

Abstract
The majority of pancreatic ductal adenocarcinomas (PDAC) are diagnosed late so that surgery is rarely curative. Earlier detection could significantly increase the likelihood of successful treatment and improve survival. The aim of the study was to provide proof of principle that point mutations in key cancer genes can be identified by sequencing circulating free DNA (cfDNA) and that this could be used to detect early PDACs and potentially, premalignant lesions, to help target early effective treatment. Targeted next generation sequencing (tNGS) analysis of mutation hotspots in 50 cancer genes was conducted in 26 patients with PDAC, 14 patients with chronic pancreatitis (CP) and 12 healthy controls with KRAS status validated by digital droplet PCR. A higher median level of total cfDNA was observed in patients with PDAC (585 ng/ml) compared to either patients with CP (300 ng/ml) or healthy controls (175 ng/ml). PDAC tissue showed wide mutational heterogeneity, whereas KRAS was the most commonly mutated gene in cfDNA of patients with PDAC and was significantly associated with a poor disease specific survival (p=0.018). This study demonstrates that tNGS of cfDNA is feasible to characterise the circulating genomic profile in PDAC and that driver mutations in KRAS have prognostic value but cannot currently be used to detect early emergence of disease. Importantly, monitoring total cfDNA levels may have utility in individuals “at risk” and warrants further investigation.
Keywords: ctDNA, cfDNA, pancreatic ductal adenocarcinomas, KRAS, liquid biopsy
INTRODUCTION
Pancreatic cancer is the eleventh most common cancer in the UK, accounting for 3% of all newly diagnosed cancers, but is the fifth leading cause of cancer-related death [1]. The incidence increases with age and worldwide the five-year survival rate is ≤7% [2]. Late presentation is a feature of the disease with only 15-20% of cancers being resectable at diagnosis due to the location of the pancreas deep within the abdomen [3]. Due to the considerably shorter median survival of locally advanced (9-11 months) and metastatic disease (6-8 months) compared to resectable disease (13-20 months), there is a growing consensus that these should be viewed as prognostically (and possibly biologically) different disease entities [4]. Pancreatic ductal adenocarcinoma (PDAC) is the most common type of pancreatic cancer, representing around 80% of all cases. These tumours often have a high stromal content creating a “fortress-like” hypovascular barrier that is thought to impair the delivery of chemotherapeutics and promote aggressive neoplastic cell behaviour [5]. Both the location and composition of the tumours explains why imaging techniques often fail to detect early lesions and have difficulty distinguishing benign from malignant disease.
The causes of pancreatic cancer are unknown but numerous studies have identified genetic and modifiable risk factors including chronic pancreatitis (CP) [6]. A meta-analysis demonstrated a relative risk of 13.3 for CP patients developing PDAC but with a ten to twenty year lag between the incidences of pancreatitis and pancreatic malignancy [7]. Therefore, it is still undetermined whether CP is a precursor disease to PDAC. A progression model has been proposed starting with the precursor disease of pancreatic intraepithelial neoplasia (PanIN) and developing in a step-wise manner through the successive accumulation of specific mutations [8]. This includes activation of the oncogene KRAS at a relatively early stage, inactivation of the tumour suppressor gene CDKN2A at an intermediate stage and at a late stage, inactivation of the tumour suppressor genes TP53 and SMAD4 [8]. However, Notta et al [9] have suggested that the switch in certain cases can be a result of simultaneous mutations in these genetic drivers and it has not been confirmed that KRAS is the gatekeeper required for neoplasia despite its occurrence in 70-90% of PDAC tissue.
Currently the diagnosis of pancreatic cancer involves tissue biopsy but this is not always possible and can give false negatives due in part to the high stromal content of many pancreatic cancers. Moreover, tissue biopsy is expensive and difficult to perform, and can result in trauma to the patients [10]. A further disadvantage is that a tissue biopsy can only give site-specific information due to sampling techniques and may prevent a more complete overview of the tumour; this is particularly pertinent when considering the high genetic heterogeneity of pancreatic cancer if mutational profiling was to be conducted clinically [11]. An alternative approach under current investigation is the use of the tumour derived fraction of circulating free DNA (cfDNA) termed circulating tumour DNA (ctDNA) as a surrogate or “liquid biopsy” of the tumour. ctDNA originates from tumour cells undergoing apoptosis or necrosis during the natural development of the cancer from the primary tumour or metastatic lesions and therefore is considered to be less impacted by intratumour heterogeneity than a single tissue biopsy [12]. This cfDNA/ctDNA has a short half-life (approx. 2 hours) allowing for the evaluation of tumour changes in hours rather than weeks to months [13] making it ideal for progression and treatment monitoring [14]. In addition, liquid biopsies are cheaper, faster, more comfortable for patients and easy to repeat. ctDNA has been detected previously in pancreatic cancer, but with a sensitivity ranging from 26-100% due to studies differing in the platforms used and the genetic markers examined [3].
The detection of circulating tumour-derived biomarkers in blood could provide a relatively non-invasive approach for earlier detection of pancreatic ductal adenocarcinoma (PDAC). Here we aimed to provide proof of principle that point mutations in key cancer genes can be identified by targeted amplicon sequencing of cfDNA isolated from blood plasma, which, with appropriate validation, may detect early PDACs and, potentially, premalignant lesions, helping target effective treatment. To achieve this we profiled and compared the cfDNA from patients with PDAC to patients with CP and healthy controls. Where available, these profiles were compared to matched FFPE tumour tissue.
RESULTS
Panel validation
We first tested the sensitivity of the Cancer Hotspot Panel v2, which comprises 207 amplicons covering approximately 2,800 COSMIC mutations from 50 cancer genes, to detect hot spot mutations in KRAS, SMAD4, CDKN2A and TP53 [15, 16]. Using 1 ng, 5 ng and 10 ng DNA derived from three pancreatic cell lines (BxPC-3, PANC-1, MIA PaCa-2) [10], expected COSMIC mutations were detected in each cell line and the mutation frequency was consistent across the three DNA concentrations (Table 1).
