

Abstract
The 5′-AMP-activated protein kinase (AMPK) is a master regulator of cellular homeostasis. Despite AMPK’s known function in physiology, its role in pathological processes such as prostate cancer is enigmatic. However, emerging evidence is now beginning to decode AMPK’s paradoxical role in cancer and therefore inform clinicians if and how AMPK could be therapeutically targeted. Here, we propose that it is the spatiotemporal regulation of AMPK complexes that govern the kinase’s role in cancer. We hypothesize that different upstream stimuli will activate select subcellular AMPK complexes. This is supported by the distinct subcellular locations of the various AMPK subunits. Each of these unique AMPK complexes regulate discrete downstream processes that can be tumor suppressive or oncogenic. It is the weighted net function of these downstream signaling events, influenced by additional prostate-specific signaling, that determines AMPK’s final biological output.
Keywords: 5′-AMP-activated protein kinase (AMPK), androgen, androgen receptor (AR), calcium/calmodulin-dependent protein kinase kinase 2 (CaMKK2), metabolism, prostate cancer
Introduction
One of the most enigmatic signaling molecules in biology is the 5′-AMP-activated protein kinase (AMPK). AMPK is an evolutionarily conserved serine/threonine protein kinase that is a master regulator of cellular homeostasis1. Classically, AMPK has been defined by its ability to help cells adapt to various energetic stresses. In this context, activation of AMPK promotes ATP-generating catabolic processes while simultaneously inhibiting ATP-depleting anabolic processes1. As such, AMPK is absolutely required for embryonic growth and development2,3. While AMPK’s function in basic physiology is established, its role in pathological processes such as cancer is far more confusing.
Initially, AMPK was described as a tumor suppressor due to its link to one of its major upstream regulators liver kinase B1 (LKB1), a bona fide tumor suppressor4–6. Correspondingly, a number of in vitro and in vivo studies have implicated an anti-cancer role for AMPK7–20. Conversely, recent reports implicate an oncogenic role for AMPK21–45. While some of these discrepancies may be attributable to the use of non-selective pharmacological modulators of AMPK, many of which have cellular effects independent of AMPK34,45–55, molecular and genetic studies do indicate AMPK can have context-dependent roles8,10,17–19,21,22,24,32–34,56–58. This paradoxical role for AMPK is no different in prostate cancer.
As described in greater detail below, much of the initial interest in AMPK in prostate cancer came from retrospective clinical studies of the use of the antidiabetic drug metformin59–62. These studies suggested that metformin decreased the risk of cancer. Accordingly, several reports using pharmacological modulators of AMPK as well as genetic knockout of one of the catalytic subunits of AMPK, PRKAA2, support a tumor suppressive role for AMPK in prostate cancer7,17,63–65. However, more recent retrospective studies did not find any link between metformin use and decreased cancer risk66–77. In fact, some studies even suggested that increased metformin use correlated with more aggressive prostate cancers66,71. Importantly, the first prospective clinical trials directly testing the impact of metformin on prostate cancer have recently been completed and found limited efficacy (78 and (NCT01433913)). Additional preclinical studies using pharmacological, molecular and genetic approaches have now identified an oncogenic role for AMPK in prostate cancer29–31,33–35,37. Further, levels of threonine-172 phosphorylated/activated AMPK (discussed below) and serine-80 of acetyl-CoA carboxylase (ACC), a canonical target of AMPK, were elevated in prostate cancer clinical samples compared to benign controls7,33,37. Phosphorylated/activated AMPK levels were found to also correlate with progression to the advanced/recurrent stages of the disease33 and higher Gleason scores7. Collectively, these findings indicate a complicated role for AMPK that is likely context dependent. This confusion has undoubtedly frustrated clinicians and researchers and thus precluded subsequent drug development efforts.
Here, we propose a mechanistic explanation to assist in understanding how AMPK works in prostate cancer and therefore determine when AMPK is functioning in an oncogenic versus tumor suppressive capacity. Specifically, we hypothesize that there are different subcellular populations of AMPK that enable compartmentalized signaling (Figure 1). The location of these AMPK complexes is influenced by factors such as subunit composition (reviewed below). Each of these subcellular populations of AMPK will be associated with unique downstream cellular processes (Figure 2). Which AMPK populations are activated is determined by the spatial and temporal regulation of diverse upstream stimuli. It is the weighted net function of these downstream signaling events, influenced by additional prostate-specific signaling, that determines AMPK’s final biological output. Below are descriptions of the factors that regulate AMPK activity, AMPK’s known functions and current/emerging approaches for targeting AMPK signaling. It is our goal that the proposed model can be leveraged to determine if, how and when AMPK could be therapeutically targeted.
Figure 1. Compartmentalized signaling.
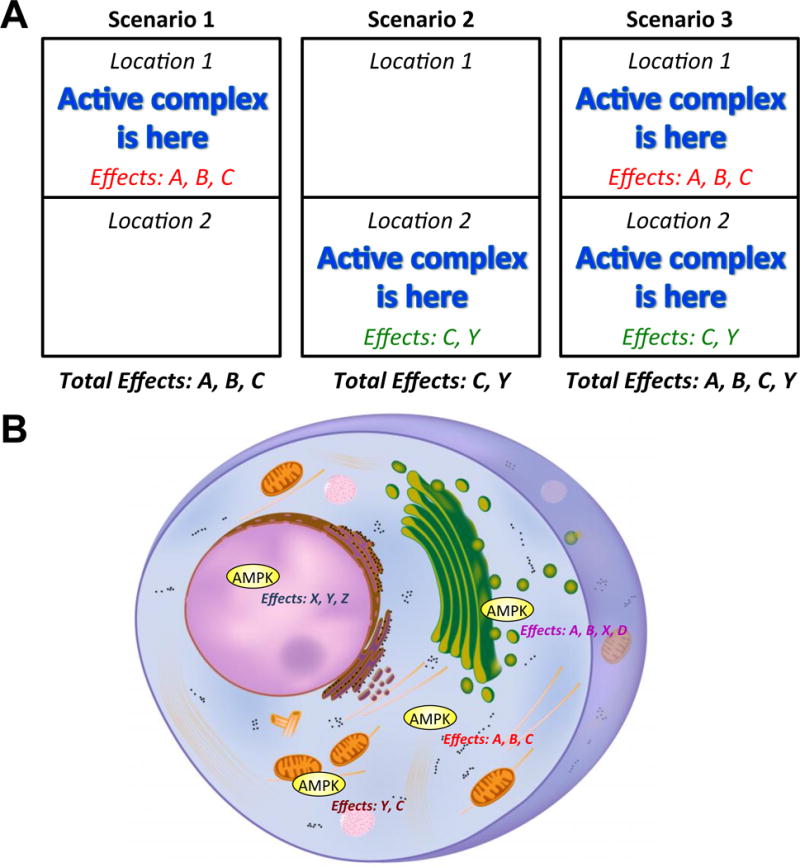
A, Example of compartmentalized signaling. In Scenario 1, a particular signaling complex is activated at a specific location (Location 1). At Location 1 are the parts needed to generate certain effects (A, B, C) that are also known to be regulated by the described active complex. Thus, when the active complex is at this site (Location 1), the associated downstream processes are regulated to produce effects A, B and C. Processes regulated by the active complex but located elsewhere, such as at Location 2 that could produce effects C and Y, will not be altered in this scenario. In contrast, when the complex is activated at Location 2 and not 1 such as in Scenario 2, then only effects C and Y are produced and not effects A and B. In Scenario 3, the complex is activated everywhere and hence, all known processes controlled by the active complex will be regulated, producing a broad range of effects (A, B, C, Y). B, Regarding AMPK-mediated cellular effects, there are different AMPK complexes located throughout the cell (ex. cytoplasmic versus nuclear). Depending on which of these complexes is activated (could be more than one), the net effect AMPK has on a cell will be the summation of the actions of all of the activated subcellular populations of AMPK and their associated downstream effector processes.
Figure 2. Upstream stimuli determine the differential regulation/activation of AMPK-mediated downstream effects.
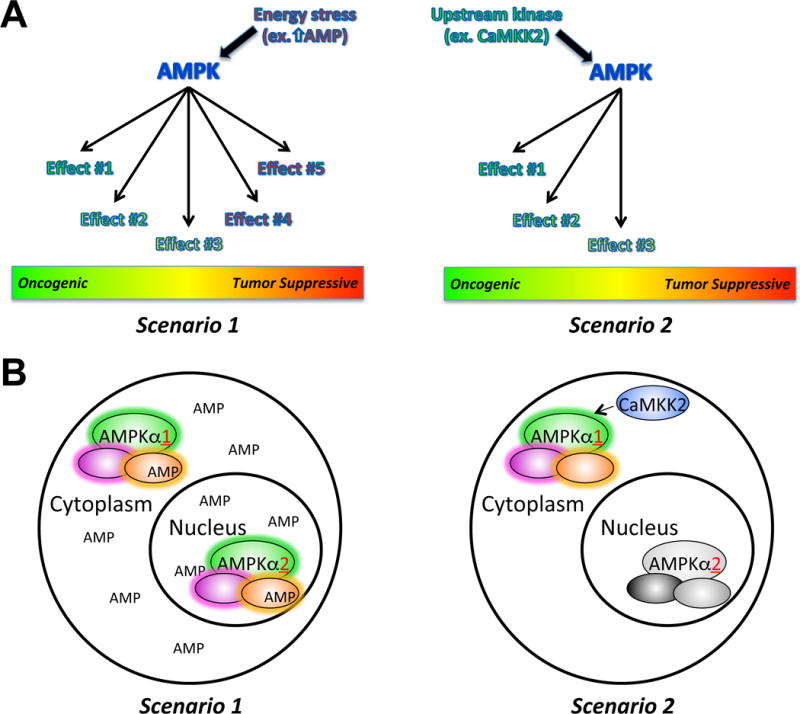
A, Depending on the particular upstream cue (ex. energy stress (Scenario 1) or phosphorylation by an upstream kinase (Scenario 2)), different subpopulations of AMPK can be activated (or inactivated). The net phenotypic effect of each type of AMPK activation will be the summation of all the regulated downstream pathways, shifting the balance between oncogenic and tumor suppressive AMPK signaling. In Scenario 1, all downstream AMPK targets (both oncogenic and tumor suppressive) are activated. Here, the tumor suppressive functions could dominate. In Scenario 2, there is a more selective activation of AMPK complexes that favor the induction of oncogenic downstream processes. B, The type of upstream stimuli and thus manner in which cellular AMPK complexes are activated is likely influenced by both the location of upstream cues and AMPK complexes, which can be influenced by amongst other aspects the subunit composition, as well as the duration of signal. In this regard, in Scenario 1, a persistent energetic stress such as high AMP (or ADP) levels would be able to activate the majority of AMPK complexes. In contrast, an upstream kinase with a more restricted location such as CaMKK2 (Scenario 2) could only phosphorylate/activate local AMPK complexes, perhaps for a limited duration. This would lead to a restricted set of downstream processes that AMPK could regulate.
Influence of AMPK’s structure and subcellular location on downstream activity
The AMPK complex is a heterotrimer consisting of a catalytic α subunit and regulatory β and γ subunits79,80. The N-terminus of the α subunits contains the serine/threonine kinase domain as well as an activation loop that requires phosphorylation at a specific threonine residue, commonly referred to at Thr172 due to its position in the original rat sequence, by upstream kinases (described below) for full AMPK activation (activity increases > 100-fold)81. The C-terminus is needed for association with the β subunit. In addition, there is a central auto-inhibitory domain. The β subunit contains a central domain that allows it to interact with glycogen and a C-terminal domain that is needed for the association with the α and γ subunits. The γ subunit contains four tandem cystathionine β-synthase (CBS) motifs, three of which can bind nucleotides (site two can not). Site four appears to constitutively bind AMP, while sites one and three bind AMP, ADP or ATP in a competitive manner82. Binding of AMP and/or ADP promotes the phosphorylation of Thr172 on the α subunit by upstream kinases while also inhibiting the dephosphorylation of this same site by phosphatases83–85. These activating actions of AMP/ADP are antagonized by ATP. AMP, but not ADP, also causes an allosteric activation (reported to be 2–5 fold but the exact fold induction is still debated86) of the phosphorylated kinase85. In fact, this allosteric activation appears to be antagonized by ATP and ADP.
In total, there are two genes (PRKAA1 and PRKAA2) that encode for the α1 and α2 catalytic subunits, two genes (PRKAB1 and PRKAB2) encoding the β1 and β2 regulatory subunits and three genes (PRKAG1, PRKAG2 and PRKAG3) encoding the γ1, γ2 and γ3 regulatory units2. The subunits are expressed to varying degrees in a cell-, tissue- and disease-specific manner2. In addition, splice variants of the subunits exist87. Thus, between the seven different subunits there are at a minimum 12 different AMPK complexes that can be formed.
An important aspect of AMPK that is often overlooked is the compartmentalization of AMPK signaling. Different isoforms of the various subunits can vary in their subcellular localization88–91. For example, the α1 subunit has a predominantly cytoplasmic localization while the α2 subunit can readily shuttle to the nucleus. Additional AMPK subunits have also been found to partly reside in the nucleus92,93. Further, the subcellular localization of subunits can be altered in response to different stimuli90,94. That said, there is evidence that under certain conditions α1 can also shuttle to the nucleus95. Whether this happens in the prostate and how this would occur are currently not known. Importantly, it has been demonstrated in yeast that the subcellular location of the β subunit directs the localization of the catalytic α subunit91. This effect has key functional consequences as it dictates which subset of downstream targets AMPK can interact with and therefore phosphorylate.
Relatively little is known regarding AMPK’s role, if any, during the development of the prostate. Since AMPKα1/AMPKα2 double knockout mice are embryonic lethal at day 10.5 post conception, it is difficult to study prostate development in this context. To our knowledge, no one has developed a prostate-specific conditional AMPKα1/AMPKα2 double knockout mouse. Both global AMPKα1 and AMPKα2 single knockout mice have been created as well as knockouts of other AMPK subunits2. However, no prostate defects have been reported in any of these animals, but this could also be because the prostate was not examined. AMPKα1 global knockout mice are subfertile due to a decreased quality of spermatozoa96. A subsequent study using AMPKα1 conditional knockout mice identified defects in the Sertoli cells as the likely culprit97. Whether there were any defects in the prostate of the AMPKα1 global knockout mice and if these could then also contribute to the decreased fertility is not known. Global knockout of Camkk2, the predominant AMPK upstream kinase in the prostate (see “Regulation of AMPK” section), in mice did not lead to any overt morphological abnormalities in the prostate (unpublished observations). Taken together, it is not clear yet whether AMPK signaling is essential for normal prostate development. Conversely, a number of recent studies point to context-dependent roles for different AMPK complexes in prostate cancer.
Unlike LKB1, the genes encoding AMPK are rarely mutated in cancer98. In contrast, the various subunits are more commonly elevated by amplifications and/or overexpression in human cancers. For example, the α1 subunit is the predominantly expressed catalytic subunit in prostate cancer33,34,99. While double catalytic subunit knockout mice (α1−/− and α2−/−) are embryonic lethal2, interestingly, mouse embryonic fibroblast (MEF) cells generated from these double knockout mice are resistant to oncogenic transformation22. In addition, spontaneous tumor formation has never been observed in α1- or α2-deficient mice, indicating that loss of AMPK itself is not sufficient to cause tumorigenesis98. However, deletion of α2 alone increased the growth of RAS-transformed MEFs8. In contrast, deletion of α1 alone decreased growth in the same cells8. While genetic deletion of the minor isoform, α2, slightly increased the incidence of prostatic intraepithelial neoplasia (PIN) in a fatty acid synthase (FAS)-transgenic model of mouse prostate hyperplasia17, knockdown of either the predominant α1 alone or double knockdown of α1 and α2 decreased prostate cancer cell growth and migration33,34. Further, increased PRKAA1 and decreased PRKAA2 expression independently predict poor prognosis in prostate cancer patients33,100. Taken together, these results suggest that the two α catalytic subunits may have opposing actions in prostate cancer, with the α1 subunit acting more oncogenic while the α2 subunit appears to have tumor suppressive properties.
Like AMPKα1, AMPKβ1 may also have oncogenic roles. The gene encoding AMPKβ1, PRKAB1, was overexpressed in metastatic prostate cancers compared to primary tumors in four separate clinical cohorts101. Correspondingly, low PRKAB1 expression predicts favourable clinical prognosis100,102,103. In addition, AMPKβ1 was identified in an unbiased RNAi screen as an essential component for prostate cancer cell survival101. These data are supported by work demonstrating increased PRKAB1 expression in colorectal cancer lesions compared to matched benign tissues104.
