

Abstract
Elevated reactive oxygen species (ROS) induce the formation of lipids in neurons that are transferred to glia where they form lipid droplets (LD). We show that glial and neuronal monocarboxylate transporters (MCTs), fatty acid transport proteins (FATP), and apolipoproteins are critical for glial LD formation. MCTs enable glia to secrete and neurons to absorb lactate, which is converted to pyruvate and acetyl-CoA in neurons. Lactate metabolites provide a substrate for synthesis of fatty acids, which are processed and transferred to glia by FATP and apolipoproteins. In the presence of high ROS, inhibiting lactate transfer or lowering FATP or apolipoprotein levels all decrease glial LD accumulation in flies and in primary mouse glial-neuronal cultures. We show that human APOE can substitute for a fly glial apolipoprotein and that APOE4, an Alzheimer’s Disease susceptibility allele, is impaired in lipid transport and promotes neurodegeneration, providing insights into disease mechanisms.
Keywords: astrocytes, APOE2, APOE3, APOE4, Alzheimer’s Disease, reactive oxygen species, ROS, ARSAL, CMT2A, Leigh Syndrome, Aats-met, MARS2, Marf, Mitofusin, sicily, NDUFAF6, Drosophila melanogaster, Mus musculus
eTOC blurb
Liu et al. unravel an evolutionarily conserved mechanism which brings neuron-glia metabolic cooperation full circle. They show that glial lactate can fuel neuronal lipogenesis in response to ROS; in turn, neuronal lipids are transported and stored in glia as lipid droplets. The inability to transport lipids to glia for lipid droplet formation leads to accelerated neurodegeneration under stress.
Introduction
Neurodegeneration is often characterized by cellular hallmarks such as mitochondrial dysfunction, oxidative stress, protein aggregates, proteasome or autophagosome dysfunction, and endolysosomal defects (Beal, 2007; Jaiswal et al., 2012; Nixon, 2013). Previously, we showed that lipid droplets (LD), a neutral lipid storing organelle, arise due to elevated levels of reactive oxygen species (ROS) and may be used as an early biomarker for the onset of neurodegeneration (Liu et al., 2015). These LD accumulate because of mitochondrial defects that lead to highly elevated ROS, which induce the production of lipids in neurons and their subsequent transfer to glia. However, the mechanisms of lipid production or transport in the nervous system and the metabolic cooperation between neurons and glia, in both health and disease, remain poorly understood.
We previously found that elevating ROS in neurons alone is sufficient for glia to accumulate LD, suggesting a transfer of lipids (or their precursors) from one cell to the other (Liu et al., 2015). The mechanism of lipid transfer in the nervous system has not been previously studied. Moreover, although the resting brain’s energy expenditure is significantly higher than that of most organs, the cellular appropriation of neuronal energy and its derivation is unclear (Raichle and Gusnard, 2002; Wang et al., 2010). In mammals, blood glucose is the brain’s primary fuel source (van Hall et al., 2009). However, metabolic intermediates, such as lactate and ketone bodies, can also be used as a source of energy (Belanger et al., 2011; van Hall et al., 2009; Zielke et al., 2009). Along these lines, the Astrocyte Neuron Lactate Shuttle (ANLS) Hypothesis (Pellerin and Magistretti, 1994) was shown to play a role of nervous system homeostasis in Drosophila (Volkenhoff et al., 2015) and mice (Funfschilling et al., 2012; Lee et al., 2012; Machler et al., 2016). In Drosophila, perineural glia possess the enzymes necessary to metabolize the sugar trehalose, whose metabolites, including lactate, are secreted by glia and thought to be taken up by neurons (Volkenhoff et al., 2015). Similarly, in mammals, spectroscopy studies have shown that circulating blood glucose is taken up by astrocytes and hypothesized to be converted to lactate (Barros, 2013; Herzog et al., 2013; Hyder et al., 2006). Given that monocarboxylate transporters (MCTs) are expressed in the Drosophila (Volkenhoff et al., 2015) and mammalian nervous systems (Funfschilling et al., 2012; Lee et al., 2012; Pierre and Pellerin, 2005), it is thought that lactate is transported from glia to neurons through these membrane carriers. However, the in vivo function of lactate, its transport and metabolism in neurons and glia, and its relationship to LD remains to be determined.
Lipid transporters are likely to play a role in the nervous system metabolic homeostasis. These include Apolipoprotein E (ApoE) and Apolipoprotein D (ApoD) (Elliott et al., 2010). ApoE assists in circulating lipoprotein formation and is assumed to function in lipid transport in the brain (Huang and Mahley, 2014). ApoE deficient mice (Apoe−/−) exhibit early onset hyperlipidemia and aortic plaque formation (Jofre-Monseny et al., 2008; Maeda, 2011). However, there are conflicting reports regarding brain morphological changes in Apoe−/− mice with respect to synaptic loss and cytoskeletal changes (Anderson et al., 1998; Masliah et al., 1995; Montine et al., 1999). Humans have three allelic variants of ApoE resulting in six possible allelic combinations that alter an individual’s likelihood of developing hyperlipoproteinemia (APOE2) or Alzheimer’s disease (AD) (APOE4) (Elliott et al., 2010; Yu et al., 2014). Indeed, 40–80% of patients with AD carry at least one copy of the APOE4 allele (Farrer et al., 1997) and homozygous carriers have above a 50% lifetime risk of developing AD (Genin et al., 2011). APOE4 is by far the most common AD susceptibility locus (Mahley et al., 2006; Yu et al., 2014). The APOE3 allele is predominant allele in the population and confers an average risk for AD, whereas APOE2 is considered “protective” as it decreases an individual’s risk for developing AD (Conejero-Goldberg et al., 2014). The importance of APOE in AD suggests that it plays a major role in facilitating proper nervous system function. However, the physiological function of ApoE in metabolism of neurons and glia is poorly characterized and its function in relation to LD metabolism has not been documented.
Although certain lipid metabolism proteins are highly conserved in flies and mammals, including Sterol Regulatory Element Binding Protein [SREBP] (Rawson, 2003), ApoE does not appear to have a direct ortholog in Drosophila. In flies, the ApoD homologs glial lazarillo (Glaz) (Sanchez et al., 2006; Walker et al., 2006) and neural lazarillo (Nlaz) (Hull-Thompson et al., 2009) are protective against stress. Loss of Glaz or Nlaz leads to elevated sensitivity to ROS and a decrease in the triglyceride content of the whole animal (Hull-Thompson et al., 2009; Sanchez et al., 2006), suggesting that ApoD provides a protection against certain stressors.
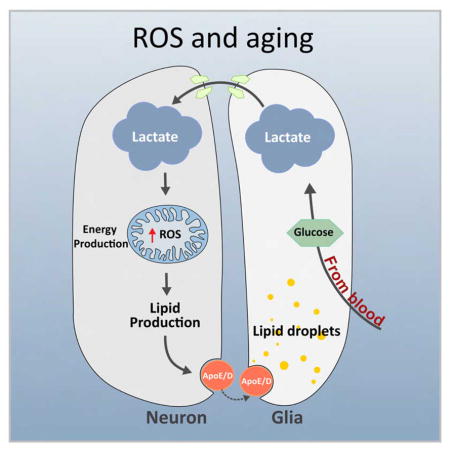
In this study, we explore the sources of energy and lipids that lead to LD accumulation in glia by selectively manipulating the expression and function of genes and proteins separately in Drosophila photoreceptor neurons and glia. We first provide evidence supporting the ANLS and demonstrate that lactate transport from glia to neuron through monocarboxylate transporters affects neuronal lipogenesis (Figure 1A). In response to elevated levels of ROS, neuronal lipids are transferred to glia where they are stored as LD. We demonstrate that lactate-derived neuronal lipid transport to glia depends on fatty acid transport proteins and apolipoproteins in flies and mice. We also document that the human APOE2 and APOE3 alleles can functionally substitute for the loss of Glaz in flies to permit lipid transport from neurons to glia, whereas APOE4 cannot. The inability of APOE4 flies to accumulate LD in response to ROS presages neuronal degeneration and loss. Likewise, when subjected to high ROS, neuron-glia co-cultured mouse primary cells that lack APOE are unable to form LD. We propose that LD formation in glia requires ApoE and that LD accumulation protects against neurodegeneration by scavenging peroxidated lipids.
Figure 1. Cell specific screen to uncover proteins involved in lipid transfer.

A) Model of the mechanism of lactate and lipid transfer and accumulation. B) ND42 RNAi under the control of the Rhodopsin promoter (Rh-ND42 IR) results in glial LD accumulation at day 1. Introducing neuronal (Elav) or glial (54C) GAL4s allows for knockdown of genes of interest (GOI) in a cell specific manner. Knockdown of proteins involved in lipid transfer should lead to a decrease in glial LD accumulation. C) A subset of proteins that may be involved in lipid transfer, their human orthologs and conservation as measured by the DIOPT score (Max score: 11). Effect of neuronal or glial protein knockdown on glial LD accumulation is depicted in a relative manner; three downward arrows (↓↓↓) indicate a dramatic decrease in LD accumulation and bar (−) indicates no significant change.
Results
A cell-specific candidate gene screen to uncover proteins required for neuron-glia lipid transfer
To assess the roles of various players in lipid production and transport in the nervous system, we selected proteins/genes implicated in lactate transport or usage as well as candidates that may affect lipid transport in neurons or glia. We induced glial LD formation by RNAi knockdown of mitochondrial complex I protein subunit ND42 (Liu et al., 2015; Zhang et al., 2013) in Drosophila photoreceptors (PR) under the control of the 5′ regulatory element of the Rhodopsin gene (Wang et al., 2008), here referred to as Rh-ND42 IR. We then examined whether decreased expression of candidate genes through neuronal or glial specific knockdowns can lead to a decrease in glial LD accumulation (Figure 1B–C). We selected candidates that are conserved and expressed in the central nervous system of flies, mice and humans. The level of homology and conservation is based on the DIOPT score (maximum score: 11, Figure 1C) (Hu et al., 2011). To ensure the robustness of our observed phenotypes, we tested two or three independent RNAis for each gene and these results were confirmed using mutant alleles for nearly all genes. The rationale for each candidate selection is outlined below.
The first candidate genes were monocarboxylate transporters (MCTs). The ANLS hypothesis posits that lactate, which is elevated in glia, is shuttled by glial MCTs from astrocytes to the extracellular space and thence to neurons that via other MCTs (Belanger et al., 2011; Machler et al., 2016; Pellerin et al., 2007). We selected fly homologs of MCTs that are expressed in the nervous system of mice and human (Uhlen et al., 2015; Zhang et al., 2014) including Silnoon (Sln) (Jang et al., 2008) and outsiders (out) (Coffman et al., 2002). We also included Basigin (Bsg), a MCT accessory protein required for the transport of many MCTs from the endoplasmic reticulum to the plasma membrane (Besse et al., 2007; Halestrap and Wilson, 2012). Expression of murine Bsg rescues the phenotypes associated with the loss bsg in the fly visual system, suggesting that they play similar functions in vivo (Curtin et al., 2005). An alternative model of lactate transport suggests that gap junctions may allow the direct cell-to-cell passage of lactate or glucose (Bosone et al., 2016; Hertz et al., 2007; Limmer et al., 2014; Tabernero et al., 2006). As Drosophila innexins are the equivalent of mammalian connexin proteins and form heteromeric and heterotypic gap junctions (Stebbings et al., 2002), we also tested shaking B (shakB), a member of the innexin family (Phelan et al., 1998; Shimohigashi and Meinertzhagen, 1998).
Subsequently, we turned to enzymes that participate in the conversion of lactate into pyruvate and the production of acetyl-CoA (AcCoA) including lactate dehydrogenase (Ldh), a pyruvate dehydrogenase subunit (Pdha) and citrate synthase (CS, called knockdown). Blocking these enzymes may decrease the levels of AcCoA and hence alter lipid production (Figure 1C).
Finally, we tested the role of SLC27A family members known to be involved in lipid activation and/or transport in a variety of cells (Kazantzis and Stahl, 2012; Mashek et al., 2007; Stahl, 2004), but whose lipid related functions have not been assessed in the nervous system. We therefore tested Fatp, the fly homolog of FATP1 (SLC27A1) and FATP4 (SLC27A4) (Dourlen et al., 2012; Dourlen et al., 2015). Finally, we assessed the function of various apolipoproteins that belong to the lipocalin family (Hauser et al., 2011; Kalaany and Mangelsdorf, 2006), such as glaz and nlaz. Although these genes are known to be expressed in the fly nervous system and important for stress tolerance, they have not been implicated in lipid transport or LD formation.
Lactate transporters are necessary for ROS-induced glial LD accumulation in Drosophila
To examine the role of MCTs in glial LD accumulation, we first induced glial LD accumulation in flies by expressing the photoreceptor specific Rh-ND42 IR in the presence of two GAL4 drivers: the 54C-GAL4 glial driver or the neuronal Elav-GAL4. These flies serve as positive controls as they do not carry a UAS-driven RNAis and should exhibit robust LD accumulation in glia (Figure 2A, a, f and quantified in Figure 2B). Our negative controls consist of a neuronal or glial knockdown of MCTs or basigin, in the absence Rh-ND42 IR. As anticipated, the photoreceptors of these flies do not exhibit morphological defects and hence the loss of these proteins does not affect PR development or induce accumulation of LDs (Figure S1A).
Figure 2. Reducing MCTs inhibit glial LD accumulation and delays neurodegeneration.

