

Abstract
The ability to regulate inflammatory pathways and host defense mechanisms is critical for maintaining homeostasis and responding to infections and tissue injury. While unbalanced inflammation is detrimental to the host; inadequate inflammation might not provide effective signals required to eliminate pathogens. On the other hand, aberrant inflammation could result in organ damage and impair host defense. The lipid mediator leukotriene B4 (LTB4) is a potent neutrophil chemoattractant and recently, its role as a dominant molecule that amplifies many arms of phagocyte antimicrobial effector function has been unveiled. However, excessive LTB4 production contributes to disease severity in chronic inflammatory diseases such as diabetes and arthritis, which could potentially be involved in poor host defense in these groups of patients. In this review we discuss the cellular and molecular programs elicited during LTB4 production and actions on innate immunity host defense mechanisms as well as potential therapeutic strategies to improve host defense.
Keywords: Leukotriene B4, innate immunity, inflammation, host defense, immune regulation, microbicidal activity
1. Introduction
The ability of innate immune cells to properly recognize, respond, and eliminate invading pathogens is a requisite for host survival. Microbial infections quickly elicit an inflammatory program that induces the recruitment of phagocytes, such as macrophages, monocytes and neutrophils. These newly migrated cells further enhance the production of pro-inflammatory mediators such as cytokines, chemokines, growth factors, and bioactive lipids in the inflammatory milieu. After exposure to pathogens, sentinel cells such as epithelial cells and phagocytes (tissue resident macrophages and dendritic cells) detect pathogen associated molecular patterns (PAMPs) via binding to pattern recognition receptors (PRRs). Activation of PRRs such as Toll like receptors (TLRs) trigger the signaling programs that culminate in the generation of inflammatory cytokines and lipid mediators to provide signals essential to the recruitment of cells involved in the control of pathogens. The bioactive lipid mediator leukotriene B4 (LTB4) is produced primarily by neutrophils and macrophages and signals through its high or low affinity receptor B leukotriene receptor (BLT) 1 or 2, respectively, to enhance phagocyte antimicrobial effector functions. However, aberrant levels of LTB4 can be detrimental to host response and may be pathogenic in inflammatory diseases.
Effective inflammatory programs induced during infections can be compromised by underlying health conditions [1]. Although inflammation is important for coordinating immune responses during infection, excessive inflammatory responses can be destructive. Patients with chronic inflammatory diseases, such as diabetes, arthritis, and atherosclerosis have dysregulated inflammatory response functions and are more prone to infections [2–4].
This review covers the current understanding of the role of LTB4 and BLT1 on host defense mechanisms and how modulating the LTB4/BLT1 pathway can be therapeutically targeted to respectively amplify or inhibit host defense in settings of immunodeficiency or aberrant inflammation.
2. LT synthesis and receptors
2.1 LT synthesis
LTs are part of a large family of lipids termed eicosanoids that are derived from the Greek word “eicosa” since eicosanoids are made of 20 carbon atoms called eicosatetraenoïc acid. The synthesis of LTs involves several rate-limiting steps that comprise activation of phospholipase A2 (PLA2) and arachidonic acid (AA) release from phospholipids in the cellular membranes. Activation of 5-LO in concert with the 5-LO activation protein (FLAP) metabolizes AA to LTA4, which is converted to LTB4 by LTA4 hydrolase. LTA4 could alternatively be modified with glutathione by LTC4 synthase to form LTC4. Further modifications of LTC4 give rise to LTD4 and LTE4. Since LTC4, D4, and E4 contain a cysteine, they are known as the cysteinyl leukotrienes (CysLTs). Even though CysLTs exert stimulatory effects on macrophages and neutrophils, this review will focus primarily on LTB4 actions. The main cellular sources of LTB4 in both murine and humans are granulocytes, monocytes, and macrophages (Table 1) [5]. However, murine (RAW264.7 and J774) and human (THP1 and U937) macrophage cell lines express low levels of 5-LO and produce barely detectable levels of LTB4 [6]. Non-immune cells are also capable to produce LTB4. Some cell types have been reported to express some but not all of the LT-synthesis enzymes, which renders these cells incapable of synthesizing leukotrienes independently. However, these cells may be able to contribute to the synthesis of LTs in a process known as transcellular biosynthesis [7]. An example of transcellular biosynthesis of LTB4 is between neutrophils-erythrocytes, and keratinocytes and endothelial cells [8–12]. Table 1 lists the cellular expression of leukotriene synthesis enzymes. Although this topic is of interest, a more comprehensive review can be found at [13].
Table 1.
Expression of LT synthesis enzymes and LTB4 receptors in immune and structural cells.
| Cell type | 5-LO | FLAP | LTA4 hydrolase | LTC4 synthase | BLT1 | BLT2 |
|---|---|---|---|---|---|---|
| Neutrophil | +++ | + | +++ | −/+ | + | + |
| Monocyte/macrophage | ++ | + | +++ | + | + | + |
| Dendritic cell | + | + | ++ | + | + | + |
| Mast cell | + | + | + | ++ | + | + |
| Eosinophil | + | + | + | ++ | + | + |
| Endothelial cell | − | − | + | + | + | + |
| Red blood cell | − | − | + | − | − | − |
| Keratinocyte | −/+ | − | + | + | + | + |
5-LO activity is dependent on various signals including calcium release and phosphorylation, which control the catalytic site and the translocation of 5-LO within the cell. In a resting cell, 5-LO location varies depending on the cell type [14]. In neutrophils and peritoneal macrophages, 5-LO is located in the cytosol whereas in alveolar macrophages and Langerhans cells, 5-LO is located within in the nucleus [15–17]. Upon cell activation, increased intracellular calcium levels induce 5-LO to translocate to the perinuclear or plasmatic membrane where it can metabolize AA into LTA4 [18]. 5-LO activity is triggered by various stimuli such as pathogens, cytokines, and immune complexes [19]. During infection, pathogens have limited abilities to increase intracellular calcium levels and therefore are poor 5-LO activators alone. However, treating infected cells with a calcium ionophore or opsonized zymosan particles are able to greatly enhance LT synthesis [20, 21]. Table 2 lists the relative levels of LTB4 produced in response to various stimuli. The molecular mechanisms that regulate 5-lipoxygenase activation are reviewed here [22, 23].
Table 2.
LTB4 generation in response to various stimuli. ND not determined.
| Cell type | Cytokines/Growth factors | Bacteria | Opsonized pathogen | Fungi | Viral |
|---|---|---|---|---|---|
| Neutrophil | ++ | ++ | +++ | ++ | ++ |
| Monocyte/macrophage | ++ | ++ | +++ | ++ | ++ |
| Dendritic cell | + | ++ | ND | + | + |
| Mast cell | + | + | ND | + | + |
| Endothelial cell | ± | ± | ± | ± | ± |
2.2 LTB4 receptors
There are two G protein coupled receptors (GPCRs) for LTB4, BLT1 and BLT2. BLT1 is a high-affinity receptor and BLT2 is a low-affinity receptor. Since other fatty acid metabolites besides LTB4 are able to activate BLT2, the effects of BLT2 signaling are not limited strictly to LTB4 effects [24]. Distribution of BLT1 and BLT2 on cells and tissues vary between mouse and human [24]. On human cells, BLT1 expression is limited to leukocytes (Table 1) and BLT2 is ubiquitously found on many cell types. On mouse cells, BLT1 expression is detected on leukocytes and BLT2 is found on intestinal epithelium and keratinocytes [24, 25]. BLT1 can be coupled to Gαi or Gαq that culminate to decrease cyclic AMP (cAMP) levels and increase intracellular calcium levels, respectively [26]. We have previously shown that BLT1 utilizes mainly Gαi to enhance antimicrobial effector functions in alveolar macrophages [26]. Figure 1 demonstrates a schematic of LTB4/BLT1-induced effector functions in innate immune cells. The low affinity receptor BLT2 is also expressed in phagocytes, but its role in host defense is poorly studied. Previously, we have shown blocking BLT2 does not influence phagocytosis and bacterial killing in alveolar macrophages [27]. However, BLT2 might be a relevant receptor for other antimicrobial effector functions in different organs [24]. Recently, Zhang and Brown have shown that in the absence of BLT1, BLT2 enhances macrophage Borrelia burgdorferi phagocytosis. It remains to be determined whether BLT2 amplifies neutrophil chemotaxis to the site of infection and whether BLT2 controls antimicrobial peptide production in the skin or in the gut. However, BLT2 regulation and actions are out of the scope of this review.