Genetic studies have revealed that the subunits are not necessarily redundant and therefore different AMPK complexes likely have unique activities2. Further, different stimuli can lead to changes in the heterotrimer composition105–107. Little is known regarding which heterotrimer complexes are preferred in cancers and if their composition changes in response to oncogenic or tumor suppressive signals. The combined effect of elevated levels of the α1 and β1 subunits with their predominantly cytoplasmic subcellular localizations suggest extranuclear AMPK complexes may play a significant role in prostate cancer. Thus, the cytoplasmic kinases (described below) that rapidly activate these AMPK complexes likely would also have important roles in prostate cancer. To that end, multiple groups have developed new molecular tools to assess AMPK activity at different subcellular locations108,109. These key studies demonstrate that diverse upstream cues can activate unique subcellular AMPK populations. We hypothesize that each one of these unique subcellular AMPK complexes will be associated with, and can therefore regulate, a specific set of downstream signalling targets. As a result, depending on the duration and type of upstream signal, very different AMPK-mediated events would be elicited. Thus, the common oversimplification of general AMPK activity could lead to numerous misunderstandings and incorrect conclusions.
Regulation of AMPK
AMPK can be activated by both allosteric modulation and posttranslational modifications (Figure 3). While still a hotly debated area, the allosteric effects may pale in comparison to the regulatory effects of posttranslational modifications such as Thr172 phosphorylation110–112. Regardless, the most well-studied mechanism of AMPK activation is activation by AMP/ADP:ATP ratio. When the AMP/ADP:ATP ratio increases in the cell, AMP/ADP binds to the γ subunit of AMPK3. This causes a conformational change in the γ subunit that, in conjunction with a β-subunit myristoylation event94, exposes the Thr172 site located on AMPK’s α-catalytic subunit. Phosphorylation of this site then activates AMPK ~100-fold81,113. Thus, the phosphorylation of Thr172 is tightly regulated by upstream kinases and phosphatases.
Figure 3. Proposed regulation of AMPK in prostate cancer.
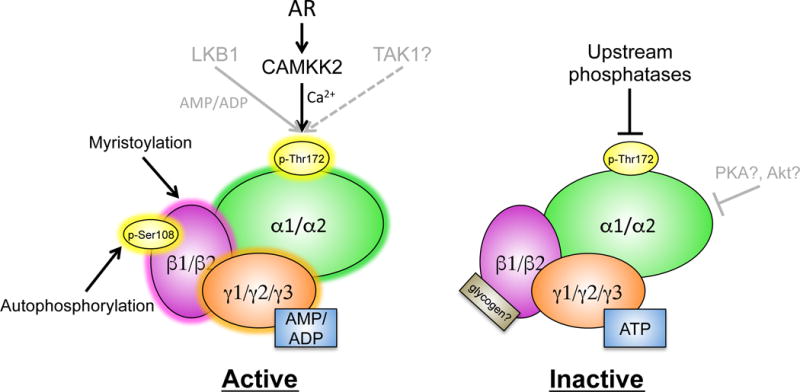
AMPK can be activated by multiple posttranslational modifications as well as energetic stress (ex. high AMP or ADP levels). In the prostate, the dominant upstream kinase of AMPK is CAMKK2, a calcium-dependent kinase whose expression is directly controlled by AR signaling. In contrast, AMPK can be inactivated by upstream phosphatases that, to date, are still ill-defined in the prostate. Further, inhibitory phosphorylation events caused by other kinases have been described but it is unclear if these modifications occur in prostate cancer. Additionally, high levels of ATP are known to inhibit AMPK. However, the inhibition of ATP may be overridden when CAMKK2 is highly expressed.
The known Thr172-targeting kinases of AMPK are LKB1, TGF-β activated kinase-1 (TAK1) and calcium/calmodulin-dependent protein kinase kinase-2 (CaMKK2 or CaMKKβ). LKB1, which activates AMPK in response to energetic stress (ex. high AMP/ADP), is a tumor suppressor that is thought to be the dominant AMPK kinase in the body. However, several lines of evidence indicate that LKB1 is not the predominant AMPK kinase in the prostate. First, while LKB1 is a known tumor suppressor for many types of cancer, prostate cancer is not one of them114. In support of this, when LKB1 was deleted in PTEN+/− mice, there was an increase in tumor incidence for many cancer types but not prostate cancer115. Second, androgens were reported to decrease LKB1 expression and subsequent AMPK phosphorylation in mouse 3T3-L1 cells116. This effect is in direct contrast to the increase in AMPK phosphorylation observed following androgen treatment in prostate cancer cells34,35. Third, LKB1 is not highly expressed or regulated by AR in prostate cancer cell models117. Forth, in a study that did suggest Lkb1 deficiency caused prostatic neoplasia in mice, the authors 1) deleted Lkb1 using a mainly gastrointestinal track-specific driven Cre rather than a prostate-specific driven Cre and more importantly 2) observed continued high levels of Thr172-phosphorylated AMPK in Lkb1-deficient prostates118. This latter data led the authors to conclude that LKB1 was not an AMPK kinase in the prostate. To that end, LKB1 phosphorylates a number of other proteins and hence, if LKB1 were to have antiproliferative effects, this could be due to AMPK-independent effects119. For example, LKB1 is known to phosphorylate and stabilize the tumor suppressor PTEN, one of the most commonly mutated/deleted tumor suppressors in prostate cancer120.
TAK1 is thought to be another activator of AMPK; however, more in depth studies are needed to understand whether this occurs in vivo and requires LKB1121,122. MAP3K7, the gene for TAK1, is often lost during development of prostate cancer123–125. But these findings appear at odds with the increased Thr172-phosphorylated AMPK levels observed during the development of prostate cancer7,33,123–125. Alternatively, functional data indicate that TAK1’s tumor suppressive effects are mediated through other stress kinases, namely p38 and c-Jun N-terminal kinase126. Taken together, these data suggest that TAK1 is not a major AMPK kinase in the prostate.
Roughly a decade ago, CaMKK2 was identified by three separate groups to be a Thr172-targeting AMPK kinase127–129. In 2011, we demonstrated that androgens, via AR, directly increased the expression of CAMKK234. CaMKK2 phosphorylated and activated AMPK, leading to increased prostate cancer cell migration and invasion. The androgen response element which we identified and showed was responsible for AR-mediated expression of CAMKK234 is one of the most robust AR binding sites in castration-resistant prostate cancer (CRPC) tissue130. Interestingly, CaMKK2 can augment AMPK activity under starvation or nutrient-rich conditions, suggesting AR-CaMKK2 may potentiate AMPK activity independent of the environmental state. Later, two independent groups confirmed our findings and also demonstrated that CaMKK2 levels were elevated in clinical samples and track with disease progression35,36. These findings correspond with the clinical data demonstrating that levels of the Thr172 phosphorylated form of AMPK are increased in prostate cancer and further increased in the advanced stages of the disease7,33,123–125. Last year, Hu et al demonstrated that the tumor suppressive microRNA, miR-224, suppressed prostate cancer cell proliferation through decreasing CAMKK2 expression131. Clinically, combined low miR-224 and high CAMKK2 expression correlated with advanced disease and shortened survival. Remarkably, in this study the authors showed a proliferative role for CaMKK2 even in AR-negative DU145 cells, indicating that in some of the most aggressive subtypes of prostate cancer CAMKK2 may be expressed and driving oncogenic processes independent of AR. Collectively, these studies suggest that CaMKK2 is the dominant AMPK kinase in prostate cancer. In addition, new oncogenic roles for CaMKK2 in other cancer types such as stomach, liver and brain have been observed132–134. While a promising target, additional work is needed to assess CaMKK2’s i) functional role at different disease stages, ii) regulation, and iii) complete mechanism(s) of action (to understand potential side effects).
AMPK can also be allosterically activated in two ways. First, AMPK can be allosterically activated by the binding of AMP, but not ADP, to the γ subunit. This direct allosteric activation by AMP does not require the β subunit myristoylation94. Second, AMPK can be pharmacologically activated by the binding of drugs such as A-769662 to the β subunit135. Though drugs like A-769662 function in part by inhibiting the dephosphorylation of Thr172, they also allosterically activate AMPK. As such, this type of activation does not necessarily require Thr172 phosphorylation of the α subunit but does typically involve the autophosphorylation of Ser108 in the β subunit, which is often required for full AMPK activity92. It is not known whether AMPK can be activated in the absence of Thr172 phosphorylation in response to endogenous signaling.
There are other ways in which AMPK could be regulated in prostate cancer. For example, DNA damaging agents like ionizing radiation and some chemotherapies activate AMPK via ataxia telangiectasia mutated (ATM), an initiator of the DNA damage response136–141. While there is still debate regarding whether LKB1 is required for this genotoxic response, this will likely have consequences for therapeutic resistance137,138,140–142. Additionally, reactive oxygen species (ROS) can increase AMPK activity38,143,144. This is significant because oxidative stress is one of the hallmarks of aggressive prostate cancers145. Although the exact mechanisms through which this occurs are still being elucidated (ex. could also include ATM142,146), this may function as a survival signal for cancer cells coping with the harsh tumor microenvironment38. Conversely, it has been reported that protein kinase B (Akt) can phosphorylate AMPK and reduce AMPK’s activation by LKB1147. This would seemingly be an important feedback mechanism in prostate cancer where most advanced stages exhibit elevated phosphoinositide 3-kinase (PI3K)-Akt signaling100. However, it is unclear if this type of regulation occurs in prostate cancer where a) levels of activated AMPK remain high even in the presence of increased Akt7,33,37 and b) LKB1 does not appear to be the major AMPK kinase (described above).
AMPK-regulated processes
One of the most pressing questions regarding AMPK signaling is “Which downstream proteins does AMPK target in cancer?” Presumably not all AMPK-modulated processes are oncogenic and/or tumor suppressive. Identification of the specific cascades that are modulated by AMPK would facilitate our understanding of whether AMPK was having oncogenic or tumor suppressive effects and therefore if AMPK should be modulated therapeutically. Further, parsing out the exact downstream processes that are true oncogenic drivers would highlight potential new therapeutic targets. Listed in Tables 1 and 2 are previously described direct18,24,26,93,95,148–203 and indirect27,198,204–211 AMPK targets, respectively. A number of the targets have been described in greater detail in several excellent reviews1,98,117,119,212,213. At this time it is not known whether many of these are regulated in prostate cancer or if they have a pathogenic role in the disease. Certainly more work is needed to elucidate their regulation and role. An important point to remember is that rarely does the regulation of signaling pathways, such as those modulated by AMPK, occur in isolation. Most cancer signaling networks are influenced by additional oncogenic cascades such as PI3K-Akt or, as is typically the case in prostate cancer, AR. Below is a description of some of the known AMPK targets that likely play a role in many prostate cancers and how they could be influenced by other events such as AR signaling.
Table 1. Direct AMPK Targets.
List of previously validated direct targets of AMPK.
| AMPK Target | Target Site (Human) | Functional Consequence | References |
|---|---|---|---|
| ACC1 | S80 | Inhibits ACC enzymatic activity leading to inhibition of de novo lipogenesis | Carlson & Kim (1973), Davies et al. (1990), Ha et al. (1994), Munday et al. (1988) |
| ACC2 | S222 | Inhibits ACC enzymatic activity leading to promotion of fatty acid oxidation | Winder et al. (1997), Chen et al. (2000), Dzamko et al. (2008), Steinberg et al. (2010) |
| AKAP1 | S107 | Facilitates mitochondrial respiration | Hoffman et al. (2015) |
| AMOTL1 | S793 | AMPK phosphorylates AMOTL1 to stimulate Lats kinase which inhibits YAP | DeRan et al. (2014) |
| Beclin-1 | S91, S94 | Induces autophagy | Kim et al. (2013) |
| BRAF | S729 | Promotes the association of BRAF with 14-3-3 proteins and disrupts its interaction with the KSR1 scaffolding protein | Shen et al. (2013) |
| CKIe (Clock) | S389 | Increases CKIe activity | Um et al. (2007) |
| CLIP-170 | S312 | Results in CLIP-170 localizing closer to the distal end of the microtubules (cell polarity), modulating cell migration | Nakano & Takashima (2010) |
| CREB | S133 | Increases CREB transcriptional activity and expression of downstream target genes | Thomson et al. (2008) |
| CRY1 Clock) | S71 | Targets CRY1 toward ubiquitin mediated degradation | Lamia et al. (2009) |
| CRTC2/TORC2 | S171 | Induces 14-3-3- interaction, blocks nuclear translocation and association/activation of CREB | Koo et al. (2005) |
| eEF2K | S398 | Activates eEF2K and blocks translation elongation by inactivating eEF2; can also induce autophagy | Browne et al. (2004), Hong-Brown et al. (2008), Leprivier et al. (2013), Xie et al. (2014) |
| FOXO3 | S413, S588 | Increases transcriptional activity of FOXO3 | Greer et al. (2007), Bodur et al. (2015) |
| GBF1 | T1337 | Suppresses GEF activity of GBF1 resulting in disassembly of the Golgi apparatus | Miyamoto et al. (2008) |
| GFAT1/GFPT1 | S261 | Inhibits enzymatic activity of GFAT1, decreasing flux through the hexosamine biosynthetic pathway | Li et al. (2007), Eguchi et al. (2009) |
| HDAC4/5/7 | S259, S498 | Induces 14-3-3 binding, cytoplasmic sequestration and inhibition of HDACs | McGee et al. (2008), Mihaylova et al. (2011) |
| H2B | S37 | Increases transcription of genes involved in cell survival | Bungard et al. (2010), |
| HMGCR | S872 | Inactivates HMGCR and thus inhibits cholesterol synthesis | Clarke & Hardie (1990) |
| HNF4 | S304 | Represses transcriptional activity of HNF4α | Hong et al. (2003) |
| IRS1 | S794 | Context-dependent regulation of PI3K-Akt signaling | Jakobsen et al. (2001), Qiao et al. (2002), Tzatsos et al. (2007) |
| MFF | S155, S172 | Induction of mitochondrial fission | Toyama et al. (2016), Ducommun et al. (2015) |
| PAK2 | S20 | Promotes PAK2 activity, leading to increased phosphorylation/inhibition of MRLC | Banko et al. (2011) |
| PFKFB2 | S466 | Increases PFKFB2 kinase activity and glycolysis | Marsin et al (2000) |
| PFKFB3 | S461 | Increases PFKFB3 kinase activity and glycolysis | Marsin et al (2002) |
| p27 | T198 | Stabilizes p27, inducing autophagy-mediated cell survival | Liang et al. (2007) |
| p300 | S89 | Inhibits its ability to interact with nuclear receptors | Leff et al. (2003), Yang et al. (2001) |
| p53 | S15 | Increases p53 activity, promotes expression of p21 and cell cycle arrest | Imamura et al. (2001), Jones et al. (2005) |
| PGC-1α | T177, S538 | Induction of PGC-1α and mitochondrial biogenesis | Jager et al. (2007) |
| PPP1R12C | S452 | Induces 14-3-3 binding and inhibition of PPP1R12C, leading to increased phosphorylation/inhibition of MRLC | Banko et al. (2011) |
| Rb | S811 | Inhibition of Rb followed by subsequent increased proliferation | Dasgupta et al. (2009), Rios et al. (2013) |
| RPTOR | S722, S792 | Induces 14-3-3 binding and inhibition of mTOR | Gwinn, D.M., et al. (2008) |
| SIRT1 | T344 | Disrupts the interaction between SIRT1 and its inhibitor DBC1 | Lau et al. (2014) |
| SREBP1 | S396 | Inhibits transcriptional activity of SREBP1 | Li et al. (2011) |
| TBC1D1 | S237, T596 | Promotes 14-3-3 binding and glucose transporter (ex. GLUT4) trafficking | Chen et al. (2008), Chavez et al. (2008), Pehmoller et al. (2009) |
| TBC1D4/AS160 | T642, S704 | Promotes 14-3-3 binding and glucose transporter (ex. GLUT4) trafficking | Treebak et al. (2006), Kramer et al. (2006), Treebak et al. (2010) |
| TSC2 | T1227, S1345 | Enhances TSC2 activity | Inoki et al (2003) |
| ULK1 | S317, S467, S556, S778 | Activates ULK1, promotes autophagy | Kim et al. (2011), Egan et al. (2011) |
| VPS34 | T163/S165 | Inhibits the non-autophagy Vps34 complex | Kim et al. (2013) |
| YAP | S94 | Disrupts YAP-TEAD interaction and leads to inhibition of YAP | Mo et al. (2014) |
Table 2.