A) a, j. Nile Red stain of whole-mount retina. a and f reveal a baseline of approximately 10 LD per ommatidium in 54C-GAL4; Rh-ND42 IR and Elav-GAL4; Rh-ND42 IR retinas. b–d. Glial knockdown of Sln (MCT), out (MCT) does not reduce glial LD accumulation in the Rh-ND42 IR background and knockdown of Bsg (MCT accessory protein) leads to a decrease of glial LD accumulation. g–i Neuronal knockdown of Sln, out and Bsg in the Rh-ND42 IR background lead to a decrease of glial LD accumulation. e,j knockdown of shakB does not alter LD accumulation. B) Quantification of A. C) Quantification of glial LD accumulation in the sicilyE and MarfB mutant clones. D) Quantification of the number of remaining rhabdomeres after aging for 5 days in sicilyE and MarfB mutant clones. All data are represented as mean ± SEM. Student’s t-tests were used to calculate all significance, n > 10 animals each (*P<0.05, **P<0.005, ***P<0.0005).
In flies expressing Rh-ND42 IR, glial knockdown of Sln or out did not affect LD accumulation (Figure 2Ab–c), suggesting they are not required in glia. However, knockdown of Bsg in glia decreases LD accumulation (Figure 2A, d, Figure 2B), therefore that an unknown MCT is likely required in glia. In contrast, neuronal knockdown of Sln, out or Bsg led to a decrease in glial LDs (Figures 2Af–j). We infer that blocking lactate transport in either cell type through MCT inhibition decreases LD accumulation. However, neither neuronal nor glial knockdown of shakB alters glial LD accumulation, suggesting that gap junctions may not be important for lactate transport in this context (Figure 2Ae,j).
To determine whether MCTs are critical for glial LD accumulation in other neurodegenerative models, we examined whether loss of MCTs would also decrease glial LD accumulation in sicily (Zhang et al., 2013) and Marf (Sandoval et al., 2014) mutant fly eyes. We created whole-eye mutant clones for sicilyE and MarfB, which exhibit severe glial LD accumulations and neurodegeneration (Liu et al., 2015). Knockdown of Sln or out, or simply introducing a single mutant allele of these genes (SlnD1/+ or Bsg1217/+) in sicilyE or MarfB eye-clones decreased glial LD accumulation (Figure 2C, S1B–D). These data show that lactate transporters are required for the accumulation of LD in glia induced by very different mitochondrial defects.
As we have shown that decreasing glial LD in flies with high levels of ROS delays neurodegeneration (Liu et al. 2015), we tested whether the above genetic manipulations can delay neurodegeneration. We examined the extent of photoreceptor loss by counting the remaining number of rhabdomeres per ommatidium upon aging for five days; the number of remaining rhabdomeres inversely correlates with the extent of neurodegeneration (Knust, 2007; Liu et al., 2015). As shown in Figure 2D and S1E, decreased MCT levels delay neurodegeneration in both sicilyE and MarfB mutant clones, indicating that lactate transport promotes neurodegeneration in the presence of high ROS.
Lactate transport is a critical component for ROS-induced glial LD accumulation in mammalian cells
Since inhibition of lactate transport by decreasing MCTs decreases glial LD accumulation in flies, we developed a primary cell culture assay using a murine co-culture system of neurons and glia to determine whether this mechanism is evolutionarily conserved. Mice that lack Ndufs4 (Kruse et al., 2008), which encodes a mitochondrial complex I subunit, exhibit glial LD accumulation in the vestibular nucleus and olfactory bulb prior to the onset of neurodegeneration (Liu et al., 2015). To model this LD accumulation in a dish, olfactory bulbs were isolated from C57BL/6J newborn pups (postnatal day 2 to 3) and plated as a monolayer co-culture for 9–11 days in vitro (DIV). This primary culture consists mostly of GFAP positive astrocytes and TUJ1 positive neurons. In addition, olfactory bulb ensheathing glia are present as well (Figure S2A) whereas IBA1 positive microglia and Olig2 positive oligodendrocytes are nearly absent (data not shown).
To determine whether a brief elevation of ROS induces glial LD accumulation in our co-culture system, we applied 2 μM of the mitochondrial complex I inhibitor, rotenone, for 24 hours. Vehicle-treated cells do not exhibit LD accumulation when stained by the neutral lipid stain, BODIPY495/505 (Figure 3Aa–b), but approximately 65% of astrocytes accumulate LDs when treated with rotenone (Figure 3Ac–d and S2D). Importantly, glial LD accumulation due to elevated ROS requires both cell types to be present. Indeed, purified astrocytes or neuronal cells did not accumulate LDs when treated with 2 μM rotenone (Figure S2C).
Figure 3. Primary neuronal-glial co-culture requires lactate transport to accumulate glial LD in response to ROS.

A) a–b. Vehicle treated cells (24 hrs) do not exhibit LD accumulation. c–d. 2 μM rotenone treatment for 24 hrs leads to glial LD accumulation (arrowhead). e–f. 24 hr treatment with 2 μM rotenone and 200 nM MCT inhibitor (MCTi, AR-C155858) leads LD to accumulate in a subset of neurons. g–h. Cells treated with 2 μM rotenone for 24 hrs and 200 nM MCTi after 12 hrs exhibit LD accumulation in neurons and glia. B) Quantification of LD accumulation. (Kruskal-Wallis, followed by Dunn’s test for post-hoc analysis. C) Quantification of TUNEL staining post treatment. (Kruskal-Wallis, followed by Dunn’s test for post-hoc analysis. n = 200 cells counted per treatment, 3 replicates) D) Percentage incorporation of 13C into palmitoleic acid in astrocytes is decreased when extracellular lactate is added (Student’s t-test. Data point represent mean +/− standard deviation. n =3 biological and 3 technical replicates). E) Rotenone treated mice (3mg/kg/day, 8 days) exhibit significant (p = 0.001) LD accumulation colocalizing with astrocyte (GFAP) and microglia (IBA1) markers compared to vehicle treated mice and accumulate LD similar to p35 Ndufs4−/− mutant mice (Student’s t-test. n = 5 per treatment. 3 sections per slice, 20 slices per animal.) All data points represent mean +/− SEM. *P<0.05, **P<0.005, ***P<0.0005. Scale bar: 50 μm.
This primary co-culture assay allows us to examine the effects of various compounds on glial LD accumulation and document mechanistic conservation. We first examined the effect of N-acetylcysteine amide (AD4)(Amer et al., 2008), a potent antioxidant that reduces LD accumulation in flies and delays neurodegeneration in Ndufs4−/− mice (Liu et al., 2015). To assess the efficacy of AD4 in our primary co-culture system, we added 1.5 mM AD4 along with 2 μM rotenone for 24 hours. AD4 treatment paired with elevated ROS leads to a significant decrease in LD accumulation (Figure S2B a–b and 3B), indicating that quenching elevated ROS efficiently suppresses LD in glia, similar to our in vivo observations for flies and mice.
To examine the role of lactate transport in glial LD, we blocked lactate transport using two MCT inhibitors, AR-C155858 (referred to here as MCTi) and SP13900 (also named AR-C122982 and now referred to as MCTi2), both developed to inhibit T cell proliferation (Murray et al., 2005). MCTi blocks the lactate binding pocket of MCT1, which is expressed in glia (Lee et al., 2012) as well as MCT2, which is expressed in neurons (Debernardi et al., 2003; Ovens et al., 2010). MCTi2 inhibits MCT1 (Doherty et al., 2014). Exposing 9DIV cells to 200 nM MCTi or 50 nM MCTi2 with 2 μM rotenone for 24 hours causes a decrease in glial LD accumulation for each inhibitor (Figure 3A e–f, 3B and S2B c–d). To determine the time course of glial LD accumulation, we separated the induction of ROS and blockage of lactate transport by first treating the culture with 2 μM rotenone for 12 hours and then introducing 200 nM MCTi. Blocking lactate transport after ROS induction, results in some LD accumulation in both neurons and astrocytes, a highly unusual feature that we did not previously observe in vivo (Figure 3Ag–h and S2D). Finally, combining AD4 and MCTis decreases glial LD accumulation in response to elevated ROS by more than either treatment alone (Figure 3B and S2B e–f). Because the synergistic effects of high ROS and glial LD accumulation lead to neurodegeneration in flies, we assayed cell death in our co-culture system using TUNEL. Vehicle-treated cells show a low number of TUNEL positive cells (~2.5%) after 24 hours whereas 20% of the rotenone-treated cells are TUNEL positive (Figure S2E and 3C). Importantly, in rotenone-treated cells, blocking lactate transport with MCTi at 0 or 12 hours after adding rotenone strongly decreases the number of apoptotic cells, suggesting that the decrease in LD accumulation that results from blocking lactate transport may ameliorate the cellular damages that lead to cell death. These results show that lactate transport is necessary for glial LD accumulation and the prolonged accumulation of LD is are the primary contributors to cell death.
To parse the effects of lactate itself compared to elevated ROS and glial LD accumulation, we assayed whether addition of excess lactate leads to cell death. Co-cultured cells treated with 11 mM exogenous L-lactate for 24 hours show a similar number of cleaved caspase 3 positive cells compared to controls (Figure S2F), suggesting that ROS and LD accumulation are the primary contributors to cell death.
Extracellular lactate incorporates into astrocyte fatty acids
The existence of an astrocyte-neuron lactate shuttle would imply that extracellular lactate could be incorporated into neuronally-synthesized fatty acids. We sought to determine whether the carbons of extracellular lactate are incorporated at significant levels into neutral lipids that accumulate in astrocyte LD in under high levels of ROS. To study the carbon source of astrocytic lipids, we harvested olfactory bulb primary neuronal/glial co-cultures and raised the cells in the presence of 28 mM uniformly 13C-labeled glucose or of 13C-glucose plus 11 mM L-lactate. Subsequently, we added 2 μM rotenone for 24 hours to induce astrocytic LD formation. After these co-cultures are sorted by fluorescent activated cell sorting (FACS) using a marker for astrocytes, neutral lipids (including triglycerides, diacylglycerol, cholesterol esters, plus a small fraction of phosphatidylcholine) were extracted, saponified and neutralized (see Methods). The fatty acids were then resolved by liquid chromatography and mass spectrometry. Fractional 13C was calculated using ion counts for the well-resolved and abundant palmitoleic acid species (see Methods), which can be synthesized from AcCoA de novo in mammalian cells (Mozaffarian et al., 2010). Labeled carbons from glucose are readily incorporated into astrocyte neutral lipid fatty acids, but addition of unlabeled lactate decreases the extent to which the glucose isotopic label was incorporated into astrocyte palmitoleic acid by 40% (from 34% to 20.5% 13C, Figure 3D and S2H). Because extracellular lactate can significantly dilute the flow of glucose carbons into fatty acids, we infer that lactate is a major source of the carbons in astrocytic fatty acids.
Inducing ROS in healthy animals leads to glial LD accumulation and neurodegeneration
ROS-induced glial LD accumulation in culture occurs within a 24-hour window. We therefore tested whether a chemically induced increase in ROS can lead to glial LD accumulation in mice. Daily intraperitoneal injection of rotenone at 3mg/kg in wildtype/C57Bl6j mice for 8 days produces obvious motor deficits as assessed by the ability to hold on to a wire (Figure S2G). Immunohistochemical analyses of the olfactory bulb glomerular layer in rotenone-treated animals reveal significant LD accumulation that co-localizes with astrocyte and microglia markers, visually consistent with P35 Ndufs4−/− mice at the onset of ataxia (Figure 3E).
In sum, a short exposure to high ROS in co-cultured neurons and glia leads to glial LD accumulation and lactate is a key substrate for lipid production in vertebrate cells as well as in Drosophila. Moreover, we provide evidence that otherwise healthy wildtype animals also accumulate glial LD when exposed to elevated levels of ROS for approximately one week. Together, our data provide evidence that glial LDs accumulate in response to elevated ROS in wild-type mice and cells.
Mitochondrial dysfunction leads to the preferential shuttling of acetyl-CoA to the lipid synthesis pathway
We sought to explore the importance of lactate/pyruvate interconversion and AcCoA production on glial LD accumulation. Ldh reversibly interconverts lactate and pyruvate, and the pyruvate dehydrogenase complex is the major source of cellular AcCoA. Decreasing Ldh will slow lactate/pyruvate interconversion (Schurr and Payne, 2007), and the loss of any component of the pyruvate dehydrogenase complex will decrease AcCoA production and lead to lactate buildup (Barnerias et al., 2010; Martin et al., 2005). By decreasing the availability of AcCoA, the first metabolite in fatty acid synthesis, we expect that such perturbations would likely diminish glial LD accumulation. However, as these enzymes are critical for life, we first verified that knockdown of Ldh or Pdha (the E1 subunit of pyruvate dehydrogenase complex) did not affect PR morphology or LD accumulation at day 1 (Figure S3A). Subsequently, we tested cell specific knockdown of Pdha (Figure 4A and S3B, b, e) or Ldh (Figure 4A and S3B c, f) in the high ROS (Rh-ND42 IR) background. Interestingly, glial knockdown of Pdha in the high ROS background does not alter LD accumulation, suggesting that glial AcCoA (and glial fatty acid synthesis) is not critical to glial LD formation. In contrast, neuronal knockdown of Pdh leads to decreased glial LD accumulation (Figure 4A), suggesting that AcCoA production in neurons is critical to glial LD formation. Either neuronal or glial knockdown of Ldh decreases glial LD formation, consistent with roles for this enzyme in both cell types: reduction of pyruvate to lactate in glia, and re-oxidation of the transported lactate to pyruvate in neurons.
Figure 4. Neuronal lactate is critical for glial LD accumulation.

A) Quantification of LD. Glial (54C-GAL4) or neuronal (Elav-GAL4) specific knockdown of Ldh and Pdha results in a decrease of LD accumulation in the Rh-ND42 IR background. B) Removal of a copy of PdhaA and KdnA reduces glial LD accumulation in the Rh-ND42 IR, Rh-Marf IR and Rh-Aats-met IR flies. C) Flies fed with dichloroacetate (DCA) exhibit a dose dependent increase in glial LD. D) Neuronal overexpression (N-Syb-GAL4) of SREBP or JNK leads to glial LD accumulation. Knockdown of MCTs (Sln, out) and metabolic enzymes (Ldh and Pdha) in the N-Syb-GAL4 overexpression background ameliorates glial LD accumulation. All data points represent mean +/− SEM (Student’s t-test. n > 10 animals each *P<0.05, **P<0.005, ***P<0.0005).