Table 1. Validation of the Cancer Hotspot Panel v2 by detecting hotspot mutations in four commonly mutated genes.
| Gene mutation; amino acid (Variant Allele Frequency) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cell line | KRAS | TP53 | SMAD4 | CDKN2A | ||||||||
| 10 ng | 5 ng | 1 ng | 10 ng | 5 ng | 1 ng | 10 ng | 5 ng | 1 ng | 10 ng | 5 ng | 1 ng | |
| BxPC-3 | Wild Type | c.659A>G; Y220C (100%) | c.659A>G; Y220C (100%) | c.659A>G; Y220C (100%) | Homozygous deletion | Homozygous deletion | ||||||
| PANC-1 | c.35G>A; p.G12D (70.5%) | c.35G>A; p.G12D (70.2%) | c.35G>A; p.G12D (72.2%) | c.818G>A; R273H (100%) | c.818G>A; R273H (100%) | c.818G>A; R273H (100%) | Wild Type | Homozygous deletion | ||||
| MIA PaCa-2 | c.34G>T; p.G12C (100%) | c.34G>T; p.G12C (100%) | c.34G>T; p.G12C (100%) | c.742C>T; R248W (100%) | c.742C>T; R248W (100%) | c.742C>T; R248W (100%) | Wild Type | Homozygous deletion | ||||
Allele frequency shown in brackets where appropriate.
Total cfDNA levels are higher in PDAC than controls
We quantified total cfDNA levels in 12 healthy controls, 14 patients with CP and 26 patients with PDAC (resectable PDAC n=6, primary non-resectable PDAC n=5 and metastatic non-resectable PDAC n=15). Table 2 shows the clinicopathological criteria for these groups with a full summary per patient in Supplementary Table 1. The total cfDNA levels ranged from a median of 175 ng/ml for health controls to 300 ng/ml for CP and 585 ng/ml for PDAC. Levels were significantly different between healthy controls and both patients with CP (p=0.003) and PDAC (p=<0.001) but not between patients with CP and PDAC (p=0.086) (Figure 1). There were 3 patients with CP (35, 81 and 88) who died during the course of the study from a non-cancer related death who had the highest total cfDNA levels overall (600, 780 and 1000 ng/ml respectively).
Table 2. Clinicopathological characteristics of cfDNA patients.
| Healthy Volunteers N (%) | Chronic Pancreatitis N (%) | PDAC N (%) | |
|---|---|---|---|
| Total number of patients | 12 | 14 | 26 |
| cfDNA (ng/ml) | |||
| Median | 175 | 300 | 585 |
| Range | 100-340 | 130-1000 | 120-4180 |
| Age (years) | |||
| Median | 55 | 56 | 69 |
| Range | 40-83 | 19-71 | 40-84 |
| Sex | |||
| Male | 2 (17) | 10 (72) | 16 (62) |
| Female | 10 (83) | 3 (21) | 10 (38) |
| Unknown | 0 (0) | 1 (7) | 0 (0) |
| Ethnicity | |||
| Caucasian | na | na | 25 (96) |
| Other | 1 (4) | ||
| TNM Staging | |||
| I/II | - | - | 7 (27) |
| III | - | - | 2 (8) |
| IV | - | - | 16 (61) |
| Unknown | - | - | 1 (4) |
| Site of Tumour | |||
| Head | - | - | 19 (73) |
| Body, Tail, Neck | - | - | 6 (23) |
| Unknown | - | - | 1 (4) |
| Ca19.9 levels (U/ml) | |||
| ≤ 37 | - | - | 3 (11) |
| > 37 | - | - | 15 (58) |
| Not Tested | - | - | 8 (31) |
| Survival (days) | |||
| Median | na | 1482 | 148 |
| Range | 101-1579 | 17-1460 | |
| Disease Specific Survival | |||
| Alive | na | 11 (79) | 1 (4) |
| Cancer death | 0 (0) | 25 (96) | |
| Non Cancer death | 3 (21) | 0 (0) |
na; data not available, -; not applicable to patient cohort.
Figure 1. Stem and Leaf plot demonstrating the differences in cfDNA levels between healthy controls, CP and PDAC patients.
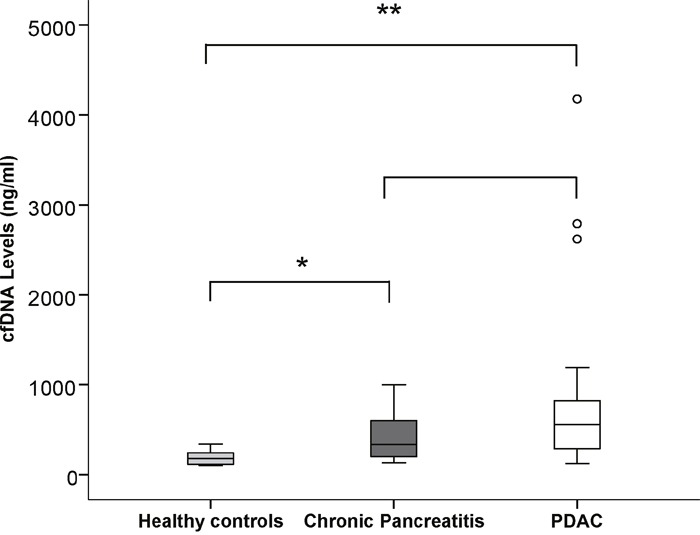
A significantly higher expression was noted in CP patients compared to healthy controls and in PDAC patients compared to healthy controls but not between CP patients and PDAC. * p<0.05, ** p<0.001.
In PDAC, the different subgroups (resectable PDAC, primary non-resectable PDAC and metastatic non-resectable PDAC) were not significantly different from one another and total cfDNA levels were not associated with disease specific survival (p=0.273, Hazard Ratio 1.000, 95% Confidence Interval 1.0-1.0), with the presence of distant metastasis (p=0.517) or with Ca19.9 levels (p=0.301).
Mutation profiling and prognostic value of somatic mutations in cfDNA
Mutation profiling was conducted using tNGS on cfDNA from 12 healthy controls, 14 patients with CP and 26 patients with PDAC. KRAS mutation status was validated by ddPCR in all cases with sufficient remaining cfDNA after tNGS. A detectable mutation was found in 1 healthy control (JAK2 p.V617F mutation, variant allelic fraction (VAF) =8%) and in 1 CP patient, multiple mutations were detected (in STK11, PDGFRA, KIT and KDR) suggesting genomic instability (Supplementary Table 2).
