

SUMMARY
Double-stranded RNA (dsRNA) is a common by-product of viral infections and acts as a potent trigger of anti-viral immunity. In the nematode C. elegans, sid-1 encodes a dsRNA transporter that is highly conserved throughout animal evolution, but the physiological role of SID-1 and its orthologs remains unclear. Here, we show that the mammalian SID-1 ortholog, SIDT2, is required to transport internalized extracellular dsRNA from endocytic compartments into the cytoplasm for immune activation. Sidt2 deficient mice exposed to extracellular dsRNA, encephalomyocarditis virus (EMCV) and herpes simplex virus 1 (HSV-1) show impaired production of anti-viral cytokines and – in the case of EMCV and HSV-1 – reduced survival. Thus, SIDT2 has retained the dsRNA transport activity of its C. elegans ortholog, and this transport is important for antiviral immunity.
eTOC blurb
Extracellular double-stranded RNA is predominantly sensed by cytosolic RLRs following endocytic uptake, but how it enters the cytoplasm is unknown. Nguyen and colleagues demonstrate that the endo-lysosomal protein SIDT2 transports double-stranded RNA into the cytoplasm for RLR signalling and is required for survival following EMCV infection.
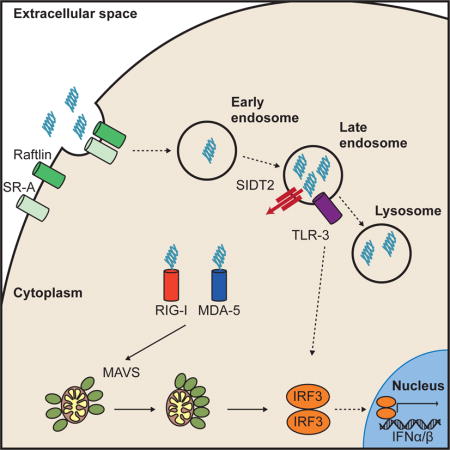
INTRODUCTION
Double-stranded RNA (dsRNA) is a common by-product of viral replication and acts as a potent pathogen associated molecular pattern (PAMP), whose intracellular detection leads to the production of type I interferons (IFN-I) and antiviral immunity. Within the cytoplasm, the RIG-I like receptor (RLR) family of proteins, including the dsRNA-binding proteins RIG-I and MDA-5, are ideally situated to detect viral dsRNA produced in infected cells. Consistent with this, RIG-I, MDA-5 and their downstream signalling adaptor protein, MAVS, are required for immunity to multiple viruses (Fredericksen and Gale, 2006; Gitlin et al., 2006; Kato et al., 2005; Sumpter et al., 2005; Sun et al., 2006; Yoneyama et al., 2005), and viruses have evolved multiple mechanisms to inhibit the RLR sensing pathway (Leung et al., 2012).
To circumvent this inhibition, infected cells can transfer viral dsRNA extracellularly to uninfected bystander cells, thereby promoting IFN-I production and limiting viral replication in vitro (Dansako et al., 2013). How such transfer occurs is yet to be determined, and the importance of this transfer to immunity in vivo – while postulated – remains unclear. What is clear is that once dsRNA is present within the extracellular milieu it can bind various cell surface receptors, such as MSR, Raftlin and CD14 (Dansako et al., 2013; DeWitte-Orr et al., 2010; Lee et al., 2006; Watanabe et al., 2011), and then be internalised via clathrin-dependent endocytosis (Itoh et al., 2008). Once endocytosed, dsRNA can be recognised by Toll-Like Receptor 3 (TLR3) within the acidic environment of the lysosome (Dansako et al., 2013; Lee et al., 2006; Schulz et al., 2005; Watanabe et al., 2011), leading to subsequent signalling via the adaptor protein, TRIF (Yamamoto et al., 2003). dsRNA that has been internalised from the extracellular environment also activates the RLR sensing pathway within the cytoplasm, and it is the cytoplasmic pathway that is responsible for the vast majority of the IFN-I response upon in vivo exposure to extracellular dsRNA (in the form of poly(I:C) (Gitlin et al., 2006).
This counterintuitive observation implies the existence of an efficient mechanism to transport extracellular dsRNA into the cytoplasm either directly across the plasma membrane or from within the endosomal compartment. Given the large size and hydrophilic nature of dsRNA and the hydrophobic barrier of the plasma and endosomal membranes, this mechanism is likely to require highly specialised proteins. To date, the identity of any such proteins is unknown. Two potential candidates are SIDT1 and SIDT2, the mammalian orthologs of the C. elegans SID-1 dsRNA transporter.
A remarkable property of RNA interference (RNAi) in plants and certain animals is its ability to spread systemically from the site of initiation (Jose and Hunter, 2007). In C. elegans, this spread requires SID-1, a broadly expressed transmembrane protein that specifically binds and imports extracellular dsRNA (Feinberg and Hunter, 2003; Jose et al., 2009; Li et al., 2015b; Winston et al., 2002), most likely following receptor-mediated endocytosis (McEwan et al., 2012). SID-1 is conserved throughout much of animal evolution, with two closely related paralogs, SIDT1 and SIDT2, present in most sequenced vertebrate genomes (Figure S1). Such conservation implies a strongly selected function, but the normal physiological role of SID-1 and its orthologs is unclear.
Here we report that SIDT2 localised to the late endo-lysosomal compartment and interacted with internalised poly(I:C) to promote its release into the cytoplasm. Loss of SIDT2 in mice resulted in endosomal dsRNA accumulation, impaired RLR signalling, and diminished type I IFN production in response to extracellular poly(I:C). Following herpes simplex virus 1 (HSV-1) infection, we observed that dsRNA was transferred to uninfected bystander cells, and that RLR activation in these cells was SIDT2-dependent. Consistent with this, loss of SIDT2 resulted in higher rates of HSV-1 infection as well as impaired anti-viral cytokine production and survival in vivo. Similarly, SIDT2-deficient mice displayed impaired anti-viral immunity and invariably succumbed to infection with encephalomyocarditis virus (EMCV), protection against which critically depends upon RLR activation (specifically, MDA-5) (Gitlin et al., 2006; Kato et al., 2006). Together, our findings demonstrate that SIDT2 has retained the dsRNA transport function of its C. elegans ortholog, and that this function is important for delivery of dsRNA to the cytoplasmic RLRs and subsequent antiviral immunity.
RESULTS
SIDT2 localises to endo-lysosomes and interacts with internalised dsRNA
Both microarray and RNAseq data indicated that Sidt2 is more broadly and more abundantly expressed than Sidt1 (data not shown) (Petryszak et al., 2014; Wu et al., 2009). Moreover, Sidt2 expression is stimulated by both type I and II IFNs (Rusinova et al., 2013). We therefore decided to focus our attention on SIDT2 and its potential role in the innate immune response to viral dsRNA. To begin, we investigated the subcellular localisation of SIDT2 using confocal microscopy. Stable expression of a SIDT2-mCherry reporter in mouse embryonic fibroblasts (MEFs) indicated that SIDT2 is present in punctate structures, and strongly co-localises with both the late endosomal marker RAB7 (Pearson’s r = 0.85) and the lysosomal marker LAMP1 (Pearson’s r = 0.64) (Figure 1A, Supplemental video 1 and 2). In contrast, minimal co-localisation was observed between SIDT2 and EEA1 (Pearson’s r = 0.25) (Figure 1A, Supplemental video 3 and 4) or RAB11 (Pearson’s r = 0.06) (data not shown), which demarcate early and recycling endosomes respectively. These data indicate that SIDT2 predominantly resides in late endosomes and lysosomes, and are consistent with previous reports localising endogenously-expressed SIDT2 to the endo-lysosomal compartment (Buschow et al., 2012; Gao et al., 2010).
Figure 1. SIDT2 localises to late endosomes and interacts with internalised poly(I:C).
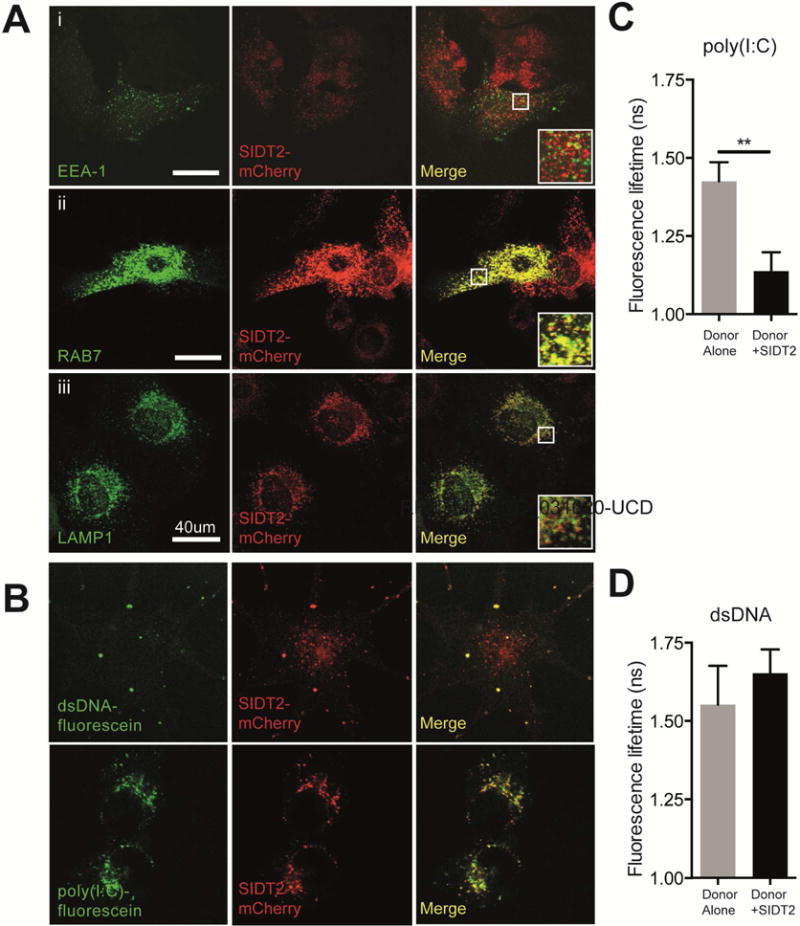
(A) MEFs stably expressing SIDT2-mCherry were transfected with markers for (i) early endosomes (EEA1-GFP), (ii) late endosomes (RAB7-GFP) and (iii) lysosomes (LAMP1-YFP), and imaged by confocal microscopy. Co-localization analysis of SIDT2-mCherry with endosomal markers was performed using FIJI software (see main text). Scale bar = 40 μm. (B) DC2.4 cells stably expressing SIDT2-mCherry were transfected with dsDNA-fluorescein or treated with poly(I:C)-fluorescein for 1 h and imaged by confocal microscopy. (C–D) Assessment of SIDT2-mCherry interactions with poly(I:C)-fluorescein and dsDNA-fluorescein in DC2.4 cells via FRET FLIM. ** P < 0.01. Data is plotted as ± SEM and are representative of 3 independent experiments. See also Supplemental videos 1–4.
Extracellular dsRNA is known to be taken up into cells via clathrin-dependent endocytosis (Itoh et al., 2008). Consistent with this, we observed that extracellular poly(I:C)-fluorescein was readily internalised by the murine dendritic cell line DC2.4 into vesicle-like structures, and subsequently co-localised with SIDT2-mCherry (Pearson’s r = 0.72) (Figure 1B). To assess interaction between SIDT2 and internalised poly(I:C), we performed fluorescence resonance energy transfer (FRET) analysis by time domain fluorescence lifetime imaging microscopy (FLIM), and observed a significant reduction in fluorescence lifetime for poly(I:C)-fluorescein in the presence of SIDT2-mCherry. This indicates a likely molecular interaction between SIDT2 and poly(I:C) (Figure 1C), and is consistent with the recent demonstration that the extracellular domain of mouse SIDT2 can bind dsRNA in vitro (Li et al., 2015a). To test the specificity of this interaction, we performed the same experiment using dsDNA, which also appeared to co-localise with SIDT2-mCherry following internalisation (Figure 1B). In contrast to poly(I:C), no reduction in fluorescence lifetime was observed between internalised dsDNA-fluorescein and SIDT2-mCherry (Figure 1D), suggesting that SIDT2 can distinguish between dsRNA and dsDNA as has previously been shown for C. elegans SID-1 (Shih and Hunter, 2011).