To further dissect the metabolites involved in lipid synthesis, we used heterozygous mutants of PdhaA (Jaiswal et al., 2015) or CS (KdnA) (Yamamoto et al., 2014) in flies that express Rh-ND42 IR, Rh-Marf-IR or Rh-Aats-met IR and accumulate numerous LD in glia. We observe a systematic decrease in glial LD accumulation when the metabolic enzymes are decreased via RNAi or incorporation of a mutant allele (Figure 4B and S3C).
Since the decrease of enzymes required for the lactate shuttle to generate neuronal AcCoA decreases glial LD accumulation, we examined whether pharmacological means of increasing AcCoA production would lead to glial LD accumulation. We raised flies on food that contained dichloroacetate (DCA) (20, 40, and 80 μg/ml), an activator of Pdh that inhibits Pdh Kinase (Michelakis et al., 2008). Flies raised on this food exhibit a dose-dependent glia LD accumulation (Figure 4C), further suggesting that lactate and pyruvate are critical players for glial LD accumulation.
Lactate transport is critical for lipid production in the absence of mitochondrial dysfunction
Our data suggest that under conditions of elevated ROS, lactate is converted into pyruvate and AcCoA to serve as building blocks for lipid production in neurons and that the lipids are shuttled to glia, where they are accumulated in LD. To determine whether lactate contributes to lipid synthesis in neurons in flies with functional mitochondria, we used the neuronal driver N-Syb-GAL4 to overexpress SREBP or JNK. This leads to glial LD accumulation without disrupting mitochondrial function (Liu et al., 2015). Interestingly, knockdown of Sln, out, Ldh or Pdha using the same GAL4 driver in the presence of elevated levels of SREBP or JNK leads to a systematic decrease in LD accumulation (Figure 4C and S3D). These data show that neuronal lactate is necessary for lipid production in the absence of mitochondrial dysfunction, demonstrating that this pathway is not solely dependent on elevated ROS, consistent with observations made with dichloroacetate supplementation.
Loss of fatty acid transport protein (FATP) inhibits glial LD accumulation
FATPs play a role in lipid metabolism, lipid transport, and lipid activation as an acyl-CoA synthetase. These proteins are localized to the ER and plasma membrane in vertebrate cells (Stahl, 2004). Drosophila Fatp is expressed in both neurons and glia (Dourlen et al., 2012) and is most similar to mammalian FATP1 and FATP4 (Dourlen et al., 2015). We reduced expression of Fatp in neurons and glia and ascertained that these manipulations do not affect PR morphology immediately after eclosion (Figure S4A). To determine whether Fatp is involved in lipid transfer and glial LD accumulation, we knocked down Fatp in a cell specific manner in the Rh-ND42 IR background, which causes a significant decrease in LD accumulation (Figure 5A and S4B). Similarly, reducing the levels of Fatp in sicilyE or MarfB (Figure S4C and S4D) reduces LD accumulation. Finally, reducing the levels of Fatp or introducing one copy of the FatpK10307 (Dourlen et al., 2012) mutant allele delays neurodegeneration in sicilyE or MarfB mutant clones (Figure 5B). Together, these findings demonstrate that Fatp is critical for both neuron and glial lipid processing and/or transport as well as glial LD accumulation.
Figure 5. Lipids are transported via Fatty acid transport proteins.

A) Decreasing Fatp levels in neuron or glia in the high ROS background (Rh-ND42 IR) led to less glial LD. B) Photoreceptor degeneration in sicilyE and MarfB mutant clones is ameliorated with whole eye (Eyeless-GAL4) knockdown of Fatp. C) Primary co-culture derived from Rosa-Cas9-eGFP were transfected at 3DIV with lentiviral packaged FATP1 and FATP4 sgRNAs and subjected to rotenone treatment at 11DIV. a–d. Vehicle treated cells do not exhibit FATP1 knockdown or LD accumulation while (e–h) 2 μM rotenone treated cells exhibit glial LD accumulation. i–l. Cells transduced with FATP1 sgRNA lead to loss of FATP1. m–p. Loss of FATP1 leads to an inability to accumulate LD when subjected to 2 μM rotenone treatment. D) Quantification of C and Figure S4F. Data are represented as mean ± SEM. (Student’s t-test. n > 10 animals each or n = 200 cells counted per treatment, 3 replicates. *P<0.05, **P<0.005, ***P<0.0005). Scale bar: 50 μm.
As FATP1 and FATP4 are both expressed in the mammalian nervous system, we first examined the expression of FATP1 and FATP4 under basal and high ROS conditions (Figure S4E). Consistent with the known expression patterns (Zhang et al., 2014), both proteins are expressed in neuron and glia. To examine the consequence of the loss of FATPs under conditions of elevated ROS in our co-culture model, we knocked out FATP1 or FATP4. We inserted sgRNAs against FATP1, FATP4 or a non-target control in a lentiviral vector and tracked the transduction efficiency with a 6× histidine sequence (histologically) and mTagBFP2 (live) (Subach et al., 2011). We transduced primary co-cultured cells derived from the olfactory bulb of constitutively expressing Cas9-eGFP (Rosa-Cas9) (Sanjana et al., 2014) mice at 3DIV and all experiments were performed at 11DIV. In cells transduced with non-target sgRNA FATP1 (Figure 5Cc, g) or FATP4 (Figure S4F c), expression is not affected and LDs accumulate in response to rotenone-induced ROS (Figure 5Cf). Cells transduced with sgRNA to knock out FATP1 (Figure 5Ci–p) or FATP4 (Figure S4C e–h) display a clear decrease of FATP1 or 4 protein levels by staining and exhibit less LD accumulation (Figure 5C–D, S4F). These in vivo and in vitro data demonstrate that Fatp and FATP1/FATP4 are involved in ROS-induced glial LD accumulation.
Drosophila ApoDs (Glaz and Nlaz) are required for lipid transfer
Dietary lipid transport in the body occurs through apolipoprotein-mediated movement of chylomicrons and high and low-density lipoproteins (Eichner et al., 2002). The mechanism of lipoprotein formation and lipid processing between cells and organs is well studied in the gut and cardiovascular system but underexplored in the brain (Ikonen, 2008; Mitchell and Hatch, 2011). In flies, two apolipoproteins are secreted proteins (Ruiz et al., 2013): Glial lazarillo (Glaz), which is expressed in glia (Sanchez et al., 2006), and Neural lazarillo (Nlaz), which expressed in neurons (Hull-Thompson et al., 2009; Sanchez et al., 2000).
To examine whether these proteins play a role in glial LD accumulation, we first verified that their knockdown does not alter photoreceptor morphology or cause LD accumulation at day 1 (Figure S5A). Subsequently, we knocked down Glaz or Nlaz with an RNAi in a cell-specific manner in Rh-ND42 IR flies. Glial knockdown of Glaz decreases glial LD accumulation whereas the neuronal knockdown of Glaz does not affect LD (Figure 6Ac–d). Similarly, neuronal knockdown of Nlaz decreases glial LD, whereas glial knockdown does not (Figure 6Ae–f). RNAi knockdown of Glaz or loss of one copy of Nlaz in the sicilyE mutant background delays neurodegeneration (Figure S5B). The cell-specific knockdown of these two proteins and the decrease in LD accumulation indicate that they play a role in lipid transport between neurons and glia in flies (Figure 6B).
Figure 6. Human APOE4 cannot functionally replace Glaz in lipid transport.

A) a–b. Nile Red stain reveals a baseline of elevated LD accumulation. c–d. Glial knockdown of Glaz in the Rh-ND42 IR background leads to decreased in glial LD accumulation but neuronal knockdown does not alter LD accumulation. e–f. Glial knockdown of Nlaz in the Rh-ND42 IR background does not alter LD accumulation but neuronal knockdown leads to a decrease in glial LD accumulation. B) Quantification of A. (Student’s t-tests, n > 10 animals each). C) MiMIC insertion in the first coding intron of Glaz allows for recombination mediated cassette exchange to insert a Trojan exon containing a splice acceptor (SA), linker, and T2A sequence and a GAL4 sequence followed by a poly A tail. The insertion of the Trojan exon produces flies with a truncated Glaz protein (mutant) and GAL4 which is used to express any UAS-gene. D) a–b. Rh-ND42 IR and Rh-Marf IR exhibit glial LD accumulation. c–d. Removing one copy of Glaz by introducing GlazT2A-GAL4 decreases glial LD accumulation in the Rh-ND42 IR and Rh-Marf IR background. e–f. Replacing the one-copy-loss of Glaz with expression of APOE2 variant restores glial LD accumulation. g–h. Substituting one-copy-loss of Glaz with expression of APOE3 variant restores glial LD accumulation. i–j. Substituting one-copy-loss of Glaz with APOE4 variant does not restore LD accumulation. E) Quantification of D. F) Flies with APOE4 expression in place of Glaz accumulate less LD compared to APOE3 expressing flies when fed rotenone (Kruskal-Wallis test followed by Dunn’s test for significance. n > 10 animals each). Data are represented as mean ± SEM. (*P<0.05, **P<0.005, ***P<0.0005.).
Glaz and APOE are functional homologs
Although APOE is not conserved in flies, lipid transport machineries may perform analogous roles. Given that Glaz is expressed in Drosophila glia and APOE is abundantly expressed in human astrocytes, we tested whether human APOE can perform the same function as Glaz in lipid transport in Drosophila. First, we created UAS transgenes expressing APOE2 (Cys112, Cys158), APOE3 (Cys112, Arg158) or APOE4 (Arg112, Cys158) to assess the role of APOE in lipid transport in flies. The APOE4 is a major risk factor for Alzheimer disease as 40–80% of patients with AD (Farrer et al., 1997) carry an APOE4 allele.
To determine whether the APOE alleles function similarly to Glaz for LD production or lipid transfer, we employed the “plug and play” method that allows for the expression of our transgene of interest in the Glaz mutant background (Diao et al., 2015). We obtained an allele of Glaz in which a versatile transposon, Minos-mediated Integration Cassette (MiMIC), is present in the first coding intron (Nagarkar-Jaiswal et al., 2015; Venken et al., 2011). We performed recombination mediated cassette exchange to introduce an artificial exon into the coding intron and create the GlazT2A-GAL4 gene trap line, in which endogenous Glaz is truncated due to the presence of the ribosomal skipping T2A sequence (Figure 6C). The GAL4 allows for expression of UAS transgenes under the control of the endogenous Glaz regulatory elements providing the proper temporal and spatial control. To confirm that GlazT2A-GAL4 is expressed in the pigment cells (glia) in the eye, we used this line to knockdown the white gene using UAS-white RNAi (Figure S5C). white is required for red pigment accumulation in pigment/glial cells. Knockdown of white leads to an obvious loss of eye pigment but this loss is not as severe as the one observed with the pigment glia driver (54C-Gal4).
To establish that GlazT2A-GAL4 expression of UAS transgenes does not lead to an overexpression phenotype, we expressed Glaz and APOE2, APOE3 or APOE4 variants without elevating ROS and find no glial LD accumulation (Figure S5D) or differences in protein levels (Figure S5E). To determine the role of APOE in lipid transport, we removed one copy of Glaz by introducing the GlazT2A-GAL4 into Rh ND42-IR flies (Figure 6Da–d); the result is a striking decrease in glial LD accumulation, providing further evidence that Glaz is required for LD accumulation. We then expressed APOE2, APOE3 or APOE4 variants using GlazT2A-GAL4 to examine whether these human proteins can functionally replace the one-copy-loss of Glaz in a high ROS background. Replacing Glaz with the APOE2 variant, which protects against AD (Yu et al., 2014), displays the highest level of LD accumulation. Expression of the APOE3 variant restores glial LD accumulation and is nearly as efficient as Glaz. However, expression of the APOE4 variant, associated with early onset of AD, poorly rescues the loss of Glaz (Figure 6De–j and 6E).
The inability of APOE4 to facilitate lipid transport leads to accelerated neurodegeneration
To further parse the role of APOE4 in lipid transfer, we pharmacologically increased levels of ROS by feeding flies with rotenone. We raised homozygous flies without a GAL4 driver (control flies = w; UAS-APOE3), Glaz null animals (GlazT2A-GAL4, UAS-eYFP) and APOE3 (GlazT2A-GAL4, UAS-APOE3) or APOE4 (GlazT2A-GAL4, UAS-APOE4) expressing flies in food supplemented with 25 μM rotenone. At one day post eclosion, control flies and flies expressing APOE3 accumulate similar numbers of glial LD. However, flies expressing APOE4 are unable to accumulate LD, comparable to Glaz null flies (Figure 6F). These data further show that APOE4 cannot promote lipid transport between neuron and glia in response to ROS.
Since APOE2 and APOE3 alleles can functionally substitute for the loss of Glaz in the process of LD accumulation, we sought to determine whether overexpression of these proteins alone was sufficient to cause a LD accumulation phenotype. Overexpression of Glaz, or APOE2 or APOE3 variants, with 54C-GAL4 leads to substantial glial LD accumulation. However, glial overexpression of the APOE4 allele induces substantially less glial LD accumulation (Figure 7A and S6A). Hence, APOE4 was less efficient at inducing LD accumulation when overexpressed, again suggesting that APOE4 is a partial loss-of-function allele for glial LD accumulation.
Figure 7. Apoe null cells and APOE4 animals cannot accumulate glial LD in response to stress.

