

Summary
Humans do not usually develop effective immunity to Staphylococcus aureus reinfection. Using a murine model that mimics human infection, we show that lack of protective immunity to S. aureus systemic reinfection is associated with robust IL-10 production and impaired protective Th17 responses. In dendritic-cell co-culture assays, priming with S. aureus promotes robust T-cell proliferation, but limits Th cells polarization and production of IL1-β and other cytokines important for Th1 and Th17 differentiation. We show that O-acetylation of peptidoglycan, a mechanism utilized by S. aureus to block bacterial cell-wall breakdown, limits the induction of pro-inflammatory signals required for optimal Th17 polarization. IL-10-deficiency in mice restores protective immunity to S. aureus infection, and adjuvancy with a staphylococcal peptidoglycan O-acetyltransferase mutant reduces IL-10, increases IL1-β, and promotes development of IL-17-dependent, Th cell-transferable protective immunity. Overall, our study suggests a mechanism whereby S. aureus modulates cytokines critical for induction of protective Th17 immunity.
eTOC
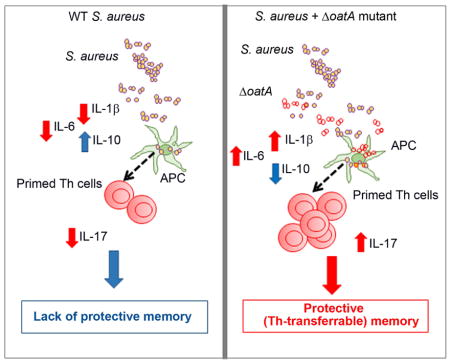
Humans do not develop robust protective immunity to S. aureus infection. Sanchez et al. show that mice, like humans, do not develop a protective Th17 response after bloodstream S. aureus infection. S. aureus, via modification of cell wall peptidoglycan, limits priming of Th17 cells and blocks development of protective immunity.
INTRODUCTION
S. aureus is a pathogen that induces significant morbidity and mortality worldwide. With the advent of methicillin-resistant S. aureus and the threat of antibiotic resistance, developing a vaccine against S. aureus has become a priority. Humans do not generally develop robust protective immunity upon S. aureus infection, as indicated by the finding that individuals can be infected with S. aureus many times throughout life (Karauzum and Datta, 2016). Although there is evidence that protective immunity could be acquired, for example, in subjects colonized nasally with S. aureus, protection is generally modest (Wertheim et al., 2004). The reason behind the lack of robust protection to S. aureus reinfection is unclear and of significant interest for the development of strategies to create efficacious vaccines against S. aureus.
Studies of the effects of S. aureus infection on T and B cells have uncovered a number of mechanisms whereby S. aureus interferes with the host adaptive immune response. Evasion factors include S. aureus superantigens that induce T cell anergy or deletion (Cantor et al., 1993), and S. aureus Protein A, which induces deletion of specific B cell populations (Goodyear and Silverman, 2003) in addition to preferential shifting of host antibody response and blocking of antibody-mediated phagocytosis (Rosenblatt et al., 1977). Additionally, S. aureus secretes an MHC-like molecule that is thought to drive Th2 differentiation (Lee et al., 2002) and toxins that induce T cell cytolysis or apoptosis (Alonzo et al., 2013). Although many of these bacterial strategies have been implicated in immune response evasion in S. aureus infection models, so far only Protein A has a confirmed role with a defined mechanism of promoting S. aureus reinfection (Falugi et al., 2013). Studies have shown a strong link between IL-17 deficiency in patients with hyper-IgE syndrome (defect in STAT3 phosphorylation) and severe S. aureus infections (Milner et al., 2008). Coupled with reports of the detrimental effects of IL-17 on S. aureus (Cho et al., 2010), the findings suggest that minimizing induction of IL-17 would be a logical strategy used by S. aureus to evade host immunity, as pathogens routinely adapt elaborate strategies to block host defenses that interfere with their survival. To date, S. aureus strategies targeting IL-17 have not been reported.
Numerous studies involving S. aureus reinfection have been published in which mice undergo repeat infections with a single strain of S. aureus and, surprisingly, develop significant protection from reinfection (Brown et al., 2015; Montgomery et al., 2014; Murphy et al., 2014; Sasaki et al., 2006). These studies have led to important insights on how the host limits S. aureus infection by promoting specific T and B cell responses. However, for the purpose of addressing why humans are not robustly protected from reinfection, these models are less informative. In the current study, we investigated potential mechanisms deployed by S. aureus to subvert host protective T cell responses.
RESULTS
Mice remain susceptible to S. aureus reinfections in a model that mimics human disease
Most humans experience S. aureus infections of varying severity throughout life, which are routinely controlled within a few days by a robust immune system or by antibiotic treatment. To mimic human infections, we used an intraperitoneal (i.p.) infection model that leads to spontaneous clearance of S. aureus after 3–4 days, and reinfected the mice 21 days after the first infection. For these experiments, we utilized a USA300 MRSA strain (LAC) which is a common cause of human infections. To enable comparison to an infection model that induces protective immunity, we performed parallel infection experiments using Group A streptococcus (GAS, M49 strain), which confers protective immunity against GAS of the same M-protein serotype in humans (Cunningham, 2000). We determined the highest inoculum of S. aureus or GAS to induce an infection without causing mortality for S. aureus LAC (1×107) and GAS M49 (5×106–1×107) and used those inocula for the remaining experiments. We confirmed that animals infected using those inocula clear the infection fully prior to reinfection on day 21 (usually 3–5 days after infection, data not shown). As shown in Figures 1A, 1B and S1A, GAS induces robust protection from reinfection as measured by CFU, body weight and LD50. S. aureus did not induce protective immunity after two or four consecutive infections in C57BL/6 mice (Figure 1A and 1C). Similar results were obtained in BALB/c mice (Figure S1B). Hence, this model mimics the observation in humans that S. aureus infection does not lead to the development of robust protective immunity.
Figure 1. Prior S. aureus infection does not induce protective immunity against reinfection.