SIDT2 is critical for antiviral immunity to EMCV
To examine a possible role for SIDT2 in the innate immune response to dsRNA, we generated mice carrying a Sidt2 gene-targeting construct (Tang et al., 2010), which successfully disrupted Sidt2 expression (Figure S2). Sidt2−/− mice were born at the expected Mendelian ratios, displayed normal viability and fertility, and weighed slightly less (~10%) than heterozygous and wild-type (WT) littermates. Given the importance of dsRNA detection for antiviral immunity, we were keen to assess the effects of disrupting Sidt2 during virus infection. To this end, we selected EMCV as a viral model, and did so for two reasons: first, it is a ssRNA virus that produces readily detectable dsRNA during replication (Weber et al., 2006); and second, it causes a lytic infection, which we theorised might be important for liberating dsRNA into the extracellular milieu.
We infected Sidt2−/− and WT mice with 50 PFU of EMCV via intraperitoneal injection (i.p.) and monitored the animals over time. All mice lacking Sidt2 succumbed to infection within 6 days post infection (p.i.) (Figure 2A) and displayed increased viral loads within the heart and peritoneal exudate cells (PECs) (Figure 2B–C) suggesting that SIDT2 is critical for the immune response to EMCV dsRNA. Consistent with these observations, Sidt2−/− mice showed a significant decrease in IFN-β in serum and peritoneal lavage fluid compared to WT at 3 days p.i. (Figure 2D–E), and also had significantly lower concentrations of other innate immune mediators such as RANTES, IL-6 and IL-12 (Figure 2F–H).
Figure 2. Loss of SIDT2 impairs innate immunity to EMCV infection.
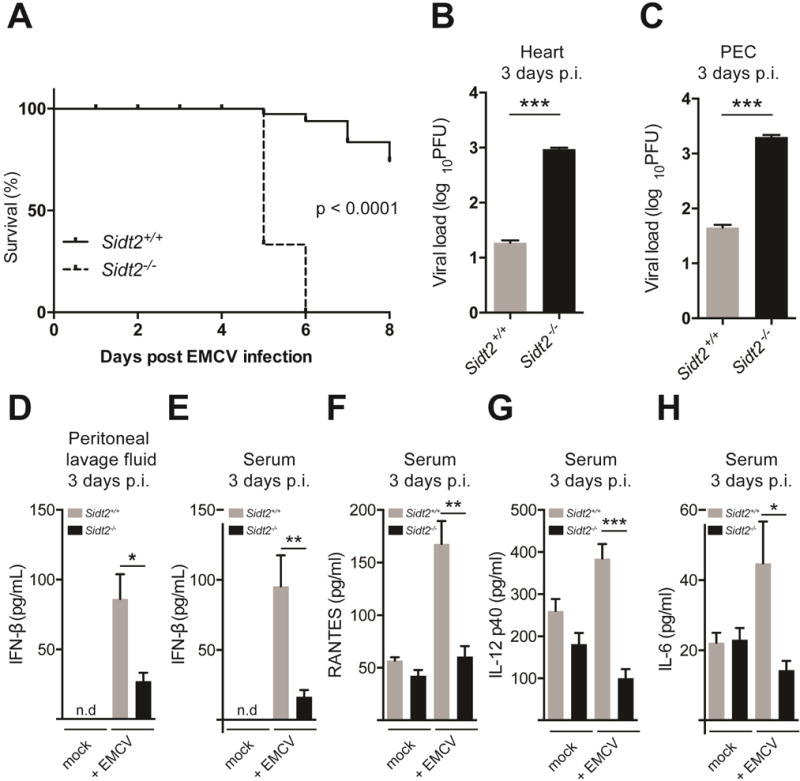
(A) Sidt2+/+ and Sidt2−/− mice (n=6–9) were infected with 50 PFU EMCV i.p. and survival monitored for 8 days. (B–C) Sidt2+/+ and Sidt2−/− mice (n=10–13) were sacrificed at 3 days p.i. and viral load was assessed in the heart and PECs by plaque assay. (D–E) Peritoneal lavage fluid and serum from Sidt2+/+ and Sidt2−/− mice (n=10–13) were collected at day 3 p.i. and IFN-β was measured via ELISA. (F–H) Serum IL-6, RANTES and IL-12 p40 from Sidt2+/+ and Sidt2−/− mice (n=10–13) at 3 days p.i. were measured using Bioplex bead assay. For panels F–H, data are plotted as mean ± SEM. * P < 0.05, *** P < 0.001, n.d = not detected.
Loss of SIDT2 impairs antiviral immunity to HSV-1
Given that dsRNA is also produced during the replication of dsDNA viruses, we wanted to know if there was a broader role for SIDT2 beyond RNA viruses such as EMCV. To this end, we chose HSV-1 as a viral model, since like EMCV it produces readily detectable amounts of dsRNA and causes a lytic infection. To initially determine whether extracellular dsRNA is relevant to HSV-1 infection, we infected Vero cells – a monkey epithelial cell line that is highly permissive for HSV-1 replication – with GFP-tagged HSV-1 for 24 h, and then stained with a monoclonal antibody (J2) that specifically recognises dsRNA (Figure 3A). We failed to detect any dsRNA within infected cells – potentially consistent with HSV-1’s ability to sequester dsRNA and suppress type I IFN production (Khoo et al., 2002; Xing et al., 2012) – but instead frequently observed dsRNA within uninfected bystander cells. To determine whether this dsRNA was of viral origin, we subsequently infected Vero cells with mCherry-tagged HSV-1, isolated HSV-1+ and HSV-1− cells via flow cytometry (Figure S3A), and then performed strand-specific RNAseq. Notably, overlapping HSV-1 RNA derived from both strands of the viral genome was readily detectable in HSV-1− bystander cells (Figure S3B), consistent with the presence of viral dsRNA. Taken together, these data suggest not only that HSV-1 dsRNA can spread extracellularly from the site of infection to non-infected bystander cells, but also that extracellular dsRNA rather than dsRNA within infected cells is the more likely trigger of innate immune activation during HSV-1 infection.
Figure 3. Loss of SIDT2 impairs innate immunity to HSV-1 infection.
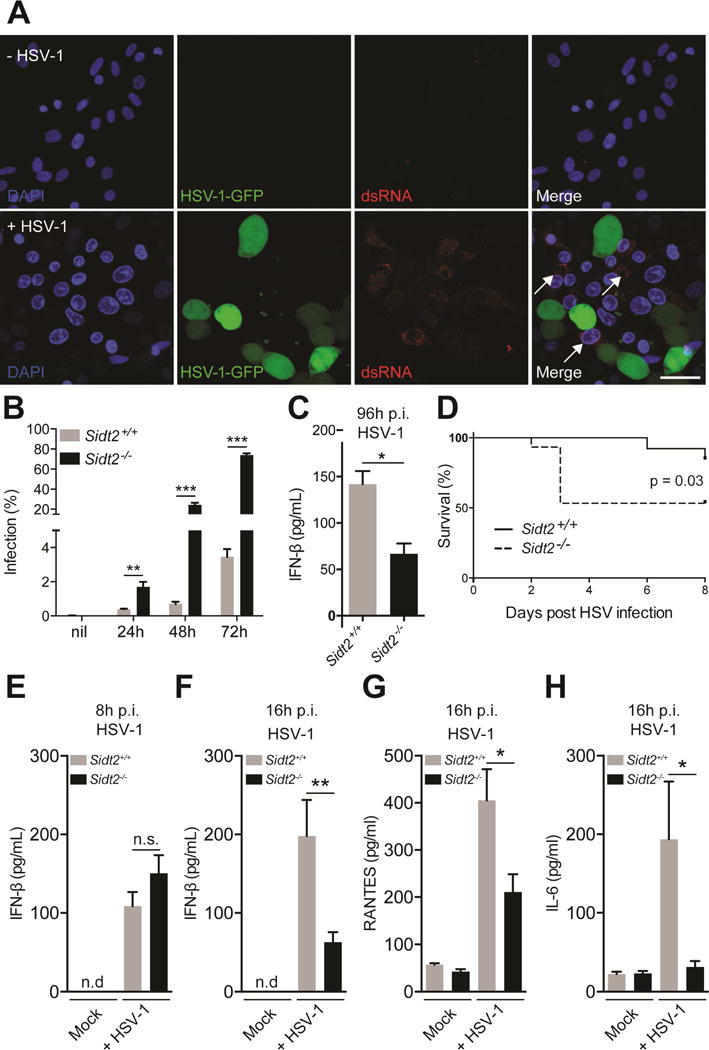
(A) Vero cells were infected with 1 MOI GFP-tagged HSV-1 virus for 24 h, stained with J2 anti-dsRNA antibody (red) and DAPI (blue), and imaged by confocal microscopy. In the absence of HSV-1 infection, no dsRNA was evident (top panel), whereas following HSV-1 challenge dsRNA was readily observed in uninfected cells (bottom panel, arrows). Data are representative of three independent experiments. Scale bar = 20 μm. (B) Sidt2+/+ and Sidt2−/− MEFs were infected with 1 MOI mCherry-tagged HSV-1 virus for the indicated times and analysed by flow cytometry. Data is representative of 3 independent experiments. Error bars represent mean ± SEM. (C) Cell culture supernatant from Sidt2+/+ and Sidt2−/− MEFs infected with 1 MOI mCherry-tagged HSV-1 was collected at 96 h p.i. and IFN-β was measured via ELISA. (D) Sidt2+/+ and Sidt2−/− mice (n=13–15) were infected with 1×107 PFU HSV-1 i.p. and survival monitored for 8 days. (E–F) Serum from Sidt2+/+ and Sidt2−/− mice (n=10–13) was collected at 8h and 16h p.i. respectively and serum IFNβ was measured via ELISA. (G–H) Serum IL-6 and RANTES from Sidt2+/+ and Sidt2−/− mice (n=10–13) at 16 h p.i. were measured using Bioplex bead assay. For panels E–H, data are plotted as mean ± SEM. * P < 0.05, *** P < 0.001, n.s. = not significant. Data represents the pooled results from 3 independent experiments. See also Figure S3.
Next, we sought to determine whether the presence of SIDT2 is important during HSV-1 infection in vitro. We therefore infected Sidt2−/− and WT MEFs with mCherry-tagged HSV-1 and assessed infection over 72 h using flow cytometry. Sidt2−/− MEFs showed much higher rates of HSV-1 infection compared to WT cells (Figure 3B), suggesting that SIDT2 might play a role in limiting HSV-1 infection in vitro. Consistent with this, we observed that Sidt2−/− MEFs produced significantly less IFN-β than WT cells following HSV-1 infection (Figure 3C).
To determine the role for SIDT2 in HSV-1 infection in vivo, we infected Sidt2−/− and WT mice with HSV-1 i.p. and observed the animals over time. While the vast majority of WT mice remained asymptomatic, ∼50% of Sidt2−/− mice developed hind-limb paralysis and died within 3 days p.i. (Figure 3D). This is a similar (albeit less penetrant) phenotype to that observed in HSV-1-infected mice lacking the type I IFN receptor (Rasmussen et al., 2007). We therefore compared the production of type I IFN in Sidt2−/− and WT mice following HSV-1 infection. As a dsDNA virus, HSV-1 induces a robust IFN response via multiple DNA sensing pathways, including the STING-cGAS axis and TLR9 (Li et al., 2013; Rasmussen et al., 2007). This DNA-dependent response occurred shortly after in vivo challenge (peaking at 4–8 h p.i.). A subsequent wave of IFN-I production (evident by 12–16 h p.i.) does not require DNA sensing (Li et al., 2013; Rasmussen et al., 2007) and appears to instead involve MAVS-dependent RNA detection pathways (Rasmussen et al., 2007). On this basis, we assessed type I IFN from the serum of HSV-1 infected mice at both 8 and 16 h p.i. to separately probe the DNA and RNA sensing pathways respectively. Consistent with the inability of SIDT2 to interact with dsDNA by FRET-FLIM (Figure 1D), we observed no difference in type I IFN at 8 h p.i. (Figure 3E). However, by 16 h p.i. Sidt2−/− mice showed a significant reduction in serum IFN-β (Figure 3F) as well as other innate immune mediators such as RANTES and IL-6 (Figure 3G–H).
