

Abstract
Ubiquitin and ubiquitin modifying enzymes play critical roles in a wide variety of intracellular signaling pathways. Inflammatory signaling cascades downstream of TNF, TLR agonists, antigen receptor cross-linking, and cytokine receptors, all rely on ubiquitination events to direct subsequent immune responses. In the past several years, inflammasome activation and subsequent signal transduction has emerged as an excellent example of how ubiquitin signals control inflammatory responses. Inflammasomes are multiprotein signaling complexes that ultimately lead to caspase activation and release of the interleukin-1 (IL-1) family members, IL-1β and IL-18. Inflammasome activation is critical for the host’s defense against pathogens, but dysregulation of inflammasomes may contribute to the pathogenesis of multiple diseases. Ultimately, understanding how various ubiquitin interacting proteins control inflammatory signaling cascades could provide new pathways for therapeutic intervention. Here we review specific ubiquitin modifying enzymes and ubiquitination events that orchestrate inflammatory responses, with an emphasis on the NLRP3 inflammasome.
Graphical abstract
INTRODUCTION
Ubiquitin signaling and the ubiquitin code
While ubiquitin modification was first described as a system of tagging proteins for degradation by the proteasome [1], here we will provide a brief overview of the ubiquitin modulation of inflammatory signaling cascades, focusing on more recent data relating to the regulation of inflammasomes. Ubiquitin (Ub) is an evolutionarily conserved 76 amino acid protein that regulates a variety of cellular processes through covalent post-translational modification of target proteins at lysine (K) or N-terminal residues. The process of ubiquitination is catalyzed by the sequential action of ubiquitin-activating (E1), ubiquitin-conjugating (E2), and ubiquitin-ligating (E3) enzymes. The specificity of ubiquitination is mainly conferred by a combination of E2 enzymes, which help to specify Ub chain linkages, and >600 E3s, which catalyze the transfer of ubiquitin from the E2 to the substrate. Deubiquitinases or DUBs can remove ubiquitin chains, providing antagonism to ubiquitination and another layer of regulation. In humans, E2s, E3s, and DUBs have been implicated in a wide variety of neurologic, cardiovascular, oncologic, and immunologic disorders [2]. Compelling genetic evidence for disease association is the identification of a mutant allele with a monogenic human disorder. Allelic variants of multiple ubiquitin modifying enzymes have been associated with monogenic autoinflammatory, immunodeficient, pathogen susceptibility, or lymphoproliferative phenotypes in humans (Table 1). There are also many other examples of monogenic disorders attributed to ubiquitin-modifying enzymes that cause epilepsy and other neurologic diseases, developmental delay, and cardiac arrhythmia. In addition, single-nucleotide polymorphisms (SNPs) identified by genome-wide association studies (GWAS) increase the list of disease-associated ubiquitin modifying enzymes significantly. As whole exome sequencing becomes more widely available, this list of disease-associated ubiquitin modifying enzymes will undoubtedly expand. These data highlight the fact that ubiquitin is a critical regulator of multiple cellular processes, including inflammatory responses.
Table 1. Ubiquitin modifying enzymes with allelic variants that have been associated with immunologic monogenic human syndromes.
Online Mendelian Inheritance in Man (OMIM) was queried for human E2s, E3s, and DUBs. Genes were included in the table above if the associated human monogenic syndrome resulted in autoinflammation, immunodeficiency, or lymphoproliferative defects. RING, Really Interesting New Gene. BTB, Broad-Complex, Tramtrack and Bric a brac. POZ, POxvirus and Zinc finger. SUMO, small ubiquitin-like (Ubl) modifier. HECT, Homologous to the E6-AP Carboxyl Terminus. OTU, ovarian tumor.
| Gene | Alias | Class/Domain | Location | Human Phenotype | Molecular Phenotype |
|---|---|---|---|---|---|
| E2s | |||||
| UBE2T | FANCT, HSPC150 | E2 | 1q32.1 | Fanconi anemia, complementation group T; pancytopenia [3] | UBE2T is an E2 that ubiquitinates FANCL and FANCD2; mutation leads to genome instability and bone marrow failure [3] |
| E3s | |||||
| BIRC4 | XIAP | RING E3 Ub ligase | Xq25 | Lymphoproliferative syndrome, X-linked [4] | Restricts apoptosis through interactions with various caspases; mutation leads to increased susceptibility to apoptotic stimuli [4] |
| CBL | C-CBL, RNF55 | RING E3 Ub ligase | 11q23.3 | Noonan syndrome-like disorder with or without juvenile myelomonocytic leukemia [5] | Ubiquitinates multiple receptor protein-tyrosine kinases; mutations lead to hyper-responsiveness to multiple cytokines and increased proliferation [5] |
| FANCL | POG | RING E3 Ub ligase | 2p16.1 | Fanconi anemia, complementation group L; pancytopenia [6] | FANCL monoubiquitinates FANCD2 in response to DNA damage; mutation leads to genome instability and bone marrow failure [6] |
| MIB1 | DIP-1 | RING E3 Ub ligase | 18q11.2 | Left-ventricular noncompaction 7 [7] | Reduced NOTCH 1 activity in peripheral blood leading to potential B and T cell defects [7,8] |
| RBCK1 | HOIL-1, HOIL-1L | RBR E3 Ub ligase | 20p13 | Polyglucosan body myopathy 1 with or without immunodeficiency [9] | Defects in LUBAC lead to NFKB dysregulation, leading to both immunodeficiency and autoinflammation [9] |
| RNF31 | HOIP | RBR E3 Ub ligase | 14q12 | Multiorgan autoinflammation, combined immunodeficiency, subclinical amylopectinosis, and systemic lymphanqiectasia [10] | Phenolype overlaps with HOIL1/RBCK1 mutated patients [10] |
| RAG1 | RNF74 | RING E3 Ub ligase | 11 p12 | Severe combined immunodeficiency, Omenn syndrome [11,12] | RAG1 is required for assembling mature antigen receptors on B & T lymphocytes; mutations in the RING domain impairs ubiquitin ligase activity and recombination [11,12] |
| RNF125 | TRAC1 | RING E3 Ub ligase | 18q12.1 | Tenorio syndrome [13] | Defects in RIG-I-IPS1-MDA5, interferon, PI3K-Akt signaling leading to a Sjögren-like syndrome [13] |
| RNF168 | RING E3 Ub ligase | 3q29 | RIDDLE syndrome; immunodeficiency [14] | RNF168 is an E3 ubiquitin ligase critical for DNA double-strand break repair; mutations lead to defective DNA repair and immunodeficiency [14] | |
| SLX4 | BTB12 | BTB/POZ domain; SUMO E3 ligase | 16p13.3 | Fanconi anemia, complementation group P; pancytopenia [15,16] | SLX4 interacts with SUMO-charged E2 UBC9 and functions as a SUMO E3 ligase; important for the local replicative DNA repair response [15,16] |
| TRAF3 | LAP1, CAP1 | RING E3 Ub ligase | 14q32.32 | Possible association with susceptibility to Herpes simplex vims (HSV) encephalitis [17] | Dominant-negative mutant TRAF3 impairs responsiveness to TLR3 agonists such as HSV [17] |
| ITCH | AIF4, AIP4, NAPP1 | HECT E3 Ub ligase | 20q 11.22 | Autoimmune disease, multisystem, with facial dysmorphism [18] | Cooperates with A20 and CYLD to terminate NFKB signaling; implicated in multiple signaling pathways, mutations lead to immune dysrequlation [18, 19] |
| DUBS | |||||
| TNFAIP3 | A20 | ZnF E3 Ub ligase; OTU domain, DUB | 6q23_3 | Autoinflammatory syndrome, familial, Behcet-like [20] | Mutations leading to truncated A20 increased NFKB activity due to defective removal of K634inked ubiquitin from RIPK1, NEMO, and TRAF6 [20] |
| OTULIN | AIPDS, FAM105B | OTU domain, DUB | 5p15.2 | Autoinflammation, panniculitis, and dermatosis syndrome [21] | OTULIN-deflcient patients have increased linear ubiquitination of ASC, NEMO, RIPK1. TNFR1, leading to severe inflammation [21] |
Ubiquitin chains can undergo many different types of linkages and modifications, resulting in a “ubiquitin code” that has been discussed in detail elsewhere [22–26]. Ubiquitin itself has 7 lysine residues (K6, K11, K27, K29, K33, K48, and K63) along with its N-terminal methionine, resulting in the potential for multiple types of ubiquitin chains. Some of these linkages can be monomeric, polymeric, linked in tandem, branched, or even unanchored, with homogeneous or heterogeneous types of linkages [22,27,28]. In contrast to phosphorylation, multiple types of ubiquitin chains can be constructed and added to target proteins, which dramatically increases the amount of signaling information that can be encoded by ubiquitination. It is important to reiterate that different ubiquitin linkages lead to different outcomes for target proteins. K48- and K63-linked chains are the most abundant linkages in cells and are also the most extensively characterized [22,29]. K48-linked polyubiquitin chains generally direct target proteins to the proteasome for degradation [22]. This is also true for K11-linked chains [22]. In contrast, K63-linked polyubiquitin chains participate in proteasome-independent signaling cascades. TRAF6 was one of the first E3 ligases described to form K63-linked polyubiquitin chains in the context of activating IkB kinase (IKK) [30,31]. K63-linked ubiquitin chains have since been described in many settings. Linear ubiquitin chains linked through their N-terminal methionine (M1), were subsequently described to modulate signaling upstream of NFκB, a critically important transcription factor involved in a vast array of immune responses [32,33]. These chains are assembled by a linear ubiquitin assembly complex (LUBAC), composed of three proteins HOIL-1L (heme-oxidized IRP2 ligase 1L), HOIP (HOIL-1 interacting protein), and SHARPIN (SHANK-associated RH domain-interacting protein) [34–37]. The K6-, K27-, K29- and K33-linked ubiquitin chains in general have fewer substrates and are less well understood [22,23].