(A) C57BL/6 mice (n=8–11) were injected i.p with 107 CFU of S. aureus (LAC USA300) GAS (M49) or PBS on d0, and re-injected on d21 with 107 CFU of either S. aureus or GAS. Bacteria from spleen and kidneys were recovered after 24hr. PBS/SA: injected with PBS on d0 and then injected with SA on d21. Dashed lines indicate limit of detection.
(B) C57BL/6 mice (n=10) were injected i.p with S. aureus (107), GAS (5×106), or PBS. The mice were inoculated after 21 days with S. aureus (4×107) or GAS (2×107) and monitored for survival.
(C) C57BL/6 mice (n=5) were infected i.p. once, twice (d0 and 21), or four times (d0, 21, 28 and 35) with S. aureus (107). Surviving CFU were enumerated 24hr after the last infection.
(D) Mice (n=5–6) were infected as in (A). Splenocytes were harvested 7 days after the last infection, stimulated with PMA, ionomycin and brefeldin A for 4hr and stained for surface CD4 and TCRβ, and intracellular IFN-γ and IL-17A. Shown are FACS plots and graphs of IFN-γ and IL-17A expression by CD4+TCRβ+ cells. Results are representative of 2 experiments.
(E) Mice (n=8–11) were infected as in (A) and splenocytes harvested 7 days after the last infection were stimulated with heat-killed S. aureus or GAS (MOI=10) for 48hr. Supernatants were analyzed by ELISA. Data are representative of 2 experiments. See also Figure S1. For (A), (C) and (D), each data point represents an individual mouse, bars denote median and dashed lines indicate the limit of detection.
For (E), data are plotted as mean ± SEM. Data analysis was performed using Mann-Whitney U-test for (A) and (D), ANOVA for (C), log rank test for (B) and Student’s t-test for (E). *p<0.05, **p<0.01, ***p<0.001.
S. aureus induces modest production of IFN-γ and IL-17A upon reinfection
We queried if protection (GAS infection) or lack of protection (S. aureus infection) in our model correlates to changes in cytokines associated with Th cell polarization in vivo. We observed that GAS reinfection enhanced production of IL-17A and IFN-γ in the spleen when compared to the initial GAS infection (Figures 1D and E). In contrast, S. aureus infection induced low levels of IL-17A and IFN-γ, which were not further increased upon reinfection, suggesting defective Th1 and Th17 memory responses. Conversely, S. aureus reinfection was associated with increased IL-10 production (Figure 1E). Further increasing the number of S. aureus reinfections did not lead to increase in IFN-γ or IL-17A expression in the spleen, consistent with impaired induction of protective immunity (Figure S1C). Protective immunity to GAS in our i.p. model was abolished when the infection was performed in IL-17−/− mice, corroborating the importance of IL17 in immunity against GAS reinfection (Figure S1D). Overall, these data suggest that GAS mounts robust protective IL17-dependent immune responses whereas S. aureus fails to induce protective immunity.
S. aureus-induced dendritic cell maturation leads to efficient proliferation but limited Th polarization in vitro
The limited Th17 and Th1 responses observed in our S. aureus reinfection model suggested the possibility that, in comparison to GAS, S. aureus would induce suboptimal priming of T cells by antigen presenting cells. Therefore, we studied initial priming of naïve CD4+ T cells isolated from OT-II transgenic Rag2−/− mice, using OVA peptide as a model antigen and bone marrow-derived dendritic cells (BMDC) infected in vitro with either GAS or S. aureus.
BMDC were incubated with either S. aureus or GAS, pulsed with OVA peptide and then co-cultured with OT-II TCR transgenic CD4+ T cells for 7 days prior to analysis. As shown in Figure 2A, OVA peptide-induced proliferation of T cells was not significantly different after DC priming with S. aureus or GAS. However, consistent with our in vivo findings, S. aureus-infected DC induced significantly lower amounts of IFN-γ and IL-17A than GAS-infected DC (Figure 2B). To determine if S. aureus and GAS differentially induce DC maturation, we measured expression of MHC II, CD80 and CD86 in S. aureus or GAS-infected DC. Expression of MHC II and CD80 was comparable in DC stimulated with GAS or S. aureus, but GAS induced higher expression of CD86 (Figure 2C), consistent with a previous study in human monocytes (Wang et al., 2012). Although CD86 has been show to favor Th17 differentiation in certain circumstances (Huang et al., 2012), at least one recent study (Kang et al., 2016) shows that Th17-inducing DC has low CD86 expression. In our in vitro co-culture system, it is likely that the reduced CD86 expression in S. aureus-infected DC did not influence Th differentiation, as we could not detect any differences in Th cell proliferation, which is required for Th cell terminal differentiation. However, this remains to be formally tested.
Figure 2. S. aureus induces suboptimal polarization of TH1 and TH17 cells associated with differential regulation of gene expression in DC.

(A)–(B) BMDC were cultured with medium only (NS – Non stimulated), S. aureus (LAC) or GAS (MOI=3) for 1hr, pulsed with OVA peptide (6hr) and then mixed with CFSE-labeled OTII transgenic CD4+ T cells. (A) CFSE dilution in gated CD4+ T cells at day 7 post stimulation and (B) IFN-γ and IL-17A in the culture supernatants after 7 days.
(C) MHCII, CD80 and CD86 expression on BMDC after 24hr of stimulation with GAS or S. aureus (MOI=10).
(D) Bone marrow-derived DC were left without any stimulation (NS) or incubated with S. aureus or GAS (MOI=3) for 8 hours and total RNA was extracted for RNAseq analysis. Top graph: Venn diagrams showing genes up- (left) or down- (right) regulated by S. aureus only (pink), GAS only (green) or both GAS and S. aureus (brown). Selected genes are listed for each group. Bottom graph: Fold change (over NS) for selected genes shown in (D).
(E) Cytokine production as measured by ELISA in the supernatant of DC treated as in (D) for 24 hours. All data are representative of at least 3 experiments except for the IL-23 ELISA which was performed twice. Data are shown as mean ± SEM and data analysis was performed using ANOVA. *p<0.05; **p<0.01. See also Figure S2 and Table S1 and S2.