From the 26 PDAC cases, only 1 of the 6 patients with primary resectable PDAC had a mutation in their cfDNA (KRAS p.G12D mutation; VAF=8.7%). None of the primary non-resectable PDAC cases had any detectable mutations (Patient 76 was not amplifiable by tNGS but was found to be wild type for KRAS during validation of cases by ddPCR) (Table 3 and Supplementary Table 3). The 15 patients with metastatic non-resectable PDAC had disease in the peritoneum, lung, liver or a combination (Supplementary Table 1). Six patients (40%) had either a KRAS p.G12R (67%) mutation or a KRAS p.G12V (33%) mutation in cfDNA with variant allele frequencies ranging from 1.4 to 62.9% (Table 3 and Supplementary Table 3). Two of the detected KRAS mutations (patients 14 and 18) had variants called below the tNGS quality score applied to this study but were confirmed by ddPCR. Only 1 patient (31) had additional mutations to KRAS in cfDNA, in TP53 p.R248Q (VAF=17.4%) and SMAD4 p.P356R (VAF=13%). The VAFs were at similar frequencies to the KRAS p.G12V mutation (VAF=16.1%) suggesting these were derived from a single predominant clone.
Table 3. Summary of the mutations identified in the cfDNA of PDAC patients by tNGS.
| Patient ID | cfDNA (ng/ml) | Gene | Mutation | Variant Allele Frequency (%) | Coverage | COSMIC | |
|---|---|---|---|---|---|---|---|
| Resectable PDAC | 63 | 540 | KRAS* | WT | -- | 606 | -- |
| 69 | 800 | KRAS* | WT | -- | 482 | -- | |
| 72 | 250 | KRAS | WT | -- | 507 | -- | |
| 92 | 220 | KRAS | WT | -- | 980 | -- | |
| 102 | 840 | KRAS* | WT | -- | 1448 | -- | |
| 104 | 450 | KRAS* | p.G12D | 8.7 | 473 | COSM521 | |
| Primary Non-Resectable PDAC | 16 | 232 | KRAS | WT | -- | 853 | -- |
| 19 | 300 | KRAS* | WT | -- | 2040 | -- | |
| 34 | 1190 | KRAS* | WT | -- | 1257 | -- | |
| 76** | 354 | KRAS | WT | na | na | na | |
| 80 | 610 | KRAS* | WT | -- | 464 | - | |
| Metastatic Non-Resectable PDAC | 12 | 120 | KRAS | WT | -- | 431 | -- |
| 13 | 270 | KRAS* | WT | -- | 456 | -- | |
| 14 | 710 | KRAS* | p.G12V | 1.4 | 1094 | COSM520 | |
| 15 | 700 | KRAS* | WT | -- | 2000 | -- | |
| 18 | 700 | KRAS* | p.G12R | 1.5 | 834 | COSM518 | |
| 21 | 2790 | KRAS* | p.G12R | 1.8 | 925 | COSM518 | |
| 24 | 300 | KRAS | WT | -- | 1155 | -- | |
| 26 | 4180 | KRAS* | WT | -- | 330 | -- | |
| 27 | 2620 | KRAS* | p.G12R | 62.9 | 705 | COSM518 | |
| 31 | 430 | KRAS* | p.G12V | 16.1 | 996 | COSM520 | |
| SMAD4 | p.P356R | 13.0 | 1642 | COSM339351 | |||
| TP53 | p.R248Q | 17.4 | 3857 | COSM10662 | |||
| 41 | 1150 | KRAS* | WT | -- | 388 | -- | |
| 53 | 970 | KRAS | p.G12R | 23.8 | 408 | COSM518 | |
| 68 | 280 | KRAS* | WT | -- | 1338 | -- | |
| 73 | 710 | KRAS* | WT | -- | 930 | -- | |
| 91 | 560 | KRAS* | WT | -- | 597 | -- |
--; not relevant as wild type, *; validated by ddPCR, **; identified by ddPCR validation, na; data not available.
Comparing the cfDNA KRAS mutation status to clinicopathological criteria for all PDAC cases (Table 4), there was no significant association between KRAS mutations and distant metastasis (p=0.190). The presence of KRAS mutations in cfDNA was significantly associated with the tumour being sited at the body, tail or neck of the pancreas (p=0.015) compared to the head of the pancreas. KRAS status did not associate with TNM stage (p=0.109) or Ca19.9 levels (p=1.00).
Table 4. Association of KRAS in the cfDNA of PDAC patients with clinicopathological criteria.
| Variable | KRAS | ||
|---|---|---|---|
| NoN (%) | YesN (%) | p value | |
| Total number of PDAC patients | 19 | 7 | |
| Site of Tumour | |||
| Head | 17 (90) | 2 (29) | 0.015 |
| Body, Tail, Neck | 2 (10) | 4 (57) | |
| Unknown | 0 (0) | 1 (14) | |
| Metastasis | |||
| No | 9 (47) | 1 (14) | 0.190 |
| Yes | 10 (53) | 6 (86) | |
| TNM Staging | |||
| I/II | 7 (37) | 0 (0) | 0.109 |
| III | 2 (10) | 0 (0) | |
| IV | 10 (53) | 6 (86) | |
| Unknown | 0 (0) | 1 (14) | |
| Ca19.9 levels (U/ml) | |||
| ≤ 37 | 2 (10) | 1 (14) | 1.000 |
| > 37 | 11 (58) | 4 (57) | |
| Unknown | 6 (32) | 2 (29) | |
The presence of KRAS mutations in the cfDNA of patients with PDAC was associated with poor disease specific survival (p=0.018, hazard ratio=2.889, 95% confidence interval (CI) =1.2-7.3) (Figure 2 and Table 5). The median survival time was 60 days for KRAS positive PDAC and 197 days for KRAS negative PDAC. In general, the presence of metastasis (p=0.013), the site of the tumour (p<0.001) and the TNM stage (p=0.015) in the PDAC cases associated with disease specific survival but Ca19.9 levels (p=0.620), gender (p=0.833) and age (p=0.655) did not (Table 5).
Figure 2. Kaplan Meier survival curve demonstrating KRAS mutation (dotted line) significantly associates with a poorer disease specific survival vs KRAS wild type (solid line) when detected in cfDNA of PDAC patients (p=0. 018).
