

Abstract
Alzheimer’s disease (AD) is a major source of morbidity and mortality, with the disease burden expected to rise as the population ages. No disease-modifying agent is currently available, but recent research suggests that nutritional and lifestyle modifications can delay or prevent the onset of AD. However, preventive nutritional interventions are not universally applicable and depend on the clinical profile of the individual patient. This article reviews existing nutritional modalities for AD prevention that act through improvement of insulin resistance, correction of dyslipidemia, and reduction of oxidative stress, and discusses how they may be modified on the basis of individual biomarkers, genetics, and behavior. In addition, we report preliminary results of clinical application of these personalized interventions at the first AD prevention clinic in the United States. The use of these personalized interventions represents an important application of precision medicine techniques for the prevention of AD that can be adopted by clinicians across disciplines.
Keywords: preclinical Alzheimer’ s disease, Alzheimer’ s prevention, clinical precision medicine, nutngenomics, non-pharmacological management, insulin resistance
Introduction
Alzheimer’ s disease (AD) is a neurodegenerative dementia characterized by early impairment of memory with progressive involvement of other cognitive domains, mood and affect, and motor function. The disease burden is immense: AD is estimated to affect one in eight individuals aged 65 years and older and nearly half of those aged 85 years and older, with the annual costs of AD care rising to above $400 billion worldwide.1
Though the initial cognitive symptoms of AD usually begin after the sixth decade, evidence indicates that the pathological amyloid plaques and neurofibrillary tangles begin to accumulate decades before the onset of clinical symptoms, in a period designated as preclinical AD.2 It is unclear how many patients with preclinical AD will progress to the symptomatic stages of mild cognitive impairment (MCI) and ultimately to Alzheimer’s dementia, but the long incubation period of the neuropathology implies that early preventive interventions may reduce both the incidence and severity of clinical AD. Myriad research biomarkers3 and clinical risk scores4 have been identified and validated to identify individuals with preclinical AD, and clinical trials are in progress to test anti-amyloid agents for preventive efficacy.5 However, successful prevention of AD will depend not only on reduction of amyloid alone but also on comprehensive targeting of other biological and cognitive risk factors.
There is a burgeoning literature on the use of nutrition for the prevention of AD in the past decade, and several systematic reviews have discussed these nutritional strategies.6–8 However, many studies have shown inconsistent results and suggest that nutritional interventions are not universally applicable but must be adapted for individual patients. Just as individual tumor characteristics drive therapeutic regimens in oncology, it is likely that a combination of the individual’ s biomarkers, genotype, and behavioral history will determine the success or failure of specific nutritional interventions. In this article, we will review the possible mechanisms of nutritional interventions, discuss the role of personalization and precision medicine for particular interventions, and finally present preliminary data on the efficacy of these interventions at the Alzheimer’ s Prevention Clinic (APC) at Weill Cornell Medical College (WCMC), the first AD prevention clinic in the United States.
Molecular biology of AD prevention
The pathology of AD is characterized by the development of extracellular senile plaques formed from amyloid-l) (Al)) peptide9 and intracellular neurofibrillary tangles formed from hyperphosphorylation of the microtubule-associated protein tau.10 Many theories have been proposed to account for the development of plaques and tangles, but three major etiologies are critical to the molecular basis of AD prevention: insulin resistance, dyslipidemia, and oxidative stress.
Insulin resistance causes both cerebral glucose hypometabolism11 and a systemic hyperinsuline- mic state.12 Glucose hypometabolism increases expression of amyloid precursor protein (APP)13 and causes cytoskeletal rearrangements associated with neurofibrillary tangles,14 while elevated systemic insulin binds to the insulin-degrading enzyme and prevents Al) degradation via the same enzyme.15 Clinically, brain glucose hypometabolism is found even at the early preclinical stages of AD,16 and therapies that enhance cerebral glucose metabolism show improvement in cognition and AD symptomatology.17 Together, these findings indicate that early correction of insulin resistance is a promising target for preventative interventions.