Differential regulation of DC gene expression by S. aureus and GAS
To compare the gene profile of DC after stimulation with GAS or S. aureus, we performed transcriptome analysis of DC stimulated with either bacteria or media alone (No stimuli, NS). As shown in Figure 2D, both S. aureus and GAS induced significant changes in DC gene expression, however GAS infection altered the expression of substantially more genes (2921 total) than S. aureus infection (426 total) (adjusted p≤0.05 and fold change ≥2). GAS-infected DC had 1409 genes up regulated and 1512 genes downregulated whereas S. aureus-infected DC showed upregulation of 329 genes and downregulation of 97.
A total of 87 genes were downregulated by both S. aureus and GAS. These included important regulators of cytokine signaling such as SOCS2 (Hu et al., 2009; Jackson et al., 2004), IRF4 which have been previously associated with the regulation of DC maturation and function (Tussiwand et al., 2015) and MYC (Fig. 2D) previously shown to function as a negative regulator of DC activation, (Kim et al., 2016) among others (Table S1 and S2). We also found that GAS but not S. aureus induced the downregulation of genes associated with inflammasome activation (NLRP1B), regulation of NF-κB (CARD11) and the activation receptor CLEC9a, whereas S. aureus specifically downregulated expression of few genes (10 total), including genes associated with regulation of cell cycle and proliferation (G1/S-Specific Cyclin-E1 CCNE1 and the GTPase Ras family member DIRAS2) and survival (RELL1) (Cusick et al., 2010). Of note, the genes encoding the inflammatory cytokines IL-1β (IL1B), TNF-α (TNF) and IL12p35 (IL12A) were amongst genes upregulated by both S. aureus and GAS, however induction by GAS infection was substantially higher than by S. aureus (Figure 2D).
S. aureus also selectively induced genes associated with DC activation (TNFRSF1B), negative regulators of DC function (IRAK3 and TNIP1) (Sumpter et al., 2011)(Callahan et al., 2013), in addition to KCTD1, a negative regulator of the canonical Wnt signaling pathway (Li et al., 2014) among others. On the other hand, genes selectively induced by GAS included several inflammatory cytokines, such as Il6, IL12p40 (IL12B), and IL23p35 (IL23A) (Figure 2D). In addition, GAS but not S. aureus infection leads to upregulation of Caspase1 (CASP1) and CD86 expression.
The preferential induction of select pro-inflammatory cytokines by GAS in comparison to S. aureus was confirmed by ELISA assays (Figure 2E). Expression of TGF-β was comparable between GAS and S. aureus infected DC, and GAS also induced significant expression of IL23, which was not detected in S. aureus-infected DC. Thus, in line with the weak induction of Th1 and Th17 memory responses to S. aureus infection, DC infection with S. aureus induced lower expression of pro-inflammatory cytokines previously associated with priming of Th1 (IL12) and Th17 (IL1β, IL-6, IL-23) cells. Pro-inflammatory cytokines (IL1α, IL-6 and IL-12, but not IL1β) were also induced at a lower level by a panel of S. aureus isolates compared to GAS, suggesting that suboptimal induction of these cytokines by S. aureus is not unique to the LAC strain (Figure S2).
S. aureus peptidoglycan O-acetyltransferase represses the induction of select innate immune genes associated with the development of Th17 immunity
The finding that S. aureus is a poor inducer of inflammatory cytokines required for development of protective T cell immunity led us to hypothesize that S. aureus limits signals required to induce protective Th cell immunity. The cell wall PGN of S. aureus is a potent inducer of pro-inflammatory signals via recognition by innate pattern recognition receptors. Degradation of PGN directly promotes the activation of the NLRP3 inflammasome in macrophages (Shimada et al., 2010), and lysis of the bacteria leads to secondary release of PAMPs (Pathogen-associated molecular patterns) that are further recognized by additional receptors (Wolf et al., 2011). O-acetylation of MurNAc on the S. aureus PGN backbone has been shown to confer protection against host lysosome degradation (Bera et al., 2005). This actively limits inflammasome activation, IL-1β beta secretion and to a lesser extent the secretion of other cytokines in S. aureus-infected macrophages (Shimada et al., 2010; Wolf et al., 2011). Based on these data, we hypothesized that O-acetylation of S. aureus PGN is responsible for limiting induction of key cytokines required for Th differentiation and induction of protective immunity from reinfection.
To address this, we tested if a S. aureus O-acetyltransferase mutant (ΔoatA) could induce production of four of the main cytokines required for the induction of Th17 differentiation, using a well characterized isogenic WT/ΔoatA mutant pair in the SA113 background (Bera et al., 2005). SA113 induces similar levels of IL-1β, IL6, IL12 and IL-1α compared to LAC (Figure S2), but ΔoatA has been more extensively studied in the SA113 background (Shimada et al., 2010; Wolf et al., 2011). As shown in Figure 3A and S3, BMDC stimulated with ΔoatA or a 1:1 mixture of WT and ΔoatA in vitro secretes higher amounts of IL1-β, IL-6 and TGF-β compared to WT S. aureus-infected DC. IL-23 was not induced by either strain (data not shown). We next infected mice with 2×107 WT S. aureus alone (SA113), or with 107 WT S. aureus together with 107 ΔoatA i.p., three times at 7 day intervals. Adjuvancy with ΔoatA led to enhanced production of IL-1β and IL-23 and lower production of IL-10 compared to infection with WT S. aureus alone (Figure 3B). Ex-vivo restimulation of total spleen cells with heat-killed S. aureus 5 days after the last infection revealed increased production of IL-17A (but not IFN-γ) and lower IL-10 levels in the cultures of WT/ΔoatA S. aureus-infected mice in comparison to cultures from WT S, aureus-infected mice (Figure 3C), suggesting enhanced differentiation of Th17 cells in these mice.
Figure 3. O-acetylation of S. aureus PGN limits the induction of pro-inflammatory cytokines critical for protective memory.

(A) BMDC were stimulated with WT (S. aureus SA113), ΔoatA (isogenic SA113 mutant) or 1:1 mixture of WT and ΔoatA at MOI of 10. Supernatants were harvested at 24h for cytokine determination. Data are representative of 3 experiments. See also Figure S3.