Taken together, the above results are consistent with our hypothesis that SIDT2 has a role in the innate immune response to extracellular viral dsRNA. However, an alternative explanation is that loss of SIDT2 causes a generalised impairment in antiviral immunity that is not specific for extracellular dsRNA. To investigate this possibility, we infected Sidt2−/− and WT mice with vesicular stomatitis virus (VSV) and lymphocytic choriomeningitis virus (LCMV). Both of these are negative strand RNA viruses, which have previously been shown to produce negligible amounts of dsRNA (Weber et al., 2006). Notably, loss of SIDT2 had no effect on clinical presentation, IFN-β production or viral titres following infection with VSV or LCMV (Figure S4), consistent with the antiviral activity of SIDT2 being dependent upon the presence of viral dsRNA.
SIDT2 facilitates trafficking of endocytosed dsRNA into the cytoplasm
Previous studies have suggested that SIDT1 promotes internalisation of extracellular dsRNAs (specifically, siRNAs) into mammalian cells (Duxbury et al., 2005; Wolfrum et al., 2007). Similarly, in mandarin fish (Siniperca chuatsi), it has been proposed that Sidt2 facilitates cellular uptake of exogenous dsRNA (Ren et al., 2011). Although our subcellular localisation data (Figure 1A) made this an unlikely possibility, we evaluated whether mammalian SIDT2 is involved in the uptake of extracellular dsRNA. Bone marrow-derived dendritic cells (BMDCs) from Sidt2−/− and WT mice were therefore incubated with extracellular poly(I:C)-rhodamine for 60 min, and intracellular rhodamine was measured via flow cytometry. Importantly, there was no difference between Sidt2−/− and WT cells in their ability to internalise poly(I:C) (Figure 4A). Identical results were obtained using 32P-labeled 500 bp dsRNA (Figure 4B). We therefore found no evidence that internalisation of extracellular dsRNA requires SIDT2.
Figure 4. Loss of SIDT2 impairs endosomal escape of internalised poly(I:C).
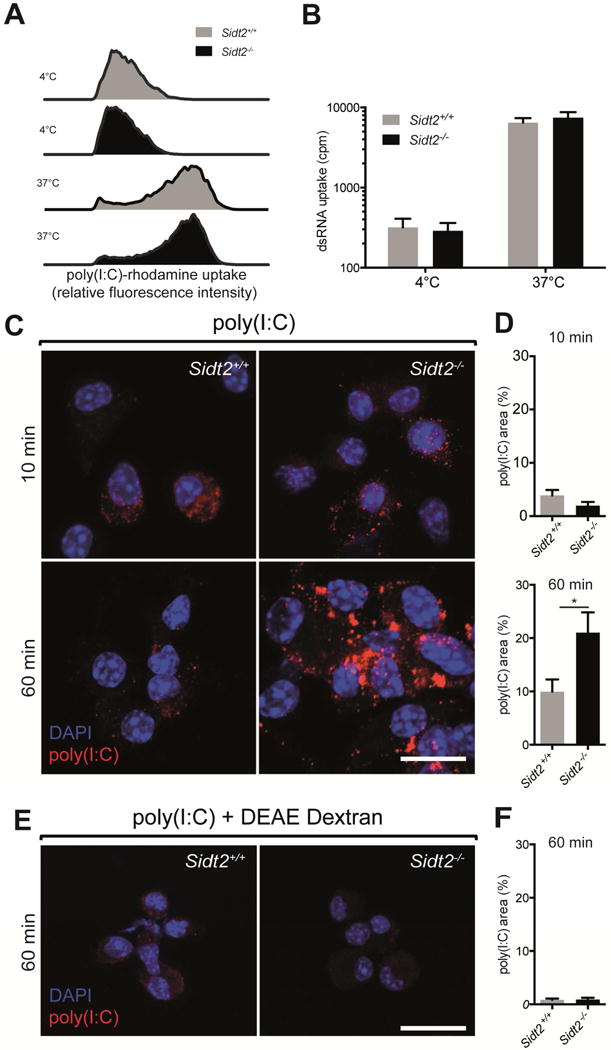
BMDCs from Sidt2+/+ and Sidt2−/− mice were treated with (A) poly(I:C)-rhodamine and (B) 32P-labeled 500bp dsRNA for 60 min at either 4°C or 37°C, and internalisation assessed via flow cytometry or radioactivity measurement respectively. Results are representative of at least 2 independent experiments. For panel B, all treatments and measurements were made in triplicate, and data are plotted as mean ± SEM. (C–D) BMDCs from Sidt2+/+ and Sidt2−/− mice were treated with poly(I:C) for either 10 or 60 min, stained with J2 anti-dsRNA antibody (red) and DAPI (blue), and imaged by confocal microscopy. The proportion of each cell occupied by punctate dsRNA staining was quantified. (E–F) Sidt2+/+ and Sidt2−/− BMDCs were treated with poly(I:C) in association with the cationic polymer DEAE-dextran for 60 min. For panels C and E, images are representative of at least three independent experiments. For panels D and F, data are plotted as mean ± SEM and between 30–150 cells were assessed per time point. * P < 0.05. Scale bar = 10 μm. See also Figure S5.
Next, we investigated the possibility that SIDT2 transports dsRNA across the endo-lysosomal membrane. To begin, we incubated Sidt2−/− and WT BMDCs with extracellular poly(I:C) for either 10 or 60 min, immunostained with J2 antibody, and compared the subcellular localisation of dsRNA via confocal microscopy (Figure 4C). After 10 min, both Sidt2−/− and WT cells displayed low punctate J2 staining, consistent with dsRNA entry into the endocytic pathway. However, after 60 min, dsRNA localisation in WT cells was predominantly diffuse, consistent with a cytoplasmic distribution; in contrast, dsRNA staining within Sidt2−/− cells was significantly more punctate and co-localised with the late endosomal marker RAB7 (Figure 4D, Figure S5A), indicating endo-lysosomal accumulation. To confirm these observations, we repeated the same experiment but delivered the poly(I:C) in conjunction with a cationic polymer transfection reagent to facilitate endosomal escape. This abrogated the punctate accumulation of poly(I:C) in Sidt2−/− cells (Figure 4E–F).
The above observations indicated that loss of SIDT2 impairs the trafficking of extracellular dsRNA from endo-lysosomes into the cytoplasm. To complement these studies, we investigated whether enhancing WT SIDT2 activity was able to augment dsRNA trafficking. We therefore generated DC2.4 cells that overexpress SIDT2-mCherry under the control of a doxycycline inducible lentiviral vector. Following exposure to extracellular poly(I:C)-fluorescein, we observed that doxycycline-treated (SIDT2-mCherry+) cells displayed a less punctate intracellular distribution of poly(I:C) than non-doxycycline-treated (SIDT2-mCherry−) cells (Figure 5A–B). Given that poly(I:C) internalisation between these two cell populations was similar (Figure 5C), these data suggest that SIDT2 actively promotes the escape of endocytosed dsRNA into the cytoplasm.
Figure 5. Overexpression of SIDT2 enhances endosomal escape of poly(I:C).
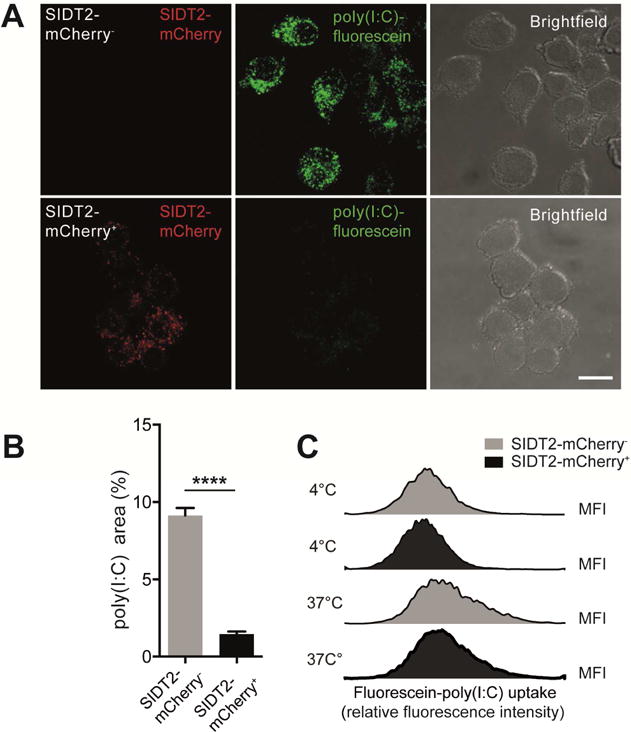
(A) Doxycycline-inducible SIDT2-mCherry DC2.4 cells were treated with poly(I:C)-fluorescein for 1 h in the presence or absence of doxycycline and imaged by confocal microscopy. Scale bar = 10 μm (B) The proportion of each cell occupied by punctate poly(I:C)-fluorescein staining was individually quantified for >60 cells across multiple fields of view, and plotted as mean ± SEM. (C) Internalisation of poly(I:C)-fluorescein by SIDT2-mCherry+ and SIDT2-mCherry− cells after 1 h at either 4°C or 37°C was assessed via flow cytometry. Data are representative of 3 independent experiments. ****P < 0.0001. See also Figure S5.
SIDT2 is required for the detection of dsRNA by the cytoplasmic RLR pathway but not endosomal TLR3
To test the role of SIDT2 in the functional response to dsRNA, we assessed whether loss of SIDT2 affects production of type I IFN following in vivo poly(I:C) challenge. Notably, Sidt2−/− mice administered poly(I:C) by i.p. injection produced significantly less IFN-β compared to WT littermates (Figure 6A), consistent with our earlier observations after EMCV and HSV-1 infection (Figure 2E and 3F). Although systemic administration of poly(I:C) is frequently perceived as a TLR3-dependent stimulus, type I IFN production following i.p. poly(I:C) injection is largely dependent on cytoplasmic RLR detection via MDA-5 rather than TLR3 (Gitlin et al., 2006). Together with our findings that SIDT2 facilitates trafficking of endocytosed dsRNA into the cytoplasm (Figures 4 and 5), this suggests that SIDT2 functions upstream of cytoplasmic RLR sensing (specifically, MDA-5) and is dispensable for endosomal TLR3 detection. Innate immune detection of picornaviruses such as EMCV strictly depends on MDA-5 but not RIG-I or TLR3 (Kato et al., 2006). That loss of SIDT2 phenocopied the reduced type I IFN response and impaired survival observed in MDA-5 deficient mice following EMCV infection (Figure 2) thus provides further support to the premise that SIDT2 facilitates MDA-5 recognition of dsRNA. To test this hypothesis further, we next performed a series of experiments designed to separately interrogate the RLR- and TLR3-dependent pathways.
Figure 6. SIDT2 is important for RLR- but not TLR-mediated IFN production in response to extracellular dsRNA.
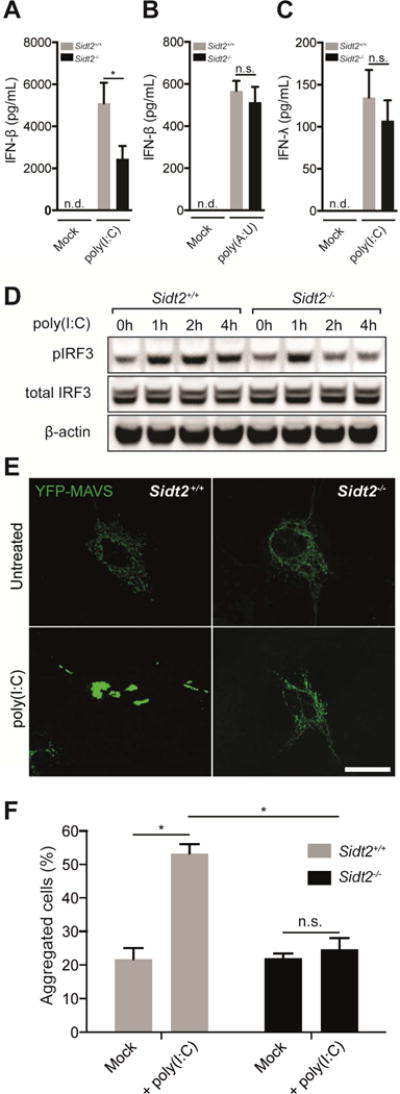
(A) Sidt2+/+ and Sidt2−/− mice were injected i.p with 50 μg poly(I:C) per 25 g body weight (n=7), and serum IFNβ measured at 3 h via ELISA. (B) Sidt2+/+ and Sidt2−/− mice (n=8) were injected i.p. with 300 μg poly(A:U) per 25 g body weight and serum IFNβ measured at 3 h via ELISA. (C) Sidt2+/+ and Sidt2−/− mice (n=8) were injected i.p. with 50 μg poly(I:C) per 25 g body weight and serum IFNλ measured at 3 h via ELISA (n=8). Data are plotted as mean ± SEM. (D) BMDMs from Sidt2+/+ and Sidt2−/− were stimulated with 10 μg/ml poly(I:C) for the indicated times and pIRF3Ser386 and total IRF3 was assessed via immunoblotting. (E) Sidt2+/+ and Sidt2−/− MEFs were transfected with MAVS-YFP, treated with poly(I:C) for 1 h, and MAVS aggregation assessed via confocal microscopy. Scale bar = 40 μm. (F) Individual cells (>150 per condition) were scored for the appearance of MAVS aggregates. Data is representative of 3 independent experiments and error bars are plotted as mean ± SEM. * P < 0.05, n.s. = not significant, n.d = not detected.