Al) is produced by cleavage of the APP at lipid rafts in the cellular membrane enriched for cholesterol and sphingomyelin;18 in the brains of AD patients, these sites demonstrate elevated levels of cholesterol, with subsequent dysfunctional Al) processing.19 Evidence that treatment with cholesterol-lowering statin medications reduces both risk of AD20 and levels of Al) in cell culture and mammalian brains21 suggests that dyslipidemia may lead to local lipid raft abnormalities and, ultimately, increased Al) production that can be prevented by early correction of lipid abnormalities.
Finally, oxidative stress induced by mitochondrial dysfunction,22 direct toxicity of Al), or inflammatory signaling23 causes direct damage to lipid membranes, DNA, and other cellular components. Oxidative stress is both a cause and a consequence of Al) aggregation, as free radicals increase expression of the APP-cleaving l)-secretase enzyme,24 and increased Al) generates free radicals both directly25 and via the NF-KB inflammatory pathway.26 Clinically, dietary intake of free radical scavengers, such as antioxidant vitamins, has been linked to reduction of AD risk, although results have been mixed and it is likely that a more individualized approach is necessary.28
In summary, insulin resistance, dyslipidemia, and oxidative stress all contribute to the generation of pathological plaques and tangles, and the nutritional preventions discussed in this article are targeted toward amelioration of these three underlying etiologies.
Interventions for insulin resistance
Peripheral insulin resistance, a known risk factor for AD,29 reduces cerebral glucose metabolism and decreases Al) clearance within the central nervous system.30 Traditionally, patients are screened for insulin resistance using three measures designed for detection of type II diabetes mellitus: glycosylated hemoglobin Au level, fasting plasma glucose level, and results of the oral glucose tolerance test.31 However, these modalities depend on abnormal serum glucose and are insensitive for subclinical insulin resistance that may lead to AD, even without development of overt hyperglycemia. Recent studies have demonstrated that alternative measurements of insulin resistance that also account for, or are associated with, insulin response are independent risk factors for AD pathology. For example, data from the Framingham Heart Study revealed that the adipokine adiponectin is an independent risk factor for AD and all-cause dementia in women,32 while insulin C-peptide levels33 and the homeostatic model assessment of insulin resistance score are both associated with cerebral amyloid burden.34 These results suggest that patients with these insulin resistance biomarkers may benefit from intervention, even in the absence of overt diabetes.
Depending on the prior lifestyle of the patient, preventive nutritional interventions can take two forms. If the patient’ s current dietary patterns are unhealthy, adoption of a Mediterranean-style diet has been demonstrated to reduce insulin resistance.35 Adherence to the Mediterranean - DASH intervention for neurodegenerative delay (MIND) diet, which emphasizes intake of berries and dark green leafy vegetables, demonstrates slower rates of cognitive decline than either a conventional Mediterranean diet or Dietary Approaches to Stop Hypertension (DASH).36 However, if the patient is already adhering to a healthy diet, additions to their meal intake have been found to reduce AD risk. In particular, consumption of cocoa flavanols has been demonstrated to reduce insulin resistance and improve cognition in older adults, suggesting that it may hold promise as a preventive nutritional intervention.37 Multiple studies have linked regular exercise to reduced insulin resistance,38 and recent evidence has revealed that exercise also reduces hippocampal atrophy in older adults, through the AD-promoting gene APOE4.39 Atrophy was only reduced with at least 30 min of athletic activity three times per week, suggesting that the presence of a high-risk genotype necessitates exercise beyond the low-intensity regimens that are effective for the prevention of metabolic syndrome.40
Interventions for dyslipidemia
Though the role of cholesterol in AD pathogenesis is controversial, dyslipidemia is an independent risk factor for AD41 that may affect production of Al) through alteration of neuronal membrane composition.42 Multiple interventions for dyslipidemia are effective for AD risk reduction, although many—including statins20—are pharmacological rather than nutritional. The two major nutritional supplements with evidence of AD prevention benefit are niacin and omega-3 fatty acids (O3FAs), but their utility depends partly on the clinical profile of the patient. Niacin has been found to reduce AD incidence and slow cognitive decline in older adults43 and significantly reduce levels of circulating apolipoprotein-B (apo-B), a component of the low- density lipoproteins (very low - density lipoprotein, intermediate-density lipoprotein, and low-density lipoprotein).44 As levels of apo-B are increased in the brains of patients with AD,45 niacin may be particularly effective for dyslipidemic patients with elevated apo-B. Similarly, O3FAs have been found to reduce incidence of AD in clinical studies46 and reduce amyloid burden in mouse models;47 given the reduction of serum triglycerides from O3FAs,48 it is possible that patients with substantial hypertriglyceridemia may selectively benefit from O3FA supplementation. To date, however, no study has examined prevention in this population, and the benefit of O3FAs likely lies in scavenging free radicals, as discussed below.