List of notable indirect targets of AMPK.
| AMPK Indirect Target | Functional Consequence | References |
|---|---|---|
| GLUT1 | Induction of GLUT1 expression, increased glucose uptake | Barnes et al. (2002), Yun et al. (2005), Wu et al. (2013) |
| MRLC | Changes in cell shape, induction of cell polarization, proper spindle pole assembly and mitosis | Lee et al. (2007), Bultot et al. (2009), Banko et al. (2011), Thaiparambil et al. (2012) |
| PRODH/POX | Increases PRODH activity, increasing flux through the pentose phosphate pathway, increasing autophagy and promoting cell survival under conditions of nutrient stress | Pandhare et al. (2009) |
| SIRT1 | Enhances SIRT1 activity by increasing NAD+ levels | Canto et al. (2009) |
| SREBP2 | Reduces levels of SREBP2 and SREBP2 downstream targets HMGCR and HMGCS, decreasing de novo cholesterol synthesis, ameliorates the SREBP2 up-regulation induced by thyroid-stimulating hormone | Liu et al. (2015) |
AMPK has been classically defined as a master regulator of cellular metabolism in which the activation of AMPK by energetic stress leads to an overall increase in catabolic processes. These catabolic reactions serve to breakdown nutrients for the generation of ATP. Simultaneously, AMPK shuts down a diverse range of anabolic processes to conserve ATP levels. Hence, many of AMPK’s direct actions lead to an inhibition of proliferation as a way to deal with the energetic stress. This would be consistent with a role for AMPK as an initial tumor suppressor. For example, AMPK can phosphorylate wild-type p53 on serine-15 to potentiate its activity as a tumor suppressor, increasing p21 levels and causing a G1/S cell cycle arrest193,194. But while sustained AMPK causes wild-type p53-mediated cellular senescence, transient AMPK activation promotes cell survival following glucose starvation, consistent with a context-dependent, oncogenic role for AMPK194. Likewise, AMPK has been shown to phosphorylate and potentiate the transcriptional activity of FOXO3179,180. FOXO3 is a transcription factor that often functions as a tumor suppressor, but can also help manage metabolic stress214–216. Further, increased AMPK activity led to the phosphorylation of threonine-198 of the cell cycle inhibitor p2726. While phosphorylation caused a stabilization of p27, it enabled survival during starvation and/or metabolic stress through the induction of autophagy. However, it is not clear whether this signaling cascade would be present in prostate cancer since it required LKB1 and, as described above, LKB1 does not appear to be the dominant AMPK kinase in the prostate. In addition, the existence of mutations and/or deletions in several of these tumor suppressors such as p53, suggest that many of these tumor suppressive signals may not even exist in advanced prostate cancer.
One of the first described activities of AMPK is its ability to regulate lipid metabolism164,217,218. This occurred by the phosphorylation and inhibition of several proteins such as acetyl Co-A carboxylase 1 (ACC1), ACC2, 3-hydroxy-3-methyl-glutaryl-CoA (HMGCR), and the lipogenic transcription factors sterol regulatory element binding proteins-1 and -2 (SREBP-1 and 2) and hepatocyte nuclear factor 4A (HNF4A)164,217–220. The exact biological effect is determined by which specific proteins are targeted. For example, phosphorylation/inhibition of ACC1 blocks de novo fatty acid synthesis, while inhibition of the related isoform ACC2 increases fatty oxidation. Recently, AMPK was demonstrated to promote cancer under conditions of matrix detachment or glucose deprivation by inhibiting ACC1 and ACC2, resulting in the maintenance and production of pro-tumorigenic NAPDH levels, respectively32. Regardless, it is not clear how AMPK signaling through any of these downstream targets such as ACC1 will impact prostate cancer. This is because many of the inhibitory effects of AMPK, such as AMPK-mediated phosphorylation and inhibition of ACC1, are overridden by AR signaling. Increased lipogenesis is one of the hallmarks of prostate cancer221. AR increases the expression of several enzymes involved in de novo lipogenesis including fatty acid synthase, ATP-citrate lyase, HMGCR, ACC and farnesyl diphosphate synthase through the increased expression of the SREBF1 (encodes SREBP-1), SREBF2 (encodes SREBP-2) and SCAP, which further activates the SREBPs221,222. Thus, when AR signaling is present, as is the case in most prostate cancers, cells can simultaneously maintain AMPK signaling and pro-tumorigenic lipogenesis.
Prostate cancer is metabolically unique compared to many other cancer types. Relative to benign prostate, prostate cancer exhibits increased fatty acid and glucose oxidation223–225. This enhanced TCA cycle flux paradoxically occurs despite the above-described accumulation of intracellular lipid levels. Increased TCA cycle flux is now know to be in part caused by decreased levels of zinc in the transformed prostate cell223,226–228. The decreased zinc leads to a derepression of the enzyme aconitase, facilitating the forward metabolism of substrates through the cycle. In prostate cancer, this process can also be augmented by the AMPK-mediated induction of peroxisome-proliferator-activated receptor γ coactivator 1α (PGC-1α), a master regulator of mitochondrial biogenesis33,172. Importantly, this entire cascade can be activated by AR33. The precise mechanism of AMPK’s induction of PGC-1α expression is not known and may involve both direct and indirect effects33,172,204. There may also be other mechanisms by which AR-mediated AMPK signaling increases fatty acid oxidation in prostate cancer28,32,178,192. Curiously, there are additional AMPK targets that are known to regulate mitochondrial turnover (ex. mitophagy) and function (ex. fragmentation), suggesting that AMPK signaling may simultaneously promote the breakdown of old mitochondria and the synthesis of new mitochondria, perhaps improving cellular function189,201,213. To what degree this occurs in prostate cancer is currently not known.
In addition to alterations in mitochondrial metabolism, AMPK can modulate other aspects of sugar metabolism. For example, AMPK can directly phosphorylate two of the four isoenzymes of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphate 2-phosphatase (PFKFB), an enzyme that represents the rate-limiting step of glycolysis40,173,174,229,230. These phosphorylation events on PFKFB2 and PFKFB3 increase their kinase activity and thus promote forward flux through glycolysis. In prostate cancer, there is likely an additional level of regulation because AR signaling increases the expression of PFKFB2231. PFKFB2 has also been demonstrated to be phosphorylated and activated at the same AMPK-target site in response to oncogenic PI3K-Akt signaling100,232. While AR-AMPK-PFKFB signaling is thought to occur in prostate cancer35, the extent each isoform is stimulated by AR and/or AMPK, how the isoforms are regulated, and their functional roles are incompletely defined.
Beyond its role in glycolysis, AMPK may also have a more general function in glucose uptake. While not yet shown, it is possible that AMPK-mediated processes identified in other tissues may have relevance in prostate cancer. For example, AMPK is known to induce the translocation of the glucose transporter, GLUT4, in muscle and fat153,156,233,234. This occurs through the direct phosphorylation and regulation of TBC1D1 and TBC1D4/AS160, molecules that control vesicle trafficking. Additionally, AMPK has been reported to increase GLUT1 levels through a variety of mechanisms209–211. But like with the regulation of PFKFB2, other oncogenic cues can influence glucose uptake, and as such need to be considered. To that end, PI3K-Akt signaling can increase glucose transporter translocation and function in other cancers235. Similarly, GLUT1 levels can be stimulated by MYC, another commonly amplified oncogene in prostate cancer100,235. In addition, both PI3K-Akt and Myc can increase the expression of HK2, the first step of glycolysis and hence may further augment glucose uptake and metabolism236,237.
Autophagy is a cellular recycling process that is increased following AMPK activation. Similar to AMPK, initial research first defined autophagy as a tumor suppressive process238,239. Also like AMPK, a number of recent studies have identified an oncogenic role for autophagy, particularly in the late stages of the disease240,241. These findings extend to prostate cancer where autophagy has been implicated in disease progression30,242–244. Despite initial indications of a functional role for autophagy in prostate cancer, it is still not clear how this process is used during the different stages of the disease or how it is regulated. For example, there are discrepancies regarding AR’s regulation of autophagy30,242,243,245. This is somewhat surprising given AR’s robust induction of AMPK in prostate cancer33–36,130. The discrepancies may be due to variations in the stimuli duration (ex. sustained versus transient), use of indirect or non-selective modulators of AMPK and autophagy such as 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR), metformin, chloroquine, etc.) and treatment conditions. Regarding the last point, most experiments that are performed to examine the effects of hormones are done using media that contains charcoal-stripped serum, a condition commonly referred to as “androgen-starved”. While the charcoal-stripping of serum indeed removes most androgens, it also removes many other steroid hormones and peptide growth factors that could affect AMPK and autophagy. To that end, it is worth noting that switching AR-negative PC-3 prostate cancer cells from regular serum-containing media to the “androgen-starved”, charcoal-stripped serum containing media activates AMPK and autophagy in an AR-independent manner (unpublished data). Clearly, additional studies are needed using more sophisticated molecular and genetic approaches to help resolve these discrepancies.
AMPK can directly increase autophagy through the phosphorylation of ULK1 and possibly ULK2188,189,246. Recently, AMPK was also shown to increase autophagy via the direct phosphorylation of VPS34 and Beclin-1169. Moreover, AMPK can indirectly increase autophagy through decreasing mTOR signaling, an inhibitor of autophagy. This inhibition of mTOR, and thus derepression of autophagy, is thought to occur predominantly through two mechanisms. First, AMPK can directly phosphorylate the mTOR adaptor protein raptor, inducing the binding of 14-3-3 to raptor and inhibiting the mTORC1 complex171. Second, AMPK can directly phosphorylate TSC2 to potentiate its repressive effects on mTORC1170. AMPK may also regulate mTOR signaling through the phosphorylation and inhibition of upstream components of the PI3K-Akt-mTOR pathway such as insulin receptor substrate-1177. However, as described above, this later regulation may not be prevalent in prostate cancer given the propensity of high PI3K/Akt signaling in advanced prostate cancers. In addition, it may be difficult to predict AMPK’s effects on mTOR signaling in prostate cancer given the unusual crosstalk between the two pathways.
In contrast to what has been described in conventional physiology, AMPK and mTOR signaling can simultaneously occur in prostate cancer33,34,247,248. Thus, caution should be used when extrapolating results from basic biology, or even other cancer types, regarding AMPK and mTOR in prostate cancer. Why some crosstalk that occurs in other tissues does not occur in prostate cancer is not entirely clear but recent evidence is beginning to address how this may mechanistically occur. In prostate cancer, AR signaling increases the expression of a splice variant of TSC2, termed TSC2A, that cannot block mTOR signaling249. This results in a unique situation in which the cancer cell can concurrently activate two normally opposing signaling pathways. Hence, it may be possible that prostate cells can enjoy the pro-cancer benefits of both pathways.
While AMPK is defined as a master regulator of cellular metabolism, new findings indicate AMPK may have several non-metabolic roles that could impact processes of pathological importance in cancer. For example, AMPK appears to have an important role in mitosis through several mechanisms including targeting components of the mitotic spindle assembly, regulating the breakdown and assembly of the Golgi apparatus, and modulating the cytoskeleton159,198,207,250–252. While these effects may at first appear contrary to the induction of tumor suppressive factors such as p53 (described above), collectively these events may function as a protective checkpoint, assuring that cell cycle progression does not occur prematurely. This type of “cautious” signaling would be in contrast to other reports of more direct oncogenic roles for AMPK such as acting as an essential downstream effector of oncogenic HRasV12 or Pten deletion that functions to directly phosphorylate and inhibit the tumor suppressor retinoblastoma (Rb) protein24. To what extent this occurs in advanced prostate cancer, where Rb is often mutated or lost, is unknown253.
Finally, one of the emerging areas of interest is AMPK’s actions in the nucleus. While early reports of AMPK functions largely pertained to cytoplasmic AMPK activity, it is now evident that this kinase has additional roles in transcription and epigenetics. To that end, AMPK can regulate the activity of several transcription factors and transcriptional coregulators such as HNF-4α, cAMP-response element binding protein (CREB), FOXO3, PGC-1α, CRTC2 and class IIa histone deacetylases33,162,172,178,179,199,200,203,219,254–258. In addition, AMPK was reported to interrupt the association of nuclear receptors with the coactivator p30093,185. This could have important implications in prostate cancer given the key role of AR in the disease. Correspondingly, AMPK was reported to inhibit AR activity15. However, this study appears to be in direct contrast to data from Karacosta et al indicating that CaMKK2 potentiates AR activity in prostate cancer36. In our hands, we have been unable to detect either agonistic or antagonistic effects of CaMKK2 and/or AMPK on AR activity with the caveat that we have only explored a limited gene set (unpublished data). Hence, differences between groups may be attributable to variations in the subsets of AR-target genes being regulated. Certainly, this is an area that needs further investigation.
The predominant view of those supporting a context-dependent role for AMPK suggest that AMPK first acts as a tumor suppressor early in tumorigenesis and then later shifts towards a more oncogenic role in the advanced stages of the disease, contributing to therapy resistance and cancer reoccurrence3,98,119,259–268. Early on, AMPK would be activated in response to inhibitory mutations in tumor suppressor genes or gain-of-function events in known oncogenic pathways. This would lead to the classic AMPK-mediated catabolic functions including inhibition of oncogenic mTOR signaling and/or lipogenesis as well as regulation of the cell cycle. As the tumor evolves, the cancer cells encounter various stresses such as hypoxia, matrix detachment and starvation in addition to chemotherapies that also increase cellular stress. At this advanced stage, AMPK is hypothesized to drive cancer progression by promoting metabolic plasticity, resistance to cellular stress and thus, cell survival.
While the early/tumor suppressive and late/oncogenic paradigm could indeed be true for a number of cancers, prostate cancer may be unique. Contrary to many other cancers, clinical data suggest that AMPK activity is increased in both early and late disease stages7,33,37,268. In addition, as described above, many of the classic tumor suppressive functions of AMPK are overridden in prostate cancer by other canonical signaling pathways. For example, AR’s ability to increase mTOR signaling and lipogenesis in the presence of AMPK signaling (described above), may negate two of the major tumor suppressive networks. However, it should be noted that prostate cancer is an extremely heterogeneous disease269. As such, broad generalizations may lead to inaccuracies. Future studies are required to better characterize AMPK’s disease stage-specific roles and whether these roles vary amongst prostate cancer subtypes.
Targeting AMPK
Several strategies are currently being employed to modulate AMPK activity in cancer. However, it is unclear whether agonists or antagonists should be used given AMPK’s context-dependent role (oncogenic versus tumor suppressive). Further, the important roles of AMPK in numerous physiological processes may ultimately preclude AMPK itself from ever being a viable therapeutic target since alteration of this key signaling molecule will likely have multiple side effects. Described below are efforts to activate or inhibit AMPK activity and special considerations that will need to be considered moving forward.
Interest in targeting AMPK started when retrospective studies of diabetic patients taking the biguanide drug metformin reported decreased risks of a variety of cancer types including prostate cancer59–62. These were extremely exciting reports since metformin was an ideal candidate drug for repurposing as it is cheap, widely available, easy to use and safe at the concentrations used to treat diabetes. But as described above, recent retrospective as well as new prospective studies have called into question the anticancer effects of metformin in prostate cancer (66–78 and (NCT01433913)). Debate continues in part because metformin’s potential anticancer mechanism of action is still not clear.
In general, it is thought that the effects of metformin on cancer can be through two mechanisms categorized under indirect or direct effects. The most obvious indirect effect would result from metformin’s ability to reduce circulating insulin levels, a known mitogen and anti-apoptotic signal for some cancers270. What is not known in this regard is whether the metformin-induced changes in insulin levels would be significant enough, particularly in nondiabetics, to alter tumor biology271. In contrast, numerous preclinical studies have shown direct tumor suppressive effects in in vitro and in vivo models272,273. One of the major concerns with ongoing clinical trials was the question of whether a high enough concentration of metformin could reach the tumor cell to have direct anti-tumor effects. This depends both on the bioavailability of the drug and cellular uptake. Doses of metformin that are used to treat diabetic patients achieve plasma concentrations in the portal vein, where the drug is first absorbed and shuttled to, between 40–70 μM274. After liver uptake (note, metformin is not metabolized in animals or humans and is eliminated by the kidneys unchanged), systemic plasma concentrations drop to ~10–40 μM275. The micromolar concentrations used to treat diabetics are clearly enough to decrease glucose production in the liver, likely functioning in part through AMPK. However, there is recent debate even in this regard276,277. An initial concern in the field was that the majority of in vitro studies required higher concentrations of metformin (i.e. millimolar range) to inhibit the respiratory chain complex 1 and have tumor suppressive effects278. This would present a problem for the model that metformin has direct tumor suppressive effects. In this case, high enough drug concentrations could not reach the tumor and would indicate any potential anti-cancer effects would have to be mediated through an indirect mechanism. If one did want direct tumor suppressive effects, new clinical trials using higher metformin doses would have to be considered. However, this might defeat one of the major benefits of repurposing metformin, its very safe clinical profile at the doses used to treat diabetics as higher doses may result in dangerous side effects such as lactic acidosis. Regardless, two brief letters were recently (April 2016) published back-to-back in Cell Metabolism that suggest the current doses being used in preclinical animal models may be sufficient to model effective clinical doses279,280.
