

Summary
We assembled genome-wide data from 16 prehistoric Africans. We show that the anciently divergent lineage that comprises the primary ancestry of the southern African San had a wider distribution in the past, contributing ~2/3 of the ancestry of Malawi hunter-gatherers ~8100–2500 years ago, and ~1/3 of Tanzanian hunter-gatherers ~1400 years ago. We document how the spread of farmers from western Africa involved complete replacement of local hunter-gatherers in some regions, and we track the spread of herders by showing that the population of a ~3100 year-old pastoralist from Tanzania contributed ancestry to people from northeast to southern Africa, including a ~1200-year-old southern African pastoralist. The deepest diversifications of African lineages were complex, involving long-distance gene flow, or a lineage more deeply diverging than that of the San contributing more to some western Africans than others. We finally leverage ancient genomes to document episodes of natural selection in southern African populations.
ETOC
The prehistory of African populations is explored by genomewide analysis of 16 human remains providing insight into lineages, admixture, and genomic adaptions.
Introduction
Africa harbors more genetic diversity than any other part of the world (Cann et al., 1987; Tishkoff et al., 2009). This is reflected both in a higher average number of differences between sub-Saharan African genomes than between non-African genomes (Cann et al., 1987; Ramachandran et al., 2005), and in the fact that the ancestry found outside of Africa is largely a subset of that within it (Tishkoff et al., 2009). Today, some of the earliest branching African lineages are present only in populations with relatively small census sizes, including the southern African Khoe-San (see STAR Methods for terminology), central African rainforest hunter-gatherers, and Hadza of Tanzania (Gronau et al., 2011; Schlebusch et al., 2012; Veeramah et al., 2012). However, the population structure of Africa prior to the expansion of food producers (pastoralists and agriculturalists) remains unknown (Busby et al., 2016; Gurdasani et al., 2015; Patin et al., 2017). Bantu-speaking agriculturalists originating in western Africa are thought to have brought farming to eastern Africa by ~2000 years calBP (calibrated radiocarbon years Before Present, defined by convention as years before 1950 CE) and to southern Africa by ~1500 BP, thereby spreading the largest single ancestry component to African genomes today (Russell et al., 2014; Tishkoff et al., 2009). Earlier migration(s), which brought ancestry related to the ancient Near East (Lazaridis et al., 2016; Pagani et al., 2012; Pickrell et al., 2014), brought herding to eastern Africa by ~4000 BP (Marshall et al., 1984), and to southern Africa by ~2000 BP (Sadr, 2015).
Results
To reconstruct African population structure prior to the spread of food production, we generated genome-wide data from 15 ancient sub-Saharan Africans (Table 1; Table S1; Table S2; STAR Methods). For three individuals from the western Cape of South Africa (~2300–1300 BP) we carried out direct shotgun sequencing to 0.7–2.0-fold coverage. For 12 individuals from eastern and south-central Africa, we used in-solution enrichment of ~1.2 million single nucleotide polymorphisms (SNPs). These included 4 individuals from the coastal region of Kenya and Tanzania (~1400–400 BP), one from interior Tanzania (~3100 BP), and 7 from Malawi (ranging over ~8100–2500 BP) (Fig. S1). All individuals had post-mortem degradation characteristic of ancient DNA (Table 1), and we confirmed that key results are unlikely to be artifacts of contamination by restricting to sequences with post-mortem degradation (Skoglund et al., 2012; Skoglund et al., 2014a) (Fig. S2). We merged the new ancient DNA data with previously reported shotgun sequence data from a ~4500 BP Ethiopian highland individual (Llorente et al., 2015), and with SNP genotypes from 584 present-day African individuals from 59 diverse populations (including new data from 34 Malawi individuals; STAR Methods) (Lazaridis et al., 2014; Patterson et al., 2012), as well as 300 high-coverage genomes from 142 worldwide populations (Mallick et al., 2016).
Table 1.
Summary of ancient DNA from 15 prehistoric individuals newly reported in this study.
| ID | Population label | Date* In calibrated years before present defined as 1950 (calBP) |
Location | Lat. | Long. | Y chromosome haplogroup | mtDNA hg | Damage rate at 5′ CpG dinucleotides | SNPs hit on 1.2 M autosomal targets |
|---|---|---|---|---|---|---|---|---|---|
| I9028 | South_Africa_2000BP | 2241–1965 calBP (2330 ± 25 BP, UGAMS-7255) | St. Helena, South Africa | −32.8 | 18.0 | A1b1b2a | L0d2c1$ | 28% | 731,098 (1.1X shotgun) |
| I9133 | South_Africa_2000BP | 2017–1748 calBP (2000 ± 50 BP, Pta-5283) | Faraoskop Rock Shelter, South Africa | −32.0 | 18.5 | A1b1b2a | L0d1b2b1b | 33% | 1,028,904 (2.3X shotgun) |
| I9134 | South_Africa_1200BP | 1282–1069 calBP (1310 ± 50 BP, Pta-4373) | Kasteelberg, South Africa | −32.8 | 17.9 | Female | L0d1a1a | 21% | 641,971 (0.8X shotgun) |
| I4427 | Malawi_Fingira_6100BP | 6175–5913 calBP (5270 ± 25 BP, UCIAMS-186346) | Fingira, Malawi | −10.8 | 33.8 | BT | L0d1b2b | 37% | 99,341 |
| I4468 | Malawi_Fingira_6100BP | 6177–5923 calBP (5290 ± 25 BP, UCIAMS-186347) | Fingira, Malawi | −10.8 | 33.8 | BT | L0d1c | 49% | 30,257 |
| I4421 | Malawi_Chencherere_5200BP | 5400–4800 calBP (radiocarbon dating was unsuccessful; the date is based on context of other materials from the same site) | Chencherere, Malawi | −14.4 | 33.8 | Female | L0k2 | 28% | 59,470 |
| I4422 | Malawi_Chencherere_5200BP | 5293–4979 calBP (4525 ± 25 BP, UCIAMS-186348) | Chencherere, Malawi | −14.4 | 33.8 | Female | L0k1 | 42% | 9,355 |
| I2966 | Malawi_Hora_8100BP | 10000–5000 calBP (radiocarbon dating was unsuccessful; the date is based on context of other materials from the same site) | Hora, Malawi | −11.7 | 33.6 | BT | L0k2 (PMDS> 3) | 54% | 610,605 |
| I2967 | Malawi_Hora_8100BP | 8173–7957 calBP (7230 ± 60 BP, PSUAMS-2535) | Hora, Malawi | −11.7 | 33.6 | Female | L0a2 | 48% | 65,686 |
| I4426 | Malawi_Fingira_2500BP | 2676–2330 calBP [2676–2343 calBP (2425 ± 20 BP, PSUAMS-1734), 2483–2330 calBP (2400 ± 20 BP, PSUAMS-1881)] | Fingira, Malawi | −10.8 | 33.8 | Female | L0f | 39% | 635,427 |
| I3726 | Tanzania_Luxmanda_3100BP | 3141–2890 calBP (2925 ± 20 BP, ISGS-A3806) | Luxmanda, Tanzania | −4.3 | 35.3 | Female | L2a1 | 65% | 845,016 |
| I0589 | Tanzania_Zanzibar_1400BP | 1370–1303 calBP (1479 ± 23 BP, OxA-31427) | Kuumbi Cave, Zanzibar Island, Tanzania | −6.4 | 39.5 | Female | L4b2a2c | 22% | 752,917 |
| I1048 | Tanzania_Pemba_1400BP | 1421–1307 calBP (1520 ± 30 BP, Beta-434912) | Makangale Cave, Pemba Island, Tanzania | −4.9 | 39.6 | Female | L0a | 42% | 168,117 |
| I2298 | Tanzania_Pemba_600BP | 639–544 calBP (623 ± 20 BP, Wk-43308) | Makangale Cave, Pemba Island, Tanzania | −4.9 | 39.6 | Female | L2a1a2 | 29% | 695,242 |
| I0595 | Kenya_400BP | 496–322 calBP (388 ± 27 BP, OxA-30803) | Panga ya Saidi, Kenya | −3.7 | 39.7 | E1b1b1b2 | L4b2a2 | 30% | 150,383 |
Table S2 provides detailed information on the direct radiocarbon dating measurements.
Consistent with previously published mtDNA sequence by (Morris et al., 2014).
An ancient cline of southern and eastern African hunter-gatherers
We used principal component analysis (PCA) (Patterson et al., 2006), and automated clustering (Alexander et al., 2009) to relate the 16 ancient individuals to present-day sub-Saharan Africans (Fig. 1). Whereas the two individuals buried in ~2000 BP hunter-gatherer contexts in South Africa share ancestry with southern African Khoe-San populations in the PCA, 11 of the 12 ancient individuals who lived in eastern and south-central Africa between ~8100–400 BP form a gradient of relatedness to the eastern African Hadza on one hand, and southern African Khoe-San on the other (Fig. 1A). The genetic cline correlates to geography, running along a north-south axis with ancient individuals from Ethiopia (~4500 BP), Kenya (~400 BP), Tanzania (both ~1400 BP) and Malawi showing increasing affinity to southern Africans (both ancient individuals and present-day Khoe-San). The seven individuals from Malawi show no significant heterogeneity, indicating a longstanding and distinctive population in ancient Malawi that persisted for at least ~5,000 years (the minimum span of our radiocarbon dates), but no longer exists today.
Figure 1. Overview of ancient genomes and African population structure.
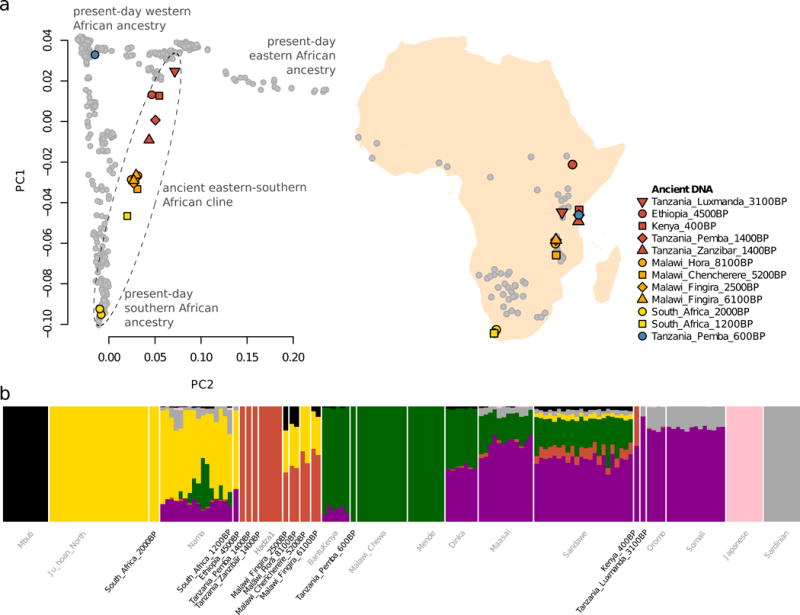
a) Map of sampling locations in Africa and principal component analysis of all individuals. Present-day individuals are indicated with gray circles. b) Automated clustering of key ancient- and present-day populations (for K = 7 cluster components). Present-day populations are labeled in gray.
