

Abstract
Bioassay-guided fractionation of a methanol extract of Magnolia grandiflora against Plasmodium falciparum yielded two new (1 and 2) and six known (3 – 8) bioactive compounds. The structures of the new compounds were assigned by mass spectrometric and 1D and 2D NMR data. Known compounds were identified by comparison of 1H NMR and MS data with literature data. The two known neolignans 3 and 4 showed moderate antiplasmodial activity with IC50 values of 2.8 ± 0.1 and 3.4 ± 0.1 μM, respectively. Weak antiplasmodial activity was recorded for compounds 1, 2, 5, 6, 7 and 8, with IC50 values of 38 ± 2, 23 ± 2, 16.5 ± 0.2, 86 ± 1, 44 ± 4 and 114 ± 9 μM, respectively.
Keywords: Antiplasmodial activity, Neolignans, Magnolia grandiflora (Magnoliaceae), 1D-and 2D-NMR, Mass spectrometry
Introduction
Malaria is one of the world’s most devastating diseases, with over 200 million people being infected every year and over 400,000 deaths in 2015 alone, most of them among children in sub-Saharan Africa.[1] Unfortunately the lethal malaria parasite, Plasmodium falciparum, has become resistant to most of the antimalarial drugs that were effective in the past, such as quinine, chloroquine, sulphadoxine-pyrimethamine, and mefloquine.[2] Drug resistance to artemisinin, which is used in combination therapy with other antimalarials as the first-line treatment for uncomplicated P. falciparum malaria, has also been observed.[3] Therefore, there is a continuing need to develop novel and more effective drugs against malaria. Various reviews have describe the current state of antimalarial drug research,[4–7] but in the words of one review “although there has been a dramatic improvement in the pipeline of new antimalarial molecules over the past decade, the glass is still rather empty”.[8]
As part of a systematic search for antimalarial agents from plant extracts from the Natural Product Discovery Institute Repository, a detanninized methanol partition fraction of the twigs and fruit of Magnolia grandiflora was found to show moderate activity against the Dd2 strain of P. falciparum. M. grandiflora is a well-investigated plant species, and is known to contain alkaloids,[9] flavonoids, phenolic compounds,[10] glycosides,[11, 12] sesquiterpene lactones,[13–15] and volatile compounds.[16] It has been reported in Traditional Chinese Medicine as a reliever of colds, headaches, and stomach ache.[9] It has not however been reported to contain any antimalarial compounds, and it was thus selected for investigation. Herein, we report the isolation and structure elucidation of two new (1 and 2) and six known (3 – 8) compounds with various degrees of antimalarial activity.
Results and Discussion
A methanol extract of M. grandiflora was subjected to liquid-liquid partitioning to eventually afford an ethyl acetate soluble fraction with an IC50 value of 10 μg/mL against the pyrimethamine, chloroquine, and mefloquine-resistant Dd2 strain of P. falciparum. Bioassay-guided isolation using solid phase extraction over C18 silica followed by C18 HPLC gave two new compounds (1 and 2) along with six known compounds (3 – 8) as shown in Figure-1. The known compounds were identified as 4′-O-methyl honokiol (3),[17] magnolol (4),[18] honokiol (5),[19] 3-methoxymagnolol (6),[18] isomagnolol (7),[20] and ketone (8)[21, 22] after comparison of their mass spectrometric and 1H NMR data (Figure S13–S18) with experimental values.
Figure 1.

Structures of compounds 1–8.
