

Abstract
Genetic perturbations of the transcription factor Forkhead Box P1 (FOXP1) are causative for severe forms of autism spectrum disorder that are often comorbid with intellectual disability. Recent work has begun to reveal an important role for FoxP1 in brain development, but the brain-region-specific contributions of Foxp1 to autism and intellectual disability phenotypes have yet to be determined fully. Here, we describe Foxp1 conditional knock-out (Foxp1cKO) male and female mice with loss of Foxp1 in the pyramidal neurons of the neocortex and the CA1/CA2 subfields of the hippocampus. Foxp1cKO mice exhibit behavioral phenotypes that are of potential relevance to autism spectrum disorder, including hyperactivity, increased anxiety, communication impairments, and decreased sociability. In addition, Foxp1cKO mice have gross deficits in learning and memory tasks of relevance to intellectual disability. Using a genome-wide approach, we identified differentially expressed genes in the hippocampus of Foxp1cKO mice associated with synaptic function and development. Furthermore, using magnetic resonance imaging, we uncovered a significant reduction in the volumes of both the entire hippocampus as well as individual hippocampal subfields of Foxp1cKO mice. Finally, we observed reduced maintenance of LTP in area CA1 of the hippocampus in these mutant mice. Together, these data suggest that proper expression of Foxp1 in the pyramidal neurons of the forebrain is important for regulating gene expression pathways that contribute to specific behaviors reminiscent of those seen in autism and intellectual disability. In particular, Foxp1 regulation of gene expression appears to be crucial for normal hippocampal development, CA1 plasticity, and spatial learning.
SIGNIFICANCE STATEMENT Loss-of-function mutations in the transcription factor Forkhead Box P1 (FOXP1) lead to autism spectrum disorder and intellectual disability. Understanding the potential brain-region-specific contributions of FOXP1 to disease-relevant phenotypes could be a critical first step in the management of patients with these mutations. Here, we report that Foxp1 conditional knock-out (Foxp1cKO) mice with loss of Foxp1 in the neocortex and hippocampus display autism and intellectual-disability-relevant behaviors. We also show that these phenotypes correlate with changes in both the genomic and physiological profiles of the hippocampus in Foxp1cKO mice. Our work demonstrates that brain-region-specific FOXP1 expression may relate to distinct, clinically relevant phenotypes.
Keywords: autism, gene expression, hippocampus, spatial learning, synaptic plasticity
Introduction
Autism spectrum disorder (ASD) is often comorbid with intellectual disability (ID) (Geschwind and State, 2015; de la Torre-Ubieta et al., 2016). Delineating the brain circuits that contribute to distinct ASD and ID symptoms could be a major first step toward improved treatments for these disorders. ASD has a strong genetic component (Geschwind and State, 2015; de la Torre-Ubieta et al., 2016) that is shared in part with ID (Vissers et al., 2016). Numerous transcription factors coordinate the expression patterns of ASD and ID risk genes during early brain development (State and Šestan, 2012; de la Torre-Ubieta et al., 2016). Therefore, investigating the role of ASD- and ID-relevant transcription factors in a brain-region-specific manner could reveal circuit-based pathways that contribute to discrete behavioral phenotypes in the two disorders.
Heterozygous mutations and deletions in the transcription factor Forkhead Box P1 (FOXP1) are causative for ASD and ID (Bacon and Rappold, 2012; Le Fevre et al., 2013; Lozano et al., 2015; Vissers et al., 2016). In addition, FOXP1 has been classified as a high-confidence ASD and ID risk gene in several recent, large-scale sequencing studies (Iossifov et al., 2014; Sanders et al., 2015; Stessman et al., 2017). FOXP1 is a member of the Forkhead Box (FOX) transcription factor family, which is denoted by a unique nomenclature (uppercase for primates, title case for rodents, and mixed case for all other species or collection of species; Kaestner et al., 2000).
Within the forebrain, Foxp1 expression is largely restricted to the pyramidal neurons of the neocortex and the CA1/CA2 hippocampal subfields, as well the medium spiny neurons of the striatum (Ferland et al., 2003; Tamura et al., 2004; Hisaoka et al., 2010). Conditional, full-brain loss of Foxp1 results in altered social behaviors, impaired learning and memory, and developmental aberrations in the striatum (Bacon et al., 2015). Neocortical knock-down of Foxp1 disrupts neuronal migration and axon formation, but the resultant behavioral phenotypes have not been assessed (Li et al., 2015). Finally, using a patient-relevant heterozygous Foxp1 knock-out mouse, we have shown that Foxp1 regulates distinct, regional transcriptional profiles in the brain and that loss of Foxp1 expression in the striatum strongly correlates with deficits in ultrasonic vocalization production (Araujo et al., 2015). Although these studies have begun to shed light on the function of Foxp1 within the brain, the specific contributions of neocortical and hippocampal Foxp1 to the ASD and ID phenotypes remain to be determined. Addressing this question is important, given that many changes to the anatomy, transcriptional profiles, and physiology of the neocortex and hippocampus are observed in ASD and ID brains (Chen et al., 2015).
To investigate the regional contributions of Foxp1 expression to ASD- and ID-relevant phenotypes, we used an Emx1.Cre mouse line (Gorski et al., 2002) to generate a Foxp1 conditional knock-out (Foxp1cKO) mouse with loss of Foxp1 in the pyramidal neurons of the neocortex and the hippocampus. In this study, we integrate behavioral profiling, electrophysiology, magnetic resonance imaging (MRI) analyses, and genomics. We show that loss of Foxp1 in the neocortex and hippocampus is sufficient to produce ASD- and ID-relevant behaviors such as hyperactivity, communication deficits, decreased sociability, and impaired spatial learning and memory. In addition, we show that the ID-like learning and memory deficits observed in Foxp1cKO mice are likely due to alterations in hippocampal function because behaviors involving broader cortical circuits are unaffected. Using RNA-sequencing (RNA-seq), we correlate these behavioral phenotypes to specific changes in the transcriptome of the Foxp1cKO hippocampus. We also use MRI to demonstrate that loss of forebrain Foxp1 expression leads to decreased hippocampal volumes. Finally, based on the genomic and morphological data, we assayed the electrophysiological properties of the Foxp1cKO hippocampus and found reduced CA1-dependent LTP maintenance. As a whole, our data suggest that certain behavioral consequences of Foxp1 loss can be attributed to disrupted gene networks within distinct regions of the brain. Therefore, these data could lead to improved treatments for specific ASD and ID symptoms.
Materials and Methods
Mice.
Homozygous-floxed Foxp1 (Foxp1flox/flox) mice (Feng et al., 2010) were backcrossed with C57BL/6J mice for at least 10 generations to obtain congenic animals. Mice hemizygous for Cre recombinase expression under the control of the Emx1 promoter (Emx1.Cre+/− mice) (Gorski et al., 2002) were purchased from The Jackson Laboratory (strain #005628) and are congenic for the C57BL/6J background. Experimental animals were generated by crossing male Emx1.Cre+/−;Foxp1flox/flox mice with female Foxp1flox/flox mice. Mice were kept in the animal facilities of the University of Texas (UT) Southwestern Medical Center under a 12 h light/dark cycle and all behavioral testing occurred during the light cycle with the experimenter blinded to genotype. Unless otherwise specified, all mice were given ad libitum access to food and water. All mouse studies were approved by the UT Southwestern Institutional Animal Care and Use Committee.
Immunoblotting.
Regional brain lysates from adult (>8 weeks old) male mice were prepared and used in immunoblotting experiments as described previously (Araujo et al., 2015).
Tissue preparation.
For immunohistochemistry, adult female and male mice were anesthetized with 80–100 mg/kg Euthasol (UT Southwestern Medical Center Animal Resources Center Veterinary Drug Services), perfused with PBS containing 10 U/ml heparin (Sigma-Aldrich) followed by fixative (4% PFA in PBS), and then immediately decapitated. Whole brains were removed and incubated in fixative for 24 h at 4°C and then incubated in 30% sucrose (made in PBS with 0.02% sodium azide) for 24–48 h at 4°C. Afterward, brains were sectioned at 35–40 μm on an SM2000 R sliding microtome (Leica). Sections were then stored in PBS containing 0.02% sodium azide until being used in immunohistochemistry.
For MRI, adult male mice were anesthetized with 80–100 mg/kg Euthasol, perfused with PBS (without Ca2+ and Mg2+) containing 10 U/ml heparin and 2 mm ProHance (Bracco Diagnostics), and then fixative (4% PFA in PBS without Ca2+ and Mg2+) containing 2 mm ProHance. Animals were then immediately decapitated and the cartilaginous nose tip, eyes, skin, lower jar, ears, and zygomatic bones were removed. Brains (encased in the remaining skull structures) were then incubated in fixative containing 2 mm ProHance and 0.02% sodium azide for 24 h at 4°C and subsequently transferred to PBS (without Ca2+ and Mg2+) containing 2 mm ProHance and 0.02% sodium azide for storage at 4°C.
Immunohistochemistry.
Floating immunohistochemistry was performed according to standard procedures. Briefly, at room temperature, sections were first washed with TBS. Next, they were incubated with 3% hydrogen peroxide for 30 min, washed with TBS, treated with 0.3 m glycine for 30 min, and washed again with TBS. Sections were then incubated with primary antibodies diluted in TBS-T (0.4% Triton X-100) containing 1% bovine serum albumin (BSA) and 3% normal donkey serum (NDS) overnight at 4°C. Next, sections were washed in TBS-T, incubated with secondary antibodies diluted in TBS-T containing 1% BSA and 3% NDS for 1 h at room temperature, and then mounted onto microscope slides. Finally, slides were washed with TBS-T and allowed to dry overnight at room temperature, mounted with ProLong Diamond Antifade Mountant with DAPI (Thermo Fisher Scientific) and a coverslip, and allowed to set overnight at room temperature. Slides were imaged using an LSM 710 Confocal Microscope (Zeiss) connected to a computer running Zen 2012 Software (Zeiss).
Magnetic resonance imaging.
A multichannel 7.0 tesla MRI scanner (Agilent Technologies) was used to image the brains within skulls. Sixteen custom-built solenoid coils were used to image the brains in parallel (Bock et al., 2005; Lerch et al., 2011). A T2-weighted 3D fast spin echo (FSE) sequence was used for the acquisition of the anatomical images. Parameters for the FSE sequence were as follows: TR of 350 ms and TEs of 12 ms per echo for 6 echoes, 2 averages, field of view of 20 × 20 × 25 mm3, and a matrix size of 504 × 504 × 630, giving an image with 40 μm isotropic resolution. K-space was acquired with a cylindrical acquisition (Nieman et al., 2005). Total imaging time was 14 h. To visualize and compare any changes in the mouse brains, the images were linearly (six parameter followed a 12 parameter) and nonlinearly registered together. All scans were then resampled with the appropriate transform and averaged to create a population atlas representing the average anatomy of the study sample. The result of the registration was to have all scans deformed into alignment with each other in an unbiased fashion. This allows for the analysis of the deformations needed to take each individual mouse's anatomy into this final atlas space, the goal being to model how the deformation fields relate to genotype (Nieman et al., 2006; Lerch et al., 2008). The Jacobian determinants of the deformation fields are then calculated as measures of volume at each voxel. Significant volume changes can then be calculated by warping a preexisting classified MRI atlas onto the population atlas, which allows for the volume of 159 segmented structures encompassing cortical lobes, large white matter structures (i.e., corpus callosum), ventricles, cerebellum, brainstem, and olfactory bulbs to be assessed in all brains. This atlas is a combination of three separate atlases (Dorr et al., 2008; Ullmann et al., 2013; Steadman et al., 2014). In addition to the regional assessment, these images can be examined on a voxelwise basis to localize the differences found within regions or across the brain. Multiple comparisons in this study were controlled for using the false discovery rate (FDR) (Genovese et al., 2002).
