

Abstract
Gas-phase ion chemistry methods that capture and characterize the degree of activation of small molecules in the active sites of homogeneous catalysts form a powerful new tool to unravel how ligand environments affect reactivity. A key roadblock in this development, however, is the ability to generate the fragile metal oxidation states that are essential for catalytic activity. Here we demonstrate the preparation of the key Ni(I) center in the widely used cyclam scaffold using ion–ion recombination as a gas-phase alternative to electrochemical reduction. The singly charged Ni+(cyclam) coordination complex is generated by electron transfer from fluoranthene and azobenzene anions to doubly charged Ni2+(cyclam), using the electron-transfer dissociation protocol in a commercial quadrupole ion trap instrument and in a custom-built octopole RF ion trap. The successful preparation of the Ni+(cyclam) cation is verified through analysis of its vibrational spectrum obtained using the infrared free electron laser FELIX.
Recent advances in mass spectrometry are creating powerful new ways to study the activation of small molecules docked in the active sites of homogeneous catalysts.1−5 One of these involves a multistep strategy in which transition-metal coordination compounds are extracted from solution using ambient ionization methods and fragmented in the gas phase to expose an open coordination site on the metal atom. This allows substrate molecules to be docked directly onto the open site using temperature-controlled condensation in an ion trap. The degree of substrate activation can then be determined through analysis of the substrate vibrational spectrum, obtained by infrared (IR) photodissociation spectroscopy.4,6−8 This approach has been recently demonstrated for N2, CO, and CO2 activation by a bis-phenoidal Ni compound,9 which serves to highlight the essential role that the oxidation state of the metal plays in controlling the degree of substrate activation. Indeed, for the case of CO2, it was observed that CO2 attachment to the Ni(II) center of the bis-phenoidal ligand was completely ineffective, while the Ni(I) variation displaced considerable charge onto the CO2 framework, as evidenced by an approximately 400 cm–1 red shift of the antisymmetric CO2 stretching mode.
Accessing the key Ni(I) species4 required a specially designed bimetallic precursor compound that supported stable Ni(I) oxidation states in both metal centers as an overall dication.10,11 The singly charged Ni(I)-based macrocycle, with an open coordination site, was then obtained by dissociation of the dicationic precursor upon injection into the mass spectrometer with an electrospray ionization (ESI) interface. While successful in demonstrating gas-phase capture and characterization of substrate activation, this early effort also highlights a key roadblock to the general application of this gas-phase method to contemporary transition-metal-based molecular catalysts, such as the Ni(cyclam) system depicted in Figure 1,12−19 which has been shown20−25 to be an effective catalyst for the reduction of CO2 because of its high selectivity toward CO (as opposed to H2) production in aqueous solution. Although the Ni(II) compound in the +2 charge state is readily prepared in routine mass spectrometric analysis using ESI, the more fragile Ni(I) analogue, Ni+(cyclam), is thought to be the catalytically active species.20,26 In the condensed phase, Ni(I) has been accessed through electrochemical reduction at mercury or glassy carbon electrodes,20 and pulse radiolysis has also been shown to reduce Ni2+(cyclam) in the presence of eaq–, CO2•–, and H•.27 Generally, these singly charged metal compounds are extremely reactive, and indeed the Ni+(cyclam) system has yet to be characterized.20 Attempts to generate the singly charged Ni+(cyclam) through manipulation of the chemistry in the ESI source have proven to be very inefficient because of the domination of the proton-transfer pathway, resulting in [Ni2+(cyclam-H)-]+ at m/z 257, as illustrated by representative mass spectra in Figure S1.
Figure 1.
Square planar structure of Ni2+(cyclam).
Here we describe an alternative method to prepare the active species based on redox manipulation in the gas phase of the readily formed Ni(II) complexes from ESI. In particular, because the Ni(II) compound is a dication, ion–ion recombination through electron transfer from an anionic agent
| 1 |
presents a promising method to rationally prepare the key Ni(I) species through one-electron reduction. Moreover, this postionization reduction method is directly compatible with the gas-phase ion chemistry characterization and spectroscopic investigation with infrared multiple photon dissociation (IRMPD) spectroscopy methods. A potential complication in the use of ion–ion collisions for single-electron reduction is that such processes are exoergic. Indeed, electron-transfer dissociation (ETD) is a widely used alternative to collision-induced dissociation used frequently in sequencing studies of biopolymers.28,29 Commercially available ETD-enabled MS platforms most often use the radical anion fluoranthene,30−34 and in fact ETD was recently coupled to vibrational spectroscopy35 for structural characterization of the ETD fragments of a tryptic peptide. Here, we demonstrate that the ETD instrumentation can be used as a means to reduce Ni2+(cyclam) to Ni+(cyclam) without dissociation (ETnoD) of the ligand framework, effectively creating an electron-transfer reduction (ETR) capability for mass spectrometry. Moreover, buffer gas cooling quenches the considerable exothermicity (∼8 eV) of the recombination reaction, allowing the product ions to be stabilized and structurally characterized using vibrational spectroscopy in combination with density functional theory calculations.