Dowling et al used liquid chromatography-mass spectrometry to demonstrate that in a xenograft model of colorectal cancer, mice given drinking water with metformin dissolved into it at a concentration of 5 mg/ml (comparable to what has previously been used in similar studies), plasma and, surprisingly, tumor concentrations of metformin reached ~30 μM, a concentration that was sufficient to increase AMPK activity (assessed by AMPK Thr172 phosphorylation)279. It was not reported whether metformin-treated mice also exhibited decreased tumors in this experiment. To achieve similar phospho-AMPK Thr172 levels in cell culture, HCT116 colon cancer cells had to be treated with 10–20 mM (>300 fold increase) metformin. These results indicate that metformin is much more readily taken up in vivo than in vitro. Interestingly, samples taken from nondiabetic breast cancer patients given metformin exhibited considerably lower plasma metformin levels (~2.8 μM) than those from diabetic patients (up to 25 μM), suggesting higher doses may be needed for anti-cancer efficacy particularly in non-diabetic patients. These results again call into question a role for metformin’s reported direct antitumor effects in previous clinical studies. If high enough concentrations could not be reached in patient tumors, how could the reported beneficial effects of metformin be due to direct actions on the tumor?
Chandel et al performed a similar study using the HCT116 colon cancer xenograft model but with a 1.25 mg/ml solution of metformin280, a concentration the investigators previously had shown to inhibit tumor growth in this model (a functional role for AMPK was never tested). Here, they detected both plasma and tumor metformin concentrations in the range of 3.2–12.4 μM, levels that can be achieved in routine diabetes treatment281. In addition, it was noted that due to its cationic nature, metformin is predicted to accumulate 100- to 500-fold in the mitochondria due to the membrane potential. Collectively, these results indicate that the doses currently being used in ongoing clinical trails are reasonable. However, this still leaves the question that if doses currently given to diabetic patients are sufficient to block mitochondrial activity in patient tumors, then why did the previous epidemiological studies not consistently demonstrate anticancer effects? More importantly, why was no survival benefit observed following the first formal blinded clinical trail of metformin even though plasma drug levels were in the micromolar range282? Certainly further work is needed in this area. It will be particularly important to determine whether isolated mitochondria from treated patients indeed do have significantly enriched metformin levels and whether additional biological factors like the expression of membrane transporters influence drug efficacy.
The hydrophilic nature of metformin likely prevents it from passively diffusing through plasma membranes. Hence, cellular uptake is controlled by cationic transporters such as OCT1, OCT2 and OCT3 (encoded by the genes SLC22A1, SLC22A2 and SLC22A3 respectively)283. OCT3/SLC22A3 is highly expressed in the prostate indicating that metformin uptake would not be an issue284. But interestingly, low SLC22A3 expression is a strong predictor of poor prognosis in prostate cancer patients285–288. Whether changes in SLC22A3 expression could in part explain some of the differences between the contrasting retrospective analyses of metformin’s effects on prostate cancer remains to be determined. One would predict that tumors with low OCT3 levels would have decreased sensitivity to metformin. As risk allele variants associated with SLC22A3 expression are known285,286,288,289, these could be screened prior to selection of patients for future clinical trials.
Despite the controversies surrounding the clinical studies, enthusiasm for metformin is still high due to preclinical studies demonstrating that the drug has potent therapeutic effects across a broad range of cancers290. Researchers have hypothesized that the drug’s anticancer effects could be mediated through AMPK because of work demonstrating that metformin can increase AMPK activity in cells20,220,291–293. Metformin, like other biguanides such as phenformin, can indirectly activate AMPK by inhibiting complex I of the electron transport chain51,63,276,278,294,295. This subsequently causes cellular energy stress, increasing the AMP/ATP ratio that could lead to an activation of AMPK complexes throughout the cell. Further studies pointed to an additional metformin-initiated, genotoxic activation of AMPK296. But the exact mechanism of this effect has been called into question297,298. Like metformin, a number of other activators of AMPK, including synthetic (ex. phenformin, AICAR, rosiglitazone, 2-deoxyglucose, etc) and natural (ex. salicylate, resveratrol, berberine, etc) compounds, can inhibit cancer cell growth, migration and/or invasion1,7,20,293,299,300. In contrast, AICAR was able to rescue the inhibition of prostate cancer cell proliferation caused by the molecular or pharmacological inhibition of CaMKK235. It is important to note that many of these compounds such as metformin and AICAR have been demonstrated to be highly nonspecific and clearly have tumor suppressive (and possibly oncogenic) properties independent of AMPK45,47,49,50,53,54,276. It is reasonable to speculate then that additional stress signaling pathways may be responsible for the decreased cellular growth following the onset of stress. In fact, induction of cell death by several AMPK “activators” can be exacerbated by knocking out AMPK, indicating that AMPK is often turned on in response to these drugs as a last ditch survival effort21–23,26,28–31,53,170,178. In this regard, use of these compounds in combination with inhibitors of AMPK could be warranted. While drugs like metformin may indeed have tumor suppressive effects under some contexts, this may be due more to systemic effects rather that direct targeting of the cancer cell itself. Genetic studies using conditional knockout and transgenic animals will be essential to parse out these mechanisms of action.
To avoid some of the uncertainties inherent to using indirect activators of AMPK, direct activators of AMPK have been identified and shown to have tumor suppressive properties in several cancers11,12,17,47. The exact AMPK binding sites for some of these compounds (ex. OSU-53, PT1) are incompletely defined11,13,14,301. Further, the off-target effects of these drugs are unclear55. One of the more recently developed direct activators, MT 63–78, was able to decrease the proliferation of prostate cancer cells in vitro as well as suppress tumor growth in vivo17. MT 63–78, similar to A-769662 and salicylates, allosterically activates AMPK by directly binding to the β1 regulatory subunit17,300,302. These results would appear at odds with the above-described clinical data and functional studies that suggest the β1 subunit is oncogenic101,104. This apparent conundrum may be due to differences in the duration of stimuli. For example, transient pulses of AMPK activity may only lead to the phosphorylation and modulation of the most sensitive downstream AMPK targets. Conversely, a sustained robust activation of AMPK, such as in the presence of pharmacological activation by MT 63–78 may hyperactivate a greater number of AMPK complexes, therefore modulating a larger set of downstream targets. In this latter case, the broad activation of downstream targets would have tumor suppressive effects such as increasing cellular stress.
In contrast to studies demonstrating AMPK activity blocks prostate cancer cell growth, antagonists of AMPK such as compound C inhibit prostate cancer cell proliferation and migration29–31,34,37. In addition to compound C, other inhibitors of AMPK have been identified. These include the kinase inhibitors sunitinib and midostaurin that have potent anticancer effects in vitro and in vivo in a variety of cancers303,304. However, similar to many of the AMPK agonists, these drugs have well known pleiotropic effects and hence likely also possess AMPK-independent tumor suppressive properties48,50,305,306.
Part of the rationale for the use of AMPK activators in cancer is AMPK’s known ability to inhibit oncogenic cellular processes such as de novo lipogenesis and mTOR signaling. However, inhibitors of lipogenesis and mTOR already exist raising the question “Could we instead target the specific oncogenic, downstream processes of AMPK signaling?” Furthermore, AMPK agonists may not have a strong effect in prostate cancer where AR and PI3K-Akt are commonly hyperactivated and increase lipogenesis and mTOR signaling and thus may override the inhibitory effects of AMPK on these processes. Conversely, inhibition of AMPK may augment lipogenesis and/or mTOR signaling. In this regard, combined treatment with inhibitors of AMPK and lipogenesis and/or mTOR may have utility. However, it remains to be determined whether such an approach would have severe side effects given the important roles of these processes throughout the body.
A key step that needs to occur in our understanding of AMPK’s actions in prostate cancer is the identification of the downstream targets of AMPK that are the drivers of the disease. Elucidation of these pathways may reveal better, and more selective, therapeutic targets. Alternatively, if we can identify the tissue- or disease-specific regulators of AMPK, these may also yield viable new targets. CaMKK2, as an upstream kinase of AMPK, may represent one such target. As highlighted above, CaMKK2 is increased in prostate cancer, has a restricted expression profile, and mediates several oncogenic processes. A cell-permeable inhibitor of CaMKK2, STO-609, has been available since 2002307. Correspondingly, STO-609 treatment reduced androgen-sensitive prostate cancer proliferation, migration and invasion as well as the growth of castration-resistant tumors34,35. Therefore, STO-609, or an STO-609-like compound with better pharmacokinetic and pharmacodynamic properties, may have therapeutic value in prostate cancer.
Conclusions
To understand AMPK’s role in prostate cancer, one must first identify 1) the type of stimulus (i.e. how AMPK is activated), 2) the composition of the heterotrimer(s) and 3) the downstream driver pathways that are being modulated. All three are interconnected. The type and duration of the stimulus will dictate which subcellular AMPK complexes are activated and for how long. The amount of AMPK complexes at each subcellular site will be influenced by the subunit composition. The location of the complexes may also be regulated by posttranslational modifications and/or additional anchoring mechanisms. At this time, it is poorly understood what determines the distribution of the complexes. Each distinct AMPK complex is going to be associated with a set of targets that can be phosphorylated when that particular complex is activated. One should also be cognizant that many of these downstream processes are influenced by other oncogenic signals such as AR and/or Myc. For instance, even though increased AMPK may lead to the phosphorylation of ACC1, an event known to block de novo lipogenesis, in most prostate cancers AR signaling likely overrides this blockade by increasing the expression of an entire network of enzymes involved in lipogenesis. Taken together, the upstream cues determine which specific AMPK complexes are activated and therefore what downstream biological processes will be modulated. An example would be the activation of AMPK by prolonged energetic stress (ex. metformin treatment) in contrast to the controlled activation of a subpopulation of AMPK by an upstream kinase such as CaMKK2. High levels of AMP caused by prolonged energetic stress would lead to the robust activation of a large number of AMPK complexes throughout the cell, modulating most of the known AMPK targets and processes. Compare this to the activation by CaMKK2, a major upstream kinase of AMPK in prostate cancer (described above). CaMKK2’s predominant cytoplasmic localization and requirement for a direct association with AMPK to phosphorylate and activate the protein indicates CaMKK2 will only increase the activity of a smaller, restricted set of AMPK complexes located primarily in the cytoplasm. This is exacerbated in prostate cancer where the α1 isoform of the catalytic subunit, which is more cytoplasmic88,89, is the predominant form33,34,99. Interestingly, to date the majority of AMPK’s tumor suppressive effects have been associated with its nuclear functions (ex. p53, p21 and p27 induction, p300 inhibition, etc). Hence, stimuli that favor the activation of non-nuclear AMPK complexes may favor the induction of more oncogenic processes.
Given AMPK’s ubiquitous and diverse roles throughout the body, we suspect that broadly targeting AMPK may not be a viable option in cancer. Further, the use of direct AMPK activators or inhibitors will likely suffer from the counterproductive activation of some AMPK-mediated oncogenic pathways and impairment of other AMPK-mediated tumor suppressive signals. We propose that the elucidation of downstream AMPK-mediated processes will 1) uncover the driver signaling events and 2) highlight new therapeutic targets. Alternatively, identification and targeting of the prostate cancer-specific upstream cascades that favor the activation of these downstream oncogenic events could have greater overall efficacy because it would impact multiple AMPK-mediated, pro-cancer processes. In addition, the tissue and disease-specific nature of the upstream signal would offer a unique therapeutic target to prostate cancer, potentially mitigating side effects. Finally, while the model of AMPK signaling outlined above is for prostate cancer, it is possible that the molecular concepts described here could be extended to explain AMPK actions in other cancers, diseases and even non-disease states.
Key points.
AMPK is a heterotrimer complex that can come in at least 12 different versions. The different AMPK complexes can have unique subcellular locations and activities.
Diverse upstream signals regulate different AMPK subcellular complexes.
While first identified as a master regulator of metabolism, AMPK may have numerous roles beyond metabolism.
AMPK can have context-dependent effects in cancer.
CaMKK2 appears to be the dominant upstream AMPK kinase in the prostate.
Most small molecule modulators of AMPK have known off-target effects.
Given its ubiquitous expression and varied roles throughout the body, directly targeting AMPK may present numerous on-target side effects.
Acknowledgments
The authors gratefully acknowledge members of the Frigo laboratory for helpful discussions and critical reading of this manuscript. This work was supported by NIH grant R01CA184208, American Cancer Society grant RSG-16-084-01 – TBE, Department of Defense/PCRP grant W81XWH-12-1-0204 and funding from the Golfers Against Cancer.
Biographies
Ayesha S. Khan is a graduate student at the Center for Nuclear Receptors and Cell Signaling at the University of Houston, Texas, USA. She received her Master of Science degree in bioengineering from Rice University, Houston, Texas, in 2011 where she was a Fulbright fellow. Her current research interests include the regulation of metabolic pathways by signal transduction in prostate cancer.
Daniel E. Frigo is an Assistant Professor within the Center for Nuclear Receptors and Cell Signaling and Department of Biology and Biochemistry at the University of Houston, Texas, USA. Prior to this position, he completed his training as a postdoctoral fellow at Duke University School of Medicine, Durham, North Carolina, USA. His laboratory is interested in molecular endocrinology, signal transduction and cancer metabolism. Specifically, his work is focused on the identification and therapeutic targeting of novel signaling pathways in prostate cancer.
Footnotes
Author contributions
A.S.K. and D.E.F. contributed equally to the manuscript. Both researched data for the manuscript, wrote the article and edited the manuscript prior to submission.
Competing interests statement
D.E.F. declares a familial association with Essa® (Essa Pharma, Inc). A.K. declares no competing interests.