Fig. 1. Host defense mechanisms enhanced by LTB4/BLT1 axis during microbial infection.
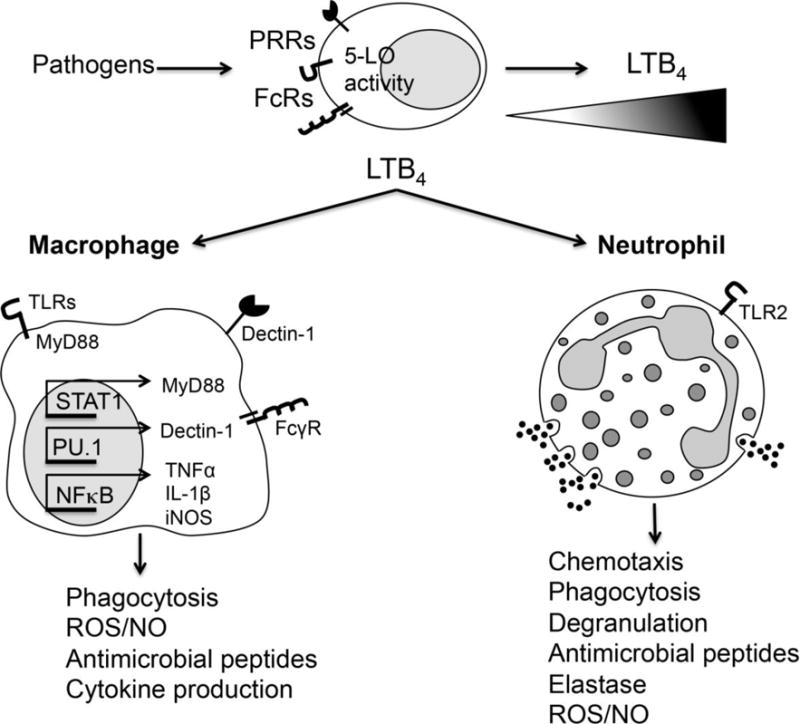
Upper panel. Upon infection, microbial recognition triggers 5-LO activation to generate LTB4 in phagocytes. Lower panel. LTB4 amplifies macrophage and neutrophil effector function by enhancing the actions of different PRRs and FcR.
3. Effects of LTB4 on host defense mechanisms
3.1 Migration and chemotaxis
Neutrophils are the first cells recruited to the site of infection or injury. There are various steps involved in successful recruitment of neutrophils: rolling, adhesion, and transendothelial migration [28]. LTB4 is well known for its role as a neutrophil chemoattractant from distant sites to the site of inflammation [29–31]. Also, LTB4 participates with chemokine gradients to further enhance neutrophil chemotaxis towards other chemoattractants such as fMLP, C5a, and heme [32–34]. Neutrophils from mice lacking BLT1 are not able to swarm and cluster to a focal damage site. BLT1−/− neutrophils have smaller neutrophil clusters than wild type neutrophils [35], demonstrating the importance of LTB4/BLT1 signaling in neutrophil accumulation. Additionally, neutrophil recruitment is not unidirectional. Reverse transendothelial migration has been reported where neutrophils reenter the blood stream after migration to the site of infection or injury [36]. In a model of ischemia-reperfusion injury, LTB4 and neutrophil elastase compose an important axis that drives reverse transendothelial migration. Neutrophils that reenter the vasculature are able to migrate to secondary organs and have the potential to cause injury [37].
3.2 Phagocytosis
We, and others, have shown that LTB4/BLT1 signaling amplifies the actions of different signaling components required for ingestion of particles. The first demonstration that LTB4 enhances phagocytosis was shown by Wirth et al using a model of Trypanosoma cruzi infection [38]. After this, Dr. Peters-Golden’s group pioneered in demonstrating the role of endogenous LTs in amplifying phagocytosis of antibody-opsonized targets [39, 40]. Both genetic and pharmacologic blockage of LTB4/BLT1 actions greatly reduces ingestion of a myriad of pathogens, including both gram-positive bacteria (Streptococcus pneumoniae) [27], gram-negative bacteria (Klebsiella pneumoniae [41]), fungi (Histoplasma capsulatum [42, 43], Candida albicans [44], and Paracoccidioides braziliensis [45]), and parasites (Leishmania braziliensis [46], L. amazonensis [47], and T. cruzi [38, 48]).
In addition to this extensive list of pathogens, the molecular mechanisms involved in LTB4-mediated amplification of phagocytosis have been studied. BLT1 signaling enhances activation of Syk, a protein tyrosine kinase, important for FcγR-mediated phagocytosis [26, 49]. When macrophages are challenged with IgG-opsonized targets, LTB4 enhances phagocytosis of IgG-RBC in an FcγR1-dependent manner. This enhancement is attributed to the association of BLT1 with lipid raft formation and heightened signaling capabilities [50]. Furthermore, LTB4 amplifies phagocytosis by increasing phosphorylation of kinases involved in the formation of a phagocytic cup, such as PKC-α, PKC-δ, PI3K, and ERK1/2 [51, 52]. However, the hierarchical role of these kinases in amplifying phagocytosis remains to be determined. Molecular studies regarding the signaling programs involved in the phagocytosis of nonopsonized pathogens are scarcer than opsonized targets. It has been shown that LTB4 enhances C. albicans phagocytosis via activation of GαI-mediated PKC-δ and PI3K activation culminating in F-actin polymerization [53]. LT enhancement of yeast phagocytosis involves the activation of PKC and PI3K, with subsequent activation of LIM Kinase, decreased cofilin-1 activation, and ultimately, F-actin assembly [53]. Furthermore, LTB4 enhances phagocytosis of fungi by increasing the expression of dectin-1, a main phagocytic receptor detecting fungal pathogens [42]. LTB4-mediated dectin-1 expression is dependent on GM-CSF production and activation of the transcription factor PU.1 [44].
3.3 Pathogen killing mechanisms
We, and others, have extensively shown that LTB4 is a potent neutrophil activator [41, 54–56]. 5-LO−/− neutrophils or neutrophils treated with leukotriene synthesis inhibitors have significantly lower ingestion of serum-opsonized K. pneumoniae [41]. Exogenous LTB4 treatment restores phagocytic capabilities in 5-LO deficient neutrophils [40, 45]. After phagocytosis, neutrophils kill pathogens by producing toxic components within granules that are released in a process known as neutrophil degranulation [57]. When human neutrophils are infected in vitro with the parasite L. amazonensis, endogenous and exogenous LTB4 promotes neutrophil degranulation [47]. Also, LTB4 induces release of myeloperoxidase [55] and elastase in neutrophils [37, 58]. LTB4 treatment of mice infected with influenza virus induced production of antimicrobial peptides β-defensin 3 and Cramp [59], the mouse ortholog of human cathelicidin LL-37. In human neutrophils, LTB4 induces the production of LL-37 in a dose-dependent manner [56].