Interventions for oxidative stress
The deleterious effects of free radicals on neuronal function are legion and include direct DNA damage, membrane destabilization, disruption of enzymatic activity,49 and Al) deposition.29 Many therapeutic interventions have been proposed to reduce cerebral oxidative stress,50 but two are particularly pertinent to personalized nutritional prevention of AD: intake of dietary antioxidants and reduction of homocysteine with B vitamin supplementation.
Dietary antioxidants that play a role in AD prevention include vitamins E and C, phenol compounds, and O3FAs. Though the role of vitamins C and E are controversial, a large prospective study from 2002 revealed decreased incidence of AD with high dietary intake of both vitamins (vitamin C from citrus fruits and certain vegetables, and vitamin E from nuts, grains, and egg yolks).27 Other prospective studies have shown no association between vitamin supplementation and AD risk,51 but this may reflect long-term consumption and different absorption patterns that confer a selective advantage of dietary intake over supplementation. Phenol compounds, particularly those found in blueberries, have demonstrated behavioral improvement in mouse models of AD52 and memory improvement in older adults,53 although no study has tested these compounds in AD prevention cohorts. The spice curcurmin also contains high levels of phenol antioxidants, but while it was found to reduce amyloid in an AD mouse model,54 its benefit in humans is unproven.55 Finally, O3FAs are found in myriad fish species and may exert AD preventative effects through their antioxidant properties rather than through improvement of dyslipidemia.56
Homocysteine is an amino acid metabolite of methionine that induces oxidative stress and apoptosis in cultured neurons,57 and B vitamin deficiency reduces metabolism of homocysteine and contributes to AD progression.58 Randomized controlled trials have shown that reduction of elevated homocysteine with B vitamin supplementation (specifically, folate and vitamins B6 and B12) reduces the rate of brain atrophy in patients with MCI.59 However, polymorphisms in the methylenetetrahy- drofolate reductase gene (MTHFR), which is associated with AD, alter metabolism of vitamin B12 and reduce the efficacy of oral cobalamin for homocysteine reduction. This suggests that the choice of cobalamin form (cobalamin, methylcobalamin, and injectable forms) for effective treatment may require individualization according to the patient’ s MTHFR status.60,61 In addition, B vitamin supplementation is beneficial regardless of homocysteine level for patients with elevated levels of the O3FA species eicosapentaenoic acid and docosahexaenoic acid.59
In summary, multiple nutritional interventions are available that may reduce the damaging effects of insulin resistance, dyslipidemia, and oxidative stress, but use of these interventions must be tailored to the individual clinical and genetic profile of the patient. Though certain biomarkers are less useful for primary care prevention of AD, full profiling and personalization of interventions may be beneficial in patients at higher risk of AD. Below, we describe preliminary results from the APC at WCMC that suggest cognitive improvement following implementation of this panoply of interventions.
Results from the Alzheimer’s Prevention Clinic
The APC at WCMC is the first AD prevention clinic in the United States and enrolls patients with a family history of AD into a clinical registry to monitor the effectiveness of personalized nutritional, lifestyle, and pharmacologic interventions delivered at the clinic. Evidence-based, multimodal nutritional interventions and pharmacologic therapies focus on the domains of insulin resistance, dyslipidemia, and oxidative stress outlined above, while the lifestyle interventions include exercise, cognitive activity, sleep hygiene, and social engagement. The results of patients’ cognitive testing, laboratory work, and blood biomarkers (associated with AD) are monitored longitudinally, and intervention plans are continuously reassessed (see Ref.62 for further details on the panoply of interventions offered at the APC).