Antibodies.
The following antibodies were used for immunoblotting (IB) or immunohistochemistry (IHC): anti-Foxp1 (Spiteri et al., 2007; rabbit; 1:5000 for IB, 1:1000 for IHC) and anti-GAPDH (mouse, Millipore; 1:5000 for IB).
RNA processing.
RNA purification was performed on tissues dissected from postnatal day 47 Foxp1cKO mice and littermate controls as described previously (Araujo et al., 2015).
qPCR.
qPCR was performed as described previously (Araujo et al., 2015). All primer sequences are available upon request.
RNA-seq library preparation.
RNA-seq library preparation was performed according to previously published methods (Takahashi et al., 2015). Briefly, mRNA was isolated from 2 μg of total RNA (at RIN values ≥8.9) harvested from adult tissues via poly(A) selection. Fifteen PCR cycles were used for cDNA amplification. Pooled libraries, each at a final concentration of 2 nm, were sequenced on a NextSeq 500 Sequencer (Illumina) by the McDermott Sequencing Core at UT Southwestern Medical Center to generate single-end 75 bp reads.
RNA-seq data analysis.
Raw reads were first filtered for phred quality and adapters using FASTQC (Andrews, 2010) and Trimmomatic (Bolger et al., 2014). Filtered reads were then aligned to the reference mouse genome mm10 (https://genome.ucsc.edu) using STAR (Dobin et al., 2013) aligner. Uniquely mapped reads were used to obtain the gene counts using HTSeq package (Anders et al., 2015) and the read counts were normalized using the CPM (counts per million) method implemented in the edgeR package (Robinson et al., 2010; McCarthy et al., 2012). For further analysis, we performed a sample-specific CPM filtering, considering genes with CPM values of 1.0 in all replicates for treatments or controls. DESeq (Anders and Huber, 2010; Love et al., 2014) was used to detect the differentially expressed genes (DEGs). We applied a filter of an adjusted p-value of ≤0.005 and absolute log fold change of ≥0.3 to identify DEGs.
DEG gene ontology (GO) analysis.
GO analysis of the significant DEGs was performed using ToppGene (https://toppgene.cchmc.org) and GO terms were reduced using REVIGO (Supek et al., 2011). GO categories were considered significant if they contained at least three genes and if they had a Benjamini–Hochberg (Benjamini–Hochberg)-corrected p-value and q-value of ≤0.05.
Weighted gene coexpression network analysis.
Weighted gene coexpression network analysis (WGCNA) was performed on 16 total RNA-seq samples (8 neocortical samples: 4 control, 4 Foxp1cKO and 8 hippocampal samples: 4 control, 4 Foxp1cKO). The R package for WGCNA (Langfelder and Horvath, 2008) was used to build gene coexpression network using filtered CPM data (CPM > = 1 across all replicates of a condition). A signed network was constructed using blockwiseModules function of the WGCNA R package. A value of 10 was chosen as Beta with highest scale-free R square (R2 = 0.8). For other parameters, we used corType = pearson, maxBlockSize = 15000, reassignThreshold = 1 × 10−6, mergeCutHeight = 0.1, deepSplit = 4, and detectCutHeight = 0.999. Visualizations of network plots were created using Cytoscape v3.4.0 (Shannon et al., 2003), representing the top 500 edges based on ranked weights.
Hippocampal electrophysiology.
An experimenter blinded to genotype performed all electrophysiology studies. Juvenile (6–7 weeks old) male mice were anesthetized briefly with isoflurane (Baxter Healthcare) and rapidly decapitated to remove the brain, which was then submerged in ice-cold ACSF containing the following (in mm): 75 sucrose, 87 NaCl, 3 KCl, 1.25 NaH2PO4, 7 MgSO4, 26 NaHCO3, 20 dextrose, and 0.5 CaCl2. Acute coronal hippocampal slices were cut 350 μm thick using a VT 1000 S vibrating microtome (Leica). To reduce recurrent excitation of CA3 neurons, a cut was made between CA3 and CA1. Slices were allowed to recover at 34°C for 15 min in normal ACSF containing the following (in mm): 124 NaCl, 5 KCl, 1.25 NaH2PO4, 2 MgCl2, 26 NaHCO3, 10 dextrose, and 1 CaCl2. Recovery continued for 45 min as slices were gradually cooled to room temperature for holding before recording. All solutions were pH 7.4 and saturated with 95% O2/5% CO2.
All recordings were performed at 33°C ± 0.5°C in ACSF containing the following (in mm): 124 NaCl, 5 KCl, 1.25 NaH2PO4, 1 MgCl2, 26 NaHCO3, 10 dextrose, and 2 CaCl2 saturated with 95% O2/5% CO2 and all data were collected using Clampex (pClamp Software Suite 10.2; Molecular Devices). Recordings were filtered at 1 kHz and digitized at 10 kHz. CA3–CA1 synapses were stimulated by a 100 μs biphasic pulse through custom-made nickel dichromate electrodes (A-M Systems) placed 400–500 μm laterally from the recording electrode and kept constant within this range for all experiments. Stimulation was controlled using a model 2200 stimulus isolator (A-M Systems). The recording electrode (1–3 MΩ) was filled with normal ACSF and placed in the stratum radiatum using a SZX7 dissecting microscope (Olympus) at 35× magnification. Sample size for all extracellular field recordings represents number of slices tested with two to four slices used per mouse. Response size was determined by fitting a straight line to the initial slope (10–40%) of the field EPSP (fEPSP) using automated analysis in Clampfit (pClamp Software Suite 10.2; Molecular Devices). For LTP, the stimulus intensity was set to generate ∼50% of the maximum fEPSP, as determined by the input/output curve. LTP was induced with 2 trains of 100 Hz stimulation for 1 s separated by 1 min. A 20 min baseline was recorded before LTP induction and followed by at least 60 min of 0.05 Hz stimulation (every 20 s).
Novel cage activity test.
Adult male and female mice were moved individually from their home cages into clean, plastic (18 × 28 cm) cages with minimal bedding. Each cage was then placed into a dark Plexiglas box. Movement was measured with Photobeam Activity System-Home Cage software (San Diego Instruments) for 2 h for each mouse. The number of beam breaks was recorded every 5 min.
Open-field test.
The open-field assay was performed on adult male and female mice as described previously (Araujo et al., 2015).
Ultrasonic vocalizations.
Ultrasonic vocalizations produced by adult male mice were assessed in a mating paradigm modified from previously published methods (Holy and Guo, 2005). Briefly, male mice were singly paired with age-matched C57BL/6J female mice for 1 week. Afterward, female mice were removed from the cages and the males were housed singly for 1 week. The next day (the test day), the male mice were allowed to habituate to the testing environment for 15 min. During habituation, food hoppers were removed and the cage lids were replaced with Styrofoam lids containing UltraSoundGate condenser microphones (Avisoft Bioacoustics) positioned at a fixed height. The condenser microphones were connected to UltraSoundGate416H hardware (Avisoft Bioacoustics) hooked up to a computer running RECORDER software (Avisoft Bioacoustics). Next, habituated, age-matched C57BL/6J female mice were placed randomly into a cage containing a male. The resultant male songs were recorded for 3 min. No female was used in >2 recording sessions per day. Analyses of ultrasonic vocalization features were performed as described previously (Araujo et al., 2015). Call duration reflects the average length of calls (in milliseconds), mean frequency denotes the average frequency of calls (in kilohertz), frequency range is the average difference between the maximum and minimum frequency at which calls are produced (in kilohertz), the fraction of calls with frequency jumps represents the ratio of calls with and without frequency breaks, and the average slope of the call reflects modulation of call frequency over time (in hertz/milliseconds).
Social interaction test.
Adult female and male mice were placed individually in an open-field environment (44 × 44 cm with walls 30 cm high) in a dimly lit room and allowed to explore for 5 min. Inside the open-field arena, a small plastic chamber (the interaction box, 8.5 × 4.5 cm) was placed along one wall of the arena. After 5 min, the test mouse was removed and a novel, unfamiliar mouse (same sex and strain as the test mouse) was placed into the interaction box. Small holes in the interaction box allow the mice to see, hear, and smell each other. The test mouse was returned to the open-field environment and allowed to explore for another 5 min. The test mouse was monitored from above by a video camera connected to a computer running Ethovision 3.0 (Noldus). Both the amount of time the test mouse spent in the interaction zone immediately adjacent to the interaction chamber (within 8 cm) and the time spent in the four corners of the arena (9 × 9 cm each) were recorded.
Nesting behavior.
Nesting behavior was gauged in adult male and female mice as described previously (Araujo et al., 2015).
Fear conditioning.
Fear conditioning was conducted on adult female and male animals using boxes containing a grid metal floor attached to a scrambled shock generator. Mice were individually trained by placing them into the box for 2 min and giving them 3 separate tone–shock pairings (30 s white noise, followed by a 2 s, 0.5 mA shock, with 1 min intervals between pairings). Context recall was assessed 24 h later by placing the mice back into the original box and recording freezing for 5 min. Cue recall was assessed 2 d after training by placing the mice in boxes that were altered with a plastic floor, an inverted roof, and a vanilla scent. Freezing was then measured for 3 min, followed by the presentation of the white noise cue and measuring freezing for an additional 3 min with Video Freeze software (Med Associates).
Morris water maze.
The Morris water maze was conducted on adult male mice using a 1.2-m-diameter circular pool filled with opaque, tempera-paint-dyed 23°C water. Stark visual cues were placed around the room containing the pool. Within the pool, a 10 cm circular Plexiglas escape platform was submerged in one of the quadrants 1 cm below the water's surface. For training, mice were placed into the pool in one of four starting locations (north, south, east, or west, with the order determined randomly) and allowed to swim until they located the escape platform. Upon finding the platform, mice were permitted to rest for 5 s before being removed. If they did not locate the platform within 60 s, mice were guided manually to the platform and given 5 s of rest before being removed. Mice received four training trials per day for 10 d and were placed in temporary cages between the training trials of a particular day. On day 12, after a day of rest on day 11, a probe test was performed in which mice were allowed to swim in the pool for 60 s, with the escape platform removed. The movements of the animals were recorded by a video camera centered above the pool and tracked using ANY-Maze software (Stoelting). The latency to reach the escape platform was quantified for each mouse during the 10 d training period. For the probe test, the number of original platform crosses was analyzed for each mouse.
T-maze.
The T-maze was constructed from gray polyvinyl, with the main array 40 cm long, the side arms 33 cm long, the walls 18.5 cm high, and the alleys 10 cm wide (Actimetrics). Before commencing any testing, adult female and male mice were subjected to food restriction until their body weight reached 85% of baseline. Then, for habituation to the T-maze, each mouse was placed into the apparatus with all of the doors open and a food pellet (20 mg; Bio-Serv) placed in a cup at the end of each arm. This was performed in 4 sessions, each 10 min long, for 2 d. After this habituation period, each mouse was used in 10 test trials per day for 10 consecutive days. For the testing phase, each trial was composed of two different runs: the sample run and the test run. In the sample run, the pellet reward was placed randomly at one end of the arms and the other arm was closed off. A mouse was then placed at the start position and allowed to travel freely to the end of the arm and consume the pellet. Immediately after consuming the pellet, the mouse was placed back in the start position, which was closed off. While the mouse was isolated in the start position, the blocked arm was opened and the entire apparatus was wiped with 10% ethanol to remove olfactory cues. A food pellet was then placed in the arm opposite from the one containing the reward in the sample run. After a 30 s delay, the test run was started. In the test run, each mouse was allowed to travel to either arm. If the mouse chose the opposite arm that was rewarded in the sample run (a correct choice), then it was allowed to consume the pellet. If the mouse chose the same arm as in the sample run (an incorrect choice), then it was blocked in the arm for 30 s as punishment. Spatial working memory was evaluated by the average success rate for each day.