In addition to homogenous polyubiquitin chains, the role of heterogeneous branched polyubiquitin chains is becoming more clear. For example, Ohtake et al recently used a unique mass spectrometry method to identify and characterize K48-K63 branched ubiquitin chains [27]. The authors showed that HUWE1 cooperates with TRAF6 to assemble branched ubiquitin chains in response to IL-1β, recruiting TAB2 and protecting against CYLD-mediated deubiquitination [27]. It is unclear if perhaps K48 linkage serves as a degradation signal later, but at least initially it appears to stabilize and enhance signaling. This is an intriguing example of how branched ubiquitin chains can cooperate to regulate NFκB.
Unanchored ubiquitin chains, those not conjugated to a specific target protein, can also act as a type of second messenger. For example, TRAF6 can synthesize unanchored K63-linked polyubiquitin chains that bind to TAB2 and activate TAK1, leading to IKK activation and NFκB signaling [38]. Similarly, the viral RNA sensor RIG-I binds unanchored K63- linked polyubiquitin chains leading to activation of the essential mitochondrial adaptor protein MAVS, leading to NFκB and IRF3 signaling [39]. In other instances, unanchored chains could be formed by cleavage after assembly on target protein. Thus, ubiquitin chains can propagate signaling events beyond directly modifying a target protein.
Another layer of information in the ubiquitin code includes additional post-translational modifications of ubiquitin itself. Phosphorylation is one such example. The disease relevance of ubiquitin phosphorylation became apparent in studies of PINK1 and PARKIN, two genes that are implicated in hereditary recessive early-onset Parkinsonism. PINK1 is a Ser/Thr kinase that phosphorylates both PARKIN and ubiquitin, leading to full activation of PARKIN E3 activity [40–42]. In addition to phosphorylation, ubiquitin can also be modified by acetylation, SUMO family members, or NEDD8, compounding the potential variations of modified ubiquitin chains [22,26,43].
Finally, one of the functions of polyubiquitin chains may be to serve as a scaffold for recruiting multiple proteins with ubiquitin binding domains, not just E3s and DUBs, to form larger signaling complexes. Two examples of this are ABIN and NEMO, which have similar ubiquitin binding domains but no ubiquitin ligase or deubiquitinating activity [44,45]. Taken as a whole, ubiquitin signaling possesses many layers of regulation and potential modification. The unique attributes of ubiquitination as a post-translational modification and second messenger account for the central role it plays in critical cellular processes, including inflammatory signaling cascades.
Ubiquitination and immune signaling
Given the extent to which ubiquitin can be used to modulate signaling cascades, it is perhaps not surprising that ubiquitin plays a role in nearly every major signaling pathway in the inflammatory response. The immune response can largely be divided into adaptive and innate immunity. The adaptive immune response involves B and T lymphocytes that undergo gene rearrangement and subsequent selective pressure to generate a highly specific immune response to a specific antigen. T cell receptor (TCR) or B cell receptor (BCR) cross-linking, along with a co-stimulatory signal, triggers a cascade of ubiquitination and phosphorylation events that ultimately lead to cell activation. The ubiquitination events important for antigen receptor signal transduction and lymphocyte activation involve E3 ligases such as Cbl-b, GRAIL, ITCH, and MIB2, as well as DUBs like Otud7b, USP9X, CYLD, and A20, which have been extensively reviewed elsewhere [46–49]. Ultimately these adaptive immune responses expand lymphocytes and generate immunologic memory.
In contrast to the exquisite specificity of the adaptive arm of the immune system, the innate immune system instead relies on a variety of germline encoded pattern-recognition receptors (PRRs) to detect pathogen-associated molecular patterns (PAMPs) or endogenous danger-associated molecular patterns (DAMPs). Toll-like receptors (TLRs) are PRRs that recognize certain invariable microbial motifs in the extracellular and endosomal compartments, for example the recognition of lipopolysaccharide (LPS) by TLR-4 (Fig 1). Ubiquitination events downstream of TLRs have been discussed in detail elsewhere and involve key E3 ligases like TRAF6, cIAP, Pellino1 (Peli1), and LUBAC, along with DUBs such as A20, CYLD, and USP18 [46,49–53]. Each PRR serves to couple the specific PAMP or DAMP to the appropriate pro-inflammatory response. The idea that the immune system has evolved a method of recognizing both pathogens as well as certain endogenous markers of non-infectious cellular stress is known as the “danger signal” hypothesis. This idea was first popularized by Polly Matzinger [54], and it presaged the subsequent description of “inflammasomes” by Tschopp and colleagues [55]. Inflammasomes are high molecular weight, multiprotein signaling complexes that link recognition of certain “danger signals” by PRRs to the activation of inflammatory caspases and the downstream release of IL-1 family members, IL-1β and IL-18.
Figure 1. Schematic of NLRP3 inflammasome activation. LPS signaling through TLR4 or other priming signal activates NFκB and upregulates NLRP3 and IL1β mRNA.
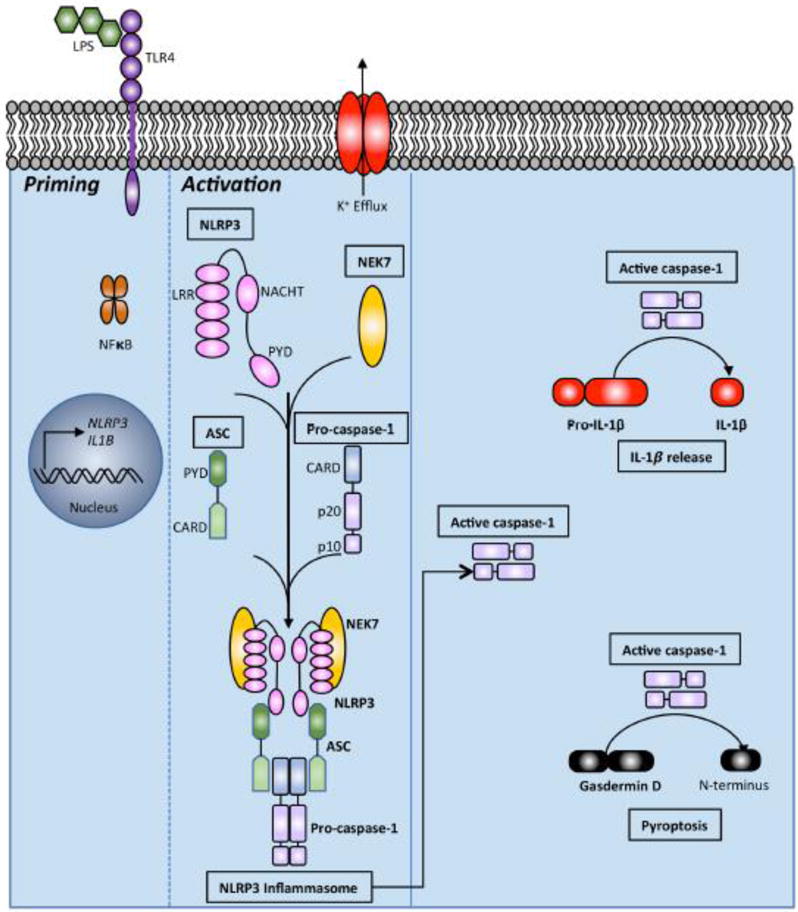
A second signal such as potassium efflux then activates the inflammasome. NLRP3, NEK7, ASC, and pro-caspase-1 assemble to form the NLRP3 inflammasome. This leads to autoproteolytic cleavage of pro-caspase-1 yielding active caspase-1. Active caspase-1 cleaves pro-IL-1β to mature IL-1β for release. Cleavage of pro-IL-18 into mature IL-18 is not pictured. Caspase-1 can also cleave Gasdermin D, releasing the N-terminal fragment that drives pyroptosis. Pyrin domain (PYD). NAIP, CIITA, HET-E and TP1 (NACHT) domain. Leucine-rich repeat (LRR) domain. Caspase recruitment domain (CARD).
Activation and regulation of inflammasomes
Inflammasomes have been extensively reviewed elsewhere [56–59]. Although there are multiple proteins involved in canonical inflammasome formation, the unifying theme is the formation of a large signaling scaffold that facilitates proximity-induced, autoproteolytic cleavage of inactive zymogen pro-caspase-1 into active caspase-1 [60] (Fig 1). Non-canonical inflammasome activation, on the other hand, results in activation of caspase-11 in mice and caspase-4 or caspase-5 in humans. One of the major pro-inflammatory consequences of active caspase-1 is conversion of pro-IL-1β and pro-IL-18 into mature IL-1β and IL-18, respectively. These mature cytokines are ultimately released from the cell, driving a pro-inflammatory response. Another important sequela of active caspase-1 is the cleavage of gasdermin D and the initiation of an inflammatory form of lytic cell death called pyroptosis (Fig 1) [56,58,61–64]. These signaling decision points are critically important. Inappropriately low activity could lead to relative immunodeficiency and/or susceptibility to certain pathogens. Conversely, hyperactivity could lead to autoimmunity or other inflammatory disorders. Inflammasome activation in fact has already been linked to multiple disorders. The link is most direct in certain autoinflammatory genetic disorders known as cryopyrinopathies [65]. These are rare inherited disorders characterized by recurrent fever, rash, joint pain, and inflammation. There are several different cryopyrinopathies of varying severity, including familial cold autoinflammatory syndrome (FCAS), Muckle-Wells syndrome (MWS), and chronic infantile cutaneous neurological articular syndrome (CINCA; also called neonatal-onset multisystem inflammatory disease, NOMID) [56–58,65,66]. MWS, as an example, results from a gain-of-function mutation in NLRP3 (R258W), a critical component of the NLRP3 inflammasome, leading to autoactivation and increased levels of IL-1β production [67]. Several IL-1 neutralizing therapeutics have been approved for treating patients with these inflammatory disorders [65]. Beyond these inherited autoinflammatory disorders, the inflammasome has been implicated in many other diseases including multiple sclerosis (MS), Alzheimer’s disease (AD), Parkinson’s disease, gout, type 2 diabetes, atherosclerosis, obesity, arthritis, and inflammatory bowel disease (IBD) [56,65,68]. As the inflammasome is both critical for host defense yet poised to drive autoinflammation or autoimmunity, it requires careful regulation to ensure homeostasis.