Currently, there are 525 patients enrolled in the AD prevention/treatment clinical precision medicine registry. The average age of this cohort is 63 years (range 25 – 96 years); 57% of the patients are female and 43% are male; 91% of patients self- identify as white, 6% as Hispanic/Latino, 5% as black, and 1% as Asian or Indian; and 24% were born outside of the United States.
Of the 525 patients, 211 are classified as prevention cases (average age = 55.8 years) and 314 as treatment cases (average age = 68 years). Prevention patients are defined as those without subjective memory complaints and are diagnosed by a consensus panel of neurologists and neuropsychologists as having either no impairment or detectable cognitive impairment (DCI), the latter construct theorized to be the earliest, measureable phase of preclinical AD.2 DCI is assigned to patients who have no or minimal subjective cognitive and functional complaints, with impairment in more than one neuropsychological test (defined as a score more than 1.5 standard deviations below the patient’ s fully adjusted crystallized intelligence score, which is estimated from measures of reading ability and vocabulary). After meeting criteria for DCI, patients are then classified as DCI due to probable neurodegenerative disease if there are noncognitive symptoms in at least one domain (mood, personality, motor, sleep, smell, or taste), and when potential vascular, traumatic, and medical etiologies are ruled out. All patients undergo cognitive testing at the time of their first visit (baseline) and after 6 months (follow-up), with implementation of the personalized dietary and lifestyle interventions in the interim. Patients continue to undergo repeat cognitive testing at a frequency determined by their physician.
At the time of writing this article, 35 of the 211 prevention patients (16.5%) had completed baseline questionnaires and assessments, including cognitive testing at baseline and 6-month follow-up. One-way analysis of variance was performed to compare cognitive test scores across this initial 6-month intervention period, which showed significant improvement in the computer-based NIH toolbox on three cognitive tests representing measures of executive function: Dimensional Change Card Sort (F(1, 37) = 4.192, P = 0.048), Flanker Inhibitory Control and Attention (F(1, 39) = 8.588, P = 0.0060), and Pattern Comparison Processing Speed (F(1, 46) = 10.419, P = 0.002) (Fig. 1). These results are preliminary and, given the absence of a control group, may partly reflect improved performance associated with practice effect; however, the 6-month interval between assessments mitigates this concern. The convergence of apparent improvement in the working memory domain provides initial evidence of cognitive benefit, and we look forward to corroborating the findings in a larger sample with appropriate controls. Additional data analyses are underway to delineate changes in blood-based biomarkers, scores on validated scales of AD risk, subjective cognitive complaints, and AD pathology on neuroimaging. In addition, we will explore differential pharmacogenomic and nutrigenomic responses and assess whether baseline serum risk markers or measures of medical comorbidities (e.g., body fat percentage, hypertension) influence responsiveness to this intervention. At this time, the early results suggest that an evidence-based precision medicine approach to AD prevention is viable in a clinical setting and that changes implemented by motivated patients can improve cognitive function.
Figure 1.
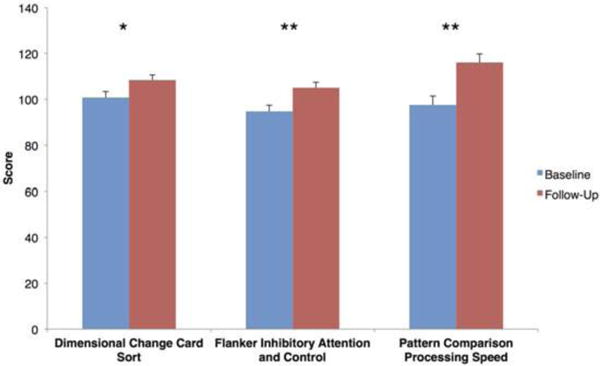
Cognitive testing results at baseline and 6-month follow-up for prevention patients at the Alzheimer’s Prevention Clinic. One-way analysis of variance was performed for 35 patients. Error bars represent standard error; one asterisk indicates P < 0.05, while two asterisks indicate P < 0.01.