Reactive oxygen and nitrogen species (ROS and RNS) production is another important antimicrobial effector function that phagocytes use to kill pathogens. LTB4 enhances the production of NADPH oxidase-dependent ROS generation [54, 60]. We, and others, have shown that LTB4 enhances ROS-dependent NADPH oxidase activation via phosphorylation of the cytosolic subunit p47phox. This activation is dependent on PKC-δ [54], ERK-1/2 and PI3K activation [61]. LTB4 is part of a positive-feedback loop in human neutrophils infected with L. amazonensis, which is important to kill parasites [47]. Similar to neutrophils, macrophages produce a variety of different microbicidal molecules such as ROS, RNS, antimicrobial peptides, and indoleamine deoxygenase (IDO). LTB4 is known to enhance the generation of most microbidicidal molecules mentioned above. LTB4-mediated ROS production in macrophages is dependent on PKC-δ-mediated p47phox and p40phox phosphorylation and membrane translocation in alveolar macrophages challenged with opsonized K. pneumoniae [52]. Furthermore, we also showed that aerosolized LTB4 increases p47phox expression and membrane translocation during Streptococcus pneumoniae lung infection [62]. Additionally, our unpublished results show that macrophages from BLT1−/− mice have impaired phagocytosis and killing of gram-positive methicillin-resistant Staphylococcus aureus (MRSA), which correlates with diminished ROS production in macrophages from the BLT1 deficient mice.
LTB4-dependent RNS production is dependent on NFκB and STAT1 activation [46]. LTB4 treatment further enhances the production of nitric oxide (NO) in murine macrophages, which improves killing of different pathogens such as L. amazonensis and T. cruzi [52, 63]. It remains to be determined whether LTB4 enhances noncanonical microbicidal effectors in macrophages such as antimicrobial peptides, tryptophan depletion (via IDO activation), GTPases, and transferrin receptor activation.
3.4 PRR activation and cytokine generation
TLRs are known to detect both PAMPs and danger associated molecular patterns (DAMPs). There are 11 TLRs in human and 13 in mouse. TLRs can be found on the cell surface of immune cells or in endosomes located within cells. The role of LTB4 in TLR activation has been suggested [64–66]. TLR signaling induces pro-inflammatory cytokine responses [67, 68]. TLR2 is a cell surface receptor that senses peptidoglycan molecules commonly found on gram-positive bacteria and TLR9 is an endosomal receptor that detects DNA [67, 68]. LTB4 stimulation upregulates the expression of TLR2 and TLR9 in human neutrophils [69]. Enhanced expression of TLRs could allow for neutrophils to better sense various pathogens and induce a stronger signaling cascade, which allows for a more potent immune response. Furthermore, LTB4 is also known to amplify the actions of different PRRs. TLR activation is dependent on different adaptors such as myeloid differentiation factor 88 (MyD88) and TIR-domain-containing adapter-inducing interferon-β (TRIF). Most TLRs except TLR3 utilize MyD88, and only TLR3 and TLR4 utilize TRIF. MyD88 activation is followed by phosphorylation of downstream components such as IRAKs and TAK-1. LTB4 enhances MyD88 expression and MyD88-dependent activation of NFκB, which are able to intensify the signaling potential of TLRs and other PPRs [64]. The mechanisms involved in LTB4-enhanced MyD88 expression relies on enhancing the activation of STAT1, the main transcription factor responsible for MyD88 expression [70]. BLT1 activation leads to mRNA degradation of suppressor of cytokine signaling 1 (SOCS1), the major STAT1 negative regulator, which contributes to enhanced MyD88 expression and subsequent enhancement of TLR-mediated macrophage activation [64]. Furthermore, it has been shown that LTB4 amplifies the phosphorylation of IRAK and TAK-1 in human neutrophils [66]. In all circumstances, through TLR and PRR activation, LTB4 amplifies the induction of cytokine production. LTB4/BLT1 enhancement of inflammatory cytokine production has been extensively studied. LTB4 is known to induce the cytokines GM-CSF, TNF-α, IL-6 and IL-1β and the chemokines KC, MCP-1, CXCL1, and CXCL2 that are thought to enhance inflammatory responses [71, 72].
Another layer of immune regulation induced by LTB4 is the expression of microRNAs (small ~22 nucleotides in length) that inhibits mRNA degradation or translation. We have shown that 5-LO deficient macrophages exhibit a decreased expression of a specific set of inflammatory microRNAs (inflammatory regulon). Among these microRNAs, we detected decreased levels of miR-155, miR-125b, and miR-146b in LT-deficient cells [73]. We have shown that these microRNAs specifically bind to SOCS1 3′UTR, restrict SOCS1 expression and enhance MyD88 expression [64]. Therefore, LTB4/BLT1 axis provides very potent and pleiotropic signals by decreasing microRNA expression and allowing inflammatory programs to be elicited in macrophages.
4. Aberrant LTB4 is detrimental to host defense
Chronic inflammatory morbidities are accompanied by aberrant LTB4 production that is thought to negatively influence disease pathogenesis. Among the diseases that LTB4 plays a detrimental role include type 1 and type 2 diabetes, arthritis, and atherosclerosis [74–80]. The bad reputation of LTB4 comes from its capacity to maintain inflammatory programs in monocytes/macrophages and prolonging neutrophil recruitment to the inflammatory foci. LTB4/BLT1 blockade is expected to dampen inflammatory responses and restore tissue homeostasis during chronic inflammation.
Besides increased production of LTB4, chronic inflammatory diseases are often associated with co-morbidities, including the major threat of increased susceptibility to infections. Although LTB4 plays a beneficial role in promoting pathogen clearance, the counterintuitive effect of LTB4 in chronic inflammatory diseases could be explained by the overwhelming inflammatory response and lack of proper phagocyte response to pathogens. Abundant LTB4 production and detrimental host defense responses have been associated in a zebrafish model of Mycobacterium marinum infection. The authors showed that LTA4 hydrolase (LTA4H), the enzyme that converts LTA4 to LTB4, is an important susceptibility factor involved in Mycobacterium infection. Zebrafish expressing hyperactive LTA4H are hypersusceptible to M. marinum infection [81]. Additionally, overexpression of LTA4H produces high levels of LTB4 that drive aberrant TNFα production and uncontrolled mycobacterial infections [82]. Furthermore, zebrafish expressing the enzyme that inactivates LTB4, leukotriene B4 dehydrogenase/prostaglandin reductase 1 (LTB4DH/PTGR1), exhibit lower bacterial numbers than WT zebrafish. The susceptibility to infection of LTB4DH mutant animals can be reversed with BLT1 antagonism, further implicating LTB4 as detrimental to host defense during Mycobacteria infection in zebrafish [83].
Exaggerated LTB4 is also detrimental to systemic infection (sepsis). 5-LO−/− mice or pharmacological inhibition of BLT1 actions protect mice during cecum ligation and puncture (CLP). 5-LO−/− and WT mice treated with a LT synthesis inhibitor have drastically lower levels of neutrophil infiltrates and lower levels of inflammatory cytokines such as IL-1β [84]. During CLP-induced sepsis, treatment with the BLT1 antagonist U-75302 decreases lung injury [85] as evidenced by decreased neutrophil recruitment. Therapeutically regulating 5-LO products or blocking BLT1 during or after sepsis may help prevent organ injury. Aberrant LTB4 production also renders diabetic mice more susceptible to sepsis than nondiabetic animals. We have shown that increased LTB4 drives MyD88 expression and mediates chronic systemic inflammation in diabetic mice. Diabetic BLT1−/− mice are protected from sepsis, which correlates with lower inflammatory cytokine production and decreased MyD88 expression.
Given the pleiotropic effects of LTB4 on amplifying the actions of immune receptors by influencing intracellular programs and gene transcription in macrophage and neutrophil activation [86], exaggerated LTB4 levels could be influencing both the actions of PRR (via increased expression of MyD88 [64, 73]), cytokine (GMCSF,TNF-a and IL1b [44, 64, 87]), and phagocytic receptors (FcRs and dectin receptor [50, 52]) and transcription factors (NFkB, AP1 and PU.1 [44, 64, 73]) to elicit inflammatory programs and cause organ injury and poor host defense.
The mechanisms involved in aberrant LTB4 production and poor systemic and localized host defense remains to be determined. However, preliminary data from our laboratory suggest that local uncontrolled LTB4 production drives a robust production of chemokines and cytokines along with overwhelming neutrophil migration to the site of infection that releases inflammatory mediators that cause tissue damage. Furthermore, exaggerated BLT1 activation seems to impair clearance of dead cells by controlling the expression of don’t eat me signals, such as CD47 [88]. When apoptotic cells are not cleared properly, these cells become necrotic and secrets many danger associated molecular patterns (DAMPs) [89, 90]. DAMP secretion might lead to LTB4 production which could be part of an amplification loop involved in chronic inflammation and lack of bacterial clearance observed in diabetes. Whether these molecular events are involved in enhanced susceptibility to skin infections in people with diabetes remains to be determined and are an active research program in our laboratory.