A) Glial (54C-GAL4) overexpression of UAS-mCD8:GFP (control) does not lead to LD accumulation. Glial overexpression of Glaz, APOE2 and APOE3 variants lead to more than 10 LDs per ommatidium whereas APOE4 variant overexpression leads to significantly less glial LD accumulation. (Student’s t-tests were used to calculate significance. n > 10 animals each). B) a–b, Vehicle treated control cells did not accumulate glial LD after 12 hrs treatment. c–d, cells treated for 12 hrs with 1.5 μM rotenone accumulate glial LDs. e–h. Apoe−/− treated for 12 hours with 1.5 μM rotenone accumulate minimal glial LDs. (Kruskal-Wallis test followed by Dunn’s test for significance. n < 200 cells per treatment, 3 replicates) C) Homozygous APOE4 expressing flies have more disrupted photoreceptors compared to APOE3 expressing flies immediately post ROS exposure. D). When aged for 10 days, Flies with APOE4 expression in place of Glaz lose a comparable number of neurons as Glaz null flies and more than APOE3 flies. (Kruskal-Wallis test followed by Dunn’s test for significance. n > 10 animals each) Data are represented as mean ± SEM. *P<0.05, **P<0.005, ***P<0.0005. Scale bar: 50 μm.
To assess the loss-of-function phenotype of ApoE in glial LD accumulation, we derived co-cultured primary cells from the olfactory bulb of Apoe−/− mice (B6.129P2-Apoetm1Unc/J), which have no detectable Apoe (Piedrahita et al., 1992). To assess the presence of glial LD accumulation using these cells, we added 1.5 μM rotenone to induce ROS. Apoe−/− cells did not display any obvious phenotypes at 10DIV, however they are very sensitive to treatment with ROS inducing reagents, similar to flies and mice that lack ApoD (Ganfornina et al., 2008). Indeed, rotenone (2 μM) treatment for 24 hours at 10DIV leads to extensive cell detachment and we were therefore unable to use our standard assay conditions. We then decreased the rotenone concentration to 1.5 μM and the exposure time to 12 hours. Under these conditions, control cells accumulate LD, although less than our previous standard conditions (Figure 7Ba–d). In contrast, Apoe−/− cells have very few LD (Figure 7B). The diminished ability for Apoe−/− cells to accumulate LD and their sensitivity to ROS suggest that Apoe is critical in facilitating lipid transport between neurons and glia and that this lipid transport affects cellular health and their ability to respond to stress.
As APOE4 is the most prominent risk factor for developing AD, we sought to determine the role of the APOE4 allele in neurodegeneration. As shown in Figure 6E, APOE4 flies exposed to rotenone accumulate few glial LD at day 1 and exhibit more disrupted photoreceptors compared to APOE3 expressing flies (Figure 7D). Furthermore, when these animals are aged for 10 days, the APOE4 expressing flies lose significantly more photoreceptors than APOE3 expressing flies, comparable to flies that lack Glaz. These data indicate that glial LD formation and accumulation triggered by elevated levels of ROS provide a protective mechanism against neurodegeneration.
DISCUSSION
Through genetic and pharmacologic studies, we have identified MCTs and lactate as critical components for LD accumulation in flies and mammalian cells. LD accumulation in glia depends on the transfer of lactate from glia to neurons. Lactate is metabolized in neurons to produce AcCoA, a key input for energy production in the TCA cycle. A surplus of AcCoA, which may be caused by a defective TCA cycle or mitochondrial dysfunction, provides the impetus to synthesize lipids, whose transport to glia depends on FATP and apolipoproteins. This leads to the accumulation of LD in glia (pigment glia in flies, astrocytes in mammals). Interestingly, loss of ApoD in fly glia can be compensated for by human APOE, which is expressed at high levels in human astrocytes. This suggests that a major role of APOE is to promote the transfer of lipids between neuron and glia for lipid storage in LD. Hence, we provide evidence that weaves together mechanisms of cell-cell communication, metabolic coupling, and neuron-glia feedback in cell death and neurodegeneration. Altogether, these observations may have important implications regarding our understanding of pathogenic mechanisms in AD.
LD accumulation in pathogenic and nonpathogenic conditions depends on processing glial lactate
Since the ANLS hypothesis was proposed, compelling evidence indicates that glia play a critical role in lactate production and release, including Drosophila perineural glia (Volkenhoff et al., 2015), mammalian astrocytes (Pellerin and Magistretti, 1994) and oligodendrocytes (Funfschilling et al., 2012). Our studies show that lactate transport from glia to neurons is critical for glial LD accumulation under normal and stress conditions. Glial lactate is transported and taken up into neurons through MCTs, providing further support for the evolutionary conservation of ANLS. Furthermore, genetic or pharmacologic inhibition of various enzymes in the metabolic pathways reduce glial LD accumulation, likely by limiting substrate available for lipid synthesis. These findings demonstrate that neuronal lipid synthesis requires lactate as a building block and that the lipids are synthesized in neurons. We provide biochemical evidence that extracellular lactate can be incorporated into glial lipids at high levels, demonstrating that lactate is an important source for lipogenesis.
The process of glial lactate transport to neurons is not solely due to elevation of ROS or mitochondrial dysfunction as restricting lactate in neurons that overexpress JNK or SREBP also reduces glial LD accumulation. This indicates that the pathway of lactate and lipid transport operates under non-pathological conditions as well. Moreover, we show that FATPs are necessary for lipid transport in neurons and LD accumulation in glial cells and that this mechanism is conserved in vertebrates. FATPs have been extensively characterized for their biochemical properties in lipid processing and uptake in vitro (Coe et al., 1999; Schaffer and Lodish, 1994; Xu et al., 2012) but until now, their role in the mammalian nervous system has not been explored. Our data show that a set of proteins expressed in neuron and glia function in a coordinated manner to provide and store lipids in the form of LD in glia when ROS are elevated.
Parallels between Glaz and APOE point to a conserved mechanism of neuron-glia lipid transport and neurodegeneration
An unanticipated discovery is the role of ApoD and ApoE in lipid transport and accumulation. ApoD is an atypical apolipoprotein that does not share significant sequence homology to other apolipoprotein family members, and it is thought to transport lipids in a manner similar to proteins in the lipocalin family (Eichinger et al., 2007; Perdomo and Dong, 2009). In flies, the ApoD homologs Glaz and Nlaz are secreted proteins (Sanchez et al., 2000). The loss of either Glaz or Nlaz in flies, or of their homolog ApoD in mice, leads to increased sensitivity to ROS (Ganfornina et al., 2008; Hull-Thompson et al., 2009; Sanchez et al., 2006). Both ApoD and ApoE are highly expressed in the mammalian nervous system (Uhlen et al., 2015; Zhang et al., 2014) and appear to have a potentially compensatory relationship. ApoD is upregulated in the circulating lipoproteins of Apoe null mice and expression of both proteins is altered in AD and after brain injuries (Elliott et al., 2010; Jansen et al., 2009; Perdomo and Dong, 2009; Terrisse et al., 1999). LD formations are lost in Rh-ND42 IR flies when there is heterozygous loss of Glaz, but LD formation is restored in when APOE2 or APOE3 is expressed under the Glaz promoter in this context. These findings show that the two variants and Glaz likely play similar roles in lipid transport and respond similarly to oxidative stress.
Overexpressing Glaz, APOE2 or APOE3 in glia results in substantial glial LD accumulation, whereas overexpression of the APOE4 variant does not. This is consistent with previous studies showing that Apoe deficient mice have less cortical fatty acids (Montine et al., 1999). Given that the APOE4 allele cannot restore lipid transport and glial LD accumulation, our data strongly suggest that APOE4 is a partial loss-of-function allele in these phenotypic assays. Several studies have shown that elevated levels of ApoD in flies or mice are neuroprotective (Ganfornina et al., 2008; Hull-Thompson et al., 2009; Terrisse et al., 1999). Interestingly, Apoe−/− mice have been linked to an altered oxidative stress response (Shea et al., 2002) and shown to exhibit increased lipid peroxidation when aged (Montine et al., 1999) but LD have not been implicated.
In the presence of low or moderate elevations of ROS, LD store peroxidated lipid, providing a protective mechanism against low levels of lipid peroxidation (Bailey et al., 2015; Listenberger et al., 2003). Indeed, our data support protective effects of glial LD accumulation in response to low ROS. Flies that express APOE3 or APOE4 under the control of the Glaz regulatory elements and are fed rotenone exhibit obvious differences: APOE4 flies are unable to accumulate glial LD and exhibit signs of neurodegeneration, whereas APOE3 flies accumulate glial LD and are comparable to wild type. Furthermore, when aged, the APOE4 flies exhibit significantly more neuronal death than APOE3 flies (Figure 6F and 7C–D). These data show that the inability to accumulate lipids into LD in the presence of elevated ROS promotes neurodegeneration.
The context of glial LD accumulation is critical in neurodegeneration. Indeed, LD accumulation itself is not detrimental, as overexpression of SREBP in the absence of ROS does not lead to neurodegeneration. Conversely, in the presence of high ROS, as observed in some mitochondrial mutants, the protective mechanisms of glial LD accumulation are overridden, damaging the glia and negatively affecting neuronal survival. In this context, LDs disappear as neurodegeneration progresses but triglyceride levels continue to rise (Liu et al., 2015). The disappearance of LDs suggest that the phospholipid monolayer of the organelle is likely no longer intact due to peroxidation, peroxidated lipids are no longer contained in LD, affecting cell health (Ayala et al., 2014; Listenberger et al., 2003).
The observation that loss of Glaz or Apoe decreases LD accumulation in flies and vertebrate cells and that these animals and cells have a compromised ability to cope with elevated levels of ROS, suggest that LD formation provides neuroprotection. Our findings are also consistent with the observations that the three human APOE alleles (E2, E3, and E4) have very different abilities to induce LD accumulation. Interestingly, the APOE2 allele, which is protective against AD (Yu et al., 2014), is the most efficient in lipid transport and in promoting LD accumulation in our assays. In contrast, the APOE4 allele, which is semi-dominantly linked to the development of AD (Genin et al., 2011), is highly inefficient in lipid transport and LD accumulation. We propose that protection from damage resulting from age-dependent progressive mitochondrial dysfunction and increased oxidative stress in neurons (Bishop et al., 2010; Floyd and Hensley, 2002) relies in part on the protection conferred by proper lipid transfer to glia, which sequestrates peroxidated lipids into LD (Liu et al., 2015).
How does the astrocyte/neuron lactate shuttle support the capacity of glial cells to protect neurons against ROS? We propose that astrocytes provide the reduced three carbon metabolite lactate to neurons as a fatty acid precursor, rather than providing fatty acids, ensuring that low-level neuronal fatty acid synthesis continually produces new, undamaged fatty acids. De novo fatty acid and lipid synthesis leads to lipid turnover, with neurons maintaining a constant lipid level by exporting the excess through an ApoE-dependent pathway that steadily removes normal and damaged lipids. Astrocytes take up these lipids and oxidize them for fuel, which provides the reducing equivalents to ensure that the astrocytes export lactate, not pyruvate, to neurons. This arrangement becomes functionally critical for neuroprotection upon exposure to ROS, due to the generation of high levels of peroxidated lipids. Neuronal induction of JNK elevates lipid production, increasing lipid turnover and locally alleviating the detrimental effects of ROS by exporting peroxidated lipids to astrocytes, where they accumulate (relatively safely) in LD. As such, LD are a lagging indicator of neuroprotection that has already occurred. After a transient ROS challenge, return to normal metabolism will allow glia to steadily deplete their LD. During ROS challenges, failure of the glia to provide neurons with lactate (via the ANLS pathway), failure of neuronal fatty acid synthesis, or failure of ApoE-dependent neuronal lipid export will block this lactate/lipid cycle and prevent neuroprotection by allowing neuronal accumulation of ROS products.
Broader implications of APOE genotypes in health and disease
Although our work focuses on the role of APOE-dependent lipid transfer between neurons and glia, the APOE alleles are associated with a susceptibility to develop other disease conditions unrelated to the nervous system. Indeed, the APOE alleles contribute most significantly to blood cholesterol variability in humans (Pedersen and Berg, 1989). For example, although APOE2 carriers are protected from AD, approximately 10% of individuals with two copies of APOE2 will develop type III hyperlipoproteinemia (Havel and Kane, 1973), leading to xanthomas in subcutaneous tissues (Davignon et al., 1988). Interestingly, the majority of APOE2 carriers have normal to low levels of circulating cholesterol, suggesting that the enhanced transport of lipids by APOE2 result in tissue specific phenotypes. Thus, it is possible that APOE2’s increased ability to transport lipids may result in excessive lipid accumulation as found in type III hyperlipoproteinemia. Meanwhile, enhanced reverse cholesterol transport due to APOE2 likely promote cholesterol clearance in the circulatory system, contributing to hypocholesterolemia (Rader et al., 2009; Zanotti et al., 2011). On the other hand, APOE4 carriers tend to have higher levels of circulating cholesterols, coronary artery disease and atherosclerosis (Eichner et al., 2002; Sparks, 1997). Our findings that APOE4 is unable to transport lipids in the CNS may be relevant for the pathogenesis of APOE in coronary health. The inability of APOE4 to efficiently transport lipids may lead to elevated blood lipid content, atherosclerosis, and increased lipid peroxidation, contributing to coronary artery disease risk (Jofre-Monseny et al., 2008; Minihane et al., 2007; Sugamura and Keaney, 2011). In sum, our findings provide key insights into the mechanism of neuron-glia metabolic cooperation and point to the broader implications of APOE allelic functional differences in systemic health and disease.