Conclusion
AD is a major public health concern that generates enormous personal, social, and economic costs globally. While evaluation of potential treatments for established AD will continue, reduction of disease burden will also depend on early prevention efforts. AD prevention must incorporate all facets of lifestyle and systemic health that affect neural function: diet, exercise, cognitive activity, social engagement, and genomics.
Central to our prevention effort is the tenet that individualized risk factors require personalized prevention strategies. Risk factor reduction has dramatically decreased the incidence of cardiovascular disease and stroke, and a similar but personalized approach may reap even larger benefits in AD prevention. We hope that the growing evidence for multimodal preventative interventions and the promising early results of the APC will encourage the adoption of AD prevention techniques in primary care and neurology settings and inspire the foundation of future AD prevention clinics that will serve a dire need for an aging population.
Footnotes
Conflicts of interest
The authors declare no conflicts of interest.
References
- 1.Wimo A, et al. The worldwide economic impact of dementia 2010. Alzheimers Dement. 2013;9:1–11. doi: 10.1016/j.jalz.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 2.Sperling RA, et al. Toward defining the preclinical stages of Alzheimer’ s disease: recommendations from the National Institute on Aging- Alzheimer’ s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tan CC, Yu J-T, Tan L. Biomarkers for preclinical Alzheimer’ s disease. J Alzheimers Dis. 2014;42:1051–1069. doi: 10.3233/JAD-140843. [DOI] [PubMed] [Google Scholar]
- 4.Anstey KJ, et al. A self-report risk index to predict occurrence of dementia in three independent cohorts of older adults: the ANU-ADRI. PLoS One. 2014;9:e86141. doi: 10.1371/journal.pone.0086141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sperling RA, et al. The A4 study: stopping AD before symptoms begin? Sci Transl Med. 2014;6:228fs13. doi: 10.1126/scitranslmed.3007941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shah R. The role of nutrition and diet in Alzheimer disease: a systematic review. J Am Med Dir Assoc. 2013;14:398–402. doi: 10.1016/j.jamda.2013.01.014. [DOI] [PubMed] [Google Scholar]
- 7.Dangour AD, Whitehouse PJ, Rafferty K, et al. B-vitamins and fatty acids in the prevention and treatment of Alzheimer’s disease and dementia: a systematic review. J Alzheimers Dis. 2010;22:205–224. doi: 10.3233/JAD-2010-090940. [DOI] [PubMed] [Google Scholar]
- 8.Middleton LE, Yaffe K. Promising strategies for the prevention of dementia. Arch Neurol. 2009;66:1210–1215. doi: 10.1001/archneurol.2009.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci. 1991;12:383–388. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- 10.Spillantini MG, Goedert M. Tau protein pathology in neurodegenerative diseases. Trends Neurosci. 1998;21:428–433. doi: 10.1016/s0166-2236(98)01337-x. [DOI] [PubMed] [Google Scholar]
- 11.Willette AA, Bendlin BB, Starks EJ, et al. Association of insulin resistance with cerebral glucose uptake in late middle–aged adults at risk for Alzheimer disease. JAMA Neurol. 2015;72:1013. doi: 10.1001/jamaneurol.2015.0613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reaven GM. Insulin resistance and compensatory hyperinsulinemia: role in hypertension, dyslipidemia, and coronary heart disease. Am Heart J. 1991;121:1283–1288. doi: 10.1016/0002-8703(91)90434-j. [DOI] [PubMed] [Google Scholar]
- 13.Blass JP, Sheu RK-F, Gibson GE. Inherent abnormalities in energy metabolism in Alzheimer disease: interaction with cerebrovascular compromise. Ann NY Acad Sci. 2000;903:204–221. doi: 10.1111/j.1749-6632.2000.tb06370.x. [DOI] [PubMed] [Google Scholar]