5. Manipulating LTB4 levels/actions as a therapeutic potential to control host defense
5.1 Enhance LTB4 effects
There are many potential advantages to using LTB4 in immunotherapeutic protocols, which include: (i) generation of proteins is relatively expensive, time consuming and subject to contamination with products of the vectors used to generate the protein, while lipids can be produced quickly and with a high degree of purity; (ii) because lipids exhibit are known to have a short half-life, it offers flexibility and precision in controlling localized or systemic actions; (iii) LTB4 is safe to be administered to the human lung in vivo; and (iv) LTB4 could amplify initial antimicrobial responses by enhancing pathogen recognition and phagocytosis, release of NADPH oxidase-derived ROS, and pro-inflammatory responses through MyD88 expression and activation of inflammatory transcription factors.
Treatment protocols employing exogenous LTB4 can be greatly beneficial to patients known to exhibit attenuated LT synthesis such as in malnutrition [91], cigarette smoking [92], vitamin D deficiency [93], HIV infection [94], and bone marrow transplantation [95]. These patients are more susceptible to infection. Therefore, adding back LTB4 to immunodeficient patients could potentially restore phagocyte response and favor appropriate host defense.
We, and others, have starting paving the way to establish treatment protocols to determine the therapeutic effects of LTB4 in host defense. Exogenous LTB4 treatment is able to boost effector functions in wild type mice. In a model of Streptococcus pneumoniae lung infections, aerosolized LTB4 treatment was effective in restoring host defense mechanisms in 5-LO−/− mice and enhancing effector functions in wild type mice [62]. Intravenous injection of LTB4 in macaques enhances plasma levels of the antimicrobial peptide α-defensins. Plasma from the treated macaques can neutralize pathogens ex vivo [96]. LTB4 treatment during influenza virus infection reduces viral titers compared to untreated mice [59]. The reduction in viral titers correlates with enhanced levels of the cathelicidin-related antimicrobial peptide (Cramp). Neutrophils are a major source of Cramp so when mice are depleted of neutrophils, exogenous LTB4 treatment during influenza infection is unable to control viral load [59].
A potential pitfall is the fact that LTB4 could lead to overwhelming recruitment of neutrophils, which may contribute to tissue injury. Also, the stability and safety of LTB4 in vivo is a concern, but its been shown that bronchoscopy instillation of LTB4 into the airways of normal human subjects elicited a marked influx of neutrophils. Its inhalation proved to be well tolerated and caused no adverse effects on blood pressure, pulmonary function, or bronchial responsiveness [97].
The importance of LTB4 during infections is clear and exogenous treatment with LTB4 may be a potential therapeutic strategy. However, high LTB4 levels during some infections or in chronic inflammatory diseases may need to be blunted to limit inflammation and alternative therapeutic strategies to prevent infection-mediated organ injury are necessary.
5.2 Preventing exaggerated LTB4 actions in host defense
Overwhelming production of inflammatory mediators and reactive oxygen species are known to be detrimental to host defense in different models of infection [98–101]. Therapeutic strategies to block the actions of inflammatory mediators could also restore protective host defense. In our laboratory, we are focusing our efforts in understanding whether preventing BLT1 and/or BLT2 actions could be an important tool to control exaggerated inflammatory response and poor host defense in people with preexisting chronic inflammatory conditions, such as diabetes, obesity, asthma, rheumatoid arthritis and elderly. It is known that people with diabetes are more susceptible to numerous infections, including systemic, respiratory, and skin infections [2–4]. Indeed, both innate and adaptive immune cells from diabetics have impaired functions including poor phagocytosis and killing of pathogens [102, 103]. We have shown that macrophages from diabetic mice produce higher levels of LTB4 than from control mice even under basal conditions [65]. Although seemingly counterintuitive, the mechanism by which people with diabetes are more susceptible to infections may operate in a similar manner to other infections where high levels of LTB4 is detrimental to host defense [81, 83]. We detected higher levels of LTB4 in the serum of septic and diabetic mice when compared to the levels of LTB4 detected in septic and nondiabetic mice. Diabetic mice treated with the 5-LO inhibitor AA-861 greatly improve survival during sepsis. The increase in survival correlates with reduction in IL-1β [65]. Since LTB4 is able to enhance IL-1β levels through inflammasome activation [104] and that LTB4/BLT1 signaling enhances MyD88 and NFκB activities [64], it is possible that LTB4 synthesis inhibition or actions prevents overwhelming Toll-interleukin receptors, such as IL1R, IL18R1 and TLR actions prevents organ damage. Therefore, a potential therapeutic opportunity to treat sepsis in diabetic mice could rely on preventing overwhelming inflammation with a BLT1 antagonist along with antibiotics to prevent bacterial growth.
Our unpublished data also show the detrimental role of aberrant LTB4 on host defense in diabetic mice in a model of local infection. Diabetic mice are more susceptible to MRSA skin infection than nondiabetic mice. Diabetic mice infected with MRSA have increased LTB4 production in the skin, which correlates with uncontrolled production of inflammatory mediators and neutrophil migration. Topical treatment with a BLT1 antagonist improves skin host defense in diabetic mice, as evidenced by decreased lesion size and bacterial numbers (data not shown).
6. Conclusion
LTB4 is a homeostatic determinant for optimal host defense in healthy individuals by driving pleiotropic actions on phagocytes that include phagocyte chemotaxis, amplifying macrophage/neutrophil antimicrobial effector functions, and production of cytokines. However, during chronic diseases characterized by aberrant LTB4 levels, excessive LTB4 could be responsible for impaired host defense mechanisms. In the era of antibiotic-resistant pathogens, new therapeutic strategies are critically needed. Modulating levels of LTB4 to either enhance or limit immune responses as appropriate is a potential strategy that could be used as a single agent or in combination with antibiotics for an added benefit. There is a great need of further research in areas of inflammation regulation under homeostatic conditions and during infections as well as understanding how chronic illnesses alter these immune responses. This will allow for the development of customized immunomodulatory therapies.
Highlights.
LTB4 triggers signaling programs necessary for effective clearance of pathogens
LTB4/BLT1 signaling enhances innate and adaptive immune receptor effector functions
LTB4 can therapeutically boost host defense in settings of host vulnerability
Aberrant LTB4 levels observed in chronic inflammation is harmful to host defense
LTB4 synthesis/actions blockage restores host defense in chronic inflammatory diseases
Acknowledgments
We acknowledge financial support from NIH Immunology and Infectious Diseases T32 AI060519 (SLB), R01 HL124159 (CHS), and R03 AI110990-01A1 (CHS).