(B)–(C) Mice (n=5) were infected with WT S. aureus (2×107) or WT + ΔoatA (107 CFU of each) at d0, 7 and 14. (B) Two days after the last infection, splenocytes were harvested from the mice for determination of cytokine levels. (C) Seven days after the last infection, splenocytes were harvested and stimulated with heat-killed WT S. aureus and cytokines were measured after 3d.
For (B) and (C), each data point represents an individual mouse, and mean ± SEM are shown for all data. Data analysis was performed using Student’s t-test (A) and using Mann-Whitney U test for (B) and (C). *p<0.05, ***p<0.001.
Adjuvancy with S. aureus O-acetyltransferase mutant or absence of IL-10 promotes the development of protective immunity against S. aureus reinfection
To determine if enhanced generation of Th17 cells leads to protection from S. aureus reinfection, we infected mice three times with WT SA113 and ΔoatA bacteria, and then challenged the mice with WT S. aureus. As shown in Figure 4A, mice initially infected with mixed ΔoatA and WT S. aureus and then challenged 10 days later with WT S. aureus showed enhanced S. aureus clearance compared to mice infected previously with WT S. aureus only. Prior infection with WT and ΔoatA S. aureus (once) or ΔoatA S. aureus (three times) did not provide significant protection against reinfection (data not shown).
Figure 4. Adjuvancy with ΔoatA or absence of IL-10 promotes development of protective immunity to S. aureus reinfection.

(A) Mice (n=6–7) were injected with PBS or infected 3 times as in (3B). Ten days after the last injection, all mice were inoculated with WT SA113 S. aureus (2×107). CFU from the kidneys were enumerated after 24h. The data are representative of two experiments.
(B) WT or IL-17A−/− mice were infected 3 times with S. aureus as in (3B). Seven days after the last infection, splenocytes were harvested and 5×107 purified total CD4+ T cells were transferred i.v. into naïve mice (n=5–6). The recipient mice were infected with WT S. aureus (2×107) the next day and kidney CFU were enumerated after 24h. See also Figure S4.
(C)–(E) WT or IL-10−/− mice (n=4–6) were infected with WT S. aureus (SA113) three times at 7 day intervals. A non-pretreated group served as a control. Seven days after the last pretreatment, all groups of mice were inoculated with WT S. aureus (2×107). (C) Spleen cytokine levels 24h after the last infection. (D) Splenocytes harvested 24h after the last infection were stimulated with heat-killed S. aureus (MOI=3), and cytokines in the supernatants were assessed after 3d. (E) CFU from splenocytes harvested 24hr after the last infection.
(F) WT or IL-10−/− mice were infected 3 times with S. aureus SA113 as in (3B). Seven days after the last infection, splenocytes were harvested and 5×107 purified total CD4+ T cells were transferred i.v. into naïve mice (n=5–6). The recipient mice were infected with WT S. aureus (2×107) the next day and kidney CFU were enumerated after 24h.
For (A), (B), (E) and (F), each data point represents an individual mouse and mean or mean ± SEM are shown. Data analysis was performed using ANOVA. *p<0.05, **p<0.01.
We also found that adoptive transfer of CD4 T cells (5 × 106 cells; purity >95%) from mice infected three times with ΔoatA/WT conferred protective immunity to S. aureus infection in naïve mice (Figure 4B), indicating that protection induced in this model can be transferred by Th cells. To determine whether, specifically, Th17 cells are important for the induced protection, we evaluated if transfer of purified CD4+ T cells from previously infected IL-17A−/− mice could induce protection in naïve mice. As shown in Figure 4B and S4, protection against S. aureus is IL-17A dependent since transfer of T cells from similarly treated IL-17A−/− mice did not confer protection.
The enhanced induction of IL-10 observed upon S. aureus reinfection (Fig. 1E) and the reduced IL-10 produced in the context of ΔoatA adjuvancy seem particularly noteworthy. Sallusto and colleagues previously described in vitro generation of human S. aureus-specific memory Th17 cells that produce IL10 upon reactivation, wherein IL-10 production could be inhibited by IL-1β (Zielinski et al., 2012). IL-10 has a well-described role in inhibition of Th17 cell development (Chaudhry et al., 2011; Frodermann et al., 2011). To address the role of IL-10 in our S. aureus reinfection model, we infected WT and IL-10−/− mice with WT S. aureus SA113 as before. As shown in Figure 4C–D, S. aureus-infected IL-10−/− mice showed enhanced production of IL-1β, IL-6, IL-17A and IFN-γ in comparison to WT mice. Infection in IL-10−/− mice also improved clearance of S. aureus during reinfection (Figure 4E). Moreover, we find that protection induced by lack of IL-10 can be transferred to naïve mice by CD4 T cells purified from IL-10−/− mice previously infected with WT S. aureus (Figure 4F).
Overall, these data suggest that O-acetylation of PGN by S. aureus limits pro-inflammatory signals and enhances IL-10 production, and the combined influence of these cytokines repress protective Th17 development and permit reinfection of the host.
DISCUSSION
Our study highlights a mechanism whereby S. aureus represses the production of cytokines needed to develop protective Th17 immunity. O-acetylation of PGN by S. aureus prevents breakdown of PGN and leads to inhibition of inflammasome activation in bone marrow-derived macrophages (Shimada et al., 2010). The modification of PGN further prevents secondary release of PAMPs trapped within the bacterial cell wall or cytoplasm (Wolf et al., 2011). Our finding was striking in that deletion of oatA provided sufficient stimulation for development of protective Th17 immunity, albeit several reinfections were required. Infection with oatA alone was not sufficient to induce immunity (data not shown), likely because ΔoatA is much more rapidly cleared during infection (Shimada et al., 2010). Our study did not explore and does not exclude other possible mechanisms underlying protection conferred by the ΔoatA strain.