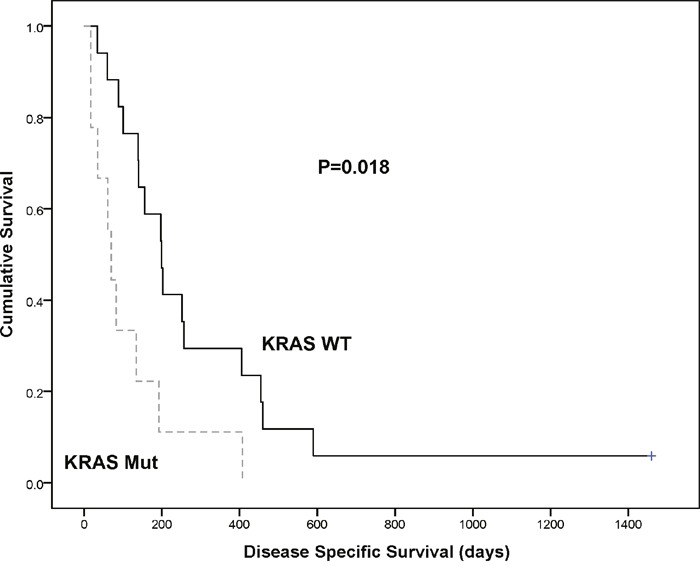
Table 5. Log rank analysis of KRAS mutation status in the cfDNA of PDAC patients and relevant clinicopathological criteria with disease specific survival.
| Variable | Disease Specific Survival | |||||
|---|---|---|---|---|---|---|
| N (%) | Median Survival (days) | X2 value | p value | Hazard Ratio | 95% Confidence Interval | |
| Total number of PDAC patients | 26 | 148 | ||||
| KRAS | ||||||
| WT | 19 (73) | 197 | 5.589 | 0.018 | 2.889 | 1.2 - 7.3 |
| Mutant | 7 (27) | 60 | ||||
| Metastasis | ||||||
| No | 10 (39) | 255 | 7.007 | 0.013 | 3.399 | 1.3 - 8.9 |
| Yes | 16 (61) | 111 | ||||
| Site of Tumour | ||||||
| Head | 19 (73) | 199 | 12.592 | <0.001 | 11.266 | 3.1 - 41.6 |
| Body, Tail, Neck | 6 (23) | 35 | ||||
| Unknown | 1 (4) | 60 | ||||
| TNM Staging | ||||||
| I/II | 7 (27) | 257 | 6.980 | 0.015 | 1.995 | 1.1 - 3.5 |
| III | 2 (8) | 421 | ||||
| IV | 16 (61) | 111 | ||||
| Unknown | 1 (4) | 60 | ||||
| Ca19.9 levels (U/ml) | ||||||
| ≤ 37 | 3 (11) | 455 | 0.261 | 0.620 | 1.385 | 0.4 - 5.0 |
| > 37 | 15 (58) | 140 | ||||
| Unknown | 8 (31) | 145 | ||||
| Gender | ||||||
| Male | 16 (61) | 195 | 0.044 | 0.833 | 1.092 | 0.5 - 2.5 |
| Female | 10 (39) | 117 | ||||
| Age (years) | ||||||
| < 65 | 7 (27) | 139 | 0.200 | 0.655 | 0.818 | 0.3 - 2.0 |
| ≥ 65 | 19 (73) | 192 | ||||
Mutation profiling of matched tumour tissue
Eleven of the cfDNA patients (4 resectable PDAC, 2 primary non-resectable PDAC and 5 metastatic non-resectable PDAC) had matching primary tumour tissue. The tissue was analysed by tNGS (Supplementary Table 4) and KRAS mutation status validated by ddPCR. No mutations were detected in the cfDNA of these patients. Eight out of the 11 tumour tissues (73%) had a KRAS mutation with p.G12D (4/8, 50%) being the predominant mutation, followed by p.G12V (3/8, 38%). Two of the KRAS mutations (patients 34 and 15) had a quality score below the tNGS threshold for variant calling in this study, but were validated by ddPCR. A further KRAS mutation (in case 72) was identified by ddPCR but not by tNGS as it was below the limit of detection for the panel (see Supplementary Table 5) (1/8, 12%).
Additional heterogeneous mutations were detected in the tissue of 5 patients. Of note was a CDKN2A p.R80* mutation in patient 41 (average VAF=31.1% based on two tissue regions, see Supplementary Table 5), detected at a similar allele frequency to the KRAS mutation (average VAF=31.2% based on two tissue regions see Supplementary Table 5). The remaining mutations were detected at a lower VAF suggesting that these were sub-clonal to mutations in KRAS. There was no pattern to the types of mutations or prevalence between the different sub groups of PDAC.
Mutational heterogeneity is seen in tumour tissues
In 8 patients with PDAC, multiple tumour tissue regions were available (Supplementary Table 5) and these were analysed by tNGS to assess intratumoural heterogeneity. For KRAS, only two patients had a mutation detected in all regions (patient 36, KRAS p.G12V and patient 41, KRAS p.G12D). In 3 patients (patients 100, 102, 51), KRAS was detected by tNGS in some regions but was below the limit of detection for the panel in other regions and therefore only identified by ddPCR.
In patient 40, region 40.2 had a KRAS p.G12C variant called below the tNGS quality score cut off but was validated by ddPCR. In region 40.1, no KRAS mutation could be detected by tNGS or ddPCR (the number of positive mutation droplets for ddPCR was below the cut off applied for the study). In patient 68, all three regions differed. Region 68.1 had a KRAS p.G12D detected by tNGS, in region 68.4 KRAS was below the limit of detection for tNGS and only identified through ddPCR validation and in region 68.3 there was no detectable KRAS by either method. In patient 72, the KRAS mutations were only detected during ddPCR validation. This demonstrates the mutational heterogeneity in different regions of pancreatic tumour tissues.
There was even greater heterogeneity with gene variants other than KRAS across all patients as the majority were solely found in only one of the tissue regions. The exceptions were CDKN2A p.R80* in patient 41, which was found in both tissue regions and the deletions in EGFR p.E746_A750delELREA, which were detected in 2 out of 3 regions in patient 51. Patient 45 was the only case that had two different KRAS mutations detectable by tNGS in the same tumour tissue region, p.G12D (VAF=17%) and p.G12C (VAF=15.4%).