Multiple proteins have been described to assemble canonical inflammasomes upon stimulation with a variety of ligands. These include the nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) family members such as NLRP1, NLRP3, and NLRC4 [57]. The NLR family of genes typically have an N-terminal pyrin domain (PYD) or caspase recruitment domain (CARD), followed by a central nucleotide-binding and oligomerization (NACHT) domain and finally a C-terminal leucine-rich repeat (LRR) (Fig 1) [57]. The DNA sensor absent in melanoma 2 (AIM2) and pyrin can also form canonical inflammasomes [58]. Each of these proteins responds to different stimuli. For example, mouse NLRP1 responds to B. anthracis lethal factor and T. gondii infection. Similarly, NLRC4 collaborates with NLR family apoptosis inhibitory proteins (NAIPs) to detect bacterial flagellin and other components of bacterial type 3 secretion systems (T3SS). NLRP3, however, responds to a diverse array of stimuli including uric acid crystals, extracellular ATP, potassium efflux, pore-forming toxins, mitochondrial reactive oxygen species (ROS), release of mitochondrial DNA, cytosolic leakage of lysosomal cathepsins, as well as multiple other viral, bacterial, and fungal pathogens [56] (Fig 1). Typically, NLRP3 activation is assayed in vitro by priming cells with LPS or another NFκB-activating stimulus (Signal 1), which upregulates NLRP3 and pro-IL-1β mRNA [56,69]. These priming stimuli involve multiple ubiquitination events downstream of TLRs, converging on NFκB [46,49–53]. After priming, cells are then treated with ATP, the pore-forming toxin nigericin, or other stimulus (Signal 2) in order to induce full NLRP3 inflammasome formation, caspase-1 activation, and IL-1β release [56,69]. Several recent studies reported that NIMA-related kinase 7 (NEK7) is essential for this process. NEK7 binds NLRP3 upon stimulation, leading to oligomerization and full activation of the NLRP3 inflammasome [58,67,70,71] (Fig 1). Ultimately, when NLRP1, NLRP3, NLRC4, AIM2, or pyrin are activated by their cognate stimulus, they oligomerize and recruit apoptosis-associated speck-like (ASC) protein. ASC assembles into a protein complex large enough to form a “speck” readily visible by fluorescence confocal microscopy. ASC contains a PYD and CARD domain that allows it to bind the various sensor molecules while simultaneously recruiting pro-caspase-1, enhancing autoproteolytic activation to caspase-1. This review will focus primarily on the NLRP3 inflammasome since it is the most extensively characterized with regard to ubiquitination events. The NLRP3 inflammasome is controlled by ubiquitin modifying enzymes at every step from initial NLRP3 activation to IL-1β release, and thus serves as an excellent example of how the ubiquitin code regulates inflammatory responses.
Regulation of inflammasomes by ubiquitination
Ubiquitination events regulating NLRP3
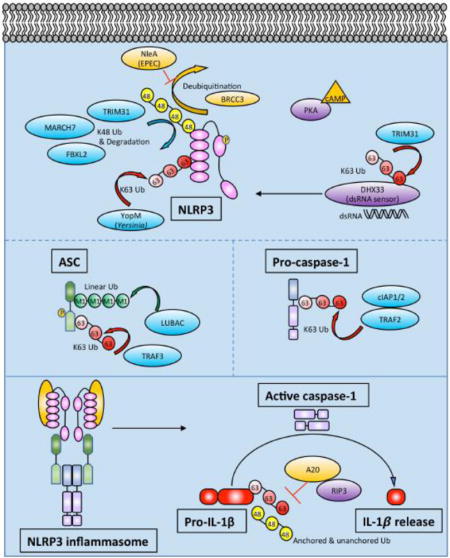
There are number of ubiquitin modifying enzymes that participate in regulating NLRP3, which is modified by a combination of K48-linked and K63-linked polyubiquitin chains (Fig 2).
Figure 2. Schematic of ubiquitin modifying enzymes and ubiquitination events regulating NLRP3 inflammasome activity.
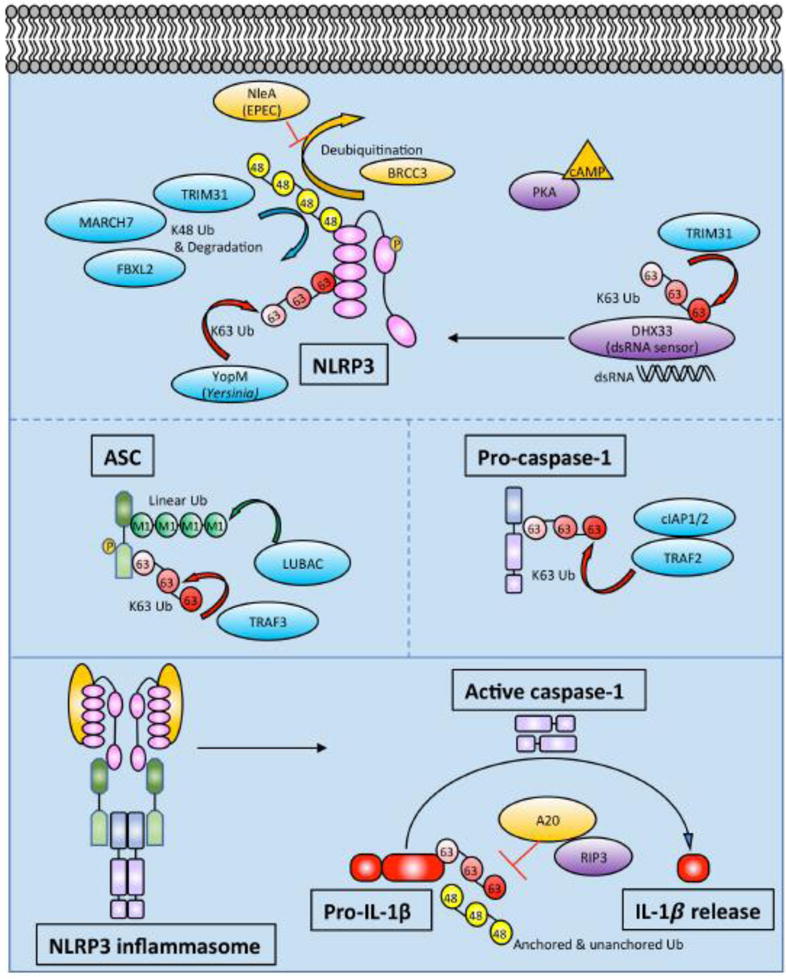
In addition to upregulating NLRP3 and IL1β mRNA, priming also leads to NLRP3 deubiquitination. NLRP3 is modified by K48- and K63-linked Ub as described in the text. Phosphorylation of NLRP3 may be linked to its ubiquitination status. Prior to NLRP3 inflammasome assembly, ASC gets phosphorylated and linearly ubiquitinated. ASC can also undergo K63-linked ubiquitination by TRAF3. Pro-caspase-1 undergoes K63-linked polyubiquitination by cIAP1, cIAP2, and TRAF2. After NLRP3 inflammasome assembly, active caspase-1 cleaves pro-IL-1β to IL-1β. Pro-IL-1β undergoes K63-linked polyubiquitination and binds unanchored Ub chains. A20 restricts polyubiquitination of pro-IL-1β in a RIPK3-dependent manner. The exact character and composition of heterogeneous ubiquitin chains in the inflammasome are not well understood.
Deubiquitination events were the first to be described. Juliana and colleagues found that wild-type, primary bone marrow-derived macrophages (BMDMs) could be primed with LPS in the absence of new protein synthesis using cycloheximide (CHX) [72]. They performed immunoprecipitation (IP) of NLRP3 and immunoblotted with anti-ubiquitin revealing a clear NLRP3 ubiquitination pattern, which was further increased by the addition of small molecule DUB inhibitors. Adding ATP or nigericin, both of which induce potassium efflux, triggered marked deubiquitination of NLRP3. Blocking deubiquitination of NLRP3 inhibited activation of caspase-1, demonstrating the importance of that step in processing. Therefore, deubiquitination of NLRP3 is apparently part of the priming step, since the authors show that priming can occur in the absence of new protein synthesis. Py et al extended these observations soon after, demonstrating that BRCA1/BRAC2 containing complex, subunit 3 (BRCC3) was the critical DUB that deubiquitinates NLRP3 [73]. They also demonstrated inhibition of NLRP3 inflammasome formation and IL-1β release in peritoneal macrophages using small molecule DUB inhibitors, observing an increase in both K48- and K63-linked ubiquitin chains on NLRP3. Screening a focused library of DUBs identified BRCC3 as the DUB which deubiquitinates NLRP3. They also showed that BRCC3 interacts specifically with the ubiquitinated LRR domain of NLRP3 (Fig 2). Since NLRC4 and AIM2 inflammasome activation was intact in the presence of DUB inhibitors, NRLP3 deubiquitination specifically seemed to be the key step, rather than deubiquitination of other inflammasome components. Deubiquitination of NLRP3 allows it to bind ASC to trigger downstream caspase-1 activation and IL-1β release.