Compound 1 was isolated as a colorless oil and was assigned the molecular formula C18H20O2 based on its sodiated molecular ion peak at m/z 291.1352 [M + Na]+ in its HRESIMS. Its 1H NMR data (Table 1) displayed three aromatic protons of an ABX system at δH 6.73 (1H, J = 8.0 Hz, H-2), 6.96 (1H, J = 8.0, 1.8 Hz, H-3), and 6.98 (1H, J = 1.8 Hz, H-5). Signals for two allyl groups were present at δH 3.34 (2H, d, J = 6.6 Hz, H-7), 2.47 – 2.51 (1H, m, H-7′a), 2.53 – 2.60 (1H, m, H-7′b), H-5.90 – 5.95 (1H, m, H-8), 5.6 – 5.76 (1H, m, H- H-8′), 5.03 – 5.12 (2H, m, H-9), and 5.04 – 5.12 (2H, m, H-9′). Proton shifts at δH 5.62 (1H, d, J = 10.0 Hz) and 5.95 – 6.00 (1H, m) were assigned to H-5′ and H-6′, respectively. Moreover, two oxygenated methine and two methylene protons were also observed at chemical shift values of δH 4.14 (1H, dq, J = 8.4, 4.5 Hz, H-1′), δH 4.71 (1H, broad t, J = 3.9 Hz, H-3′), δH 2.00 – 2.08 (1H, m, H-2′a) and δH 2.41 – 2.48 (1H, m, H-2′b). Its 13C NMR spectrum (Table 1) showed the presence of 18 carbon signals. An HSQC experiment in combination with 13C NMR data identified five methylenes at δC 31.6, 39.8, 41.4, 115.6 and 118.6, two sp3-methines at δC 62.2 and 85.3, and seven sp2-methines at δC 110.1, 123.2, 128.0, 128.6, 131.4, 133.2, and 137.8. The remaining 4 carbon signals were classified as quaternary carbons with resonances at δC 47.8, 133.2 (2C), and 156.0. The NMR data (Table 1) of 1 is similar to that of ketone (8)[21, 22] except for the presence of a hydroxymethine at δC 62.2 (C-1′) instead of the ketone carbonyl at δC 195.2 (C-1′). An HMBC experiment on 1 (Figure 2) indicated correlations of H-7 (δH 3.34) with C-4 (δC 133.2), C-3 (δC 128.6) and C-5 (δC 123.2) confirming the presence of an allyl group on the aromatic ring. The presence of a second allyl group at C-4′ was validated from correlations between H-7′ (δH 2.56) to C-4′ (δC 47.8), and C-3′ (δC 85.3). The position of the double bond was fixed between C-5′ and C-6′ by the correlations of H-5′ (δH 5.62) with C-4′, C-1′, and C-3′. The position of C-3′ (δC 85.3) was confirmed on the basis of its correlations to H-5′ (δH 5.62) and H-7′ (δH 2.56). A COSY spectrum (Figure 2) was also obtained for compound 1, and cross peaks were located for H-1′ to H-2′a/b and H-6′ and H-2′a/b to H-3′, indicating the presence of a cyclohexene ring (C-1′ through C-6′) in 1. Compound 1 was thus identified as 4,4′-diallyl-1,2,6,4′-tetrahydrodibenzo[b,d]furan-3′-ol.
Table 1.
1H- and 13C NMR data of compounds 1 and 2.