- 14.Cheng B, Mattson MP. Glucose deprivation elicits neurofibrillary tangle-like antigenic changes in hippocampus neurons: prevention by NGF and bFGF. Exp Neurol. 1992;117:114–123. doi: 10.1016/0014-4886(92)90120-f. [DOI] [PubMed] [Google Scholar]
- 15.Farris W, Mansourian S, Chang Y, et al. Insulindegrading enzyme regulates the levels of insulin, amyloid l)-protein, and the l)-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci USA. 2003;100:4162–4167. doi: 10.1073/pnas.0230450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mosconi L, Pupi A, De Leon MJ. Brain glucose hypometabolism oxidative stress in preclinical Alzheimer’ s disease. Ann NY Acad Sci. 2008;1147:180–195. doi: 10.1196/annals.1427.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Costantini LC, et al. Hypometabolism as a therapeutic target in Alzheimer’s disease. BMC Neurosci. 2008;9(Suppl 2):S16. doi: 10.1186/1471-2202-9-S2-S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ehehalt R, Keller P, Haass C, et al. Amyloidogenic processing of the Alzheimer l)-amyloid precursor protein depends on lipid rafts. J Cell Biol. 2003;160:113–123. doi: 10.1083/jcb.200207113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cutler RG, Kelly J, Storie K, et al. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc Natl Acad Sci USA. 2004;101:2070–2075. doi: 10.1073/pnas.0305799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wolozin B. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch Neurol. 2000;57:1439. doi: 10.1001/archneur.57.10.1439. [DOI] [PubMed] [Google Scholar]
- 21.Fassbender K, Simons M, Bergmann C, et al. Simvastatin strongly reduces levels of Alzheimer’s disease l)-amyloid peptides Al)42 and Al)40 in vitro and in vivo. Proc Natl Acad Sci USA. 2001;98:5856–5861. doi: 10.1073/pnas.081620098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 23.Christen Y. Oxidative stress and Alzheimer disease. Am J Clin Nutr. 2000;71:621S–629S. doi: 10.1093/ajcn/71.2.621s. [DOI] [PubMed] [Google Scholar]
- 24.Tamagno E, Parola M, Bardini P, et al. l)-Site APP cleaving enzyme up-regulation induced by 4-hydroxynonenal is mediated by stress-activated protein kinases pathways. J Neurochem. 2005;92:628–636. doi: 10.1111/j.1471-4159.2004.02895.x. [DOI] [PubMed] [Google Scholar]
- 25.Pappolla MA, Chyan YJ, Omar TR, et al. Evidence of oxidative stress and in vivo neurotoxicity of l)-amyloid in a transgenic mouse model of Alzheimer’s disease: a chronic oxidative paradigm for testing antioxidant therapies in vivo. Am J Pathol. 1998;152:871–877. [PMC free article] [PubMed] [Google Scholar]
- 26.Yan SD, Chen X, Fu J, et al. RAGE and amyloid- l) peptide neurotoxicity in Alzheimer’s disease. Nature. 1996;382:685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- 27.Engelhart MJ. Dietary intake of antioxidants and risk of Alzheimer disease. JAMA. 2002;287:3223. doi: 10.1001/jama.287.24.3223. [DOI] [PubMed] [Google Scholar]
- 28.Luchsinger JA, Tang M-X, Shea S, et al. Antioxidant vitamin intake and risk of Alzheimer disease. Arch Neurol. 2003;60:203. doi: 10.1001/archneur.60.2.203. [DOI] [PubMed] [Google Scholar]
- 29.Craft S. Insulin resistance and Alzheimer’ s disease pathogenesis: potential mechanisms and implications for treatment. Curr Alzheimer Res. 2007;4:147–152. doi: 10.2174/156720507780362137. [DOI] [PubMed] [Google Scholar]
- 30.Craft S. Insulin resistance syndrome and Alzheimer’s disease: age- and obesity-related effects on memory, amyloid, and inflammation. Neurobiol Aging. 2005;26:65–69. doi: 10.1016/j.neurobiolaging.2005.08.021. [DOI] [PubMed] [Google Scholar]