We constructed a model where ancient and present-day African populations trace their ancestry to a putative set of nine ancestral populations. As proxies for these populations we used three different ancient Near Eastern populations and six African populations that, according to our analyses, harbor substantial ancestry related to major lineages present in Africa today. The Mende from Sierra Leone are used in this model to represent a component of ancestry that exists in high proportions in western African populations, the ancient southern African genomes (South_Africa_2000BP) are used to represent the ancestry of southern Africa before agriculture, the Ethiopian individual (Ethiopia_4500BP) is used to represent northeastern African ancestry before agriculture, the Mbuti are used to represent central African rainforest hunter-gatherer ancestry, the individual from an eastern African pastoralist context (Tanzania_Luxmanda_3100BP) is used to represent an early pastoralist lineage from eastern Africa (see below), and the Dinka (from Sudan) are used to represent distinctive ancestry found in Nilotic speakers today. The ancient Near Eastern populations were representative of Anatolia, the Levant, and Iran, respectively (Lazaridis et al., 2016; Mathieson et al., 2015). We used qpAdm (Haak et al., 2015), a generalization of f4 symmetry statistics, to successively test 1-source, 2-source, or 3-source models and admixture proportions for all other ancient and present-day African populations, with a set of 10 non-African populations as outgroups (STAR Methods).
We find that ancestry closely related to the ancient southern Africans was present much farther north and east in the past than is apparent today. This ancient southern African ancestry comprises up to 91% of the ancestry of Khoe-San groups today (Table S3), and also 31% ± 3% of the ancestry of Tanzania_Zanzibar_1400BP, 60% ± 6% of the ancestry of Malawi_Fingira_6100BP, and 65% ± 3% of the ancestry of Malawi_Fingira_2500BP (Fig. 2A). Notably, the Khoe-San-related ancestry in ancient individuals from Malawi and Tanzania is symmetrically related to the two previously identified lineages present in the San (Z < 2; Fig. S2), estimated to have diverged at least 20,000 years ago (Mallick et al., 2016; Pickrell et al., 2012; Schlebusch et al., 2012), implying that this was an ancient divergent branch of this group that lived in eastern Africa at least until 1400 BP. However, it was not present in all eastern Africans, as we do not detect it in the ~400-year old individual from coastal Kenya, nor in the present-day Hadza.
Figure 2. Ancestral components in eastern and southern Africa.
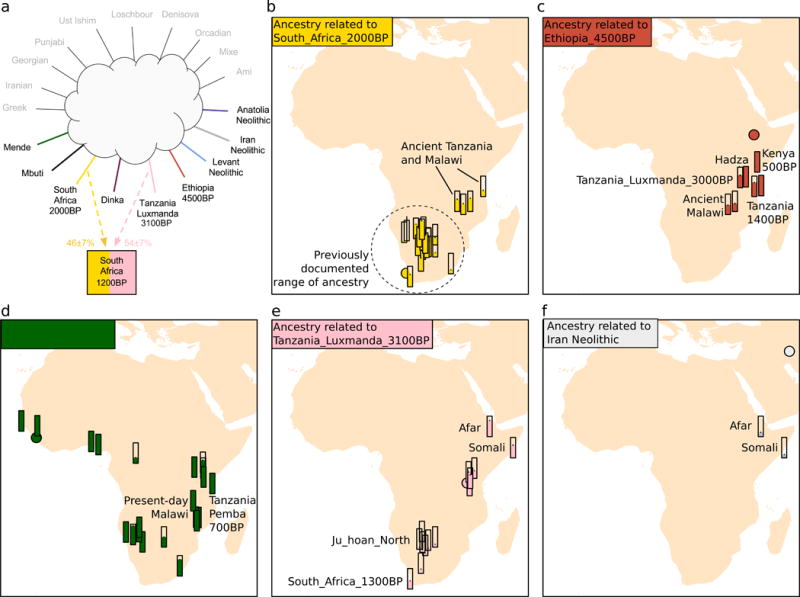
We show bar plots with the proportions inferred for the best model for each target population. We used a model that inferred the ancestry of each target population as 1-source, 2-source, or 3-source mixture of a set of potential source populations. In (a) we show an example of the inferred model for South_Africa_1200BP, an early pastoralist. A filled circle symbol in each panel indicates the geographic location of the sample that we use as a representative of the source population. We show five sources: b) South_Africa_2000BP representing forager populations in southern Africa and a component of prehistoric Malawi and Tanzania that is no longer extant; c) Ethiopia_4500BP which is today found in the Hadza but in the past was characteristic of eastern African hunter-gatherers; d) the Mende from Sierra Leone which is related deeply to the western African ancestry that was spread with the Bantu expansion of agriculturalists; e) the Savanna Pastoral Neolithic sample Tanzania_Luxmanda_3100BP which provides a missing link of the pastoralist population that brought ancestry most closely related to the ancient Levant to southern Africa, and which is also closely but not exclusively related to present-day Cushitic speakers; and f) ancestry more closely related to the Iran Neolithic than what is found in Tanzania_Luxmanda_3100BP, and which may have entered the Horn of Africa in later migrations.
Displacement of forager populations in eastern Africa
Both unsupervised clustering (Fig. 1B) and formal ancestry estimation (Fig. 2B) suggests that individuals from the Hadza group in Tanzania can be modeled as deriving all their ancestry from a lineage related deeply to ancient eastern Africans such as the Ethiopia_4500BP individual (Fig. 3A; Table S3). However, this lineage appears to have contributed little ancestry to present-day Bantu-speakers in eastern Africa, who instead trace their ancestry to a lineage related to present-day western Africans, with additional components related to the Nilotic-speaking Dinka and to the Tanzania_Luxmanda_3100BP pastoralist (see below and Fig. 2). The Sandawe, another population that like the Hadza uses click consonants in their spoken language, are modeled as having ancestry similar to the Hadza but also admixture related to that of neighboring populations (Fig. 3A; Table S3) consistent with previous findings (Henn et al., 2011; Tishkoff et al., 2009). Population replacement by incoming food-producers appears to have been nearly complete in Malawi, where we detect little if any ancestry from the ancient individuals who lived ~8100–2500 BP. Instead, present-day Malawian individuals are consistent with deriving all their ancestry from the Bantu expansion of ultimate western African origin (Fig. 3).
Figure 3. Mixture events in the deeper population history of continental African lineages.
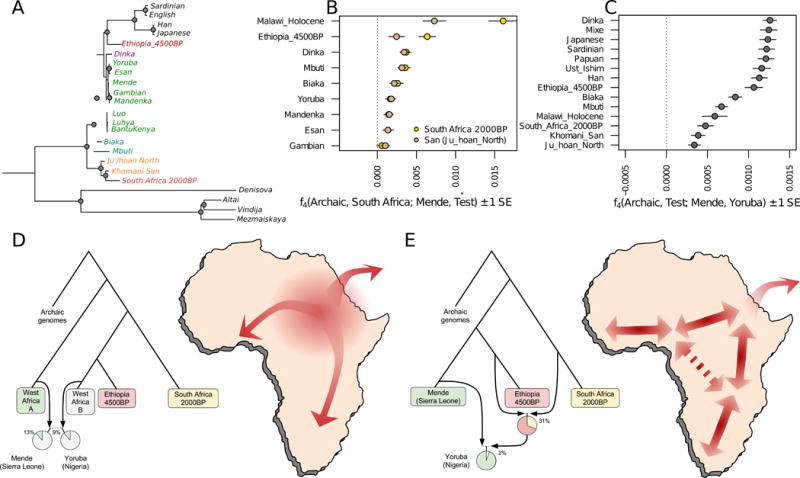
A) Maximum likelihood tree of genome sequences from present-day and ancient populations, excluding populations with evidence of asymmetrical allele sharing with non-Africans indicative of recent gene flow (Table S5). Nodes with bootstrap support > 95% are indicated with a circle. B) A symmetry test of the hypothesis that ancient southern Africans are an outgroup lineage to other African populations, which can be rejected for most pairs. C) Asymmetry between western African Mende and Yoruba in the 1000 Genomes Project data is maximized in the Yoruba’s excess affinity to eastern Africans and non-Africans, but highly significant also for groups as distant as southern Africans. D) Admixture Graph solution where Mende from Sierra Leone and Yoruba from Nigeria have ancestry from a basal western African lineage. The other source of western African ancestry is most closely related to eastern Africans and non-Africans (Fig. S5D), which could be consistent with an expansion from eastern Africa. Note that the exact proportion ‘West Africa A’ ancestry is not well constrained by the model, but the difference between Yoruba and Mende is highly significant (panel C). E) Admixture graph solution where the Yoruba have gene flow from a population related to both southern and eastern Africa, which could be consistent with a more complex pattern of isolation-by-distance in the continent.
Among the ancient individuals analyzed here, only a ~600 BP individual from the Zanzibar archipelago has a genetic profile similar to present-day Bantu-speakers (Fig. 1). Notably, this individual is consistent with having even more western African-related ancestry than the present-day Bantu-speakers we analyzed from Kenya, who also derive some of their ancestry from lineages related to Dinka and Tanzania_Luxmanda_3100BP (Fig. 1B). Using linkage disequilibrium, we estimate that this admixture between western and eastern African related lineages occurred an average of 800–400 years ago (STAR Methods). This suggests a scenario of genetic isolation between early farmers and previously established foragers during the initial phase of the Bantu expansion into eastern Africa (Crowther et al., 2017; Ribot et al., 2010), a barrier that broke down over time as mixture occurred. This parallels the patterns previously observed in genomic analyses of the Neolithic expansion into Europe (Haak et al., 2015; Skoglund et al., 2012), and the East Asian farming expansion into Remote Oceania (Skoglund et al., 2016). However, this process of delayed admixture did not always apply in Africa, as is evident in the absence of admixture from previously established hunter-gatherers in present-day Malawians.
Early Levantine farmer-related admixture in a ~3100-year-old pastoralist from Tanzania
Western Eurasian-related ancestry is pervasive in eastern Africa today (Pagani et al., 2012; Tishkoff et al., 2009), and the timing of this admixture has been estimated to be ~3000 BP on average (Pickrell et al., 2014). We found that the ~3100 BP individual (Tanzania_Luxmanda_3100BP), associated with a Savanna Pastoral Neolithic archeological tradition, could be modeled as having 38 ± 1% of her ancestry related to the nearly 10,000 year old pre-pottery farmers of the Levant (Lazaridis et al., 2016), and we can exclude source populations related to early farmer populations in Iran and Anatolia. These results could be explained by migration into Africa from descendants of pre-pottery Levantine farmers, or alternatively by a scenario in which both pre-pottery Levantine farmers and Tanzania_Luxmanda_3100BP descend from a common ancestral population that lived thousands of years earlier in Africa or the Near East. We fit the remaining approximately 2/3 of Tanzania_Luxmanda_3100BP as most closely related to the Ethiopia_4500BP (P = 0.029) or, allowing for 3-way mixture also from a source closely related to the Dinka (P = 0.18; the Levantine-related ancestry in this case was 39 ± 1%) (Table S3).
While these findings show that a Levant Neolithic-related population made a critical contribution to the ancestry of present-day eastern Africans (Lazaridis et al., 2016), present-day Cushitic-speakers such as the Somali cannot be fit simply as being of Tanzania_Luxmanda_3100BP ancestry. The best fitting model for the Somali includes Tanzania_Luxmanda_3100BP ancestry, Dinka-related ancestry, and 16% ± 3% Iranian Neolithic-related ancestry (P = 0.015). This suggests that ancestry related to the Iranian Neolithic appeared in eastern Africa after earlier gene flow related to Levant Neolithic populations, a scenario that is made more plausible by the genetic evidence of admixture of Iranian Neolithic-related ancestry throughout the Levant by the time of the Bronze Age (Lazaridis et al., 2016) and in ancient Egypt by the Iron Age (Schuenemann et al., 2017).