| Position | 1 | 2 | ||
|---|---|---|---|---|
|
| ||||
| 1H NMR (J in Hz) | 13C NMR | 1H NMR (J in Hz) | 13C NMR | |
| 1 | - | 156.2 | - | 128.0 |
| 2 | 6.73, d, J = 8.0 | 110.3 | 7.02, d, J = 2.3 | 130.3 |
| 3 | 6.96, dd, J = 8.0, 1.8 | 128.6 | - | 132.2 |
| 4 | - | 133.4 | - | 151.0 |
| 5 | 6.98, d, J = 1.8 | 123.4 | 6.89, d, J = 8.8 | 115.4 |
| 6 | - | 133.2 | 7.03, dd, J = 8.8, 2.3 | 128.5 |
| 7 | 3.34, d, J = 6.6 | 39.9 | 3.34, dt, J = 6.7, 1.7 | 39.6 |
| 8 | 5.90 – 5.95, m | 138.0 | 5.97, ddt, J = 16.8, 10.0, 6.7 | 138.0 |
| 9 | 5.03 – 5.12, m | 115.8 | 5.04, dq, J = 10.0, 1.7 5.08, dq, J = 17.0, 1.7 |
115.6 |
| 1′ | 4.14, dq, J = 8.4, 4.5 | 62.4 | - | 125.3 |
| 2′/a or b | 2.00 – 2.08, m, 2.41 – 2.48, m | 31.8 | 7.11, d, J = 2.2 | 130.6 |
| 3′ | 4.71, br t, J = 3.9 | 85.6 | - | 124.8 |
| 4′ | - | 47.9 | - | 146.1 |
| 5′ | 5.62, d, J = 10.0 | 131.6 | 6.77, d, J = 8.2 | 111.2 |
| 6′ | 5.95–6.00, m | 128.2 | 7.24, dd, J = 8.2, 2.2 | 128.5 |
| 7′/a or b | 2.47 – 2.51, m, 2.53 – 2.60, m | 41.6 | 3.21, dt, J = 6.3, 1.7 | 36.6 |
| 8′ | 5.66 – 5.76, m | 133.4 | 5.82, ddt, J = 16.6, 10.2, 6.3 | 135.5 |
| 9′ | 5.04 – 5.12, m | 118.7 | 4.99, dq, J = 17.0, 1.7 5.01, dq, J = 10.2, 1.7 |
116.8 |
| 1″ | - | - | - | 131.3 |
| 2″ | - | - | 7.11, d, J = 8.4 | 130.1 |
| 3″ | - | - | 6.80, d, J = 8.4 | 115.6 |
| 4″ | - | - | - | 154.3 |
| 5″ | - | - | 6.80, d, J = 8.4 | 115.6 |
| 6″ | - | - | 7.11, d, J = 8.4 | 130.1 |
| 7″ | 2.90, t, J = 6.9 | 34.6 | ||
| 8″ | 3.41, dt, J = 6.9, 6.9 | 45.1 | ||
| NH | 3.85, br s | |||
| OH | 5.21, s | |||
| OH | 4.70, s | |||
Figure 2.
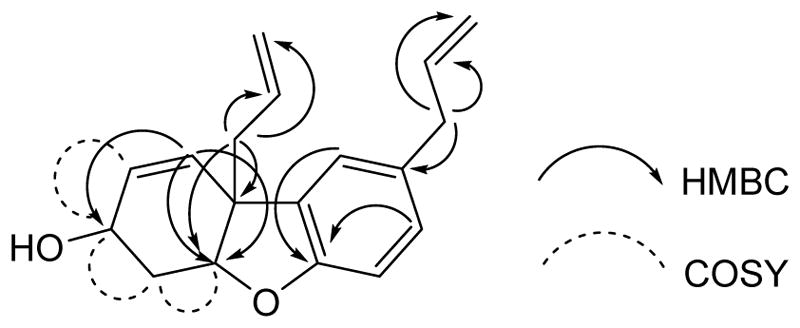
Key HMBC and COSY correlations of compound 1
Compound 2, also a colorless oil, was assigned the molecular formula C26H27NO2 (m/z 386.2107 [M + H]+, calcd. 386.2115) on the basis of its HRESIMS. 