To illustrate the generality and versatility of ETR, we carried out the measurements on two different instruments using two different anionic reduction agents: the fluoranthene and azobenzene radical anions with structures indicated in panels d and f in Figure 2, respectively. For fluoranthene, we used the modified 3D quadrupole ion trap (QIT, Bruker, AmaZon Speed ETD) at the FELIX Laboratory, where Ni2+(cyclam) ions were generated by ESI from a solution containing 10–6 M cyclam and 10–5 M Ni(NO3)2 as shown in Figure 2a. The doubly charged coordination complex containing the 58Ni isotope at m/z = 129 was isolated (colored blue in Figure 2b) and then charge-reduced using the ETD option of the QIT MS, as further described in the Supporting Information. A reaction time of 200–300 ms was found to give optimal conversion to the charge-reduced species, as illustrated in the product mass distribution displayed in Figure 2b. The expanded region in the important mass range (Figure 2d) reveals efficient generation of ions with m/z = 258 (red), which is expected for nondissociative electron transfer to Ni2+(cyclam). In addition to the formation of Ni+(cyclam), the singly charged [Ni2+(cyclam-H)−]+ cation (gold peak in Figure 2d) at m/z = 257 is also generated. This ion likely corresponds to the Ni(II) ion coordinated to the deprotonated ligand, which was exclusively formed in previous attempts to generate singly charged Ni+(cyclam) using reductive chemistry in the ESI solution (as seen in Figure S1). Note that the isolation of the 58Ni ion prior to charge reduction in the QIT instrument allows us to uniquely identify the one-electron reduction product against background arising from H-loss fragments involving heavier Ni isotopes
Figure 2.

QIT mass spectra of (a) Ni2+(cyclam) at m/z = 129 and (b) the ETD reaction of isolated monoisotopic Ni2+(cyclam) with fluoranthene (top structure), which shows a peak corresponding to the formation of Ni+(cyclam) at m/z = 258. Trace d expands the crucial region of trace b. CID on m/z = 258 results in the mass spectrum in trace c. Traces e and f show the analogous reaction of Ni2+(cyclam) with the azobenzene anion (bottom structure).
To establish how the properties of the reagent anion affect the ETR process, we explored a second, commonly used ETD reagent, azobenzene,36 this time using a Sciex QTRAP 4001 hybrid triple quadrupole linear ion trap mass spectrometer at Purdue, which was modified to perform ion–ion reactions in mutual storage mode.37 Interestingly, carrying out the reduction with azobenzene produced an even larger relative yield of m/z = 258 corresponding to the stoichiometry of Ni+(cyclam) as compared to the H-loss byproduct at m/z = 257 (see Figure 2e,f). We note that the reaction time for optimal yield was much shorter (10 ms for azobenzene in the linear quadrupole vs 200 ms for fluoranthene in the 3D QIT). Concomitantly, the reaction with azobenzene also resulted in a much lower abundance of dissociation products, as shown by the much reduced intensity of the mass peaks at intermediate m/z (compare panels b and e of Figure 2).
The production of the m/z = 258 ion is a necessary step in the application of ETR to the preparation of the key Ni(I)(cyclam)+ species, but this method of preparation raises the important question of whether the organic scaffold survives the substantial exoergicity, ΔE, inherent to the bimolecular reaction:
| 2 |
| 3 |
A rough estimate of ΔE based on the calculated ionization energy (IE) of Ni+(cyclam) of about 8.5 eV and the adiabatic electron affinities (AEA) of the anions (0.2 and 1.6 eV for fluoranthene and azobenene, respectively)38,39 sets limiting values of ΔE in the range of 8.3 and 6.9 eV for the two anions (using ΔE = IE – AEA). Interestingly, we note that the more exothermic fluoranthene reaction yields more fragmentation peaks arising from eq 3, as expected for the usual ETD processes. These degradation products (interloper peaks in Figure 2b) largely arise from sequential neutral losses of 30 and 43 mass units, which are consistent with ejection of C2H6 and C2H5N groups. To evaluate whether these are characteristic of an excited [Ni(cyclam)*]+ ion, we measured the decomposition pathways of the m/z = 258 parent ion using collision-induced dissociation (CID), with the result presented as the inverted trace in Figure 2c. The overall similarity in the breakup patterns upon CID and ETD with fluoranthene establishes that a significant fraction of the nascent ETR ions decompose before they can be cooled in the trap. We note that related work involving charge reduction of multiply charged ions in high-energy collisions with alkali metal vapors40−42 has also established partial survival of the nascent ions with considerable internal energy content.