The next question is which ubiquitin E3 ligases polyubiquitinate NLRP3. Song et al showed that TRIM31 directly binds to NLRP3 and promotes K48-linked polyubiquitination and proteasomal degradation [74]. TRIM31 is a tripartite motif (TRIM) family member. TRIM proteins belong to the RING domain class of E3 ligases. Many TRIM family members have been shown to play a role in innate immune responses [75,76]. TRIM31 deficient mouse peritoneal macrophages exhibit enhanced IL-1β secretion and caspase-1 cleavage in response to LPS and ATP, nigericin, or alum, despite normal levels of IL-1β mRNA. Overexpression of TRIM31 in 293T cells reduced NLRP3 protein levels. Other TRIM family members have been shown to play a role in targeting proteins for proteasomal degradation, so the authors used the proteasome inhibitor MG-132 to demonstrate reversal of TRIM31-mediated degradation of NLRP3. Interestingly, this NLRP3 stabilizing effect was not seen with the autophagy inhibitor 3-MA. They went on to show that the second C-terminal coiled-coil domain of TRIM31 interacts with the PYD of NLRP3. Using overexpression in 293T cells, TRIM31 was found to selectively promote K48 polyubiquitination of NLRP3. Supporting this model, TRIM31-deficient macrophages had normal levels of K63-linked ubiquitin chains on NLRP3 but significantly reduced K48-linked ubiquitin chains. Deleting the RING domain or making cysteine mutants abrogated the E3 ligase activity of TRIM31 and prevented K48-linked polyubiquitination of NLRP3. The net effects of TRIM31 deficiency are decreased K48-linked Ub and increased levels of NLRP3, leading to enhanced IL-1β secretion in response to LPS both in vitro and in vivo. As one would predict, enhanced inflammasome activity led to worse alum-induced peritonitis. Interestingly, in the dextran sodium sulfate (DSS) mouse model of colitis, a model of human IBD, TRIM31 deficiency ameliorated disease. This is consistent with prior studies suggesting that NLRP3, ASC, and caspase-1 deficient mice also exhibited increased susceptibility to DSS colitis [77]. The DSS colitis data argue that adequate inflammasome activation is required to maintain intestinal epithelial homeostasis. TRIM31 also plays a role in K63-linked polyubiquitination of the signaling adaptor MAVS, promoting aggregation and activation of the downstream antiviral response [78]. Thus, through K63-linked polyubiquitination of MAVS and K48-linked polyubiquitination of NLRP3, TRIM31 is an E3 ligase that plays a role in host defense through multiple innate immune signaling pathways. Additionally, another TRIM family member, TRIM33, was reported to ubiquitinate DHX33 with K63-linked polyubiquitin chains. DHX33 was previously identified as the dsRNA sensor for the NLRP3 inflammasome [79], and TRIM33 polyubiquitination of DHX33 is essential for DHX33-NLRP3 inflammasome formation [80] (Fig 2). Therefore, multiple TRIM family members influence inflammasome activation through ubiquitin-mediated signaling.
In addition to TRIM31, other proteins have been reported to polyubiquitinate NLRP3. Han et al found that FBXL2 ubiquitinates NLRP3 using a combination of co-IP, overexpression, and siRNA knock-down experiments, as well as in vitro ubiquitination assays [81]. They used UbPred software [82] to predict potential ubiquitination sites, followed by a site-directed mutagenesis approach to show that Lys689 as the major ubiquitination acceptor site, and that Trp73 on NLRP3 is the molecular recognition site for FBXL2 binding. Finally, since FBXO3 is another E3 ligase that is known to target FBXL2 for proteasomal degradation, they used a small molecule FBXO3 inhibitor BC-1215 to show that pharmacologic inhibition of FBXO3 led to increased FBXL2 levels, which in turn reduced NLRP3 inflammasome activation and IL-1β release. Interestingly, a naturally occurring loss-of-function variant allele in human FBXO3 (V221I) lead to increased FBXL2 levels and reduced IL-1β release from peripheral blood mononuclear cells (PBMCs) in vitro [83]. Therefore, FBXL2 appears to be able to polyubiquitinate NLRP3 and target it for degradation (Fig 2).
Investigation the neurotransmitter dopamine (DA) led to the identification of MARCH7, another ubiquitin E3 ligase that targets NLRP3 for degradation (Fig 2). Yan et al showed that DA signaling through the Dopamine D1 Receptor (DRD1) on BMDMs led to NLRP3 ubiquitination and degradation, thereby inhibiting inflammasome activation [84]. DRD1 is known to signal through the second messenger cAMP, and the authors went on to show that cAMP inhibited NLRP3 inflammasome activation by promoting ubiquitination and degradation. Interestingly, neither Protein Kinase A (PKA) nor exchange protein activated by cAMP (EPAC), the two known intracellular sensors for cAMP, appeared to be involved in promoting NLRP3 ubiquitination and degradation. The authors then showed that an anti-cAMP antibody immunoprecipitated NLRP3 after DA stimulation, suggesting that cAMP might directly bind NLRP3. They further showed that the ubiquitin-mediated degradation could be reversed with the autophagy inhibitor 3-MA, but not the proteasome inhibitor MG132. Mass spectrometry identified potential E3 ligases that co-IP with NRLP3 and siRNA of these hits ultimately identified MARCH7 as the E3 ligase that induces NLRP3 degradation downstream of DRD1. Consistent with previous work, they also mapped K48-linked ubiquitination and degradation of NLRP3 to the LRR domain. They then used a drug-induced mouse model of Parkinson’s Disease (PD) and showed that Drd1−/− mice exhibited more severe disease compared to wild-type. This effect was rescued in Drd1−/−Nlrp3−/− mice, suggesting that DRD1 signaling can prevent neuroinflammation via NRLP3 inflammasome inhibition. It is unclear why MARCH7-mediated K48-linked polyubiquitination of NLRP3 would lead to degradation via autophagosomes as opposed to the proteasome as in prior studies. This finding could relate to prior work showing that large inflammasome complexes are directed to autophagosomes for degradation [85].
Phosphorylation of NLRP3 may also its influence ubiquitination. The evidence for phosphorylation of NLRP3 came from studying the bile acid membrane receptor TGR5. Bile acids are cholesterol-derived molecules synthesized by the liver to aid in digestion, but they also play a role in multiple pathways related to metabolic homeostasis. Guo et al assessed the effect of bile acids on inflammasome activity [86]. They observed that bile acid signaling through TGR5 negatively regulated the NLRP3 inflammasome. From a disease standpoint, they showed that TGR5-mediated inhibition of the NLRP3 inflammasome reduced LPS and alum-induced inflammation in vivo, as well as preventing insulin resistance on a high-fat diet. Investigating the mechanism of NLRP3 inhibition, they found that elevated levels of cAMP, the known second messenger of TGR5, led to NLRP3 degradation in a PKA-dependent manner (Fig 2). Although the stimuli are different, the PKA-dependent NLRP3 degradation seen here contrasts with the model proposed by Yan et al, where cAMP directly binds to NLRP3 upon DRD1 stimulation [84]. Interestingly, kinase-dead PKA impaired both phosphorylation and ubiquitination of NLRP3 when ectopically expressed in 293T cells. To further support the role of PKA in this setting, alanine substitution of the primary phosphorylation site Ser291 in the NACHT domain of NLRP3 reduced both phosphorylation and ubiquitination. Stable reconstitution of NLRP3 deficient THP-1 cells with phosphomimetic substitutions, using aspartic or glutamic acid substitutions for serine, significantly impaired IL-1β release. These data support a model in which phosphorylation, either through conformational change or some induced protein-protein interaction, leads to both K48- and K63-linked polyubiquitination and degradation. This mechanism could be similar to IκBα, a classic example of phosphorylation-dependent ubiquitination and degradation.
Pathogen subversion of NLRP3 ubiquitination
Although NLRP3 ubiquitination events have only recently been described, there are already several examples of pathogens hijacking this system to subvert host defense. Yen et identified a bacterial protein that exploits this pathway to inhibit inflammasome formation by inhibiting the deubiquitination of NLRP3 [87]. Enteropathogenic Escherichia coli (EPEC) cause an infectious colitis characterized by watery or bloody diarrhea. Using EPEC deletion mutants, they found that NleA could inhibit the inflammasome in PMA-differentiated human myeloid THP-1 cells, without affecting NFκB activity. This is in contrast to NleE, another EPEC gene product that has been found to antagonize NFκB signaling [88,89]. Using an IP approach, they went on to show that NleA interacts with NLRP3 by binding both the LRR and PYD domains. More importantly, they found that NleA binds ubiquitinated NRLP3 and prevents its deubiquitination both in cellular overexpression assays and in vitro ubiquitination assays. These data support a model wherein NleA, by preventing deubiquitination of NRLP3, suppresses caspase-1 activation and IL-1β secretion, leading to a clear advantage for the invading pathogen (Fig 2).
Another pathogen-associated effector that affects NLRP3 ubiquitination is YopM. YopM is part of the T3SS “delivery system” for bacterial effectors of Yersinia pestis. YopM has previously been reported to be an inflammasome inhibitor, either by acting as a pseudosubstrate for caspase-1 or by inhibiting IQGAP1-dependent NLRP3 inflammasome formation in response to Yersinia infections [90,91]. More recently, Wei et al reported that YopM possesses E3 ligase activity [92]. Using a combination of in vitro ubiquitination assays, a yeast two-hybrid screen, and cellular overexpression experiments, they showed that YopM directly binds NLRP3 and ubiquitinates it with K63-linked polyubiquitin chains. This K63 ubiquitination stabilizes NLRP3 and induces inflammasome activation, somehow predisposing to necrotic rather than pyroptotic cell death. In this setting, the inhibition of caspase-1 by YopM may prevent pyroptosis, and instead predispose to necrotic cell death (Fig 2). Both pyroptosis and necroptosis are inflammatory forms of cell death, so stimulating either cell death pathway would contribute to the virulence of Y. pestis.
A third example of pathogens modifying ubiquitin includes the Shigella IpaH7.8 E3 ubiquitin ligase [93]. IpaH7.8 is a virulence factor that was found to induce pyroptotic cell death in murine BMDMs by activating both NLRP3 and NLRC4. The authors did not show a direct interaction between IpaH7.8 and NLRP3 or NLRC4, but instead they identified glomulin/flagellar-associated protein 68 (GLMN) as an IpaH7.8 target by a yeast two-hybrid screen, followed by GST pull-down and in vitro ubiquitination assays. GLMN has been described previously as a Cullin ring ligase inhibitor, and these authors showed that GLMN negatively regulates NLRP3 and NLRC4 inflammasome activation and pyroptosis, though the details of that mechanism are still unclear. Despite the fact that bacteria do not possess endogenous Ub systems, virulence factors acting as ubiquitin modifying enzymes is an emerging paradigm for combating host defense mechanisms.