1H NMR data (Table 1) exhibited the presence of two ABX substituted benzene rings and a 1,4-disubstituted benzene ring. Protons of the two ABX aromatic rings were assigned chemical shift values of δH 7.11 (1H, m, overlapped, H-2′), 7.02 (1H, m, overlapped, H-2), 6.89 (1H, m, H-5), 6.77 (1H, d, J = 8.2 Hz, H-5′), 7.03 (1H,dd, J = 8.8, 2.3 Hz, H-6), and 7.24 (1H, dd, J = 8.2, 2.2 Hz, H-6′). The 1,4-disubstituted third aromatic ring had proton signals at δH 7.11 (2H, d, J = 8.4 Hz, H-2″ and H-6″) and 6.80 (2H, d, J = 8.4 Hz, H-3″ and H-5″). Chemical shifts of 3.34 (2H, dt, J = 6.7, 1.7 Hz, H-7), 3.21 (2H, dt, J = 6.3, 1.7 Hz, H-7′), 5.97 (1H, ddt, J = 16.8, 10.0, 6.7 Hz, H-8), 5.82 (1H, ddt, J = 16.6, 10.2, 6.3 Hz, H-8′), 5.04 (1H, dq, J = 10.0, 1.7, H-9a), 5.08 (1H, dq, J = 17.0, 1.7, H-9b), 4.99 (1H, dq, J = 17.0, 1.7, H-9a′) and 5.01 (1H, dq, J = 10.2, 1.7, H-9b′) were assigned to two allyl groups on the separate aromatic rings. Additional signals characteristic of an ethylamino group[23] were observed at δH 2.90 (2H, t, J = 6.9 Hz, H-7″) and 3.41 (2H, dt J = 6.9, 6.9 Hz, H-8″). 13C NMR data (Table 1) for compound 2 displayed a total of 26 carbon atoms. An HSQC NMR experiment in combination with 13C NMR data classified these carbon atoms into 6 methylenes (δC 34.6, 36.6, 39.6, 45.1, 115.6, and 116.8), 12 methines (δC 111.2, 115.4, 115.6 (2C), 128.4, 128.5 (2C), 130.1 (3C), 135.5, and 138.0) and 7 quaternary carbons (δC 124.8, 128.0, 130.2, 130.6, 131.4, and 154.3 (2C). Assignment of the 13C NMR spectrum of 2 was made on the basis of its HMQC spectrum and by comparison with the 13C NMR spectra of honokiol analogs.[24] An HMBC NMR spectrum (Figure 3) showed key correlations for 2, including cross-peaks for the three-bond correlation H-2″/H-6″ to C-7″ (δC 34.6), for the two-bond correlation H-7″ to C-8″ (δC 45.1), and for the three bond correlations H-2 to C-1′ (δC 125.3) and to C-4 (δC 151.1), H-7 to C-2 (δC 130.3), and H-7′ to C-2′ (δC 130.6) and C-4′ (δC 146.1). These correlations established the structure of 2 as 3,3′-diallyl-4′-((4-hydroxyphenethyl)amino)-[1,1′-biphenyl]-4-ol.
Figure 3.
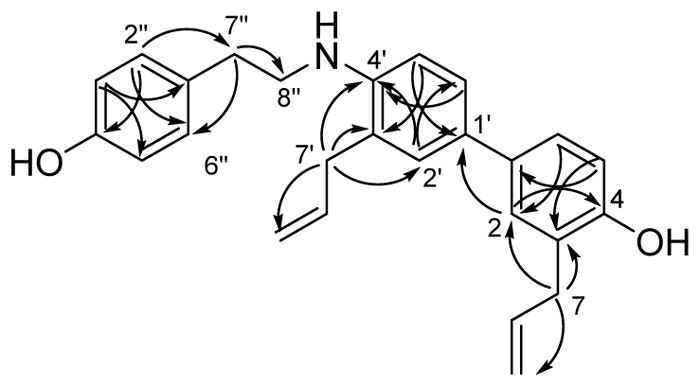
Key HMBC correlations of compound 2
Compound 2 is most probably formed by oxidative coupling of 2-allyphenol followed by condensation of the intermediate with p-hydroxyphenethylamine. Figure 4 shows an example of one plausible pathway.