At a basic level, our model posits that O-acetylation of PGN by S. aureus limits the production of inflammatory cytokines, including IL-1β, IL-6, and TFG-β, and thereby hampers the development of a protective Th17 response. The importance of IL-1β and IL-6 in Th17 development is well-established (Korn et al., 2009). Less well-understood are the characteristics of Th17 cells that confer or do not confer protection to S. aureus reinfection. In their landmark study, Sallusto and colleagues highlight the generation of different subtypes of Th17 cells in response to infections (Zielinski et al., 2012). Using human naïve T cells and autologous monocytes pulsed with dead S. aureus or Candida, they showed that S. aureus preferentially induce development of Th17 cells that co-produce IL-17A and IL-10. In comparison, Candida-pulsed monocytes induced Th17 cells that secrete IL-17 and IFN-γ, but not IL-10. They further demonstrated that addition of exogenous IL-1β can drive S. aureus-primed Th17 cells to increase IL-17A and IFN-γ and reduce IL-10 produced upon reactivation. What was not clear from that study were the functional implications of those Th17 cell subtypes in vivo. Although our study was not designed to dovetail with that report, our results are consistent with their findings, as we demonstrate that S. aureus induces, in vivo, a modest Th17 response associated with enhanced IL-10 production. That Th17 response proves not to be protective against reinfection, but coinfection with a ΔOat mutant that significantly boosted IL-1β and other selective cytokines fostered Th17 responses and reduced IL-10 expression, thus leading to IL-17-dependent Th cell-mediated protective immunity. Using a genetic approach, we showed that deletion of IL10 in vivo increases IL-1β production, enhances Th17 responses and promote protection against reinfection. IL-10 production in response to S. aureus has been shown to be associated with S. aureus regulation of Th17 cells in vitro (Frodermann et al., 2011). It is not clear if the increase in IL-1β or other cytokines, reduction of IL-10 or other possible alterations of the Th17 subtype are the primary drivers of the phenotypic change from a non-protective to a protective immune response in vivo. Our model should prove useful for future studies of the role of specific cytokines and Th17 responses in protective immune memory to S. aureus infection.
Our findings further advance the current understanding of how the host responds to S. aureus reinfection in mice. Many localized reinfection models (peritoneal or skin) reported the development of protective immunity following S. aureus infection (Brown et al., 2015; Montgomery et al., 2014; Murphy et al., 2014). Protective mechanisms identified include the induction of Th1 cells and IL-17-producing γδT cells from the peritoneum and antibody and Th17 responses following subcutaneous infection (Brown et al., 2015; Montgomery et al., 2014; Murphy et al., 2014). We and others (Kim et al., 2011) have observed in contrast that upon systemic bloodstream infection (i.p. or i.v.) protective immunity is not induced. Interestingly, studies of human PBMC have reported that S. aureus-specific Th1 responses are more readily demonstrated than Th17 responses following bloodstream infection (Brown et al., 2015). It is likely that mucosal environments, which favor development of Th17 immunity (Korn et al., 2009), contribute significantly to tissue-specific difference in S. aureus infection outcome. Different host compartments could have additional effects on virulence gene expression and the involvement of these factors in pathogenesis, and therefore could impact the induction of a protective Th response.
Overall, our study suggests a mechanism by which S. aureus interferes with development of an effective Th17 response against reinfection and therefore provides an explanation for the conundrum that both human and mice fail to mount a robust protective response to systemic infection. Our study also suggests that induction of IL-17-producing Th cells may require additional context to predict protection, since the quantity of IL-17A secreted, the subtype of Th17 cells, and other parameters that govern Th17 development are all potentially important. Therefore, future vaccines that aim to induce a protective Th17 response need to consider these subtleties of Th17 biology.
STARS METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, George Liu MD PhD (George.liu@cshs.org)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Microbes
GAS M49, S. aureus LAC, COL, Newman, MW2, SA113, and ΔoatA SA113 and complemented ΔoatA SA113 (courtesy of Dr. Fritz Goetz) were routinely cultured in Brain Heart Infusion, Todd Hewitt broth or Todd Hewitt broth supplemented with 0.5% Yeast Extract. Mid-log phase bacteria sub-cultured from overnight cultures or overnight cultures were used for experiments. Bacteria were routinely washed twice in PBS prior to use and inocula were confirmed by determination of CFU on agar plates. Heat-killed S. aureus was prepared by incubating a washed overnight culture of S. aureus for 1h at 60°C. Sterility of killed bacteria was confirmed by enumeration of CFU on an agar plate.
Primary cells
DC from 8–12 week old female C57BL/6 mice were derived by incubating murine bone marrow cells with in RPMI 1640 media containing Pen/Strep and supplemented with 10% FBS and 20ng/mL of mGM-CSF (PeproTech). After 9 days, the differentiated cells were used for in vitro stimulation assays. CD4 T cells were isolated from spleen and lymph nodes of 8–12 week old female C57BL/6 mice infected with S. aureus using a negative selection kit (STEMCELL). Purity of CD4 T cells was confirmed by flow cytometry (>95%).
Animals
C57BL/6 and BALB/c mice were purchased from Jackson Lab. IL-17A−/− (Courtesy of Dr. Yoichiro Iwakura), IL-10−/− and OT-II transgenic/Rag−/− mice were maintained at Cedars-Sinai. All mice were housed in specific-pathogen free facilities and 8 to 12-week old age-matched female mice were used for in vitro and in vivo experiments. Because of sex-related differences in susceptibility to S. aureus (Yanke et al., 2000), only female mice were used in this study. This study was performed under strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals. CSMC is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC), and in compliance with NIH guideline of laboratory animal care and use. The protocol was approved by the institutional animal use and care committee of the Cedars-Sinai Medical Center.
METHOD DETAILS
S. aureus and GAS reinfection models
Reinfection (two-infection) model - Mice were inoculated with S. aureus or GAS via intra-peritoneal injection. After 21 days, the mice were reinfected i.p. with the same inoculum of S. aureus or GAS unless indicated otherwise. Mice were killed 1–2 days after reinfection and spleen and kidneys were harvested, homogenized and plated on blood agar plates. CFU were counted after overnight incubation at 37°C.
Multiple reinfection model (WT SA113 and ΔoatA SA113) – Mice were infected with 2×107 CFU of WT S. aureus (SA113) or with 107 SA113 + 107 SA113 ΔoatA every 7 days for a total of 3 reinfections. Seven or ten days after the last infection, mice were challenged 2×107 WT SA113 S. aureus. After 1–2 days, the mice were sacrificed and spleen and kidneys were harvested for further analyses. For the multiple reinfection experiment performed in Figure 1 using S. aureus LAC, the experiment was performed as described in the figure legend.