To assess TLR signalling, Sidt2−/− and WT mice were first injected with poly(A:U), a dsRNA mimic that specifically activates TLR but not RLR signalling (Perrot et al., 2010; Sugiyama et al., 2008). As would be predicted if endocytosed dsRNA is detected within endosomes independently of SIDT2 transporter activity, Sidt2−/− mice showed no difference in poly(A:U)-induced type I IFN production (Figure 6B). Similarly, when we measured serum IFN-λ, a type III IFN that is produced in a TLR3-dependent manner in response to poly(I:C) (Lauterbach et al., 2010), Sidt2−/− mice responded similarly compared to WT after poly(I:C) injection (Figure 6C). This suggests that the loss of SIDT2 does not affect dsRNA-dependent TLR signalling. In keeping with these observations, Sidt2−/− BMDMs also showed reduced IRF3 activation in response to poly(I:C) treatment as assessed by phospho-IRF3 immunoblot (Figure 6D), although activation was similar to WT cells early on presumably due to unaffected TLR3-dependent TRIF signalling.
To specifically assess RLR signalling, we measured aggregation of mitochondrial antiviral-signalling (MAVS) protein via confocal microscopy. During viral infection, the RLRs, RIG-I and MDA-5, detect viral RNAs within the cytoplasm and trigger aggregation of MAVS, which subsequently activates the transcription factor IRF3 to induce type I IFNs. In this way, aggregation of MAVS serves as a specific indicator of RLR pathway activation (Hou et al., 2011; Seth et al., 2005). We therefore transfected Sidt2−/− and WT MEFs with a MAVS-YFP construct and compared MAVS aggregation following poly(I:C) treatment (Figure 6E). After 60 min of poly(I:C) exposure, WT MEFs displayed a robust increase in MAVS aggregation but Sidt2−/− cells did not (Figure 6F), consistent with a requirement for SIDT2 to transport endocytosed dsRNA into the cytoplasm for RLR recognition.
SIDT2 facilitates the endosomal escape of dsRNA and subsequent RLR sensing during HSV-1 infection
Finally, we wanted to better understand the mechanisms underlying the defective immunity against HSV-1 that we observed in Sidt2−/− mice (Figure 3). In light of our observations that SIDT2 promotes the escape of endocytosed poly(I:C) into the cytoplasm (Figure 4), we infected Sidt2−/− and WT mice with HSV-1 i.p. and immunostained PECs 16 h p.i. with J2 antibody to compare the intracellular distribution of dsRNA (Figure 7A). Similar to our in vitro data with poly(I:C) (Figure 4D), PECs from HSV-1 infected Sidt2−/− mice showed a significant increase in punctate dsRNA staining compared to WT (Figure 7B). Importantly, this punctate dsRNA staining was observed in uninfected bystander cells rather than HSV-1 infected cells, as revealed by the use of a GFP-tagged HSV-1 (Figure S5B). This suggested that SIDT2 facilitated the endosomal escape of viral dsRNAs in bystander cells, and predicted that immune activation of these cells via cytoplasmic RLR signalling would be impaired in the absence of SIDT2.
Figure 7. SIDT2 is required for RLR-induced MAVS activation in bystander cells during HSV-1 infection.
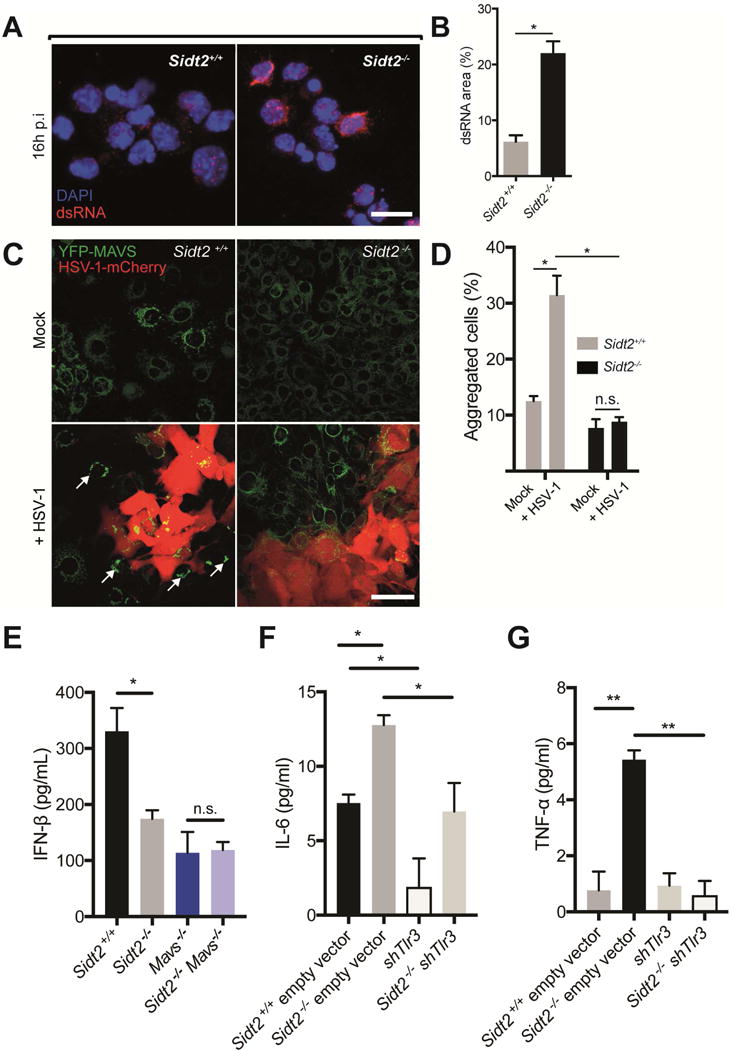
(A) PECs from Sidt2+/+ and Sidt2−/− mice infected with 1×107 PFU at 16 h p.i. were stained with J2 dsRNA antibody (red) and DAPI (blue) and imaged by confocal microscopy. Scale bar = 10 μm. (B) The proportion of each cell occupied by punctate dsRNA staining was quantified using FIJI software. (C) Sidt2+/+ and Sidt2−/− MEFs stably expressing MAVS-YFP were infected with 1 MOI HSV-1-mCherry for 48 h, and imaged via confocal microscopy to assess for MAVS aggregation (arrows) in uninfected, bystander cells. Scale bar = 80 μm. (D) Individual cells were segmented and HSV-1-mCherry infected cells were excluded using FIJI software. Uninfected bystander cells (250 cells per condition) were scored for the appearance of MAVS aggregates. (E) Sidt2+/+ (Cas9 only), Sidt2−/− (Cas9 only), MAVS−/− and Sidt2−/− MAVS−/− MEFs were infected with 1 MOI mCherry-tagged HSV-1, and IFNβ was measured in cell culture supernatant at 96 h p.i. via ELISA. (F) IL-6 and (G) TNF-α were measured in cell culture supernatant from shTLR3 and Sidt2−/− shTLR3 MEFs infected with 1 MOI mCherry-tagged HSV-1 96h p.i. Sidt2+/+ and Sidt2−/− MEFs transduced with retroviral vector lacking shRNA were used as controls. IFN-β, IL-6 and TNF-α in non-infected cells were below the limit of assay detection. Data is representative of results from 2–3 independent experiments and expressed as the mean ± SEM of triplicate wells. * P < 0.05, ** P < 0.01, n.s. = not significant. See also Figure S6.
To test this, we determined whether loss of SIDT2 resulted in defective MAVS signalling in bystander cells during HSV-1 infection. To do so, we stably expressed MAVS-YFP in Sidt2−/− and WT MEFs, and then compared MAVS aggregation following infection with mCherry-tagged HSV-1 using a low multiplicity of infection (MOI) to ensure a mixed population of infected and uninfected cells (Figure 7C). While uninfected WT bystander MEFs contained readily detectable MAVS aggregates, uninfected Sidt2−/− bystander cells completely failed to activate MAVS (Figure 7D).
To formally assess whether SIDT2 is required for RLR- but not TLR3-dependent immune signalling during HSV-1 infection, we used CRISPR-Cas9 technology and shRNA retroviral constructs to reduce, respectively, MAVS and TLR3 expression in Sidt2−/− and WT MEF cells (Figure S6A–B). We then measured IFN-β production in response to HSV-1 infection (Figure 7E). Consistent with SIDT2 and MAVS functioning in the same signalling pathway, loss of either MAVS or SIDT2 impaired IFN-β production to a similar extent, while Sidt2−/−Mavs−/− doubly deficient MEFs produced comparable IFN-β to Sidt2−/− cells (Figure 7E). In contrast, TLR3 activity had no influence on IFN-β production (Figure S6C). To determine whether SIDT2 is required for TLR3-dependent signalling, we therefore examined TLR3-dependent IL-6 expression (Kato et al., 2006) in response to HSV-1 infection. Notably, IL-6 production was impaired by siRNA-mediated silencing of TLR3 in both WT and Sidt2−/− MEFs (Figure 7F), indicating that SIDT2 is not required for TLR3 activity. Indeed, Sidt2−/− MEFs actually displayed a small but significant increase in IL-6 production (Figure 7F), suggesting that loss of SIDT2 might enhance TLR3 activation. This aligns with our earlier observation that Sidt2−/− cells accumulate endosomal dsRNA (Figure S5), and implies that the endosomal export of dsRNA by SIDT2 may serve to limit the amount of substrate available for TLR3 recognition. Consistent with this, we also observed that TNF-α production following HSV-1 infection was detectable only in Sidt2−/− MEFs and that this production required intact TLR3 signalling (Figure 7G). Taken together, our results therefore indicate that SIDT2 helps to promote RLR but not TLR3 activity during HSV-1 infection (Figure S7).
DISCUSSION
Previous observations that MDA-5 is the dominant in vivo sensor of systemically administered poly(I:C) (Gitlin et al., 2006) directly imply the existence of an efficient mechanism by which extracellular dsRNA gains entry into the cytoplasm. This mechanism has been postulated to involve a transmembrane transporter either at the cell surface or within endosomes (Gitlin et al., 2006; Nellimarla and Mossman, 2014). Our data indicate that SIDT2, a predicted nine-pass transmembrane protein (Gao et al., 2010), fulfils this role by transporting internalised dsRNA across the endo-lysosomal membrane to engage the cytoplasmic RLRs and thereby induce type I IFN production.
By extension, our data also suggest an important role for extracellular dsRNA in anti-viral immunity. Specifically, we observed that dsRNA triggers RLR signalling within uninfected bystander cells in a SIDT2-dependent manner during HSV-1 infection, and that the absence of this bystander activation due to loss of SIDT2 is associated with impaired anti-viral cytokine production and reduced survival. At first, this might seem surprising since immunity within HSV-1-infected cells presumably remains intact in the absence of SIDT2. However, HSV-1 is well-known for its extensive ability to suppress immunity in cells that it infects (Melchjorsen et al., 2009), so SIDT2-mediated bystander activation – whereby uninfected host cells take up, detect, and respond to extracellular HSV-1 dsRNA released from infected cells – may represent an important counter strategy for host immunity. Along similar lines, recent in vitro experiments have demonstrated a role for dsRNA-based activation of bystander cells in limiting hepatitis C virus (HCV) infection (Dansako et al., 2013), but our results provide evidence that this activation is important in vivo and highlight a physiological role for extracellular RNA at a time when this is a matter of considerable interest and debate (Leslie, 2013).