Direct evidence of migration bringing pastoralism to eastern and southern Africa
In contrast to the Malawi and Zanzibar individuals, all three ancient southern Africans show affinities to the ancestry predominant in present-day Tuu speakers in the southern Kalahari more than to present-day Ju’|hoan speakers in the northern Kalahari (Fig. S2B; Fig. S2C). However, the ~1200 BP sample from the western Cape that is found in a pastoralist context has a specific similarity in clustering analyses to present-day Khoe-Khoe-speaking pastoralist populations such as the Nama (Fig. 1B), and like them has affinity to three groups: Khoe-San, western Eurasians and eastern Africans. This supports the hypothesis that a non-Bantu-related population carried eastern African and Levantine ancestry to southern Africa by at least around 1200 BP, providing direct evidence for claims previously made based on analysis of present-day populations (Pickrell et al., 2014).
We used our modeling framework to show that the South_Africa_1200BP pastoralist individual from the Western Cape is consistent with being a mixture of just two streams of ancestry relative to non-southern African populations, with 40.3% ± 2.3%, ancestry related to the Tanzania_Luxmanda_3100BP individual (54% ± 7% when restricting to sequences with postmortem damage), and the remainder being related to the South_Africa_2000BP hunter-gatherers (Table S3). This supports the hypothesis that the Savanna Pastoral Neolithic archaeological tradition in eastern Africa is a plausible source for the spread of herding to southern Africa. Even the Ju_hoan_North, the San individuals with the least genetic affinity to eastern Africans, have 9% ± 1% of their ancestry most closely related to Tanzania_Luxmanda_3100BP, consistent with previous findings that the ancestries of all present-day San and Khoe were affected by agro-pastoralist migrations in the last two millennia (Pickrell et al., 2014).
The earliest divergences among modern human populations
Previous studies have suggested that the primary ancestry in the San is from a lineage that separated from all other lineages represented in modern humans today, before the latter separated from each other (Gronau et al., 2011; Veeramah et al., 2012). Such a model emerges when we automatically fit a tree without admixture to the data (Fig. 3A), but we also find that a tree-like representation is a poor fit (Fig. S4A), in the sense that ancient southern Africans who lived ~2000 BP were not strictly an outgroup to extant lineages in other parts of sub-Saharan Africa. In particular, we find that ancient southern Africans, who have none of the eastern African admixture that is ubiquitous today, share significantly more alleles with present-day and ancient eastern Africans (including Dinka, Hadza and Ethiopia_4500BP), than they do with present-day western Africans (Fig. 3B). Even within present-day western Africans, the genetic differences between Yoruba from Nigeria and the Mende from Sierra Leone are inconsistent with descent from a homogeneous ancestral population isolated from ancient southern Africans. The asymmetry between Yoruba and Mende is also observed with non-Africans, but no stronger than in eastern Africans (the most closely related Africans to the ancestral out-of-Africa population), and thus these signals are not driven by admixture from outside Africa, and instead likely reflect demographic events entirely within Africa (Fig. 3C).
We carried out admixture graph modeling of the allele frequency correlations and found two parsimonious models that fit the data. The first posited that present-day western Africans harbor ancestry from a basal African lineage that contributed more to the Mende than it did to the Yoruba, with the other source of western African ancestry being related to eastern Africans and non-Africans (Fig. 3D; Fig. S4; Fig. S5; Table S6). The second model posited that long-range and long-standing gene-flow has connected southern and eastern Africa to some groups in western Africa (e.g. the ancestors of the Yoruba) to a greater extent than to other groups in western Africa (e.g. the ancestors of the Mende) (Fig. 3E) (Pleurdeau et al., 2012). The possible basal western African population lineage would represent the earliest known divergence of a modern human lineage that contributed a major proportion of ancestry to present-day humans. Such a lineage must have separated before the divergence of San ancestors, which is estimated to have begun on the order of 200 to 300 thousand years ago (Scally and Durbin, 2012). Such a model of basal western African ancestry might support the hypothesis that there has been ancient structure in the ancestry of present-day Africans, using a line of evidence independent from previous findings based on long haplotypes with deep divergences from other human haplotypes (Hammer et al., 2011; Lachance et al., 2012; Plagnol and Wall, 2006). One scenario consistent with this result could involve ancestry related to eastern Africans (and the out-of-Africa population) expanding into western Africa and mixing there with more basal lineages. Our genetic data do not support the theory that this putative basal lineage diverged prior to the ancestors of Neandertals, since the African populations we analyze here are approximately symmetrically related to Neandertals (Mallick et al., 2016; Prufer et al., 2014).
A selective sweep targeting a taste receptor locus in southern Africa
The availability of ancient African genomes provides an opportunity to search for genomic footprints of natural selection manifested as regions of greater allele frequency differentiation between ancient and present-day populations than predicted by the genome-wide background. We compared to the two ancient southern African ~2000 BP shotgun sequence genomes to six present-day high-coverage San genomes with minimal recent mixture. The small number of ancient individuals does not permit inference of changing allele frequencies at single loci, so we performed a scan for high allele frequency differentiation in 500 kb windows with a step size of 10 kb. Using ~500 windows spaced at least 5 million base pairs apart as a null distribution, we found that the most differentiated locus was 15 standard deviations from the observed genome-wide mean and overlapped a cluster of eight taste-receptor genes on chromosome 12 (Fig. 4A; Table 2). Taste receptor genes have previously been identified as targets of natural selection in humans, as they modulate the ability to detect poisonous compounds in plants (Campbell et al., 2011).
Figure 4. Ancient genomes provide evidence of natural selection in present-day southern African San populations.
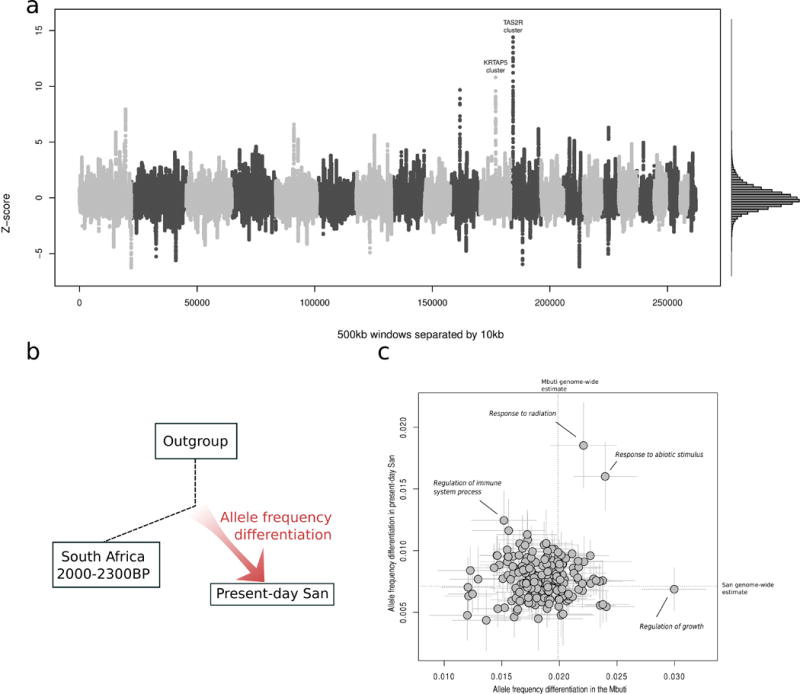
We computed branch-specific allele frequency differentiation in 6 present-day high-coverage San genomes compared to a pool of two ~2000 BP South African genomes as an outgroup, using two approaches. A) We computed the statistic in windows of 500 kb separated by 10 kb. We also estimated genome-wide average and standard deviation of the statistic using windows separated by at least 5 Mb, and transformed the genome wide distribution of the sliding windows to be approximately normal (right panel). We observe outliers 15 standard deviations from the mean in a taste receptor gene cluster on chromosome 12, and a secondary peak in the Keratin Associated Protein 4 gene cluster. The outgroup used was 4 Central African Mbuti genomes. See Table 2 for details on all major outlying regions. B) Illustration of the branch-specific allele frequency differentiation approach. C) We computed the statistic and block jackknife standard errors for 208 gene ontology categories with at least 50 genes each (y-axis). The outgroup used was western Africans. As a control to confirm that outlier categories do not show larger magnitudes of allele frequency differentiation across populations, we replaced the present-day San with the central African Mbuti (x-axis).
Table 2.
Top five candidate regions identified in genome-wide scan for selective sweeps in present-day San populations in southern Africa compared to ancient genomes.
| Rank | Chrom. | start-end | f3-statistic | Z-score | Genes in top 500 kb window in peak region |
|---|---|---|---|---|---|
| 1 | 12 | 11,123,548–11,623,548 | 0.163 | 14.3 | TAS2R43, PRH1-PRR4, TAS2R20, TAS2R50, TAS2R42, TAS2R46, TAS2R30, TAS2R31, PRB1, PRB2, PRB3, PRB4, LOC100129361, TAS2R19 |
| 2 | 11 | 71,208,258–71,708,258 | 0.125 | 10.8 | LOC100129216, KRTAP5-7, DEFB108B, KRTAP5-8, NADSYN1, KRTAP5-9, FAM86C1, RNF121, ALG1L9P, LOC100133315, KRTAP5-10, KRTAP5-11 |
| 3 | 10 | 46,069,893–46,569,893 | 0.113 | 9.7 | DQ577099, PTPN20B, PTPN20A, ZFAND4, AGAP4, DQ588224, FAM21C |
| 4 | 1 | 224,960,062–225,460,062 | 0.095 | 8.0 | DNAH14 |
| 5 | 5 | 82,375,629–82,875,629 | 0.08 | 6.6 | VCAN, XRCC4 |
Polygenic adaptation
Natural selection on phenotypic traits in humans is expected to only occasionally take the form of sweeps on a single locus, instead acting on multiple genes simultaneously to drive phenotypic adaptation (Coop et al., 2009). While a lack of genome-wide association studies in eastern and southern Africans has left the genetic basis of phenotypic traits far less well documented than it is for other populations, a variety of studies have linked broad functional classes of genes to phenotypic traits. To test for evidence of selection on specific functional categories of genes in present-day San since the divergence of the two ancient genomes from southern Africa (Fig. 4B), we estimated allele frequency differentiation for 208 gene ontology categories with 50 or more genes in each, and computed weighted block jackknife standard errors. The functional category that displays the most extreme allele frequency differentiation between present-day San and ancient southern Africans is “response to radiation” (Z = 3.3 compared to the genome-wide average). To control for the possibility that genes in this category show an inflated allele frequency differentiation in general, we computed the same statistic for the Mbuti central African rainforest hunter-gatherer group, but found no evidence for selection affecting the “response to radiation” category (Fig. 4C). Instead, the top category for the Mbuti is “response to growth”, suggesting the possibility that the small stature of rainforest hunter-gatherer populations such as the Mbuti may be an acquired adaptation (although we have no ancient central African genome and thus no information about the time frame of selection). We speculate that the signal for selection in the “response to radiation” category in the San could be due to exposure to sunlight associated with the life of ‡Khomani and Ju’|hoan North in the Kalahari Basin, which has harbored a larger proportion of San populations in the last millenia due to encroachment from pastoralist and agriculturalist groups (Morris, 2002).