Adoptive Transfer of CD4+ T cells - For adoptive transfer experiments, 5 × 106 CD4+ T cells were injected i.v. into recipient mice.
Mice were divided into cages by comparative medicine staff with no involvement in study design, and cages were randomly assigned to experimental groups. The investigators were not blinded to the group allocation during the experiment or when assessing the outcome.
ELISA and flow cytometry
Enzyme linked immunosorbent assays (ELISA) were performed according to the manufacturers’ instructions.
FACS analysis – Cells were incubated in FACS buffer (PBS containing 3% FBS and 0.1% sodium azide) in the presence of purified neutralizing monoclonal antibodies against CD16:CD32 (Fc Block; eBiosciences) for 20 minutes at 4°C prior to staining. Specific antibodies (1:100 dilution) in FACS buffer were then added for 30 min at 4°C in the dark. Samples were then analyzed using a FACS LSRII Cytometer or Fortessa (BDbiosciences) and FlowJo 7.6.1 software (TreeStar, Co).
For intracellular cytokine staining, cell suspensions were incubated in ice-cold PBS supplemented with 3% (v/v) FBS cells containing anti-TCRβ and anti-CD4, and then fixed and resuspended in permeabilization buffer containing anti–IFN-γ or anti-IL-17A antibody. Data were acquired on a LSRII Flow cytometer and analyzed with FlowJo Software (Treestar, Ashland, OR).
Ex vivo stimulation of splenocytes
Splenocytes were isolated at indicated time points after infection. Red blood cells were lysed and leukocytes were resuspended in RPMI 1640 media containing Pen/Strep and supplemented with 10% FBS. For specific restimulation, 5 × 105 cells were cultured with heat-killed S. aureus or GAS at an MOI of 10. Supernatants were collected after 48–72 h for analysis of cytokines by ELISA. For intracellular cytokine analysis, the splenocytes were stimulated with phorbol 12-myristate 13-acetate (500 ng/ml), ionomycin (50ng/mL), and GolgiStop for 4 h. Intracellular cytokines were then analyzed by flow cytometry.
BMDC stimulation and DC-TC co-culture assays
BMDC were plated in 48-well plates at a density of 2 × 105/ml and infected with S. aureus or GAS at the indicated MOI. Infected DC were incubated for 1 h in antibiotic-free medium, washed twice to remove unbound bacteria and further incubated in RPMI media supplemented with 10% FBS and 100 μg/ml of Gentamicin for 12 hr. After washing, cells were resuspended in fresh media containing 100 μg/ml Gentamicin and further incubated for 6 h with 5μg/mL of OVA peptide. CD4+ T cells were isolated from spleen and lymph nodes of OT-II mice using a negative selection kit (STEMCELL). Cells were labeled with CFSE and added (1 × 106/well) to the DCs and then incubated for up to 7 days at 37°C.
RNA isolation and quantitative Reverse Transcription PCR
BMDC were infected with bacteria at an MOI of 3 for 1hr, and washed with gentamicin containing media to kill the extracellular. Total RNA was isolated using an RNeasy Mini-kit (Qiagen, Valencia, CA) according to the manufacturer’s protocol. Briefly, at 24 hr, cells were centrifuged and lysed in RLT lysis buffer (Qiagen) with 10μl/mL of 2-ME. RNA was isolated following manufacturer’s instructions. The concentration and the RNA integrity number were measured using an ND-1000 spectrophotometer (NanoDrop Technologies) and a Bioanalyzer, and only RNA samples with a concentration >1μg and a 260/280 ratio ≥ 2.0 were retained for further analysis. Total RNA was treated with DNase (Life Technologies) and used as template for cDNA synthesis using the High Capacity cDNA Reverse Transcription Kit (Invitrogen).
Library Construction and RNA Sequencing
Total RNA was extracted as described for RT-QPCR analysis above. mRNA was purified from one microgram of total RNA using the Ambion Dynabeads® mRNA DIRECT™ Micro Purification Kit (Austin, TX) per manufacturer’s recommendations. Purified mRNA was fragmented, adapters ligated, reverse transcribed to make cDNA, and cDNA amplified with Ion Total RNA-Seq Kit v2, Ion Torrent™, per manufacturer’s recommendations. Samples were individually barcoded using Ion Xpress™ RNA-Seq Barcode 1–16 Kit, Ion Torrent™, before their cDNA was amplified. RNAseq libraries were assessed for concentration and length using Invitrogen Qubit® dsDNA HS Assay Kit (Carlsbad, CA) and Agilent DNA 1000 Kit respectively (Carlsbad, CA). Samples were multiplexed to obtain 10 million reads each for sequencing. The pooled libraries were amplified onto Ion Sphere™ particles using Ion PI™ Template OT2 200 Kit, Ion Torrent™ per manufacturer’s recommendations. Ion Sphere™ particles were purified per manufacturer’s recommendations and prepared for sequencing using the Ion PI™ Sequencing 200 Kit, Ion Torrent™ per manufacturer’s recommendations. Sequence data was then pre-processed using the Torrent Suite software and the FASTX-toolkit (Hannon laboratory, CSHL). Quality control was performed using FastQC.
Sequencing data analysis
Single-end RNA-seq reads were aligned to the transcriptome of mouse genome GRCm38/mm10 using the Kallisto package with the quant command. An estimated average fragment size of 200 bp was used, with an estimated standard deviation of 20 bp. Sequence based bias correction was also performed during alignment and quantification of transcripts. For each sample, BAM files, containing all the alignments, were obtained along with the estimated counts for each transcript; these were then summed up for each gene. The genic counts for all the samples were assembled, quantile normalized, before getting the counts per million to account for the library sizes. These normalized counts were log2 transformed, then input to the R package limma’s lmFit function - and using empirical Bayes methods to borrow information between genes along with correcting for mean-variance trend - to get Differentially Expressed Genes (DEGs) for SA vs NS (S. aureus vs Non Stimulated) and GAS vs NS. The p-values obtained using t-tests were adjusted using the Benjamini-Hochberg procedure. Log 2 Fold Changes (FCs) were also returned, with positive values indicating over-expression compared to the Non Stimulated condition, and negative values indicating under-expression with SA or GAS. DEGs were called using adjusted p-value < 0.05 and actual FCs > 2 in either direction.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are expressed as mean ± standard error (SEM). We assumed a Gaussian distribution for our data. Accordingly, statistical significance was determined by two tailed unpaired t-test or 1-way ANOVA with Bonferroni correction for multiple comparisons. In vivo experiments frequently follow a non-Gaussian distribution. Therefore, in vivo experiments were analyzed using non-parametric Mann–Whitney U-test or Kruskal-Wallis test in the case of missing normality. Log rank test was used for analysis of mouse survival. With few exceptions, all in vitro studies were done with at least three sets of independent experiments. GraphPad Prism or Excel were used for all analyses. All statistical details of experiments including the number of replicates can be found in the figure legends. p values less than 0.05 are considered significant.