References
- 1.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Viollet B, et al. AMPK: Lessons from transgenic and knockout animals. Front Biosci (Landmark Ed) 2009;14:19–44. doi: 10.2741/3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dasgupta B, Chhipa RR. Evolving Lessons on the Complex Role of AMPK in Normal Physiology and Cancer. Trends Pharmacol Sci. 2016;37:192–206. doi: 10.1016/j.tips.2015.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hawley SA, et al. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28. doi: 10.1186/1475-4924-2-28. This is one of three papers that first linked the tumor suppressor LKB1 to AMPK, thus implicating AMPK as a potential tumor suppressor. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Woods A, et al. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003;13:2004–2008. doi: 10.1016/j.cub.2003.10.031. This is one of three papers that first linked the tumor suppressor LKB1 to AMPK, thus implicating AMPK as a potential tumor suppressor. [DOI] [PubMed] [Google Scholar]
- 6.Shaw RJ, et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A. 2004;101:3329–3335. doi: 10.1073/pnas.0308061100. This is one of three papers that first linked the tumor suppressor LKB1 to AMPK, thus implicating AMPK as a potential tumor suppressor. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choudhury Y, et al. AMP-activated protein kinase (AMPK) as a potential therapeutic target independent of PI3K/Akt signaling in prostate cancer. Oncoscience. 2014;1:446–456. doi: 10.18632/oncoscience.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Phoenix KN, Devarakonda CV, Fox MM, Stevens LE, Claffey KP. AMPKalpha2 Suppresses Murine Embryonic Fibroblast Transformation and Tumorigenesis. Genes Cancer. 2012;3:51–62. doi: 10.1177/1947601912452883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Audet-Walsh E, et al. The PGC-1alpha/ERRalpha Axis Represses One-Carbon Metabolism and Promotes Sensitivity to Anti-folate Therapy in Breast Cancer. Cell Rep. 2016;14:920–931. doi: 10.1016/j.celrep.2015.12.086. [DOI] [PubMed] [Google Scholar]
- 10.Faubert B, et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 2013;17:113–124. doi: 10.1016/j.cmet.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee KH, et al. Targeting energy metabolic and oncogenic signaling pathways in triple-negative breast cancer by a novel adenosine monophosphate-activated protein kinase (AMPK) activator. J Biol Chem. 2011;286:39247–39258. doi: 10.1074/jbc.M111.264598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chou CC, et al. AMPK reverses the mesenchymal phenotype of cancer cells by targeting the Akt-MDM2-Foxo3a signaling axis. Cancer Res. 2014;74:4783–4795. doi: 10.1158/0008-5472.CAN-14-0135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Valtorta S, et al. A novel AMPK activator reduces glucose uptake and inhibits tumor progression in a mouse xenograft model of colorectal cancer. Invest New Drugs. 2014;32:1123–1133. doi: 10.1007/s10637-014-0148-8. [DOI] [PubMed] [Google Scholar]
- 14.Tripodi F, et al. Synthesis and biological evaluation of 1,4-diaryl-2-azetidinones as specific anticancer agents: activation of adenosine monophosphate activated protein kinase and induction of apoptosis. J Med Chem. 2012;55:2112–2124. doi: 10.1021/jm201344a. [DOI] [PubMed] [Google Scholar]
- 15.Jurmeister S, Ramos-Montoya A, Neal DE, Fryer LG. Transcriptomic analysis reveals inhibition of androgen receptor activity by AMPK in prostate cancer cells. Oncotarget. 2014;5:3785–3799. doi: 10.18632/oncotarget.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou J, et al. Inactivation of AMPK alters gene expression and promotes growth of prostate cancer cells. Oncogene. 2009;28:1993–2002. doi: 10.1038/onc.2009.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zadra G, et al. A novel direct activator of AMPK inhibits prostate cancer growth by blocking lipogenesis. EMBO Mol Med. 2014;6:519–538. doi: 10.1002/emmm.201302734. In this study, pharmacological activation of AMPK complexes by a novel direct AMPK agonist suppressed prostate cancer cell growth. In addition, genetic knockout of AMPKα2 lead to increased proliferation in a mouse model of prostatic hyperplasia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mo JS, et al. Cellular energy stress induces AMPK-mediated regulation of YAP and the Hippo pathway. Nat Cell Biol. 2015;17:500–510. doi: 10.1038/ncb3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang W, et al. AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat Cell Biol. 2015;17:490–499. doi: 10.1038/ncb3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Brien AJ, et al. Salicylate activates AMPK and synergizes with metformin to reduce the survival of prostate and lung cancer cells ex vivo through inhibition of de novo lipogenesis. Biochem J. 2015;469:177–187. doi: 10.1042/BJ20150122. [DOI] [PubMed] [Google Scholar]
- 21.Saito Y, Chapple RH, Lin A, Kitano A, Nakada D. AMPK Protects Leukemia-Initiating Cells in Myeloid Leukemias from Metabolic Stress in the Bone Marrow. Cell Stem Cell. 2015;17:585–596. doi: 10.1016/j.stem.2015.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laderoute KR, et al. 5′-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol Cell Biol. 2006;26:5336–5347. doi: 10.1128/MCB.00166-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song X, et al. AMP-activated protein kinase is required for cell survival and growth in HeLa-S3 cells in vivo. IUBMB Life. 2014;66:415–423. doi: 10.1002/iub.1279. [DOI] [PubMed] [Google Scholar]
- 24.Rios M, et al. AMPK activation by oncogenesis is required to maintain cancer cell proliferation in astrocytic tumors. Cancer Res. 2013;73:2628–2638. doi: 10.1158/0008-5472.CAN-12-0861. This study demonstrated that AMPK had oncogenic roles in Ras-driven brain tumors by phosphorylating and inactivating the tumor suppressor Rb, leading to increased cancer cell proliferation. [DOI] [PubMed] [Google Scholar]
- 25.Mendoza EE, et al. Control of Glycolytic Flux by AMP-Activated Protein Kinase in Tumor Cells Adapted to Low pH. Transl Oncol. 2012;5:208–216. doi: 10.1593/tlo.11319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liang J, et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9:218–224. doi: 10.1038/ncb1537. [DOI] [PubMed] [Google Scholar]
- 27.Pandhare J, Donald SP, Cooper SK, Phang JM. Regulation and function of proline oxidase under nutrient stress. J Cell Biochem. 2009;107:759–768. doi: 10.1002/jcb.22174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zaugg K, et al. Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev. 2011;25:1041–1051. doi: 10.1101/gad.1987211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jung SN, et al. Down-regulation of AMP-activated protein kinase sensitizes DU145 carcinoma to Fas-induced apoptosis via c-FLIP degradation. Exp Cell Res. 2009;315:2433–2441. doi: 10.1016/j.yexcr.2009.05.018. [DOI] [PubMed] [Google Scholar]
- 30.Chhipa RR, Wu Y, Ip C. AMPK-mediated autophagy is a survival mechanism in androgen-dependent prostate cancer cells subjected to androgen deprivation and hypoxia. Cell Signal. 2011;23:1466–1472. doi: 10.1016/j.cellsig.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chhipa RR, Wu Y, Mohler JL, Ip C. Survival advantage of AMPK activation to androgen-independent prostate cancer cells during energy stress. Cell Signal. 2010;22:1554–1561. doi: 10.1016/j.cellsig.2010.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jeon SM, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012;485:661–665. doi: 10.1038/nature11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tennakoon JB, et al. Androgens regulate prostate cancer cell growth via an AMPK-PGC-1alpha-mediated metabolic switch. Oncogene. 2014;33:5251–5261. doi: 10.1038/onc.2013.463. This article demonstrates that androgen-mediated AMPK signaling supports prostate cancer proliferation by increasing glycolysis, glucose and fatty acid oxidation and that AMPK activity tracks with clinical prostate cancer progression. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frigo DE, et al. CaM kinase kinase beta-mediated activation of the growth regulatory kinase AMPK is required for androgen-dependent migration of prostate cancer cells. Cancer Res. 2011;71:528–537. doi: 10.1158/0008-5472.CAN-10-2581. This was the first study to describe a potential oncogenic role for the CaMKK2-AMPK signaling cascade in prostate cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Massie CE, et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011;30:2719–2733. doi: 10.1038/emboj.2011.158. In this article, CaMKK2 was identified as a major modulator of the androgen-dependent changes in prostate cancer metabolism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karacosta LG, Foster BA, Azabdaftari G, Feliciano DM, Edelman AM. A regulatory feedback loop between Ca2+/calmodulin-dependent protein kinase kinase 2 (CaMKK2) and the androgen receptor in prostate cancer progression. J Biol Chem. 2012;287:24832–24843. doi: 10.1074/jbc.M112.370783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park HU, et al. AMP-activated protein kinase promotes human prostate cancer cell growth and survival. Mol Cancer Ther. 2009;8:733–741. doi: 10.1158/1535-7163.MCT-08-0631. This study demonstrated that AMPK increased prostate cancer cell proliferation and that increased AMPK activity correlated with prostate cancer in patients. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hart PC, et al. MnSOD upregulation sustains the Warburg effect via mitochondrial ROS and AMPK-dependent signalling in cancer. Nat Commun. 2015;6:6053. doi: 10.1038/ncomms7053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nardo G, et al. Glycolytic phenotype and AMP kinase modify the pathologic response of tumor xenografts to VEGF neutralization. Cancer Res. 2011;71:4214–4225. doi: 10.1158/0008-5472.CAN-11-0242. [DOI] [PubMed] [Google Scholar]
- 40.Domenech E, et al. AMPK and PFKFB3 mediate glycolysis and survival in response to mitophagy during mitotic arrest. Nat Cell Biol. 2015;17:1304–1316. doi: 10.1038/ncb3231. [DOI] [PubMed] [Google Scholar]
- 41.Laderoute KR, et al. 5′-AMP-activated protein kinase (AMPK) supports the growth of aggressive experimental human breast cancer tumors. J Biol Chem. 2014;289:22850–22864. doi: 10.1074/jbc.M114.576371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ng TL, et al. The AMPK stress response pathway mediates anoikis resistance through inhibition of mTOR and suppression of protein synthesis. Cell Death Differ. 2012;19:501–510. doi: 10.1038/cdd.2011.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fernandez MR, Henry MD, Lewis RE. Kinase suppressor of Ras 2 (KSR2) regulates tumor cell transformation via AMPK. Mol Cell Biol. 2012;32:3718–3731. doi: 10.1128/MCB.06754-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Possik E, et al. Folliculin regulates ampk-dependent autophagy and metabolic stress survival. PLoS Genet. 2014;10:e1004273. doi: 10.1371/journal.pgen.1004273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vincent EE, et al. Differential effects of AMPK agonists on cell growth and metabolism. Oncogene. 2015;34:3627–3639. doi: 10.1038/onc.2014.301. This study demonstrated that several known AMPK activators impair cell proliferation independently of AMPK. Conversely, this study also reported that the direct AMPK activator, A-769662, increased proliferation in an AMPK-dependent manner. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guigas B, et al. Beyond AICA riboside: in search of new specific AMP-activated protein kinase activators. IUBMB Life. 2009;61:18–26. doi: 10.1002/iub.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu X, et al. Discrete mechanisms of mTOR and cell cycle regulation by AMPK agonists independent of AMPK. Proc Natl Acad Sci U S A. 2014;111:E435–444. doi: 10.1073/pnas.1311121111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu X, Chhipa RR, Nakano I, Dasgupta B. The AMPK inhibitor compound C is a potent AMPK-independent antiglioma agent. Mol Cancer Ther. 2014;13:596–605. doi: 10.1158/1535-7163.MCT-13-0579. This study demonstrated that the commonly used antagonist of AMPK, compound C, has significant off-target effects. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Santidrian AF, et al. AICAR induces apoptosis independently of AMPK and p53 through up-regulation of the BH3-only proteins BIM and NOXA in chronic lymphocytic leukemia cells. Blood. 2010;116:3023–3032. doi: 10.1182/blood-2010-05-283960. [DOI] [PubMed] [Google Scholar]
- 50.Bain J, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hardie DG. Neither LKB1 nor AMPK are the direct targets of metformin. Gastroenterology. 2006;131:973. doi: 10.1053/j.gastro.2006.07.032. author reply 974–975. [DOI] [PubMed] [Google Scholar]
- 52.Akinyeke T, et al. Metformin targets c-MYC oncogene to prevent prostate cancer. Carcinogenesis. 2013;34:2823–2832. doi: 10.1093/carcin/bgt307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Griss T, et al. Metformin Antagonizes Cancer Cell Proliferation by Suppressing Mitochondrial-Dependent Biosynthesis. PLoS Biol. 2015;13:e1002309. doi: 10.1371/journal.pbio.1002309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kalender A, et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010;11:390–401. doi: 10.1016/j.cmet.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moreno D, Knecht E, Viollet B, Sanz P. A769662, a novel activator of AMP-activated protein kinase, inhibits non-proteolytic components of the 26S proteasome by an AMPK-independent mechanism. FEBS Lett. 2008;582:2650–2654. doi: 10.1016/j.febslet.2008.06.044. [DOI] [PubMed] [Google Scholar]
- 56.Liu L, et al. Deregulated MYC expression induces dependence upon AMPK-related kinase 5. Nature. 2012;483:608–612. doi: 10.1038/nature10927. [DOI] [PubMed] [Google Scholar]
- 57.Yan M, et al. The tumor suppressor folliculin regulates AMPK-dependent metabolic transformation. J Clin Invest. 2014;124:2640–2650. doi: 10.1172/JCI71749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Daurio NA, et al. AMPK activation and metabolic reprogramming by tamoxifen through estrogen receptor-independent mechanisms suggests new uses for this therapeutic modality in cancer treatment. Cancer Res. 2016 doi: 10.1158/0008-5472.CAN-15-2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Margel D, et al. Metformin use and all-cause and prostate cancer-specific mortality among men with diabetes. J Clin Oncol. 2013;31:3069–3075. doi: 10.1200/JCO.2012.46.7043. [DOI] [PubMed] [Google Scholar]
- 60.Wright JL, Stanford JL. Metformin use and prostate cancer in Caucasian men: results from a population-based case-control study. Cancer Causes Control. 2009;20:1617–1622. doi: 10.1007/s10552-009-9407-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Murtola TJ, Tammela TL, Lahtela J, Auvinen A. Antidiabetic medication and prostate cancer risk: a population-based case-control study. Am J Epidemiol. 2008;168:925–931. doi: 10.1093/aje/kwn190. [DOI] [PubMed] [Google Scholar]
- 62.Spratt DE, et al. Metformin and prostate cancer: reduced development of castration-resistant disease and prostate cancer mortality. Eur Urol. 2013;63:709–716. doi: 10.1016/j.eururo.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ben Sahra I, et al. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008;27:3576–3586. doi: 10.1038/sj.onc.1211024. [DOI] [PubMed] [Google Scholar]
- 64.Xiang X, Saha AK, Wen R, Ruderman NB, Luo Z. AMP-activated protein kinase activators can inhibit the growth of prostate cancer cells by multiple mechanisms. Biochem Biophys Res Commun. 2004;321:161–167. doi: 10.1016/j.bbrc.2004.06.133. [DOI] [PubMed] [Google Scholar]
- 65.Zakikhani M, Dowling RJ, Sonenberg N, Pollak MN. The effects of adiponectin and metformin on prostate and colon neoplasia involve activation of AMP-activated protein kinase. Cancer Prev Res (Phila) 2008;1:369–375. doi: 10.1158/1940-6207.CAPR-08-0081. [DOI] [PubMed] [Google Scholar]
- 66.Allott EH, et al. Metformin does not affect risk of biochemical recurrence following radical prostatectomy: results from the SEARCH database. Prostate Cancer Prostatic Dis. 2013;16:391–397. doi: 10.1038/pcan.2013.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rieken M, et al. Association of diabetes mellitus and metformin use with biochemical recurrence in patients treated with radical prostatectomy for prostate cancer. World J Urol. 2013 doi: 10.1007/s00345-013-1171-7. [DOI] [PubMed] [Google Scholar]
- 68.Wang SY, et al. Metformin and the incidence of cancer in patients with diabetes: a nested case-control study. Diabetes Care. 2013;36:e155–156. doi: 10.2337/dc13-0708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Soranna D, et al. Cancer risk associated with use of metformin and sulfonylurea in type 2 diabetes: a meta-analysis. Oncologist. 2012;17:813–822. doi: 10.1634/theoncologist.2011-0462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Azoulay L, Dell’Aniello S, Gagnon B, Pollak M, Suissa S. Metformin and the incidence of prostate cancer in patients with type 2 diabetes. Cancer Epidemiol Biomarkers Prev. 2011;20:337–344. doi: 10.1158/1055-9965.EPI-10-0940. [DOI] [PubMed] [Google Scholar]
- 71.Bensimon L, Yin H, Suissa S, Pollak MN, Azoulay L. The use of metformin in patients with prostate cancer and the risk of death. Cancer Epidemiol Biomarkers Prev. 2014;23:2111–2118. doi: 10.1158/1055-9965.EPI-14-0056. [DOI] [PubMed] [Google Scholar]