Set-shifting task.
With minor modifications, the set-shifting test was conducted as described previously (Cho et al., 2015). Before testing began, adult male and female mice were subjected to food restriction until their body weight reached 85% of baseline. Afterward, animals were housed individually and then presented with two bowls in their home cage until they began digging in one bowl. Each of these bowls contained a different odor and a different digging medium and the odor–medium combinations were altered and counterbalanced from trial to trial. The digging media were white calcium sand and pine wood shavings mixed with an odorant (ground garlic or clove 0.01% by volume) and peanut butter chip powder (0.1% by volume). The reward, a 5–10 mg piece of a peanut butter chip, was buried in the medium in both of the food bowls.
Testing began once mice reached their target weight. The testing procedure consisted of three phases (training, initial association, and rule-shift), with each phase lasting 1 d for a total of 3 consecutive days. On day 1 (the training day), mice were given 10 consecutive trials in which they were allowed to dig among two bowls containing two different mediums to learn that they could reliably find a food reward in only one of the bowls. On day 2 (the initial association day), mice learned that a specific cue (an odor or medium) predicted the presence of food reward by being presented with different odor–medium combinations (which were switched and counterbalanced) during each trial. This predictive cue remained constant over the whole day, with each mouse being randomly assigned their own cue. On day 3 (the rule-shift day), the dimension (odor or medium) signaling the reward was changed. If the initial association paired a specific odor with the food reward, then the rule-shift phase paired a certain digging medium with reward and the mice needed to learn this new rule to obtain a reward.
In the initial association and the rule-shift phases, the mice were considered to have learned the association between stimulus and reward if they made 10 consecutive correct choices. The phase ended when they met this criterion. When the mice made a correct choice on a trial, they were allowed to consume the food reward before the next trial. Between trials, mice were transferred from their home cage to a holding cage while new bowls were set up. After making an error on a trial, the mice were transferred to the holding cage for 1 min as punishment. In addition, in the rule-shift phase, two types of error were analyzed: perseverative errors, when a choice was consistent with the rule of the initial association phase, and random errors, when a choice was inconsistent with both the rules of the initial association and the rule-shift phases.
Experimental design and statistical analysis.
All experiments reported in this study were designed to examine genotype-based effects between Foxp1cKO and littermate control mice. Effects on Foxp1 protein levels in the brain (see Fig. 1A–C) were examined using adult (≥8 weeks old) male mice (3 mice/genotype). Effects on activity in a novel cage activity test (see Fig. 2A,B) were tested using adult female and male mice (7–9 mice/genotype). Effects on activity and anxiety in an open-field assay (see Fig. 2C,D) were examined in adult male and female mice (8 mice/genotype). Adult male mice (12–15 mice/genotype) were used to test genotype-based effects on ultrasonic vocalization (USV or song) production (see Fig. 3A–F). Genotype effects on nest building (see Fig. 3G,H) were assessed in adult male and female mice (7–8 mice/genotype). Effects on sociability (see Fig. 3I) were determined using adult female and male mice (12–16 mice/genotype). Adult male mice (10–12 mice/genotype) were used to evaluate effects on performance in the Morris water maze (see Fig. 4A–C). Effects on performance in an alternating T-maze (see Fig. 4D–F) were tested using adult male and female mice (10 mice/genotype). Effects on performance in a fear-conditioning test (see Fig. 5A,B) were determined using adult female and male mice (12–16 mice/genotype). Adult male and female mice (9–10 mice/genotype) were used to examine effects on performance in a set-shifting task (see Fig. 5C,D). Adult male mice (10 mice/genotype) were used to determine the effects of genotype of relative regional brain volumes (see Fig. 6A,B). Effects on hippocampal electrophysiological properties (see Fig. 8A–C) were determined using juvenile (6–7 weeks old) male mice (15–20 recordings/genotype). Effects on gene expression via qPCR (see Figs. 7F, 8E) were tested using adult male mice (3/genotype). Except as noted for genomic analyses, Student's t tests (2-tailed, type 2) were performed for analyses of data in Figures 1B; 2, B–D; 3, A–E, F, H, and I; 4C, D, and F; 5B; 6B; 7F; and 8B and E and two-way ANOVAs (all with a Sidak's multiple-comparisons test) were performed for analyses of data in Figures 2A; 4, A and E; 4A, C, and D; and 5C. FDR was used for Figure 6B. p-values were calculated using Prism 7 software (GraphPad) and significance was assigned to values ≤0.05. More detailed statistical information can be found in the Results section for each figure.
Figure 1.

Generation of Foxp1cKO mice. A, Representative immunoblot displaying reduced Foxp1 protein levels in the CTX and HIP, but not the STR, of Foxp1cKO mice compared with littermate control mice. GAPDH was used as a loading control. B, Quantification of Foxp1 expression in adult Foxp1cKO mouse brains. Data are represented as means ± SEM. n = 3 control mice; n = 3 Foxp1cKO mice. **p = 0.002; ***p = 0.0002, Student's t test, compared with control levels normalized to GAPDH. C, Representative immunohistochemistry images showing that Foxp1 protein (green) expression is ablated in the projection neurons of the CTX and the CA1/CA2 subfields of the HIP, but preserved in the STR of Foxp1cKO mice. cc, Corpus callosum; DG, dentate gyrus; CPu, caudate/putamen. Scale bars indicate 100 μm.
Figure 2.

Foxp1cKO mice display hyperactivity and anxiety-like behaviors. A, B, Foxp1cKO mice are hyperactive, as indicated by their increased activity in a novel cage environment. A, Foxp1cKO mice display sustained, increased activity in a novel cage. Data are represented as means ± SEM) n = 9 control mice; n = 7 Foxp1cKO mice. p < 0.0001, two-way ANOVA, compared between genotypes. B, As measured by their average activity over the course of 2 h, Foxp1cKO mice are hyperactive. Data are represented as means ± SEM. n = 9 control mice; n = 7 Foxp1cKO mice. **p = 0.002, Student's t test, compared between genotypes. C, Foxp1cKO mice are hyperactive, as determined by their total distance moved in the open field test. Data are represented as means ± SEM. n = 8 control mice; n = 8 Foxp1cKO mice. *p = 0.02, Student's t test, compared between genotypes. D, Foxp1cKO mice are anxious, as determined by the amount of time they spend in the border of the open-field apparatus. Data are represented as means ± SEM. n = 8 control mice; n = 8 Foxp1cKO mice. *p = 0.02, Student's t test, compared between genotypes.
Figure 3.

Impaired social communication in Foxp1cKO mice. A, Adult Foxp1cKO male mice produce fewer total numbers of USVs in a mating paradigm. Data are represented as means ± SEM. n = 13 control mice; n = 15 Foxp1cKO mice. **p = 0.0019, Student's t test, compared between genotypes. B, Foxp1cKO mice exhibit a significant reduction in their mean call duration. Data are represented as means ± SEM. n = 13 control mice; n = 14 Foxp1cKO mice. *p = 0.011, Student's t test, compared between genotypes. C, Foxp1cKO mice show no differences in their mean call frequencies. Data are represented as means ± SEM. n = 13 control mice; n = 14 Foxp1cKO mice. p = 0.45, Student's t test, compared between genotypes. D, Adult Foxp1cKO male mice produce USVs with smaller frequency ranges. Data are represented as means ± SEM. n = 13 control mice; n = 14 Foxp1cKO mice. *p = 0.019, Student's t test, compared between genotypes. E, Foxp1cKO mice produce a smaller fraction of USVs with frequency jumps. Data are represented as means ± SEM. n = 13 control mice; n = 15 Foxp1cKO mice. *p = 0.025, Student's t test, compared between genotypes. F, Foxp1cKO mice show a significant difference in the average slope of their USVs. Data are represented as means ± SEM. n = 13 control mice; n = 14 Foxp1cKO mice. ***p = 0.0003, Student's t test, compared between genotypes. G, Representative photographs of the nests produced by littermate control and Foxp1cKO mice. H, Foxp1cKO mice produce nests with low-quality scores. Data are represented as means ± SEM. n = 8 control mice; n = 7 Foxp1cKO mice. ****p < 0.0001, Student's t test, compared between genotypes. I, Foxp1cKO mice are less social than their littermate controls, as determined by the decreased time they spend interacting with a sex-matched conspecific (time in interaction zone). Data are represented as means ± SEM. n = 16 control mice; n = 12 Foxp1cKO mice. **p < 0.01, ***p < 0.001, Student's t test, compared between genotypes.
Figure 4.

Foxp1cKO mice display impairments in spatial learning. A, Foxp1cKO mice display poor learning via their escape latency in the training phase of the Morris water maze (MWM). Data are represented as means ± SEM. n = 12 control mice; n = 10 Foxp1cKO mice. p < 0.0001, two-way ANOVA, compared between genotypes. B, C, Foxp1cKO show poor memory via the number of platform crosses that they make during the MWM spatial probe. B, Representative trace of swimming paths taken by Foxp1cKO and control littermate mice on a spatial probe day. Roman numerals designate different quadrants. The original location of the hidden platform is indicated by a circle in quadrant I. C, Quantification of the number of platform crosses made by Foxp1cKO and control mice on a spatial probe day. Data are represented as means ± SEM. n = 12 control mice; n = 10 Foxp1cKO mice. **p = 0.002, Student's t test, compared between genotypes. D, Foxp1cKO mice display no difference in their ability to locate a raised platform during a visual probe day in the MWM. Data are represented as means ± SEM. n = 12 control mice; n = 10 Foxp1cKO mice. p = 0.34, Student's t test, compared between genotypes. E, Foxp1cKO mice demonstrate poor learning and memory, as measured by their percentage of successful trials during training in the T-maze. Dashed line represents success based on chance. Data are represented as means ± SEM. n = 10 control mice; n = 9 Foxp1cKO mice. p < 0.0001, two-way ANOVA, compared between genotypes. F, As measured by their average performance during training, Foxp1cKO mice display impaired learning in the T-maze. Data are represented as means ± SEM. n = 10 control mice; n = 10 Foxp1cKO mice. ****p < 0.0001, Student's t test, compared between genotypes. The main effects for genotype and postnatal day and their interactions are presented in A and E.
Figure 5.

Foxp1cKO mice do not display deficits in generalized learning and memory. A, B, Foxp1cKO mice show no deficiencies in associative fear-memory tasks, as displayed by their performance in both the cue-dependent (A) and context-dependent (B) portions of a fear conditioning (FC) paradigm. Data are represented as means ± SEM. n = 16 control mice; n = 12 Foxp1cKO mice. p = 0.27, two-way ANOVA compared between genotypes (A), p = 0.12, Student's t test compared between genotypes (B). C, D, Foxp1cKO show no deficits in cognitive flexibility, as measured by the number of trials that they needed to reach criterion during both the initial association (IA) or the rule-shift (RS) portion of training for the set-shifting task (SST) (C) or the number of perseverative (Pers) or random (Rand) errors that they made in the RS portion of the SST (D). Data are represented as means ± SEM. n = 10 control; n = 9 Foxp1cKO mice. p = 0.65 (C) and p = 0.43 (D), two-way ANOVA, compared between genotypes.
Figure 6.