DATA AVAILABILITY
RNAseq data has been submitted to the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=qfcjueckdzyxnuv&acc=GSE96086).
Supplementary Material
HIGHLIGHTS.
Protective T cell memory is not induced after systemic S. aureus infections
S. aureus induces proliferation but limits polarization of Th1 and Th17 cells
O-acetylation of peptidoglycan represses cytokines required for Th17 polarization
Adjuvancy with ΔoatA S. aureus promotes protective memory to reinfection
Acknowledgments
Funding for this work was provided by NIH research grant R01AI103542 and R21AI083948 (to G.A.M.), R01AI127406 (to G.Y.L., G.A.M. and D.M.U) and by the F. Widjaja Foundation IBIRI Institute, Cedars-Sinai Medical Center (to G.A.M.)
Footnotes
AUTHOR CONTRIBUTIONS
G.A.M. and G.Y.L conceived and directed the project. M. S., S. L. K., S. M., M.A., D.M.U., G. A. M. and G.Y.L. designed experiments, and prepared the manuscript. M. S., S. L. K., S. M., C. R., D.C., R.S., G.A.M. and G.Y.L. conducted most of the experiments and analyzed the data. A.J.W. and C.O. performed some of the in vitro molecular and cellular experiments.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alonzo F, 3rd, Kozhaya L, Rawlings SA, Reyes-Robles T, DuMont AL, Myszka DG, Landau NR, Unutmaz D, Torres VJ. CCR5 is a receptor for Staphylococcus aureus leukotoxin ED. Nature. 2013;493:51–55. doi: 10.1038/nature11724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bera A, Herbert S, Jakob A, Vollmer W, Gotz F. Why are pathogenic staphylococci so lysozyme resistant? The peptidoglycan O-acetyltransferase OatA is the major determinant for lysozyme resistance of Staphylococcus aureus. Molecular microbiology. 2005;55:778–787. doi: 10.1111/j.1365-2958.2004.04446.x. [DOI] [PubMed] [Google Scholar]
- Brown AF, Murphy AG, Lalor SJ, Leech JM, O’Keeffe KM, Mac Aogain M, O’Halloran DP, Lacey KA, Tavakol M, Hearnden CH, et al. Memory Th1 Cells Are Protective in Invasive Staphylococcus aureus Infection. PLoS pathogens. 2015;11:e1005226. doi: 10.1371/journal.ppat.1005226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan JA, Hammer GE, Agelides A, Duong BH, Oshima S, North J, Advincula R, Shifrin N, Truong HA, Paw J, et al. Cutting edge: ABIN-1 protects against psoriasis by restricting MyD88 signals in dendritic cells. Journal of immunology. 2013;191:535–539. doi: 10.4049/jimmunol.1203335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor H, Crump AL, Raman VK, Liu H, Markowitz JS, Grusby MJ, Glimcher LH. Immunoregulatory effects of superantigens: interactions of staphylococcal enterotoxins with host MHC and non-MHC products. Immunological reviews. 1993;131:27–42. doi: 10.1111/j.1600-065x.1993.tb01528.x. [DOI] [PubMed] [Google Scholar]
- Chaudhry A, Samstein RM, Treuting P, Liang Y, Pils MC, Heinrich JM, Jack RS, Wunderlich FT, Bruning JC, Muller W, et al. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity. 2011;34:566–578. doi: 10.1016/j.immuni.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JS, Pietras EM, Garcia NC, Ramos RI, Farzam DM, Monroe HR, Magorien JE, Blauvelt A, Kolls JK, Cheung AL, et al. IL-17 is essential for host defense against cutaneous Staphylococcus aureus infection in mice. The Journal of clinical investigation. 2010;120:1762–1773. doi: 10.1172/JCI40891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham MW. Pathogenesis of group A streptococcal infections. Clinical microbiology reviews. 2000;13:470–511. doi: 10.1128/cmr.13.3.470-511.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusick JK, Mustian A, Goldberg K, Reyland ME. RELT induces cellular death in HEK 293 epithelial cells. Cellular immunology. 2010;261:1–8. doi: 10.1016/j.cellimm.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falugi F, Kim HK, Missiakas DM, Schneewind O. Role of protein A in the evasion of host adaptive immune responses by Staphylococcus aureus. mBio. 2013;4:e00575–00513. doi: 10.1128/mBio.00575-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frodermann V, Chau TA, Sayedyahossein S, Toth JM, Heinrichs DE, Madrenas J. A modulatory interleukin-10 response to staphylococcal peptidoglycan prevents Th1/Th17 adaptive immunity to Staphylococcus aureus. The Journal of infectious diseases. 2011;204:253–262. doi: 10.1093/infdis/jir276. [DOI] [PubMed] [Google Scholar]
- Goodyear CS, Silverman GJ. Death by a B cell superantigen: In vivo VH-targeted apoptotic supraclonal B cell deletion by a Staphylococcal Toxin. The Journal of experimental medicine. 2003;197:1125–1139. doi: 10.1084/jem.20020552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Winqvist O, Flores-Morales A, Wikstrom AC, Norstedt G. SOCS2 influences LPS induced human monocyte-derived dendritic cell maturation. PloS one. 2009;4:e7178. doi: 10.1371/journal.pone.0007178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G, Wang Y, Chi H. Regulation of TH17 cell differentiation by innate immune signals. Cellular & molecular immunology. 2012;9:287–295. doi: 10.1038/cmi.2012.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SH, Yu CR, Mahdi RM, Ebong S, Egwuagu CE. Dendritic cell maturation requires STAT1 and is under feedback regulation by suppressors of cytokine signaling. Journal of immunology. 2004;172:2307–2315. doi: 10.4049/jimmunol.172.4.2307. [DOI] [PubMed] [Google Scholar]