- 31.Siu AL, U.S. Preventive Services Task Force Screening for abnormal blood glucose and type 2 diabetes mellitus: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2015;163:861–868. doi: 10.7326/M15-2345. [DOI] [PubMed] [Google Scholar]
- 32.Van Himbergen TM, et al. Biomarkers for insulin resistance and inflammation and the risk for all-cause dementia and Alzheimer disease: results from the Framingham Heart Study. Arch Neurol. 2012;69:594–600. doi: 10.1001/archneurol.2011.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kiddle SJ, et al. Plasma based markers of [11C] PiB- PET brain amyloid burden. PLoS One. 2012;7:e44260. doi: 10.1371/journal.pone.0044260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsuzaki T, et al. Insulin resistance is associated with the pathology of Alzheimer disease: the Hisayama study. Neurology. 2010;75:764–770. doi: 10.1212/WNL.0b013e3181eee25f. [DOI] [PubMed] [Google Scholar]
- 35.Ryan M. Diabetes and the Mediterranean diet: a beneficial effect of oleic acid on insulin sensitivity, adipocyte glucose transport and endothelium-dependent vasoreactivity. QJM. 2000;93:85–91. doi: 10.1093/qjmed/93.2.85. [DOI] [PubMed] [Google Scholar]
- 36.Morris MC, Tangney CC, Wang Y, et al. MIND diet score more predictive than DASH or Mediterranean diet scores. Alzheimers Dement. 2014;10:P166. [Google Scholar]
- 37.Mastroiacovo D, et al. Cocoa flavanol consumption improves cognitive function, blood pressure control, and metabolic profile in elderly subjects: the Cocoa, Cognition, and Aging (CoCoA) Study—a randomized controlled trial. Am J Clin Nutr. 2015;101:538–548. doi: 10.3945/ajcn.114.092189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hawley JA. Exercise as a therapeutic intervention for the prevention and treatment of insulin resistance. Diabetes Metab Res Rev. 2004;20:383–393. doi: 10.1002/dmrr.505. [DOI] [PubMed] [Google Scholar]
- 39.Smith JC, Nielson KA, Woodard JL, et al. Physical activity reduces hippocampal atrophy in elders at genetic risk for Alzheimer’s disease. Front Aging Neurosci. 2014;6:61. doi: 10.3389/fnagi.2014.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johnson JL, Slentz CA, Houmard JA, et al. Exercise training amount and intensity effects on metabolic syndrome (from studies of a targeted risk reduction intervention through defined exercise) Am J Cardiol. 2007;100:1759–1766. doi: 10.1016/j.amjcard.2007.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kivipelto M. Apolipoprotein E s4 allele, elevated midlife total cholesterol level, and high midlife systolic blood pressure are independent risk factors for late-life Alzheimer disease. Ann Intern Med. 2002;137:149. doi: 10.7326/0003-4819-137-3-200208060-00006. [DOI] [PubMed] [Google Scholar]
- 42.Di Paolo G, Kim T-W. Linking lipids to Alzheimer’s disease: cholesterol and beyond. Nat Rev Neurosci. 2011;12:284–296. doi: 10.1038/nrn3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morris MC. Dietary niacin and the risk of incident Alzheimer’s disease and of cognitive decline. J Neurol Neurosurg Psychiatry. 2004;75:1093–1099. doi: 10.1136/jnnp.2003.025858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kamanna VS, Kashyap ML. Mechanism of action of niacin. Am J Cardiol. 2008;101:20B–26B. doi: 10.1016/j.amjcard.2008.02.029. [DOI] [PubMed] [Google Scholar]
- 45.Caramelli P, Nitrini R, Maranhao R, et al. Increased apolipoprotein B serum concentration in Alzheimer’s disease. Acta Neurol Scand. 1999;100:61–63. doi: 10.1111/j.1600-0404.1999.tb00724.x. [DOI] [PubMed] [Google Scholar]
- 46.Fotuhi M, Mohassel P, Yaffe K. Fish consumption, long-chain omega-3 fatty acids and risk of cognitive decline or Alzheimer disease: a complex association. Nat Clin Pract Neurol. 2009;5:140–152. doi: 10.1038/ncpneuro1044. [DOI] [PubMed] [Google Scholar]