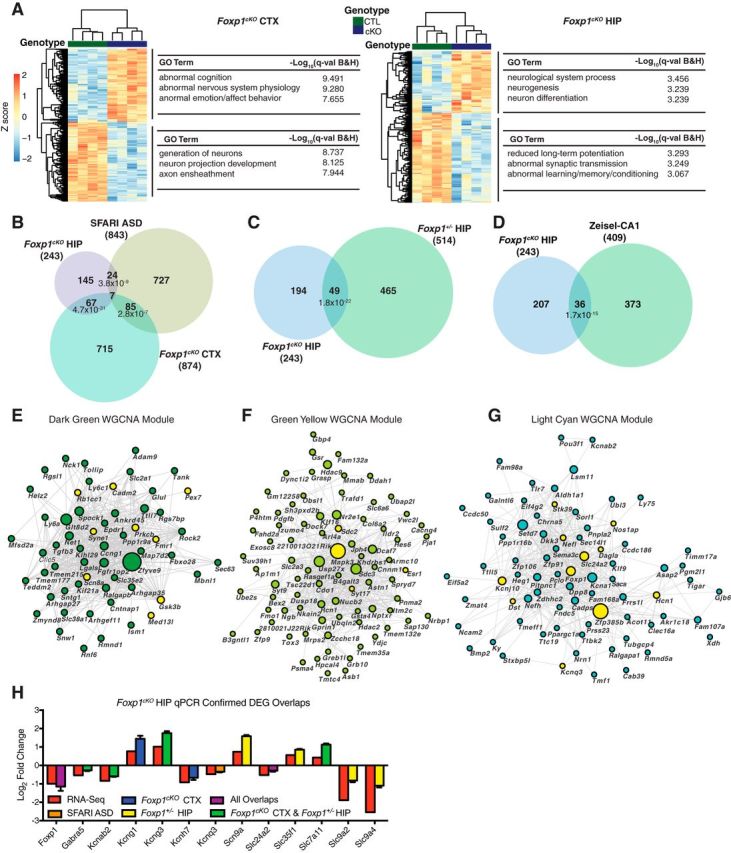
Altered transcriptional programs in Foxp1cKO brains. A, Heat map showing that, based on the DEGs of either region, the CTX or HIP, segregate by genotype: Foxp1cKO (cKO) and control (CTL). Color indicates a Z score from −2 to 2 for each gene. Significantly enriched GO terms (https://toppgene.cchmc.org; GO enrichment is the negative log of a Benjamini–Hochberg-corrected p-value, q-value) associated with groups of genes are highlighted next to their respective DEG clusters. Representative GO categories for both the Foxp1cKO neocortex and hippocampus can be found in Figure 6-1. B, Significant overlaps between DEGs in Foxp1cKO mouse CTX and HIP and ASD-associated genes (SFARI-ASD genes; https://sfari.org/resources/sfari-gene). A total of 92 genes overlapped between Foxp1cKO CTX and SFARI-ASD genes (p = 2.8 × 10−7; hypergeometric test) and 31 genes overlapped between Foxp1cKO HIP and SFARI-ASD genes (p = 3.8 × 10−9; hypergeometric test). C, Significant overlaps between Foxp1cKO HIP DEGs and heterozygous Foxp1 knock-out (Foxp1+/−) HIP DEGs (Araujo et al., 2015). The Foxp1+/− HIP dataset was filtered using the same cutoffs for identifying DEGs (an adjusted p-value of ≤0.005 and an absolute log fold change of ≥0.3) that were applied to the Foxp1cKO datasets. A total of 49 genes overlapped between Foxp1+/− HIP and Foxp1cKO HIP (p = 1.8 × 10−22; hypergeometric test). D, Significant overlaps between Foxp1cKO HIP DEGs and CA1 pyramidal neuron single-cell sequencing data (Zeisel et al., 2015). A total of 36 genes overlapped between Foxp1cKO HIP and Zeisel CA1 data (p = 1.7 × 10−15; hypergeometric test). The enrichment of DEGs in other single-cell categories are in Figure 6-2 (available at 10.1523/JNEUROSCI.1005-17.2017.f6-2). Visualization of the top 500 connections in the hippocampus-specific dark green module (E), the green yellow module (F), and the light cyan module (G). ASD-SFARI genes are highlighted in yellow. H, Confirmation of salient gene targets in independent Foxp1cKO hippocampal samples using qPCR. Red bars indicate RNA-seq-based log2-fold changes in expression. Colored bars represent the category of gene (SFARI-ASD and/or Foxp1cKO neocortex and/or Foxp1+/− hippocampus) that these Foxp1cKO hippocampal DEGs overlap with. Data are represented as means ± SEM. n = 3 control mice; n = 3 Foxp1cKO mice. All qPCR values are significant at p < 0.05 (Student's t test, compared with control levels, normalized to β-actin).
Figure 8.

Altered hippocampal synaptic plasticity in Foxp1cKO mice. A, B, In response to high-frequency stimulation (HFS), there is no difference in the initial magnitude of LTP in Foxp1cKO CA1 neurons (A), but there is a difference in the LTP response during the last 10 min of stimulation (B). Data are represented as means ± SEM. n = 15 control recordings; n = 20 Foxp1cKO recordings, Student's t test, compared between genotypes. C, Basal synaptic transmission is unchanged between Foxp1cKO and littermate control mice as measured by input/output curves comparing stimulus intensity to fEPSP slope in CA1 pyramidal neurons. Data are represented as means ± SEM. n = 15 control recordings; n = 19 Foxp1cKO recordings. p = 0.63, two-way ANOVA, compared between genotypes. D, Significant overlaps between Foxp1cKO HIP DEGs and LTP maintenance DEGs (Ryan et al., 2012). A total of 12 genes overlapped between the Foxp1cKO HIP and LTP maintenance datasets (p = 2.32 × 10−4; hypergeometric test). E, Confirmation of genes that overlapped between the Foxp1cKO hippocampal dataset and LTP maintenance genes in independent Foxp1cKO hippocampal samples using qPCR. Box insert highlights Foxp1. With the exception of Ccnd1, Dusp5, and Sorcs3, all qPCR values are significant at p < 0.05 (Student's t test, compared with control levels, normalized to β-actin). Data are represented as means ± SEM. n = 3 control mice; n = 3 Foxp1cKO mice.
Figure 7.
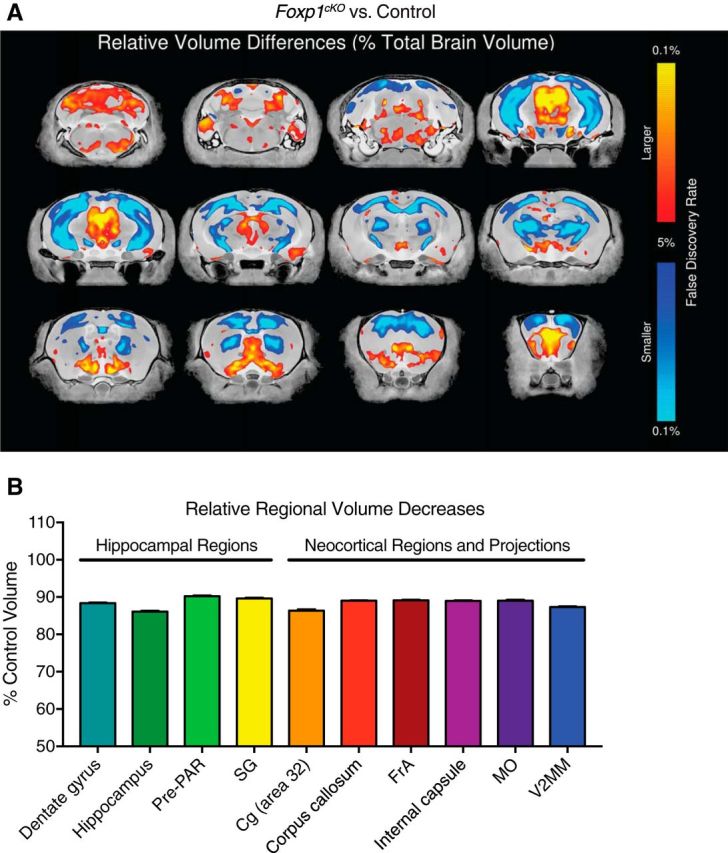
Altered regional brain volumes in Foxp1cKO mice. A, Fly-through of representative coronal slices of the Foxp1cKO brain highlighting average relative differences in regions with larger (red) or smaller (blue) volumes. B, Representation of the average relative volume decreases in several of the most significantly (in terms of percentage decreases from control 100% levels) affected hippocampal and neocortical areas in the Foxp1cKO mouse brain. Dashed line represents control levels. Data are represented as means ± SEM. All values are significant at p < 0.05, Student's t test, and FDR < 0.05. Cg, Cingulate cortex; FrA, frontal association cortex; MO, medial orbital cortex; Pre-PAR, pre-para subiculum; V2MM, secondary visual cortex mediomedial area. The top 20 increases and decreases in relative regional brain volumes (in terms of percentage difference from control volumes) are in Figure 7-1 (available at 10.1523/JNEUROSCI.1005-17.2017.f7-1).
Gene Expression Omnibus (GEO) accession information.
The National Center for Biotechnology Information GEO accession number for the RNA-seq data reported in this study is GSE97181.
Results
Foxp1 is expressed within the pyramidal neurons of the neocortex and the CA1/CA2 hippocampal subfields (Ferland et al., 2003; Hisaoka et al., 2010). However, the distinct ASD- and ID-related behavioral phenotypes governed by Foxp1 within these regions are undetermined. To examine the neocortical and hippocampal contributions of Foxp1 to ASD- and ID-relevant behaviors, we characterized Foxp1 conditional knock-out (Emx1.Cre+/−;Foxp1flox/flox) mice (hereafter called Foxp1cKO mice) compared with littermate control (Foxp1flox/flox) mice.
There are four murine isoforms of Foxp1 and two of them (Foxp1A and Foxp1D) are highly expressed within the mouse brain (Wang et al., 2003; Araujo et al., 2015). Consistent with the forebrain expression pattern of Cre recombinase under the Emx1 locus (Gorski et al., 2002), Foxp1cKO mice displayed near total loss of Foxp1 protein isoforms A and D in the neocortex (CTX) and hippocampus (HIP; Fig. 1A,B; Student's t test; CTX: F(2,2) = 4.21, **p = 0.002; HIP: F(2,2) = 9.59 ***p = 0.0002). As a negative control, we demonstrated that Foxp1 protein expression is preserved in the striatum (STR) of these animals (Fig. 1A,B; Student's t test, F(2,2) = 2.67, p = 0.7). Consistent with these results, we observed that Foxp1 expression is ablated in the projection neurons of the neocortex and the CA1/CA2 hippocampal subfields of Foxp1cKO mice (Fig. 1C). Because neuroglia and interneurons do not express Foxp1 (Hisaoka et al., 2010; Precious et al., 2016), we believe that this decrease in protein levels is due to a loss of Foxp1 specifically in forebrain pyramidal neurons. Adult Foxp1cKO mice are viable, superficially healthy, and exhibit no differences in body weight (Student's t test, F(14,14) = 2.45, p = 0.96, n = 15 mice/genotype). Therefore, these animals represent a model with which to examine the neocortical- and hippocampal-based contributions of Foxp1 to ASD- and ID-relevant phenotypes.
Symptoms common between ASD and ID include hyperactivity and anxiety (van Steensel et al., 2011; Ageranioti-Bélanger et al., 2012; Leitner, 2014; de la Torre-Ubieta et al., 2016). Therefore, we first assessed the baseline activity of adult (≥8 weeks old) Foxp1cKO mice. Foxp1cKO mice exhibit sustained, increased activity over a 2 h period in a novel cage (Fig. 2A; two-way ANOVA; genotype effect: F(1,336) = 163.6, ****p < 0.0001; time effect: F(23,336) = 22.44, p < 0.0001; interaction effect: F(23,336) = 2.2, p = 0.001; and Fig. 2B; Student's t test, F(6,8) = 3.6, **p = 0.002). In addition, in an open-field assay, Foxp1cKO mice cover more distance (Fig. 2C; Student's t test, F(7,7) = 2.62, *p = 0.02) and spend less time in the center of the arena, with a corresponding increase in time spent around the border of the arena (Fig. 2D; Student's t test; time in center: F(7,7) = 2.79, *p = 0.02; time in border: F(7,7) = 2.7, *p = 0.02). Together, these findings suggest a phenotype of hyperactivity and increased anxiety in Foxp1cKO mice.