- 72.Currie CJ, Poole CD, Gale EA. The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia. 2009;52:1766–1777. doi: 10.1007/s00125-009-1440-6. [DOI] [PubMed] [Google Scholar]
- 73.Kaushik D, et al. Effect of metformin on prostate cancer outcomes after radical prostatectomy. Urol Oncol. 2014;32:43 e41–47. doi: 10.1016/j.urolonc.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Margel D, et al. Association between metformin use and risk of prostate cancer and its grade. J Natl Cancer Inst. 2013;105:1123–1131. doi: 10.1093/jnci/djt170. [DOI] [PubMed] [Google Scholar]
- 75.Patel T, Hruby G, Badani K, Abate-Shen C, McKiernan JM. Clinical outcomes after radical prostatectomy in diabetic patients treated with metformin. Urology. 2010;76:1240–1244. doi: 10.1016/j.urology.2010.03.059. [DOI] [PubMed] [Google Scholar]
- 76.Tsilidis KK, et al. Metformin does not affect cancer risk: a cohort study in the U.K. Clinical Practice Research Datalink analyzed like an intention-to-treat trial. Diabetes Care. 2014;37:2522–2532. doi: 10.2337/dc14-0584. [DOI] [PubMed] [Google Scholar]
- 77.Feng T, et al. Metformin use and risk of prostate cancer: results from the REDUCE study. Cancer Prev Res (Phila) 2015;8:1055–1060. doi: 10.1158/1940-6207.CAPR-15-0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Joshua AM, et al. A pilot ‘window of opportunity’ neoadjuvant study of metformin in localised prostate cancer. Prostate Cancer Prostatic Dis. 2014;17:252–258. doi: 10.1038/pcan.2014.20. [DOI] [PubMed] [Google Scholar]
- 79.Stapleton D, et al. Mammalian AMP-activated protein kinase subfamily. J Biol Chem. 1996;271:611–614. doi: 10.1074/jbc.271.2.611. [DOI] [PubMed] [Google Scholar]
- 80.Woods A, et al. Characterization of AMP-activated protein kinase beta and gamma subunits. Assembly of the heterotrimeric complex in vitro. J Biol Chem. 1996;271:10282–10290. doi: 10.1074/jbc.271.17.10282. [DOI] [PubMed] [Google Scholar]
- 81.Suter M, et al. Dissecting the role of 5′-AMP for allosteric stimulation, activation, and deactivation of AMP-activated protein kinase. J Biol Chem. 2006;281:32207–32216. doi: 10.1074/jbc.M606357200. [DOI] [PubMed] [Google Scholar]
- 82.Xiao B, et al. Structural basis of AMPK regulation by small molecule activators. Nat Commun. 2013;4:3017. doi: 10.1038/ncomms4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hawley SA, et al. 5′-AMP activates the AMP-activated protein kinase cascade, and Ca2+/calmodulin activates the calmodulin-dependent protein kinase I cascade, via three independent mechanisms. J Biol Chem. 1995;270:27186–27191. doi: 10.1074/jbc.270.45.27186. [DOI] [PubMed] [Google Scholar]
- 84.Davies SP, Helps NR, Cohen PT, Hardie DG. 5′-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2C alpha and native bovine protein phosphatase-2AC. FEBS Lett. 1995;377:421–425. doi: 10.1016/0014-5793(95)01368-7. [DOI] [PubMed] [Google Scholar]
- 85.Oakhill JS, et al. AMPK is a direct adenylate charge-regulated protein kinase. Science. 2011;332:1433–1435. doi: 10.1126/science.1200094. [DOI] [PubMed] [Google Scholar]
- 86.Hardie DG. AMPK–sensing energy while talking to other signaling pathways. Cell Metab. 2014;20:939–952. doi: 10.1016/j.cmet.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Towler MC, Hardie DG. AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res. 2007;100:328–341. doi: 10.1161/01.RES.0000256090.42690.05. [DOI] [PubMed] [Google Scholar]
- 88.Turnley AM, et al. Cellular distribution and developmental expression of AMP-activated protein kinase isoforms in mouse central nervous system. J Neurochem. 1999;72:1707–1716. doi: 10.1046/j.1471-4159.1999.721707.x. [DOI] [PubMed] [Google Scholar]
- 89.Salt I, et al. AMP-activated protein kinase: greater AMP dependence, and preferential nuclear localization, of complexes containing the alpha2 isoform. Biochem J. 1998;334(Pt 1):177–187. doi: 10.1042/bj3340177. This study showed that the two different AMPK alpha catalytic subunits have distinct subcellular locations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.McGee SL, et al. Exercise increases nuclear AMPK alpha2 in human skeletal muscle. Diabetes. 2003;52:926–928. doi: 10.2337/diabetes.52.4.926. [DOI] [PubMed] [Google Scholar]
- 91.Schmidt MC, McCartney RR. beta-subunits of Snf1 kinase are required for kinase function and substrate definition. EMBO J. 2000;19:4936–4943. doi: 10.1093/emboj/19.18.4936. This study demonstrated in yeast that the AMPK beta subunits can define which downstream targets AMPK will phosphorylate/regulate. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Warden SM, et al. Post-translational modifications of the beta-1 subunit of AMP-activated protein kinase affect enzyme activity and cellular localization. Biochem J. 2001;354:275–283. doi: 10.1042/0264-6021:3540275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Leff T. AMP-activated protein kinase regulates gene expression by direct phosphorylation of nuclear proteins. Biochem Soc Trans. 2003;31:224–227. doi: 10.1042/bst0310224. [DOI] [PubMed] [Google Scholar]
- 94.Oakhill JS, et al. beta-Subunit myristoylation is the gatekeeper for initiating metabolic stress sensing by AMP-activated protein kinase (AMPK) Proc Natl Acad Sci U S A. 2010;107:19237–19241. doi: 10.1073/pnas.1009705107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lamia KA, et al. AMPK regulates the circadian clock by cryptochrome phosphorylation and degradation. Science. 2009;326:437–440. doi: 10.1126/science.1172156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tartarin P, et al. Inactivation of AMPKalpha1 induces asthenozoospermia and alters spermatozoa morphology. Endocrinology. 2012;153:3468–3481. doi: 10.1210/en.2011-1911. [DOI] [PubMed] [Google Scholar]
- 97.Bertoldo MJ, et al. Specific deletion of AMP-activated protein kinase (alpha1AMPK) in mouse Sertoli cells modifies germ cell quality. Mol Cell Endocrinol. 2016;423:96–112. doi: 10.1016/j.mce.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 98.Liang J, Mills GB. AMPK: a contextual oncogene or tumor suppressor? Cancer Res. 2013;73:2929–2935. doi: 10.1158/0008-5472.CAN-12-3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Berglund L, et al. A genecentric Human Protein Atlas for expression profiles based on antibodies. Mol Cell Proteomics. 2008;7:2019–2027. doi: 10.1074/mcp.R800013-MCP200. [DOI] [PubMed] [Google Scholar]
- 100.Taylor BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ros S, et al. Functional metabolic screen identifies 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 4 as an important regulator of prostate cancer cell survival. Cancer Discov. 2012;2:328–343. doi: 10.1158/2159-8290.CD-11-0234. This article described the identification of the AMPKβ1 subunit as an essential component for prostate cancer cell-specific survival from an unbiased siRNA functional screen. [DOI] [PubMed] [Google Scholar]
- 102.Cerami E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gao J, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Vetvik KK, et al. Globular adiponectin and its downstream target genes are up-regulated locally in human colorectal tumors: ex vivo and in vitro studies. Metabolism. 2014;63:672–681. doi: 10.1016/j.metabol.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 105.Wojtaszewski JF, et al. 5′AMP activated protein kinase expression in human skeletal muscle: effects of strength training and type 2 diabetes. J Physiol. 2005;564:563–573. doi: 10.1113/jphysiol.2005.082669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Birk JB, Wojtaszewski JF. Predominant alpha2/beta2/gamma3 AMPK activation during exercise in human skeletal muscle. J Physiol. 2006;577:1021–1032. doi: 10.1113/jphysiol.2006.120972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Treebak JT, et al. AS160 phosphorylation is associated with activation of alpha2beta2gamma1- but not alpha2beta2gamma3-AMPK trimeric complex in skeletal muscle during exercise in humans. Am J Physiol Endocrinol Metab. 2007;292:E715–722. doi: 10.1152/ajpendo.00380.2006. [DOI] [PubMed] [Google Scholar]
- 108.Miyamoto T, et al. Compartmentalized AMPK signaling illuminated by genetically encoded molecular sensors and actuators. Cell Rep. 2015;11:657–670. doi: 10.1016/j.celrep.2015.03.057. This work described the development of an improved AMPK activity reporter that could be localized to different subcellular compartments. This study was also demonstrated that various upstream signalings could activate distinct subcellular AMPK populations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tsou P, Zheng B, Hsu CH, Sasaki AT, Cantley LC. A fluorescent reporter of AMPK activity and cellular energy stress. Cell Metab. 2011;13:476–486. doi: 10.1016/j.cmet.2011.03.006. This paper describes the development of the first AMPK activity reporter that could be localized to different subcellular compartments. This study was also one of the first to demonstrate that various upstream signalings could activate distinct subcellular AMPK populations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gowans GJ, Hawley SA, Ross FA, Hardie DG. AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab. 2013;18:556–566. doi: 10.1016/j.cmet.2013.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Carling D, Thornton C, Woods A, Sanders MJ. AMP-activated protein kinase: new regulation, new roles? Biochem J. 2012;445:11–27. doi: 10.1042/BJ20120546. [DOI] [PubMed] [Google Scholar]
- 112.Oakhill JS, Scott JW, Kemp BE. AMPK functions as an adenylate charge-regulated protein kinase. Trends Endocrinol Metab. 2012;23:125–132. doi: 10.1016/j.tem.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 113.Li X, et al. Structural basis of AMPK regulation by adenine nucleotides and glycogen. Cell Res. 2015;25:50–66. doi: 10.1038/cr.2014.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sanchez-Cespedes M. A role for LKB1 gene in human cancer beyond the Peutz-Jeghers syndrome. Oncogene. 2007;26:7825–7832. doi: 10.1038/sj.onc.1210594. [DOI] [PubMed] [Google Scholar]
- 115.Huang X, et al. Important role of the LKB1-AMPK pathway in suppressing tumorigenesis in PTEN-deficient mice. Biochem J. 2008;412:211–221. doi: 10.1042/BJ20080557. [DOI] [PubMed] [Google Scholar]
- 116.McInnes KJ, Brown KA, Hunger NI, Simpson ER. Regulation of LKB1 expression by sex hormones in adipocytes. Int J Obes (Lond) 2012;36:982–985. doi: 10.1038/ijo.2011.172. [DOI] [PubMed] [Google Scholar]
- 117.Popovics P, Frigo DE, Schally AV, Rick FG. Targeting the 5′-AMP-activated protein kinase and related metabolic pathways for the treatment of prostate cancer. Expert Opin Ther Targets. 2015;19:617–632. doi: 10.1517/14728222.2015.1005603. [DOI] [PubMed] [Google Scholar]
- 118.Pearson HB, McCarthy A, Collins CM, Ashworth A, Clarke AR. Lkb1 deficiency causes prostate neoplasia in the mouse. Cancer Res. 2008;68:2223–2232. doi: 10.1158/0008-5472.CAN-07-5169. [DOI] [PubMed] [Google Scholar]
- 119.Faubert B, Vincent EE, Poffenberger MC, Jones RG. The AMP-activated protein kinase (AMPK) and cancer: many faces of a metabolic regulator. Cancer Lett. 2015;356:165–170. doi: 10.1016/j.canlet.2014.01.018. [DOI] [PubMed] [Google Scholar]
- 120.Shen MM, Abate-Shen C. Molecular genetics of prostate cancer: new prospects for old challenges. Genes Dev. 2010;24:1967–2000. doi: 10.1101/gad.1965810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Herrero-Martin G, et al. TAK1 activates AMPK-dependent cytoprotective autophagy in TRAIL-treated epithelial cells. EMBO J. 2009;28:677–685. doi: 10.1038/emboj.2009.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Xie M, et al. A pivotal role for endogenous TGF-beta-activated kinase-1 in the LKB1/AMP-activated protein kinase energy-sensor pathway. Proc Natl Acad Sci U S A. 2006;103:17378–17383. doi: 10.1073/pnas.0604708103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Konishi N, et al. Genetic mapping of allelic loss on chromosome 6q within heterogeneous prostate carcinoma. Cancer Sci. 2003;94:764–768. doi: 10.1111/j.1349-7006.2003.tb01516.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Liu W, et al. Deletion of a small consensus region at 6q15, including the MAP3K7 gene, is significantly associated with high-grade prostate cancers. Clin Cancer Res. 2007;13:5028–5033. doi: 10.1158/1078-0432.CCR-07-0300. [DOI] [PubMed] [Google Scholar]
- 125.Kluth M, et al. Genomic deletion of MAP3K7 at 6q12-22 is associated with early PSA recurrence in prostate cancer and absence of TMPRSS2:ERG fusions. Mod Pathol. 2013;26:975–983. doi: 10.1038/modpathol.2012.236. [DOI] [PubMed] [Google Scholar]
- 126.Wu M, et al. Suppression of Tak1 promotes prostate tumorigenesis. Cancer Res. 2012;72:2833–2843. doi: 10.1158/0008-5472.CAN-11-2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hawley SA, et al. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. This is one of three papers that first described CaMKK2 as an alternative upstream kinase of AMPK. [DOI] [PubMed] [Google Scholar]
- 128.Woods A, et al. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005;2:21–33. doi: 10.1016/j.cmet.2005.06.005. This is one of three papers that first described CaMKK2 as an alternative upstream kinase of AMPK. [DOI] [PubMed] [Google Scholar]
- 129.Hurley RL, et al. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem. 2005;280:29060–29066. doi: 10.1074/jbc.M503824200. This is one of three papers that first described CaMKK2 as an alternative upstream kinase of AMPK. [DOI] [PubMed] [Google Scholar]
- 130.Sharma NL, et al. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell. 2013;23:35–47. doi: 10.1016/j.ccr.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 131.Fu H, et al. MicroRNA-224 and its target CAMKK2 synergistically influence tumor progression and patient prognosis in prostate cancer. Tumour Biol. 2015;36:1983–1991. doi: 10.1007/s13277-014-2805-0. [DOI] [PubMed] [Google Scholar]
- 132.Subbannayya Y, et al. Calcium calmodulin dependent kinase kinase 2 - a novel therapeutic target for gastric adenocarcinoma. Cancer Biol Ther. 2015;16:336–345. doi: 10.4161/15384047.2014.972264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Liu DM, et al. CAMKK2, Regulated by Promoter Methylation, is a Prognostic Marker in Diffuse Gliomas. CNS Neurosci Ther. 2016 doi: 10.1111/cns.12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lin F, et al. The camKK2/camKIV relay is an essential regulator of hepatic cancer. Hepatology. 2015;62:505–520. doi: 10.1002/hep.27832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Scott JW, et al. Small molecule drug A-769662 and AMP synergistically activate naive AMPK independent of upstream kinase signaling. Chem Biol. 2014;21:619–627. doi: 10.1016/j.chembiol.2014.03.006. [DOI] [PubMed] [Google Scholar]
- 136.Fu X, Wan S, Lyu YL, Liu LF, Qi H. Etoposide induces ATM-dependent mitochondrial biogenesis through AMPK activation. PLoS One. 2008;3:e2009. doi: 10.1371/journal.pone.0002009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sapkota GP, et al. Ionizing radiation induces ataxia telangiectasia mutated kinase (ATM)-mediated phosphorylation of LKB1/STK11 at Thr-366. Biochem J. 2002;368:507–516. doi: 10.1042/BJ20021284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol. 2013;14:197–210. [PubMed] [Google Scholar]
- 139.Sanli T, et al. Ionizing radiation activates AMP-activated kinase (AMPK): a target for radiosensitization of human cancer cells. Int J Radiat Oncol Biol Phys. 2010;78:221–229. doi: 10.1016/j.ijrobp.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 140.Sanli T, Steinberg GR, Singh G, Tsakiridis T. AMP-activated protein kinase (AMPK) beyond metabolism: a novel genomic stress sensor participating in the DNA damage response pathway. Cancer Biol Ther. 2014;15:156–169. doi: 10.4161/cbt.26726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sanli T, et al. Ionizing radiation regulates the expression of AMP-activated protein kinase (AMPK) in epithelial cancer cells: modulation of cellular signals regulating cell cycle and survival. Radiother Oncol. 2012;102:459–465. doi: 10.1016/j.radonc.2011.11.014. [DOI] [PubMed] [Google Scholar]
- 142.Alexander A, et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci U S A. 2010;107:4153–4158. doi: 10.1073/pnas.0913860107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Hawley SA, et al. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 2010;11:554–565. doi: 10.1016/j.cmet.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Emerling BM, et al. Hypoxic activation of AMPK is dependent on mitochondrial ROS but independent of an increase in AMP/ATP ratio. Free Radic Biol Med. 2009;46:1386–1391. doi: 10.1016/j.freeradbiomed.2009.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Paschos A, Pandya R, Duivenvoorden WC, Pinthus JH. Oxidative stress in prostate cancer: changing research concepts towards a novel paradigm for prevention and therapeutics. Prostate Cancer Prostatic Dis. 2013;16:217–225. doi: 10.1038/pcan.2013.13. [DOI] [PubMed] [Google Scholar]
- 146.Zhang J, et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat Cell Biol. 2015;17:1259–1269. doi: 10.1038/ncb3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Hawley SA, et al. Phosphorylation by Akt within the ST loop of AMPK-alpha1 down-regulates its activation in tumour cells. Biochem J. 2014;459:275–287. doi: 10.1042/BJ20131344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Winder WW, et al. Phosphorylation of rat muscle acetyl-CoA carboxylase by AMP-activated protein kinase and protein kinase A. J Appl Physiol (1985) 1997;82:219–225. doi: 10.1152/jappl.1997.82.1.219. [DOI] [PubMed] [Google Scholar]