Abbreviations
- LT
leukotriene
- PAMP
pathogen associated molecular patterns
- PRR
pattern recognition receptor
- AA
arachidonic acid
- LO
lipoxygenase
- cPLA2
cytosolic phospholipase A2
- FLAP
5-lipoxygenase-activating protein
- cysLT
cysteinyl LT
- BLT
B leukotriene receptor
- cAMP
cyclic adenosine monophosphate
- PKC
protein kinase C
- ERK
extracellular signal-related kinase
- PI3K
phosphoinositide 3-kinase
- NFκB
nuclear factor kappa B
- AP1
activator protein 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dhainaut JF, Claessens YE, Janes J, Nelson DR. Underlying disorders and their impact on the host response to infection. Clin Infect Dis. 2005;41(Suppl 7):S481–9. doi: 10.1086/432001. [DOI] [PubMed] [Google Scholar]
- 2.Aswani SM, Chandrashekar U, Shivashankara K, Pruthvi B. Clinical profile of urinary tract infections in diabetics and non-diabetics. Australas Med J. 2014;7(1):29–34. doi: 10.4066/AMJ.2014.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lipsky BA, Tabak YP, Johannes RS, Vo L, Hyde L, Weigelt JA. Skin and soft tissue infections in hospitalised patients with diabetes: culture isolates and risk factors associated with mortality, length of stay and cost. Diabetologia. 2010;53(5):914–23. doi: 10.1007/s00125-010-1672-5. [DOI] [PubMed] [Google Scholar]
- 4.Casqueiro J, Casqueiro J, Alves C. Infections in patients with diabetes mellitus: A review of pathogenesis. Indian J Endocrinol Metab. 2012;16(Suppl 1):S27–36. doi: 10.4103/2230-8210.94253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Werz O. 5-lipoxygenase: cellular biology and molecular pharmacology. Curr Drug Targets Inflamm Allergy. 2002;1(1):23–44. doi: 10.2174/1568010023344959. [DOI] [PubMed] [Google Scholar]
- 6.Nolfo R, Rankin JA. U937 and THP-1 cells do not release LTB4, LTC4, or LTD4 in response to A23187. Prostaglandins. 1990;39(2):157–65. doi: 10.1016/0090-6980(90)90072-4. [DOI] [PubMed] [Google Scholar]
- 7.Sala A, Folco G, Murphy RC. Transcellular biosynthesis of eicosanoids. Pharmacol Rep. 2010;62(3):503–10. doi: 10.1016/s1734-1140(10)70306-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McGee JE, Fitzpatrick FA. Erythrocyte-neutrophil interactions: formation of leukotriene B4 by transcellular biosynthesis. Proc Natl Acad Sci U S A. 1986;83(5):1349–53. doi: 10.1073/pnas.83.5.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iversen L, Fogh K, Ziboh VA, Kristensen P, Schmedes A, Kragballe K. Leukotriene B4 formation during human neutrophil keratinocyte interactions: evidence for transformation of leukotriene A4 by putative keratinocyte leukotriene A4 hydrolase. J Invest Dermatol. 1993;100(3):293–8. doi: 10.1111/1523-1747.ep12469865. [DOI] [PubMed] [Google Scholar]
- 10.Breton J, Woolf D, Young P, Chabot-Fletcher M. Human keratinocytes lack the components to produce leukotriene B4. J Invest Dermatol. 1996;106(1):162–7. doi: 10.1111/1523-1747.ep12329890. [DOI] [PubMed] [Google Scholar]
- 11.Janssen-Timmen U, Vickers PJ, Wittig U, Lehmann WD, Stark HJ, Fusenig NE, Rosenbach T, Radmark O, Samuelsson B, Habenicht AJ. Expression of 5-lipoxygenase in differentiating human skin keratinocytes. Proc Natl Acad Sci U S A. 1995;92(15):6966–70. doi: 10.1073/pnas.92.15.6966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feinmark SJ. The role of the endothelial cell in leukotriene biosynthesis. Am Rev Respir Dis. 1992;146(5 Pt 2):S51–5. doi: 10.1164/ajrccm/146.5_Pt_2.S51. [DOI] [PubMed] [Google Scholar]
- 13.Capra V, Rovati GE, Mangano P, Buccellati C, Murphy RC, Sala A. Transcellular biosynthesis of eicosanoid lipid mediators. Biochim Biophys Acta. 2015;1851(4):377–82. doi: 10.1016/j.bbalip.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 14.Peters-Golden M, Brock TG. 5-Lipoxygenase and the Nucleus: Where, When, How, and Why? In: Samuelsson B, Paoletti R, Folco GC, Granström E, Nicosia S, editors. Advances in Prostaglandin and Leukotriene Research: Basic Science and New Clinical Applications. Springer; Netherlands, Dordrecht: 2001. pp. 9–15. [Google Scholar]
- 15.Glover S, de Carvalho MS, Bayburt T, Jonas M, Chi E, Leslie CC, Gelb MH. Translocation of the 85-kDa phospholipase A2 from cytosol to the nuclear envelope in rat basophilic leukemia cells stimulated with calcium ionophore or IgE/antigen. J Biol Chem. 1995;270(25):15359–67. doi: 10.1074/jbc.270.25.15359. [DOI] [PubMed] [Google Scholar]
- 16.Peters-Golden M, McNish RW. Redistribution of 5-lipoxygenase and cytosolic phospholipase A2 to the nuclear fraction upon macrophage activation. Biochem Biophys Res Commun. 1993;196(1):147–53. doi: 10.1006/bbrc.1993.2227. [DOI] [PubMed] [Google Scholar]
- 17.Woods JW, Coffey MJ, Brock TG, Singer II, Peters-Golden M. 5-Lipoxygenase is located in the euchromatin of the nucleus in resting human alveolar macrophages and translocates to the nuclear envelope upon cell activation. J Clin Invest. 1995;95(5):2035–46. doi: 10.1172/JCI117889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peters-Golden M. Cell biology of the 5-lipoxygenase pathway. Am J Respir Crit Care Med. 1998;157(6 Pt 2):S227–31. discussion S231–2, S247–8. [PubMed] [Google Scholar]
- 19.Peters-Golden M, Brock TG. 5-Lipoxygenase and FLAP, Prostaglandins. Leukotrienes and Essential Fatty Acids. 2003;69(2–3):99–109. doi: 10.1016/s0952-3278(03)00070-x. [DOI] [PubMed] [Google Scholar]
- 20.Gosselin J, Borgeat P. Epstein-Barr virus modulates 5-lipoxygenase product synthesis in human peripheral blood mononuclear cells. Blood. 1997;89(6):2122–30. [PubMed] [Google Scholar]
- 21.Grone M, Scheffer J, Konig W. Modulation of leukotriene generation by invasive bacteria. Immunology. 1992;77(3):400–7. [PMC free article] [PubMed] [Google Scholar]
- 22.Brock TG, Peters-Golden M. Activation and regulation of cellular eicosanoid biosynthesis. Scientific World Journal. 2007;7:1273–84. doi: 10.1100/tsw.2007.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Radmark O, Samuelsson B. 5-Lipoxygenase: mechanisms of regulation. J Lipid Res. 2009;50(Suppl):S40–5. doi: 10.1194/jlr.R800062-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yokomizo T. Two distinct leukotriene B4 receptors, BLT1 and BLT2. J Biochem. 2015;157(2):65–71. doi: 10.1093/jb/mvu078. [DOI] [PubMed] [Google Scholar]
- 25.Tager AM, Luster AD. BLT1 and BLT2: the leukotriene B(4) receptors. Prostaglandins Leukot Essent Fatty Acids. 2003;69(2–3):123–34. doi: 10.1016/s0952-3278(03)00073-5. [DOI] [PubMed] [Google Scholar]
- 26.Peres CM, Aronoff DM, Serezani CH, Flamand N, Faccioli LH, Peters-Golden M. Specific leukotriene receptors couple to distinct G proteins to effect stimulation of alveolar macrophage host defense functions. J Immunol. 2007;179(8):5454–61. doi: 10.4049/jimmunol.179.8.5454. [DOI] [PubMed] [Google Scholar]