Because decreased social communication is one of the core features of ASD (Fakhoury, 2015; Park et al., 2016; de la Torre-Ubieta et al., 2016), we examined both the social and communicative behaviors of Foxp1cKO mice. The USVs produced by adult male mice in response to the presence of female mice are a well characterized form of mouse communication (Holy and Guo, 2005; Portfors and Perkel, 2014). We therefore assayed the USVs, which are known as “songs,” produced by adult male Foxp1cKO mice during courtship encounters with age-matched females. Foxp1cKO mice produce fewer numbers of songs (Fig. 3A; Student's t test, F(14,12) = 1.12, **p = 0.002). In addition, the songs of Foxp1cKO mice were shorter (Fig. 3B; Student's t test, F(12,13) = 3.75, *p = 0.01), covered a smaller frequency range (Fig. 3D; Student's t test, F(12,13) = 1.94, *p = 0.02), and were less complex, as revealed by both a reduction in the number of songs with frequency jumps (Fig. 3E; Student's t test, F(12,14) = 1.73, *p = 0.02) and an alteration in the average call slope (Fig. 3F; Student's t test, F(13,12) = 1.72, ***p = 0.0003). Notably, the mean frequency of Foxp1cKO songs was not altered (Fig. 3C; Student's t test, F(13,12) = 1.52, p = 0.45). (See the Materials and Methods section for details on call parameters.) We then tested nest building in Foxp1cKO mice because it is an important behavior for communal animals such as rodents (Deacon, 2006; Silverman et al., 2010). Foxp1cKO animals consistently produced low-quality nests and, in most cases, never interacted with the provided nesting material (Fig. 3G,H; Student's t test, F(6,7) = 2.29, ****p < 0.0001). In a social interaction paradigm, Foxp1cKO mice exhibited a social retreat phenotype, as indicated by their decreased time in the interaction zone (Fig. 3I; Student's t test, F(15,11) = 1.91, **p < 0.01) and their increased time in the corners of the testing arena (Fig. 3I, Student's t test, F(15,11) = 2.68, ***p < 0.001). The increased preference for the corners of the sociability apparatus suggests that the decreased social interaction displayed by Foxp1cKO mice was not simply due to hyperactivity. Together, our results indicate that neocortical and hippocampal loss of Foxp1 protein expression is sufficient to lead to ASD-relevant deficits in communication and sociability.
Because both ASD patients and patients with FOXP1 haploinsufficiency frequently present with ID (Le Fevre et al., 2013; Geschwind and State, 2015; Lozano et al., 2015; de la Torre-Ubieta et al., 2016), we next examined the learning and memory capabilities of Foxp1cKO mice. Over the course of 10 d of training in the Morris water maze, Foxp1cKO mice never learn to find the submerged platform (Fig. 4A; two-way ANOVA; genotype effect: F(1,200) = 199.6, ****p < 0.0001; training day effect: F(9,200) = 6.98, p < 0.0001; interaction effect: F(9,200) = 3.25, p = 0.001), indicating poor spatial learning (Vorhees and Williams, 2006). In addition, on a probe day 48 h after the last day of training, Foxp1cKO mice made fewer numbers of platform crosses (Fig. 4B,C; Student's t test, F(11,9) = 2.78, **p = 0.002). This latter result was not unexpected, given that the Foxp1cKO mice showed no demonstrable learning. These deficits in the Morris water maze were not due to problems in visual acuity because Foxp1cKO mice were able to escape the maze just as quickly as control littermates on a visual probe day (Fig. 4D; Student's t test, F(11,9) = 3.88, p = 0.34). Moreover, these deficits were not due to changes in swim speeds because Foxp1cKO mice showed no differences in their average swimming velocity during training (two-way ANOVA, genotype effect: F(1,120) = 3.35 p = 0.07). In a T-maze, Foxp1cKO mice do no better than chance during the course of a 10-day training period (Fig. 4E; two-way ANOVA; genotype effect: F(1,170) = 123.6, ****p < 0.0001; training day effect: F(9,170) = 0.59, p = 0.81; interaction effect: F(9,170) = 1.11, p = 0.36; and Fig. 4F; Student's t test, F(8,9) = 1.3, ****p < 0.0001), implying deficits in spatial working memory in these animals (Shoji et al., 2012). Learning in the Morris water maze and the T-maze both rely on hippocampal function (Vorhees and Williams, 2006; Shoji et al., 2012; Yamamoto et al., 2014), so these results support altered hippocampal mechanisms in Foxp1cKO animals.
We next investigated whether the decreased performance of Foxp1cKO mice in learning and memory tasks is due to impairments in broad cortical circuits or if it is restricted to hippocampal-based spatial memory. We first used cue/contextual fear conditioning because it involves hippocampal, neocortical, and amygdala-based circuits in associative learning and memory (Puzzo et al., 2014; Tovote et al., 2015). We observed no differences in the fear response of Foxp1cKO mice as measured by their performance in a cue-dependent fear-learning paradigm (Fig. 5A; two-way ANOVA; genotype effect: F(1,52) = 1.23, p = 0.27; tone effect: F(1,52) = 242, p < 0.0001; interaction effect: F(1,52) = 0.44, p = 0.51). We also observed no differences in a context-dependent fear-learning paradigm (Fig. 5B; Student's t test, F(11,15) = 2.63, p = 0.12). Foxp1cKO mice were able to hear the conditioned stimulus (the tone before the shock) because they demonstrated no differences in freezing when presented with the stimulus during training (p = 0.77, two-way ANOVA). Foxp1cKO mice were also able to perceive the unconditioned stimulus (the shock itself) just as well as their littermate controls because they showed no difference in the stimulus strengths needed to induce jumping, flinching, or vocalizing (Student's t test; jumping: F(11,15) = 1.44, p = 0.53; flinching: F(11,15) = 1.51, p = 0.40; vocalizing: F(11,15) = 2.0, p = 0.45). Given that contextual fear conditioning is heavily dependent on hippocampal function (Puzzo et al., 2014; Tovote et al., 2015), the intact context-dependent fear conditioning that we observed in Foxp1cKO mice was surprising. This pointed to a specific deficit in complex hippocampal-based spatial tasks. To test this hypothesis, we examined the performance of Foxp1cKO animals in a set-shifting paradigm, a complex task that is largely reliant on prefrontal neocortical function (Cho et al., 2015; Heisler et al., 2015). We saw no differences in the behavior of Foxp1cKO mice compared with controls during the set-shifting task, as measured by the number of trials needed to reach criterion for training or the number of errors made during testing (Fig. 5C; two-way ANOVA; genotype effect: F(1,9) = 0.219, p = 0.65; test phase effect: F(1,9) = 14.74, p = 0.004; interaction effect: F(1,9) = 1.05, p = 0.33; and Fig. 5D; two-way ANOVA; genotype effect: F(1,18) = 0.65, p = 0.43; error type effect: F(1,18) = 33.51, p < 0.0001; interaction effect: F(1,18) = 0.11, p = 0.74). Together, these results indicate that the learning and memory deficits seen in Foxp1cKO mice were restricted to complex, spatial, hippocampal-based processes.
The behavioral deficits that we observed in Foxp1cKO mice are associated with many molecular mechanisms in the hippocampus (Lynch, 2004; Kumar, 2011). To determine the processes governing the behavioral phenotypes in Foxp1cKO mice, we ascertained transcriptional changes due to Foxp1 loss by carrying out RNA-seq on tissue samples from the neocortex and hippocampus of both Foxp1cKO mice and littermate controls. DEGs were identified by applying an adjusted p-value of ≤0.005 and an absolute log fold change of ≥0.3 (see “GEO accession information” section in Materials and Methods). By clustering the same number of top DEGs (based on fold change) in both the neocortex and hippocampus, we found transcriptional signatures (gene clusters) that differentiated the two brain regions by genotype (Fig. 6A). In addition, these upregulated and downregulated gene clusters are enriched for specific GO categories (https://toppgene.cchmc.org; Fig. 6A and Fig. 6-1). For the hippocampus, these GO categories include terms such as reduced LTP, abnormal synaptic transmission, and abnormal learning/memory/conditioning (Fig. 6A; Benjamini–Hochberg-corrected p-value, q-value; reduced LTP, q = 5.09 × 10−4; abnormal synaptic transmission, q = 5.64 × 10−4, abnormal learning/memory/conditioning, q = 8.57 × 10−4).
Representative Foxp1cKO neocortex and hippocampus DEG REVIGO GO categories. Using the REVIGO web tool (see Materials and Methods), we identified enrichment for gene ontology (GO) categories among the up-regulated and down-regulated differentially expressed genes (DEGs) in the Foxp1cKO neocortex and hippocampus. GO category enrichment was determined using a Benjamini and Hochberg corrected p-value, q-value, ≤ 0.05. Download Figure 6-1, XLSX file (11.8KB, xlsx)
Overlaps between Foxp1cKO DEGs and cell-type-specific genes. Foxp1cKO neocortical (CTX) DEGs and hippocampal (HIP) DEGs significantly overlap with cell-type-specific genes (Zeisel et al., 2015). The number of genes for each Foxp1cKO DEG category and overlap are reported. Download Figure 6-2, XLSX file (11.3KB, xlsx)
To characterize the relevance of Foxp1cKO hippocampal and neocortical DEGs with regard to ASD pathophysiology, we overlapped these lists with those genes included on the Simons Foundation Autism Research Initiative (SFARI) website (843 genes; https://sfari.org/resources/sfari-gene). The SFARI gene list represents ASD risk genes that have been curated manually from the scientific literature. We found that both the neocortical and hippocampal Foxp1cKO DEGs overlapped significantly with the ASD-SFARI genes (Fig. 6B; hypergeometric test; overlap between Foxp1cKO CTX and SFARI-ASD genes, p = 2.8 × 10−7; overlap between Foxp1cKO HIP and SFARI-ASD genes, p = 3.8 × 10−9). When we excluded ASD-SFARI genes from categories 5 and 6 (hypothesized and not supported, respectively) from this analysis, we obtained a similar result (hypergeometric test; 17 genes (p = 0.003) for Foxp1cKO hippocampal DEGs and 49 genes (p = 0.0002) for Foxp1cKO neocortical DEGs). Finally, accounting for directional consistency, we found a significant overlap between the Foxp1cKO neocortical and hippocampal DEG datasets (Fig. 6B; hypergeometric test, overlap between Foxp1cKO HIP and Foxp1cKO CTX genes, p = 4.7 × 10−31). The genes included in this directional overlap are enriched for GO categories such as potassium channel activity (Benjamini–Hochberg-corrected p-value, q = 3.3 × 10−3) and calcium ion transmembrane transporter activity (Benjamini–Hochberg-corrected p-value, q = 2.6 × 10−2). Combined with our previous report (Araujo et al., 2015), these data suggest that, although Foxp1 regulates distinct targets within different neuronal populations, it has an overall role in regulating the expression of ASD risk genes and ion receptor genes throughout the brain.
