 18. Sarkar B. (2000) Copper transport and its defect in Wilson disease: characterization of the copper-binding domain of Wilson disease ATPase. J. Inorg. Biochem. 79, 187–191 [DOI] [PubMed] [Google Scholar]
- 19. Braiterman L., Nyasae L., Guo Y., Bustos R., Lutsenko S., and Hubbard A. (2009) Apical targeting and Golgi retention signals reside within a 9-amino acid sequence in the copper-ATPase, ATP7B. Am. J. Physiol. Gastrointest. Liver Physiol 296, G433–G444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Arnesano F., Banci L., Bertini I., Ciofi-Baffoni S., Molteni E., Huffman D. L., and O'Halloran T. V. (2002) Metallochaperones and metal-transporting ATPases: a comparative analysis of sequences and structures. Genome Res. 12, 255–271 [DOI] [PubMed] [Google Scholar]
- 21. Wernimont A. K., Huffman D. L., Lamb A. L., O'Halloran T. V., and Rosenzweig A. C. (2000) Structural basis for copper transfer by the metallochaperone for the Menkes/Wilson disease proteins. Nat. Struct. Biol. 7, 766–771 [DOI] [PubMed] [Google Scholar]
- 22. Achila D., Banci L., Bertini I., Bunce J., Ciofi-Baffoni S., and Huffman D. L. (2006) Structure of human Wilson protein domains 5 and 6 and their interplay with domain 4 and the copper chaperone HAH1 in copper uptake. Proc. Natl. Acad. Sci. U.S.A. 103, 5729–5734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cater M. A., Forbes J., La Fontaine S., Cox D., and Mercer J. F. (2004) Intracellular trafficking of the human Wilson protein: the role of the six N-terminal metal-binding sites. Biochem. J. 380, 805–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Huster D., and Lutsenko S. (2003) The distinct roles of the N-terminal copper-binding sites in regulation of catalytic activity of the Wilson's disease protein. J. Biol. Chem. 278, 32212–32218 [DOI] [PubMed] [Google Scholar]
- 25. Argüello J. M., Eren E., and González-Guerrero M. (2007) The structure and function of heavy metal transport P1B-ATPases. Biometals 20, 233–248 [DOI] [PubMed] [Google Scholar]
- 26. Sitsel O., Grønberg C., Autzen H. E., Wang K., Meloni G., Nissen P., and Gourdon P. (2015) Structure and function of Cu(I)- and Zn(II)-ATPases. Biochemistry 54, 5673–5683 [DOI] [PubMed] [Google Scholar]
- 27. González-Guerrero M., and Argüello J. M. (2008) Mechanism of Cu+-transporting ATPases: soluble Cu+ chaperones directly transfer Cu+ to transmembrane transport sites. Proc. Natl. Acad. Sci. U.S.A. 105, 5992–5997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Banci L., Bertini I., Cantini F., Rosenzweig A. C., and Yatsunyk L. A. (2008) Metal binding domains 3 and 4 of the Wilson disease protein: solution structure and interaction with the copper(I) chaperone HAH1. Biochemistry 47, 7423–7429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yu C. H., Lee W., and Dmitriev O. Y. (2016) NMR structure of metal-binding domain 1 of ATP7B, RCSB Protein Data Bank deposition 2N7Y [Google Scholar]
- 30. Banci L., Bertini I., Del Conte R., D'Onofrio M., and Rosato A. (2004) Solution structure and backbone dynamics of the Cu(I) and apo forms of the second metal-binding domain of the Menkes protein ATP7A. Biochemistry 43, 3396–3403 [DOI] [PubMed] [Google Scholar]
- 31. Banci L., Bertini I., Cantini F., DellaMalva N., Herrmann T., Rosato A., and Wüthrich K. (2006) Solution structure and intermolecular interactions of the third metal-binding domain of ATP7A, the Menkes disease protein. J. Biol. Chem. 281, 29141–29147 [DOI] [PubMed] [Google Scholar]
- 32. DeSilva T. M., Veglia G., and Opella S. J. (2005) Solution structures of the reduced and Cu(I) bound forms of the first metal binding sequence of ATP7A associated with Menkes disease. Proteins 61, 1038–1049 [DOI] [PubMed] [Google Scholar]
- 33. Gitschier J., Moffat B., Reilly D., Wood W. I., and Fairbrother W. J. (1998) Solution structure of the fourth metal-binding domain from the Menkes copper-transporting ATPase. Nat. Struct. Biol. 5, 47–54 [DOI] [PubMed] [Google Scholar]
- 34. Dominguez C., Boelens R., and Bonvin A. M. (2003) HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 125, 1731–1737 [DOI] [PubMed] [Google Scholar]
- 35. Banci L., Bertini I., Cantini F., Massagni C., Migliardi M., and Rosato A. (2009) An NMR study of the interaction of the N-terminal cytoplasmic tail of the Wilson disease protein with copper(I)-HAH1. J. Biol. Chem. 284, 9354–9360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yatsunyk L. A., and Rosenzweig A. C. (2007) Cu(I) binding and transfer by the N terminus of the Wilson disease protein. J. Biol. Chem. 282, 8622–8631 [DOI] [PubMed] [Google Scholar]
- 37. Tsay M. J., Fatemi N., Narindrasorasak S., Forbes J. R., and Sarkar B. (2004) Identification of the “missing domain” of the rat copper-transporting ATPase, ATP7B: insight into the structural and metal binding characteristics of its N-terminal copper-binding domain. Biochim. Biophys. Acta 1688, 78–85 [DOI] [PubMed] [Google Scholar]