- 27.Soares EM, Mason KL, Rogers LM, Serezani CH, Faccioli LH, Aronoff DM. Leukotriene B4 enhances innate immune defense against the puerperal sepsis agent Streptococcus pyogenes. J Immunol. 2013;190(4):1614–22. doi: 10.4049/jimmunol.1202932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choi EY, Santoso S, Chavakis T. Mechanisms of neutrophil transendothelial migration. Front Biosci (Landmark Ed) 2009;14:1596–605. doi: 10.2741/3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Samuelsson B, Dahlen SE, Lindgren JA, Rouzer CA, Serhan CN. Leukotrienes and lipoxins: structures, biosynthesis, and biological effects. Science. 1987;237(4819):1171–6. doi: 10.1126/science.2820055. [DOI] [PubMed] [Google Scholar]
- 30.Yokomizo T, Izumi T, Chang K, Takuwa Y, Shimizu T. A G-protein-coupled receptor for leukotriene B4 that mediates chemotaxis. Nature. 1997;387(6633):620–4. doi: 10.1038/42506. [DOI] [PubMed] [Google Scholar]
- 31.Woo CH, Yoo MH, You HJ, Cho SH, Mun YC, Seong CM, Kim JH. Transepithelial migration of neutrophils in response to leukotriene B4 is mediated by a reactive oxygen species-extracellular signal-regulated kinase-linked cascade. J Immunol. 2003;170(12):6273–9. doi: 10.4049/jimmunol.170.12.6273. [DOI] [PubMed] [Google Scholar]
- 32.Afonso PV, Janka-Junttila M, Lee YJ, McCann CP, Oliver CM, Aamer KA, Losert W, Cicerone MT, Parent CA. LTB4 is a signal-relay molecule during neutrophil chemotaxis. Dev Cell. 2012;22(5):1079–91. doi: 10.1016/j.devcel.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Monteiro AP, Pinheiro CS, Luna-Gomes T, Alves LR, Maya-Monteiro CM, Porto BN, Barja-Fidalgo C, Benjamim CF, Peters-Golden M, Bandeira-Melo C, Bozza MT, Canetti C. Leukotriene B4 mediates neutrophil migration induced by heme. J Immunol. 2011;186(11):6562–7. doi: 10.4049/jimmunol.1002400. [DOI] [PubMed] [Google Scholar]
- 34.Allendorf DJ, Yan J, Ross GD, Hansen RD, Baran JT, Subbarao K, Wang L, Haribabu B. C5a-mediated leukotriene B4-amplified neutrophil chemotaxis is essential in tumor immunotherapy facilitated by anti-tumor monoclonal antibody and beta-glucan. J Immunol. 2005;174(11):7050–6. doi: 10.4049/jimmunol.174.11.7050. [DOI] [PubMed] [Google Scholar]
- 35.Lammermann T, Afonso PV, Angermann BR, Wang JM, Kastenmuller W, Parent CA, Germain RN. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature. 2013;498(7454):371–5. doi: 10.1038/nature12175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hirano Y, Aziz M, Wang P. Role of reverse transendothelial migration of neutrophils in inflammation. Biol Chem. 2016;397(6):497–506. doi: 10.1515/hsz-2015-0309. [DOI] [PubMed] [Google Scholar]
- 37.Colom B, Bodkin JV, Beyrau M, Woodfin A, Ody C, Rourke C, Chavakis T, Brohi K, Imhof BA, Nourshargh S. Leukotriene B4-Neutrophil Elastase Axis Drives Neutrophil Reverse Transendothelial Cell Migration In Vivo. Immunity. 2015;42(6):1075–86. doi: 10.1016/j.immuni.2015.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wirth JJ, Kierszenbaum F. Stimulatory effects of leukotriene B4 on macrophage association with and intracellular destruction of Trypanosoma cruzi. J Immunol. 1985;134(3):1989–93. [PubMed] [Google Scholar]
- 39.Bailie MB, Standiford TJ, Laichalk LL, Coffey MJ, Strieter R, Peters-Golden M. Leukotriene-deficient mice manifest enhanced lethality from Klebsiella pneumonia in association with decreased alveolar macrophage phagocytic and bactericidal activities. J Immunol. 1996;157(12):5221–4. [PubMed] [Google Scholar]
- 40.Mancuso P, Standiford TJ, Marshall T, Peters-Golden M. 5-Lipoxygenase reaction products modulate alveolar macrophage phagocytosis of Klebsiella pneumoniae. Infect Immun. 1998;66(11):5140–6. doi: 10.1128/iai.66.11.5140-5146.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mancuso P, Nana-Sinkam P, Peters-Golden M. Leukotriene B4 augments neutrophil phagocytosis of Klebsiella pneumoniae. Infect Immun. 2001;69(4):2011–6. doi: 10.1128/IAI.69.4.2011-2016.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Secatto A, Rodrigues LC, Serezani CH, Ramos SG, Dias-Baruffi M, Faccioli LH, Medeiros AI. 5-Lipoxygenase deficiency impairs innate and adaptive immune responses during fungal infection. PLoS One. 2012;7(3):e31701. doi: 10.1371/journal.pone.0031701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Secatto A, Soares EM, Locachevic GA, Assis PA, Paula-Silva FW, Serezani CH, de Medeiros AI, Faccioli LH. The leukotriene B(4)/BLT(1) axis is a key determinant in susceptibility and resistance to histoplasmosis. PLoS One. 2014;9(1):e85083. doi: 10.1371/journal.pone.0085083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Serezani CH, Kane S, Collins L, Morato-Marques M, Osterholzer JJ, Peters-Golden M. Macrophage dectin-1 expression is controlled by leukotriene B4 via a GM-CSF/PU.1 axis. J Immunol. 2012;189(2):906–15. doi: 10.4049/jimmunol.1200257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Santos PC, Santos DA, Ribeiro LS, Fagundes CT, Paula TPde, Avila TV, Lde MBaltazar, Madeira MM, Rde CCruz, Dias AC, Machado FS, Teixeira MM, Cisalpino PS, Souza DG. The pivotal role of 5-lipoxygenase-derived LTB4 in controlling pulmonary paracoccidioidomycosis. PLoS Negl Trop Dis. 2013;7(8):e2390. doi: 10.1371/journal.pntd.0002390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morato CI, da Silva IA, Jr, Borges AF, Dorta ML, Oliveira MA, Jancar S, Serezani CH, Ribeiro-Dias F. Essential role of leukotriene B4 on Leishmania (Viannia) braziliensis killing by human macrophages. Microbes Infect. 2014;16(11):945–53. doi: 10.1016/j.micinf.2014.08.015. [DOI] [PubMed] [Google Scholar]
- 47.Tavares NM, Araujo-Santos T, Afonso L, Nogueira PM, Lopes UG, Soares RP, Bozza PT, Bandeira-Melo C, Borges VM, Brodskyn C. Understanding the mechanisms controlling Leishmania amazonensis infection in vitro: the role of LTB4 derived from human neutrophils. J Infect Dis. 2014;210(4):656–66. doi: 10.1093/infdis/jiu158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Talvani A, Machado FS, Santana GC, Klein A, Barcelos L, Silva JS, Teixeira MM. Leukotriene B(4) induces nitric oxide synthesis in Trypanosoma cruzi-infected murine macrophages and mediates resistance to infection. Infect Immun. 2002;70(8):4247–53. doi: 10.1128/IAI.70.8.4247-4253.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Canetti C, Hu B, Curtis JL, Peters-Golden M. Syk activation is a leukotriene B4-regulated event involved in macrophage phagocytosis of IgG-coated targets but not apoptotic cells. Blood. 2003;102(5):1877–83. doi: 10.1182/blood-2003-02-0534. [DOI] [PubMed] [Google Scholar]
- 50.Serezani CH, Aronoff DM, Sitrin RG, Peters-Golden M. FcgammaRI ligation leads to a complex with BLT1 in lipid rafts that enhances rat lung macrophage antimicrobial functions. Blood. 2009;114(15):3316–24. doi: 10.1182/blood-2009-01-199919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Campos MR, Serezani CH, Peters-Golden M, Jancar S. Differential kinase requirement for enhancement of Fc gammaR-mediated phagocytosis in alveolar macrophages by leukotriene B4 vs. D4. Mol Immunol. 2009;46(6):1204–11. doi: 10.1016/j.molimm.2008.11.024. [DOI] [PubMed] [Google Scholar]