The isolated compounds 1–8 were evaluated for their antiplasmodial activity against the Dd2 strain of P. falciparum. Compounds 3 and 4 showed moderate antimalarial activity with IC50 values of 2.8 ± 0.06 and 3.4 ± 0.08 μM, respectively. Compounds 1, 2, 5, 6, 7, and 8 had weaker activities with IC50 values of 37.5 ± 2.00, 22.7 ± 1.81, 16.6 ± 0.2, 86.1 ± 0.6, 44.4 ± 4.1 and 114 ± 9 μM, respectively.
The fact that simple lignans show moderate antiplasmodial activity is not unprecedented. Thus several lignans with antiplasmodial activity from African medicinal plants are reported in the review by Ntie-Kang et al.,[6] and lignans with antiplasmodial activity have been reported from Asparagus africanus[25] and Carrisa edulis.[26] Neolignans have also shown good antiplasmodial activity.[27] One point of interest is that antiplasmodial activity appears to be strongly dependent on the substitution pattern of simple lignans. Thus compounds 3 and 4 were active at the single digit micromolar level, but compound 5, isomeric with 4, was fivefold less potent, and compound 6, with an extra hydroxyl group, was thirtyfold less potent than 3.
Experimental Part
General Experimental Procedures
UV spectra were measured on Shimadzu a UV-1201 spectrophotometer. 1H- and 13C NMR spectra were recorded on a Bruker Avance 500 spectrometer in CDCl3 with TMS as internal standard. An Agilent 6220 mass spectrometer was used to obtain high resolution mass spectra. Solid phase extraction was performed using RP-18 silica gel (40–63μm, EM Science, Germany). Semi-preparative HPLC was performed on a semipreparative Phenomenex C18 column (5μm, 250 × 10 mm), using Shimadzu LC-10AT pumps coupled with a Shimadzu SPD M10A diode array detector, and a SCL-10A system controller.
Antiplasmodial Bioassay
The effect of each fraction or compound on parasite growth of the Dd2 strain of P. falciparum was measured in a 72 h growth assay in the presence of compound as descibed previously [28, 29]. Briefly, ring stage parasite cultures (100 μL per well, with 1% hematocrit and 1% parasitemia) were grown for 72 h in the presence of increasing concentration of the drug in a 5% CO2, 5% O2, and 90% N2 gas mixture at 37 °C. After 72 h in culture, parasite viability was determined by DNA quantitation using SYBR Green I assay [29]. The half-maximum inhibitory concentration (IC50) calculation was performed with KaleidaGraph software using a nonlinear regression curve fitting. IC50 values are the average of three independent determinations with each determination in duplicate and are expressed ± SEM. Artemisinin was used as the positive control with an IC50 of 6 ± 2 nM.
Plant Material
The plant material was collected in Santa Barbara, CA on September 11, 1995 by Cori Morenberg and Jan Wienpahl (NYBG) in the Santa Barbara Alice Keck Memorial Park, along Arrelaga St. 34°25′ N, 119°42′ W. http://sweetgum.nybg.org/science/vh/specimen_details.php?irn=284841. Voucher specimen CM00144c.
Extraction and Initial Fractionation
The dried and powdered twigs and fruit of M. grandiflora (175 g) were exhaustively extracted with MeOH in two 24-hour percolation steps followed by partition into hexane, methylene chloride and an aqueous methanolic fractions. The latter was detanninized by passage through a column of polyvinyl pyrrolidone to give the active methanolic fraction 0038279-03C, X-4568 (about 10 g). For purposes of fractionation and purification, 1.5 g of extract was shipped to Virginia Tech for bioassay-guided isolation.