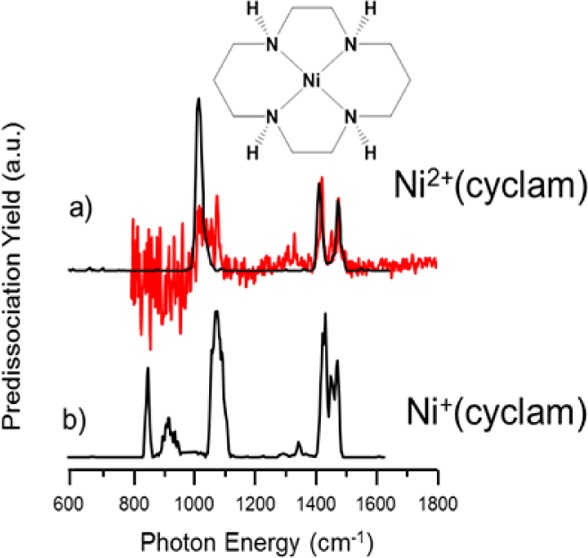
The fact that ETR necessarily occurs with deposition of considerable internal energy into the target ion raises the important question of whether the ligand scaffold can survive intact when the ion is cooled through collisions with a buffer gas. A powerful method to establish the structure of ionic species is through analysis of their vibrational spectrum, which can be obtained through action spectroscopy methods using tunable IR lasers such as the FELIX free electron laser employed here.35,43,44 The 600–1650 cm–1 region of the IRMPD vibrational spectrum for the mass-isolated m/z = 258 ion was recorded by irradiating the trapped ions for 1.5 s with the FEL operating at a repetition rate of 10 Hz and producing 6–10 μs long macropulses with energies up to 100 mJ per pulse. When the frequency of the FEL is resonant with a vibrational mode of the trapped ions, multiple photons are absorbed and the internal energy of the ions increases to above the dissociation threshold so that unimolecular dissociation occurs along the pathway with the lowest energy barrier. We generate IR spectra by relating the fraction of dissociation (yield = ∑(fragment ions)/∑(precursor + fragment ions)) to the IR wavelength as the laser frequency is scanned.44,45 The yield is linearly corrected for the wavelength dependence of the laser pulse energy, and the wavelength is calibrated (online) using a grating spectrometer. The resulting IRMPD spectrum of Ni(cyclam)+ generated by ETR is shown in Figure 3c and is dominated by three relatively sharp transitions near 850, 1000, and 1450 cm–1, which are attributed to the CH rocking, CN stretching, and CH bending fundamentals, respectively.
Figure 3.
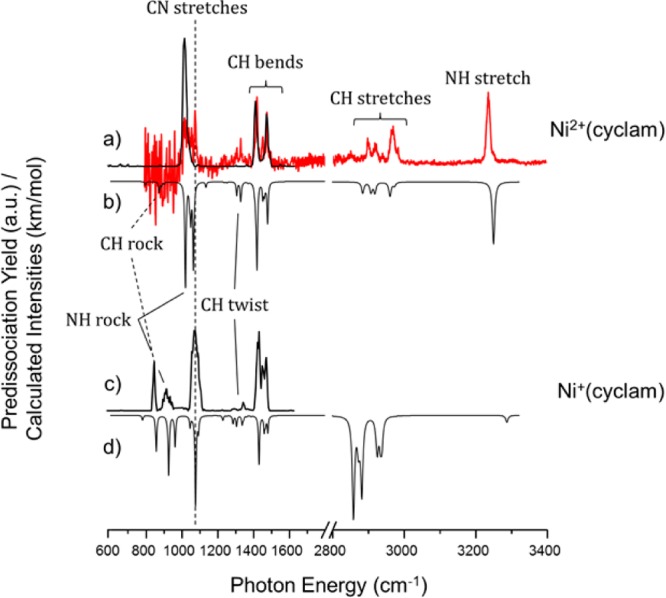
IRMPD spectra of (a) Ni2+(cyclam) and (c) Ni+(cyclam). The single-photon N2 predissociation spectrum of Ni2+(cyclam), red trace in panel a, taken at 30 K shows good agreement with the IRMPD data while revealing more detailed fine structure in the spectral signatures. The inverted traces display the computed harmonic spectra for (b) Ni2+(cyclam) and (d) Ni+(cyclam). Theoretical frequencies are scaled by 0.975 below 2200 cm–1 and 0.95 above 2200 cm–1.