Although HSV-1 appears to be an informative model for studying the role of SIDT2 in dsRNA-induced bystander activation, the requirement for SIDT2 in mounting a protective immune response against HSV-1 is only partial. This is presumably since multiple DNA and RNA sensing pathways are important for anti-HSV-1 immunity, and most of these (e.g. STING, TLR) would be expected to remain functional in the absence of SIDT2. In contrast, protection against EMCV infection wholly depends upon intact dsRNA sensing by MDA-5 (Gitlin et al., 2006; Kato et al., 2006). To this end, our observation that SIDT2 is absolutely required for survival following EMCV infection – and phenocopies loss of MDA-5 – was notable for two reasons. First, together with our data indicating that SIDT2 transports dsRNA into the cytoplasm, it strongly suggested that the main role of SIDT2 during EMCV infection is to deliver dsRNA for MDA-5 sensing. Second, it indirectly implied that the dsRNA substrates recognised by MDA-5 during EMCV infection are predominantly extracellular in origin. Given that EMCV employs multiple strategies to disrupt RLR signalling within infected host cells (Barral et al., 2009; Ng et al., 2013) and also causes lytic infection, bystander activation via the SIDT2-dependent trafficking of extracellular dsRNA would make intuitive sense. Developing experimental reagents (e.g. recombinant fluorescent virus) that enable discrimination between EMCV infected and uninfected cells should enable this proposed bystander activation to be directly tested.
Looking ahead, it will also be interesting to define the mechanism by which viral dsRNA is transferred to bystander cells, since efforts to either augment or inhibit this process (and thereby boost or dampen type I IFN responses) may be relevant to not only improving immunity but also limiting immunopathology (Davidson et al., 2015). Cell lysis – either as a direct result of viral infection or via NK cell-mediated destruction in vivo – might be one potential route. Another is exosomes, which have been previously reported to contain RNAs from multiple viruses, including HSV-1, HCV, HIV and EBV (Dansako et al., 2013; Dreux et al., 2012; Narayanan et al., 2013; Pegtel et al., 2010). At the same time, it will also be interesting to determine whether SIDT2 has a protective role against viruses other than EMCV and HSV-1. Since dsRNA production seems to be a general feature of most viruses (with the exception of negative sense RNA viruses) (Weber et al., 2006), this seems plausible. However, if lytic replication is important for the extracellular release of dsRNA, then SIDT2 may only be relevant to lytic viruses such as EMCV and HSV-1. In either case, the highly conserved nature of SIDT2 at a sequence level suggests that it may provide protection against viruses in multiple species, which is consistent with a previous report in mandarin fish (Ren et al., 2011).
Although Sidt2−/− mice exposed to poly(I:C), EMCV and HSV-1 showed impaired type I IFN production, these defects were incomplete. Notwithstanding the role for intact DNA sensing pathways in HSV-1 as discussed earlier, there are at least two possible explanations. First, TLR3 sensing of endosomal dsRNA, which is not dependent on SIDT2, likely contributes to residual IFN production and antiviral activity. Second, alternative mechanisms might facilitate the cytoplasmic entry of extracellular dsRNA. In this regard, it is notable that the other mammalian SID-1 ortholog, Sidt1, is strongly induced following dsRNA challenge, and has been previously reported to be both an IFN-stimulated gene and upregulated following HSV-1 infection (Miyazaki et al., 2011; Rusinova et al., 2013). Future studies to determine whether SIDT1 has a role in dsRNA trafficking during antiviral immunity and to examine possible redundancy and/or synergy between the two mammalian SID-1 orthologs may therefore be revealing.
Finally, it will be important to learn in which cells SIDT2 functions and to define its RNA substrate specificity. It remains unknown what cell types respond to extracellular dsRNA to activate RLRs in vivo whether in the context of systemic poly(I:C) administration, EMCV or HSV-1 infection. Our data indicate that the relevant cells in both these cases must express Sidt2. Given the broad expression of Sidt2, many cell types could be involved. Since chemical modifications (e.g. 5′ PPP) of viral RNAs facilitate the discrimination between self and non-self by host RNA sensors such as RIG-I, it will also be interesting to investigate whether any modifications facilitate SIDT2-dependent RNA transport. Similarly, dsRNA length is a key determinant of recognition by RIG-I, MDA-5 and TLR3 (Kato et al., 2008; Leonard et al., 2008), and SIDT2 was recently reported as having a higher binding affinity for longer dsRNAs (300–700 bp) (Li et al., 2015a). Functional investigations to determine whether SIDT2 can transport shorter substrates such as siRNAs may shed light on the development of more effective RNAi therapeutics, whose delivery continues to be hindered by poor endosomal escape (Dominska and Dykxhoorn, 2010).
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ken Pang (ken.pang@mcri.edu.au).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Mice were bred and maintained in the animal facilities at the Walter and Eliza Hall Institute of Medical Research (WEHI) and at the Department of Molecular and Cellular Biology, Harvard University, according to national and institutional guidelines for animal care. Mice were housed in individual ventilated cages at 19 – 24°C and maintained on a 1 4 h light to 10 h dark cycle with continuous access to Barastoc Custom Mixed Ration (irradiated) and acidified and filtered water. All experimental procedures were approved by the relevant animal ethics committees at the WEHI and Harvard University. Targeted Sidt2 alleles were generated as part of a mouse knockout library for transmembrane proteins as described previously (Tang et al., 2010), and mice were obtained from the Knock-Out Mouse Project (KOMP) Repository (www.komp.org). These mice were originally on a 129 background but were subsequently backcrossed 6 times onto a C57BL/6. Age-matched female mice were used for infection experiments.
Cell lines and reagents
Sidt2+/+ and Sidt2−/− MEFs were generated from littermates at E13.5 and cultured at 5% CO2 in DMEM with 10% foetal bovine serum (FBS) using standard methods (Sun et al., 2007). Dendritic cell-like cell line, DC2.4 (Shen et al., 1997), was cultured at 37°C and 5% CO2 in DMEM with 10% foetal bovine serum (FBS). Primary BMDCs were generated and cultured as previously described (Pang et al., 2009; Pang et al., 2006). Briefly, bone marrow was flushed from the hind legs of 6 – 9 week old Sidt2+/+ and Sidt2−/− mice and incubated in 10% X-63 (GM-CSF) supernatant for 7 days. Cells were then harvested by aspiration and used for experiments. Primary BMDMs were generated and cultured as previously described (Davis, 2013). Briefly bone marrow was flushed from the hind legs of 6 – 9 week old Sidt2+/+ and Sidt2−/− mice and incubated in 20% L929 (G-CSF) supernatant for 6 days. Cells were then harvested by scraping and used for experiments. Vero cells (a kind gift from C. Jones) were cultured at 37°C a nd 5% CO2 in DMEM supplemented with 10% FBS. To generate cell lines with stably inducible SIDT2-mCherry or MAVS-YFP, cells were infected with relevant lentiviral constructs (see below) and selected in puromycin. For SIDT2-mCherry constructs, full-length mouse SIDT2 (a kind gift from L. Attardi, Stanford University) was amplified via PCR, cloned into a lentiviral vector in which expression of the target gene is under the control of a reverse tetracycline-controlled transactivator (rtTA) (Kelly et al., 2014) (a kind gift from T Okamoto and D. Huang, WEHI), ligated to mCherry at the C-terminus, and then verified by sequencing. For YFP-MAVS constructs, full-length human MAVS-YFP (a kind gift from Q. Sun, Institute of Zoology, Chinese Academy of Sciences) was amplified via PCR, and cloned into the same lentiviral vector as described above.
METHOD DETAILS
SID-1 family members: phylogenetic analysis and mammalian expression
The radial tree display in Fig. S1A was assembled using the multiprotein alignment tool COBALT (Papadopoulos and Agarwala, 2007), accessed via NCBI, using the proteins listed in Supplemental Table 1. The radial tree display used neighbour joining, Grishin distance matrix, and max_seq = 0.85. For Fig. S2, publicly available microarray data for mouse Sidt1 and Sidt2 from the GNF Gene Expression Atlas were downloaded from BioGPS (Wu et al., 2009), while RNAseq data for human, mouse and rat Sidt1 and Sidt2 from the GTEX consortium and others (Barbosa-Morais et al., 2012; Consortium, 2013; Merkin et al., 2012) were downloaded via the EMBL-EBI Expression Atlas (Petryszak et al., 2014). For display purposes, expression thresholds of 10 relative fluorescence units (for microarray data) and 0.5 FPKM (for RNAseq data) were used.
SIDT2 subcellular localisation
The following constructs were transiently transfected into cells in association with FuGENE HD (Promega) according to manufacturer’s instructions: EEA-1-GFP and LAMP1-YFP (kind gifts from A. Irving, Monash Micro Imaging, Monash University); RAB7-eGFP and RAB11-eGFP (kind gifts from P. Gleeson, University of Melbourne). Briefly, 7×103 Sidt2-mCherry transduced MEF cells were seeded in μ-Slide 8 Well (Ibidi) and treated with 1 μg/mL doxycycline for 32 h to induce SIDT2-mCherry expression. Cells were subsequently transiently transfected with 0.25 μg of either EEA-1-GFP, LAMP1-YFP, RAB7-eGFP or RAB11-eGFP plasmid using FuGENE HD transfection reagent for 48 h at 37°C, 5% CO2. Cells were then imaged live every 30 secs for 20 min on a Leica SP8 laser scanning confocal microscope. Experiments were repeated at least 2 times.
FRET FLIM
5×103 SIDT2-mCherry transduced DC2.4 cells were seeded in μ-Slide 8 Well (Ibidi) and treated with 1 μg/mL doxycycline for 32 h to induce SIDT2-mCherry expression. Cells were subsequently treated with either 1μg/mL poly(I:C)-fluorescein or transfected with 100 nM dsDNA-fluorescein complexed with FuGENE HD for 1 h at 37°C. dsDNA-Fluorescein (FAM) was purchased from Bioneer Pacific, and corresponded to the following sequence: 5′ GCA TCA AGG TGA ACT TCA A(TT), 3′ TTG AAG TTC ACC TTG ATG C(TT). Cells were then washed, and fixed in 4% paraformaldehyde for 10 min on ice. FLIM data was recorded using an Olympus FV1000 microscope equipped with a PicoHarp300 FLIM extension and a 485 nm pulsed laser diode from PicoQuant (Berlin, Germany). Cells were first imaged for green and red fluorescence by confocal microscopy using the Olympus FV100 system with a 60× oil immersion objective to verify presence of both donor (fluorescein) and acceptor (mCherry) dyes. Subsequently corresponding FLIM images of green fluorescence was recorded using the PicoHarp extension. Pixel integration time for FLIM images was kept at 40 ms per pixel and fluorescence lifetime histograms were accumulated to at least 10,000 counts in the maximum to ensure sufficient statistics for FLIM-FRET analysis. Photon count rates were kept below 5% of the laser repetition rate to prevent pileup. FLIM-FRET analysis was performed using the SymPhoTime 64 software (PicoQuant). Poly(I:C)-fluorescein or dsDNA-fluorescein positive compartments of individual cells were chosen as regions of interest (ROIs), then the fluorescence lifetime decay of each ROI was de-convolved with the measured instrument response function and fitted with a biexponential decay. The amplitude weighted average lifetime was extracted from each fit and averaged over all values of one sample condition. P-values were determined to assess the significance of donor lifetime changes in absence and presence of the SIDT2-mCherry acceptor. Experiments were repeated at least 3 times.
In vivo viral infections and dsRNA challenge
For EMCV infections, 6-week-old female mice were infected with 50 PFU via i.p. injection. For survival studies, animals were followed for 8 days; for cytokine analysis, serum and peritoneal lavage fluid were harvested at 3 days p.i. infection and for viral titres, whole heart homogenate and peritoneal exudate cells (PECs) from mice 3 days p.i. were assessed by viral plaque assay using Vero cells. For HSV-1 infections, 6–8 week old female mice were infected with 2 × 107 PFU HSV-1 (KOS strain) via i.p. injection (Smith et al., 2004). For survival studies, animals were followed for 8 days; for cytokine analysis, serum was harvested at 8 and 16 h post infection; and for dsRNA localisation studies, peritoneal cells were isolated at 16 h post infection via peritoneal lavage. For VSV infections, 3-month-old mice were infected with 106 PFU VSV (Indiana strain, a kind gift from A. Moseman) via footpad injection, and draining popliteal lymph nodes were harvested at 6 h post infection for Ifn-β mRNA quantification. For LCMV infections, 6–8 week old mice were injected i.v. with 2×106 PFU LCMV (Docile strain) (Ebert et al., 2015), and serum was subsequently collected at 24 h post infection for cytokine analysis. For viral titres, lung, liver and brain were collected and homogenised at 8 and 28 days p.i. and assessed via plaque assay. For dsRNA challenge, age-matched female mice were injected i.p. with 50 μg poly(I:C) per 25g body weight or 300 μg poly(A:U) per 25g body weight, and serum was collected 3 h later for cytokine analysis. Experiments were repeated at least 3 times.