Discussion
This study, which multiplies by 16-fold the number of individuals with genome-wide ancient DNA data from sub-Saharan Africa, highlights the power of ancient African genomes to provide insights into prehistoric events that are difficult to discern based solely on analysis of present-day genomes. We reveal the presence of a hitherto unknown cline of geographically structured hunter-gatherer populations stretching from Ethiopia to South Africa, which we show existed prior to the great populations transformations that occurred in the last few thousand years in association with the spread of herders and farmers. We also document deeper structure in western Africa, possibly predating the divergence of the ancestors of southern African hunter-gatherers from other population lineages. We finally provide case examples of how populations in eastern and southern Africa were transformed by the spread of food producers, and show how the process gave rise to interactions with the previously established hunter-gatherers, with the outcomes ranging from no detectable mixture in present-day populations to substantial mixture. Our documentation of a radically different landscape of human populations before and after the spread of food producers highlights the difficulty of reconstructing the African past based solely on analysis of present-day populations and the importance of using ancient DNA to study deep African population history in an era in which technological improvements have now made it feasible. It is clear that ancient DNA studies with larger sample sizes and covering a broader chronological and geographic range have the potential to make major progress in improving our understanding of African prehistory.
STAR Methods text
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, David Reich (reich@genetics.med.harvard.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
We generated new genome-wide data from skeletal remains of 15 prehistoric individuals: 5 from eastern Africa, 7 from south-central Africa, and 3 from southern Africa (Table 1; Table S1; Table S2). One of these individuals, from St. Helena Bay and directly dated to ~2100 BP, previously yielded a complete mitochondrial genome (Morris et al., 2014). We directly dated a second South African individual buried in a hunter-gatherer context from Faraoskop to ~2000 BP, and a third individual buried in a pastoralist context from Kasteelberg to ~1,200 BP. We also directly dated and used in-solution enrichment to obtain genome-wide DNA from four individuals from coastal eastern Africa: From the cave site of Panga ya Saidi in the coastal region of southeastern Kenya (~400 BP), Kuumbi Cave in the southeast of Zanzibar Island (Tanzania; ~1,400 BP), and Makangale Cave in the northwest of Pemba Island (Tanzania; ~1,400 BP and ~600 BP). We also obtained genome-wide data from a ~3100 BP individual from a pastoralist context in north-central Tanzania, and ~8100–2500 year old individuals from Malawi.
Terminology
There is no widely accepted term with neutral connotations for indigenous communities in southern Africa (Schlebusch, 2010). In this manuscript, we follow San council recommendations in using population-specific terms whenever possible, and alternatively the terms San for Tuu and K’xaa speaking hunter-gatherer groups and Khoe for Khoe-khoe speakers. When necessary we collectively refer to groups with southern Africa-specific ancestry as Khoe-San, or as having San-related ancestry.
Panga ya Saidi Cave, Kilifi County, Kenya (n = 1)
Panga ya Saidi is a large limestone cave complex formed within an escarpment c. 15 km from the Indian Ocean coast in southern Kenya. Excavated in multiple Sealinks Project campaigns, the cave’s long and complex depositional sequence spans, discontinuously, more than 76,000 years, and contains mainly Later Stone Age (LSA) deposits, overlain by Middle Iron Age (MIA) and Later Iron Age (LIA) deposits dating to the last two thousand years (Helm et al., 2012). The sampled specimen (I0595, Kenya_400BP) is a phalanx recovered from an in situ burial (context 403) and directly AMS radiocarbon dated to 496–322 calBP (388 ± 27 BP, OxA-30803). The individual was a tall, robust young adult male. He was buried in a shallow grave in a crouched position with two hands and one foot in the small of the back and the skull disarticulated and placed by the knees. The individual was buried by sediment containing marine shell beads, small knapped stone tools, and Tana Tradition potsherds. The associated faunal remains are exclusively wild, with the exception of a single possible caprine bone. Large numbers of remains of birds, rodents, and other microfauna suggest that the cave may have only been sporadically occupied when the human remains were deposited. We infer from the material culture and fauna that the cave was occupied by foragers during the time the individual was buried, although food producers were present at nearby settlements such as Mtsengo and Mbuyuni (Helm, 2000). Crop remains of African sorghum, pearl millet and finger millet found at the site suggest these foragers had access to agricultural resources.
Makangale Cave, Pemba, Tanzania (n = 2)
This limestone cave at the northern end of Pemba Island in the Zanzibar archipelago has been excavated in multiple campaigns, the most recent two seasons conducted by the ERC-funded Sealinks Project at Oxford University in 2012 and then the Max Planck Institute for the Science of Human History in 2016 (unpublished; see also (Chami et al., 2009)). The sequence shows clear evidence of human occupation beginning around 1400 BP with an escargotière layer of giant African land snail shells, pottery, and disarticulated human remains. Above this layer, the sequence shows regular human use of the cave into the last thousand years. The first sampled specimen (I0589, Tanzania_Pemba_1400BP) is a sacral vertebra from context 204 (Sealinks Project faunal catalog no. 15336), directly dated to 1421–1307 calBP (1520 ± 30 BP, Beta-434912). The second specimen (I2298, Tanzania_Pemba_600BP) is a lower molar from context 301 (Sealinks Project faunal catalog no. 15624), which lies just below the surface and was dated to 639–544 calBP (623 ± 20 BP, Wk-43308). Both specimens are associated with a highly unusual faunal assemblage, dominated by fragmented crocodile (Crocodylus cf. niloticus) remains and diverse microfauna, including Rattus rattus (Asian black rat), a nonnative rodent that must have arrived to the area via maritime exchange routes. There are no taphonomic indicators in the faunal assemblage of hunting by humans, nor of crocodile predation on humans. During both of the occupational phases targeted in this study, there were nearby settlements occupied by farmers whose ancestors likely came from the mainland, for example at the sites of Tumbe (c. 1400–1000 BP) and Chwaka (1000–400 BP) (Fleisher and LaViolette, 2013).
Kuumbi Cave, Zanzibar, Tanzania (n = 1)
Kuumbi Cave is a limestone solutional cave excavated in multiple campaigns (Chami, 2009; Sinclair et al., 2006). Sealinks Project excavations in 2012 documented a complex depositional sequence stretching discontinuously over 20,000 years, containing evidence of LSA and MIA occupations in five discernible phases (Shipton et al., 2016). The analyzed specimen (I0589, Tanzania_Zanzibar_1400BP) is a complete second phalanx of an adult (Sealinks Project faunal catalog no. 4353). It was recovered from context 1011, in association with local Tana Tradition ceramics typical of the MIA, moderately-sized limestone lithic artifacts, and diverse wild game animals, but no additional human remains (Prendergast et al., 2016). The specimen is directly dated to 1370–1303 calBP (1479 ± 23 BP, OxA-31427), thus placing it at the beginning of the MIA phase. While Kuumbi Cave is interpreted as a forager site, elsewhere on the island at this time, large settlements such as Unguja Ukuu emerge, occupied by farmers whose origins are likely on the mainland (Crowther et al., 2015; Juma, 2004).
Luxmanda, Babati District, Tanzania (n = 1)
Luxmanda is an open-air settlement sitting atop the Rift Valley escarpment (1878 m above sea level) at the southern edge of the Mbulu Plateau, just north of Lake Balangida and Mount Hanang. Excavations in 2012, 2013, and 2015 by the RAPT project (Research on the Archaeology of Pastoralism in Tanzania) have shown Luxmanda to be the largest and southernmost known settlement site of the Pastoral Neolithic (PN), the era corresponding to the spread of mobile livestock herding in eastern Africa (Prendergast et al., 2013) (Grillo, Prendergast et al. forthcoming). Despite its isolated location, Luxmanda shows strong material culture affinities to sites of southern Kenya classified as Savanna Pastoral Neolithic (SPN), in particular the Narosura type-site (Odner, 1972); Luxmanda’s ties to SPN sites are further supported by sourcing of obsidian stone tools to the Naivasha Basin in the Central Rift Valley of Kenya. Faunal remains from Luxmanda indicate a diet almost exclusively focused on sheep, goat, and cattle; botanical remains are currently under study. A suite of eleven radiocarbon dates provides a tightly constrained window of occupation c. 3000–2900 calBP, which is at the early end of the range for SPN sites. The analyzed specimen (Tanzania_Luxmanda_3100BP) is a petrous bone from a perinatal infant. The infant was found complete and buried just to the west of, and c. 35 cm below, a burnt earth feature, interpreted as a hearth. The burnt earth feature was then overlain by domestic refuse. Collagen from the same petrous bone was AMS radiocarbon dated to 3141–2890 calBP (2925 ± 20 BP, ISGS-A3806), a date nearly identical to those of charcoal samples taken from the overlying burned earth feature and domestic refuse.
Hora, Malawi (n = 2)
The Hora 1 and Hora 2 skeletons from Malawi were excavated from the Hora 1 site in the Mzimba District of Malawi, located on the northeastern side of Mount Hora. Mount Hora lies south of the Nyika Plateau and west of the Viphya Mountains, where miombo vegetation grades southwest into edaphic grasslands (DeBusk, 1997). The region divides the Luangwa River Basin in Zambia from Lake Malawi ~130 km to the east, which receives all water from the district via the South Rukuru River (Fig. S1). The South Rukuru flows south-to-north along the Zambian border before turning east to intersect the Kasitu, a major interior waterway that flows past Mount Hora and divides the mountains to the east from plains to the west. It is notable that in the Hora region, rock art, stone tools, and burial practices all have substantial differences from those even in the nearby Luangwa Valley of Zambia, suggesting cultural subdivisions across relatively small areas (Clark, 1959; Phillipson, 1976; Sandelowsky, 1972).
Hora is a distinctive granite-gneiss inselberg that rises 110 m from superficial floodplain deposits. Hora 1 is a large overhang at the base (1,420 m AMSL) that covers ~80 m2. Although the shelter has no surviving rock art, at least four other shelters on the inselberg contain paintings that include white stars and abstract red “gridirons” or “nets”, both of which are motifs replicated at other sites on Mount Hora and nearby localities (Clark, 1956; Cole-King). Hora 1 was excavated by Clark in 1950, and produced a 2.2 m cultural sequence containing pottery and iron slag at the top, faunal remains and LSA lithic assemblages with mollusk shell beads below this, and two human burials approximately 70 cm below the surface (Clark, 1956). The first burial to be revealed was Hora 1 (UCT-242), a short-statured male in his thirties or forties at death, who may have been buried with a flaked stone axe (Morris and Ribot, 2006). The skeleton was left partially in situ during the original excavation and later exhumed by Rangeley (Clark, 1956). About 3 m to the south, the skeleton of a female was recovered (Hora 2, UCT-243); she was of similar stature to the male and died in her early twenties (Morris and Ribot, 2006). The female was found in a flexed position on her left side, and most of the bones of the hands and feet were missing or had been displaced – suggesting a degree of exposure prior to burial (Clark, 1956).