To quantify the spectral signature of the ligand and to establish the efficacy of theoretical methods used to characterize this system, we also recorded the spectrum of the dicationic Ni2+(cyclam) precursor using both IRMPD spectroscopy at FELIX as well as cryogenic ion spectroscopy at Yale to establish the sensitivity of this spectrum to ion internal energy. The spectrum of the 300 K ion obtained in the QIT MS at FELIX covers the 600–1800 cm–1 region. The 30 K ion spectrum obtained via N2 tagging at Yale using a LaserVision OPO/OPA system covers the 800–3400 cm–1 range. The N2 tag is calculated to attach to the NH groups of the Ni2+(cyclam) ligand and is not believed to be “activated” by the Ni(II) ion.4 The N2-tagged spectrum is obtained in a linear action regime and interrogates vibrationally cold ions. Availability of both spectra enables us to confirm that the IRMPD spectra are hardly affected by the higher internal energy of the ions or by the multiple-photon nature of excitation in IRMPD,46 nor is the N2-tagged spectrum affected by the presence of the tag. Figure 3a compares the N2-tagged Ni2+(cyclam) spectrum (red) with that recorded for the 300 K ion using the IRMPD approach (black trace). Although there are minor differences in the multiplet structure of the strong features, two strong bands at similar frequencies dominate both spectra. Figure S4 displays additional vibrational spectra recorded at 300 K using the FTICR-MS at Nijmegen, where some additional spectral features are recovered as a result of the lower background pressure and the inherently increased efficiency of multiple-photon excitation. Most importantly, analogous features are also found in the m/z = 258 product ion from ETR, providing strong evidence that the structure of the ligand is retained in spite of the exoergicity involved in the gas-phase one-electron reduction.
The fine structure on the bands in the fingerprint region, combined with the CH and NH band patterns in the higher-energy range obtained with N2 tagging, provides the most useful benchmarks for structure determination by comparison with computed spectra for various local minimum geometries. DFT calculations were performed at the B3LYP/6-31++G(d,p) level of theory and recovered the lowest-energy structure of the Ni2+(cyclam) ion displayed in Figure 4a, with the corresponding (scaled) harmonic spectrum displayed in Figure 3b. In particular, the fine structure associated with the dominant bands at 1000 and 1450 cm–1, as well as the weaker feature at 1320 cm–1 (which were not evident in the IRMPD spectrum measured in the QIT, Figure 3a, but are evident in the FTICR/FELIX shown in Figure S4), are accurately reproduced by the predicted spectrum. Note that this structure (Figure 4a) features two of the proximal NH groups on the three carbon ring oriented in the same direction, roughly orthogonal to the plane containing the Ni atom and the four N atoms. This isomer, denoted trans-III, is the most abundant of the two isomers (the other being trans-I with all four NH groups pointing in the same direction) in solution, 85%, as characterized by NMR;47 isomerization between the trans-III and trans-I isomers was determined to occur with a rate constant of 121 ± 21 M–1 s–1 in solution.47 The trans notation refers to the distortion of the alkyl chains relative to the N atom plane, as illustrated in the two views of the trans-III structure in Figure 4. The overall spin state of the trans-III isomer is a singlet with a d6 low-spin electron configuration on the Ni center where, by NPA analysis (described in detail in the Supporting Information), the largest positive charge per atom lies on the metal center (9% on the Ni, 5% each on the NH protons, with the residual distributed among the CH protons of the ligand (about ∼3% of the fundamental charge on each)). The absence of the trans-I form in the gas-phase experiments reported here is confirmed by the single sharp NH transition at 3239 cm–1, which is predicted to be split into a doublet spaced by about 45 cm–1 in the trans-I isomer. Supplementary Figure S2 presents a summary of the five isomers of Ni2+(cyclam), along with the scaled harmonic spectra for the trans-I and trans-III isomers in Figure S3 and energetics for all five isomers in Tables S1 and S2.
Figure 4.
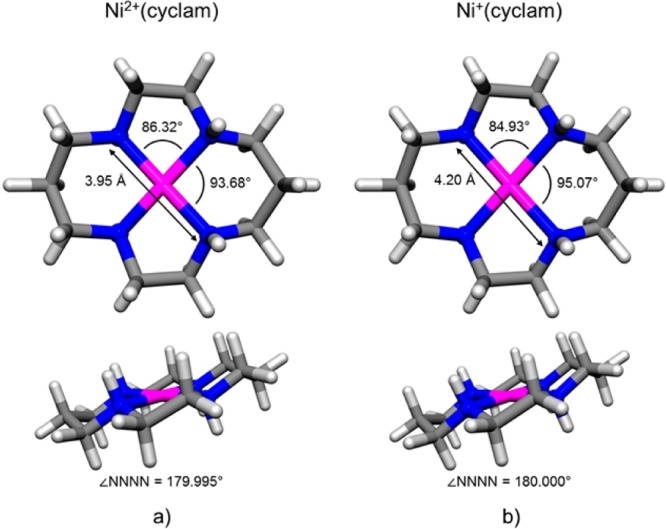
Calculated minimum energy structures of the trans-III isomers of (a) Ni2+(cyclam) and (b) Ni+(cyclam) revealing a lengthening of the N–N bond and a change of the N–Ni–N angles upon reduction of the metal center. The four nitrogen atoms of the cyclam macrocycle remain square planar. Calculations were performed at the B3LYP/6-31++G(d,p) level of theory.