STAR METHODS
CONTACT FOR REAGENTS AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Hugo Bellen at hbellen@bcm.edu.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Drosophila
Female flies were used for all experiments. Flies were raised on molasses based food at 22 °C with con stant light unless otherwise noted. The full list of genotypes of the flies used can be found in the Key Resources Table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER (RRID) |
|---|---|---|
| Antibodies | ||
| Mouse anti-beta III tubulin (801201) | Biolegend | RRID:AB_2313773 |
| Mouse Anti-Glial Fibrillary Acid Protein (G3893) | Sigma | RRID:AB_2313773 |
| Rabbit Anti-Glial Fibrillary Acid Protein | EnCor Biotechnology | RRID:AB_2572310 |
| Goat anti-FATP1 | Santa Cruz Biotechnology | RRID:AB_2239414 |
| Goat anti-FATP4 (SC-5835) | Santa Cruz Biotechnology | RRID:AB_2190629 |
| Mouse Anti-6x-His Epitope Tag (HIS.H8) | ThermoFisher | RRID:AB_2313773 |
| Rabbit Anti-Human APOE | Abcam | RRID:AB_867703 |
| Mouse Anti-Actin (C4) | EMD Millipore | RRID:AB_2223041 |
| Rabbit P75-NTR | Biolegend | RRID:AB_2565441 |
| Rabbit Anti-active Caspase 3(Asp175) | Cell Signaling | RRID:AB_443014 |
| Bacterial and Virus Strains | ||
| Lentiguide-Puro-BFP | This manuscript | Backbone obtained from addgene: Plasmid # 52963 Plasmid #34632 |
| Stbl3 competent cells | Thermo Fisher | Cat # C737303 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Nile Red | Sigma | Cat # 72485 |
| BODIPY (493/503) | Thermo Fisher | Cat # D3922 |
| SR 13800 (MCTi2) | Tocris | Cat # 5431 |
| AR-C155858 (MCTi) | Tocris | Cat # 4960 |
| Rotenone | Sigma | Cat # R8875 |
| Sodium L-Lactate | Sigma | Cat # 71718 |
| Dichloroacetic acid (PESTANAL® analytical standard) | Aldrich | Cat # 36545 |
| U-13C-d-Glucose | Cambridge Isotopes | Cat # CLM-1396-PK |
| TrypLE | ThermoFisher | Cat # 12605010 |
| SytoxRed | ThermoFisher | Cat # S34859 |
| Critical Commercial Assays | ||
| In Situ Cell Death Detection Kit, Fluorescein | Roche | Cat # 11684795910 |
| SuperSignal™ West Pico Chemiluminescent Substrate | ThermoFisher | Cat # 34080 |
| Experimental Models: Organisms/Strains | ||
| y w*; P{GAL4-elav.L}2 | Drosophila melanogaster | BDSC_8765 |
| y1 w*; P{w[+m*]=GAL4}54C | Drosophila melanogaster | BDSC_27328 |
| y1 w*; P{nSyb-GAL4.S}3 | Drosophila melanogaster | BDSC_51635 |
| y1 w*; P{w+mC=UAS-mCD8::GFP.L}LL5 | Drosophila melanogaster | BDSC_5137 |
| w*; P{w+mC = UAS-bsk.B}2 | Drosophila melanogaster | BDSC_9310 |
| y w; P{UAS-SREBP.K}2/CyO | Drosophila melanogaster | BDSC_38396 |
| P{NinaE-GD6220} (Rh-ND42 RNAi) | Drosophila melanogaster | (Liu et al., 2015) |
| P{NinaE-GD11094} (Rh-Marf RNAi) | Drosophila melanogaster | (Liu et al., 2015) |
| P{NinaE-KK108492} Rh-Aats-Met RNAi) | Drosophila melanogaster | (Liu et al., 2015) |
| P{KK104306}VIE-260B (Sln RNAi) (Volkenhoff et al., 2015) | Drosophila melanogaster | FlyBase_FBst0481152 |
| w1118; P{GD1940}v4607 (Sln RNAi) | Drosophila melanogaster | FlyBase_FBst0466501 |
| w1118; P{GD3448}v51157 (out RNAi) (Volkenhoff et al., 2015) | Drosophila melanogaster | FlyBase_FBst0469314 |
| P{KK104187}VIE-260B (out RNAi) (Volkenhoff et al., 2015) | Drosophila melanogaster | FlyBase_FBst0480175 |
| w[1118]; P{GD12666}v26802/CyO (shakB RNAi) (Pezier et al., 2016) | Drosophila melanogaster | FlyBase_FBst0456596 |
| y1 sc* v1; P{y+t7.7 v+t1.8=TRiP.HMC04895}attP2 (shakB RNAi)(Pezier et al., 2016) | Drosophila melanogaster | BDSC_57706 |
| y1 sc* v1; P{TRiP.HMC03111}attP2 (Fatp RNAi) | Drosophila melanogaster | BDSC_50709 |
| y1 sc* v1; P{TRiP.HMC03960}attP40 (Fatp RNAi) | Drosophila melanogaster | BDSC_55273 |
| y1 sc* v1; P{TRiP.HMC04206}attP2 (Fatp RNAi) | Drosophila melanogaster | BDSC_55919 |
| P{KK104809}VIE-260B (Fatp RNAi) (Dourlen et al., 2012) | Drosophila melanogaster | FlyBase_FBst0471998 |
| y1 v1; P{TRiP.HMS00039}attP2 (Ldh RNAi) | Drosophila melanogaster | BDSC_33640 |
| P{KK107553}VIE-260B (Nlaz RNAi) | Drosophila melanogaster | FlyBase_FBst0473194 |
| w1118; P{GD12709}v35558 (Nlaz RNAi) | Drosophila melanogaster | FlyBase_FBst0461214 |
| P{KK107553}VIE-260B (Nlaz RNAi) | Drosophila melanogaster | FlyBase_FBst0473194 |
| w1118; P{GD4806}v15387/TM3 (Glaz RNAi) | Drosophila melanogaster | FlyBase_FBst0451831 |
| w1118; P{GD4806}v15389/TM3 (Glaz RNAi) | Drosophila melanogaster | FlyBase_FBst0451832 |
| P{KK106377}VIE-260B (Glaz RNAi) | Drosophila melanogaster | FlyBase_FBst0479254 |
| P{KK101856}VIE-260B (Pdha RNAi) | Drosophila melanogaster | FlyBase_FBst0479031 |
| w1118; P{GD12103}v40410 (Pdha RNAi) | Drosophila melanogaster | FlyBase_FBst0463545 |
| w1118; P{GD15718}v43306 (Bsg RNAi) | Drosophila melanogaster | FlyBase_FBst0465019 |
| w1118; P{GD15718}v43307(Bsg RNAi) | Drosophila melanogaster | FlyBase_FBst0465020 |
| w1118; P{GD989}v2789 (Bsg RNAi) | Drosophila melanogaster | FlyBase_FBst0457179 |
| P{w+mC=GMR-hid}SS1, y1 w* P{ry+t7.2=neoFRT}19A; P{ w+m*=GAL4-ey.H}SS5, P{w+mC=UAS-FLP1.D}JD2 | Drosophila melanogaster | BDSC_5248 |
| y1 w* sicilyE P{ry+t7.2=neoFRT}19A/FM7c, P{w+mC=GAL4-Kr.C}DC1, P{w+mC=UAS-GFP.S65T}DC5, sn+ | Drosophila melanogaster | BDSC_52394 |
| y1 w* MarfB P{ry+t7.2=neoFRT}19A/FM7c, P{w+mC=GAL4-Kr.C}DC1, P{w+mC=UAS-GFP.S65T}DC5, sn+ | Drosophila melanogaster | BDSC_67154 |
| P{lacW}BsgSH1217P{neoFRT}40A/CyO | Drosophila melanogaster | (Besse et al., 2007) |
| y1 w*; Mi{Trojan-GAL4.no-pA.0}GLazMI02243 | Drosophila melanogaster | This paper |
| w1118; NlazNW5 | Drosophila melanogaster | (Hull-Thompson et al., 2009) |
| w*; SlnD1/CyO | Drosophila melanogaster | (Jang et al., 2008) |
| y1 w* l(1)G0334A P{neoFRT}19A/FM7c, P{GAL4-Kr.C}DC1, P{UAS-GFP.S65T}DC5, sn+ | Drosophila melanogaster | BDSC_52370 |
| y1 w* kdnA P{neoFRT}19A/FM7c, P{GAL4-Kr.C}DC1, P{UAS-GFP.S65T}DC5, sn+ | Drosophila melanogaster | BDSC_52364 |
| yd2 w1118 P{ey-FLP.N}2 P{GMR-lacZ.C(38.1)}TPN1; P{lacW}Fatpk10307 P{neoFRT}40A/CyO y+ | Drosophila melanogaster | BDSC_10988 |
| y1 w*; PBac{UAS-APOE2.C112, C158}VK00033 | Drosophila melanogaster | This paper |
| y1 w*; PBac{UAS-APOE3.C112, R158}VK00037 | Drosophila melanogaster | This paper |
| y1 w*; PBac{UAS-APOE4.R112, R158}VK00037 | Drosophila melanogaster | This paper |
| B6.129P2-Apoetm1Unc/J | Mus musculus | IMSR_JAX:002052 |
| Gt(ROSA)26Sortm1.1(CAG-cas9*,-EGFP)Fezh/J | Mus musculus | IMSR_JAX:024858 |
| Tg(Aldh1l1-EGFP,-DTA)D8Rth | Mus musculus | IMSR_JAX:026033 |
| C57BL/6J | Mus musculus | IMSR_JAX:000664 |
| FATP1 | CACCGGTCGTTTGCCACCGTTAGAGTGG | n/a |
| FATP1 | CACCGTCGTTTGCCACCGTTAGAGTGGG | n/a |
| FATP4 | CACCGAGGTAGCGTTGTGCCTCTACCGG | n/a |
| FATP4 | CACCGTATGTCCCCACGACGAGGCTTGG | n/a |
| Nontarget | CACCGGCGAGGTATTCGGCTCCGCGTGG | n/a |
| Recombinant DNA | ||
| APOE3 | Harvard Human cDNA collection | HsCD00323815 |
| Software and Algorithms | ||
| ImageJ | NIH | n/a |
| Illustrator CS6 | Adobe | n/a |
| Photoshop CS5 | Adobe | n/a |
| GraphPad Prism 7 | GraphPad software | n/a |
| Microsoft Excel 2016 | Microsoft Corporation | n/a |
Primary Cells
Production of primary olfactory bulb neuron glia culture cells were derived from C57BL/6J pups for all experiments except for lentiviral knockdown experiments (derived from B6J.129 (B6N)-Gt(ROSA)26Sortm1(CAG-cas9*,-EGFP)Fezh/J, the Jackson Laboratory) and Apoe experiment (B6.129P2-Apoetm1Unc/J mice in a C57BL/6J background). Animals used for cell culture production were treated in compliance with the US Department of Health and Human Services and Baylor College of Medicine IUCAC guidelines. Gender was not considered in the derivation of primary cells; the sex of the cells is unknown.
Primary olfactory bulb (OB) neuron and glia were obtained from P2–3 C57BL/6J pups. Cortical tissue was dissected into cold dissection medium (DM) (Hanks’ balanced saline solution [HBSS] [Invitrogen], 15 mM HEPES, pH 7.4). OB were added to 10 mL of DM on ice. After dissection, supernatant was aspirated, and 5 ml of 37ºC 0.25% trypsin-EDTA (Sigma) was added. The bulbs were incubated for 10 minutes at 37°C, and the supernatant was removed. 5 ml of FBS was added for trypsin inactivation. Subsequently, the pellet was washed twice with ice-cold DM and re-suspended in 1mL of fresh DM. OB were dissociated by triturating with 1 ml pipette tip followed by a 25-gauge syringe. After adding 4 ml of DM, cells were centrifuged at 100 rpm for 10 minutes to form a cell pellet. After removal of supernatant, 1 ml of growth media (GM, EMEM [Lonza], 5% Glutamax, 15mM HEPES, 1% pyruvate, 28mM glucose, 10% fetal bovine serum, 5% horse serum, 1% penicillin/streptomycin) was added and filtered through a 70 μM nylon cell strainer (EMD Millipore). Cells were counted and seeded at a density of 2 × 104 onto poly-D-lysine coated coverslips (Neuvitro). 50% of the GM was changed every other day until ~9–11DIV.
Purified culture preparation
Astrocytes were isolated from OB in a similar manner as described above and isolated in as described in (Schildge et al., 2013). Neuronal culture was obtained by adding the mitotic inhibitor 5-fluorodeoxyuridine at 3DIV.
Mice
Male mice were used in vivo experiments. All animal experiments were approved by the Animal Care and Use Committee at Baylor College of Medicine. Mice were maintained with rodent diet (5053; Picolab) and water available ad libitum with 12-h light–dark cycle at 22 °C. The full list of genotypes of the mice used can be found in the Key Resources Table.
METHOD DETAILS
Drosophila Genetics and Analysis
The mutants Aats-metFB, sicilyE and MarfB were isolated via EMS mutagenesis as described previously (Bayat et al., 2012; Sandoval et al., 2014; Zhang et al., 2013)
The genotype of the three mutants are y w; FRT82B Aats-metFB/TM3, Kr>GFP; FRT19A sicilyE/FM7, Kr>GFP and FRT19A MarfB/FM7, Kr>GFP. Mitotic clones were generated using y w, FRT19A, GMR-hid/FM7a; ey>Flp for X chromosome mutants and y w; ey>Flp; FRT82B GMR-hid cl/TM2 for 3rd chromosome mutants for Nile Red staining (Kr>GFP = Kr-GAL4 UAS-GFP; ey>FLP = ey-GAL4, UAS-FLP).
Generation of Transgenic Flies
For the generation of the UAS-APOE2 and UAS-APOE4 mutant constructs, the sequences of APOE2 and APOE4 were synthesized through Sigma gene synthesis services. The UAS-APOE3 construct was retrieved from cDNA clone HsCD00323815 from the Harvard Human cDNA collection. The coding sequences were retrieved and subcloned into the pUASattB vector using EcoRI and XbaI sites. A Kozak consensus sequence was added to 5′ of the CDS.
Production of lentiviral constructs
Lentiviral constructs were based on the LentiGuide-Puro (Addgene plasmid # 52963) (Sanjana et al., 2014). To visualize the transduction efficiency, a mTagBFP2 (Subach et al., 2011) sequence was inserted between the BsiWI sites, after the Ef1-α promoter and before the puromycin resistance cassette.