A previous study (Clark, 1956) reports three major stratigraphic units at Hora 1, and places the burials in the upper part of the second unit in association with the “Nachikufan II” – an industry with a type site ~300 km to the east in Zambia. Clark does not consider the burials intrusive, noting that they are overlain exclusively by hunter-gatherer material culture. The earliest occupation layer at the site is “reddish brown earth” that begins at about 1.7 m depth, and contains a lithic assemblage assigned by Clark to the Nachikufu I. On the basis of typology, the earliest deposits at the site may date to between 16000–11000 BP, the burials to between 10000–5000 BP, and materials in the uppermost unit are likely as recent as the last few hundred years (Miller, 1971).
As this age range is imprecise, new pilot excavations were conducted in 2016. These confirmed that there is no pottery, slag, or other indication of non-hunter-gatherer material culture within the 0.5 m overlying the depth of the burials. Unfortunately, attempts to directly date the Hora 1 skeletons through 14C AMS failed at two labs because of lack of preserved collagen. However, the other Malawi specimens reported here were all directly dated (Table 1; Table S2). The Hora 1 and Hora 2 genetic data show that these specimens align genetically with the other ancient individuals from Malawi, which cover a time period from approximately 6200–2300 calBP. Their cultural associations, state of preservation, and genetic affinities therefore together place the Hora specimens in the late Holocene of Malawi.
Fingira, Malawi (n = 3)
The three Fingira samples derived from ex situ human remains (2 adult femora and one subadult femur) recovered in 2016 from Fingira Rock, a large shelter located within the boundaries of Nyika National Park in northern Malawi. The climate is cool and moist compared to the surrounding regions (~1,600 mm annual rainfall, and average daily highs 10 – 20° C). Fingira Rock is an isolated inselberg at ~2100 meters above sea level, near the upper limit of the miombo woodland. Within this inselberg is set a single deep rock shelter with 160 m2 of deposit (Fig. S1). First excavated in 1966, Fingira yielded fragmentary remains of at least 15 human individuals (subadults and adults), plus one more complete burial near the front, in association with rich lithic, archaeofaunal, and archaeobotanical assemblages. All human remains previously recovered from Fingira were studied by Brothwell, and reported in in a previous study (Sandelowsky, 1972). They are curated at the Natural History Museum in London.
In addition to human remains, the Fingira deposits contained bone tools, pigments, and ornaments (Robinson and Sandelowsky, 1968; Sandelowsky, 1972). Two conventional 14C ages obtained near the base of the ca. 50 cm-deep excavation were returned in stratigraphic sequence as 3,260 ± 80 BP and 3,430 ± 80 BP (Sandelowsky 1972). Geometric rock paintings at the site exhibit white overpainting on red, which has been interpreted elsewhere in Malawi as re-use by food producers (Smith, 1995; Zubieta, 2016). Ceramics are rare but present at the site, where the large deposit exhibits predominately Later Stone Age material culture. This was confirmed during 2016 pilot excavations, permitted by the Departments of Antiquities and National Parks and Wildlife.
The deposits at Fingira had not been backfilled after the 1966 excavation, and had undergone extensive erosion and slumping. As the site is accessible to the public, piles of materials had been pulled from the section and placed on fallen rocks. Two of those specimens comprised the two adult femoral specimens reported here. Using original site plans, we identified the extent of the 1966 excavations prior to slumping, including the relative positions of originally-recovered human remains. A large part of this area had been covered with a central termite mound, and it was within this that the partial skeleton of a neonate was recovered. This comprised the subadult sample from Fingira. Direct 14C AMS ages on these three specimens are reported in Table S2, and show that LSA people were using the site as a cemetery for at least 3,700 years, from 6177–5923 calBP (5290 ± 25 BP, UCIAMS-186347) to 2676–2330 calBP [2676–2343 calBP (2425 ± 20 BP, PSUAMS-1734), 2483–2330 calBP (2400 ± 20 BP, PSUAMS-1881)]. The two adult specimens therefore significantly pre-date the earliest known age in Malawi for the Bantu expansion, which derives from the Kasitu Valley (containing Hora Mountain) at 1,750 ± 60 BP (Robinson, 1982). If the Bantu expansion into Malawi began more than ~700 years before what is currently known, then the neonate from Fingira could potentially overlap in time with it. However, the overall antiquity of these specimens makes admixture attributable to the Bantu expansion highly unlikely in light of current knowledge.
Chencherere II (n = 2)
Mwana wa Chencherere II is a painted rock shelter set in a granitic inselberg at ca. 1,700 meters above sea level in the Chongoni Rock Art Region (Smith, 1995). Its relatively high altitude results in cool, moist year-round conditions between 10 – 20° C. It was excavated by Clark in 1972 (Clark, 1972; Clark and Clerk, 1973), and the faunal assemblage published in detail by Crader (Crader, 1984), who also reports one adult male burial and the fragmentary remains of seven other individuals (adult and subadult). The site contained large lithic and faunal assemblages, bone and shell tools and ornaments, and increasing abundances of pottery and other evidence of interaction with food-producers over time. The youngest reported date from the site is a conventional 14C age on charcoal from the top of Level 3, at 800 ± 50 BP. The oldest date is from near the base of Level 4, and is 2,480 ± 200 BP.
All human remains are reported from Levels 2, 3, and 4 – with most in Level 3. The original description of human remains was by Brothwell, and Crader (1984b:Appendix 2) later listed several more individuals that had been discovered within the faunal material. All human remains recovered from Chencherere II were thought to be held at the Natural History Museum in London, but during a 2016 visit to the Malawi National Repository in Nguludi (near Blantyre), six additional specimens were identified. Five of these derived from a cluster in square A3 (Fig. S1): right ilium, left femur (sampled), and 3 partial ribs. These were inferred to belong to the same individual, a subadult aged 3–5. An upper right incisor (sampled) was labeled as deriving from square E2, and therefore although of similar ontogenetic age it was deemed likely to be from a different individual.
Although the genetic analysis confirms that these are two different individuals, insufficient material remained from the incisor root of the second individual for a direct age. The first individual returned a direct 14C AMS age of 5293–4979 calBP (4525 ± 25 BP, UCIAMS-186348). As with Fingira, the direct AMS ages on the human remains are substantially older than the conventional charcoal ages suggested, indicating either intrusive charcoal or problems with the original dates.
St. Helena Bay, South Africa (n = 1)
In June 2010, an intact skeleton was excavated by Andrew B Smith along the southwest coastal region of South Africa at St. Helena Bay. The skeleton is stored in the Department of Human Biology at the University of Cape Town under the accession number UCT-606. The body had been placed on an impermeable consolidated dune surface, on its right side in a fully flexed position. The bones originate from a single male who stood no more than 1.5 m in height. Dental wear and significant areas of osteoarthritis suggest that he was at least 50 years of age at time of death. Lack of any evidence of tooth decay and excessive occlusal wear suggests a diet typical of hunter-gatherer subsistence. The presence of abnormal bone growths in the right auditory meatus (ear canal opening) caused a condition known as “surfer’s ear” (auditory exostosis) and provides evidence that this individual most likely spent considerable time in the cold coastal waters sourcing food No obvious cause of death was evident. The results of carbon-14 to stable carbon-13 isotope ratio analysis of a rib provided a date of 2241–1965 calBP (2330 ± 25 BP, UGAMS-7255) years before present (UGAMS-7255) with a d13C value of −14.6%. Although the minimum date falls right on the edge of the arrival of pastoralism in the Western Cape, anatomical and archaeological analysis of this skeleton and the associated burial site clearly defines this individual as an indigenous Southern African, predating pastoral arrival into the region. This individual has previously been sampled for mtDNA analysis (Morris et al., 2014).
Faraoskop, South Africa (n = 1)
The site of Faraoskop is a rock shelter about 30 kilometres inland from Elands Bay on the west coast of the Western Cape Province of South Africa. The shelter is on the highest ridge of a small hill at an altitude of 300 metres about the surrounding plain. Seven skeletons were collected by a local farmer in 1984, but the site was subsequently excavated under controlled conditions in 1987 and 1988 (Manhire, 1993) and five more skeletons were collected including the one referred to here as UCT-386. The shelter has no rock paintings but there is a rich assemblage of Later Stone Age artefacts including stone tools, ostrich eggshell beads, worked marine shell, leather and twine (Manhire, 1993). No pottery is associated with the site. Six skeletons have been C14 dated with resulting dates ranging from 2300 years BP to 1900 years BP (Manhire, 1993; Sealy et al., 1992) but all the dates overlap at the 2nd standard deviation. UCT-386 is the skeleton of a female about 40–50 years at death. A bone sample provided a date of 2017–1748 calBP (2000 ± 50 BP, Pta-5283) with a d13C value of −16.8% (Manhire, 1993). A recent re-evaluation of the site indicates the possibility that all of the individuals died in one event. Not only do all of the dates overlap, but the excavation data suggest no separate grave shafts and at least two of individuals show signs of perimortem injury and violent death (Parkington and Dlamini, 2015). The Faraoskop human skeletons are stored in the Department of Human Biology at the University of Cape Town.
Kasteelberg, South Africa (n = 1)
The site of Kasteelberg is on a granite hill on the Vredenberg Peninsula about 4 kilometres from the coastal village of Paternoster, approximately 150 kilometres north of Cape Town (Smith, 1992a). There are several sites on the hill including a small rockshelter, but the human skeleton was excavated from square 22 extension at KBB on the eastern side of the base of the hill. UCT-437 is the nearly complete skeleton of a child of about 4 years of age excavated by Lita Webley in 1986. The body was in a shallow pit about 1.5 metres deep and with no stone cover. The specimen has a date of 1282–1069 calBP (1310 ± 50 BP, Pta-4373). There were no grave goods in association with the skeleton. The earliest sites on top of the hill provide evidence of domestic sheep at around 2100 years ago. The KBB site at the base of the hill is dated to the latter part of the occupation but present the first appearance of cattle in the region (Smith, 1992b). Overall, the Later Stone Age occupation of the Kasteelberg indicates the presence of herder-foragers who practiced seasonal economic systems, sometimes relying on domestic stock while at other times hunting seals (Sadr et al., 2003). The human skeleton from Kasteelberg is stored in the Department of Human Biology at the University of Cape Town.
METHOD DETAILS
Direct AMS 14C Bone Dates
We report new direct AMS 14C bone dates in this study from multiple laboratories. In general, bone samples were manually cleaned and demineralized in weak HCl and, in most cases (PSU, UCIAMS, OxA, ISGS), soaked in an alkali bath (NaOH) at room temperature to remove contaminating soil humates. Samples were then rinsed to neutrality in Nanopure H2O and gelatinized in HCL (Longin, 1971). The resulting gelatin was lyophilized and weighed to determine percent yield as a measure of collagen preservation (% crude gelatin yield).
Collagen was then directly AMS 14C dated (ISGS, Pta) or further purified using ultrafiltration (PSUAMS, UCIAMS, OxA, Wk, Beta) (Brown et al., 1988; Kennett et al., 2017) or a modified XAD method (Lohse et al., 2014; Stafford et al., 1991). It is standard in some laboratories (PSUAMS, UCIAMS, Wk, OxA) to use stable carbon and nitrogen isotopes as an additional quality control measure. For these samples, the %C, %N and C:N ratios were evaluated before AMS 14C dating. C/N ratios for well-preserved samples fall between 2.9 and 3.6, indicating good collagen preservation (Van Klinken, 1999). Additional quality control work was carried out on the samples from Malawa using FTIR spectra (Fig. 1E).