Given that the B3LYP/6-31++G(d,p) level accurately recovers both the cold and IRMPD spectra of the Ni2+(cyclam) ion, we next extend this method to consider the structural implications of the IRMPD spectrum of the Ni+(cyclam) prepared by ETR (Figure 3c). The computed lowest-energy structure is presented in Figure 4b, with its computed spectrum in Figure 3d, indeed capturing all the strong features in the experimental IRMPD spectrum (Figure 3c). Note that there are significant differences between the IRMPD spectra for the two charge states, indicating that there is significant distortion of the ligand as a result of charge reduction of the metal center. In particular, new features appear near 800 and 900 cm–1 in the Ni+(cyclam) system, which are accurately recovered in the calculated spectrum and are attributed to NH and CH rocking modes of the ligand scaffold. The structure in Figure 4b is also derived from a trans conformation with substantial elongation (0.123 Å) of the Ni–N bonds. The spin state of this structure is a doublet with a d9 electron configuration on the Ni center, where the partial positive charge is now quite similar for the metal center and the NH protons. In fact, the NH protons have slightly more positive charge per atom than the Ni atom (6% for each NH proton, 5.7% for Ni, 3% for each of the remaining CH protons).
The spectroscopic measurements establish that ETR indeed provides a straightforward synthetic path for the preparation of the critical Ni(I) oxidation state with retention of the cyclam coordination environment. It is therefore of interest to elucidate the features of the reaction that optimize nondestructive electron attachment to the dication precursor. That is, the anionic reagents used here were optimized for dissociative electron transfer, while suppressing the branching to proton transfer, to provide as a general means to break up biopolymers for sequence analysis.28,29 In the case of ETR, we also seek to suppress proton transfer but strive for less exothermic processes in order to preserve the ligand environment. The observation that azobenzene dramatically reduces the degradation byproducts (compare panels c and d of Figure 2) indicates that lowering the exoergicity by 1.4 eV has a profound effect on the degree of fragmentation. Part of this enhancement likely also results from the strong geometry change in the azobenzene anion framework upon electron detachment, which has been quantified through negative ion photoelectron spectroscopy.38,39 This is evident by the degree of vibrational excitation in the neutral upon vertical electron detachment, which in the case of azobenzene deposits considerable (∼1.6 eV) additional energy in the neutral azobenzene moiety, thus reducing the energy partitioned to the nascent Ni+(cyclam) ion. It would therefore be valuable to refine the ETR approach by identifying higher electron affinity electron donors as well as systems which feature large geometry changes upon charge transfer.
In conclusion, we have demonstrated how the key Ni+(cyclam) oxidation state can be prepared in vacuum through nondissociative electron transfer from two molecular anions, fluoranthene and azobenzene, to the stable Ni2+(cyclam) ion in an ion–ion recombination process carried out in RF ion traps. The structure of both the dicationic precursor and the Ni+(cyclam) product ions were established with vibrational spectroscopy, verifying that the ligand structure is retained despite the large exoergicities of the electron-transfer reactions. This chemistry was carried out using the ETD capability of a modified commercial mass spectrometer (Bruker, AmaZon Speed ETD) as well as in a modified triple quadrupole mass spectrometer. The latter instrument is readily adapted to cryogenic ion processing methods which can stabilize small molecules in the active sites of Ni(I) reduction catalysts, and these directions are presently under study.
Acknowledgments
M.U.M., G.B., J.M., and J.O. gratefully acknowledge the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO) for the support of the FELIX Laboratory. Financial support for this project was provided by NWO Chemical Sciences under VICI Project No. 724.011.002 awarded to J.O. We also thank NWO Physical Sciences and the SARA Supercomputer Centre for providing the computational time and resources (grant 15408). S.M.C. and M.A.J. thank the Air Force Office of Scientific Research (Grant FA9550-13-1-0007).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpclett.7b02223.