The final vector, named Lentiguide-Puro-BFP was digested with BsmBI for gRNA insertion. All constructs were transformed into Stbl3 chemically competent cells (ThermoFisher). Lentiviral packaging was performed through the IDDRC Neuroconnectivity Core.
Lentiviral transduction of Rosa-Cas9-eGFP cells were plated and transduced at 3DIV. Transduced cells recovered for 8 additional days before mitochondrial stress assays were performed as described below. Transduction was performed after brief (1hr) serum starvation (DMEM with glucose [Gibco], 5% glutamax, 15mM HEPES, 1% penicillin/streptomycin) and the virus was diluted in the same medium. Media was removed after 18 hours and replaced with GM with B27 (Gibco). Viral transduction was performed at approximately MOI = 0.1 to 0.4 depending on the initial knockout efficiency.
Protein extraction and immunoblotting
Protein was extracted from 3rd instar larvae with Da-GAL4 overexpression of human APOE2, APOE3 or APOE4. Crosses were all set and kept at 25C in standard fly food. Fifteen third instar larvae were washed in 1× PBS and homogenized in SDS sample buffer (277.8 mM Tris-HCl, pH 6.8, 4.4% LDS, 44.4% (w/v) glycerol, 0.02% bromophenol blue – Bio-Rad) to a total of 150 μl. Samples were boiled for 10 min, subjected to SDS-PAGE and transferred onto nitrocellulose membranes. Transferred membranes were blocked with 5% (wt/volume) non-fat dry milk in Tris-buffered saline containing 0.1% Tween-20 at room temperature for 1h and then incubated with anti-APOE (Abcam, EP1373Y) (1:1000) and anti-actin (EMD Millipore, clone C4) (1:10,000) overnight at 4°C. After washing, blots w ere incubated with secondary antibodies and developed using an ECL detection system (Supersignal WestPico; Pierce).
Immunostaining and imaging
Drosophila
Nile Red and phalloidin staining of Drosophila eyes were performed as described in Liu et al., 2015 on adult female flies. Experimenters were blinded during data analysis.
Cell culture
Cells were fixed with 4% paraformaldehyde in PBS for 15 minutes on ice and washed 3× with PBS. Cells were cryo-protected with 30% sucrose and subsequently subjected to freeze/thaw cycles for detergent-free membrane permeabilization for immunocytochemistry experiments. Cells were blocked for 1hr with either 10% normal goat serum or 10% normal donkey serum with 0.1% cold water fish gelatin – primary antibody staining was conducted at 4ºC overnight and secondary antibody staining was conducted at room temperature for 2 hours followed by PBS washes, mounting (ProLong Diamond [ThermoFisher]) and visualization. Immunofluorescence for cells that did not include a stain for LDs were blocked in the same serum block as listed above with 0.1% triton-x. Images were acquired using a Leica SP8 confocal microscope.
Cell Death
Cell death in primary cell culture was performed using the In Situ Cell Death Detection Kit (see Key Resource Table). Experimenters were blinded during data analysis.
Wire hang assay
Mice were allowed to acclimate to the procedure room for 30 minutes prior to behavioral experiments. Holding a mouse by the tail, allow it to grasp, with its forepaw, the wire is suspended one feet above a plastic covered foam pad. Gently release the mouse. Latency to fall is measured within a 120 second cutoff. Experimenters were blinded during the experiment and data analysis.
Drug Studies
Mice
Two to three months old wildtype (C57BL/6J) mice were administered rotenone (3mg/kg, 300mg/ml concentration) or vehicle (Miglyol 812N, IOI Oleo GmbH) via intraperitoneal injections at 24-hour intervals for 8 days. The protocol was adapted for mice from (Cannon et al., 2009). Wildtype vehicle and rotenone treated (n=5 each) were assessed for the onset of ataxic phenotypes using the wire hang assay (see behavioral assay). Rotenone stock solution was prepared every two days and stored in the dark at 4C°. All treatme nts and behavioral assays were performed in parallel. Experimenters were blinded during the experiment and data analysis.
Cell culture
Antioxidant
The antioxidant, AD4 (N-acetylcysteine amide) was dissolved PBS at 150 mM and diluted to 1.5μM in OB growth media. The equivalent amount of PBS was added to OB growth media as control. AD4 was freshly prepared from powder for each experiment to retain potency.
MCT inhibition
The monocarboxylate transporter inhibitor (AR-C155858, Tocris) known here as MCTi was solubilized to 100mM in DMSO and subsequently diluted to 200 nM in OB growth media. The monocarboxylate transporter inhibitor (SR 13800, also known as AR-C122982, Tocris) is known here as MCTi-2 was solubilized to 50mM in DMSO and subsequently diluted to 50nM in OB growth media. MCT inhibitors was aliquoted and stored at -80ºC for each separate experiment.
ROS
For mitochondrial ROS and lactate inhibition assays, rotenone (Sigma) was used to inhibit mitochondrial complex I to induce elevated levels of ROS. Rotenone was solubilized to 10 mM in DMSO and diluted to 1.5 or 2 μM in olfactory bulb growth media. Vehicle control contained a similar amount of DMSO alone. Cells were treated for 12 or 24 hours at 9–10 DIV and subsequently fixed in 4% paraformaldehyde. Rotenone was freshly prepared from powder for each experiment to retain potency.
Lactate
To determine whether increased lactate is detrimental to neuronal/glial health. Sodium L-lactate, ≥99.0% (Aldrich) was dissolved in OB growth media and subsequently filtered through a 0.22 μm filter. Lactate media was incorporated at 5DIV and cells were fixed for and processed for immunocytochemistry at 11 DIV.
Drosophila
Activating Pdh
Dichloroacetic acid (PESTANAL® analytical standard, Sigma Aldrich) was first in 100% ethanol and subsequently diluted into warm liquid fly food at 20μg/ml, 40μg/ml and 80μg/ml. Ethanol without DCA was added to food for flies fed with control food. All vials were used within one week after preparation.
ROS
Rotenone (Sigma-Aldrich), was first solubilized to 10mM in DMSO and then diluted to 25μM into warm liquid fly food. Food was protected from light and used within 1 week of preparation. Rotenone was freshly prepared from powder for each experiment to retain potency.
Rotenone feeding assay
Adult mated flies were placed into vials containing comparable amount of DMSO (vehicle) or food with 25μM rotenone. All vials were protected from light to prevent rotenone degradation. Adult flies that raised on vehicle/rotenone food were dissected at 1-day post eclosion for Nile Red or Phalloidin fluorescent staining. Flies collected for the 10-day aging experiment were collected and transferred to normal food.
Carbon labeled lactate competition assay workflow
Mouse: we derived primary co-culture (neuron and glia) from over 80 BAC Aldh1L1-GFP mice pups (Heintz, 2004). Aldh1l1 is a folate metabolism enzyme that is also a well-established astrocyte marker (Yang et al., 2011).
Co-cultured cells were separated into two experimental groups, one group was cultured using OB growth media supplemented with U-13C-D-glucose (Cambridge Isotopes) while the other group was cultured using OB growth media supplemented with 28 mM U-13C-D-glucose and 11 mM sodium L-lactate (Aldrich).
We then added rotenone (2 μM) for 24 hours to the cells to induce glial LD accumulation.
These co-cultured cells were processed for live cell sorting to isolate GFP+ and live cells using a live-dead indicator (Sytox Red, ThermoFisher).
The GFP+ live cells (astrocytes) were spun down at 0.4G and flash frozen in liquid nitrogen.
Thawed cells were extracted with chloroform/methanol (17:1 v/v) to recover neutral lipids (Ejsing et al., 2009). Dried neutral lipids were dissolved in ethanolic KOH (1M KOH in 95% ethanol), saponified at 60°C for one hour in capped tubes, neutralized with HCl, and dried under vacuum. Fatty acids were dissolved in 1 mM HCl, extracted into hexanes, and dried for LC-MS analysis by the BCM Metabolomics Advanced Technology Core. Ion counts for all expected fatty acid species (M+0 to M+16 for palmitoleic acid) were extracted from the high resolution QTOF data and used to calculate fractional 13C.
Cell harvesting
After 24-hour treatment of co-cultured primary cells with rotenone, cells were washed with 1X PBS and enzymatically released from the plate using TrypLE (ThermoFisher) at 37°C for approximately 8 minutes until cells are released from the plate with agitation. OB growth media was added to resuspend the cells. Cells and media were subsequently transferred to a 15ml conical tube and centrifuged at 0.4G for 5 minutes. Cell pellets were isolated, washed, re-spun down and finally and resuspended in fresh room temperature cell sorting media (1× PBS, no calcium no magnesium, 1mM EDTA, 25mM HEPES, 2% FBS, pH 7.4). Cells were counted and diluted to a concentration of 1×106 ml and kept on ice before sorting.
Cell sorting
Fluorescent cells were isolated using the BD FACSAriaII (BD biosciences). Cell catch buffer is made with EMEM (Lonza), 5% glutamax, 25mM HEPES, 1% pyruvate, 28mM glucose, 10% fetal bovine serum, 5% horse serum, and 1% penicillin/streptomycin.
QUANTIFICATION AND STATISTICAL ANALYSIS
Image Analysis
Quantifications performed in the Drosophila visual system were performed manually using cell counter on ImageJ due to the presence of round rhabdomeres that interfere with automated analysis. Quantifications of cell culture experiments and mice were performed using semi-automatic methods using threshold and particle count.
Statistical analysis
All data sets were organized and analyzed in Microsoft excel 2016 and Prism. Statistical tests are listed in the figure legends. Normality and homogeneity of variance were used to determine whether the data met the assumptions of the statistical test used. All data sets were assumed to be independent. Data sets with unequal variance were analyzed using the Kruskal-Wallis test followed by Dunn’s test for post-hoc analysis for significance due to unequal sample sizes. All other datasets were quantified for significance using Student’s t-test. Significance is defined as P<0.05 and error bars are shown as standard error of the mean (SEM) unless otherwise noted. No outliers were found in any data set and no animals or data were excluded from statistical analysis. For fly experiments, more than 10 flies were used for each individual experiment and all crosses were performed at least twice. For cell experiments, all studies were conducted in parallel with vehicle controls in the neighboring well for at least 3 wells (biological replicates) and also three sets of pups (technical replicates).
Supplementary Material
Highlights.