- 52.Serezani CH, Aronoff DM, Jancar S, Mancuso P, Peters-Golden M. Leukotrienes enhance the bactericidal activity of alveolar macrophages against Klebsiella pneumoniae through the activation of NADPH oxidase. Blood. 2005;106(3):1067–75. doi: 10.1182/blood-2004-08-3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morato-Marques M, Campos MR, Kane S, Rangel AP, Lewis C, Ballinger MN, Kim SH, Peters-Golden M, Jancar S, Serezani CH. Leukotrienes target F-actin/cofilin-1 to enhance alveolar macrophage anti-fungal activity. J Biol Chem. 2011;286(33):28902–13. doi: 10.1074/jbc.M111.235309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Serezani CH, Aronoff DM, Jancar S, Peters-Golden M. Leukotriene B4 mediates p47phox phosphorylation and membrane translocation in polyunsaturated fatty acid-stimulated neutrophils. J Leukoc Biol. 2005;78(4):976–84. doi: 10.1189/jlb.1004587. [DOI] [PubMed] [Google Scholar]
- 55.Widegren H, Andersson M, Borgeat P, Flamand L, Johnston S, Greiff L. LTB4 increases nasal neutrophil activity and conditions neutrophils to exert antiviral effects. Respir Med. 2011;105(7):997–1006. doi: 10.1016/j.rmed.2010.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wan M, Sabirsh A, Wetterholm A, Agerberth B, Haeggstrom JZ. Leukotriene B4 triggers release of the cathelicidin LL-37 from human neutrophils: novel lipid-peptide interactions in innate immune responses. FASEB J. 2007;21(11):2897–905. doi: 10.1096/fj.06-7974com. [DOI] [PubMed] [Google Scholar]
- 57.Kobayashi Y. Neutrophil biology: an update. EXCLI J. 2015;14:220–7. doi: 10.17179/excli2015-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yoshimura K, Nakagawa S, Koyama S, Kobayashi T, Homma T. Roles of neutrophil elastase and superoxide anion in leukotriene B4-induced lung injury in rabbit. J Appl Physiol (1985) 1994;76(1):91–6. doi: 10.1152/jappl.1994.76.1.91. [DOI] [PubMed] [Google Scholar]
- 59.Gaudreault E, Gosselin J. Leukotriene B4 induces release of antimicrobial peptides in lungs of virally infected mice. J Immunol. 2008;180(9):6211–21. doi: 10.4049/jimmunol.180.9.6211. [DOI] [PubMed] [Google Scholar]
- 60.Wymann MP, von Tscharner V, Deranleau DA, Baggiolini M. The onset of the respiratory burst in human neutrophils. Real-time studies of H2O2 formation reveal a rapid agonist-induced transduction process. J Biol Chem. 1987;262(25):12048–53. [PubMed] [Google Scholar]
- 61.Woo CH, You HJ, Cho SH, Eom YW, Chun JS, Yoo YJ, Kim JH. Leukotriene B(4) stimulates Rac-ERK cascade to generate reactive oxygen species that mediates chemotaxis. J Biol Chem. 2002;277(10):8572–8. doi: 10.1074/jbc.M104766200. [DOI] [PubMed] [Google Scholar]
- 62.Mancuso P, Lewis C, Serezani CH, Goel D, Peters-Golden M. Intrapulmonary administration of leukotriene B4 enhances pulmonary host defense against pneumococcal pneumonia. Infect Immun. 2010;78(5):2264–71. doi: 10.1128/IAI.01323-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Serezani CH, Perrela JH, Russo M, Peters-Golden M, Jancar S. Leukotrienes Are Essential for the Control of Leishmania amazonensis Infection and Contribute to Strain Variation in Susceptibility. The Journal of Immunology. 2006;177(5):3201–3208. doi: 10.4049/jimmunol.177.5.3201. [DOI] [PubMed] [Google Scholar]
- 64.Serezani CH, Lewis C, Jancar S, Peters-Golden M. Leukotriene B4 amplifies NF-kappaB activation in mouse macrophages by reducing SOCS1 inhibition of MyD88 expression. J Clin Invest. 2011;121(2):671–82. doi: 10.1172/JCI43302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Filgueiras LR, Brandt SL, Wang S, Wang Z, Morris DL, Evans-Molina C, Mirmira RG, Jancar S, Serezani CH. Leukotriene B4-mediated sterile inflammation promotes susceptibility to sepsis in a mouse model of type 1 diabetes. Sci Signal. 2015;8(361):ra10. doi: 10.1126/scisignal.2005568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gaudreault E, Paquet-Bouchard C, Fiola S, Le Bel M, Lacerte P, Shio MT, Olivier M, Gosselin J. TAK1 contributes to the enhanced responsiveness of LTB(4)-treated neutrophils to Toll-like receptor ligands. Int Immunol. 2012;24(11):693–704. doi: 10.1093/intimm/dxs074. [DOI] [PubMed] [Google Scholar]
- 67.Dowling JK, Mansell A. Toll-like receptors: the swiss army knife of immunity and vaccine development. Clin Transl Immunology. 2016;5(5):e85. doi: 10.1038/cti.2016.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Leifer CA, Medvedev AE. Molecular mechanisms of regulation of Toll-like receptor signaling. J Leukoc Biol. 2016 doi: 10.1189/jlb.2MR0316-117RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gaudreault E, Gosselin J. Leukotriene B4 potentiates CpG signaling for enhanced cytokine secretion by human leukocytes. J Immunol. 2009;183(4):2650–8. doi: 10.4049/jimmunol.0804135. [DOI] [PubMed] [Google Scholar]
- 70.Croker BA, Kiu H, Nicholson SE. SOCS regulation of the JAK/STAT signalling pathway. Semin Cell Dev Biol. 2008;19(4):414–22. doi: 10.1016/j.semcdb.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huang L, Zhao A, Wong F, Ayala JM, Struthers M, Ujjainwalla F, Wright SD, Springer MS, Evans J, Cui J. Leukotriene B4 strongly increases monocyte chemoattractant protein-1 in human monocytes. Arterioscler Thromb Vasc Biol. 2004;24(10):1783–8. doi: 10.1161/01.ATV.0000140063.06341.09. [DOI] [PubMed] [Google Scholar]
- 72.Saiwai H, Ohkawa Y, Yamada H, Kumamaru H, Harada A, Okano H, Yokomizo T, Iwamoto Y, Okada S. The LTB4-BLT1 axis mediates neutrophil infiltration and secondary injury in experimental spinal cord injury. Am J Pathol. 2010;176(5):2352–66. doi: 10.2353/ajpath.2010.090839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang Z, Filgueiras LR, Wang S, Serezani AP, Peters-Golden M, Jancar S, Serezani CH. Leukotriene B4 enhances the generation of proinflammatory microRNAs to promote MyD88-dependent macrophage activation. J Immunol. 2014;192(5):2349–56. doi: 10.4049/jimmunol.1302982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Martinez-Clemente M, Claria J, Titos E. The 5-lipoxygenase/leukotriene pathway in obesity, insulin resistance, and fatty liver disease. Curr Opin Clin Nutr Metab Care. 2011;14(4):347–53. doi: 10.1097/MCO.0b013e32834777fa. [DOI] [PubMed] [Google Scholar]
- 75.Soumaya K. Molecular mechanisms of insulin resistance in diabetes. Adv Exp Med Biol. 2012;771:240–51. doi: 10.1007/978-1-4614-5441-0_19. [DOI] [PubMed] [Google Scholar]
- 76.Spite M, Hellmann J, Tang Y, Mathis SP, Kosuri M, Bhatnagar A, Jala VR, Haribabu B. Deficiency of the leukotriene B4 receptor, BLT-1, protects against systemic insulin resistance in diet-induced obesity. J Immunol. 2011;187(4):1942–9. doi: 10.4049/jimmunol.1100196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ahmadzadeh N, Shingu M, Nobunaga M, Tawara T. Relationship between leukotriene B4 and immunological parameters in rheumatoid synovial fluids. Inflammation. 1991;15(6):497–503. doi: 10.1007/BF00923346. [DOI] [PubMed] [Google Scholar]