Collectively, there are multiple ubiquitin modifying enzymes that modify NLRP3 with multiple of types of polyubiquitin chains to enhance or restrict inflammasome activation (Fig 2). There is still a lot to learn regarding the ubiquitination of NLRP3. For example, NLRP3 and other components of the inflammasome undergo polyubiquitination with both K48- and K63-linked polyubiquitin chains. It is still unclear if these are homogeneous, heterogeneous, or branched chains. Other signaling molecules are also known to be modified by heterogeneous Ub chains, but the significance of this modification for NLRP3 remains to be determined. It is also unclear which endogenous E3 ligases add K63-linked polyubiquitin chains to NLRP3. While this perspective focuses primarily on ubiquitination events and ubiquitin modifying enzymes that regulate NLRP3, it is important to note that proteins with ubiquitin binding domains may play important roles even if they lack ubiquitin ligase or deubiquitinase activity. For example, Histone deacetylase 6 (HDAC6) inhibits the NLRP3 inflammasome through its direct association with ubiquitinated NLRP3, independent of its deacetylase activity [94]. HDAC6 appears to associate with ubiquitinated NLRP3 through its ubiquitin-binding domain. Other proteins with ubiquitin-binding domains could play similar roles. Clearly there is more ubiquitin-related biology to discover regarding NLRP3. Also, we have chosen to emphasis the NLRP3 inflammasome because it is the best example of ubiquitination events that regulate inflammasome signaling activity. There is evidence, however, that other PRRs undergo similar ubiquitin-mediated processing. For example, there is evidence that NLRC4 also undergoes ubiquitination which may contribute to a caspase-8-mediated apoptotic death, rather than pyroptosis [95,96]. Since NLRP3 and other PRRs are at the top of the inflammasome signaling cascade, perhaps it makes teleological sense that there are multiple levels of ubiquitin-related regulation.
Ubiquitination events regulating ASC, caspase-1, and IL-1β
In addition to ubiquitination events regulating NLRP3, ubiquitination of ASC has been described as well. ASC is an adaptor that links multiple inflammasome sensors to caspase-1 activation (Fig 2). As mentioned previously, linear ubiquitin chains are assembly by the LUBAC consisting of HOIL-1L, HOIP, and SHARPIN [34–37]. HOIL-1L-deficient BMDMs exhibit reduced NLRP3 inflammasome activation in response to a wide variety of stimuli, as measured by IL-1β release, confocal microscopy, and caspase-1 activation [97]. To identify ASC as a LUBAC substrate, Rodgers et al used a combination of in vitro ubiquitination assays, overexpression of LUBAC and inflammasome components in 293T cells, and IP from BMDMs. Additionally, a DUB specific for linear ubiquitin, Otulin, abolished ubiquitination on ASC, further confirming that ASC is modified by linear ubiquitin. HOIL-1L deficiency reduced inflammasome activation in mice using endotoxic shock and peritonitis as models. These data collectively showed that linear ubiquitination of ASC is critical for inflammasome activation in vivo. With regard to other components of LUBAC and inflammasome activation, SHARPIN-deficient macrophages exhibit expected defects in transcriptional priming of the inflammasome, but defects in linear ubiquitination of ASC have not yet been described [98]. The role of LUBAC in ubiquitinating ASC is intriguing given that humans with loss-of-function mutations in HOIL-1L and HOIP exhibit autoinflammation and immunodeficiency (Table 1). Mutations in Otulin also lead to an early onset autoinflammatory phenotype (Table 1). While LUBAC plays a role in multiple inflammatory signaling cascades, dysregulated inflammasome activity may contribute to the observed phenotypes.
In addition to modification by linear ubiquitin, ASC also appears to be modified by K63-linked polyubiquitin chains. Guan et al. noted ubiquitination of ASC after infection of THP-1 cells with an RNA virus [99]. They screened a small library of E3 ligases and identified TRAF3 as the critical E3 ligase for K63-linked polyubiquitination of ASC. They mapped ubiquitination to Lys174 on ASC and showed that an arginine substitution at this residue completely abrogated ASC speck formation. Interestingly, MAVS engagement of TRAF3 was critical for ASC speck formation and subsequent NLRP3 inflammasome activation. A recent report further suggested that USP50 binds ASC and deubiquitinates K63-linked polyubiquitin chains on ASC (citation). Counter-intuitively, in this study deubiquitination of K63-linked chains by USP50 positively regulated inflammasome activation, so the exact role of K63-polyubiquitination of ASC is still somewhat sunclear. Finally, it is worth noting that phosphorylation of ASC has previously been shown to control inflammasome formation [100], but unlike the phosphorylation and ubiquitination of NLRP3, it is unclear at this point if ASC phosphorylation is directly linked to its ubiquitination (Fig 2).
After NLRP3 and ASC are activated, they recruit pro-capase-1 and facilitate its autoproteolytic activation to caspase-1. A key step in this process turns out to be the addition of K63-linked polyubiquitin chains in a complex containing the E3 ligases cIAP1, cIAP2 and TRAF2 [101]. Since cIAP1 and cIAP2 are known to play central roles in NFκB signaling downstream of the TNF Receptor 1 (TNFR1), and more importantly have been described to ubiquitinate several caspases, Labbe et al. investigated their role in inflammasome assembly and caspase-1 activation. They observed that deficiency in cIAP1 or cIAP2 impaired caspase-1 activation in LPS-stimulated BMDMs. They then showed that cIAP1, cIAP2, and TRAF2 could co-IP with caspase-1 in a cell-free system, in transiently transfected 293T cells, and human THP-1 cells. Using overexpression, the authors showed K63-linked polyubiquitination of caspase-1 in the presence of cIAP2. They also detected K63-linked polyubiquitination of endogenous pro-caspase-1 after LPS+ATP stimulation in wild-type (WT) BMDMs but not cIAP2-deficient BMDMs. As expected, cIAP2-deficient mice exhibited blunted inflammasome activation in vivo after intraperitoneal injection of monosodium urate (MSU) crystals.
The major consequence of canonical inflammasome formation is the cleavage of pro-IL-1β by activated caspase-1, followed by IL-1β release (Fig 2). It has been shown that IL-1β undergoes polyubiquitination and proteasomal degradation in bone marrow-derived dendritic cells (BMDCs) [102]. Consistent with that observation, a recent study demonstrated that the E2 conjugating enzyme UBE2L3 promotes K48-polyubiquitination of pro-IL-1β protein complexes, thereby decreasing mature IL-1β release (citation). This pathway may even be exploited by the Human Papilloma Virus (HPV) E6 oncoprotein which associates with the endogenous E3 ligase E6AP to polyubiquitinate IL-1β, targeting it for proteasomal degradation [103]. It is unclear if E6AP (also known as UBE3A) plays a role in this process in the absence of HPV infection. Our lab found that pro-IL-1β undergoes K63-linked polyubiquitination and binds K48-linked unanchored ubiquitin as part of its proteolytic processing during inflammasome activation [69]. A20, also known as TNFAIP3, is known to restrict multiple signaling pathways [104–108]. With regard to the inflammasome, deletion of A20 leads to increased caspase-1 activation, IL-1β release, and pyroptosis in response to NLRP3 inflammasome stimulation [69,109]. This spontaneous NLRP3 inflammasome activation appears to be due to both increased TLR signaling and regulation of pro-IL-1β ubiquitination [69,109]. Using various linkage-specific DUBs and linkage-specific antibodies, we found that the pro-IL-1β complex contains K63-linked polyubiquitin chains, as well as unanchored K48-linked polyubiquitin chains (Fig 2). A20-deficient macrophages exhibited markedly increased ubiquitination of pro-IL-1β. A20 has a deubiquitinating OTU domain, but it is unclear if A20 directly deubiquitinates pro-IL-1β or if it restricts ubiquitination of pro-IL-1β through a non-enzymatic mechanism. Interestingly, RIPK3 deficiency restricted IL-1β release in A20-deficient cells, and in fact RIPK3-deficient cells exhibited significantly less pro-IL-1β ubiquitination than RIPK3+/+ cells. Finally, anti-G-G antibody assisted mass spectrometry identified K133 as the primary ubiquitination site on pro-IL-1β. Mutation of that lysine markedly decreased ubiquitination and cleavage of pro-IL-1β. Interestingly, K133 is present in cleaved, mature IL-1β and overlaps with a KFERQ-motif implicated in IL-1β secretion [110]. In summary, A20 restricts polyubiquitination of pro-IL-1β and reduces the amount of mature IL-1β released.
SUMMARY AND FUTURE DIRECTIONS
In summary, the inflammasome provides a case study of how ubiquitin modifying enzymes coordinate downstream signaling cascades. NLRP3 regulation by ubiquitin is the best characterized, but clearly every step in assembling the inflammasome from NLRP3 interacting with ASC, to caspase-1 activation, pro-IL-1β cleavage, and IL-1β release, involves ubiquitination. There are still many questions, however, related to ubiquitin modifying enzymes in inflammasome activation. For instance, NEK7 binds NLRP3 to assemble the inflammasome, but so far NEK7 has not been characterized with regard to ubiquitination. Similarly, Gasdermin D drives pyroptosis yet it has not been associated with ubiquitination events. Further, other inflammasome sensors besides NLRP3 are likely regulated by ubiquitin. The types of ubiquitin chains associated with individual inflammasome components will also need to be characterized further. Whether these ubiquitin chains are anchored at a single site or multiple sites, or whether they are branched versus linked in tandem is still unclear. Certainly there are more E3 ligases and DUBs to discover in this pathway. There may also be additional roles for ubiquitin scaffolds in inflammasome activation. Recent work has employed cryoelectron microscopy and NMR spectroscopy to form a more detailed structural template of long ASC filaments [111,112]. Given the filamentous structure of polymerized ASC, coupled with the potential for ubiquitin to form polymeric chains, it is tempting to imagine that the mature NLRP3 inflammasome could take advantage of ubiquitin’s scaffolding function to assemble large ASC specks.