- 149.Chen ZP, et al. AMPK signaling in contracting human skeletal muscle: acetyl-CoA carboxylase and NO synthase phosphorylation. Am J Physiol Endocrinol Metab. 2000;279:E1202–1206. doi: 10.1152/ajpendo.2000.279.5.E1202. [DOI] [PubMed] [Google Scholar]
- 150.Dzamko N, et al. AMPK-independent pathways regulate skeletal muscle fatty acid oxidation. J Physiol. 2008;586:5819–5831. doi: 10.1113/jphysiol.2008.159814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Steinberg GR, et al. Whole body deletion of AMP-activated protein kinase {beta}2 reduces muscle AMPK activity and exercise capacity. J Biol Chem. 2010;285:37198–37209. doi: 10.1074/jbc.M110.102434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Li Y, et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011;13:376–388. doi: 10.1016/j.cmet.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Chen S, et al. Complementary regulation of TBC1D1 and AS160 by growth factors, insulin and AMPK activators. Biochem J. 2008;409:449–459. doi: 10.1042/BJ20071114. [DOI] [PubMed] [Google Scholar]
- 154.Chavez JA, Roach WG, Keller SR, Lane WS, Lienhard GE. Inhibition of GLUT4 translocation by Tbc1d1, a Rab GTPase-activating protein abundant in skeletal muscle, is partially relieved by AMP-activated protein kinase activation. J Biol Chem. 2008;283:9187–9195. doi: 10.1074/jbc.M708934200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Pehmoller C, et al. Genetic disruption of AMPK signaling abolishes both contraction- and insulin-stimulated TBC1D1 phosphorylation and 14-3-3 binding in mouse skeletal muscle. Am J Physiol Endocrinol Metab. 2009;297:E665–675. doi: 10.1152/ajpendo.00115.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Treebak JT, et al. Identification of a novel phosphorylation site on TBC1D4 regulated by AMP-activated protein kinase in skeletal muscle. Am J Physiol Cell Physiol. 2010;298:C377–385. doi: 10.1152/ajpcell.00297.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Treebak JT, et al. AMPK-mediated AS160 phosphorylation in skeletal muscle is dependent on AMPK catalytic and regulatory subunits. Diabetes. 2006;55:2051–2058. doi: 10.2337/db06-0175. [DOI] [PubMed] [Google Scholar]
- 158.Kramer HF, et al. Distinct signals regulate AS160 phosphorylation in response to insulin, AICAR, and contraction in mouse skeletal muscle. Diabetes. 2006;55:2067–2076. doi: 10.2337/db06-0150. [DOI] [PubMed] [Google Scholar]
- 159.Miyamoto T, et al. AMP-activated protein kinase phosphorylates Golgi-specific brefeldin A resistance factor 1 at Thr1337 to induce disassembly of Golgi apparatus. J Biol Chem. 2008;283:4430–4438. doi: 10.1074/jbc.M708296200. [DOI] [PubMed] [Google Scholar]
- 160.Li Y, et al. Identification of a novel serine phosphorylation site in human glutamine:fructose-6-phosphate amidotransferase isoform 1. Biochemistry. 2007;46:13163–13169. doi: 10.1021/bi700694c. [DOI] [PubMed] [Google Scholar]
- 161.Eguchi S, et al. AMP-activated protein kinase phosphorylates glutamine : fructose-6-phosphate amidotransferase 1 at Ser243 to modulate its enzymatic activity. Genes Cells. 2009;14:179–189. doi: 10.1111/j.1365-2443.2008.01260.x. [DOI] [PubMed] [Google Scholar]
- 162.Thomson DM, et al. AMP-activated protein kinase phosphorylates transcription factors of the CREB family. J Appl Physiol. 2008;104:429–438. doi: 10.1152/japplphysiol.00900.2007. [DOI] [PubMed] [Google Scholar]
- 163.Lau AW, Liu P, Inuzuka H, Gao D. SIRT1 phosphorylation by AMP-activated protein kinase regulates p53 acetylation. Am J Cancer Res. 2014;4:245–255. [PMC free article] [PubMed] [Google Scholar]
- 164.Carlson CA, Kim KH. Regulation of hepatic acetyl coenzyme A carboxylase by phosphorylation and dephosphorylation. J Biol Chem. 1973;248:378–380. [PubMed] [Google Scholar]
- 165.Davies SP, Sim AT, Hardie DG. Location and function of three sites phosphorylated on rat acetyl-CoA carboxylase by the AMP-activated protein kinase. Eur J Biochem. 1990;187:183–190. doi: 10.1111/j.1432-1033.1990.tb15293.x. [DOI] [PubMed] [Google Scholar]
- 166.Ha J, Daniel S, Broyles SS, Kim KH. Critical phosphorylation sites for acetyl-CoA carboxylase activity. J Biol Chem. 1994;269:22162–22168. [PubMed] [Google Scholar]
- 167.Munday MR, Campbell DG, Carling D, Hardie DG. Identification by amino acid sequencing of three major regulatory phosphorylation sites on rat acetyl-CoA carboxylase. Eur J Biochem. 1988;175:331–338. doi: 10.1111/j.1432-1033.1988.tb14201.x. [DOI] [PubMed] [Google Scholar]
- 168.Marin TL, et al. Identification of AMP-activated protein kinase targets by a consensus sequence search of the proteome. BMC Syst Biol. 2015;9:13. doi: 10.1186/s12918-015-0156-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kim J, et al. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell. 2013;152:290–303. doi: 10.1016/j.cell.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 171.Gwinn DM, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Marsin AS, et al. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr Biol. 2000;10:1247–1255. doi: 10.1016/s0960-9822(00)00742-9. [DOI] [PubMed] [Google Scholar]
- 174.Marsin AS, Bouzin C, Bertrand L, Hue L. The stimulation of glycolysis by hypoxia in activated monocytes is mediated by AMP-activated protein kinase and inducible 6-phosphofructo-2-kinase. J Biol Chem. 2002;277:30778–30783. doi: 10.1074/jbc.M205213200. [DOI] [PubMed] [Google Scholar]
- 175.Jakobsen SN, Hardie DG, Morrice N, Tornqvist HE. 5′-AMP-activated protein kinase phosphorylates IRS-1 on Ser-789 in mouse C2C12 myotubes in response to 5-aminoimidazole-4-carboxamide riboside. J Biol Chem. 2001;276:46912–46916. doi: 10.1074/jbc.C100483200. [DOI] [PubMed] [Google Scholar]
- 176.Qiao LY, Zhande R, Jetton TL, Zhou G, Sun XJ. In vivo phosphorylation of insulin receptor substrate 1 at serine 789 by a novel serine kinase in insulin-resistant rodents. J Biol Chem. 2002;277:26530–26539. doi: 10.1074/jbc.M201494200. [DOI] [PubMed] [Google Scholar]
- 177.Tzatsos A, Tsichlis PN. Energy depletion inhibits phosphatidylinositol 3-kinase/Akt signaling and induces apoptosis via AMP-activated protein kinase-dependent phosphorylation of IRS-1 at Ser-794. J Biol Chem. 2007;282:18069–18082. doi: 10.1074/jbc.M610101200. [DOI] [PubMed] [Google Scholar]
- 178.Bungard D, et al. Signaling kinase AMPK activates stress-promoted transcription via histone H2B phosphorylation. Science. 2010;329:1201–1205. doi: 10.1126/science.1191241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Greer EL, et al. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J Biol Chem. 2007;282:30107–30119. doi: 10.1074/jbc.M705325200. [DOI] [PubMed] [Google Scholar]
- 180.Bodur C, Karakas B, Timucin AC, Tezil T, Basaga H. AMP-activated protein kinase couples 3-bromopyruvate-induced energy depletion to apoptosis via activation of FoxO3a and upregulation of proapoptotic Bcl-2 proteins. Mol Carcinog. 2015 doi: 10.1002/mc.22411. [DOI] [PubMed] [Google Scholar]
- 181.Browne GJ, Finn SG, Proud CG. Stimulation of the AMP-activated protein kinase leads to activation of eukaryotic elongation factor 2 kinase and to its phosphorylation at a novel site, serine 398. J Biol Chem. 2004;279:12220–12231. doi: 10.1074/jbc.M309773200. [DOI] [PubMed] [Google Scholar]
- 182.Hong-Brown LQ, Brown CR, Huber DS, Lang CH. Lopinavir impairs protein synthesis and induces eEF2 phosphorylation via the activation of AMP-activated protein kinase. J Cell Biochem. 2008;105:814–823. doi: 10.1002/jcb.21882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Leprivier G, et al. The eEF2 kinase confers resistance to nutrient deprivation by blocking translation elongation. Cell. 2013;153:1064–1079. doi: 10.1016/j.cell.2013.04.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Xie CM, Liu XY, Sham KW, Lai JM, Cheng CH. Silencing of EEF2K (eukaryotic elongation factor-2 kinase) reveals AMPK-ULK1-dependent autophagy in colon cancer cells. Autophagy. 2014;10:1495–1508. doi: 10.4161/auto.29164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Yang W, et al. Regulation of transcription by AMP-activated protein kinase: phosphorylation of p300 blocks its interaction with nuclear receptors. J Biol Chem. 2001;276:38341–38344. doi: 10.1074/jbc.C100316200. [DOI] [PubMed] [Google Scholar]
- 186.Um JH, et al. Activation of 5′-AMP-activated kinase with diabetes drug metformin induces casein kinase Iepsilon (CKIepsilon)-dependent degradation of clock protein mPer2. J Biol Chem. 2007;282:20794–20798. doi: 10.1074/jbc.C700070200. [DOI] [PubMed] [Google Scholar]
- 187.Nakano A, et al. AMPK controls the speed of microtubule polymerization and directional cell migration through CLIP-170 phosphorylation. Nat Cell Biol. 2010;12:583–590. doi: 10.1038/ncb2060. [DOI] [PubMed] [Google Scholar]
- 188.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Egan DF, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Shen CH, et al. Phosphorylation of BRAF by AMPK impairs BRAF-KSR1 association and cell proliferation. Mol Cell. 2013;52:161–172. doi: 10.1016/j.molcel.2013.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.DeRan M, et al. Energy stress regulates hippo-YAP signaling involving AMPK-mediated regulation of angiomotin-like 1 protein. Cell Rep. 2014;9:495–503. doi: 10.1016/j.celrep.2014.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Hoffman NJ, et al. Global Phosphoproteomic Analysis of Human Skeletal Muscle Reveals a Network of Exercise-Regulated Kinases and AMPK Substrates. Cell Metab. 2015;22:922–935. doi: 10.1016/j.cmet.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Imamura K, Ogura T, Kishimoto A, Kaminishi M, Esumi H. Cell cycle regulation via p53 phosphorylation by a 5′-AMP activated protein kinase activator, 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside, in a human hepatocellular carcinoma cell line. Biochem Biophys Res Commun. 2001;287:562–567. doi: 10.1006/bbrc.2001.5627. [DOI] [PubMed] [Google Scholar]
- 194.Jones RG, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18:283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 195.Hong YH, Varanasi US, Yang W, Leff T. AMP-activated protein kinase regulates HNF4alpha transcriptional activity by inhibiting dimer formation and decreasing protein stability. J Biol Chem. 2003;278:27495–27501. doi: 10.1074/jbc.M304112200. [DOI] [PubMed] [Google Scholar]
- 196.Clarke PR, Hardie DG. Regulation of HMG-CoA reductase: identification of the site phosphorylated by the AMP-activated protein kinase in vitro and in intact rat liver. EMBO J. 1990;9:2439–2446. doi: 10.1002/j.1460-2075.1990.tb07420.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Dasgupta B, Milbrandt J. AMP-activated protein kinase phosphorylates retinoblastoma protein to control mammalian brain development. Dev Cell. 2009;16:256–270. doi: 10.1016/j.devcel.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Banko MR, et al. Chemical genetic screen for AMPKalpha2 substrates uncovers a network of proteins involved in mitosis. Mol Cell. 2011;44:878–892. doi: 10.1016/j.molcel.2011.11.005. This study described the identification of AMPK functions beyond metabolism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.McGee SL, et al. AMP-activated protein kinase regulates GLUT4 transcription by phosphorylating histone deacetylase 5. Diabetes. 2008;57:860–867. doi: 10.2337/db07-0843. [DOI] [PubMed] [Google Scholar]
- 200.Mihaylova MM, et al. Class IIa histone deacetylases are hormone-activated regulators of FOXO and mammalian glucose homeostasis. Cell. 2011;145:607–621. doi: 10.1016/j.cell.2011.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Toyama EQ, et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science. 2016;351:275–281. doi: 10.1126/science.aab4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Ducommun S, et al. Motif affinity and mass spectrometry proteomic approach for the discovery of cellular AMPK targets: identification of mitochondrial fission factor as a new AMPK substrate. Cell Signal. 2015;27:978–988. doi: 10.1016/j.cellsig.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 203.Koo SH, et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature. 2005;437:1109–1111. doi: 10.1038/nature03967. [DOI] [PubMed] [Google Scholar]
- 204.Canto C, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Lee JH, et al. Energy-dependent regulation of cell structure by AMP-activated protein kinase. Nature. 2007;447:1017–1020. doi: 10.1038/nature05828. [DOI] [PubMed] [Google Scholar]
- 206.Bultot L, et al. Myosin light chains are not a physiological substrate of AMPK in the control of cell structure changes. FEBS Lett. 2009;583:25–28. doi: 10.1016/j.febslet.2008.11.022. [DOI] [PubMed] [Google Scholar]
- 207.Thaiparambil JT, Eggers CM, Marcus AI. AMPK regulates mitotic spindle orientation through phosphorylation of myosin regulatory light chain. Mol Cell Biol. 2012;32:3203–3217. doi: 10.1128/MCB.00418-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Liu S, et al. AICAR-Induced Activation of AMPK Inhibits TSH/SREBP-2/HMGCR Pathway in Liver. PLoS One. 2015;10:e0124951. doi: 10.1371/journal.pone.0124951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Barnes K, et al. Activation of GLUT1 by metabolic and osmotic stress: potential involvement of AMP-activated protein kinase (AMPK) J Cell Sci. 2002;115:2433–2442. doi: 10.1242/jcs.115.11.2433. [DOI] [PubMed] [Google Scholar]
- 210.Yun H, Lee M, Kim SS, Ha J. Glucose deprivation increases mRNA stability of vascular endothelial growth factor through activation of AMP-activated protein kinase in DU145 prostate carcinoma. J Biol Chem. 2005;280:9963–9972. doi: 10.1074/jbc.M412994200. [DOI] [PubMed] [Google Scholar]
- 211.Wu N, et al. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol Cell. 2013;49:1167–1175. doi: 10.1016/j.molcel.2013.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Canto C, Auwerx J. AMP-activated protein kinase and its downstream transcriptional pathways. Cell Mol Life Sci. 2010;67:3407–3423. doi: 10.1007/s00018-010-0454-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Cornforth AN, Davis JS, Khanifar E, Nastiuk KL, Krolewski JJ. FOXO3a mediates the androgen-dependent regulation of FLIP and contributes to TRAIL-induced apoptosis of LNCaP cells. Oncogene. 2008;27:4422–4433. doi: 10.1038/onc.2008.80. [DOI] [PubMed] [Google Scholar]
- 215.Kops GJ, et al. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419:316–321. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- 216.Chiacchiera F, Simone C. The AMPK-FoxO3A axis as a target for cancer treatment. Cell Cycle. 2010;9:1091–1096. doi: 10.4161/cc.9.6.11035. [DOI] [PubMed] [Google Scholar]
- 217.Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 218.Hardie DG, Pan DA. Regulation of fatty acid synthesis and oxidation by the AMP-activated protein kinase. Biochem Soc Trans. 2002;30:1064–1070. doi: 10.1042/bst0301064. [DOI] [PubMed] [Google Scholar]
- 219.Leclerc I, et al. Hepatocyte nuclear factor-4alpha involved in type 1 maturity-onset diabetes of the young is a novel target of AMP-activated protein kinase. Diabetes. 2001;50:1515–1521. doi: 10.2337/diabetes.50.7.1515. [DOI] [PubMed] [Google Scholar]
- 220.Zhou G, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Swinnen JV, Brusselmans K, Verhoeven G. Increased lipogenesis in cancer cells: new players, novel targets. Curr Opin Clin Nutr Metab Care. 2006;9:358–365. doi: 10.1097/01.mco.0000232894.28674.30. [DOI] [PubMed] [Google Scholar]
- 222.Heemers HV, Verhoeven G, Swinnen JV. Androgen activation of the sterol regulatory element-binding protein pathway: Current insights. Mol Endocrinol. 2006;20:2265–2277. doi: 10.1210/me.2005-0479. [DOI] [PubMed] [Google Scholar]
- 223.Costello LC, Franklin RB, Feng P. Mitochondrial function, zinc, and intermediary metabolism relationships in normal prostate and prostate cancer. Mitochondrion. 2005;5:143–153. doi: 10.1016/j.mito.2005.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Stoss O, et al. Transcriptional profiling of transurethral resection samples provides insight into molecular mechanisms of hormone refractory prostate cancer. Prostate Cancer Prostatic Dis. 2008;11:166–172. doi: 10.1038/sj.pcan.4501001. [DOI] [PubMed] [Google Scholar]
- 225.Sotgia F, Martinez-Outschoorn UE, Lisanti MP. Cancer metabolism: new validated targets for drug discovery. Oncotarget. 2013;4:1309–1316. doi: 10.18632/oncotarget.1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Costello LC, Franklin RB. Novel role of zinc in the regulation of prostate citrate metabolism and its implications in prostate cancer. Prostate. 1998;35:285–296. doi: 10.1002/(sici)1097-0045(19980601)35:4<285::aid-pros8>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 227.Costello LC, Liu Y, Franklin RB, Kennedy MC. Zinc inhibition of mitochondrial aconitase and its importance in citrate metabolism of prostate epithelial cells. J Biol Chem. 1997;272:28875–28881. doi: 10.1074/jbc.272.46.28875. This article reports for the first time that elevated levels of zinc in prostate epithelial cells suppress the oxidation of citrate via the inhibition of m-aconitase. [DOI] [PubMed] [Google Scholar]