DISCUSSION
In this study we aimed to provide proof of principle that point mutations in key cancer genes could be identified by sequencing cfDNA and provide a relatively non-invasive approach to detecting early PDACs and potentially, premalignant lesions, to help target early effective treatment. Only 1 CP patient had detectable somatic mutations in cfDNA, and of note was the mutation in the tumour suppressor gene, STK11, that encodes for a serine/threonine kinase, a known genetic risk factor for PDAC [6]. Therefore, it may have been of value to monitor this patient through serial blood samples although the patient did not develop PDAC during the course of the study. Previous studies have detected KRAS mutations in the cfDNA of patients with CP, which were not detectable in this study. However, these studies followed the patients for a period of time (ranging from 6 to 64 months), and none of the patients developed pancreatic cancer [17, 18]. Due to the difficulty in diagnosing PDAC especially in patients with CP, a separate study [7] looked at the relationship between Ca19.9 combined with KRAS mutation and suggested that in a CP patient with normal Ca19.9 levels and no KRAS mutations, the diagnosis of pancreatic cancer can be excluded with almost certainty. A study to validate this finding is warranted due to the inadequacy of standard imaging and tissue biopsies and we would also recommend inclusion of the preneoplastic lesions, PanINs 1a/b, 2 and 3 [8]. The JAK2 p.V617F mutation found in the cfDNA of one healthy control is a pathogenic risk factor but is not directly related to PDAC [19]. The lack of significance between the total cfDNA levels in CP patients and PDAC (p=0.086) may be explained by the inflammatory nature of chronic pancreatitis: Inflammation causes cell death, with ensuing release of cfDNA into the blood stream [20].
In the cfDNA of patients with PDAC, KRAS was the most frequently detected mutation. Castells et al were the first to detect circulating mutant KRAS genes in plasma from patients with PDAC in 1999 [17] and mutations in KRAS are reported to be among the earliest to occur during carcinogenesis [21, 22] and therefore may be founder mutations [8]. Overall 27% (7/26) of patients with PDAC had a detectable KRAS mutation in plasma cfDNA by tNGS, which is in the range reported in other studies (27-81%) (reviewed in [23]). This range of KRAS mutation detection is mainly due to the following factors: different stages of pancreatic cancer examined (i.e. late stage unresectable 81% [24] vs earlier stage primary operable 27% [17]) and differences in methods and sensitivity of these methods. This study was designed as a proof of concept, to include all the different stages of PDAC, and to assess an up to date, diagnostically relevant technique, tNGS.
We found that the presence of mutant KRAS was associated with a poor disease specific survival (p=0.018) (60 days vs 197 days). This has been observed in previous studies [17, 21, 23, 25] and interestingly Hadano et al [26] found that the presence of mutant KRAS was only significantly associated with a poor prognosis if it was detected in the plasma samples but not if the expression was only analysed in the matching tissue samples. Another study found no statistical significance [18] but as stated there is a large variability in previous studies in patient groups, technical approaches and sample preparation. In this study, the presence of KRAS mutations was also significantly associated with the tumour being sited at the body, tail or neck of the pancreas (p=0.015), known to have an inferior survival outcome compared to the tumour being sited in the head of the pancreas [27].
In the cfDNA of a metastatic non-resectable PDAC patient, a SMAD4 (VAF=13%) and TP53 mutation (VAF=17.4%) was detected at a similar frequency to the KRAS mutation (VAF=16.1%) but there was no CDK2NA mutation. This differs to the progression model proposed by Hruban et al [8], however, the model is based on progression from PanIN and it is not known if the PDAC lesions in this study originated from PanIN or other preneoplastic conditions.
In the 11 patients for whom matching cfDNA and tumour tissue samples were available, no mutations were detected in the cfDNA despite detection of mutations in the tissue. PDAC is known to be a hypovascular tumour and it is thought that due to this, DNA may not be released into the circulation as readily, especially in the earlier stages of tumour development [21]. This may explain the low number of patients with mutations in cfDNA overall and compounding this, PDAC cancers have a low median tumour cellularity of around 30% [22] due to a high stromal content [5]. The sequencing depth at the KRAS locus also varies due to the efficiency of the PCR reaction at that locus and because both cfDNA and FFPE samples can vary in quality. The more fragmented a sample is, the more difficult it is to amplify.
In the PDAC tissue samples, a lot of intertumoral heterogeneity was observed. A previous comprehensive genetic analysis of 24 pancreatic cancers [11], showed that the genetic basis of pancreatic cancer is extremely complex and heterogeneous with an average of 63 relevant genetic abnormalities per tumour, which was organized into 12 functional cancer relevant pathways.
We also observed intratumoral heterogeneity between different regions within the same tumour, which is an important considerations for any future tissue based biomarker analysis as only taking samples from one part of the tumour is likely to miss this. In later stages of the disease, sampling purely at the primary tumour site will also not be able to take into account the intermetastatic heterogeneity between metastatic sites within the patient, which would affect the ability to personalise treatment. With the additional complication of the inaccessibility of pancreatic tumours, the less invasive liquid biopsy approach is still a viable alternative to overcome these limitations in PDAC.
From the important mutations known to occur in PDAC [28], TP53 was the most frequently observed mutation in the tissue in association with KRAS but was generally at a lower frequency. Only one metastatic case had a detectable CDK2NA mutation. Further somatic mutations of note were the EGFR deletions. Erlotinib is the only targeted agent that has demonstrated a statistically significant effect on overall survival when combined with Gemcitabine in advanced pancreatic cancer [29]. Detection of deletions via tNGS is not optimal and therefore future studies would use ddPCR validation. Unfortunately all of the cases with an EGFR mutation also had a KRAS mutation and it has been observed in colorectal cancer (although inconclusive in NSCLC) that a high frequency of KRAS mutations limits the benefits of EGFR inhibitors [30] as it leaves the KRAS protein turned “on” and signalling within the cancer cell continues regardless of the fact that the EGFR receptor is blocked.