The next question is how to exploit our understanding of the inflammasome, and specifically ubiquitin biology, to move toward more targeted therapies in various diseases. Regarding the inflammasome, there have been multiple reports of therapeutic interventions targeting different aspects of inflammasome biology. IL-1 neutralizing therapies are one successful approach in the treatment of cryopyrinopathies or systemic juvenile idiopathic arthritis [56,65]. There are also now several small molecules that have been described to inhibit the inflammasome [56,113]. The only compound that seems to inhibit the inflammasome by targeting the ubiquitin signaling pathway is BC-1215 [81,83]. BC-1215 was originally identified as an inhibitor of FBXO3, and it was found to block the interaction with FBXL2, thereby preventing ubiquitination and degradation of FBXL2. Stabilizing FBXL2, as described earlier, leads to increased ubiquitination and degradation of NLRP3. Since FBXL2 also mediates degradation of TRAFs, BC-1215 may have immunosuppressive effects outside of the inflammasome pathway, which could affect its therapeutic potential. This example nevertheless highlights the possibility of targeting an E3 ligase to alter inflammation. Unfortunately, targeting ubiquitin modifying enzymes is challenging. Intracellular protein-protein interactions are generally not as conducive to small molecule inhibition, when compared to targeting the catalytic pocket of a kinase for example. There are some prominent examples of drugs that interfere with ubiquitin related processes in the oncology literature [2]. High-throughput identification of ubiquitin variants that act as specific inhibitors of DUBs or E3 ligases could be the foothold needed to develop targeted therapeutics in a meaningful way [114–117]. Additionally, if knock-down or knock-out methods evolve into more broadly applicable clinical tools, then certainly E3 ligases or DUBs could be targeted genetically, but those approaches have a separate set of challenges. Our understanding of ubiquitin modifying enzymes in regulating inflammatory responses is still in its infancy. The technology to better characterize ubiquitin chains is still developing, and we have only recently begun to appreciate the complexity of the ubiquitin code. As we learn more about ubiquitin modifying enzymes and the nuances of ubiquitination in inflammation, we will undoubtedly glean new therapeutic opportunities.
RESEARCH HIGHLIGHTS.
Ubiquitin modifying enzymes play critical roles in inflammatory signaling cascades
The NLRP3 inflammasome is a prime example of ubiquitin-mediated regulation
Each component of the NLRP3 inflammasome undergoes ubiquitin modification
Despite challenges, ubiquitin modifying enzymes could be important drug targets
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hershko A, Ciechanover A. The ubiquitin system for protein degradation. Annu Rev Biochem. 1992;61:761–807. doi: 10.1146/annurev.bi.61.070192.003553. [DOI] [PubMed] [Google Scholar]
- 2.Huang X, Dixit VM. Drugging the undruggables: exploring the ubiquitin system f or drug development. Nature Publishing Group. 2016;26:484–498. doi: 10.1038/cr.2016.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hira A, Yoshida K, Sato K, Okuno Y, Shiraishi Y, Chiba K, et al. Mutations in the gene encoding the E2 conjugating enzyme UBE2T cause Fanconi anemia. Am J Hum Genet. 2015;96:1001–1007. doi: 10.1016/j.ajhg.2015.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rigaud S, Fondanèche MC, Lambert N, Pasquier B, Mateo V, Soulas P, et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature. 2006;444:110–114. doi: 10.1038/nature05257. [DOI] [PubMed] [Google Scholar]
- 5.Pérez B, Mechinaud F, Galambrun C, Ben Romdhane N, Isidor B, Philip N, et al. Germline mutations of the CBL gene define a new genetic syndrome with predisposition to juvenile myelomonocytic leukaemia. J Med Genet. 2010;47:686–691. doi: 10.1136/jmg.2010.076836. [DOI] [PubMed] [Google Scholar]
- 6.Meetei AR, de Winter JP, Medhurst AL, Wallisch M, Waisfisz Q, van de Vrugt HJ, et al. A novel ubiquitin ligase is deficient in Fanconi anemia. Nat Genet. 2003;35:165–170. doi: 10.1038/ng1241. [DOI] [PubMed] [Google Scholar]
- 7.Song R, Kim YW, Koo BK, Jeong HW, Yoon MJ, Yoon KJ, et al. Mind bomb 1 in the lymphopoietic niches is essential for T and marginal zone B cell development. J Exp Med. 2008;205:2525–2536. doi: 10.1084/jem.20081344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luxán G, Casanova JC, Martínez-Poveda B, Prados B, D’Amato G, MacGrogan D, et al. Mutations in the NOTCH pathway regulator MIB1 cause left ventricular noncompaction cardiomyopathy. Nature Medicine. 2013;19:193–201. doi: 10.1038/nm.3046. [DOI] [PubMed] [Google Scholar]
- 9.Boisson B, Laplantine E, Prando C, Giliani S, Israelsson E, Xu Z, et al. Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nat Immunol. 2012;13:1178–1186. doi: 10.1038/ni.2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boisson B, Laplantine E, Dobbs K, Cobat A, Tarantino N, Hazen M, et al. Human HOIP and LUBAC deficiency underlies autoinflammation, immunodeficiency, amylopectinosis, and lymphangiectasia. J Exp Med. 2015;212:939–951. doi: 10.1084/jem.20141130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simkus C, Anand P, Bhattacharyya A, Jones JM. Biochemical and folding defects in a RAG1 variant associated with Omenn syndrome. The Journal of Immunology. 2007;179:8332–8340. doi: 10.4049/jimmunol.179.12.8332. [DOI] [PubMed] [Google Scholar]
- 12.Schwarz K, Gauss GH, Ludwig L, Pannicke U, Li Z, Lindner D, et al. RAG mutations in human B cell-negative SCID. Science. 1996;274:97–99. doi: 10.1126/science.274.5284.97. [DOI] [PubMed] [Google Scholar]
- 13.Tenorio J, Mansilla A, Valencia M, Martínez-Glez V, Romanelli V, Arias P, et al. A new overgrowth syndrome is due to mutations in RNF125. Hum Mutat. 2014;35:1436–1441. doi: 10.1002/humu.22689. [DOI] [PubMed] [Google Scholar]
- 14.Devgan SS, Sanal O, Doil C, Nakamura K, Nahas SA, Pettijohn K, et al. Homozygous deficiency of ubiquitin-ligase ring-finger protein RNF168 mimics the radiosensitivity syndrome of ataxia-telangiectasia. Cell Death Differ. 2011;18:1500–1506. doi: 10.1038/cdd.2011.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stoepker C, Hain K, Schuster B, Hilhorst-Hofstee Y, Rooimans MA, Steltenpool J, et al. SLX4, a coordinator of structure-specific endonucleases, is mutated in a new Fanconi anemia subtype. Nat Genet. 2011;43:138–141. doi: 10.1038/ng.751. [DOI] [PubMed] [Google Scholar]
- 16.Guervilly JH, Takedachi A, Naim V, Scaglione S, Chawhan C, Lovera Y, et al. The SLX4 complex is a SUMO E3 ligase that impacts on replication stress outcome and genome stability. Molecular Cell. 2015;57:123–137. doi: 10.1016/j.molcel.2014.11.014. [DOI] [PubMed] [Google Scholar]
- 17.Pérez de Diego R, Sancho-Shimizu V, Lorenzo L, Puel A, Plancoulaine S, Picard C, et al. Human TRAF3 adaptor molecule deficiency leads to impaired Toll-like receptor 3 response and susceptibility to herpes simplex encephalitis. Immunity. 2010;33:400–411. doi: 10.1016/j.immuni.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lohr NJ, Molleston JP, Strauss KA, Torres-Martinez W, Sherman EA, Squires RH, et al. Human ITCH E3 ubiquitin ligase deficiency causes syndromic multisystem autoimmune disease. Am J Hum Genet. 2010;86:447–453. doi: 10.1016/j.ajhg.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Venuprasad K, Zeng M, Baughan SL, Massoumi R. Multifaceted role of the ubiquitin ligase Itch in immune regulation. Immunol Cell Biol. 2015;93:452–460. doi: 10.1038/icb.2014.118. [DOI] [PubMed] [Google Scholar]
- 20.Zhou Q, Wang H, Schwartz DM, Stoffels M, Park YH, Zhang Y, et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat Genet. 2015;48:67–73. doi: 10.1038/ng.3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou Q, Yu X, Demirkaya E, Deuitch N, Stone D, Tsai WL, et al. Biallelic hypomorphic mutations in a linear deubiquitinase define otulipenia, an early-onset autoinflammatory disease. Proc Natl Acad Sci USA. 2016;113:10127–10132. doi: 10.1073/pnas.1612594113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81:203–229. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- 23.Ohtake F, Tsuchiya H. The emerging complexity of ubiquitin architecture. J Biochem. 2016:mvw088–9. doi: 10.1093/jb/mvw088. [DOI] [PubMed] [Google Scholar]
- 24.Husnjak K, Dikic I. Ubiquitin-binding proteins: decoders of ubiquitin-mediated cellular functions. Annu Rev Biochem. 2012;81:291–322. doi: 10.1146/annurev-biochem-051810-094654. [DOI] [PubMed] [Google Scholar]
- 25.Swatek KN, Komander D. Ubiquitin modifications. Cell Res. 2016;26:1–24. doi: 10.1038/cr.2016.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yau R, Rape M. The increasing complexity of the ubiquitin code. Nat Cell Biol. 2016;18:1–8. doi: 10.1038/ncb3358. [DOI] [PubMed] [Google Scholar]
- 27.Ohtake F, Saeki Y, Ishido S, Kanno J, Tanaka K. The K48-K63 Branched Ubiquitin Chain Regulates NFκB Signaling. Molecular Cell. 2016;64:251–266. doi: 10.1016/j.molcel.2016.09.014. [DOI] [PubMed] [Google Scholar]
- 28.Ohtake F, Tsuchiya H. The emerging complexity of ubiquitin architecture. J Biochem. 2016:mvw088–9. doi: 10.1093/jb/mvw088. [DOI] [PubMed] [Google Scholar]
- 29.Ohtake F, Saeki Y, Ishido S, Kanno J, Tanaka K. The K48-K63 Branched Ubiquitin Chain Regulates NF-κB Signaling. Molecular Cell. 2016;64:251–266. doi: 10.1016/j.molcel.2016.09.014. [DOI] [PubMed] [Google Scholar]