- 228.Franz MC, et al. Zinc transporters in prostate cancer. Mol Aspects Med. 2013;34:735–741. doi: 10.1016/j.mam.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Almeida A, Moncada S, Bolanos JP. Nitric oxide switches on glycolysis through the AMP protein kinase and 6-phosphofructo-2-kinase pathway. Nat Cell Biol. 2004;6:45–51. doi: 10.1038/ncb1080. [DOI] [PubMed] [Google Scholar]
- 230.Bando H, et al. Phosphorylation of the 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase/PFKFB3 family of glycolytic regulators in human cancer. Clin Cancer Res. 2005;11:5784–5792. doi: 10.1158/1078-0432.CCR-05-0149. [DOI] [PubMed] [Google Scholar]
- 231.Moon JS, et al. Androgen stimulates glycolysis for de novo lipid synthesis by increasing the activities of hexokinase 2 and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 2 in prostate cancer cells. Biochem J. 2011;433:225–233. doi: 10.1042/BJ20101104. [DOI] [PubMed] [Google Scholar]
- 232.Ros S, Schulze A. Balancing glycolytic flux: the role of 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatases in cancer metabolism. Cancer Metab. 2013;1:8. doi: 10.1186/2049-3002-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Kurth-Kraczek EJ, Hirshman MF, Goodyear LJ, Winder WW. 5′ AMP-activated protein kinase activation causes GLUT4 translocation in skeletal muscle. Diabetes. 1999;48:1667–1671. doi: 10.2337/diabetes.48.8.1667. [DOI] [PubMed] [Google Scholar]
- 234.Sakamoto K, Holman GD. Emerging role for AS160/TBC1D4 and TBC1D1 in the regulation of GLUT4 traffic. Am J Physiol Endocrinol Metab. 2008;295:E29–37. doi: 10.1152/ajpendo.90331.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Barron CC, Bilan PJ, Tsakiridis T, Tsiani E. Facilitative glucose transporters: Implications for cancer detection, prognosis and treatment. Metabolism. 2016;65:124–139. doi: 10.1016/j.metabol.2015.10.007. [DOI] [PubMed] [Google Scholar]
- 236.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Dang CV, Le A, Gao P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin Cancer Res. 2009;15:6479–6483. doi: 10.1158/1078-0432.CCR-09-0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12:401–410. doi: 10.1038/nrc3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res. 2009;15:5308–5316. doi: 10.1158/1078-0432.CCR-07-5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Galluzzi L, et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015;34:856–880. doi: 10.15252/embj.201490784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Mathew R, White E. Autophagy, Stress, and Cancer Metabolism: What Doesn’t Kill You Makes You Stronger. Cold Spring Harbor symposia on quantitative biology. 2012 doi: 10.1101/sqb.2012.76.011015. [DOI] [PubMed] [Google Scholar]
- 242.Shi Y, et al. Androgens promote prostate cancer cell growth through induction of autophagy. Mol Endocrinol. 2013;27:280–295. doi: 10.1210/me.2012-1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Nguyen HG, et al. Targeting autophagy overcomes Enzalutamide resistance in castration-resistant prostate cancer cells and improves therapeutic response in a xenograft model. Oncogene. 2014;33:4521–4530. doi: 10.1038/onc.2014.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Santanam U, et al. Atg7 cooperates with Pten loss to drive prostate cancer tumor growth. Genes Dev. 2016;30:399–407. doi: 10.1101/gad.274134.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Bennett HL, Fleming JT, O’Prey J, Ryan KM, Leung HY. Androgens modulate autophagy and cell death via regulation of the endoplasmic reticulum chaperone glucose-regulated protein 78/BiP in prostate cancer cells. Cell Death Dis. 2010;1:e72. doi: 10.1038/cddis.2010.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Alers S, Loffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol. 2012;32:2–11. doi: 10.1128/MCB.06159-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Tsouko E, et al. Regulation of the pentose phosphate pathway by an androgen receptor-mTOR-mediated mechanism and its role in prostate cancer cell growth. Oncogenesis. 2014;3:e103. doi: 10.1038/oncsis.2014.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Xu Y, Chen SY, Ross KN, Balk SP. Androgens induce prostate cancer cell proliferation through mammalian target of rapamycin activation and post-transcriptional increases in cyclin D proteins. Cancer Res. 2006;66:7783–7792. doi: 10.1158/0008-5472.CAN-05-4472. [DOI] [PubMed] [Google Scholar]
- 249.Munkley J, et al. A novel androgen-regulated isoform of the TSC2 tumour suppressor gene increases cell proliferation. Oncotarget. 2014;5:131–139. doi: 10.18632/oncotarget.1405. This article described an AR-regulated splice variant of TSC2 that when expressed blocked AMPK’s ability to inhibit mTOR signaling, thus allowing the two signaling cascades (AMPK and mTOR) to simultaneously exist. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. The active form of the metabolic sensor: AMP-activated protein kinase (AMPK) directly binds the mitotic apparatus and travels from centrosomes to the spindle midzone during mitosis and cytokinesis. Cell Cycle. 2009;8:2385–2398. doi: 10.4161/cc.8.15.9082. [DOI] [PubMed] [Google Scholar]
- 251.Mao L, et al. AMPK phosphorylates GBF1 for mitotic Golgi disassembly. J Cell Sci. 2013;126:1498–1505. doi: 10.1242/jcs.121954. [DOI] [PubMed] [Google Scholar]
- 252.Lee IJ, Lee CW, Lee JH. CaMKKbeta-AMPKalpha2 signaling contributes to mitotic Golgi fragmentation and the G2/M transition in mammalian cells. Cell Cycle. 2015;14:598–611. doi: 10.4161/15384101.2014.991557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Aparicio A, Den RB, Knudsen KE. Time to stratify? The retinoblastoma protein in castrate-resistant prostate cancer. Nat Rev Urol. 2011;8:562–568. doi: 10.1038/nrurol.2011.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.van der Linden AM, Nolan KM, Sengupta P. KIN-29 SIK regulates chemoreceptor gene expression via an MEF2 transcription factor and a class II HDAC. EMBO J. 2007;26:358–370. doi: 10.1038/sj.emboj.7601479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Chang S, Bezprozvannaya S, Li S, Olson EN. An expression screen reveals modulators of class II histone deacetylase phosphorylation. Proc Natl Acad Sci U S A. 2005;102:8120–8125. doi: 10.1073/pnas.0503275102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Dequiedt F, et al. New role for hPar-1 kinases EMK and C-TAK1 in regulating localization and activity of class IIa histone deacetylases. Mol Cell Biol. 2006;26:7086–7102. doi: 10.1128/MCB.00231-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Mihaylova MM, Shaw RJ. Metabolic reprogramming by class I and II histone deacetylases. Trends Endocrinol Metab. 2013;24:48–57. doi: 10.1016/j.tem.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Wang B, et al. A hormone-dependent module regulating energy balance. Cell. 2011;145:596–606. doi: 10.1016/j.cell.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Bonini MG, Gantner BN. The multifaceted activities of AMPK in tumor progression–why the “one size fits all” definition does not fit at all? IUBMB Life. 2013;65:889–896. doi: 10.1002/iub.1213. [DOI] [PubMed] [Google Scholar]
- 260.Brown KA, Samarajeewa NU, Simpson ER. Endocrine-related cancers and the role of AMPK. Mol Cell Endocrinol. 2013;366:170–179. doi: 10.1016/j.mce.2012.06.016. [DOI] [PubMed] [Google Scholar]
- 261.Choudhury Y, Salt IP, Leung HY. AMPK-friend or foe for targeted therapy? Cell Cycle. 2015;14:1761–1762. doi: 10.1080/15384101.2015.1022066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Hardie DG. The LKB1-AMPK pathway-friend or foe in cancer? Cancer Cell. 2013;23:131–132. doi: 10.1016/j.ccr.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 263.Hardie DG. Molecular Pathways: Is AMPK a Friend or a Foe in Cancer? Clin Cancer Res. 2015;21:3836–3840. doi: 10.1158/1078-0432.CCR-14-3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Jeon SM, Hay N. The dark face of AMPK as an essential tumor promoter. Cell Logist. 2012;2:197–202. doi: 10.4161/cl.22651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Jeon SM, Hay N. The double-edged sword of AMPK signaling in cancer and its therapeutic implications. Arch Pharm Res. 2015;38:346–357. doi: 10.1007/s12272-015-0549-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Svensson RU, Shaw RJ. Cancer metabolism: Tumour friend or foe. Nature. 2012;485:590–591. doi: 10.1038/485590a. [DOI] [PubMed] [Google Scholar]
- 267.Viollet B, et al. AMPK inhibition in health and disease. Crit Rev Biochem Mol Biol. 2010;45:276–295. doi: 10.3109/10409238.2010.488215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Zadra G, Batista JL, Loda M. Dissecting the Dual Role of AMPK in Cancer: From Experimental to Human Studies. Mol Cancer Res. 2015;13:1059–1072. doi: 10.1158/1541-7786.MCR-15-0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Cancer Genome Atlas Research N. The Molecular Taxonomy of Primary Prostate Cancer. Cell. 2015;163:1011–1025. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Cusi K, Consoli A, DeFronzo RA. Metabolic effects of metformin on glucose and lactate metabolism in noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab. 1996;81:4059–4067. doi: 10.1210/jcem.81.11.8923861. [DOI] [PubMed] [Google Scholar]
- 271.Foretz M, Guigas B, Bertrand L, Pollak M, Viollet B. Metformin: from mechanisms of action to therapies. Cell Metab. 2014;20:953–966. doi: 10.1016/j.cmet.2014.09.018. [DOI] [PubMed] [Google Scholar]
- 272.Pollak MN. Investigating metformin for cancer prevention and treatment: the end of the beginning. Cancer Discov. 2012;2:778–790. doi: 10.1158/2159-8290.CD-12-0263. [DOI] [PubMed] [Google Scholar]
- 273.Viollet B, et al. Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond) 2012;122:253–270. doi: 10.1042/CS20110386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.He L, Wondisford FE. Metformin action: concentrations matter. Cell Metab. 2015;21:159–162. doi: 10.1016/j.cmet.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 275.Wilcock C, Bailey CJ. Accumulation of metformin by tissues of the normal and diabetic mouse. Xenobiotica. 1994;24:49–57. doi: 10.3109/00498259409043220. [DOI] [PubMed] [Google Scholar]
- 276.Foretz M, et al. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest. 2010;120:2355–2369. doi: 10.1172/JCI40671. This study challenged the notion that metformin’s primary antidiabetic effects were mediated through AMPK. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Cao J, et al. Low concentrations of metformin suppress glucose production in hepatocytes through AMP-activated protein kinase (AMPK) J Biol Chem. 2014;289:20435–20446. doi: 10.1074/jbc.M114.567271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.El-Mir MY, et al. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. 2000;275:223–228. doi: 10.1074/jbc.275.1.223. This study, along with Owen et al were the first reports demonstrating that metformin did not directly activate AMPK, but rather indirectly stimulated AMPK by inhibiting the respiratory chain complex I. This inhibition leads to an increase in AMP/ATP ratios which can, among other effects, increase AMPK activity. [DOI] [PubMed] [Google Scholar]
- 279.Dowling RJ, et al. Metformin Pharmacokinetics in Mouse Tumors: Implications for Human Therapy. Cell Metab. 2016;23:567–568. doi: 10.1016/j.cmet.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 280.Chandel NS, et al. Are Metformin Doses Used in Murine Cancer Models Clinically Relevant? Cell Metab. 2016;23:569–570. doi: 10.1016/j.cmet.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 281.Graham GG, et al. Clinical pharmacokinetics of metformin. Clin Pharmacokinet. 2011;50:81–98. doi: 10.2165/11534750-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 282.Kordes S, et al. Metformin in patients with advanced pancreatic cancer: a double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol. 2015;16:839–847. doi: 10.1016/S1470-2045(15)00027-3. [DOI] [PubMed] [Google Scholar]
- 283.Quinn BJ, Kitagawa H, Memmott RM, Gills JJ, Dennis PA. Repositioning metformin for cancer prevention and treatment. Trends Endocrinol Metab. 2013;24:469–480. doi: 10.1016/j.tem.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 284.Verhaagh S, Schweifer N, Barlow DP, Zwart R. Cloning of the mouse and human solute carrier 22a3 (Slc22a3/SLC22A3) identifies a conserved cluster of three organic cation transporters on mouse chromosome 17 and human 6q26-q27. Genomics. 1999;55:209–218. doi: 10.1006/geno.1998.5639. [DOI] [PubMed] [Google Scholar]
- 285.Chen L, et al. Genetic and epigenetic regulation of the organic cation transporter 3, SLC22A3. Pharmacogenomics J. 2013;13:110–120. doi: 10.1038/tpj.2011.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Grisanzio C, et al. Genetic and functional analyses implicate the NUDT11, HNF1B, and SLC22A3 genes in prostate cancer pathogenesis. Proc Natl Acad Sci U S A. 2012;109:11252–11257. doi: 10.1073/pnas.1200853109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Yeh HY, et al. Identifying significant genetic regulatory networks in the prostate cancer from microarray data based on transcription factor analysis and conditional independency. BMC Med Genomics. 2009;2:70. doi: 10.1186/1755-8794-2-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Hoffmann TJ, et al. A large multiethnic genome-wide association study of prostate cancer identifies novel risk variants and substantial ethnic differences. Cancer Discov. 2015;5:878–891. doi: 10.1158/2159-8290.CD-15-0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Hao Q, et al. Systematic meta-analyses of gene-specific genetic association studies in prostate cancer. Oncotarget. 2016 doi: 10.18632/oncotarget.7926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Chae YK, et al. Repurposing metformin for cancer treatment: current clinical studies. Oncotarget. 2016 doi: 10.18632/oncotarget.8194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Pollak M. Metformin and other biguanides in oncology: advancing the research agenda. Cancer Prev Res (Phila) 2010;3:1060–1065. doi: 10.1158/1940-6207.CAPR-10-0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006;66:10269–10273. doi: 10.1158/0008-5472.CAN-06-1500. [DOI] [PubMed] [Google Scholar]
- 293.Hardie DG. AMPK: a target for drugs and natural products with effects on both diabetes and cancer. Diabetes. 2013;62:2164–2172. doi: 10.2337/db13-0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000;348(Pt 3):607–614. This study, along with El-Mir et al were the first reports demonstrating that metformin did not directly activate AMPK, but rather indirectly stimulated AMPK by inhibiting the respiratory chain complex I. This inhibition leads to an increase in AMP/ATP ratios which can, among other effects, increase AMPK activity. [PMC free article] [PubMed] [Google Scholar]
- 295.Ben Sahra I, et al. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011;71:4366–4372. doi: 10.1158/0008-5472.CAN-10-1769. [DOI] [PubMed] [Google Scholar]
- 296.Zhou K, et al. The role of ATM in response to metformin treatment and activation of AMPK. Nat Genet. 2012;44:361–362. doi: 10.1038/ng.2234. [DOI] [PubMed] [Google Scholar]
- 297.Woods A, Leiper JM, Carling D. The role of ATM in response to metformin treatment and activation of AMPK. Nat Genet. 2012;44:360–361. doi: 10.1038/ng.2235. [DOI] [PubMed] [Google Scholar]
- 298.Yee SW, Chen L, Giacomini KM. The role of ATM in response to metformin treatment and activation of AMPK. Nat Genet. 2012;44:359–360. doi: 10.1038/ng.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Fryer LG, Parbu-Patel A, Carling D. The Anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J Biol Chem. 2002;277:25226–25232. doi: 10.1074/jbc.M202489200. [DOI] [PubMed] [Google Scholar]
- 300.Hawley SA, et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science. 2012;336:918–922. doi: 10.1126/science.1215327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Pang T, et al. Small molecule antagonizes autoinhibition and activates AMP-activated protein kinase in cells. J Biol Chem. 2008;283:16051–16060. doi: 10.1074/jbc.M710114200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Scott JW, et al. Thienopyridone drugs are selective activators of AMP-activated protein kinase beta1-containing complexes. Chem Biol. 2008;15:1220–1230. doi: 10.1016/j.chembiol.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 303.Laderoute KR, Calaoagan JM, Madrid PB, Klon AE, Ehrlich PJ. SU11248 (sunitinib) directly inhibits the activity of mammalian 5′-AMP-activated protein kinase (AMPK) Cancer Biol Ther. 2010;10:68–76. doi: 10.4161/cbt.10.1.12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Borgdorff V, et al. A chemical biology approach identifies AMPK as a modulator of melanoma oncogene MITF. Oncogene. 2014;33:2531–2539. doi: 10.1038/onc.2013.185. [DOI] [PubMed] [Google Scholar]
- 305.Gan HK, Seruga B, Knox JJ. Sunitinib in solid tumors. Expert Opin Investig Drugs. 2009;18:821–834. doi: 10.1517/13543780902980171. [DOI] [PubMed] [Google Scholar]
- 306.Peter B, et al. Target interaction profiling of midostaurin and its metabolites in neoplastic mast cells predicts distinct effects on activation and growth. Leukemia. 2016;30:464–472. doi: 10.1038/leu.2015.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Tokumitsu H, et al. STO-609, a specific inhibitor of the Ca(2+)/calmodulin-dependent protein kinase kinase. J Biol Chem. 2002;277:15813–15818. doi: 10.1074/jbc.M201075200. [DOI] [PubMed] [Google Scholar]