All 14C ages were δ13C-corrected for mass dependent fractionation with measured 13C/12C values (Stuiver and Polach, 1977) and calibrated with OxCal version 4.3 using the southern hemisphere calibration curve (SHCal13). Given their proximity to the equator, AMS 14C dates for sites in coastal Kenya and Tanzania were calibrated using OxCal v. 4.3 (Bronk Ramsey, 2009) at 95.4% probability employing a mixed curve that combines the SHCal13 (Hogg et al., 2013) and IntCal13 (Reimer et al., 2013) curves at ratios of 70:30 to account for the differential effects of the intertropical convergence zone.
Ancient DNA sample processing in Leipzig: St. Helena Bay sample
DNA extraction and library preparation
30.4 mg of bone powder was removed from the internal root canal of the tooth (SP2809) using a sterile dentistry drill in the clean room facilities of the Max Planck Institute for Evolutionary Anthropology in Leipzig, Germany. A DNA extract (E649) was prepared with a silica-based method, described in detail previously (Rohland and Hofreiter, 2007). 15 μL (15% of the total volume) of the extract was converted into a single-stranded DNA library (A5354) using a modified version of the single-stranded DNA library preparation protocol (Gansauge and Meyer, 2013; Korlević et al., 2015). Library positive and negative controls were carried throughout the library preparation process. Library A5354 was pre-treated with the USER enzyme, a mixture of uracil-DNA glycosylase (UDG) and endonuclease VIII, in order to remove uracils from the internal parts of ancient DNA molecules (Briggs et al., 2010; Meyer et al., 2012). The number of DNA molecules in the library (Table S1) was determined by digital droplet PCR (Bio-Rad QX200), using 1 μL of a 5,000-fold dilution of the library in EBT buffer (10 mM Tris-HCl pH 8.0, 0.05% Tween 20) as template in an Eva Green assay (Bio-Rad) with primers IS7 and IS8 (Meyer and Kircher, 2010). The library was amplified into the PCR plateau in a 100 μL reaction with AccuPrime Pfx DNA polymerase (Dabney and Meyer, 2012) using a pair of primers with two unique index sequences according to a double indexing scheme described in detail elsewhere (Kircher et al., 2011). 50 μL of amplification products were purified using the MinElute PCR Purification Kit (Qiagen) and eluted in 30 μL TE buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA). The DNA concentration of the indexed library (A5369) was determined using a NanoDrop 1000 Spectrophotometer.
Size fractionation for shotgun sequencing
From the amplified library A5369, 1 μl was taken as template for a second round of amplification in a 100 μl PCR reaction using primers IS5 and IS6 (Meyer and Kircher, 2010) with Herculase II Fusion DNA polymerase (Agilent Technologies) under the conditions described in detail previously (Dabney and Meyer, 2012). The concentration of the final library was determined on a Bioanalyzer 2100 instrument (Agilent Technologies) using a DNA-1000 chip. Library A5369 was pooled and sequenced with libraries from another experiment, occupying 33% of one lane of a flow cell on the Illumina HiSeq 2500 platform in rapid mode, using a double index configuration (2 × 76 + 2 × 76) (Kircher et al., 2011).
For a more effective use of sequencing capacity, 10 μL of the library A5369 was additionally separated on a Criterion Precast polyacrylamide gel (10% TBE, BioRad), and the fraction of library molecules with insert sizes larger than 40 bp was gel-excised as described in detail previously (Meyer et al., 2012). Gel-excised library molecules were subjected to a second round of amplification and the concentration of the final library (A5386) was determined using a DNA 1000 chip on the Bioanalyzer 2100.
Ancient DNA sample processing in Tübingen: Faraoskop and Kasteelberg samples
Sampling and extraction
Sampling took place in the clean room facilities of the Institute for Archaeological Sciences at the University of Tübingen. Both samples were irradiated with UV light for 10 minutes from all sides to remove surface contamination. The tooth from the South African forager from Faraoskop (UCT386) was sawed apart transversally at the border of crown and root, and dentine from inside the crown was sampled using a sterile dentistry drill, resulting in 56 mg dentine powder. For the femur fragment from the South African pastoralist from Kasteelberg (UCT437), the surface layer from the sampling area was removed with a dentistry drill prior to obtaining four aliquots between 51 and 80 mg of bone powder from the inside of the bone by drilling.
Extraction was performed following a protocol optimized for the recovery of small ancient DNA molecules (Dabney et al., 2013), resulting in 100 μl of DNA extract per sample. Three of the bone powder aliquots from UCT437 underwent a 10 minute pre-digestion step after which the extraction buffer was removed (pre-digest) and replaced by fresh extraction buffer followed by over-night digestion (ON-digest), the powder from UCT386 and one aliquot of UCT437 were extracted without the pre-digestion step (full-digest). All eight resulting extracts were taken along for further library preparation. Negative controls were included in the extraction and taken along for all further processing steps.
Screening
Two double-indexed libraries were produced from an aliquot of 20 μl of the full-digest extractions of UCT386 and UCT437 (Kircher et al., 2011; Meyer and Kircher, 2010). Positive and negative controls were included in library preparation and taken along into sequencing. Libraries were enriched for human mitochondrial DNA (Maricic et al., 2010) and both enriched and shotgun libraries were sequenced on a HiSeq2500 with 2 × 101 + 8 cycles. Processing by the EAGER pipeline (Peltzer et al., 2016) and schmutzi (Renaud et al., 2015) resulted in an endogenous DNA content of 39% and 8% and an estimated mitochondrial contamination of 0–2% and 1–3% for UCT386 and UCT437, respectively.
Library preparation for shotgun sequencing
For UCT386 four more libraries were produced from an aliquot of 20 ul of full-digest extract each, including a DNA repair step with UDG and endonuclease VIII to remove deaminated bases (Briggs and Heyn, 2012). For UCT437, six additional UDG-treated libraries were produced from 20 ul each of extract from the three pre-digest and the three ON-digest extracts. After indexing PCR (Kircher et al., 2011), aliquots of the UDG-treated libraries were size selected on a PAGE gel to remove fragments of sizes below 35 and above 80 bp (Meyer et al., 2012).
Ancient DNA sample processing in Dublin: Malawi samples
Sampling took place in ancient DNA-dedicated clean room facilities at University College Dublin. The petrous part of the temporal bone was selected for analysis from each individual (n = 2). Each complete petrous was UV irradiated for 10 minutes on each side prior to processing to reduce surface contamination. Any remaining sediment was removed using a Renfert Basic Classic Sandblaster (Renfert GmbH) at low power. Bone powder was retrieved from the petrous of UCT242 (Hora 1) by drilling a small hole on the superior surface of the petrous with a 4.8 mm High Speed Cutter (Dremel) until the cochlea was accessible. Bone powder was then collected directly from the cochlea using a 3.2 mm Tungsten Carbide Cutter (Dremel). The petrous from UCT243 (Hora 2) was cut from anterior to posterior using a Dremel drill at a location that transected the cochlea. The powder was collected directly from the cochlea using a 3.2 mm Tungsten Carbide Cutter (Dremel). Powder aliquots from both samples were then UV irradiated for 5 minutes and placed in 2.0 mL Eppendorf tubes.
Ancient DNA sample processing in Boston: Tanzania samples, Kenya samples, and Malawi sample powder
Sampling and DNA extraction
In a dedicated ancient DNA facility at Harvard Medical School, samples were UV-irradiated for 10 minutes in a UVP crosslinker. At the chosen part of each sample (root for the tooth and compact part for bones) the surface was removed with a sanding disk. About 75 mg (± 10 mg) of fine powder was obtained by drilling into the physically cleaned part with a sterile dentist drill bit and collected for DNA extraction (Table S1). In the case of KC-10-1011(4353) (I0589), additional bone powder was collected for a second DNA extraction attempt. The seven Malawi_Holocene samples arrived in the Boston laboratory as powders prepared in Dublin, Ireland. Starting from the sample powder, we followed the Dabney et al. 2013 extraction protocol for all samples, but replaced the funnel/MinElute assemblage with the pre-assembled Roche columns (Korlević et al., 2015) and eluted two times in 45 μl for a total of 90 μl DNA extract.
Initial library preparation
One initial barcoded library (L1) was prepared from 30 μl DNA extracts for all but two samples (Hora1 and Hora2), which were discolored and we reduced the volume to 3 μl (reducing the volume seems to mitigate library preparation inhibition, which we often find to be associated with discolored DNA extracts) following protocols published previously (Rohland et al., 2015) (Table S1). For three samples (I0589, I0595, I1048) the initial library was UDG-treated (Briggs et al., 2010) following a modification from Rohland et al. 2015 (partial UDG treatment) that is tailored to inefficiently remove terminal Uracils therefore leaving the aDNA authenticity signal in the terminal bases while efficiently removing miscoding damage within the molecules. The initial libraries for the other samples (I2298, I2966, I2967) were not UDG-treated. The last step of the library preparation, the amplification with universal primers, was set up in the cleanroom, but the thermal cycling happened in another laboratory physically separated from the cleanroom. The final products of our barcoded libraries cannot be sequenced right away; an additional PCR step is needed to finalize the adapter sites. This is advantageous in that we can incorporate a second set of barcodes through dual indexing to differentiate two or more experiments done with the same barcoded library within the same sequencing run (see below).
Screening
Each initial library underwent screening that consisted, first, of shallow shotgun sequencing after an indexing PCR (that adds dual indices to each library, (Kircher et al., 2011), and second, target capture enrichment for mitochondrial DNA and a varying number of nuclear loci to assess mitochondrial haplogroup, mitochondrial contamination, aDNA authenticity and nuclear complexity (Meyer et al., 2014; Rohland et al., 2015). This experiment is finished by adding unique index combinations to each captured library, which is then subsequently pooled with the shotgun indexing PCR product for sequencing. Sequencing was done on an Illumina NextSeq500 with 2 × 76cycles + 2 × 7cycles.
We demultiplexed reads to be sample-specific requiring that that the 7 bp P5 and P7 indices matched (allowing one mismatch). Sample identification was further ensured by requiring that additional 7 bp internal barcodes matched, again allowing one mismatch. We merged with a modified form of SeqPrep (github.com/jstjohn/SeqPrep) (the modification ensures that the highest quality base is retained in the overlap region), requiring at least 15 bp overlap between forward and reverse reads, allowing one mismatch, retaining only reads of length greater than or equal to 30 base pairs, generating single ended reads.
Reads were then aligned using the samse algorithm of BWA (version 0.6.1) (Li and Durbin, 2009) using ancient parameters to allow an increased mismatch rate, and to disable seeding (“−n 0.01 −0 2 −l 16500”). Multiple sequencing runs were run to increase coverage, and merged together. Duplicates were then removed by identifying clusters of reads which have the same start and stop position, and the same mapped orientation. The highest base quality representative of each set is used to represent the cluster. The mix of mitochondrial and nuclear loci necessitates two different references for the alignment process: for mitochondrial analysis, we use the RSRS (Behar et al., 2012) mitochondrial genome; for nuclear analysis we use the hg19/GRCh37, 1000 Genomes release reference genome.
Additional libraries and processing
To collect more nuclear data for a subset of the samples, we prepared additional barcoded libraries (I0589, I0595, I1048, I3726) without UDG-treatment from existing DNA extracts (L2-L5). For sample I0589 we collected more bone powder than necessary for one extraction, and therefore prepared three additional libraries from a newly prepared DNA extract (E2). Four additional libraries for one sample (I3726) were prepared on an Agilent Bravo Workstation using an automated protocol based on the partial UDG protocol that replaced the MinElute cleanups with magnetic bead cleanups.