Mass spectra recorded at the Yale tandem time-of-flight mass spectrometer; structures, energetics, and computed spectra for alternative conformations of the coordination complexes; IRMPD spectrum of Ni2+(cyclam) recorded in the Fourier-Transform Ion Cyclotron Resonance MS at FELIX (PDF)
Author Contributions
# M.U.M. and S.M.C. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Chen P. Electrospray Ionization Tandem Mass Spectrometry in High-Throughput Screening of Homogeneous Catalysts. Angew. Chem., Int. Ed. 2003, 42, 2832–2847. 10.1002/anie.200200560. [DOI] [PubMed] [Google Scholar]
- Lang S. M.; Frank A.; Bernhardt T. M. Activation and Catalytic Dehydrogenation of Methane on Small Pdx+ and PdxO+ Clusters. J. Phys. Chem. C 2013, 117, 9791–9800. 10.1021/jp312852r. [DOI] [Google Scholar]
- Miller G. B. S.; Esser T. K.; Knorke H.; Gewinner S.; Schöllkopf W.; Heine N.; Asmis K. R.; Uggerud E. Spectroscopic Identification of a Bidentate Binding Motif in the Anionic Magnesium-CO2 Complex ([ClMgCO2]−). Angew. Chem., Int. Ed. 2014, 53, 14407–14410. 10.1002/anie.201409444. [DOI] [PubMed] [Google Scholar]
- Menges F. S.; Craig S. M.; Totsch N.; Bloomfield A.; Ghosh S.; Kruger H. J.; Johnson M. A. Capture of CO2 by a Cationic Nickel(I) Complex in the Gas Phase and Characterization of the Bound, Activated CO2 Molecule by Cryogenic Ion Vibrational Predissociation Spectroscopy. Angew. Chem., Int. Ed. 2016, 55, 1282–1285. 10.1002/anie.201507965. [DOI] [PubMed] [Google Scholar]
- Schwarz H. Metal-Mediated Activation of Carbon Dioxide in the Gas Phase: Mechanistic Insight Derived from a Combined Experimental/Computational Approach. Coord. Chem. Rev. 2017, 334, 112–123. 10.1016/j.ccr.2016.03.009. [DOI] [Google Scholar]
- Knurr B. J.; Weber J. M. Solvent-Driven Reductive Activation of Carbon Dioxide by Gold Anions. J. Am. Chem. Soc. 2012, 134, 18804–18808. 10.1021/ja308991a. [DOI] [PubMed] [Google Scholar]
- Zhao Z.; Kong X. T.; Yang D.; Yuan Q. Q.; Xie H.; Fan H. J.; Zhao J. J.; Jiang L. Reactions of Copper and Silver Cations with Carbon Dioxide: An Infrared Photodissociation Spectroscopic and Theoretical Study. J. Phys. Chem. A 2017, 121, 3220–3226. 10.1021/acs.jpca.7b01320. [DOI] [PubMed] [Google Scholar]
- Iskra A.; Gentleman A. S.; Kartouzian A.; Kent M. J.; Sharp A. P.; Mackenzie S. R. Infrared Spectroscopy of Gas-Phase M+(CO2)n (M = Co, Rh, Ir) Ion–Molecule Complexes. J. Phys. Chem. A 2017, 121, 133–140. 10.1021/acs.jpca.6b10902. [DOI] [PubMed] [Google Scholar]
- Craig S. M.; Menges F. S.; Johnson M. A. Application of Gas Phase Cryogenic Vibrational Spectroscopy to Characterize the CO2, CO, N2 and N2O Interactions with the Open Coordination Site on a Ni(I) Macrocycle using Dual Cryogenic Ion Traps. J. Mol. Spectrosc. 2017, 332, 117–123. 10.1016/j.jms.2016.11.015. [DOI] [Google Scholar]
- Kruger H. J. Spin Transition in Octahedral Metal Complexes Containing Tetraazamacrocyclic Ligands. Coord. Chem. Rev. 2009, 253, 2450–2459. 10.1016/j.ccr.2009.06.004. [DOI] [Google Scholar]
- Koch W. O.; Kruger H. J. A Highly Reactive and Catalytically Active Model System for Intradiol-Cleaving Catechol Dioxygenases: Structure and Reactivity of Iron(III) Catecholate Complexes of N,N′-dimethyl-2,11-diaza[3.3](2,6)pyridinophane. Angew. Chem., Int. Ed. Engl. 1996, 34, 2671–2674. 10.1002/anie.199526711. [DOI] [Google Scholar]
- Yao S. L.; Driess M. Lessons from Isolable Nickel(I) Precursor Complexes for Small Molecule Activation. Acc. Chem. Res. 2012, 45, 276–287. 10.1021/ar200156r. [DOI] [PubMed] [Google Scholar]
- Perry R. H.; Brownell K. R.; Chingin K.; Cahill T. J.; Waymouth R. M.; Zare R. N. Transient Ru-methyl Formate Intermediates Generated with Bifunctional Transfer Hydrogenation Catalysts. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 2246–2250. 10.1073/pnas.1118934109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider J.; Jia H. F.; Muckerman J. T.; Fujita E. Thermodynamics and Kinetics of CO2, CO, and H+ Binding to the Metal Centre of CO2 Reduction Catalysts. Chem. Soc. Rev. 2012, 41, 2036–2051. 10.1039/C1CS15278E. [DOI] [PubMed] [Google Scholar]