Isolation of Bioactive Constituents
The crude extract (1.3 g) was first detanninized again by dissolving in MeOH and passing it through a polyamide column. A total of 932 mg detanninized extract was collected after elution and evaporation. It was dissolved in 200 mL of 90% aquous methanol and extracted with hexanes (200 × 5 mL). A total of 69 mg hexanes fraction was obtained. The 90% aqeous methanol extact was evaporated, suspended in 200 mL water, and then extracted with EtOAc (200 × 5 mL); evaporation of this EtOAc-soluble fraction gave 317 mg residue. The water layer was concentrated to a brown reside (530 mg). The EtOAc fraction had the highest antiplasmodial activity, with an IC50 value between 5 and 2.5 μg/mL. It was separated into seven sub-fractions (F1–F7) by open column chromatography over RP-18 silica gel with a MeOH/H2O gradient. Sub-fraction F5 (eluted with 90% MeOH/H2O) was the most active fraction with an IC50 value between 2.5 and 1.5 μg/mL. Semi-preparative C18 HPLC (MeOH/H2O sovent system) on F5 yielded nine subfractions (F5-1 to F5-9). Fraction F5-8 was further purified on HPLC using a C18 column with a MeOH/H2O solvent system to give compound 3 (3.2 mg, tR 19.2 min). Fraction F5-6 was subjected to HPLC purification on a C18 column using MeCN/H2O as solvent system to afford compounds 4 (2.0 mg, tR 12.76 min) and 6 (0.3 mg, tR 14.5 min). F5-5 was purified by HPLC on a C18 column using the MeCN/H2O solvent system to obtain compound 1 (0.5 mg, tR 12.1 min). The seventh fraction F5-7 yieded compounds 2 (0.6 mg, tR 14.2 min) and 7 (0.3 mg, tR 18.8 min) on HPLC separation on a C18 column using the MeCN/H2O solvent system. HPLC of fraction F5-3 on a C18 column (MeCN/H2O solvent system) resulted in the isolation of compounds 5 (2.7 mg, tR 10.1 min) and 8 (0.3 mg, tR 14.4 min).
Structural Identification
Characterization data of compounds 1 and 2 are shown below and their 1H NMR, 13C NMR, HSQC, and HMBC spectra are reproduced in the Supporting Information. The 1H NMR spectra of compounds 3 – 8 are also reproduced in the Supporting Information.
4,4′-Diallyl-1,2,6,4′-tetrahydrodibenzo[b,d]furan-3′-ol; 1
Colorless oil. [α]D21-33.8 (c = 0.020, MeOH), LC-UV [(acetonitrile in H2O)] λmax 225, 285 nm. 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz), see Table 1. ESI-HRMS m/z 291.1352 [M + Na]+; C18H20O2Na+ (calc. 291.1361).
3,3′-Diallyl-4′-((4-hydroxyphenethyl)amino)-[1,1′-biphenyl]-4-ol; 2
LC-UV [(acetonitrile in H2O)] λmax 219, 280 nm. 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz), see Table 1. ESI-HRMS m/z 386.2107 [M + H]+; C26H28NO2+ (calc. 386.2120).
Supplementary Material
Scheme 1.

Plausible biosynthetic pathway for compound 2
Acknowledgments
This project was supported by the National Center for Complementary and Integrative Health (NCCIH) under award 1 R01 AT008088. This support is gratefully acknowledged. This work was also supported by the National Science Foundation under Grant No. CHE-0619382 for purchase of the Bruker Avance 500 NMR spectrometer and Grant No. CHE-0722638 for the purchase of the Agilent 6220 mass spectrometer. We thank William Bebout for obtaining the mass spectra.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Author Contributions Statement
David Kingston conceived and designed the experiments, Abdul Latif isolated the compounds, and Abdul Latif and Yongle Du determined their structures. Seema Dalal, Maria Fernández-Murga, and Emilio Merino carried out the antiplasmodial bioassays under the direction of Maria Cassera, and Michael Goetz provided the crude extract.