All libraries underwent the same procedure as outlined, screening (see above) and nuclear target enrichment (see below), with the exception that up to 4 libraries from the same sample were pooled in equimolar concentrations before screening and nuclear target capture (Table S1). Preprocessing and alignment for nuclear data used the same procedure as performed in screening, without requiring the mitochondrial alignments.
Shotgun genome sequencing
Shotgun sequencing of the ancient South African from St. Helena Bay was performed at the Max Planck Institute in Leipzig, Germany, on four lanes of the Illumina HiSeq 2500 platform in rapid mode, using a double index configuration (2 × 76 + 2 × 76) (Kircher et al., 2011). An indexed ΦX174 control library was spiked in prior to sequencing. Base calling was done with the machine-learning algorithm freeIBIS (Renaud et al., 2013). Overlapping pair-end reads were merged (Kircher, 2012) and mapped to the human reference genome (hg19/GRCh37, 1000 Genomes release) using the Burrows-Wheeler Aligner (BWA) (Li and Durbin, 2009). BWA parameters were adjusted for ancient DNA sequences (“−n 0.01 −o 2 −l 16500”), to allow for more mismatches and indels and to turn off the seeding (Meyer et al., 2012). A total of 64,128,220 raw sequences were obtained from the first shotgun sequencing of the library A5369. Another 800,205,849 raw sequences were generated from the size-selected library using four lanes of the Illumina HiSeq 2500. Only mapped sequences longer than 35 bp were retained and duplicates removed (bam-rmdup; https://github.com/udo-stenzel/biohazard), leaving 9,880,908 sequences from the first shotgun run and 52,551,348 sequences from sequencing the size-fractionated library. Duplication rates were 1.02 and 1.06, respectively, indicating that both libraries were not sequenced to exhaustion. The proportion of sequences ≥ 35 bp that mapped to the human reference genome was ~28%.
Shotgun sequencing of the ancient South African from St. Helena Bay (UCT386) and the ancient South African from Kasteelberg (UCT437) was performed at the University of Tuebingen using two lanes of an Illumina HiSeq2500 instrument for 2 × 101 + 8 cycles (UCT386 non-UDG-treated library), on 50% of two lanes of an Illumina NextSeq500 instrument for 2 × 151 + 8 cycles (UCT437 non-UDG-treated library), on a complete run of an Illumina HiSeq2500 instrument for 2 × 125 + 8 cycles (four UCT386 UDG-treated and size-selected libraries and six UCT437 UDG-treated and size-selected libraries), and on 5.5 lanes of an Illumina HiSeq2500 instrument for 2 × 125 + 8 cycles (four UCT386 UDG-treated libraries without size-selection and three UCT437 UDG-treated full-digest libraries without size-selection). The samples were processed using the EAGER pipeline (Peltzer et al., 2016), clipping adapters and merging reads subsequently with an overlap of 10 bp. Resulting reads were then mapped within the pipeline against the human reference genome GrCh37 and using BWA 0.7.5 (Li and Durbin, 2009) for further downstream analysis.
Shotgun sequencing of the Malawi_Hora_8100BP samples was performed at Harvard Medical School using an Illumina NextSeq500 instrument. Preprocessing and alignment used the same procedure as performed in screening.
In-solution nuclear target enrichment
After libraries passed screening QC (that is, there was evidence of authentic ancient DNA), we performed nuclear target enrichment of the short (but barcoded) libraries following (Fu et al., 2015) aiming to enrich for about 1.24 M SNPs in total (Fu et al., 2015; Haak et al., 2015; Mathieson et al., 2015) using a semi-automated approach on a Perkin Elmer Evolution P3 liquid handler. For two libraries (S0589.E1.L1 and S2595.E1.L1) the desired 1.24 M targeted SNPs were captured in two independent reactions by enriching, first, for about 0.39 M SNPs, and second, for 0.84 M SNPs. The other first libraries (L1) were enriched in one single reaction (1240 k). After indexing PCRs with dual indices and equimolar pooling sequencing was performed on an Illumina NextSeq500 with 2 × 76cycles + 2 × 7cycles. Preprocessing and alignment used the same procedure as performed in screening, without requiring the mitochondrial alignments.
Genotyping and initial processing of 34 present-day individuals from Malawi
We newly report data from 34 present-day individuals from Malawi, genotyped on the Affymetrix Human Origins SNP array (Patterson et al., 2012). Quality control of the data prior to merging involved screening for outlier individuals, excess missingness, as well as deviations from Hardy-Weinberg equilibrium, and was performed in a manner similar to what has previously been described (Lazaridis et al., 2014).
Data processing and preparation
We extracted genotypes from the ancient genomes by drawing a random sequence read at each position, ignoring the first and last 3 bp of every read and any read containing insertions or deletions in their alignment to the human reference genome. If the randomly drawn haploid genotype of an ancient individual did not match either of the alleles of the biallelic SNP in the reference panel, we set the genotype of the ancient individual as missing.
We added these pseudohaploid genotypes to 17 million dinucleotide transversion SNPs identified between present-day genomes from the Simons Genome Diversity Panel (which includes human-fixed differences to chimpanzee). We also added the ancient genotypes to 550 individuals from 56 African populations genotyped on the Affymetrix Human Origins array (Lazaridis et al., 2014; Patterson et al., 2012; Pickrell et al., 2012; Pickrell et al., 2014; Skoglund et al., 2015). To all these datasets we added diploid genotypes from two archaic human genomes – a Neanderthal and a Denisovan (Meyer et al., 2012; Prufer et al., 2014). The populations shown in Fig. S2 are individuals from the Affymetrix Human Origins array, when we in the text refer to Khomani_San, they are the individuals from the Simons Genome Diversity panel and so are not shown in the legend in Fig. S2.
QUANTIFICATION AND STATISTICAL ANALYSIS
Population genetic approaches that quantify shared genetic drift, such as f-statistics and admixture graph fitting, are maximally robust when ascertainment of SNPs are performed in an outgroup (Patterson et al., 2012; Wang and Nielsen, 2012), such that there is no bias in allele frequencies between the analyzed populations and the polymorphism that appeared by mutation in the ancestral population of all analyzed populations. Whereas the Human Origins Array comprises 13 different panels ascertained in modern humans (Patterson et al., 2012), none of these can be regarded as outgroup-ascertained for the purpose of African populations. To obtain an outgroup-ascertained set of SNPs for African populations, we identified 814,242 transversion SNPs polymorphic between the archaic Denisovan (Meyer et al., 2012) and Neanderthal (Prufer et al., 2014) genomes (together labeled as ‘Archaic’ here). Since the ancestors of Denisovans and Neanderthals are consistent with having diverged from sub-Saharan lineages before those lineages separated from each other (Green et al., 2010; Mallick et al., 2016; Meyer et al., 2012; Prufer et al., 2014; Reich et al., 2010), the ascertained SNPs that were also present as polymorphisms in sub-Saharan Africa were highly likely to have been polymorphic before the African populations diversified. We extracted these positions from the 1000 genomes project MSL (Mende from Sierra Leone; 81 unrelated individuals), and YRI (Yoruba from Ibadan, Nigeria; 107 unrelated individuals), to increase power. These 1000 genomes project sequences were processed by sampling a random sequence at each position as for the ancient data, setting the genotype as missing if it did not match either of the two alleles in the ascertained SNP set.
Principal component analysis and ADMIXTURE clustering analyses
We used smartpca (Patterson et al., 2006) to compute principal components using all transversion and transitions SNPs, and the present-day populations shown in Fig. 1 and Fig. S2. We projected the ancient individuals the option lsqproject: YES, on eigenvectors computed using the present-day populations. To deal with the confounder factor of recent admixture with western Eurasian-related populations on the PCA, we removed all northern Africans, eastern African Cushitic speakers, Nama, and 8 Khomani individuals that had 5% or more cluster membership in the shared with Europeans in an ADMIXTURE analysis.
For our main ADMIXTURE clustering analysis (Alexander et al., 2009) (Fig. 1B; Fig. S3) we excluded 166,439 SNPs that were in a CpG context and thus retain postmortem damage, and used 431,134 SNPs and 208 selected ancient- and present-day individuals genotyped on the Human Origins Array. In Fig. 1B, we show only eight representative individuals for the non-African Japanese (originally n = 29), and Sardinian (n = 27) populations. For the authentication analysis investigating evidence of contamination, we used PMDtools (Skoglund et al., 2014a) to isolate sequences from each sample that had clear evidence of contamination according the postmortem damage score (PMD score > 3, using only based with phred-scaled quality of at least 30 to compute the score), and performed clustering analysis only on 111,208 transversion SNPs (Fig. S3). The exclusion of transition SNPs is due to the PMD score approach enriching for C > T and G > A substitutions indicative of ancient DNA.
Symmetry statistics and admixture tests
D-statistics, f4-statistics, and f3-statistics (Patterson et al., 2012; Reich et al., 2009) were computed with POPSTATS (Skoglund et al., 2015). f4-statistics test whether two pairs of populations are symmetric with respect to one another, and quantify any asymmetry arising from admixture. More specifically, if p1, p2, p3, and p4 are the derived allele frequencies at a biallelic SNP locus in population 1, population 2, population 3, and population 4, we can estimate f4 as a sum over all SNP loci f4 = Σ(p1−p2)(p3−p4) (Reich et al., 2009). D-statistics (Green et al., 2010), which are also used in this study, are a version of f4-statistics with a denominator to normalize for heterozygosity, but in practice both statistics have similar power to detect deviations from the null model, and f4-statistics have the additional advantage of being directly informative about admixture proportions and shared genetic drift (Patterson et al., 2012).
For the statistics in Fig. 3B and Fig. 3C, we used 814,242 transversion SNPs polymorphic between the archaic Denisovan (Meyer et al., 2012) and Neanderthal (Prufer et al., 2014) genomes (together labeled as ‘Archaic’ here). We extracted these loci from the 1000 genomes project MSL (Mende from Sierra Leone; 81 unrelated individuals), and YRI (Yoruba from Ibadan, Nigeria; 107 unrelated individuals). The statistics in Fig. S2C used either complete genome sequences from the Simons Genome Diversity Panel (Mallick et al., 2016), or panel 5 of the Human Origins Array, which comprises 119,413 SNPs that were originally ascertained as polymorphic positions in a single Yoruba individual. We used this set to test whether the ancient individuals were closer to one of two San groups because some of the SNPs on the Human Origins array were ascertained in one of the San groups, potentially affecting the statistic. In Table S5 we report multiple D-statistics for different configurations of populations using transversion SNPs in complete genomes from the Simons Genome Diversity Project and ancient shotgun sequences.
Y-chromosomal and mitochondrial haplogroups
For Y-chromosome haplogroup calling, we filtered reads with mapping quality < 30 and bases with base quality < 30, and determined the most derived mutation for each sample using the tree of the International Society of Genetic Genealogy (http://www.isogg.org) version 11.110 (21 April 2016). We also used Yfitter (Jostins et al., 2014) to confirm the haplogroups of the male Faraoskop and St. Helena Bay individuals using the entire shotgun sequence data, with identical haplogroup calls as the other approach.