- Knickelbein M. B. Reactions of Transition Metal Clusters with Small Molecules. Annu. Rev. Phys. Chem. 1999, 50, 79–115. 10.1146/annurev.physchem.50.1.79. [DOI] [PubMed] [Google Scholar]
- Jun C. H. Transition Metal-Catalyzed Carbon-Carbon Bond Activation. Chem. Soc. Rev. 2004, 33, 610–618. 10.1039/B308864M. [DOI] [PubMed] [Google Scholar]
- Shilov A. E.; Shul’pin G. B. Activation of C-H Bonds by Metal Complexes. Chem. Rev. 1997, 97, 2879–2932. 10.1021/cr9411886. [DOI] [PubMed] [Google Scholar]
- Cokoja M.; Bruckmeier C.; Rieger B.; Herrmann W. A.; Kuhn F. E. Transformation of Carbon Dioxide with Homogeneous Transition-Metal Catalysts: A Molecular Solution to a Global Challenge?. Angew. Chem., Int. Ed. 2011, 50, 8510–8537. 10.1002/anie.201102010. [DOI] [PubMed] [Google Scholar]
- Fujita E.; Brunschwig B. S.; Ogata T.; Yanagida S. Toward Photochemical Carbon-Dioxide Activation by Transition-Metal Complexes. Coord. Chem. Rev. 1994, 132, 195–200. 10.1016/0010-8545(94)80040-5. [DOI] [Google Scholar]
- Froehlich J. D.; Kubiak C. P. Homogeneous CO2 Reduction by Ni(cyclam) at a Glassy Carbon Electrode. Inorg. Chem. 2012, 51, 3932–3934. 10.1021/ic3001619. [DOI] [PubMed] [Google Scholar]
- Song J. S.; Klein E. L.; Neese F.; Ye S. F. The Mechanism of Homogeneous CO2 Reduction by Ni(cyclam): Product Selectivity, Concerted Proton-Electron Transfer and C-O Bond Cleavage. Inorg. Chem. 2014, 53, 7500–7507. 10.1021/ic500829p. [DOI] [PubMed] [Google Scholar]
- Benson E. E.; Kubiak C. P.; Sathrum A. J.; Smieja J. M. Electrocatalytic and Homogeneous Approaches to Conversion of CO2 to Liquid Fuels. Chem. Soc. Rev. 2009, 38, 89–99. 10.1039/B804323J. [DOI] [PubMed] [Google Scholar]
- Beley M.; Collin J. P.; Ruppert R.; Sauvage J. P. Electrocatalytic Reduction of CO2 by Ni Cyclam2+ in Water - Study of the Factors Affecting the Efficiency and the Selectivity of the Process. J. Am. Chem. Soc. 1986, 108, 7461–7467. 10.1021/ja00284a003. [DOI] [PubMed] [Google Scholar]
- Beley M.; Collin J. P.; Ruppert R.; Sauvage J. P. Nickel(I) Cyclam - an Extremely Selective Electrocatalyst for Reduction of CO2 in Water. J. Chem. Soc., Chem. Commun. 1984, 1315–1316. 10.1039/c39840001315. [DOI] [Google Scholar]
- Craig C. A.; Spreer L. O.; Otvos J. W.; Calvin M. Photochemical Reduction of Carbon-Dioxide Using Nickel Tetraazamacrocycles. J. Phys. Chem. 1990, 94, 7957–7960. 10.1021/j100383a038. [DOI] [Google Scholar]
- Balazs G. B.; Anson F. C. The Adsorption of Ni(Cyclam)+ at Mercury-Electrodes and Its Relation to the Electrocatalytic Reduction of CO2. J. Electroanal. Chem. 1992, 322, 325–345. 10.1016/0022-0728(92)80086-J. [DOI] [Google Scholar]
- Kelly C. A.; Blinn E. L.; Camaioni N.; D’Angelantonio M.; Mulazzani Q. G. Mechanism of CO2 and H+ Reduction by Ni(cyclam)+ in Aqueous Solution: A Pulse and Continuous Radiolysis Study. Inorg. Chem. 1999, 38, 1579–1584. 10.1021/ic980902p. [DOI] [Google Scholar]
- Kim M. S.; Pandey A. Electron Transfer Dissociation Mass Spectrometry in Proteomics. Proteomics 2012, 12, 530–542. 10.1002/pmic.201100517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLuckey S. A.; Huang T. Y. Ion/Ion Reactions: New Chemistry for Analytical MS. Anal. Chem. 2009, 81, 8669–8676. 10.1021/ac9014935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubarev R. A.; Kelleher N. L.; McLafferty F. W. Electron Capture Dissociation of Multiply Charged Protein Cations: A Nonergodic Process. J. Am. Chem. Soc. 1998, 120, 3265–3266. 10.1021/ja973478k. [DOI] [Google Scholar]
- Parker M. L.; Gronert S. Investigating Reduced Metal Species via Sequential Ion/Ion and Ion/Molecule Reactions: The Reactions of Transition Metal Phenanthrolines with Allyl Iodide. Int. J. Mass Spectrom. 2017, 418, 73–78. 10.1016/j.ijms.2016.11.018. [DOI] [Google Scholar]