IFNβ and cytokine analysis
Assessment of serum IFN-β and IFN-λ was performed via ELISA according to the manufacturer’s instructions (PBL Assay Science (Piscataway, NJ) and R&D Systems respectively). For HSV-1 in vivo infection, mice that did not produce detectable IFNβ at 8 h p.i. were excluded from the experiment. To assess IL-6, RANTES and IL-12 p40, serum was analysed via a Bio-Plex cytokine assay (Bio-Rad) according to manufacturer’s instructions. To compare Ifnβ mRNA, total RNA was isolated using an RNeasy kit (QIAGEN), treated with DNase I (Roche), reverse transcribed with ThermoScript (Life Technologies), and quantitative PCR performed on a DNA Engine Opticon system (Bio-Rad) using QuantiTect SYBR Green PCR reagents (QIAGEN) and the following primers for IFNβ (F: CCCTATGGAGATGACGGAGA; R: CTGTCTGCTGGTGGAGTTCA), VSV (F: TGATACAGTACAATTATTTTGGGAC; R:GAGACTTTCTGTTACGGGATCTGG) and Gapdh as a normalisation control (F: CAACTTTGTCAAGCTCATTTCCTG; R: CCTCTCTTGCTCAGTGTCCTT) (Chevrier et al., 2011; Sjolinder et al., 2012; Yen and Ganea, 2009). Assessment of cell culture supernatant IL-6 and TNF-α was performed via ELISA according to manufacturer’s instructions (eBioscence).
Western blotting
For immunoblotting, equivalent numbers of Sidt2+/+ and Sidt2−/− MEFs were permeabilised in 0.025% digitonin (with 20 mM HEPES-NaOH pH 7.5, 100 nM sucrose, 2.5mM MgCl2, 100 mM KCl, and protease inhibitors (Roche)) for 10 min and centrifuged at >10,000 g. The resulting pellets were lysed in KALB buffer and denatured in 4X SDS PAGE sample buffer. For detection of SIDT2, a monoclonal antibody recognising mouse SIDT2 (amino acids 29–40: SQKDAEFERTYA) was generated by immunising BALB/C mice and screening the resultant hybridomas (Abmart, Shanghai, China) for suitable clones using a flow-cytometry based method as previously described (O’Reilly et al., 1998). For SIDT2 immunoblotting, lysates were first subjected to immunoprecipitation with anti- SIDT2 Ab-conjugated, NHS-activated sepharose (GE Healthcare) overnight at 4°C; beads were subsequently washed in KALB buffer, and bound proteins eluted in 4x SDS-PAGE sample buffer at 95°C. For pIRF3 and IRF3 immunoblotting, 2×106 Sidt2+/+ and Sidt2−/− BMDMs were lysed in RIPA buffer supplemented with protease inhibitor and PhosSTOP (Roche) and denatured in 4x SDS PAGE sample buffer at 95°C. Proteins were separated on NuPAGE Novex 4–12% Bis-Tris Gels (Life Technologies), and transferred electrophoretically to Hybond ECL nitrocellulose membrane, blocked in 5% skim milk, incubated with the relevant primary antibodies, and washed again. Membranes were then incubated for 1 h with horseradish peroxidase-conjugated secondary antibodies, washed, and treated with Luminata Forte Western HRP substrate (Millipore), and visualised on the ChemiDoc MP system (Bio-Rad). Experiments were repeated at least 3 times.
qRT-PCR
For Sidt2 RNA analysis, total RNA was isolated and cDNA synthesised from Sidt2+/+ and Sidt2−/− MEFs as described above, then 30 cycles of PCR performed for Sidt2 using the following primers (F: CAGAAGGAGGCTGTTGTGTC; R: CAGGGTAGACACGTCCACAT). For Tlr3 RNA analysis, total RNA was isolated from MEFs and analysed by qRT-PCR using the following primers: (F: CCC CCT TTG AAC TCC TCT TC and R: TTT CGG CTT CTT TTG ATG CT) (Martinez et al., 2010). For Sidt1 RNA analysis, total RNA was isolated from WT BMDMs and analysed by RT-PCR as described above using primers purchased from QIAGEN (cat no. QT00147252). Experiments were repeated at least 2 times.
dsRNA internalisation and subcellular localisation
To assess poly(I:C) internalisation, cells were treated with 1 μg/mL of fluorescein- or rhodamine-conjugated poly(I:C) at either 37°C or 4°C (the latter served as a negative control) for 1 h, washed and analysed on an LSR II flow cytometer (BD Biosciences). Internalisation of 32P-labeled 500bp dsRNA was assessed for replicates of 1 million BMDCs pre-incubated at assay temperature for 10 minutes prior to resuspension in media containing 10 ng/ml 32P-labeled 500bp dsRNA. Following 1 hour of incubation cells were processed as previously described (Feinberg and Hunter, 2003). To characterise the subcellular localisation of dsRNA, different methods were employed depending on the cell populations being assessed and the nature of the dsRNA under investigation. In the first method, doxycycline treated, SIDT2-mCherry transduced DC2.4 cells were treated for 1 h with 1 μg/mL poly(I:C)-fluorescein at 37°C in 8 well chamber μ-s lides washed in PBS, and fixed in 4% paraformaldehyde (PFA) on ice, and then imaged on a Zeiss LSM 780 confocal microscope. To assess the proportion of poly(I:C) within the endocytic compartment, the poly(I:C)-fluorescein signal was thresholded using the FIJI software package (Schindelin et al., 2012) to ensure that only those FITCbright punctate regions within the cell were evident; for each individual cell, the total area occupied by these regions was then calculated as a proportion of the entire cell area; an average % was then derived across the entire cell population. In the second method, BMDCs were incubated for the indicated times with 2 μg/mL poly(I:C) ± 50 μg/mL DEAE-dextran, washed in PBS, fixed for 10 min in 4% PFA on ice, permeabilised using 0.1% Tween/PBS, blocked in 5% normal goat serum for 1 h, stained with J2 primary antibody and then anti-mouse Alexa-594 secondary antibody, mounted onto glass slides using a Cytospin centrifuge (ThermoScientific), and finally imaged on a Zeiss LSM 780 confocal microscope. The proportion of poly(I:C) within the endocytic compartment was then calculated as described above, but thresholding was performed on the basis of Alexa-594 rather than FITC signal. In the third method, Vero were grown in 8 well chamber slides, infected with HSV-1 at an MOI of 1, and J2 staining performed as described above after 48 h. Similarly, following HSV-1 i.p. infection in vivo, peritoneal lavage cells were harvested at 16 h, stained with J2 Ab as described above, and mounted onto glass slides using a Cytospin centrifuge. Experiments were repeated at least 3 times.
Assessment of MAVS activation in MEFs
To assess MAVS activation in response to poly(I:C), Sidt2−/− and WT MEFs were grown in 8 well chamber slides and transfected with MAVS-YFP using Fugene HD according to manufacturer’s instructions. After 24 h, cells were washed in PBS, re-plated in fresh DMEM with 10% FBS for 4 h, treated with 2 μg/mL poly(I:C) for 1 h at 37°C, fixed in 4% PFA for 10 min on ice, and imaged on a Zeiss LSM 780 confocal microscope. To determine the proportion of cells displaying MAVS activation, individual YFP+ cells were scored for the presence or absence of MAVS-YFP aggregation across multiple fields, and an average % calculated. To assess MAVS activation in response to HSV-1 infection, MAVS-YFP transduced Sidt2−/− and WT MEFs were grown in 8 well chamber slides and infected with HSV-1 mCherry at an MOI of 1. After 24 h, the cells were treated with 0.5 μg/mL doxycycline for a further 24 h to induce MAVS-YFP expression, fixed in 4% PFA for 10 min on ice, and imaged on a Zeiss LSM 780 confocal microscope. Experiments were repeated 3 times and the data pooled across the 3 independent experiments.
Generation of Mavs−/− MEFs using CRIPSR/Cas9
Sidt2+/+ and Sidt2−/− MEFs were infected with lentiCas9-Blast. Cells were selected using Blasticidin (10μg/ml) for 48 h at 37°C, 5% Co2. Specific sgRNA against mMAVS (F: TCCCGTTGTCTCGGATATACTTAT R: AAACATAAGTATATCCGAGACAAC) was ligated into the BsmB1 restriction site of the inducible sgRNA lentiviral vector (FgH1t_UTG). Lentivirus particles were produced by transient transfection of HEK293T cells with passenger DNA (10 μg), packaging vectors pMDL (5μg) and pRSV-rev (2.5μg) and envelope vector pVSV-G (3μg) using Lipofectamine 2000 (Life Technologies). The media was changed to DMEM + 10% FCS at 24 h post transfection and viral supernatant was collected at 48 h – 72 h. A total of 1 × 105 Sidt2+/+ and Sidt2−/− MEFs were infected with 2 ml viral supernatant supplemented with 8 μg/ml polybrene and centrifuged at 840 × g for 3 h at 32° C, before incubation at 37° C overnight. The process was repeated the following day, and viral supernatant was replaced with fresh media following centrifugation. Cells positive for eGFP were sorted and CRISPR Cas9 targeting was induced with doxycycline as previously described (Aubrey et al., 2015).
Generation of Tlr3 knockdown MEFs using shRNA
A miR-30-based shRNA against TLR3 was synthesised as 97 bp oligo by GeneWorks (TGCTGTTGACAGTGAGCGCAAGGGTGTTCCTCTTATCTAATAGTGAAGCCACAGATGTATTAGATAAGAGGAACACCCTTTTGCCTACTGCCTCGGA), PCR-amplified, cloned into LMS vector (Dickins et al., 2005) and verified by sequencing. Retrovirus particles were produced by transient transfection of 293T cells with shRNA against TLR3 or LMS empty vector (20μg), packaging vector gag-pol (10μg) and envelope vector VSVg (1μg) using Lipofectamine 2000 (Life Technologies). The media was changed to DMEM + 10% FCS at 24 h post transfection and viral supernatant was collected at 48 h – 72 h. A total of 1 × 105 Sidt2+/+ and Sidt2−/− MEFs were infected with 2 ml viral supernatant supplemented with 4 μg/ml polybrene and centrifuged at 840 × g for 3 h at 32 °C, before incubation at 37°C overnight. The process was repeated the following day, and viral supernatant was replaced with fresh media following centrifugation. Cells positive for GFP were sorted and used for experiments as stated.
RNAseq
1×106 Vero cells were infected with 0.5 MOI HSV-1-mCherry (Russell et al., 2015) for 48 h and HSV-1+ and HSV-1− cells were sorted based on mCherry fluorescence. Uninfected cells were used as a negative control. Three independent biological replicates of each condition were obtained. RNA was extracted using the RNeasy Plus mini kit (QIAGEN) followed by quantification and quality control using TapeStation 2200 (Agilent Technologies). RNA sequencing libraries were prepared using the Illumina TruSeq stranded total RNA library prep kit and sequenced on an Illumina NextSeq 500. On average, 45.4 million 75 base pair paired-end reads were obtained for each sample. To distinguish Green Monkey and HSV-1 (KOS strain) RNA, an index was built containing both genomes using the Rsubread package (Liao et al., 2013). The reference genome and gene annotation for both the Green Monkey and HSV-1 (KOS) strain was obtained via NCBI (assembly Chlorocebus_sabeus 1.1 and version JQ673480.1 respectively). All reads were aligned to this combined genome using the Rsubread aligner (Liao et al., 2013). Coverage plots were generated using Integrated Genome Brower (IGB) (Freese et al., 2016). For visualization, the aligned samples were sorted and indexed using Rsamtools version 1.24.0. RNAseq data is publicly available at Gene Expression Omnibus (Accession GSE101960).
QUANTIFICATION AND STATISTICAL ANALYSIS
Graphpad Prism (version 7) was used for all statistical analyses. Statistical analyses were performed using unpaired, two-tailed Student’s t-tests or two-way ANOVAs, except in the case of the HSV-1 and EMCV survival analysis, where a generalised Wilcoxon (Gehan–Breslow) test was used to compare survival curves. P-values < 0.05 were considered statistically significant. Sample sizes (n) are provided in the respective figure legends.