- 30.Deng L, Wang C, Spencer E, Yang L, Braun A, You J, et al. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103:351–361. doi: 10.1016/s0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- 31.Kanayama A, Seth RB, Sun L, Ea CK, Hong M, Shaito A, et al. TAB2 and TAB3 Activate the NF-κB Pathway through Binding to Polyubiquitin Chains. Molecular Cell. 2004;15:535–548. doi: 10.1016/j.molcel.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 32.Kirisako T, Kamei K, Murata S, Kato M, Fukumoto H, Kanie M, et al. A ubiquitin ligase complex assembles linear polyubiquitin chains. Embo J. 2006;25:4877–4887. doi: 10.1038/sj.emboj.7601360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sasaki K, Iwai K. Roles of linear ubiquitinylation, a crucial regulator of NF-κB and cell death, in the immune system. Immunol Rev. 2015;266:175–189. doi: 10.1111/imr.12308. [DOI] [PubMed] [Google Scholar]
- 34.Tokunaga F, Sakata SI, Saeki Y, Satomi Y, Kirisako T, Kamei K, et al. Involvement of linear polyubiquitylation of NEMO in NF-kappaB activation. Nat Cell Biol. 2009;11:123–132. doi: 10.1038/ncb1821. [DOI] [PubMed] [Google Scholar]
- 35.Tokunaga F, Nakagawa T, Nakahara M, Saeki Y, Taniguchi M, Sakata SI, et al. SHARPIN is a component of the NF-κB-activating linear ubiquitin chain assembly complex. Nature. 2011;471:633–636. doi: 10.1038/nature09815. [DOI] [PubMed] [Google Scholar]
- 36.Gerlach B, Cordier SM, Schmukle AC, Emmerich CH, Rieser E, Haas TL, et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature. 2011;471:591–596. doi: 10.1038/nature09816. [DOI] [PubMed] [Google Scholar]
- 37.Ikeda F, Deribe YL, Skånland SS, Stieglitz B, Grabbe C, Franz-Wachtel M, et al. SHARPIN forms a linear ubiquitin ligase complex regulating NF-κB activity and apoptosis. Nature. 2011;471:637–641. doi: 10.1038/nature09814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xia ZP, Sun L, Chen X, Pineda G, Jiang X, Adhikari A, et al. Direct activation of protein kinases by unanchored polyubiquitin chains. Nature. 2009;461:114–119. doi: 10.1038/nature08247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zeng W, Sun L, Jiang X, Chen X, Hou F, Adhikari A, et al. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell. 2010;141:315–330. doi: 10.1016/j.cell.2010.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014;510:162–166. doi: 10.1038/nature13392. [DOI] [PubMed] [Google Scholar]
- 41.Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, et al. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol. 2014;205:143–153. doi: 10.1083/jcb.201402104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, et al. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem J. 2014;460:127–139. doi: 10.1042/BJ20140334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Swatek KN, Komander D. Ubiquitin modifications, Nature Publishing Group. 2016;26:399–422. doi: 10.1038/cr.2016.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wagner S, Carpentier I, Rogov V, Kreike M, Ikeda F, Löhr F, et al. Ubiquitin binding mediates the NF-kappaB inhibitory potential of ABIN proteins. Oncogene. 2008;27:3739–3745. doi: 10.1038/sj.onc.1211042. [DOI] [PubMed] [Google Scholar]
- 45.Oshima S, Turer EE, Callahan JA, Chai S, Advincula R, Barrera J, et al. ABIN-1 is a ubiquitin sensor that restricts cell death and sustains embryonic development. Nature. 2009;457:906–909. doi: 10.1038/nature07575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hu H, Sun SC. Ubiquitin signaling in immune responses, Nature Publishing Group. 2016;26:457–483. doi: 10.1038/cr.2016.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. 2009;27:591–619. doi: 10.1146/annurev.immunol.021908.132706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu YC. Ubiquitin Ligases and the Immune Response. Annu Rev Immunol. 2004;22:81–127. doi: 10.1146/annurev.immunol.22.012703.104813. [DOI] [PubMed] [Google Scholar]
- 49.Wertz IE, Dixit VM. Signaling to NF-κB: Regulation by Ubiquitination. Cold Spring Harbor Perspectives in Biology. 2010;2:a003350–a003350. doi: 10.1101/cshperspect.a003350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li J, Chai QY, Liu CH. The ubiquitin system: a critical regulator of innate immunity and pathogen-dash;host interactions. Cell Mol Immunol. 2016;13:560–576. doi: 10.1038/cmi.2016.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.West AP, Koblansky AA, Ghosh S. Recognition and signaling by toll-like receptors. Annu Rev Cell Dev Biol. 2006;22:409–437. doi: 10.1146/annurev.cellbio.21.122303.115827. [DOI] [PubMed] [Google Scholar]
- 52.Skaug B, Jiang X, Chen ZJ. The role of ubiquitin in NF-kappaB regulatory pathways. Annu Rev Biochem. 2009;78:769–796. doi: 10.1146/annurev.biochem.78.070907.102750. [DOI] [PubMed] [Google Scholar]
- 53.Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. 2014;5:461. doi: 10.3389/fimmu.2014.00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 55.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Molecular Cell. 2002;10:417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 56.Guo H, Callaway JB, Ting JPY. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nature Medicine. 2015;21:677–687. doi: 10.1038/nm.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schroder K, Tschopp J. The Inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 58.Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nature Reviews Immunology. 2016;16:407–420. doi: 10.1038/nri.2016.58. [DOI] [PubMed] [Google Scholar]
- 59.Bednash JS, Mallampalli RK. Regulation of inflammasomes by ubiquitination. Cell Mol Immunol. 2016;13:722–728. doi: 10.1038/cmi.2016.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang X, Chang HY, Baltimore D. Autoproteolytic activation of pro-caspases by oligomerization. Molecular Cell. 1998;1:319–325. doi: 10.1016/s1097-2765(00)80032-5. [DOI] [PubMed] [Google Scholar]
- 61.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–665. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 62.Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–671. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- 63.Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111–116. doi: 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
- 64.Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, et al. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci USA. 2016;113:7858–7863. doi: 10.1073/pnas.1607769113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hoffman HM, Wanderer AA. Inflammasome and IL-1beta-mediated disorders. Curr Allergy Asthma Rep. 2010;10:229–235. doi: 10.1007/s11882-010-0109-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet. 2001;29:301–305. doi: 10.1038/ng756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.He Y, Zeng MY, Yang D, Motro B, Núñez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature. 2016;530:354–357. doi: 10.1038/nature16959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.de Souza HSP, Fiocchi C. Immunopathogenesis of IBD: current state of the art. Nat Rev Gastroenterol Hepatol. 2016;13:13–27. doi: 10.1038/nrgastro.2015.186. [DOI] [PubMed] [Google Scholar]
- 69.Duong BH, Onizawa M, Oses-Prieto JA, Advincula R, Burlingame A, Malynn BA, et al. A20 Restricts Ubiquitination of Pro-Interleukin-1β; Protein Complexes and Suppresses NLRP3 Inflammasome Activity. Immunity. 2015;42:55–67. doi: 10.1016/j.immuni.2014.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shi H, Wang Y, Li X, Zhan X, Tang M, Fina M, et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol. 2015;17:250–258. doi: 10.1038/ni.3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schmid-Burgk JL, Chauhan D, Schmidt T, Ebert TS, Reinhardt J, Endl E, et al. A Genome-wide CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) Screen Identifies NEK7 as an Essential Component of NLRP3 Inflammasome Activation. J Biol Chem. 2016;291:103–109. doi: 10.1074/jbc.C115.700492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Juliana C, Fernandes-Alnemri T, Kang S, Farias A, Qin F, Alnemri ES. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem. 2012;287:36617–36622. doi: 10.1074/jbc.M112.407130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Py BF, Kim MS, Vakifahmetoglu-Norberg H, Yuan J. Deubiquitination of NLRP3 by BRCC3 Critically Regulates Inflammasome Activity, Molecular. Cell. 2013;49:331–338. doi: 10.1016/j.molcel.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 74.Song H, Liu B, Huai W, Yu Z, Wang W, Zhao J, et al. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nature Communications. 2016;7:1–11. doi: 10.1038/ncomms13727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tomar D, Singh R. TRIM family proteins: emerging class of RING E3 ligases as regulator of NF-κB pathway. Biol Cell. 2015;107:22–40. doi: 10.1111/boc.201400046. [DOI] [PubMed] [Google Scholar]
- 76.Ozato K, Shin DM, Chang TH, Morse HC. TRIM family proteins and their emerging roles in innate immunity. Nature Reviews Immunology. 2008;8:849–860. doi: 10.1038/nri2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity. 2010;32:379–391. doi: 10.1016/j.immuni.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu B, Zhang M, Chu H, Zhang H, Wu H, Song G, et al. The ubiquitin E3 ligase TRIM31 promotes aggregation and activation of the signaling adaptor MAVS through Lys63-linked polyubiquitination. Nat Immunol. 2016:1–13. doi: 10.1038/ni.3641. [DOI] [PubMed] [Google Scholar]