To compare the KRAS mutation frequencies to other studies, all PDAC patients were taken into account (15 cfDNA only, 11 cfDNA and tissue, 7 tissue only), therefore overall 27% (7/26) of PDAC had a detectable KRAS mutation in plasma with 78% percent (14/18) in tissue. Castells et al [17] reported a similar ratio with 72% (28/39) in the primary tumour and 32% (9/28) showing an identical alteration in the corresponding plasma. From the 20 PDAC patients with known KRAS mutation types, 35% had a p.G12D mutation (7/20), 35% a p.G12V (7/20), 20% a p.G12R (4/20) and 15% a p.G12C (3/20) representing the four most common KRAS mutations at codon 12 (patient 45 had a p.G12C and p.G12D mutation). In Europe and China, the most common alterations in the KRAS oncogene are reported to be in the order of p.G12D (c.35G>A), p.G12V (c.35G>T), p.G12R (c.34G>C) and p.G12C (c.34G>T), whereas in the USA the order has been reported to be p.G12D (c.35G>A), p.G12V (c.35G>T), p.G12C (c.34G>T) and p.G12R (c.34G>C) (reviewed in [23] and [31]). In a separate study of a Japanese cohort, p.G12V had the highest prevalence (37.3%), correlating with a shorter survival [32] and found to be more invasive in vitro [33] but this relationship has differed in other studies [34]. We also found one case that had two KRAS mutations coexisting at similar frequencies (p.G12D VAF=17% and p.G12C VAF=15.4%) something that has been mainly observed in pancreatic cancer with associated PanIN [35].
In conclusion, we demonstrated mutational heterogeneity in primary tumour tissue of patients with PDAC, and found circulating tumour derived DNA in 27% of patients based on detection of mutations in the KRAS gene. These results demonstrate that point mutations in key cancer genes can be identified by tNGS of cfDNA. Moreover, monitoring the levels of cfDNA in PDAC patients for driver mutations in the KRAS gene has prognostic value warranting a larger study. Unfortunately, this study was not able to help detect early PDACs or premalignant lesions of pancreatic cancer but since this study, there have been more advances in tNGS, allowing for better detection of low frequency variants, and combined with the recommendations for standardization of study cohorts and extraction methods, this may help with future validation of cfDNA biomarkers.
MATERIALS AND METHODS
Patients and samples
A total of 59 patients were analysed for the study; 51 had amplifiable cfDNA from plasma and 18 had amplifiable DNA from formalin fixed paraffin embedded (FFPE) tissue samples. Full details including clinicopathological characteristics are presented in Supplementary Table 1. Blood sample collection was conducted in accordance with the Declaration of Helsinki and all samples were taken prior to treatment. The study protocol was approved by the Leicestershire, Rutland and Northamptonshire Research Ethics Committee (University Hospitals of Leicester NHS Trust; REC reference number: 7176). All eligible patients during the course of the study gave written informed consent prior to participation. Blood was taken by venepuncture into K2 EDTA-containing collection tubes (BD Biosciences, San Jose, CA, USA) and processed as described previously [36]. The median age for all patients was 66 with a range of 19-84. The median survival was 1482 days for patients with CP and 150 days for patients with PDAC.
Cell culture
Pancreatic cancer cell lines (PANC-1, BxPC-3 and MIA PaCa-2) obtained from ATCC (Manassas, Virginia, USA) were used for assay development and validation. Cells were cultured in DMEM + 10% FBS (Sigma-Aldrich, St Louis, Mo, USA). Prior to extraction cells were trypsinised and washed in 1X PBS (Sigma). Cells were pelleted and stored at −80°C. Cell pellets were reconstituted in 200μl of 1X PBS before extraction.
Extraction and quantitation of DNA
cfDNA was extracted from plasma using the QIAamp Circulating Nucleic Acid kit (Qiagen, Hilden, Germany) as per the manufacturer's instructions. DNA was extracted from pancreatic cancer cell lines using the DNA blood mini kit (Qiagen) according to the manufacturer's instructions. FFPE tissue blocks with matching H&E were retrieved from the histopathology archive and reviewed by a consultant Histopathologist. DNA from FFPE samples was extracted using the GeneRead DNA FFPE Kit (Qiagen). DNA was quantified using the Qubit 2.0 fluorometer according to manufacturers’ instructions.
Targeted next generation sequencing (tNGS)
Reactions were set up using the Ion AmpliseqTM Cancer Hotspot Panel V2 (4475346, Thermo Fisher Scientific, Waltham, Mass., USA) using 10 ng of cfDNA or 20 ng FFPE DNA. Samples were sequenced on the Ion Personal Genome Machine (Ion PGMTM) using Ion 316TM v2 chips according to manufacturer's instructions. Sequencing data was analysed through the Torrent SuiteTM v4.2.0. Reads were aligned against the human genome (hg19) using Alignment v4.0-r77189 and variants called using the coverageAnalysis v4.0-r77897 and variantCaller v4.0-r76860, respectively [10]. COSMIC IDs were obtained using COSMIC v76 (http://cancer.sanger.ac.uk/cosmic, last accessed 2nd May 2016). All mutations with a quality score below 20 were omitted and all variants detected in the first or last 10 bases of an amplicon were omitted as likely mispriming events [37]. All variants detected were also manually confirmed across all samples using the Integrated Genomics Viewer 5.01. The mutant variant allele frequency (VAF) was calculated as the proportion of total reads at a site, which contained the variant allele.
Droplet digital PCR (ddPCR)
Droplet digital PCR was used to validate the KRAS mutational status of patient samples where possible. 5 ng of cfDNA and 10 ng of FFPE DNA was used in conjunction with Bio-Rad's ddPCRTM KRAS G12/G13 Screening kit according to manufacturers’ instructions (dHsaMDV2510586, dHsaMDV2510584, dHsaMDV2510596, dHsaMDV2510590, dHsaMDV25 10588, dHsaMDV2510592, dHsaMDV2510598, Bio-Rad, Hercules, Calif., USA) using a Bio-Rad QX200 digital droplet PCR system as previously described [38]. For all assays, no template controls were run to determine lack of contamination. All assays with ≥4 mutant droplets were considered positive for the KRAS mutation. Raw fluorescence amplitude was analysed using the Quantasoft version 1.6.6.0320 software to obtain the fractional abundance (FA) of mutant DNA alleles to total (mutant plus wild type) DNA alleles.