Glia derived lactate fuels neuronal lipid production
Elevated levels of ROS promote lipid production in neurons
Neuronal lipids are transported to glia via apolipoproteins E and D
APOE4’s inability to transport lipids for LD formation leads to neurodegeneration
Acknowledgments
We thank K. Schulze, Hsiao-Tuan Chao and Shinya Yamamoto for comments; Amber T. Levine for help with mice experiments; J. Chung (SlnD1 and UAS-Sln), H. Jasper (NlazNW5), B. Mollereau (FatpK10307), the Bloomington Drosophila Stock Center, the Vienna Drosophila Resource Center, the TRiP at Harvard Medical School (NIH/NIGMS R01-GM084947), for providing stocks and reagents. The IDDRC Microscopy Core, Neurobehavior core and Neuroconnectivity core supported in part by IDDRC grant number 1U54 HD083092 from the Eunice Kennedy Shriver National Institute of Child Health & Human Development. This project was supported by the Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the NIH (P30 AI036211, P30 CA125123, and S10 RR024574) and by the Metabolomics Core at Baylor College of Medicine with funding from the NIH (P30 CA125123), CPRIT Proteomics and Metabolomics Core Facility (D.P.E.), (RP120092), and Dan L. Duncan Cancer Center. L.L. was supported by the Neuroscience graduate program training grant (5T32GM008507-18) from 2011 to 2012 and the Rush and Helen Record Fellowship in 2016. We acknowledge support of the NIH R01GM120033-01 to M.M.S. and 5R01GM067858 to H.J.B., the Robert A. and Renee E. Belfer Family Foundation, the Huffington Foundation and Target ALS to H.J.B. H.J.B. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Author contribution
L.L. and H.J.B. conceived and designed the project. L.L. conducted the experiments and analyzed the data. L.L. and K.R.M. designed, performed and analyzed the cell based lactate and lipid metabolomics experiments. M.M-S. helped with the mice and metabolomics experiments. M.M-S. and K.R.M. edited the manuscript. L.L. and H.J.B. wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amer J, Atlas D, Fibach E. N-acetylcysteine amide (AD4) attenuates oxidative stress in beta-thalassemia blood cells. Biochim Biophys Acta. 2008;1780:249–255. doi: 10.1016/j.bbagen.2007.11.009. [DOI] [PubMed] [Google Scholar]
- Anderson R, Barnes JC, Bliss TV, Cain DP, Cambon K, Davies HA, Errington ML, Fellows LA, Gray RA, Hoh T, et al. Behavioural, physiological and morphological analysis of a line of apolipoprotein E knockout mouse. Neuroscience. 1998;85:93–110. doi: 10.1016/s0306-4522(97)00598-8. [DOI] [PubMed] [Google Scholar]
- Ayala A, Munoz MF, Arguelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev. 2014;2014:360438. doi: 10.1155/2014/360438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey AP, Koster G, Guillermier C, Hirst EM, MacRae JI, Lechene CP, Postle AD, Gould AP. Antioxidant Role for Lipid Droplets in a Stem Cell Niche of Drosophila. Cell. 2015;163:340–353. doi: 10.1016/j.cell.2015.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnerias C, Saudubray JM, Touati G, De Lonlay P, Dulac O, Ponsot G, Marsac C, Brivet M, Desguerre I. Pyruvate dehydrogenase complex deficiency: four neurological phenotypes with differing pathogenesis. Dev Med Child Neurol. 2010;52:e1–9. doi: 10.1111/j.1469-8749.2009.03541.x. [DOI] [PubMed] [Google Scholar]
- Barros LF. Metabolic signaling by lactate in the brain. Trends Neurosci. 2013;36:396–404. doi: 10.1016/j.tins.2013.04.002. [DOI] [PubMed] [Google Scholar]
- Bayat V, Thiffault I, Jaiswal M, Tetreault M, Donti T, Sasarman F, Bernard G, Demers-Lamarche J, Dicaire MJ, Mathieu J, et al. Mutations in the mitochondrial methionyl-tRNA synthetase cause a neurodegenerative phenotype in flies and a recessive ataxia (ARSAL) in humans. PLoS Biol. 2012;10:e1001288. doi: 10.1371/journal.pbio.1001288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal MF. Mitochondria and neurodegeneration. Novartis Found Symp. 2007;287:183–192. doi: 10.1002/9780470725207.ch13. [DOI] [PubMed] [Google Scholar]
- Belanger M, Allaman I, Magistretti P. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell metabolism. 2011;14:724–738. doi: 10.1016/j.cmet.2011.08.016. [DOI] [PubMed] [Google Scholar]
- Besse F, Mertel S, Kittel RJ, Wichmann C, Rasse TM, Sigrist SJ, Ephrussi A. The Ig cell adhesion molecule Basigin controls compartmentalization and vesicle release at Drosophila melanogaster synapses. J Cell Biol. 2007;177:843–855. doi: 10.1083/jcb.200701111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop NA, Lu T, Yankner BA. Neural mechanisms of ageing and cognitive decline. Nature. 2010;464:529–535. doi: 10.1038/nature08983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosone C, Andreu A, Echevarria D. GAP junctional communication in brain secondary organizers. Dev Growth Differ. 2016;58:446–455. doi: 10.1111/dgd.12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon JR, Tapias V, Na HM, Honick AS, Drolet RE, Greenamyre JT. A highly reproducible rotenone model of Parkinson’s disease. Neurobiol Dis. 2009;34:279–290. doi: 10.1016/j.nbd.2009.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coe NR, Smith AJ, Frohnert BI, Watkins PA, Bernlohr DA. The fatty acid transport protein (FATP1) is a very long chain acyl-CoA synthetase. J Biol Chem. 1999;274:36300–36304. doi: 10.1074/jbc.274.51.36300. [DOI] [PubMed] [Google Scholar]
- Coffman CR, Strohm RC, Oakley FD, Yamada Y, Przychodzin D, Boswell RE. Identification of X-linked genes required for migration and programmed cell death of Drosophila melanogaster germ cells. Genetics. 2002;162:273–284. doi: 10.1093/genetics/162.1.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conejero-Goldberg C, Gomar JJ, Bobes-Bascaran T, Hyde TM, Kleinman JE, Herman MM, Chen S, Davies P, Goldberg TE. APOE2 enhances neuroprotection against Alzheimer’s disease through multiple molecular mechanisms. Mol Psychiatry. 2014;19:1243–1250. doi: 10.1038/mp.2013.194. [DOI] [PubMed] [Google Scholar]
- Curtin KD, Meinertzhagen IA, Wyman RJ. Basigin (EMMPRIN/CD147) interacts with integrin to affect cellular architecture. J Cell Sci. 2005;118:2649–2660. doi: 10.1242/jcs.02408. [DOI] [PubMed] [Google Scholar]
- Davignon J, Gregg RE, Sing CF. Apolipoprotein E polymorphism and atherosclerosis. Arteriosclerosis. 1988;8:1–21. doi: 10.1161/01.atv.8.1.1. [DOI] [PubMed] [Google Scholar]
- Debernardi R, Pierre K, Lengacher S, Magistretti PJ, Pellerin L. Cell-specific expression pattern of monocarboxylate transporters in astrocytes and neurons observed in different mouse brain cortical cell cultures. J Neurosci Res. 2003;73:141–155. doi: 10.1002/jnr.10660. [DOI] [PubMed] [Google Scholar]
- Diao F, Ironfield H, Luan H, Diao F, Shropshire WC, Ewer J, Marr E, Potter CJ, Landgraf M, White BH. Plug-and-play genetic access to drosophila cell types using exchangeable exon cassettes. Cell Rep. 2015;10:1410–1421. doi: 10.1016/j.celrep.2015.01.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty JR, Yang C, Scott KE, Cameron MD, Fallahi M, Li W, Hall MA, Amelio AL, Mishra JK, Li F, et al. Blocking lactate export by inhibiting the Myc target MCT1 Disables glycolysis and glutathione synthesis. Cancer Res. 2014;74:908–920. doi: 10.1158/0008-5472.CAN-13-2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dourlen P, Bertin B, Chatelain G, Robin M, Napoletano F, Roux M, Mollereau B. Drosophila Fatty Acid transport protein regulates rhodopsin-1 metabolism and is required for photoreceptor neuron survival. PLoS Genet. 2012;8 doi: 10.1371/journal.pgen.1002833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dourlen P, Sujkowski A, Wessells R, Mollereau B. Fatty acid transport proteins in disease: New insights from invertebrate models. Prog Lipid Res. 2015;60:30–40. doi: 10.1016/j.plipres.2015.08.001. [DOI] [PubMed] [Google Scholar]
- Eichinger A, Nasreen A, Kim HJ, Skerra A. Structural insight into the dual ligand specificity and mode of high density lipoprotein association of apolipoprotein D. J Biol Chem. 2007;282:31068–31075. doi: 10.1074/jbc.M703552200. [DOI] [PubMed] [Google Scholar]
- Eichner JE, Dunn ST, Perveen G, Thompson DM, Stewart KE, Stroehla BC. Apolipoprotein E polymorphism and cardiovascular disease: a HuGE review. Am J Epidemiol. 2002;155:487–495. doi: 10.1093/aje/155.6.487. [DOI] [PubMed] [Google Scholar]
- Ejsing CS, Sampaio JL, Surendranath V, Duchoslav E, Ekroos K, Klemm RW, Simons K, Shevchenko A. Global analysis of the yeast lipidome by quantitative shotgun mass spectrometry. Proc Natl Acad Sci U S A. 2009;106:2136–2141. doi: 10.1073/pnas.0811700106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott DA, Weickert CS, Garner B. Apolipoproteins in the brain: implications for neurological and psychiatric disorders. Clin Lipidol. 2010;51:555–573. doi: 10.2217/CLP.10.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N, van Duijn CM. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. Jama. 1997;278:1349–1356. [PubMed] [Google Scholar]
- Floyd RA, Hensley K. Oxidative stress in brain aging. Implications for therapeutics of neurodegenerative diseases. Neurobiol Aging. 2002;23:795–807. doi: 10.1016/s0197-4580(02)00019-2. [DOI] [PubMed] [Google Scholar]
- Funfschilling U, Supplie LM, Mahad D, Boretius S, Saab AS, Edgar J, Brinkmann BG, Kassmann CM, Tzvetanova ID, Mobius W, et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature. 2012;485:517–521. doi: 10.1038/nature11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganfornina MD, Do Carmo S, Lora JM, Torres-Schumann S, Vogel M, Allhorn M, Gonzalez C, Bastiani MJ, Rassart E, Sanchez D. Apolipoprotein D is involved in the mechanisms regulating protection from oxidative stress. Aging Cell. 2008;7:506–515. doi: 10.1111/j.1474-9726.2008.00395.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genin E, Hannequin D, Wallon D, Sleegers K, Hiltunen M, Combarros O, Bullido MJ, Engelborghs S, De Deyn P, Berr C, et al. APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Mol Psychiatry. 2011;16:903–907. doi: 10.1038/mp.2011.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP, Wilson MC. The monocarboxylate transporter family--role and regulation. IUBMB Life. 2012;64:109–119. doi: 10.1002/iub.572. [DOI] [PubMed] [Google Scholar]
- Hauser PS, Narayanaswami V, Ryan RO. Apolipoprotein E: from lipid transport to neurobiology. Prog Lipid Res. 2011;50:62–74. doi: 10.1016/j.plipres.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havel RJ, Kane JP. Primary dysbetalipoproteinemia: predominance of a specific apoprotein species in triglyceride-rich lipoproteins. Proc Natl Acad Sci U S A. 1973;70:2015–2019. doi: 10.1073/pnas.70.7.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintz N. Gene expression nervous system atlas (GENSAT) Nat Neurosci. 2004;7:483. doi: 10.1038/nn0504-483. [DOI] [PubMed] [Google Scholar]
- Hertz L, Peng L, Dienel GA. Energy metabolism in astrocytes: high rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J Cereb Blood Flow Metab. 2007;27:219–249. doi: 10.1038/sj.jcbfm.9600343. [DOI] [PubMed] [Google Scholar]
- Herzog RI, Jiang L, Herman P, Zhao C, Sanganahalli BG, Mason GF, Hyder F, Rothman DL, Sherwin RS, Behar KL. Lactate preserves neuronal metabolism and function following antecedent recurrent hypoglycemia. J Clin Invest. 2013;123:1988–1998. doi: 10.1172/JCI65105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Flockhart I, Vinayagam A, Bergwitz C, Berger B, Perrimon N, Mohr SE. An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinformatics. 2011;12:357. doi: 10.1186/1471-2105-12-357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Mahley RW. Apolipoprotein E: structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol Dis. 2014;72(Pt A):3–12. doi: 10.1016/j.nbd.2014.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull-Thompson J, Muffat J, Sanchez D, Walker D, Benzer S, Ganfornina M, Jasper H. Control of metabolic homeostasis by stress signaling is mediated by the lipocalin NLaz. PLoS Genet. 2009;5 doi: 10.1371/journal.pgen.1000460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyder F, Patel AB, Gjedde A, Rothman DL, Behar KL, Shulman RG. Neuronal-glial glucose oxidation and glutamatergic-GABAergic function. J Cereb Blood Flow Metab. 2006;26:865–877. doi: 10.1038/sj.jcbfm.9600263. [DOI] [PubMed] [Google Scholar]
- Ikonen E. Cellular cholesterol trafficking and compartmentalization. Nat Rev Mol Cell Biol. 2008;9:125–138. doi: 10.1038/nrm2336. [DOI] [PubMed] [Google Scholar]
- Jaiswal M, Haelterman NA, Sandoval H, Xiong B, Donti T, Kalsotra A, Yamamoto S, Cooper TA, Graham BH, Bellen HJ. Impaired Mitochondrial Energy Production Causes Light-Induced Photoreceptor Degeneration Independent of Oxidative Stress. PLoS Biol. 2015;13:e1002197. doi: 10.1371/journal.pbio.1002197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal M, Sandoval H, Zhang K, Bayat V, Bellen HJ. Probing mechanisms that underlie human neurodegenerative diseases in Drosophila. Annu Rev Genet. 2012;46:371–396. doi: 10.1146/annurev-genet-110711-155456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang C, Lee G, Chung J. LKB1 induces apical trafficking of Silnoon, a monocarboxylate transporter, in Drosophila melanogaster. J Cell Biol. 2008;183:11–17. doi: 10.1083/jcb.200807052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen PJ, Lutjohann D, Thelen KM, von Bergmann K, van Leuven F, Ramaekers FC, Monique M. Absence of ApoE upregulates murine brain ApoD and ABCA1 levels, but does not affect brain sterol levels, while human ApoE3 and human ApoE4 upregulate brain cholesterol precursor levels. J Alzheimers Dis. 2009;18:319–329. doi: 10.3233/JAD-2009-1150. [DOI] [PubMed] [Google Scholar]
- Jofre-Monseny L, Minihane AM, Rimbach G. Impact of apoE genotype on oxidative stress, inflammation and disease risk. Mol Nutr Food Res. 2008;52:131–145. doi: 10.1002/mnfr.200700322. [DOI] [PubMed] [Google Scholar]
- Kalaany NY, Mangelsdorf DJ. LXRS and FXR: the yin and yang of cholesterol and fat metabolism. Annu Rev Physiol. 2006;68:159–191. doi: 10.1146/annurev.physiol.68.033104.152158. [DOI] [PubMed] [Google Scholar]