Next, to understand the role of Foxp1 specifically within the hippocampus, we compared the transcriptional targets in the hippocampus of the Foxp1cKO mice with DEGs we identified previously in a whole-body heterozygous Foxp1 knock-out (Foxp1+/−) mouse (Araujo et al., 2015). We were unable to compare neocortical data because there were no DEGs in the neocortex of the heterozygous Foxp1 mice (Araujo et al., 2015). When we applied the same DEG cutoffs to these two datasets, we found a significant overlap of directionally consistent hippocampal target genes in these two Foxp1 mouse models (Fig. 6C; hypergeometric test, p = 1.8 × 10−22). This overlap represented ∼20% of the Foxp1cKO DEGs, but only ∼9% of the Foxp1+/− DEGs. That a relatively small proportion of Foxp1+/− hippocampal DEGs overlapped with the DEGs in the Foxp1cKO mouse hippocampus is an important finding because the only major behavioral phenotype seen in Foxp1+/− mice was an alteration in USV production (Araujo et al., 2015). Furthermore, we did not observe a learning and memory deficit in the Foxp1+/− mice using a novel object recognition task (data not shown). Together, these results indicate that complete and partial loss of Foxp1 produce unique alterations in signaling pathways in the hippocampus. Moreover, these data suggest that the nonoverlapping genes in the Foxp1cKO hippocampus are likely important for the observed learning and memory deficits seen in Foxp1cKO mice.
Next, we sought to determine the extent to which Foxp1cKO hippocampal DEGs affect signaling processes involved in hippocampal neuron identity. Specifically, we wanted to assess whether loss of Foxp1 perturbs the expression of genes specific to CA1 pyramidal neurons. To accomplish this, we overlapped the Foxp1cKO hippocampal DEGs with CA1-specific genes identified by single-cell sequencing of the mouse CA1 (Zeisel et al., 2015). We found a significant overlap between these two gene expression datasets (Fig. 6D; hypergeometric test, p = 1.7 × 10−15; Fig. 6-2). The relatively small size of this overlap (∼15% of the Foxp1cKO hippocampal DEGs) was most likely due to the fact that our RNA-seq data captured direct and indirect (as well as cell-autonomous and non-cell-autonomous) gene expression changes due to loss of Foxp1 in both CA1 and CA2 neurons throughout the entirety of the hippocampus.
We then used WGCNA to further prioritize Foxp1cKO hippocampal DEGs with respect to ASD and ID (Langfelder and Horvath, 2008). WGCNA allows for the identification of networks (or modules) of genes with high coexpression. Three modules were genotype specific (Fig. 6E–G). The “dark green” module was hippocampus specific (Fig. 6E) and contained several ASD-SFARI genes such as Cadm2, Prkcb, Scn8a, and Syne1. The dark green module also contained Fmr1, which when disrupted in humans leads to Fragile X syndrome, which frequently includes the presentation of ID (Hernandez et al., 2009). In addition, both the “green yellow” and “light cyan” modules also included a number of ASD-SFARI genes (Fig. 6F,G). The light cyan module not only included Foxp1, but also several ASD-related genes encoding ion channels such as Kcnj10, Kcnq3, and Slc24a2 (Fig. 6G). Finally, a number of Foxp1cKO hippocampal DEGs that overlapped with each of the relevant gene lists discussed above (SFARI-ASD genes and/or Foxp1cKO neocortical DEGs and/or Foxp1+/− hippocampal DEGs) were chosen at random and confirmed in independent Foxp1cKO hippocampal samples via qPCR (Fig. 6H; Student's t test, p < 0.05 for all genes). In summary, these genomic data identify transcriptional programs downstream of Foxp1 that may drive the regulation of hippocampal function.
The genomic data indicated a role for Foxp1 in the regulation of both neocortical and hippocampal development (Fig. 6A,D). To assess neuroanatomical alterations due to forebrain Foxp1 loss that could explain the observed phenotypes, we analyzed the brains of Foxp1cKO mice via MRI, which has been used on other ASD mouse models (Ellegood et al., 2015). We chose to focus our analyses on relative regional volumes (normalized to total volumes) because there was a significant decrease in the overall volume of Foxp1cKO mouse brains (−12%, p < 0.0001, Student's t test and FDR < 1%). In summary, we found reductions and increases in the relative volumes of numerous brain regions in Foxp1cKO mice (Fig. 7A,B; p < 0.05, Student's t test, and FDR < 0.05 for all regions; Fig. 7-1). Brain regions with decreased relative volumes constituted neuronal populations expressing Emx1 (including many neocortical areas) or white matter tracts originating from populations expressing Emx1 (Fig. 7A,B and Fig. 7-1). Interestingly, the most affected region (in terms of percentage decrease) was the hippocampus (Fig. 7B). Moreover, hippocampal subfields and parahippocampal regions such as the dentate gyrus, stratum granulosum, and the pre-para subiculum were also significantly reduced in volume (Fig. 7A,B and Fig. 7-1, available at 10.1523/JNEUROSCI.1005-17.2017.f7-1). Brain regions with increased relative volumes mostly constituted cerebellar nuclei, cerebellar white matter tracts, and subcortical nuclei (Fig. 7A and Fig. 7-1). Taken as a whole, these data demonstrate that loss of Foxp1 expression in the forebrain leads to alterations in regional volumes throughout the brain, with hippocampal structures being affected drastically.
Top 20 increases and decreases to regional brain volumes in Foxp1cKO mice. Regions with relative volume increases are characterized by Emx1-expressing neuronal subpopulations, whereas regions with relative volume decreases are characterized by cerebellar nuclei, white matter tracts, and subcortical nuclei (FDR < 0.05). The percent difference (% Diff) from control volumes are reported for each region. Download Figure 7-1, XLSX file (11.3KB, xlsx)
The significant decrease in hippocampal volumes exhibited by Foxp1cKO mice suggested alterations in the functional properties of this region (Fig. 7A,B). In addition, plasticity in general and LTP in particular were GO categories enriched in the Foxp1cKO HIP DEGs (Fig. 6A). LTP is a well studied process by which excitatory synapses are strengthened in response to neuronal stimulation (Lynch, 2004; Kumar, 2011). Hippocampal LTP mediated by projections from CA3 pyramidal neurons to CA1 pyramidal neurons (Schaffer collateral projections) is thought to underlie the encoding of spatial memory (Lynch, 2004; Kumar, 2011). Therefore, we chose to examine hippocampal LTP in Foxp1cKO mice. Given that Foxp1 expression in the hippocampus is restricted to CA1/2 pyramidal neurons (Ferland et al., 2003), we recorded LTP in area CA1 in response to Schaffer collateral stimulation in Foxp1cKO mice. Although the initial magnitude of CA1 LTP was normal in Foxp1cKO mice (Fig. 8A, arrows), the mean magnitude of LTP during the last 10 min of stimulation was reduced significantly (Fig. 8A,B; Student's t test, F(19,14) = 2.16, ****p < 0.0001), indicative of impaired plasticity maintenance. This result was not due to differential baseline synaptic transmission in the Foxp1cKO mice because we found no difference in the fEPSP slope relative to current stimulation intensity (Fig. 8C; two-way ANOVA; genotype effect: F(1,236) = 0.23, p = 0.63; time effect: F(7,236) = 11.97, p < 0.0001; interaction effect: F(7,236) = 0.04, p > 0.99). To characterize the Foxp1cKO hippocampal RNA-seq data with regard to genes involved in LTP maintenance, we overlapped our gene list with a dataset generated from microarrays performed on hippocampi 5 h after undergoing LTP-inducing high-frequency stimulation (Ryan et al., 2012). These two datasets overlapped significantly (Fig. 8D; hypergeometric test, p = 2.32 × 10−4). We then tested all 12 of the overlapping genes (which included Foxp1) via qPCR and were able to confirm differential expression for 9 of them in independent Foxp1cKO hippocampal samples (Fig. 8E; Student's t test, p < 0.05 for all genes except Ccnd1, Dusp5, and Sorcs3). Of the 12 genes overlapping between the LTP maintenance dataset and the Foxp1cKO hippocampus dataset, seven (Ccnd1, Dsp, Gnb4, Grin3a, Rasd1, Runx1t1, and Sorcs3) are not included in the Foxp1+/− hippocampus dataset (Araujo et al., 2015). These seven genes therefore represent mechanisms that are uniquely disrupted in the Foxp1cKO hippocampus and could explain the physiological and behavioral phenotypes displayed by Foxp1cKO mice. Indeed, several of these genes (Rasd1, Gnb4, and Grin3a) have been implicated directly in hippocampal-based learning and memory (de Quervain and Papassotiropoulos, 2006; Vilches et al., 2014; Carlson et al., 2016). These data suggest that control of genes involved in hippocampal LTP maintenance is disrupted in Foxp1cKO mice and that this may contribute to their learning and memory deficits.
Discussion
Elucidating molecular mechanisms important for learning and memory is an ambitious undertaking, especially in terms of connecting these mechanisms to disorders such as ASD and ID. Numerous genes have been linked to an increased risk for both of these disorders and this suggests common mechanisms between them (Santini and Klann, 2014; Plummer et al., 2016). However, only a few of these genes have been categorized as high-confidence risk genes (Iossifov et al., 2014; Sanders et al., 2015; Mullins et al., 2016; Vissers et al., 2016; de la Torre-Ubieta et al., 2016; Stessman et al., 2017). FOXP1 is among this list of high-confidence ASD and ID risk genes (Iossifov et al., 2014; Sanders et al., 2015; Vissers et al., 2016; Stessman et al., 2017), yet relatively little is known about the function of FOXP1 in the brain. Previously, we demonstrated that an ASD- and ID-patient-relevant haploinsufficient Foxp1 mouse model exhibits vocalization deficits with alterations in striatal function and gene expression (Araujo et al., 2015). In addition, a mouse model with complete loss of Foxp1 in the brain demonstrates a number of behavioral deficits and functional alterations in several brain regions (Bacon et al., 2015). Therefore, these previously published data do not address the requirement for Foxp1 in specific brain regions and how such a requirement might be important for specific ASD- and ID-relevant phenotypes.
To investigate the functional role of Foxp1 in a brain-region-specific manner, we generated Foxp1cKO mice with complete loss of Foxp1 in the hippocampus and neocortex (Fig. 1A,B). We observe a number of striking behavioral deficits in these mice, most notably altered social interactions (Fig. 3I) and an almost total absence of spatial learning (Fig. 4A–F). Because the neural circuits for social behaviors are not understood fully and because the hippocampus has been studied in depth for its relationship to learning and memory, we chose to focus on the potential role of Foxp1 in hippocampal-based functions in Foxp1cKO mice. Importantly, the majority of documented patients with ASD-causing mutations in FOXP1 also have a diagnosis of ID (Le Fevre et al., 2013; Lozano et al., 2015; Vissers et al., 2016), making a mechanistic study of Foxp1 function in the hippocampus disease relevant.
Foxp1cKO mice possess deficits in spatial learning because they perform poorly in both the alternating T-maze and the Morris water maze (Fig. 4A–F). Conversely, Foxp1cKO mice have intact learning in contextual fear conditioning (Fig. 5A,B). Such results are seemingly in conflict with one another. However, when performed before testing, lesions of the hippocampus impair learning in the Morris water maze but preserve contextual fear conditioning in mice (Cho et al., 1999). Indeed, other mouse models of neuropsychiatric disorders have also shown that these two processes can be dissociated (Kubota et al., 2001; Huynh et al., 2009). Another explanation could be that hippocampal function in Foxp1cKO mice is sufficient to allow for associations between discrete, easily recognizable elements of the contextual conditioning environment and the unconditioned stimulus (Maren, 2001). Finally, these opposing results could be explained by the simple idea that Foxp1cKO mice might not be able to meet the cognitive load necessary for complex spatial tasks, but are able to meet the cognitive load required for fear-based tasks, which involve more salient conditioning. The amygdala is critically involved in cue-based fear learning (Puzzo et al., 2014; Tovote et al., 2015) and there is limited expression of Foxp1 in the amygdala (Ferland et al., 2003). However, the intact cue-dependent fear conditioning in Foxp1cKO mice (Fig. 5A,B) suggests that the role the hippocampal–amygdala circuit plays in this task is spared with loss of Foxp1.