References
- 1.Anonymous. ‘World Malaria Report 2016’. World Health Organization; Geneva: 2016. [Google Scholar]
- 2.Farooq U, Mahajan RC. Drug Resistance in Malaria. J Vect Borne Dis. 2004;41:45–53. [PubMed] [Google Scholar]
- 3.Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, Lwin KM, Ariey F, Hanpithakpong W, Lee SJ, Ringwald P, Silamut K, Imwong M, Chotivanich K, Lim P, Herdman T, An SS, Yeung S, Singhasivanon P, Day NPJ, Lindegardh N, Socheat D, White NJ. Artemisinin Resistance in Plasmodium falciparum Malaria. New Engl J Med. 2009;361:455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benelli G, Maggi F, Petrelli R, Canale A, Nicoletti M, Rakotosaona R, Rasoanaivo P. Not Ordinary Antimalarial Drugs: Madagascar Plant Decoctions Potentiating the Chloroquine Action Against Plasmodium parasites. Ind Crops Prod. 2017;103:19–38. [Google Scholar]
- 5.Fernandez-Alvaro E, Hong WD, Nixon GL, O’Neill PM, Calderon F. An timalarial Chemotherapy: Natural Product Inspired Development of Preclinical and Clinical Candidates with Diverse Mechanisms of Action. J Med Chem. 2016;59:5587–5603. doi: 10.1021/acs.jmedchem.5b01485. [DOI] [PubMed] [Google Scholar]
- 6.Ntie-Kang F, Onguéné PA, Lifongo LL, Ndom JC, Sippl W, Mbaze LMa. The Potential of Anti-malarial Compounds Derived from African Medicinal Plants, Part II: A Pharmacological Evaluation of Non-alkaloids and Non-terpenoids. Malaria J. 2014;13:81. doi: 10.1186/1475-2875-13-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Onguéné PA, Ntie-Kang F, Lifongo LL, Ndom JC, Sippl W, Mbaze LM. The Potential of Anti-malarial Compounds Derived from African Medicinal Plants. Part I: A Pharmacological Evaluation of Alkaloids and Terpenoids. Malaria J. 2013;12:449. doi: 10.1186/1475-2875-12-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wells TNC, Hooft van Huijsduijnen R, Van Voorhis WC. Malaria Medicines: A Glass Half Full? Nat Rev Drug Discovery. 2015;14:424–442. doi: 10.1038/nrd4573. [DOI] [PubMed] [Google Scholar]
- 9.Wu Z, Zhou T, Xiao P. Compendium of Xinhua Herbal Book. 1988. [Google Scholar]
- 10.Clark AM, El-Feraly AS, Li WS. Antimicrobial Activity of Phenolic Constituents of Magnolia grandiflora L. J Pharm Sci. 1981;70:951–952. doi: 10.1002/jps.2600700833. [DOI] [PubMed] [Google Scholar]
- 11.Rao KV. Glycosides of Magnolia grandiflora. Planta Med. 1975;27:24–30. [PubMed] [Google Scholar]
- 12.Rao KV, Wu WN. Glycosides of Magnolia III. Structural Elucidation of Magnolenin C. Lloydia. 1978;41:56–62. [Google Scholar]
- 13.Li HM, Zhao SR, Huo Q, Ma T, Liu H, Lee JK, Hong YS, Wu CZ. A New Dimeric Neolignan from Magnolia grandiflora L. Seeds. Arch Pharm Res. 2015;38:1066–1071. doi: 10.1007/s12272-014-0476-4. [DOI] [PubMed] [Google Scholar]
- 14.El-Feraly FS. Melampolides from Magnolia grandiflora. Phytochemistry. 1984;23:2372–2374. [Google Scholar]
- 15.Yang M, Blunden G, Patel A, O’Neill M, Lewis J. Coumarins and Sesquiterpene Lactones from Magnolia grandiflora Leaves. Planta Med. 1994;60:390–390. doi: 10.1055/s-2006-959515. [DOI] [PubMed] [Google Scholar]