- 78.Chen M, Lam BK, Kanaoka Y, Nigrovic PA, Audoly LP, Austen KF, Lee DM. Neutrophil-derived leukotriene B4 is required for inflammatory arthritis. J Exp Med. 2006;203(4):837–42. doi: 10.1084/jem.20052371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mathis S, Jala VR, Haribabu B. Role of leukotriene B4 receptors in rheumatoid arthritis. Autoimmun Rev. 2007;7(1):12–7. doi: 10.1016/j.autrev.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sanchez-Galan E, Gomez-Hernandez A, Vidal C, Martin-Ventura JL, Blanco-Colio LM, Munoz-Garcia B, Ortega L, Egido J, Tunon J. Leukotriene B4 enhances the activity of nuclear factor-kappaB pathway through BLT1 and BLT2 receptors in atherosclerosis. Cardiovasc Res. 2009;81(1):216–25. doi: 10.1093/cvr/cvn277. [DOI] [PubMed] [Google Scholar]
- 81.Tobin DM, Vary JC, Jr, Ray JP, Walsh GS, Dunstan SJ, Bang ND, Hagge DA, Khadge S, King MC, Hawn TR, Moens CB, Ramakrishnan L. The lta4h locus modulates susceptibility to mycobacterial infection in zebrafish and humans. Cell. 2010;140(5):717–30. doi: 10.1016/j.cell.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tobin DM, Roca FJ, Oh SF, McFarland R, Vickery TW, Ray JP, Ko DC, Zou Y, Bang ND, Chau TT, Vary JC, Hawn TR, Dunstan SJ, Farrar JJ, Thwaites GE, King MC, Serhan CN, Ramakrishnan L. Host genotype-specific therapies can optimize the inflammatory response to mycobacterial infections. Cell. 2012;148(3):434–46. doi: 10.1016/j.cell.2011.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tobin DM, Roca FJ, Ray JP, Ko DC, Ramakrishnan L. An enzyme that inactivates the inflammatory mediator leukotriene b4 restricts mycobacterial infection. PLoS One. 2013;8(7):e67828. doi: 10.1371/journal.pone.0067828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Monteiro AP, Soledade E, Pinheiro CS, Dellatorre-Teixeira L, Oliveira GP, Oliveira MG, Peters-Golden M, Rocco PR, Benjamim CF, Canetti C. Pivotal role of the 5-lipoxygenase pathway in lung injury after experimental sepsis. Am J Respir Cell Mol Biol. 2014;50(1):87–95. doi: 10.1165/rcmb.2012-0525OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li XJ, Fu HY, Yi WJ, Zhao YJ, Wang J, Li JB, Wang JF, Deng XM. Dual role of leukotriene B4 receptor type 1 in experimental sepsis. J Surg Res. 2015;193(2):902–8. doi: 10.1016/j.jss.2014.09.013. [DOI] [PubMed] [Google Scholar]
- 86.Peters-Golden M, Canetti C, Mancuso P, Coffey MJ. Leukotrienes: underappreciated mediators of innate immune responses. J Immunol. 2005;174(2):589–94. doi: 10.4049/jimmunol.174.2.589. [DOI] [PubMed] [Google Scholar]
- 87.Peters-Golden M, Henderson WR., Jr Leukotrienes N Engl J Med. 2007;357(18):1841–54. doi: 10.1056/NEJMra071371. [DOI] [PubMed] [Google Scholar]
- 88.Kojima Y, Volkmer JP, McKenna K, Civelek M, Lusis AJ, Miller CL, Direnzo D, Nanda V, Ye J, Connolly AJ, Schadt EE, Quertermous T, Betancur P, Maegdefessel L, Matic LP, Hedin U, Weissman IL, Leeper NJ. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature. 2016;536(7614):86–90. doi: 10.1038/nature18935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Piccinini AM, Midwood KS. DAMPening inflammation by modulating TLR signalling. Mediators Inflamm. 2010;2010 doi: 10.1155/2010/672395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pittman K, Kubes P. Damage-associated molecular patterns control neutrophil recruitment. J Innate Immun. 2013;5(4):315–23. doi: 10.1159/000347132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Skerrett SJ, Henderson WR, Martin TR. Alveolar macrophage function in rats with severe protein calorie malnutrition. Arachidonic acid metabolism, cytokine release, and antimicrobial activity. J Immunol. 1990;144(3):1052–61. [PubMed] [Google Scholar]
- 92.Balter MS, Toews GB, Peters-Golden M. Multiple defects in arachidonate metabolism in alveolar macrophages from young asymptomatic smokers. J Lab Clin Med. 1989;114(6):662–73. [PubMed] [Google Scholar]
- 93.Coffey MJ, Wilcoxen SE, Phare SM, Simpson RU, Gyetko MR, Peters-Golden M. Reduced 5-lipoxygenase metabolism of arachidonic acid in macrophages rrom 1,25-dihydroxyvitamin D3-deficient rats. Prostaglandins. 1994;48(5):313–29. doi: 10.1016/0090-6980(94)90031-0. [DOI] [PubMed] [Google Scholar]
- 94.Coffey MJ, Phare SM, George S, Peters-Golden M, Kazanjian PH. Granulocyte colony-stimulating factor administration to HIV-infected subjects augments reduced leukotriene synthesis and anticryptococcal activity in neutrophils. J Clin Invest. 1998;102(4):663–70. doi: 10.1172/JCI2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ballinger MN, McMillan TR, Moore BB. Eicosanoid regulation of pulmonary innate immunity post-hematopoietic stem cell transplantation. Arch Immunol Ther Exp (Warsz) 2007;55(1):1–12. doi: 10.1007/s00005-007-0001-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Flamand L, Tremblay MJ, Borgeat P. Leukotriene B4 Triggers the In Vitro and In Vivo Release of Potent Antimicrobial Agents. The Journal of Immunology. 2007;178(12):8036–8045. doi: 10.4049/jimmunol.178.12.8036. [DOI] [PubMed] [Google Scholar]
- 97.Martin TR, Pistorese BP, Chi EY, Goodman RB, Matthay MA. Effects of leukotriene B4 in the human lung. Recruitment of neutrophils into the alveolar spaces without a change in protein permeability. J Clin Invest. 1989;84(5):1609–19. doi: 10.1172/JCI114338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Puchta A, Naidoo A, Verschoor CP, Loukov D, Thevaranjan N, Mandur TS, Nguyen PS, Jordana M, Loeb M, Xing Z, Kobzik L, Larche MJ, Bowdish DM. TNF Drives Monocyte Dysfunction with Age and Results in Impaired Anti-pneumococcal Immunity. PLoS Pathog. 2016;12(1):e1005368. doi: 10.1371/journal.ppat.1005368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yin F, Banerjee R, Thomas B, Zhou P, Qian L, Jia T, Ma X, Ma Y, Iadecola C, Beal MF, Nathan C, Ding A. Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice. J Exp Med. 2010;207(1):117–28. doi: 10.1084/jem.20091568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Headley AS, Tolley E, Meduri GU. Infections and the inflammatory response in acute respiratory distress syndrome. Chest. 1997;111(5):1306–21. doi: 10.1378/chest.111.5.1306. [DOI] [PubMed] [Google Scholar]
- 101.Ng HP, Zhou Y, Song K, Hodges CA, Drumm ML, Wang G. Neutrophil-mediated phagocytic host defense defect in myeloid Cftr-inactivated mice. PLoS One. 2014;9(9):e106813. doi: 10.1371/journal.pone.0106813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Geerlings SE, Hoepelman AI. Immune dysfunction in patients with diabetes mellitus (DM) FEMS Immunol Med Microbiol. 1999;26(3–4):259–65. doi: 10.1111/j.1574-695X.1999.tb01397.x. [DOI] [PubMed] [Google Scholar]
- 103.Lecube A, Pachon G, Petriz J, Hernandez C, Simo R. Phagocytic activity is impaired in type 2 diabetes mellitus and increases after metabolic improvement. PLoS One. 2011;6(8):e23366. doi: 10.1371/journal.pone.0023366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Amaral FA, Costa VV, Tavares LD, Sachs D, Coelho FM, Fagundes CT, Soriani FM, Silveira TN, Cunha LD, Zamboni DS, Quesniaux V, Peres RS, Cunha TM, Cunha FQ, Ryffel B, Souza DG, Teixeira MM. NLRP3 inflammasome-mediated neutrophil recruitment and hypernociception depend on leukotriene B(4) in a murine model of gout. Arthritis Rheum. 2012;64(2):474–84. doi: 10.1002/art.33355. [DOI] [PubMed] [Google Scholar]