- Kang JO, Lee JB, Chang J. Cholera Toxin Promotes Th17 Cell Differentiation by Modulating Expression of Polarizing Cytokines and the Antigen-Presenting Potential of Dendritic Cells. PloS one. 2016;11:e0157015. doi: 10.1371/journal.pone.0157015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karauzum H, Datta SK. Adaptive Immunity Against Staphylococcus aureus. Current topics in microbiology and immunology. 2016 doi: 10.1007/82_2016_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HK, Kim HY, Schneewind O, Missiakas D. Identifying protective antigens of Staphylococcus aureus, a pathogen that suppresses host immune responses. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2011;25:3605–3612. doi: 10.1096/fj.11-187963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TW, Hong S, Lin Y, Murat E, Joo H, Kim T, Pascual V, Liu YJ. Transcriptional Repression of IFN Regulatory Factor 7 by MYC Is Critical for Type I IFN Production in Human Plasmacytoid Dendritic Cells. Journal of immunology. 2016;197:3348–3359. doi: 10.4049/jimmunol.1502385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annual review of immunology. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- Lee LY, Miyamoto YJ, McIntyre BW, Hook M, McCrea KW, McDevitt D, Brown EL. The Staphylococcus aureus Map protein is an immunomodulator that interferes with T cell-mediated responses. The Journal of clinical investigation. 2002;110:1461–1471. doi: 10.1172/JCI16318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Chen C, Wang F, Huang W, Liang Z, Xiao Y, Wei K, Wan Z, Hu X, Xiang S, et al. KCTD1 suppresses canonical Wnt signaling pathway by enhancing beta-catenin degradation. PloS one. 2014;9:e94343. doi: 10.1371/journal.pone.0094343. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, Kanno Y, Spalding C, Elloumi HZ, Paulson ML, et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature. 2008;452:773–776. doi: 10.1038/nature06764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery CP, Daniels M, Zhao F, Alegre ML, Chong AS, Daum RS. Protective immunity against recurrent Staphylococcus aureus skin infection requires antibody and interleukin-17A. Infection and immunity. 2014;82:2125–2134. doi: 10.1128/IAI.01491-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy AG, O’Keeffe KM, Lalor SJ, Maher BM, Mills KH, McLoughlin RM. Staphylococcus aureus infection of mice expands a population of memory gammadelta T cells that are protective against subsequent infection. Journal of immunology. 2014;192:3697–3708. doi: 10.4049/jimmunol.1303420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblatt J, Zeltzer PM, Portaro J, Seeger RC. Inhibition of antibody-dependent cellular cytotoxicity by protein A from Staphylococcus aureus. Journal of immunology. 1977;118:981–985. [PubMed] [Google Scholar]
- Sasaki S, Tagawa Y, Iwakura Y, Nakane A. The role of gamma interferon in acquired host resistance against Staphylococcus aureus infection in mice. FEMS immunology and medical microbiology. 2006;46:367–374. doi: 10.1111/j.1574-695X.2005.00037.x. [DOI] [PubMed] [Google Scholar]
- Shimada T, Park BG, Wolf AJ, Brikos C, Goodridge HS, Becker CA, Reyes CN, Miao EA, Aderem A, Gotz F, et al. Staphylococcus aureus evades lysozyme-based peptidoglycan digestion that links phagocytosis, inflammasome activation, and IL-1beta secretion. Cell host & microbe. 2010;7:38–49. doi: 10.1016/j.chom.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumpter TL, Packiam V, Turnquist HR, Castellaneta A, Yoshida O, Thomson AW. DAP12 promotes IRAK-M expression and IL-10 production by liver myeloid dendritic cells and restrains their T cell allostimulatory ability. Journal of immunology. 2011;186:1970–1980. doi: 10.4049/jimmunol.1000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tussiwand R, Everts B, Grajales-Reyes GE, Kretzer NM, Iwata A, Bagaitkar J, Wu X, Wong R, Anderson DA, Murphy TL, et al. Klf4 expression in conventional dendritic cells is required for T helper 2 cell responses. Immunity. 2015;42:916–928. doi: 10.1016/j.immuni.2015.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Roderiquez G, Norcross MA. Control of adaptive immune responses by Staphylococcus aureus through IL-10, PD-L1, and TLR2. Sci Rep. 2012;2:606. doi: 10.1038/srep00606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertheim HF, Vos MC, Ott A, van Belkum A, Voss A, Kluytmans JA, van Keulen PH, Vandenbroucke-Grauls CM, Meester MH, Verbrugh HA. Risk and outcome of nosocomial Staphylococcus aureus bacteraemia in nasal carriers versus non-carriers. Lancet. 2004;364:703–705. doi: 10.1016/S0140-6736(04)16897-9. [DOI] [PubMed] [Google Scholar]
- Wolf AJ, Arruda A, Reyes CN, Kaplan AT, Shimada T, Shimada K, Arditi M, Liu G, Underhill DM. Phagosomal degradation increases TLR access to bacterial ligands and enhances macrophage sensitivity to bacteria. Journal of immunology. 2011;187:6002–6010. doi: 10.4049/jimmunol.1100232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanke SJ, Olson ME, Davies HD, Hart DA. A CD-1 mouse model of infection with Staphylococcus aureus: influence of gender on infection with MRSA and MSSA isolates. Canadian journal of microbiology. 2000;46:920–926. [PubMed] [Google Scholar]
- Zielinski CE, Mele F, Aschenbrenner D, Jarrossay D, Ronchi F, Gattorno M, Monticelli S, Lanzavecchia A, Sallusto F. Pathogen-induced human TH17 cells produce IFN-gamma or IL-10 and are regulated by IL-1beta. Nature. 2012;484:514–518. doi: 10.1038/nature10957. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNAseq data has been submitted to the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=qfcjueckdzyxnuv&acc=GSE96086).