- 16.Morshedloo MR, Quassinti L, Bramucci M, Lupidi G, Maggi F. Chemical Composition, Antioxidant Activity and Cytotoxicity on Tumour Cells of the Essential Oil from Flowers of Magnolia grandiflora Cultivated in Iran. Nat Prod Res. 2017;31 doi: 10.1080/14786419.14782017.11303699. [DOI] [PubMed] [Google Scholar]
- 17.Denton RM, Scragg JT, Saska J. A Concise Synthesis of 4′-O-Methyl Honokiol. Tetrahedron Lett. 2011;52:2554–2556. [Google Scholar]
- 18.Kijjoa A, Pinto MMM, Tantisewie B, Herz W. A Biphenyl Type Neolignan and a Biphenyl Ether from Magnolia henryi. Phytochemistry. 1989;28:1284–1286. [Google Scholar]
- 19.Denton RM, Scragg JT, Galofré AM, Gui X, Lewis W. A Concise Synthesis of Honokiol. Tetrahedron. 2010;66:8029–8035. [Google Scholar]
- 20.El-Feraly FS, Cheatham SF, Breedlove RL. Antimicrobial Neolignans of Sassafras randaiense Roots. J Nat Prod. 1983;46:493–498. [Google Scholar]
- 21.Sy L-K, Brown GD. Biomimetic Synthesis of Illicium Oligomeric Neolignans. J Chem Res, Synop. 1998:476–477. [Google Scholar]
- 22.Tzeng SC, Liu YC. Peroxidase-catalyzed Synthesis of Neolignan and its Anti-inflammatory Activity. J Mol Catal B: Enzym. 2004;32:7–13. [Google Scholar]
- 23.Liu Y, Chen L, Ji Y. Quantification and Structural Elucidation of Potential Impurities in Agomelatine Active Pharmaceutical Ingredient. J Pharm Biomed Anal. 2013;81–82:193–201. doi: 10.1016/j.jpba.2013.04.016. [DOI] [PubMed] [Google Scholar]
- 24.Lin JM, Gowda ASP, Sharma AK, Amin S. In Vitro Growth Inhibition of Human Cancer Cells by Novel Honokiol Analogs. Bioorg Med Chem. 2012;20:3202–3211. doi: 10.1016/j.bmc.2012.03.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oketch-Rabah HA, Dossaji SF, Christensen SB, Frydenvang K, Lemmich E, Cornett C, Olsen CE, Chen M, Kharazmi A, Theander T. Antiprotozoal Compounds from Asparagus africanus. Journal of Natural Products. 1997;60:1017–1022. doi: 10.1021/np970217f. [DOI] [PubMed] [Google Scholar]
- 26.Kebenei JS, Ndalut PK, Sabah AO. Anti-plasmodial Activity of Nortrachelogenin from the Root Bark of Carissa edulis (Vahl) Int J Appl Res Nat Prod. 2011;4:21. [Google Scholar]
- 27.Rakotondraibe HL, Graupner PR, Xiong Q, Olson M, Wiley JD, Krai P, Brodie P, Callmander JMW, Rakotobe E, Ratovoson F, Rasamison VE, Cassera MB, Hahn DR, Kingston DGI, Fotso S. Neolignans and Other Metabolites from Ocotea cymosa from the Madagascar Rain Forest and their Biological Activities. J Nat Prod. 2015;78:431–440. doi: 10.1021/np5008153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bennett TN, Paguio M, Gligorijevic B, Seudieu C, Kosar AD, Davidson E, Roepe PD. Novel, Rapid, and Inexpensive Cell-based Quantification of Antimalarial Drug Efficacy. Antimicrob Agents Chemother. 2004;48:1807–1810. doi: 10.1128/AAC.48.5.1807-1810.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smilkstein M, Sriwilaijaroen N, Kelly JX, Wilairat P, Riscoe M. Simple and Inexpensive Fluorescence-based Technique for High-throughput Antimalarial Drug Screening. Antimicrob Agents Chemother. 2004;48:1803–1806. doi: 10.1128/AAC.48.5.1803-1806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.