The intact set-shifting learning that we observed in Foxp1cKO mice (Fig. 5C,D) suggests that, whereas Foxp1 expression is almost completely absent in the neocortex of these animals, this expression loss does not affect other, broader types of learning. In addition, the spared set-shifting learning in Foxp1cKO mice (Fig. 5C,D) could represent compensation by subcortical circuits in which Foxp1 expression is preserved. Regardless, these results need to be interpreted carefully because we cannot fully rule out other learning defects due to neocortical loss of Foxp1 that were not tested in this study. Together, our results suggest that Foxp1cKO mice exhibit a specific deficit in complex, hippocampal-based spatial tasks.
Mice with full-brain loss of Foxp1 also display hyperactivity, impaired spatial learning, and impaired social behaviors (Bacon et al., 2015). Conversely, unlike the results presented here (Fig. 8A,B), hippocampal neuron excitability is reduced and hippocampal LTP is unaltered in full-brain Foxp1 knock-out mice (Bacon et al., 2015). However, the study of full-brain Foxp1 knock-out mice used a weaker protocol for LTP induction (Bacon et al., 2015) than our study, so it is possible that such a protocol may not have recruited the same plasticity maintenance mechanisms recruited in this study.
The Foxp1cKO hippocampus dataset possessed fewer DEGs than the Foxp1+/− hippocampus RNA-seq dataset (Fig. 6C). This result was surprising to us because we expected complete loss of Foxp1 to yield greater disruptions to the hippocampal transcriptome than Foxp1 haploinsufficiency. The larger number of hippocampal DEGs observed in Foxp1+/− mice could be explained by improper signaling to the hippocampus from subcortical regions that also experience reduced Foxp1 levels. Using an available single-cell RNA-seq dataset (Zeisel et al., 2015), we found that Foxp1cKO hippocampal DEGs were most significantly enriched for genes expressed in CA1 and Foxp1cKO neocortical DEGs were most significantly enriched for genes expressed in pyramidal neurons of somatosensory cortex (Fig. 6-2). Therefore, our data suggest that loss of Foxp1 leads to disruptions in the expression of genes important for cellular identity in both brain regions. The DEGs that result from loss of Foxp1 in the hippocampus also indicate changes in pathways known to be important in hippocampal LTP, synaptic signaling, and spatial memory (Fig. 6A). We observed no differences in the basal synaptic transmission in the Foxp1cKO mouse hippocampus (Fig. 8C), suggesting that the diminished LTP maintenance that we recorded in the Foxp1cKO CA1 region (Fig. 8A,B) was due to the dysregulation of downstream signaling networks and not to alterations to baseline synaptic function (Kotaleski and Blackwell, 2010). This is supported by the finding that our Foxp1cKO hippocampal RNA-seq dataset overlapped significantly with a hippocampal maintenance gene list (Fig. 8D; Ryan et al., 2012).
Future studies that test directly the involvement of genes regulated by Foxp1 in LTP maintenance would be compelling, especially because disrupted synaptic signaling has a widely known role in explaining the etiology of ASD and ID (Santini and Klann, 2014). Nevertheless, it is unclear whether restoration of any one downstream gene (or combination of genes) would be sufficient to rescue both the physiological and behavioral deficits seen in Foxp1cKO mice. The generation of additional brain-region-specific Foxp1 knock-out mice will address whether the expression of Foxp1 in the hippocampus is required for the observed behavioral deficits. Moreover, Foxp1 expression in the striatum is intact in the Foxp1cKO mice detailed here (Fig. 1A,B), yet there are robust striatal deficits in heterozygous Foxp1 knock-out mice (Araujo et al., 2015) and brain-wide Foxp1 knock-out mice (Bacon et al., 2015). Therefore, a Foxp1 knock-out mouse that primarily targets the striatum will be important for understanding the contributions of Foxp1 to the complex behavioral phenotypes associated with ASD and ID.
In summary, this study is an important step toward completing our understanding of the region-specific roles of FoxP1 within the brain. Because FOXP1 is among the most salient ASD and ID genes, any knowledge of FOXP1 function should contribute to our understanding of ASD and ID pathophysiology. Indeed, an in-depth understanding of the basic mechanisms of brain development and function as it pertains to certain neurodevelopmental disease-relevant genes such as FOXP1 will be critical to designing effective therapeutics for the resultant conditions.
Footnotes
This work was supported by the National Science Foundation (Graduate Research Fellowship Program Grant 2013162469 to D.J.A.); the Program for Advancing Strategic International Networks to Accelerate the Circulation of Talented Researchers (Grant S2603 to K.T.); the National Institutes of Health (National Institute of Mental Health Training Grant T32-MH76690 to A.G.A.; Grants R01DC014702 and R01MH102603 to G.K.; and Grant R01HD069560 to C.M.P.); the Uehara Memorial Foundation (N.U.); the Ontario Brain Institute (J.P.L.), Brain Canada (J.P.L.), and the Canadian Institute for Health Research (J.P.L.); the University of Texas Southwestern Jon Heighten Scholar in Autism Research Endowment (G.K.); and the Simons Foundation Autism Research Initiative (Project 401220 to G.K.). We thank Drs. Courtney Lane, Irene Masiulis Bowen, and Joachim Herz for assistance with data collection and experimental design and Drs. Amelia Eisch, Lenora Volk, and Jiang Wu for providing valuable comments on this study.
The authors declare no competing financial interests.
References
- Ageranioti-Bélanger S, Brunet S, D'Anjou G, Tellier G, Boivin J, Gauthier M (2012) Behaviour disorders in children with an intellectual disability. Paediatr Child Health 17:84–88. 10.1093/pch/17.2.84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11:R106. 10.1186/gb-2010-11-10-r106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W (2015) HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31:166–169. 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S. (2010) FastQC: a quality control tool for high throughput sequence data. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
- Araujo DJ, Anderson AG, Berto S, Runnels W, Harper M, Ammanuel S, Rieger MA, Huang HC, Rajkovich K, Loerwald KW, Dekker JD, Tucker HO, Dougherty JD, Gibson JR, Konopka G (2015) FoxP1 orchestration of ASD-relevant signaling pathways in the striatum. Genes Dev 29:2081–2096. 10.1101/gad.267989.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacon C, Rappold GA (2012) The distinct and overlapping phenotypic spectra of FOXP1 and FOXP2 in cognitive disorders. Hum Genet 131:1687–1698. 10.1007/s00439-012-1193-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacon C, Schneider M, Le Magueresse C, Froehlich H, Sticht C, Gluch C, Monyer H, Rappold GA (2015) Brain-specific Foxp1 deletion impairs neuronal development and causes autistic-like behaviour. Mol Psychiatry 20:632–639. 10.1038/mp.2014.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock NA, Nieman BJ, Bishop JB, Mark Henkelman R (2005) In vivo multiple-mouse MRI at 7 tesla. Magn Reson Med 54:1311–1316. 10.1002/mrm.20683 [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson GC, Lin RE, Chen Y, Brookshire BR, White RS, Lucki I, Siegel SJ, Kim SF (2016) Dexras1 a unique ras-GTPase interacts with NMDA receptor activity and provides a novel dissociation between anxiety, working memory and sensory gating. Neuroscience 322:408–415. 10.1016/j.neuroscience.2016.02.063 [DOI] [PubMed] [Google Scholar]
- Chen JA, Peñagarikano O, Belgard TG, Swarup V, Geschwind DH (2015) The emerging picture of autism spectrum disorder: genetics and pathology. Annu Rev Pathol 10:111–144. 10.1146/annurev-pathol-012414-040405 [DOI] [PubMed] [Google Scholar]
- Cho KK, Hoch R, Lee AT, Patel T, Rubenstein JL, Sohal VS (2015) Gamma rhythms link prefrontal interneuron dysfunction with cognitive inflexibility in Dlx5/6(+/-) mice. Neuron 85:1332–1343. 10.1016/j.neuron.2015.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YH, Friedman E, Silva AJ (1999) Ibotenate lesions of the hippocampus impair spatial learning but not contextual fear conditioning in mice. Behav Brain Res 98:77–87. [DOI] [PubMed] [Google Scholar]
- Deacon RM. (2006) Assessing nest building in mice. Nat Protoc 1:1117–1119. 10.1038/nprot.2006.170 [DOI] [PubMed] [Google Scholar]
- de la Torre-Ubieta L, Won H, Stein JL, Geschwind DH (2016) Advancing the understanding of autism disease mechanisms through genetics. Nat Med 22:345–361. 10.1038/nm.4071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Quervain DJ, Papassotiropoulos A (2006) Identification of a genetic cluster influencing memory performance and hippocampal activity in humans. Proc Natl Acad Sci U S A 103:4270–4274. 10.1073/pnas.0510212103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorr AE, Lerch JP, Spring S, Kabani N, Henkelman RM (2008) High resolution three-dimensional brain atlas using an average magnetic resonance image of 40 adult C57Bl/6J mice. Neuroimage 42:60–69. 10.1016/j.neuroimage.2008.03.037 [DOI] [PubMed] [Google Scholar]
- Ellegood J, et al. (2015) Clustering autism: using neuroanatomical differences in 26 mouse models to gain insight into the heterogeneity. Mol Psychiatry 20:118–125. 10.1038/mp.2014.98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakhoury M. (2015) Autistic spectrum disorders: A review of clinical features, theories and diagnosis. Int J Dev Neurosci 43:70–77. 10.1016/j.ijdevneu.2015.04.003 [DOI] [PubMed] [Google Scholar]
- Feng X, Ippolito GC, Tian L, Wiehagen K, Oh S, Sambandam A, Willen J, Bunte RM, Maika SD, Harriss JV, Caton AJ, Bhandoola A, Tucker PW, Hu H (2010) Foxp1 is an essential transcriptional regulator for the generation of quiescent naive T cells during thymocyte development. Blood 115:510–518. 10.1182/blood-2009-07-232694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferland RJ, Cherry TJ, Preware PO, Morrisey EE, Walsh CA (2003) Characterization of Foxp2 and Foxp1 mRNA and protein in the developing and mature brain. J Comp Neurol 460:266–279. 10.1002/cne.10654 [DOI] [PubMed] [Google Scholar]
- Genovese CR, Lazar NA, Nichols T (2002) Thresholding of statistical maps in functional neuroimaging using the false discovery rate. Neuroimage 15:870–878. 10.1006/nimg.2001.1037 [DOI] [PubMed] [Google Scholar]
- Geschwind DH, State MW (2015) Gene hunting in autism spectrum disorder: on the path to precision medicine. Lancet Neurol 14:1109–1120. 10.1016/S1474-4422(15)00044-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, Jones KR (2002) Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci 22:6309–6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heisler JM, Morales J, Donegan JJ, Jett JD, Redus L, O'Connor JC (2015) The attentional set shifting task: a measure of cognitive flexibility in mice. J Vis Exp 96:51944. 10.3791/51944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez RN, Feinberg RL, Vaurio R, Passanante NM, Thompson RE, Kaufmann WE (2009) Autism spectrum disorder in fragile X syndrome: a longitudinal evaluation. Am J Med Genet A 149A:1125–1137. 10.1002/ajmg.a.32848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisaoka T, Nakamura Y, Senba E, Morikawa Y (2010) The forkhead transcription factors, Foxp1 and Foxp2, identify different subpopulations of projection neurons in the mouse cerebral cortex. Neuroscience 166:551–563. 10.1016/j.neuroscience.2009.12.055 [DOI] [PubMed] [Google Scholar]