Statistical analysis
SPSS version 22 statistical software package was used. A stem and leaf plot was constructed to demonstrate the difference in cfDNA levels between the different groups of patients and the Independent Samples Mann-Whitney U test. For PDAC patients, disease specific survival was calculated from the date of the blood sample until 15th September 2015 when any remaining survivors were censored. The Mann Whitney U test was used to examine the levels of cfDNA between PDAC patients with and without distant metastasis and to the Ca19.9 levels. The association between the KRAS status and metastasis was assessed using a Fisher's exact test due to there being less than five cases in some cells. Survival analysis comparing age, gender or KRAS status with disease specific survival for the PDAC patients was conducted using the log rank test and Kaplan-Meier curves. Hazard ratios and 95% confidence intervals where determined using Cox regression analysis as was the relationship between cfDNA levels and disease specific survival. The correlation between KRAS and distant metastasis was confirmed using Spearman rank-order.
Abbreviations
- PDAC
Pancreatic Ductal Adenocarcinoma
- cfDNA
Circulating free DNA
- ctDNA
Circulating tumour DNA
- CP
Chronic Pancreatitis
- tNGS
Targeted Next Generation Sequencing
- ddPCR
Droplet Digital PCR
- PanIN
Pancreatic Intraepithelial Neoplasia
SUPPLEMENTARY MATERIALS TABLES
Acknowledgments
We thank Pancreatic Cancer UK and Hope Against Cancer for funding the research and the Cancer Research UK Leicester Centre for bioinformatics support. We thank the University of Leicester Core Biotechnology Histology staff (A Gillies and L Potter) for supporting tissue processing.
Author contributions
All authors have confirmed that they have contributed to the intellectual content of this paper in one or more of the following three requirements: (a) significant contributions to the conception and design, acquisition of data, or analysis and interpretation of data; (b) drafting or revising the article for intellectual content; and (c) final approval of the published article.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to report.
FUNDING
This study was supported by grants from Pancreatic Cancer UK and from Hope Against Cancer.
REFERENCES
- 1.Cancer RU. Pancreatic cancer statistics. 2016 http://www.cancerresearchukorg/health-professional/cancer-statistics/statistics-by-cancer-type/pancreatic-cancer (01/11/2016)
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 3.Lewis AR, Valle JW, McNamara MG. Pancreatic cancer: are “liquid biopsies” ready for prime-time? World J Gastroenterol. 2016;22:7175–7185. doi: 10.3748/wjg.v22.i32.7175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heinemann V, Haas M, Boeck S. Systemic treatment of advanced pancreatic cancer. Cancer Treat Rev. 2012;38:843–853. doi: 10.1016/j.ctrv.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 5.Neesse A, Michl P, Frese KK, Feig C, Cook N, Jacobetz MA, Lolkema MP, Buchholz M, Olive KP, Gress TM, Tuveson DA. Stromal biology and therapy in pancreatic cancer. Gut. 2011;60:861–868. doi: 10.1136/gut.2010.226092. [DOI] [PubMed] [Google Scholar]
- 6.Becker AE, Hernandez YG, Frucht H, Lucas AL. Pancreatic ductal adenocarcinoma: risk factors, screening, and early detection. World J Gastroenterol. 2014;20:11182–11198. doi: 10.3748/wjg.v20.i32.11182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raimondi S, Lowenfels AB, Morselli-Labate AM, Maisonneuve P, Pezzilli R. Pancreatic cancer in chronic pancreatitis; aetiology, incidence, and early detection. Best Pract Res Clin Gastroenterol. 2010;24:349–358. doi: 10.1016/j.bpg.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 8.Hruban RH, Wilentz RE, Kern SE. Genetic progression in the pancreatic ducts. Am J Pathol. 2000;156:1821–1825. doi: 10.1016/S0002-9440(10)65054-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Notta F, Chan-Seng-Yue M, Lemire M, Li Y, Wilson GW, Connor AA, Denroche RE, Liang SB, Brown AM, Kim JC, Wang T, Simpson JT, Beck T, et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature. 2016;538:378–382. doi: 10.1038/nature19823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guttery DS, Page K, Hills A, Woodley L, Marchese SD, Rghebi B, Hastings RK, Luo J, Pringle JH, Stebbing J, Coombes RC, Ali S, Shaw JA. Noninvasive detection of activating estrogen receptor 1 (ESR1) mutations in estrogen receptor-positive metastatic breast cancer. Clin Chem. 2015;61:974–982. doi: 10.1373/clinchem.2015.238717. [DOI] [PubMed] [Google Scholar]
- 11.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, Hong SM, Fu B, Lin MT, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diaz LA, Jr, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol. 2014;32:579–586. doi: 10.1200/JCO.2012.45.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, Thornton K, Agrawal N, Sokoll L, Szabo SA, Kinzler KW, Vogelstein B, Diaz LA., Jr Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14:985–990. doi: 10.1038/nm.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wan JC, Massie C, Garcia-Corbacho J, Mouliere F, Brenton JD, Caldas C, Pacey S, Baird R, Rosenfeld N. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer. 2017;17:223–238. doi: 10.1038/nrc.2017.7. [DOI] [PubMed] [Google Scholar]
- 15.Deer EL, Gonzalez-Hernandez J, Coursen JD, Shea JE, Ngatia J, Scaife CL, Firpo MA, Mulvihill SJ. Phenotype and genotype of pancreatic cancer cell lines. Pancreas. 2010;39:425–435. doi: 10.1097/MPA.0b013e3181c15963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sausen M, Phallen J, Adleff V, Jones S, Leary RJ, Barrett MT, Anagnostou V, Parpart-Li S, Murphy D, Kay Li Q, Hruban CA, Scharpf R, White JR, et al. Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nat Commun. 2015;6:7686. doi: 10.1038/ncomms8686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Castells A, Puig P, Mora J, Boadas J, Boix L, Urgell E, Sole M, Capella G, Lluis F, Fernandez-Cruz L, Navarro S, Farre A. K-ras mutations in DNA extracted from the plasma of patients with pancreatic carcinoma: diagnostic utility and prognostic significance. J Clin Oncol. 1999;17:578–584. doi: 10.1200/JCO.1999.17.2.578. [DOI] [PubMed] [Google Scholar]