- 38. Guo Y., Nyasae L., Braiterman L. T., and Hubbard A. L. (2005) NH2-terminal signals in ATP7B Cu-ATPase mediate its Cu-dependent anterograde traffic in polarized hepatic cells. Am. J. Physiol. Gastrointest. Liver Physiol 289, G904–G916 [DOI] [PubMed] [Google Scholar]
- 39. Dmitriev O., Tsivkovskii R., Abildgaard F., Morgan C. T., Markley J. L., and Lutsenko S. (2006) Solution structure of the N-domain of Wilson disease protein: distinct nucleotide-binding environment and effects of disease mutations. Proc. Natl. Acad. Sci. U.S.A. 103, 5302–5307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Toyoshima C., and Inesi G. (2004) Structural basis of ion pumping by Ca2+-ATPase of the sarcoplasmic reticulum. Annu. Rev. Biochem. 73, 269–292 [DOI] [PubMed] [Google Scholar]
- 41. Fatemi N., Korzhnev D. M., Velyvis A., Sarkar B., and Forman-Kay J. D. (2010) NMR characterization of copper-binding domains 4–6 of ATP7B. Biochemistry 49, 8468–8477 [DOI] [PubMed] [Google Scholar]
- 42. Bartee M. Y., Ralle M., and Lutsenko S. (2009) The loop connecting metal-binding domains 3 and 4 of ATP7B is a target of a kinase-mediated phosphorylation. Biochemistry 48, 5573–5581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Walker J. M., Huster D., Ralle M., Morgan C. T., Blackburn N. J., and Lutsenko S. (2004) The N-terminal metal-binding site 2 of the Wilson's disease protein plays a key role in the transfer of copper from Atox1. J. Biol. Chem. 279, 15376–15384 [DOI] [PubMed] [Google Scholar]
- 44. LeShane E. S., Shinde U., Walker J. M., Barry A. N., Blackburn N. J., Ralle M., and Lutsenko S. (2010) Interactions between copper-binding sites determine the redox status and conformation of the regulatory N-terminal domain of ATP7B. J. Biol. Chem. 285, 6327–6336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rosenzweig A. C., Huffman D. L., Hou M. Y., Wernimont A. K., Pufahl R. A., and O'Halloran T. V. (1999) Crystal structure of the Atx1 metallochaperone protein at 1.02 A resolution. Structure 7, 605–617 [DOI] [PubMed] [Google Scholar]
- 46. Tsivkovskii R., MacArthur B. C., and Lutsenko S. (2001) The Lys1010–Lys1325 fragment of the Wilson's disease protein binds nucleotides and interacts with the N-terminal domain of this protein in a copper-dependent manner. J. Biol. Chem. 276, 2234–2242 [DOI] [PubMed] [Google Scholar]
- 47. Gupta A., Bhattacharjee A., Dmitriev O. Y., Nokhrin S., Braiterman L., Hubbard A. L., and Lutsenko S. (2011) Cellular copper levels determine the phenotype of the Arg875 variant of ATP7B/Wilson disease protein. Proc. Natl. Acad. Sci. U.S.A. 108, 5390–5395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yu C. H., Dolgova N. V., and Dmitriev O. Y. (2017) Dynamics of the metal binding domains and regulation of the human copper transporters ATP7B and ATP7A. IUBMB Life 69, 226–235 [DOI] [PubMed] [Google Scholar]
- 49. Banci L., Bertini I., Ciofi-Baffoni S., Kozyreva T., Zovo K., and Palumaa P. (2010) Affinity gradients drive copper to cellular destinations. Nature 465, 645–648 [DOI] [PubMed] [Google Scholar]
- 50. Bunce J., Achila D., Hetrick E., Lesley L., and Huffman D. L. (2006) Copper transfer studies between the N-terminal copper binding domains one and four of human Wilson protein. Biochim. Biophys. Acta 1760, 907–912 [DOI] [PubMed] [Google Scholar]
- 51. Kay K. L., Zhou L., Tenori L., Bradley J. M., Singleton C., Kihlken M. A., Ciofi-Baffoni S., and Le Brun N. E. (2017) Kinetic analysis of copper transfer from a chaperone to its target protein mediated by complex formation. Chem. Commun. (Camb.) 53, 1397–1400 [DOI] [PubMed] [Google Scholar]
- 52. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 53. Johnson B. A., and Blevins R. A. (1994) Nmr View: a computer-program for the visualization and analysis of NMR data. J. Biomol. NMR 4, 603–614 [DOI] [PubMed] [Google Scholar]
- 54. Petoukhov M. V., Franke D., Shkumatov A. V., Tria G., Kikhney A. G., Gajda M., Gorba C., Mertens H. D., Konarev P. V., and Svergun D. I. (2012) New developments in the program package for small-angle scattering data analysis. J. Appl. Crystallogr. 45, 342–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Franke D., and Svergun D. I. (2009) DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 42, 342–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Petoukhov M. V., and Svergun D. I. (2005) Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys. J. 89, 1237–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lanzetta P. A., Alvarez L. J., Reinach P. S., and Candia O. A. (1979) An improved assay for nanomole amounts of inorganic phosphate. Anal. Biochem. 100, 95–97 [DOI] [PubMed] [Google Scholar]
- 58. Gourdon P., Liu X. Y., Skjørringe T., Morth J. P., Møller L. B., Pedersen B. P., and Nissen P. (2011) Crystal structure of a copper-transporting PIB-type ATPase. Nature 475, 59–64 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.