- 79.Mitoma H, Hanabuchi S, Kim T, Bao M, Zhang Z, Sugimoto N, et al. The DHX33 RNA helicase senses cytosolic RNA and activates the NLRP3 inflammasome. Immunity. 2013;39:123–135. doi: 10.1016/j.immuni.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Weng L, Mitoma H, Trichot C, Tricot C, Bao M, Liu Y, et al. The E3 ubiquitin ligase tripartite motif 33 is essential for cytosolic RNA-induced NLRP3 inflammasome activation. J Immunol. 2014;193:3676–3682. doi: 10.4049/jimmunol.1401448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Han S, Lear TB, Jerome JA, Rajbhandari S, Snavely CA, Gulick DL, et al. Lipopolysaccharide Primes the NALP3 Inflammasome by Inhibiting Its Ubiquitination and Degradation Mediated by the SCF FBXL2E3 Ligase. J Biol Chem. 2015;290:18124–18133. doi: 10.1074/jbc.M115.645549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Radivojac P, Vacic V, Haynes C, Cocklin RR, Mohan A, Heyen JW, et al. Identification, analysis, and prediction of protein ubiquitination sites. Proteins. 2010;78:365–380. doi: 10.1002/prot.22555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen BB, Coon TA, Glasser JR, McVerry BJ, Zhao J, Zhao Y, et al. A combinatorial F box protein directed pathway controls TRAF adaptor stability to regulate inflammation. Nat Immunol. 2013;14:470–479. doi: 10.1038/ni.2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yan Y, Jiang W, Liu L, Wang X, Ding C, Tian Z, et al. Dopamine Controls Systemic Inflammation through Inhibition of NLRP3 Inflammasome. Cell. 2015;160:62–73. doi: 10.1016/j.cell.2014.11.047. [DOI] [PubMed] [Google Scholar]
- 85.Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, et al. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13:255–263. doi: 10.1038/ni.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Guo C, Xie S, Chi Z, Zhang J, Liu Y, Zhang L, et al. Bile Acids Control Inflammation and Metabolic Disorder through Inhibition of NLRP3 Inflammasome. Immunity. 2016;45:1–16. doi: 10.1016/j.immuni.2016.10.009. [DOI] [PubMed] [Google Scholar]
- 87.Hilo Yen NSTT. Enteropathogenic Escherichia coli Uses NleA to Inhibit NLRP3 Inflammasome Activation. PLoS Pathog. 2015;11:1–23. doi: 10.1371/journal.ppat.1005121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nadler C, Baruch K, Kobi S, Mills E, Haviv G, Farago M, et al. The type III secretion effector NleE inhibits NF-kappaB activation. PLoS Pathog. 2010;6:e1000743. doi: 10.1371/journal.ppat.1000743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Newton HJ, Pearson JS, Badea L, Kelly M, Lucas M, Holloway G, et al. The type III effectors NleE and NleB from enteropathogenic E. coli and OspZ from Shigella block nuclear translocation of NF-kappaB p65. PLoS Pathog. 2010;6:e1000898. doi: 10.1371/journal.ppat.1000898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chung LK, Philip NH, Schmidt VA, Koller A, Strowig T, Flavell RA, et al. IQGAP1 is important for activation of caspase-1 in macrophages and is targeted by Yersinia pestis type III effector YopM. MBio. 2014;5:e01402–14. doi: 10.1128/mBio.01402-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.LaRock CN, Cookson BT. The Yersinia virulence effector YopM binds caspase-1 to arrest inflammasome assembly and processing. Cell Host and Microbe. 2012;12:799–805. doi: 10.1016/j.chom.2012.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wei C, Wang Y, Du Z, Guan K, Cao Y, Yang H, et al. The Yersinia Type III secretion effector YopM Is an E3 ubiquitin ligase that induced necrotic cell death by targeting NLRP3. Cell Death and Disease. 2016;7:1–12. doi: 10.1038/cddis.2016.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Suzuki S, Mimuro H, Kim M, Ogawa M, Ashida H, Toyotome T, et al. Shigella IpaH7.8 E3 ubiquitin ligase targets glomulin and activates inflammasomes to demolish macrophages. Proc Natl Acad Sci USA. 2014;111:E4254–63. doi: 10.1073/pnas.1324021111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hwang I, Lee E, Jeon SA, Yu JW. Histone deacetylase 6 negatively regulates NLRP3 inflammasome activation. Biochemical and Biophysical Research Communications. 2015;467:973–978. doi: 10.1016/j.bbrc.2015.10.033. [DOI] [PubMed] [Google Scholar]
- 95.Raghawan AK, Sripada A, Gopinath G, Pushpanjali P, Kumar Y, Radha V, et al. A disease-associated mutant of NLRC4 shows enhanced interaction with SUG1 leading to constitutive FADD dependent caspase-8 activation and cell death. J Biol Chem. 2016 doi: 10.1074/jbc.M116.763979. jbc.M116.763979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kumar Y, Radha V, Swarup G. Interaction with Sug1 enables Ipaf ubiquitination leading to caspase 8 activation and cell death. Biochem J. 2010;427:91–104. doi: 10.1042/BJ20091349. [DOI] [PubMed] [Google Scholar]
- 97.Rodgers MA, Bowman JW, Fujita H, Orazio N, Shi M, Liang Q, et al. The linear ubiquitin assembly complex (LUBAC) is essential for NLRP3 inflammasome activation. Journal of Experimental Medicine. 2014;211:1333–1347. doi: 10.1084/jem.20132486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gurung P, Lamkanfi M, Kanneganti TD. Cutting edge: SHARPIN is required for optimal NLRP3 inflammasome activation. J Immunol. 2015;194:2064–2067. doi: 10.4049/jimmunol.1402951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Guan K, Wei C, Zheng Z, Song T, Wu F, Zhang Y, et al. MAVS Promotes Inflammasome Activation by Targeting ASC for K63-Linked Ubiquitination via the E3 Ligase TRAF3. The Journal of Immunology. 2015;194:4880–4890. doi: 10.4049/jimmunol.1402851. [DOI] [PubMed] [Google Scholar]
- 100.Hara H, Tsuchiya K, Kawamura I, Fang R, Hernandez-Cuellar E, Shen Y, et al. Phosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck-like aggregates and inflammasome activity. Nat Immunol. 2013;14:1247–1255. doi: 10.1038/ni.2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Labbé K, McIntire CR, Doiron K, Leblanc PM, Saleh M. Cellular Inhibitors of Apoptosis Proteins cIAP1 and cIAP2 Are Required for Efficient Caspase-1 Activation by the Inflammasome. Immunity. 2011;35:897–907. doi: 10.1016/j.immuni.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 102.Ainscough JS, Frank Gerberick G, Zahedi-Nejad M, Lopez-Castejon G, Brough D, Kimber I, et al. Dendritic cell IL-1α and IL-1β are polyubiquitinated and degraded by the proteasome. J Biol Chem. 2014;289:35582–35592. doi: 10.1074/jbc.M114.595686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Niebler M, Qian X, Höfler D, Kogosov V, Kaewprag J, Kaufmann AM, et al. Post-translational control of IL-1β via the human papillomavirus type 16 E6 oncoprotein: a novel mechanism of innate immune escape mediated by the E3-ubiquitin ligase E6-AP and p53. PLoS Pathog. 2013;9:e1003536. doi: 10.1371/journal.ppat.1003536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ma A, Malynn BA. A20: linking a complex regulator of ubiquitylation to immunity and human disease. Nature Reviews Immunology. 2012:1–12. doi: 10.1038/nri3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289:2350–2354. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lu TT, Onizawa M, Hammer GE, Turer EE, Yin Q, Damko E, et al. Dimerization and Ubiquitin Mediated Recruitment of A20, a Complex Deubiquitinating Enzyme. Immunity. 2013:1–10. doi: 10.1016/j.immuni.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hammer GE, Turer EE, Taylor KE, Fang CJ, Advincula R, Oshima S, et al. Expression of A20 by dendritic cells preserves immune homeostasis and prevents colitis and spondyloarthritis. Nat Immunol. 2011;12:1184–1193. doi: 10.1038/ni.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Boone DL, Turer EE, Lee EG, Ahmad RC, Wheeler MT, Tsui C, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol. 2004;5:1052–1060. doi: 10.1038/ni1110. [DOI] [PubMed] [Google Scholar]
- 109.Walle LV, Van Opdenbosch N, Jacques P, Fossoul A, Verheugen E, Vogel P, et al. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature. 2014;512:69–73. doi: 10.1038/nature13322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhang M, Kenny SJ, Ge L, Xu K, Schekman R. Translocation of interleukin-1β into a vesicle intermediate in autophagy-mediated secretion. eLife. 2015;4:1463. doi: 10.7554/eLife.11205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sborgi L, Ravotti F, Dandey VP, Dick MS, Mazur A, Reckel S, et al. Structure and assembly of the mouse ASC inflammasome by combined NMR spectroscopy and cryo-electron microscopy. Proc Natl Acad Sci USA. 2015;112:13237–13242. doi: 10.1073/pnas.1507579112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell. 2014;156:1193–1206. doi: 10.1016/j.cell.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shao BZ, Xu ZQ, Han BZ, Su DF, Liu C. NLRP3 inflammasome and its inhibitors: a review. Front Pharmacol. 2015;6:262. doi: 10.3389/fphar.2015.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhang W, Wu KP, Sartori MA, Kamadurai HB, Ordureau A, Jiang C, et al. System-Wide Modulation of HECT E3 Ligases with Selective Ubiquitin Variant Probes. Molecular Cell. 2016;62:1–48. doi: 10.1016/j.molcel.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ernst A, Avvakumov G, Tong J, Fan Y, Zhao Y, Alberts P, et al. A strategy for modulation of enzymes in the ubiquitin system. Science. 2013;339:590–595. doi: 10.1126/science.1230161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gorelik M, Orlicky S, Sartori MA, Tang X, Marcon E, Kurinov I, et al. Inhibition of SCF ubiquitin ligases by engineered ubiquitin variants that target the Cul1 binding site on the Skp1-F-box interface. Proc Natl Acad Sci USA. 2016;113:3527–3532. doi: 10.1073/pnas.1519389113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Skaar JR, Pagan JK, Pagano M. SCF ubiquitin ligase-targeted therapies. Nat Rev Drug Discov. 2014;13:889–903. doi: 10.1038/nrd4432. [DOI] [PMC free article] [PubMed] [Google Scholar]