- 18.Maire F, Micard S, Hammel P, Voitot H, Levy P, Cugnenc PH, Ruszniewski P, Puig PL. Differential diagnosis between chronic pancreatitis and pancreatic cancer: value of the detection of KRAS2 mutations in circulating DNA. Br J Cancer. 2002;87:551–554. doi: 10.1038/sj.bjc.6600475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kocher HM, Mears L, Lea NC, Raj K, Mufti GJ. JAK V617F missense mutation is absent in pancreatic cancer. Gut. 2007;56:1174–1175. doi: 10.1136/gut.2006.117754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gornik I, Wagner J, Gasparovic V, Lauc G, Gornik O. Free serum DNA is an early predictor of severity in acute pancreatitis. Clin Biochem. 2009;42:38–43. doi: 10.1016/j.clinbiochem.2008.09.121. [DOI] [PubMed] [Google Scholar]
- 21.Takai E, Totoki Y, Nakamura H, Morizane C, Nara S, Hama N, Suzuki M, Furukawa E, Kato M, Hayashi H, Kohno T, Ueno H, Shimada K, et al. Clinical utility of circulating tumor DNA for molecular assessment in pancreatic cancer. Sci Rep. 2015;5:18425. doi: 10.1038/srep18425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Song S, Nones K, Miller D, Harliwong I, Kassahn KS, Pinese M, Pajic M, Gill AJ, Johns AL, Anderson M, Holmes O, Leonard C, Taylor D, et al. qpure: a tool to estimate tumor cellularity from genome-wide single-nucleotide polymorphism profiles. PLoS One. 2012;7:e45835. doi: 10.1371/journal.pone.0045835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen H, Tu H, Meng ZQ, Chen Z, Wang P, Liu LM. K-ras mutational status predicts poor prognosis in unresectable pancreatic cancer. Eur J Surg Oncol. 2010;36:657–662. doi: 10.1016/j.ejso.2010.05.014. [DOI] [PubMed] [Google Scholar]
- 24.Mulcahy HE, Lyautey J, Lederrey C, qi Chen X, Anker P, Alstead EM, Ballinger A, Farthing MJ, Stroun M. A prospective study of K-ras mutations in the plasma of pancreatic cancer patients. Clin Cancer Res. 1998;4:271–275. [PubMed] [Google Scholar]
- 25.Tjensvoll K, Lapin M, Buhl T, Oltedal S, Steen-Ottosen Berry K, Gilje B, Soreide JA, Javle M, Nordgard O, Smaaland R. Clinical relevance of circulating KRAS mutated DNA in plasma from patients with advanced pancreatic cancer. Mol Oncol. 2016;10:635–643. doi: 10.1016/j.molonc.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hadano N, Murakami Y, Uemura K, Hashimoto Y, Kondo N, Nakagawa N, Sueda T, Hiyama E. Prognostic value of circulating tumour DNA in patients undergoing curative resection for pancreatic cancer. Br J Cancer. 2016;115:59–65. doi: 10.1038/bjc.2016.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Artinyan A, Soriano PA, Prendergast C, Low T, Ellenhorn JD, Kim J. The anatomic location of pancreatic cancer is a prognostic factor for survival. HPB (Oxford) 2008;10:371–376. doi: 10.1080/13651820802291233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, Miller DK, Wilson PJ, Patch AM, Wu J, Chang DK, Cowley MJ, Gardiner BB, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405. doi: 10.1038/nature11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, Campos D, Lim R, Ding K, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25:1960–1966. doi: 10.1200/JCO.2006.07.9525. [DOI] [PubMed] [Google Scholar]
- 30.Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605–1617. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- 31.Earl J, Garcia-Nieto S, Martinez-Avila JC, Montans J, Sanjuanbenito A, Rodriguez-Garrote M, Lisa E, Mendia E, Lobo E, Malats N, Carrato A, Guillen-Ponce C. Circulating tumor cells (Ctc) and kras mutant circulating free Dna (cfdna) detection in peripheral blood as biomarkers in patients diagnosed with exocrine pancreatic cancer. BMC Cancer. 2015;15:797. doi: 10.1186/s12885-015-1779-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kinugasa H, Nouso K, Miyahara K, Morimoto Y, Dohi C, Tsutsumi K, Kato H, Matsubara T, Okada H, Yamamoto K. Detection of K-ras gene mutation by liquid biopsy in patients with pancreatic cancer. Cancer. 2015;121:2271–2280. doi: 10.1002/cncr.29364. [DOI] [PubMed] [Google Scholar]
- 33.Al-Mulla F, MacKenzie EM. Differences in in vitro invasive capacity induced by differences in Ki-Ras protein mutations. J Pathol. 2001;195:549–556. doi: 10.1002/path.995. [DOI] [PubMed] [Google Scholar]
- 34.Ogura T, Yamao K, Hara K, Mizuno N, Hijioka S, Imaoka H, Sawaki A, Niwa Y, Tajika M, Kondo S, Tanaka T, Shimizu Y, Bhatia V, et al. Prognostic value of K-ras mutation status and subtypes in endoscopic ultrasound-guided fine-needle aspiration specimens from patients with unresectable pancreatic cancer. J Gastroenterol. 2013;48:640–646. doi: 10.1007/s00535-012-0664-2. [DOI] [PubMed] [Google Scholar]
- 35.Laghi L, Orbetegli O, Bianchi P, Zerbi A, Di Carlo V, Boland CR, Malesci A. Common occurrence of multiple K-RAS mutations in pancreatic cancers with associated precursor lesions and in biliary cancers. Oncogene. 2002;21:4301–4306. doi: 10.1038/sj.onc.1205533. [DOI] [PubMed] [Google Scholar]
- 36.Page K, Guttery DS, Zahra N, Primrose L, Elshaw SR, Pringle JH, Blighe K, Marchese SD, Hills A, Woodley L, Stebbing J, Coombes RC, Shaw JA. Influence of plasma processing on recovery and analysis of circulating nucleic acids. PLoS One. 2013;8:e77963. doi: 10.1371/journal.pone.0077963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCall CM, Mosier S, Thiess M, Debeljak M, Pallavajjala A, Beierl K, Deak KL, Datto MB, Gocke CD, Lin MT, Eshleman JR. False positives in multiplex PCR-based next-generation sequencing have unique signatures. J Mol Diagn. 2014;16:541–549. doi: 10.1016/j.jmoldx.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, Bright IJ, Lucero MY, Hiddessen AL, Legler TC, Kitano TK, Hodel MR, Petersen JF, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011;83:8604–8610. doi: 10.1021/ac202028g. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.