Supplementary Material
Table S1. Accession number and description of proteins used in Figure S1.
MEFs stably expressing SIDT2-mCherry were transfected with a late endosomal marker (RAB7-eGFP) and imaged live every 30 sec for 20 min on a Leica SP8 laser scanning confocal microscope.
MEFs stably expressing SIDT2-mCherry were transfected with a lysosomal marker (LAMP-1-YFP) and imaged live every 30 sec for 20 min on a Leica SP8 laser scanning confocal microscope.
MEFs stably expressing SIDT2-mCherry were transfected with a lysosomal marker (EEA-1-GFP) and imaged live every 30 sec for 20 min on a Leica SP8 laser scanning confocal microscope
MEFs stably expressing SIDT2-mCherry were transfected with a recycling endosome marker (RAB11-eGFP) and imaged live every 30 sec for 20 min on a Leica SP8 laser scanning confocal microscope.
Highlights.
SIDT2 is in endo-lysosomes and interacts with internalised double-stranded RNA
SIDT2 promotes escape of endosomal dsRNA & cytoplasmic RLR signalling
During HSV-1 infection, RLR signalling in bystander cells requires SIDT2
Loss of SIDT2 impairs IFN-β production & survival after HSV-1 & EMCV infection
Acknowledgments
We wish to thank C. Jones and A. Moseman for gifts of HSV-1 and VSV virus respectively, L. Attardi, T Okamoto, D. Huang, A. Irving, and P. Gleeson, A. Janic, J. Bernardini for gifts of plasmids, as well as J. Botros, F. Song, N. Nakatsuka, L. Mackiewicz, R. Crawley, L. Scott, C. Hay, and L. Inglis for technical assistance. We also wish to thank W. Chen, M. Zhao, N. Hacohen, N. Chevrier, M. Fleming, A. Lew, D. Miller, K. Rodgers, D. Segal and all members of the Hunter, Masters and Wicks laboratories for helpful discussions and comments. K.C.P., T.A.N., B.R.S., and M.B. are supported by the Australian NHMRC (ID 520574 and 1064591), Royal Australasian College of Physicians, Menzies Foundation, CASS Foundation and Reid Family Trust. M.D.T was supported by the Australian NHMRC (Fellowship #1123319). M.P. was supported by Australian NHMRC (Grants 1006592, 1045549 and 1065626), The Sylvia & Charles Viertel Senior Medical Research Fellowship, the Victorian State Government Operational Infrastructure Support and the Independent Research Institutes Infrastructure Support Scheme (NHMRC). S.L.M receives funding from GlaxoSmithKline. A.S.W. and C.P.H. are supported by the US NIH (1R01GM089795 and U19CA179513).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
K.C.P., T.A.N., B.R.S., M.D.T., A.G., C.P.H., G.T.B., K.E., M.P., M.O., I.P.W., S.L.M., designed experiments and/or analysed the data. K.C.P., T.A.N., B.R.S., M.D.T., C.P.H., P.J.B, G.T.B, M.B., K.E., A.S.W., S.P., R.L. and S.L.M. performed experiments. K.C.P., C.P.H and T.A.N. wrote the manuscript. K.C.P., S.L.M. and C.P.H supervised the project.
The authors declare no competing financial interests.
References
- Aubrey BJ, Kelly GL, Kueh AJ, Brennan MS, O’Connor L, Milla L, Wilcox S, Tai L, Strasser A, Herold MJ. An inducible lentiviral guide RNA platform enables the identification of tumor-essential genes and tumor-promoting mutations in vivo. Cell Rep. 2015;10:1422–1432. doi: 10.1016/j.celrep.2015.02.002. [DOI] [PubMed] [Google Scholar]
- Barbosa-Morais NL, Irimia M, Pan Q, Xiong HY, Gueroussov S, Lee LJ, Slobodeniuc V, Kutter C, Watt S, Colak R, et al. The evolutionary landscape of alternative splicing in vertebrate species. Science. 2012;338:1587–1593. doi: 10.1126/science.1230612. [DOI] [PubMed] [Google Scholar]
- Barral PM, Sarkar D, Fisher PB, Racaniello VR. RIG-I is cleaved during picornavirus infection. Virology. 2009;391:171–176. doi: 10.1016/j.virol.2009.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buschow SI, Lasonder E, Szklarczyk R, Oud MM, de Vries IJ, Figdor CG. Unraveling the human dendritic cell phagosome proteome by organellar enrichment ranking. Journal of proteomics. 2012;75:1547–1562. doi: 10.1016/j.jprot.2011.11.024. [DOI] [PubMed] [Google Scholar]
- Chevrier N, Mertins P, Artyomov MN, Shalek AK, Iannacone M, Ciaccio MF, Gat-Viks I, Tonti E, DeGrace MM, Clauser KR, et al. Systematic discovery of TLR signaling components delineates viral-sensing circuits. Cell. 2011;147:853–867. doi: 10.1016/j.cell.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium, G. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dansako H, Yamane D, Welsch C, McGivern DR, Hu F, Kato N, Lemon SM. Class A scavenger receptor 1 (MSR1) restricts hepatitis C virus replication by mediating toll-like receptor 3 recognition of viral RNAs produced in neighboring cells. PLoS pathogens. 2013;9 doi: 10.1371/journal.ppat.1003345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson S, Maini MK, Wack A. Disease-promoting effects of type I interferons in viral, bacterial, and coinfections. Journal of Interferon & Cytokine research. 2015;35:252–264. doi: 10.1089/jir.2014.0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BK. Isolation, culture, and functional evaluation of bone marrow-derived macrophages. Methods Mol Biol. 2013;1031:27–35. doi: 10.1007/978-1-62703-481-4_3. [DOI] [PubMed] [Google Scholar]
- DeWitte-Orr S, Collins S, Bauer C, Bowdish D, Mossman K. An accessory to the ‘Trinity’: SR-As are essential pathogen sensors of extracellular dsRNA, mediating entry and leading to subsequent type I IFN responses. PLoS pathogens. 2010;6 doi: 10.1371/journal.ppat.1000829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickins RA, Hemann MT, Zilfou JT, Simpson DR, Ibarra I, Hannon GJ, Lowe SW. Probing tumor phenotypes using stable and regulated synthetic microRNA precursors. Nat Genet. 2005;37:1289–1295. doi: 10.1038/ng1651. [DOI] [PubMed] [Google Scholar]
- Dominska M, Dykxhoorn D. Breaking down the barriers: siRNA delivery and endosome escape. J Cell Sci. 2010;123:1183–1189. doi: 10.1242/jcs.066399. [DOI] [PubMed] [Google Scholar]
- Dreux M, Garaigorta U, Boyd B, Décembre E, Chung J, Whitten-Bauer C, Wieland S, Chisari FV. Short-range exosomal transfer of viral RNA from infected cells to plasmacytoid dendritic cells triggers innate immunity. Cell host & microbe. 2012;12:558–570. doi: 10.1016/j.chom.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duxbury MS, Ashley SW, Whang EE. RNA interference: a mammalian SID-1 homologue enhances siRNA uptake and gene silencing efficacy in human cells. Biochem Biophys Res Commun. 2005;331:459–463. doi: 10.1016/j.bbrc.2005.03.199. [DOI] [PubMed] [Google Scholar]
- Ebert G, Preston S, Allison C, Cooney J, Toe JG, Stutz MD, Ojaimi S, Scott HW, Baschuk N, Nachbur U, et al. Cellular inhibitor of apoptosis proteins prevent clearance of hepatitis B virus. Proc Natl Acad Sci U S A. 2015;112:5797–5802. doi: 10.1073/pnas.1502390112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg EH, Hunter CP. Transport of dsRNA into cells by the transmembrane protein SID-1. Science. 2003;301:1545–1547. doi: 10.1126/science.1087117. [DOI] [PubMed] [Google Scholar]
- Fredericksen BL, Gale M. West Nile virus evades activation of interferon regulatory factor 3 through RIG-I-dependent and-independent pathways without antagonizing host defense signaling. Journal of virology. 2006;80:2913–2923. doi: 10.1128/JVI.80.6.2913-2923.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freese NH, Norris DC, Loraine AE. Integrated genome browser: visual analytics platform for genomics. Bioinformatics. 2016;32:2089–2095. doi: 10.1093/bioinformatics/btw069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Gu X, Zhang H. SID1 transmembrane family, member 2 (Sidt2): A novel lysosomal membrane protein. Biochem Biophys Res Commun. 2010;402:588–594. doi: 10.1016/j.bbrc.2010.09.133. [DOI] [PubMed] [Google Scholar]
- Gilchrist MJ, Zorn AM, Voigt J, Smith JC, Papalopulu N, Amaya E. Defining a large set of full-length clones from a Xenopus tropicalis EST project. Dev Biol. 2004;271:498–516. doi: 10.1016/j.ydbio.2004.04.023. [DOI] [PubMed] [Google Scholar]
- Gitlin L, Barchet W, Gilfillan S, Cella M, Beutler B, Flavell RA, Diamond MS, Colonna M. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc Natl Acad Sci U S A. 2006;103:8459–8464. doi: 10.1073/pnas.0603082103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. 2011;146:448–461. doi: 10.1016/j.cell.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh K, Watanabe A, Funami K, Seya T, Matsumoto M. The clathrin-mediated endocytic pathway participates in dsRNA-induced IFN-beta production. Journal of immunology (Baltimore, Md: 1950) 2008;181:5522–5529. doi: 10.4049/jimmunol.181.8.5522. [DOI] [PubMed] [Google Scholar]
- Jose AM, Hunter CP. Transport of sequence-specific RNA interference information between cells. Annu Rev Genet. 2007;41:305–330. doi: 10.1146/annurev.genet.41.110306.130216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jose AM, Smith JJ, Hunter CP. Export of RNA silencing from C. elegans tissues does not require the RNA channel SID-1. Proc Natl Acad Sci U S A. 2009 doi: 10.1073/pnas.0809760106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, Tsujimura T, Takeda K, Fujita T, Takeuchi O, et al. Cell type-specific involvement of RIG-I in antiviral response. Immunity. 2005;23:19–28. doi: 10.1016/j.immuni.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, Matsushita K, Hiiragi A, Dermody TS, Fujita T, Akira S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. The Journal of experimental medicine. 2008;205:1601–1610. doi: 10.1084/jem.20080091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- Kelly GL, Grabow S, Glaser SP, Fitzsimmons L, Aubrey BJ, Okamoto T, Valente LJ, Robati M, Tai L, Fairlie WD, et al. Targeting of MCL-1 kills MYC-driven mouse and human lymphomas even when they bear mutations in p53. Genes Dev. 2014;28:58–70. doi: 10.1101/gad.232009.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoo D, Perez C, Mohr I. Characterization of RNA determinants recognized by the arginine-and proline-rich region of Us11, a herpes simplex virus type 1-encoded double-stranded RNA binding protein that prevents PKR activation. Journal of virology. 2002;76:11971–11981. doi: 10.1128/JVI.76.23.11971-11981.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauterbach H, Bathke B, Gilles S, Traidl-Hoffmann C, Luber CA, Fejer G, Freudenberg MA, Davey GM, Vremec D, Kallies A, et al. Mouse CD8alpha+ DCs and human BDCA3+ DCs are major producers of IFN-lambda in response to poly IC. Journal of Experimental Medicine. 2010;207:2703–2717. doi: 10.1084/jem.20092720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HKK, Dunzendorfer S, Soldau K, Tobias PS. Double-stranded RNA-mediated TLR3 activation is enhanced by CD14. Immunity. 2006;24:153–163. doi: 10.1016/j.immuni.2005.12.012. [DOI] [PubMed] [Google Scholar]
- Leonard JN, Ghirlando R, Askins J, Bell JK, Margulies DH, Davies DR, Segal DM. The TLR3 signaling complex forms by cooperative receptor dimerization. Proc Natl Acad Sci U S A. 2008;105:258–263. doi: 10.1073/pnas.0710779105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie M. Cell Biology. NIH effort gambles on mysterious extracellular RNAs. Science. 2013;341:947. doi: 10.1126/science.341.6149.947. [DOI] [PubMed] [Google Scholar]