- Holy TE, Guo Z (2005) Ultrasonic songs of male mice. PLoS Biol 3:e386. 10.1371/journal.pbio.0030386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh DP, Maalouf M, Silva AJ, Schweizer FE, Pulst SM (2009) Dissociated fear and spatial learning in mice with deficiency of ataxin-2. PLoS One 4:e6235. 10.1371/journal.pone.0006235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, et al. (2014) The contribution of de novo coding mutations to autism spectrum disorder. Nature 515:216–221. 10.1038/nature13908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaestner KH, Knochel W, Martinez DE (2000) Unified nomenclature for the winged helix/forkhead transcription factors. Genes Dev 14:142–146. [PubMed] [Google Scholar]
- Kotaleski JH, Blackwell KT (2010) Modelling the molecular mechanisms of synaptic plasticity using systems biology approaches. Nat Rev Neurosci 11:239–251. 10.1038/nrn2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota M, Murakoshi T, Saegusa H, Kazuno A, Zong S, Hu Q, Noda T, Tanabe T (2001) Intact LTP and fear memory but impaired spatial memory in mice lacking Ca(v)2.3 (alpha(IE)) channel. Biochem Biophys Res Commun 282:242–248. 10.1006/bbrc.2001.4572 [DOI] [PubMed] [Google Scholar]
- Kumar A. (2011) Long-term potentiation at CA3-CA1 hippocampal synapses with special emphasis on aging, disease, and stress. Front Aging Neurosci 3:7. 10.3389/fnagi.2011.00007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Horvath S (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9:559. 10.1186/1471-2105-9-559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Fevre AK, Taylor S, Malek NH, Horn D, Carr CW, Abdul-Rahman OA, O'Donnell S, Burgess T, Shaw M, Gecz J, Bain N, Fagan K, Hunter MF (2013) FOXP1 mutations cause intellectual disability and a recognizable phenotype. Am J Med Genet A 161A:3166–3175. 10.1002/ajmg.a.36174 [DOI] [PubMed] [Google Scholar]
- Leitner Y. (2014) The co-occurrence of autism and attention deficit hyperactivity disorder in children: what do we know? Front Hum Neurosci 8:268. 10.3389/fnhum.2014.00268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerch JP, Carroll JB, Spring S, Bertram LN, Schwab C, Hayden MR, Henkelman RM (2008) Automated deformation analysis in the YAC128 Huntington disease mouse model. Neuroimage 39:32–39. 10.1016/j.neuroimage.2007.08.033 [DOI] [PubMed] [Google Scholar]
- Lerch JP, Sled JG, Henkelman RM (2011) MRI phenotyping of genetically altered mice. Methods Mol Biol 711:349–361. 10.1007/978-1-61737-992-5_17 [DOI] [PubMed] [Google Scholar]
- Li X, Xiao J, Fröhlich H, Tu X, Li L, Xu Y, Cao H, Qu J, Rappold GA, Chen JG (2015) Foxp1 regulates cortical radial migration and neuronal morphogenesis in developing cerebral cortex. PLoS One 10:e0127671. 10.1371/journal.pone.0127671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozano R, Vino A, Lozano C, Fisher SE, Deriziotis P (2015) A de novo FOXP1 variant in a patient with autism, intellectual disability and severe speech and language impairment. Eur J Hum Genet 23:1702–1707. 10.1038/ejhg.2015.66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch MA. (2004) Long-term potentiation and memory. Physiol Rev 84:87–136. 10.1152/physrev.00014.2003 [DOI] [PubMed] [Google Scholar]
- Maren S. (2001) Neurobiology of Pavlovian fear conditioning. Annu Rev Neurosci 24:897–931. 10.1146/annurev.neuro.24.1.897 [DOI] [PubMed] [Google Scholar]
- McCarthy DJ, Chen Y, Smyth GK (2012) Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucl Acids Res 40:4288–4297. 10.1093/nar/gks042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins C, Fishell G, Tsien RW (2016) Unifying views of autism spectrum disorders: a consideration of autoregulatory feedback loops. Neuron 89:1131–1156. 10.1016/j.neuron.2016.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieman BJ, Bock NA, Bishop J, Sled JG, Josette Chen X, Mark Henkelman R (2005) Fast spin-echo for multiple mouse magnetic resonance phenotyping. Magn Reson Med 54:532–537. 10.1002/mrm.20590 [DOI] [PubMed] [Google Scholar]
- Nieman BJ, Flenniken AM, Adamson SL, Henkelman RM, Sled JG (2006) Anatomical phenotyping in the brain and skull of a mutant mouse by magnetic resonance imaging and computed tomography. Physiol Genomics 24:154–162. [DOI] [PubMed] [Google Scholar]
- Park HR, Lee JM, Moon HE, Lee DS, Kim BN, Kim J, Kim DG, Paek SH (2016) A short review on the current understanding of autism spectrum disorders. Exp Neurobiol 25:1–13. 10.5607/en.2016.25.1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plummer JT, Gordon AJ, Levitt P (2016) The genetic intersection of neurodevelopmental disorders and shared medical comorbidities: relations that translate from bench to bedside. Front Psychiatry 7:142. 10.3389/fpsyt.2016.00142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portfors CV, Perkel DJ (2014) The role of ultrasonic vocalizations in mouse communication. Curr Opin Neurobiol 28:115–120. 10.1016/j.conb.2014.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Precious SV, Kelly CM, Reddington AE, Vinh NN, Stickland RC, Pekarik V, Scherf C, Jeyasingham R, Glasbey J, Holeiter M, Jones L, Taylor MV, Rosser AE (2016) FoxP1 marks medium spiny neurons from precursors to maturity and is required for their differentiation. Exp Neurol 282:9–18. 10.1016/j.expneurol.2016.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puzzo D, Lee L, Palmeri A, Calabrese G, Arancio O (2014) Behavioral assays with mouse models of Alzheimer's disease: practical considerations and guidelines. Biochem Pharmacol 88:450–467. 10.1016/j.bcp.2014.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan MM, Ryan B, Kyrke-Smith M, Logan B, Tate WP, Abraham WC, Williams JM (2012) Temporal profiling of gene networks associated with the late phase of long-term potentiation in vivo. PLoS One 7:e40538. 10.1371/journal.pone.0040538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, et al. (2015) Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 87:1215–1233. 10.1016/j.neuron.2015.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Klann E (2014) Reciprocal signaling between translational control pathways and synaptic proteins in autism spectrum disorders. Sci Signal 7:re10. 10.1126/scisignal.2005832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13:2498–2504. 10.1101/gr.1239303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoji H, Hagihara H, Takao K, Hattori S, Miyakawa T (2012) T-maze forced alternation and left-right discrimination tasks for assessing working and reference memory in mice. J Vis Exp 60: pii: 3300. 10.3791/3300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman JL, Yang M, Lord C, Crawley JN (2010) Behavioural phenotyping assays for mouse models of autism. Nat Rev Neurosci 11:490–502. 10.1038/nrn2851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiteri E, Konopka G, Coppola G, Bomar J, Oldham M, Ou J, Vernes SC, Fisher SE, Ren B, Geschwind DH (2007) Identification of the transcriptional targets of FOXP2, a gene linked to speech and language, in developing human brain. Am J Hum Genet 81:1144–1157. 10.1086/522237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- State MW, Šestan N (2012) Neuroscience: the emerging biology of autism spectrum disorders. Science 337:1301–1303. 10.1126/science.1224989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steadman PE, Ellegood J, Szulc KU, Turnbull DH, Joyner AL, Henkelman RM, Lerch JP (2014) Genetic effects on cerebellar structure across mouse models of autism using a magnetic resonance imaging atlas. Autism Res 7:124–137. 10.1002/aur.1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stessman HA, et al. (2017) Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat Genet 49:515–526. 10.1038/ng.3792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F, Bošnjak M, Škunca N, Šmuc T (2011) REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 6:e21800. 10.1371/journal.pone.0021800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi JS, Kumar V, Nakashe P, Koike N, Huang HC, Green CB, Kim TK (2015) ChIP-seq and RNA-seq methods to study circadian control of transcription in mammals. Methods Enzymol 551:285–321. 10.1016/bs.mie.2014.10.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura S, Morikawa Y, Iwanishi H, Hisaoka T, Senba E (2004) Foxp1 gene expression in projection neurons of the mouse striatum. Neuroscience 124:261–267. 10.1016/j.neuroscience.2003.11.036 [DOI] [PubMed] [Google Scholar]
- Tovote P, Fadok JP, Lüthi A (2015) Neuronal circuits for fear and anxiety. Nat Rev Neurosci 16:317–331. 10.1038/nrn3945 [DOI] [PubMed] [Google Scholar]
- Ullmann JF, Watson C, Janke AL, Kurniawan ND, Reutens DC (2013) A segmentation protocol and MRI atlas of the C57BL/6J mouse neocortex. Neuroimage 78:196–203. 10.1016/j.neuroimage.2013.04.008 [DOI] [PubMed] [Google Scholar]
- van Steensel FJ, Bögels SM, Perrin S (2011) Anxiety disorders in children and adolescents with autistic spectrum disorders: a meta-analysis. Clin Child Fam Psychol Rev 14:302–317. 10.1007/s10567-011-0097-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilches N, Spichiger C, Mendez N, Abarzua-Catalan L, Galdames HA, Hazlerigg DG, Richter HG, Torres-Farfan C (2014) Gestational chronodisruption impairs hippocampal expression of NMDA receptor subunits Grin1b/Grin3a and spatial memory in the adult offspring. PLoS One 9:e91313. 10.1371/journal.pone.0091313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissers LE, Gilissen C, Veltman JA (2016) Genetic studies in intellectual disability and related disorders. Nat Rev Genet 17:9–18. 10.1038/nrg3999 [DOI] [PubMed] [Google Scholar]
- Vorhees CV, Williams MT (2006) Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc 1:848–858. 10.1038/nprot.2006.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Lin D, Li C, Tucker P (2003) Multiple domains define the expression and regulatory properties of Foxp1 forkhead transcriptional repressors. J Biol Chem 278:24259–24268. 10.1074/jbc.M207174200 [DOI] [PubMed] [Google Scholar]
- Yamamoto J, Suh J, Takeuchi D, Tonegawa S (2014) Successful execution of working memory linked to synchronized high-frequency gamma oscillations. Cell 157:845–857. 10.1016/j.cell.2014.04.009 [DOI] [PubMed] [Google Scholar]
- Zeisel A, Muñoz-Manchado AB, Codeluppi S, Lönnerberg P, La Manno G, Juréus A, Marques S, Munguba H, He L, Betsholtz C, Rolny C, Castelo-Branco G, Hjerling-Leffler J, Linnarsson S (2015) Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347:1138–1142. 10.1126/science.aaa1934 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Representative Foxp1cKO neocortex and hippocampus DEG REVIGO GO categories. Using the REVIGO web tool (see Materials and Methods), we identified enrichment for gene ontology (GO) categories among the up-regulated and down-regulated differentially expressed genes (DEGs) in the Foxp1cKO neocortex and hippocampus. GO category enrichment was determined using a Benjamini and Hochberg corrected p-value, q-value, ≤ 0.05. Download Figure 6-1, XLSX file (11.8KB, xlsx)
Overlaps between Foxp1cKO DEGs and cell-type-specific genes. Foxp1cKO neocortical (CTX) DEGs and hippocampal (HIP) DEGs significantly overlap with cell-type-specific genes (Zeisel et al., 2015). The number of genes for each Foxp1cKO DEG category and overlap are reported. Download Figure 6-2, XLSX file (11.3KB, xlsx)
Top 20 increases and decreases to regional brain volumes in Foxp1cKO mice. Regions with relative volume increases are characterized by Emx1-expressing neuronal subpopulations, whereas regions with relative volume decreases are characterized by cerebellar nuclei, white matter tracts, and subcortical nuclei (FDR < 0.05). The percent difference (% Diff) from control volumes are reported for each region. Download Figure 7-1, XLSX file (11.3KB, xlsx)