- Kazantzis M, Stahl A. Fatty acid transport proteins, implications in physiology and disease. Biochim Biophys Acta. 2012;1821:852–857. doi: 10.1016/j.bbalip.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knust E. Photoreceptor morphogenesis and retinal degeneration: lessons from Drosophila. Curr Opin Neurobiol. 2007;17:541–547. doi: 10.1016/j.conb.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Kruse SE, Watt WC, Marcinek DJ, Kapur RP, Schenkman KA, Palmiter RD. Mice with mitochondrial complex I deficiency develop a fatal encephalomyopathy. Cell Metab. 2008;7:312–320. doi: 10.1016/j.cmet.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Morrison B, Li Y, Lengacher S, Farah M, Hoffman P, Liu Y, Tsingalia A, Jin L, Zhang PW, et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature. 2012;487:443–448. doi: 10.1038/nature11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limmer S, Weiler A, Volkenhoff A, Babatz F, Klambt C. The Drosophila blood-brain barrier: development and function of a glial endothelium. Front Neurosci. 2014;8:365. doi: 10.3389/fnins.2014.00365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Listenberger LL, Han X, Lewis SE, Cases S, Farese RV, Jr, Ory DS, Schaffer JE. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci U S A. 2003;100:3077–3082. doi: 10.1073/pnas.0630588100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Zhang K, Sandoval H, Yamamoto S, Jaiswal M, Sanz E, Li Z, Hui J, Graham BH, Quintana A, et al. Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell. 2015;160:177–190. doi: 10.1016/j.cell.2014.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machler P, Wyss MT, Elsayed M, Stobart J, Gutierrez R, von Faber-Castell A, Kaelin V, Zuend M, San Martin A, Romero-Gomez I, et al. In Vivo Evidence for a Lactate Gradient from Astrocytes to Neurons. Cell Metab. 2016;23:94–102. doi: 10.1016/j.cmet.2015.10.010. [DOI] [PubMed] [Google Scholar]
- Maeda N. Development of apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2011;31:1957–1962. doi: 10.1161/ATVBAHA.110.220574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc Natl Acad Sci U S A. 2006;103:5644–5651. doi: 10.1073/pnas.0600549103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin E, Rosenthal RE, Fiskum G. Pyruvate dehydrogenase complex: metabolic link to ischemic brain injury and target of oxidative stress. J Neurosci Res. 2005;79:240–247. doi: 10.1002/jnr.20293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashek DG, Li LO, Coleman RA. Long-chain acyl-CoA synthetases and fatty acid channeling. Future Lipidol. 2007;2:465–476. doi: 10.2217/17460875.2.4.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Ge N, Alford M, Veinbergs I, Roses AD. Neurodegeneration in the central nervous system of apoE-deficient mice. Exp Neurol. 1995;136:107–122. doi: 10.1006/exnr.1995.1088. [DOI] [PubMed] [Google Scholar]
- Michelakis ED, Webster L, Mackey JR. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer. 2008;99:989–994. doi: 10.1038/sj.bjc.6604554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minihane AM, Jofre-Monseny L, Olano-Martin E, Rimbach G. ApoE genotype, cardiovascular risk and responsiveness to dietary fat manipulation. Proc Nutr Soc. 2007;66:183–197. doi: 10.1017/S0029665107005435. [DOI] [PubMed] [Google Scholar]
- Mitchell RW, Hatch GM. Fatty acid transport into the brain: of fatty acid fables and lipid tails. Prostaglandins Leukot Essent Fatty Acids. 2011;85:293–302. doi: 10.1016/j.plefa.2011.04.007. [DOI] [PubMed] [Google Scholar]
- Montine TJ, Montine KS, Olson SJ, Graham DG, Roberts LJ, 2nd, Morrow JD, Linton MF, Fazio S, Swift LL. Increased cerebral cortical lipid peroxidation and abnormal phospholipids in aged homozygous apoE-deficient C57BL/6J mice. Exp Neurol. 1999;158:234–241. doi: 10.1006/exnr.1999.7067. [DOI] [PubMed] [Google Scholar]
- Mozaffarian D, Cao H, King IB, Lemaitre RN, Song X, Siscovick DS, Hotamisligil GS. Circulating palmitoleic acid and risk of metabolic abnormalities and new-onset diabetes. Am J Clin Nutr. 2010;92:1350–1358. doi: 10.3945/ajcn.110.003970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray CM, Hutchinson R, Bantick JR, Belfield GP, Benjamin AD, Brazma D, Bundick RV, Cook ID, Craggs RI, Edwards S, et al. Monocarboxylate transporter MCT1 is a target for immunosuppression. Nat Chem Biol. 2005;1:371–376. doi: 10.1038/nchembio744. [DOI] [PubMed] [Google Scholar]
- Nagarkar-Jaiswal S, Lee PT, Campbell ME, Chen K, Anguiano-Zarate S, Gutierrez MC, Busby T, Lin WW, He Y, Schulze KL, et al. A library of MiMICs allows tagging of genes and reversible, spatial and temporal knockdown of proteins in Drosophila. Elife. 2015;4 doi: 10.7554/eLife.05338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19:983–997. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- Ovens MJ, Davies AJ, Wilson MC, Murray CM, Halestrap AP. AR-C155858 is a potent inhibitor of monocarboxylate transporters MCT1 and MCT2 that binds to an intracellular site involving transmembrane helices 7–10. Biochem J. 2010;425:523–530. doi: 10.1042/BJ20091515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen JC, Berg K. Interaction between low density lipoprotein receptor (LDLR) and apolipoprotein E (apoE) alleles contributes to normal variation in lipid level. Clin Genet. 1989;35:331–337. doi: 10.1111/j.1399-0004.1989.tb02953.x. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Bouzier-Sore AK, Aubert A, Serres S, Merle M, Costalat R, Magistretti PJ. Activity-dependent regulation of energy metabolism by astrocytes: an update. Glia. 2007;55:1251–1262. doi: 10.1002/glia.20528. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci U S A. 1994;91:10625–10629. doi: 10.1073/pnas.91.22.10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdomo G, Dong H. Apolipoprotein D in lipid metabolism and its functional implication in atherosclerosis and aging. Aging (Albany NY) 2009;1:17–27. doi: 10.18632/aging.100004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan P, Stebbings LA, Baines RA, Bacon JP, Davies JA, Ford C. Drosophila Shaking-B protein forms gap junctions in paired Xenopus oocytes. Nature. 1998;391:181–184. doi: 10.1038/34426. [DOI] [PubMed] [Google Scholar]
- Piedrahita JA, Zhang SH, Hagaman JR, Oliver PM, Maeda N. Generation of mice carrying a mutant apolipoprotein E gene inactivated by gene targeting in embryonic stem cells. Proc Natl Acad Sci U S A. 1992;89:4471–4475. doi: 10.1073/pnas.89.10.4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierre K, Pellerin L. Monocarboxylate transporters in the central nervous system: distribution, regulation and function. J Neurochem. 2005;94:1–14. doi: 10.1111/j.1471-4159.2005.03168.x. [DOI] [PubMed] [Google Scholar]
- Rader DJ, Alexander ET, Weibel GL, Billheimer J, Rothblat GH. The role of reverse cholesterol transport in animals and humans and relationship to atherosclerosis. J Lipid Res. 2009;50(Suppl):S189–194. doi: 10.1194/jlr.R800088-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raichle ME, Gusnard DA. Appraising the brain’s energy budget. Proc Natl Acad Sci U S A. 2002;99:10237–10239. doi: 10.1073/pnas.172399499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawson RB. The SREBP pathway--insights from Insigs and insects. Nat Rev Mol Cell Biol. 2003;4:631–640. doi: 10.1038/nrm1174. [DOI] [PubMed] [Google Scholar]
- Ruiz M, Sanchez D, Correnti C, Strong RK, Ganfornina MD. Lipid-binding properties of human ApoD and Lazarillo-related lipocalins: functional implications for cell differentiation. FEBS J. 2013;280:3928–3943. doi: 10.1111/febs.12394. [DOI] [PubMed] [Google Scholar]
- Sanchez D, Ganfornina M, Torres-Schumann S, Speese S, Lora J, Bastiani M. Characterization of two novel lipocalins expressed in the Drosophila embryonic nervous system. The International journal of developmental biology. 2000;44:349–359. [PubMed] [Google Scholar]
- Sanchez D, Lopez-Arias B, Torroja L, Canal I, Wang X, Bastiani M, Ganfornina M. Loss of glial lazarillo, a homolog of apolipoprotein D, reduces lifespan and stress resistance in Drosophila. Current biology: CB. 2006;16:680–686. doi: 10.1016/j.cub.2006.03.024. [DOI] [PubMed] [Google Scholar]
- Sandoval H, Yao CK, Chen K, Jaiswal M, Donti T, Lin YQ, Bayat V, Xiong B, Zhang K, David G, et al. Mitochondrial fusion but not fission regulates larval growth and synaptic development through steroid hormone production. Elife. 2014;3 doi: 10.7554/eLife.03558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer JE, Lodish HF. Expression cloning and characterization of a novel adipocyte long chain fatty acid transport protein. Cell. 1994;79:427–436. doi: 10.1016/0092-8674(94)90252-6. [DOI] [PubMed] [Google Scholar]
- Schildge S, Bohrer C, Beck K, Schachtrup C. Isolation and culture of mouse cortical astrocytes. J Vis Exp. 2013 doi: 10.3791/50079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurr A, Payne RS. Lactate, not pyruvate, is neuronal aerobic glycolysis end product: an in vitro electrophysiological study. Neuroscience. 2007;147:613–619. doi: 10.1016/j.neuroscience.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Shea TB, Rogers E, Ashline D, Ortiz D, Sheu MS. Apolipoprotein E deficiency promotes increased oxidative stress and compensatory increases in antioxidants in brain tissue. Free Radic Biol Med. 2002;33:1115–1120. doi: 10.1016/s0891-5849(02)01001-8. [DOI] [PubMed] [Google Scholar]
- Shimohigashi M, Meinertzhagen IA. The shaking B gene in Drosophila regulates the number of gap junctions between photoreceptor terminals in the lamina. J Neurobiol. 1998;35:105–117. doi: 10.1002/(sici)1097-4695(199804)35:1<105::aid-neu9>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Sparks DL. Coronary artery disease, hypertension, ApoE, and cholesterol: a link to Alzheimer’s disease? Ann. N Y Acad Sci. 1997;826:128–146. doi: 10.1111/j.1749-6632.1997.tb48466.x. [DOI] [PubMed] [Google Scholar]
- Stahl A. A current review of fatty acid transport proteins (SLC27) Pflugers Arch. 2004;447:722–727. doi: 10.1007/s00424-003-1106-z. [DOI] [PubMed] [Google Scholar]
- Stebbings LA, Todman MG, Phillips R, Greer CE, Tam J, Phelan P, Jacobs K, Bacon JP, Davies JA. Gap junctions in Drosophila: developmental expression of the entire innexin gene family. Mech Dev. 2002;113:197–205. doi: 10.1016/s0925-4773(02)00025-4. [DOI] [PubMed] [Google Scholar]
- Subach OM, Cranfill PJ, Davidson MW, Verkhusha VV. An enhanced monomeric blue fluorescent protein with the high chemical stability of the chromophore. PLoS One. 2011;6:e28674. doi: 10.1371/journal.pone.0028674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugamura K, Keaney JF., Jr Reactive oxygen species in cardiovascular disease. Free Radic Biol Med. 2011;51:978–992. doi: 10.1016/j.freeradbiomed.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabernero A, Medina JM, Giaume C. Glucose metabolism and proliferation in glia: role of astrocytic gap junctions. J Neurochem. 2006;99:1049–1061. doi: 10.1111/j.1471-4159.2006.04088.x. [DOI] [PubMed] [Google Scholar]
- Terrisse L, Seguin D, Bertrand P, Poirier J, Milne R, Rassart E. Modulation of apolipoprotein D and apolipoprotein E expression in rat hippocampus after entorhinal cortex lesion. Brain Res Mol Brain Res. 1999;70:26–35. doi: 10.1016/s0169-328x(99)00123-0. [DOI] [PubMed] [Google Scholar]
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- van Hall G, Stromstad M, Rasmussen P, Jans O, Zaar M, Gam C, Quistorff B, Secher NH, Nielsen HB. Blood lactate is an important energy source for the human brain. J Cereb Blood Flow Metab. 2009;29:1121–1129. doi: 10.1038/jcbfm.2009.35. [DOI] [PubMed] [Google Scholar]
- Venken K, Schulze K, Haelterman N, Pan H, He Y, Evans-Holm M, Carlson J, Levis R, Spradling A, Hoskins R, et al. MiMIC: a highly versatile transposon insertion resource for engineering Drosophila melanogaster genes. Nat Methods. 2011;8:737–743. doi: 10.1038/nmeth.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkenhoff A, Weiler A, Letzel M, Stehling M, Klambt C, Schirmeier S. Glial Glycolysis Is Essential for Neuronal Survival in Drosophila. Cell Metab. 2015;22:437–447. doi: 10.1016/j.cmet.2015.07.006. [DOI] [PubMed] [Google Scholar]
- Walker DW, Muffat J, Rundel C, Benzer S. Overexpression of a Drosophila homolog of apolipoprotein D leads to increased stress resistance and extended lifespan. Curr Biol. 2006;16:674–679. doi: 10.1016/j.cub.2006.01.057. [DOI] [PubMed] [Google Scholar]
- Wang Z, Ying Z, Bosy-Westphal A, Zhang J, Schautz B, Later W, Heymsfield SB, Muller MJ. Specific metabolic rates of major organs and tissues across adulthood: evaluation by mechanistic model of resting energy expenditure. Am J Clin Nutr. 2010;92:1369–1377. doi: 10.3945/ajcn.2010.29885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu N, Zhang SO, Cole RA, McKinney SA, Guo F, Haas JT, Bobba S, Farese RV, Jr, Mak HY. The FATP1-DGAT2 complex facilitates lipid droplet expansion at the ER-lipid droplet interface. J Cell Biol. 2012;198:895–911. doi: 10.1083/jcb.201201139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S, Jaiswal M, Charng WL, Gambin T, Karaca E, Mirzaa G, Wiszniewski W, Sandoval H, Haelterman NA, Xiong B, et al. A drosophila genetic resource of mutants to study mechanisms underlying human genetic diseases. Cell. 2014;159:200–214. doi: 10.1016/j.cell.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Vidensky S, Jin L, Jie C, Lorenzini I, Frankl M, Rothstein JD. Molecular comparison of GLT1+ and ALDH1L1+ astrocytes in vivo in astroglial reporter mice. Glia. 2011;59:200–207. doi: 10.1002/glia.21089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JT, Tan L, Hardy J. Apolipoprotein E in Alzheimer’s disease: an update. Annu Rev Neurosci. 2014;37:79–100. doi: 10.1146/annurev-neuro-071013-014300. [DOI] [PubMed] [Google Scholar]
- Zanotti I, Pedrelli M, Poti F, Stomeo G, Gomaraschi M, Calabresi L, Bernini F. Macrophage, but not systemic, apolipoprotein E is necessary for macrophage reverse cholesterol transport in vivo. Arterioscler Thromb Vasc Biol. 2011;31:74–80. doi: 10.1161/ATVBAHA.110.213892. [DOI] [PubMed] [Google Scholar]
- Zhang K, Li Z, Jaiswal M, Bayat V, Xiong B, Sandoval H, Charng WL, David G, Haueter C, Yamamoto S, et al. The C8ORF38 homologue Sicily is a cytosolic chaperone for a mitochondrial complex I subunit. J Cell Biol. 2013;200:807–820. doi: 10.1083/jcb.201208033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34:11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielke HR, Zielke CL, Baab PJ. Direct measurement of oxidative metabolism in the living brain by microdialysis: a review. J Neurochem. 2009;109(Suppl 1):24–29. doi: 10.1111/j.1471-4159.2009.05941.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.