- Leung DW, Basler CF, Amarasinghe GK. Molecular mechanisms of viral inhibitors of RIG-I-like receptors. Trends in microbiology. 2012;20:139–146. doi: 10.1016/j.tim.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Koutmou KS, Leahy DJ, Li M. Systemic RNA Interference Deficiency-1 (SID-1) extracellular domain selectively binds long double-stranded RNA and is required for RNA transport by SID-1. The Journal of biological chemistry. 2015a doi: 10.1074/jbc.M115.658864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Koutmou KS, Leahy DJ, Li M. Systemic RNA Interference Deficiency-1 (SID-1) extracellular domain selectively binds long double-stranded RNA and is required for RNA transport by SID-1. The Journal of biological chemistry. 2015b;290:18904–18913. doi: 10.1074/jbc.M115.658864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XDD, Wu J, Gao D, Wang H, Sun L, Chen ZJ. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science (New York, NY) 2013;341:1390–1394. doi: 10.1126/science.1244040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013;41:e108. doi: 10.1093/nar/gkt214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez J, Huang X, Yang Y. Direct TLR2 signaling is critical for NK cell activation and function in response to vaccinia viral infection. PLoS Pathog. 2010;6:e1000811. doi: 10.1371/journal.ppat.1000811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwan DL, Weisman AS, Hunter CP. Uptake of extracellular double-stranded RNA by SID-2. Mol Cell. 2012;47:746–754. doi: 10.1016/j.molcel.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchjorsen J, Matikainen S, Paludan SR. Activation and evasion of innate antiviral immunity by herpes simplex virus. Viruses. 2009;1:737–759. doi: 10.3390/v1030737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkin J, Russell C, Chen P, Burge CB. Evolutionary dynamics of gene and isoform regulation in Mammalian tissues. Science. 2012;338:1593–1599. doi: 10.1126/science.1228186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki D, Haruki T, Takeda S, Sasaki S, Yakura K, Terasaka Y, Komatsu N, Yamagami S, Touge H, Touge C, et al. Herpes simplex virus type 1-induced transcriptional networks of corneal endothelial cells indicate antigen presentation function. Invest Ophthalmol Vis Sci. 2011;52:4282–4293. doi: 10.1167/iovs.10-6911. [DOI] [PubMed] [Google Scholar]
- Narayanan A, Iordanskiy S, Das R, Van Duyne R, Santos S, Jaworski E, Guendel I, Sampey G, Dalby E, Iglesias-Ussel M, et al. Exosomes derived from HIV-1-infected cells contain trans-activation response element RNA. The Journal of biological chemistry. 2013;288:20014–20033. doi: 10.1074/jbc.M112.438895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nellimarla S, Mossman KL. Extracellular dsRNA: its function and mechanism of cellular uptake. Journal of interferon & cytokine research. 2014;34:419–426. doi: 10.1089/jir.2014.0002. [DOI] [PubMed] [Google Scholar]
- Ng CS, Jogi M, Yoo JS, Onomoto K, Koike S, Iwasaki T, Yoneyama M, Kato H, Fujita T. Encephalomyocarditis virus disrupts stress granules, the critical platform for triggering antiviral innate immune responses. J Virol. 2013;87:9511–9522. doi: 10.1128/JVI.03248-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly LA, Cullen L, Moriishi K, O’Connor L, Huang DC, Strasser A. Rapid hybridoma screening method for the identification of monoclonal antibodies to low-abundance cytoplasmic proteins. BioTechniques. 1998;25:824–830. doi: 10.2144/98255st03. [DOI] [PubMed] [Google Scholar]
- Pang KC, Dinger ME, Mercer TR, Malquori L, Grimmond SM, Chen W, Mattick JS. Genome-wide identification of long noncoding RNAs in CD8+ T cells. J Immunol. 2009;182:7738–7748. doi: 10.4049/jimmunol.0900603. [DOI] [PubMed] [Google Scholar]
- Pang KC, Sanders MT, Monaco JJ, Doherty PC, Turner SJ, Chen W. Immunoproteasome Subunit Deficiencies Impact Differentially on Two Immunodominant Influenza Virus-Specific CD8+ T Cell Responses. J Immunol. 2006;177:7680–7688. doi: 10.4049/jimmunol.177.11.7680. [DOI] [PubMed] [Google Scholar]
- Papadopoulos JS, Agarwala R. COBALT: constraint-based alignment tool for multiple protein sequences. Bioinformatics. 2007;23:1073–1079. doi: 10.1093/bioinformatics/btm076. [DOI] [PubMed] [Google Scholar]
- Pegtel DM, Cosmopoulos K, Thorley-Lawson DA, van Eijndhoven MA, Hopmans ES, Lindenberg JL, de Gruijl TD, Wurdinger T, Middeldorp JM. Functional delivery of viral miRNAs via exosomes. Proc Natl Acad Sci U S A. 2010;107:6328–6333. doi: 10.1073/pnas.0914843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrot I, Deauvieau F, Massacrier C, Hughes N, Garrone P, Durand I, Demaria O, Viaud N, Gauthier L, Blery M, et al. TLR3 and Rig-like receptor on myeloid dendritic cells and Rig-like receptor on human NK cells are both mandatory for production of IFN-gamma in response to double-stranded RNA. Journal of Immunology. 2010;185:2080–2088. doi: 10.4049/jimmunol.1000532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petryszak R, Burdett T, Fiorelli B, Fonseca NA, Gonzalez-Porta M, Hastings E, Huber W, Jupp S, Keays M, Kryvych N, et al. Expression Atlas update–a database of gene and transcript expression from microarray- and sequencing-based functional genomics experiments. Nucleic Acids Res. 2014;42:D926–932. doi: 10.1093/nar/gkt1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SB, Sørensen LN, Malmgaard L, Ank N, Baines JD, Chen ZJ, Paludan SR. Type I interferon production during herpes simplex virus infection is controlled by cell-type-specific viral recognition through Toll-like receptor 9, the mitochondrial antiviral signaling protein pathway, and novel recognition systems. Journal of virology. 2007;81:13315–13324. doi: 10.1128/JVI.01167-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren R, Xu X, Lin T, Weng S, Liang H, Huang M, Dong C, Luo Y, He J. Cloning, characterization, and biological function analysis of the SidT2 gene from Siniperca chuatsi. Developmental and comparative immunology. 2011;35:692–701. doi: 10.1016/j.dci.2011.02.003. [DOI] [PubMed] [Google Scholar]
- Rusinova I, Forster S, Yu S, Kannan A, Masse M, Cumming H, Chapman R, Hertzog PJ. Interferome v2.0: an updated database of annotated interferon-regulated genes. Nucleic acids research. 2013;41:6. doi: 10.1093/nar/gks1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell TA, Stefanovic T, Tscharke DC. Engineering herpes simplex viruses by infection-transfection methods including recombination site targeting by CRISPR/Cas9 nucleases. J Virol Methods. 2015;213:18–25. doi: 10.1016/j.jviromet.2014.11.009. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz O, Diebold SS, Chen M, Näslund TI, Nolte MA, Alexopoulou L, Azuma YTT, Flavell RA, Liljeström P, Reise Sousa C. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature. 2005;433:887–892. doi: 10.1038/nature03326. [DOI] [PubMed] [Google Scholar]
- Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Shen Z, Reznikoff G, Dranoff G, Rock KL. Cloned dendritic cells can present exogenous antigens on both MHC class I and class II molecules. J Immunol. 1997;158:2723–2730. [PubMed] [Google Scholar]
- Shih J, Hunter C. SID-1 is a dsRNA-selective dsRNA-gated channel. RNA. 2011;17:1057–1065. doi: 10.1261/rna.2596511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjolinder M, Altenbacher G, Wang X, Gao Y, Hansson C, Sjolinder H. The meningococcal adhesin NhhA provokes proinflammatory responses in macrophages via toll-like receptor 4-dependent and -independent pathways. Infection and immunity. 2012;80:4027–4033. doi: 10.1128/IAI.00456-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CM, Wilson NS, Waithman J, Villadangos JA, Carbone FR, Heath WR, Belz GT. Cognate CD4(+) T cell licensing of dendritic cells in CD8(+) T cell immunity. Nat Immunol. 2004;5:1143–1148. doi: 10.1038/ni1129. [DOI] [PubMed] [Google Scholar]
- Sugiyama T, Hoshino K, Saito M, Yano T, Sasaki I, Yamazaki C, Akira S, Kaisho T. Immunoadjuvant effects of polyadenylic:polyuridylic acids through TLR3 and TLR7. International immunology. 2008;20:1–9. doi: 10.1093/intimm/dxm112. [DOI] [PubMed] [Google Scholar]
- Sumpter R, Loo YM, Foy E, Li K, Yoneyama M. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J Virol. 2005;79:2689–2699. doi: 10.1128/JVI.79.5.2689-2699.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Gulbagci NT, Taneja R. Analysis of growth properties and cell cycle regulation using mouse embryonic fibroblast cells. Methods in molecular biology. 2007;383:311–319. doi: 10.1007/978-1-59745-335-6_20. [DOI] [PubMed] [Google Scholar]
- Sun Q, Sun L, Liu HHH, Chen X, Seth RB, Forman J, Chen ZJ. The specific and essential role of MAVS in antiviral innate immune responses. Immunity. 2006;24:633–642. doi: 10.1016/j.immuni.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Tang T, Li L, Tang J, Li Y, Lin WY, Martin F, Grant D, Solloway M, Parker L, Ye W, et al. A mouse knockout library for secreted and transmembrane proteins. Nature biotechnology. 2010;28:749–755. doi: 10.1038/nbt.1644. [DOI] [PubMed] [Google Scholar]
- Watanabe A, Tatematsu M, Saeki K, Shibata S, Shime H, Yoshimura A, Obuse C, Seya T, Matsumoto M. Raftlin is involved in the nucleocapture complex to induce poly(I:C)-mediated TLR3 activation. The Journal of biological chemistry. 2011;286:10702–10711. doi: 10.1074/jbc.M110.185793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber F, Wagner V, Rasmussen SB, Hartmann R, Paludan SR. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. Journal of virology. 2006;80:5059–5064. doi: 10.1128/JVI.80.10.5059-5064.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winston WM, Molodowitch C, Hunter CP. Systemic RNAi in C. elegans requires the putative transmembrane protein SID-1. Science. 2002;295:2456–2459. doi: 10.1126/science.1068836. [DOI] [PubMed] [Google Scholar]
- Wolfrum C, Shi S, Jayaprakash KN, Jayaraman M, Wang G, Pandey RK, Rajeev KG, Nakayama T, Charrise K, Ndungo EM, et al. Mechanisms and optimization of in vivo delivery of lipophilic siRNAs. Nat Biotechnol. 2007;25:1149–1157. doi: 10.1038/nbt1339. [DOI] [PubMed] [Google Scholar]
- Wu C, Orozco C, Boyer J, Leglise M, Goodale J, Batalov S, Hodge CL, Haase J, Janes J, Huss JW, 3rd, et al. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009;10:R130. doi: 10.1186/gb-2009-10-11-r130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing J, Wang S, Lin R, Mossman KL, Zheng C. Herpes simplex virus 1 tegument protein US11 downmodulates the RLR signaling pathway via direct interaction with RIG-I and MDA-5. Journal of virology. 2012;86:3528–3540. doi: 10.1128/JVI.06713-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- Yen JH, Ganea D. Interferon beta induces mature dendritic cell apoptosis through caspase-11/caspase-3 activation. Blood. 2009;114:1344–1354. doi: 10.1182/blood-2008-12-196592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneyama M, Fujita T, Lemon SM. Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc Natl Acad Sci U S A. 2005;102:2986–2991. doi: 10.1073/pnas.0408707102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Accession number and description of proteins used in Figure S1.
MEFs stably expressing SIDT2-mCherry were transfected with a late endosomal marker (RAB7-eGFP) and imaged live every 30 sec for 20 min on a Leica SP8 laser scanning confocal microscope.
MEFs stably expressing SIDT2-mCherry were transfected with a lysosomal marker (LAMP-1-YFP) and imaged live every 30 sec for 20 min on a Leica SP8 laser scanning confocal microscope.
MEFs stably expressing SIDT2-mCherry were transfected with a lysosomal marker (EEA-1-GFP) and imaged live every 30 sec for 20 min on a Leica SP8 laser scanning confocal microscope
MEFs stably expressing SIDT2-mCherry were transfected with a recycling endosome marker (RAB11-eGFP) and imaged live every 30 sec for 20 min on a Leica SP8 laser scanning confocal microscope.