- 47.Lim GP. A diet enriched with the omega-3 fatty acid docosahexaenoic acid reduces amyloid burden in an aged Alzheimer mouse model. J Neurosci. 2005;25:3032–3040. doi: 10.1523/JNEUROSCI.4225-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harris WS, Miller M, Tighe AP, et al. Omega- 3 fatty acids and coronary heart disease risk: clinical and mechanistic perspectives. Atherosclerosis. 2008;197:12–24. doi: 10.1016/j.atherosclerosis.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 49.Coyle J, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:689–695. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- 50.Uttara B, Singh A, Zamboni P, et al. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol. 2009;7:65–74. doi: 10.2174/157015909787602823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Masaki KH, Losonczy KG, Izmirlian G, et al. Association of vitamin E and C supplement use with cognitive function and dementia in elderly men. Neurology. 2000;54:1265–1272. doi: 10.1212/wnl.54.6.1265. [DOI] [PubMed] [Google Scholar]
- 52.Joseph JA, Arendash G, Gordon M, et al. Blueberry supplementation enhances signaling and prevents behavioral deficits in an Alzheimer disease model. Nutr Neurosci. 2003;6:153–162. doi: 10.1080/1028415031000111282. [DOI] [PubMed] [Google Scholar]
- 53.Krikorian R, Shidler MD, Nash TA, et al. Blueberry supplementation improves memory in older adults. J Agric Food Chem. 2010;58:3996–4000. doi: 10.1021/jf9029332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lim GP, Chu T, Yang F, et al. The curry spice cur- cumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J Neurosci. 2001;21:8370–8377. doi: 10.1523/JNEUROSCI.21-21-08370.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baum L, Lam CWK, Cheung SKK, et al. Six- month randomized, placebo-controlled, double-blind, pilot clinical trial of curcumin in patients with Alzheimer disease. J Clin Psychopharmacol. 2008;28:110–113. doi: 10.1097/jcp.0b013e318160862c. [DOI] [PubMed] [Google Scholar]
- 56.Cole GM, Lim GP, Yang F, et al. Prevention of Alzheimer’ s disease: omega-3 fatty acid and phenolic antioxidant interventions. Neurobiol Aging. 2005;26:133–136. doi: 10.1016/j.neurobiolaging.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 57.Ho PI, Ortiz D, Rogers E, Shea TB. Multiple aspects of homocysteine neurotoxicity: glutamate excitotoxicity, kinase hyperactivation and DNA damage. J Neurosci Res. 2002;70:694–702. doi: 10.1002/jnr.10416. [DOI] [PubMed] [Google Scholar]
- 58.Morris MS. Homocysteine and Alzheimer’ s disease. Lancet Neurol. 2003;2:425–428. doi: 10.1016/s1474-4422(03)00438-1. [DOI] [PubMed] [Google Scholar]
- 59.Smith AD, et al. Homocysteine-lowering by B vitamins slows the rate of accelerated brain atrophy in mild cognitive impairment: a randomized controlled trial. PLoS One. 2010;5:e12244. doi: 10.1371/journal.pone.0012244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Peng Q, et al. The MTHFR C677T polymorphism contributes to increased risk of Alzheimer’s disease: evidence based on 40 case–control studies. Neurosci Lett. 2015;586:36–42. doi: 10.1016/j.neulet.2014.11.049. [DOI] [PubMed] [Google Scholar]
- 61.Hustad S, et al. The methylenetetrahydrofolate reductase 677C^ T polymorphism as a modulator of a B vitamin network with major effects on homocysteine metabolism. Am J Hum Genet. 2007;80:846–855. doi: 10.1086/513520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Seifan A, Isaacson RS. The Alzheimer’ s prevention clinic at Weill Cornell Medical College/New York- Presbyterian Hospital: risk stratification and personalized early intervention. J Prev Alzheimers Dis. 2015;2:254–266. doi: 10.14283/jpad.2015.81. [DOI] [PMC free article] [PubMed] [Google Scholar]