- Syka J. E. P.; Coon J. J.; Schroeder M. J.; Shabanowitz J.; Hunt D. F. Peptide and Protein Sequence Analysis by Electron Transfer Dissociation Mass Spectrometry. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 9528–9533. 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawardena H. P.; Gorenstein L.; Erickson D. E.; Xia Y.; McLuckey S. A. Electron Transfer Dissociation of Multiply Protonated and Fixed Charge Disulfide Linked Polypeptides. Int. J. Mass Spectrom. 2007, 265, 130–138. 10.1016/j.ijms.2007.01.017. [DOI] [Google Scholar]
- Martens J.; Berden G.; Oomens J. Structures of Fluoranthene Reagent Anions Used in Electron Transfer Dissociation and Proton Transfer Reaction Tandem Mass Spectrometry. Anal. Chem. 2016, 88, 6126–6129. 10.1021/acs.analchem.6b01483. [DOI] [PubMed] [Google Scholar]
- Martens J.; Grzetic J.; Berden G.; Oomens J. Structural Identification of Electron Transfer Dissociation Products in Mass Spectrometry using Infrared Ion Spectroscopy. Nat. Commun. 2016, 7, 11754. 10.1038/ncomms11754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y.; Han H.; McLuckey S. A. Activation of Intact Electron-Transfer Products of Polypeptides and Proteins in Cation Transmission Mode Ion/Ion Reactions. Anal. Chem. 2008, 80, 1111–1117. 10.1021/ac702188q. [DOI] [PubMed] [Google Scholar]
- Xia Y.; Wu J.; McLuckey S. A.; Londry F. A.; Hager J. W. Mutual Storage Mode Ion/Ion Reactions in a Hybrid Linear Ion Trap. J. Am. Soc. Mass Spectrom. 2005, 16, 71–81. 10.1016/j.jasms.2004.09.017. [DOI] [PubMed] [Google Scholar]
- Frey W. F.; Compton R. N.; Naff W. T.; Schweinler H. C. Electron Impact Studies of Some Cyclic Hydrocarbons. Int. J. Mass Spectrom. Ion Phys. 1973, 12, 19–32. 10.1016/0020-7381(73)80083-X. [DOI] [Google Scholar]
- Paik D. H.; Baskin J. S.; Kim N. J.; Zewail A. H. Ultrafast Vectorial and Scalar Dynamics of Ionic Clusters: Azobenzene Solvated by Oxygen. J. Chem. Phys. 2006, 125, 133408. 10.1063/1.2205855. [DOI] [PubMed] [Google Scholar]
- Byskov C. S.; Nielsen S. B. On the Formation, Stability, and Dissociation of Peptide Radicals after Femtosecond Electron Transfer from Alkali Metal Atoms. Int. J. Mass Spectrom. 2015, 390, 2–13. 10.1016/j.ijms.2015.05.014. [DOI] [Google Scholar]
- Byskov C. S.; Weber J. M.; Nielsen S. B. Gas-phase Spectroscopy of Singly Reduced Tris(bipyridine)ruthenium Ions, Ru(bipy)3+. Phys. Chem. Chem. Phys. 2015, 17, 5561–5564. 10.1039/C4CP05477F. [DOI] [PubMed] [Google Scholar]
- Turecek F.; Holm A. I. S.; Panja S.; Nielsen S. B.; Hvelplund P. Transition Metals as Electron Traps. II. Structures, Energetics and Electron Transfer Dissociations of Ternary Co, Ni and Zn-Peptide Complexes in the Gas Phase. J. Mass Spectrom. 2009, 44, 1518–1531. 10.1002/jms.1642. [DOI] [PubMed] [Google Scholar]
- Martens J.; Berden G.; Gebhardt C. R.; Oomens J. Infrared Ion Spectroscopy in a Modified Quadrupole Ion Trap Mass Spectrometer at the FELIX Free Electron Laser Laboratory. Rev. Sci. Instrum. 2016, 87, 103108. 10.1063/1.4964703. [DOI] [PubMed] [Google Scholar]
- Oomens J.; Sartakov B. G.; Meijer G.; Helden G. v. Gas Phase Infrared Multiple Photon Dissociation Spectroscopy of Mass Selected Molecular Ions. Int. J. Mass Spectrom. 2006, 254, 1–19. 10.1016/j.ijms.2006.05.009. [DOI] [Google Scholar]
- Rijs A. M.; Oomens J. IR Spectroscopic Techniques to Study Isolated Biomolecules. Top. Curr. Chem. 2014, 364, 1–42. 10.1007/128_2014_621. [DOI] [PubMed] [Google Scholar]
- Yacovitch T. I.; Heine N.; Brieger C.; Wende T.; Hock C.; Neumark D. M.; Asmis K. R. Vibrational Spectroscopy of Bisulfate/Sulfuric Acid/Water Clusters: Structure, Stability, and Infrared Multiple-Photon Dissociation Intensities. J. Phys. Chem. A 2013, 117, 7081–7090. 10.1021/jp400154v. [DOI] [PubMed] [Google Scholar]
- Connolly P. J.; Billo E. J. 1H NMR Evidence for the R,S,R,S Isomer of the (1,4,8,11-Tetraazacyclotetradecane) Nickel(II) Ion. Inorg. Chem. 1987, 26, 3224–3226. 10.1021/ic00266a033. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.