For mitochondrial DNA haplogroups, we used Haplogrep2 (Weissensteiner et al., 2016) with Phylotree 17 (Van Oven and Kayser, 2009), restricting to sites with base quality 10, and depth 1. These relatively permissive thresholds were used to maximize coverage on the mitogenome. For sample I2966, which was found to have mitochondrial contamination, we first restricted to damaged reads using a PMD score threshold of 3 (Skoglund et al., 2014a).
Ancestry model and estimates with qpAdm
Clustering analyses and PCA are sensitive to genetic drift, such as the genetic drift that occurs in a population after the time ancient individuals lived (Skoglund et al., 2014b), and may thus not provide an accurate view of shared ancestry between ancient and present-day individuals. We employed a framework for estimating ancestry proportions that is based on f4-symmetry statistics, taking advantage of the fact that f4-statistics are proportional to admixture proportions and genetic drift. In the well-documented case of Neanderthal admixture into non-African populations, for example, the statistic f4(chimpanzee, Neanderthal; African, non-African) is proportional to αx, where α is the proportion of Neanderthal-related ancestry (approximately 2%) and x is proportional to the amount of genetic drift that occurred from the divergence of Neanderthal ancestors and African ancestors, to the divergence of the sampled Neanderthal genome and the Neanderthal population that admixed with non-Africans. By analyzing many such f4-statistics, (Lazaridis et al., 2014) and (Haak et al., 2015) showed that it is possible to estimate admixture proportions for a target population without detailed assumptions about population phylogeny, and also to perform hypothesis tests for whether a particular mixture model fits the data (Reich et al., 2012) and to estimate standard errors for admixture proportions with a weighted block jackknife procedure over large segments over the genome (in this study 5 cM). This has been implemented as the qpAdm algorithm in the ADMIXTOOLS package and requires the proposal of a set of source populations as well as a set of outgroups that are proposed to not share drift with the target population more recently than the source populations. In other words, appropriate source populations do not need to be the true source populations but instead, need only be more closely related to the true source populations than they are to any of the outgroups. Violations of these assumptions can be detected as an increase in rank in the matrix of f4-statistics computed (Reich et al., 2012). We also analyze a statistic using fitted allele frequencies predicted using the estimated mixture proportions, f4(Target population, Fitted Target population; Mbuti, Test). Deviations of this statistic from 0 are informative about whether some outgroups have an excess, or deficiency, of shared drift with the Target population under the fitted mixture proportions.
Here, we used a model with 19 populations (Mbuti, Dinka, Mende, South_Africa_2000BP, Tanzania_Luxmanda_3100BP, Ethiopia_4500BP, Levant_Neolithic (PPNB), Anatolia_Neolithic, Iran_Neolithic, Denisova, Loschbour, Ust_Ishim, Georgian, Iranian, Greek, Punjabi, Orcadian, Ami, and Mixe), using previously published complete genomes (Fu et al., 2014; Lazaridis et al., 2014; Mallick et al., 2016; Meyer et al., 2012) and ancient DNA data enriched using the 1240 k SNP set (Lazaridis et al., 2016; Mathieson et al., 2015) to maximize the power to infer admixture proportions for the ancient African populations. These populations, and in particular the ones from Africa, were chosen to capture major strands of ancestry and extremes in population differentiation found in sub-Saharan Africa (Fig. 1)
We then successively moved a set of candidate source populations (Mende, Dinka, Mbuti, South_Africa_2000BP, Tanzania_Luxmanda_3100BP, Ethiopia_4500BP, PPNB Anatolia_Neolithic, Iran_Neolithic) from the outgroup set to test if they fit as sources in admixture models. Using these 9 candidate sources populations, for each target population we thus tested 9 one-source ancestry models, two-source admixture models, and three source admixture models, for a total of 9 + 36 + 84 = 129 models. In Fig. S2 and Table S3, we show admixture proportions for the model with the lowest chi-square score (or highest p-value), if that model had a p-value > 0.01. If a one-source model did not fulfill this criterion, we considered two-source models, and then subsequently three-source models if no two-source model fulfilled the criteria.
We successfully obtained mixture models for 55 Target populations, comprising both ancient populations (we excluded Malawi_Chencherere_5200BP due to low SNP coverage) and populations genotyped on the Affymetrix Human Origins array, all shown in Table S3. In one analysis, Tanzania_Luxmanda_3100BP was also used as a target population, and in these analyses it was dropped from the outgroup set. We highlight some notable mixture models inferred here:
Kenya_400BP, Tanzania_Pemba_1400BP and Hadza1 are all fitted as having ~100% Ethiopia_4500BP-related ancestry. The other group of Hadza samples are fitted as having 19% ± 8% Dinka-related ancestry (the remainder being Ethiopia_4500BP-related).
Tanzania_Pemba_600BP, Malawi_Chewa, Malawi_Ngoni, Malawi_Tumbuka, Malawi_Yao, Yoruba, Esan, Gambian, Luo, BantuKenya, BantuSA_Ovambo, Himba, Wambo, BantuSA_Herero are all fitted as consistent with having ~100% Mende-related western African-related ancestry. The Mandenka, from the western African coast, are fitted as having 2.8% ± 0.6% Levant Neolithic-related ancestry (PPNB).
The Luhya, an eastern Bantu-speaking group, are fitted as having 40% ± 6% Dinka-related ancestry, with the remainder being western African Mende-related ancestry.
The Biaka, a western rainforest hunter-gatherer Pygmy group in Cameroon, is fitted as having 72% ± 2% Mbuti-related ancestry (the Mbuti are an eastern rainforest hunter-gatherer Pygmy group), with the remainder being western African Mende-related ancestry.
The minimum indigenous southern African ancestry observed in Khoe-groups and Bantu-speakers in southern Africa is ~8% ± 2% in the Damara, and the remainder is western African-related.
The maximum indigenous southern African ancestry observed in the present-day populations is the 91% ± 1% inferred for the Ju_hoan_North, with the remainder being related to Tanzania_Luxmanda_3100BP.
Some populations in northern and eastern Africa are fitted as having large proportions of Tanzania_Luxmanda_3100BP related ancestry. This includes the Maasai (49% ± 2%) and Datog (66% ± 3%) who have ancestry also related to the Dinka; the Kikuyu (63% ± 2%) who also have ancestry related to the Mende; and finally, the Afar (79% ± 3%) and Somali (62% ± 6%) who have large amounts of inferred Tanzania_Luxmanda_3100BP-related ancestry in addition to ancestry related to the Iran Neolithic.
Maximum likelihood tree model
We used the four ancient African shotgun genomes together with complete genomes from African populations in the Simons Genome Diversity project (Mallick et al., 2016), excluding populations with evidence of asymmetrical allele sharing with non-Africans indicative of gene flow (Table S5), to reconstruct a maximum likelihood tree using Treemix v1.12 (Pickrell and Pritchard, 2012). We performed 100 bootstrap replicates to assess the uncertainty of the fitted model. While this tree is not an adequate representation of human population history in Africa, we found 100% bootstrap support for the Ethiopian_4500BP Mota being most closely related to the ancestral population of all non-Africans (Fig. 3A).
Testing a tree-like model of African population history
The maximum-likelihood tree based on allele frequency covariance between the ancient African genomes and complete genomes from the SGDP panel (Fig. 3A) recapitulates many aspects of previous analyses of African populations (Pickrell et al., 2012; Schlebusch et al., 2012). When African populations are forced into a tree (not allowing for mixture), southern Africans diverge first, followed by Pygmies (e.g. Mbuti), West Africans, ancient and present-day eastern Africans (Dinka, Hadza), then non-Africans.
To scrutinize this tree-like model in more detail, we computed all 35 D-statistics that include the outgroup and have an expected value of 0 if a tree-like model (chimpanzee, (South_Africa_2000BP, (Mbuti, (Mende, (Dinka, Ethiopia_4500BP))))) is true. We find that most statistics computed are inconsistent with the null model (Fig. S4A). Notably, we reject the hypothesis that the ancient South Africans are an outgroup to other African populations for several pairs of present-day populations (Table S5; Fig. S4A). For instance, with genome sequence data we find that the Ethiopian_4500BP Mota genome shares more derived alleles with the ancient South Africans than with present-day western Africans (D[chimpanzee, South_Africa_2000BP; Yoruba, Ethiopia_4500BP] > 0). This is also true when West Africans are compared to Mbuti (D[chimpanzee, South_Africa_2000BP; Yoruba, Mbuti] > 0). Similar excess of shared derived alleles is observed for the eastern Pygmies (Mbuti) compared to western Pygmies, and even when contrasting western African populations such as the Yoruba and Mende (Fig. 3B; Fig. 3C; Table S5; Fig. S4). This could be explained in two ways: 1) there has been gene flow between ancient southern Africans and a broad set of other populations that has resulted in a gradient of southern Africans relatedness, or 2) there is a gradient of ancestry in western Africans that is basal to southern Africans, causing an attraction to the outgroup (chimpanzee in this case) (see below).
Testing admixture graph models of African population history
To reconstruct admixture graph models relating the histories of African populations, we used South_Africa_2000BP (South Africa), Mende (MSL; West Africa), and Ethiopia_4500BP (East Africa) to represent major lineages contributing to present-day Africans. In addition, we sought to explain one of the most surprising observations in our data, that the Mende and Yoruba West African populations are not symmetrically related to South_Africa_2000BP, and so we also included the Yoruba (YRI) in these analyses. For all admixture graph analyses, we used 814,242 transversion SNPs polymorphic between the archaic Denisovan (Meyer et al., 2012) and Neanderthal (Prufer et al., 2014) genomes (together labeled as ‘Archaic’ here).
A tree-like model does not fit the data
We first tested a strict tree-like model with no admixture edges, hypothesizing that the topology obtained from basic tree reconstructions where southern Africans are the earliest diverging lineage, and Yoruba and Mende are a clade to the exclusion of the Ethiopia_4500BP, is true (Fig. S4B). We found that this model is strongly rejected by the data with 19 predicted f4-statistics deviating from the empirically observed data by |Z| > 3. The most deviating f4-statistic was f4 (Archaic, Ethiopia_4500BP; MSL, YRI), which is predicted to be 0 in the tree-like model but is empirically observed to be f4 = 0.000427, Z = 9.157. Insight into the imperfect fit can be obtained by inspecting all four significant f4-statistics that are predicted to be zero in the model:
f4(Archaic, Ethiopia_4500BP; MSL, YRI) = 0.000427, Z = 9.157
f4(Archaic, South_Africa_2000BP; MSL, YRI) = 0.000193, Z = 4.742
f4(South_Africa_2000BP, Ethiopia_4500BP; MSL, YRI) = 0.000235, Z = 4.583
f4(Archaic, South_Africa_2000BP; Ethiopia_4500BP, MSL) = −0.000728, Z = −3.068
The three most significant deviations all test the (MSL, YRI) clade. The deviations thus indicate shared history either between MSL and the outgroup Archaics, or between YRI and South_Africa_2000BP/Ethiopia_4500BP. These observations could be parsimoniously explained by any of the following gene flow events
Gene flow from a basal human lineage (separating from the ancestors of all sub-Saharan Africans before their separation from each other) into the ancestors of MSL to a greater extent than YRI (there is no evidence of specifically Neanderthal/Denisovan gene flow since f4(chimpanzee, Archaics; MSL, YRI) = −1.6 in this data)
Gene flow related to YRI into the ancestors of Ethiopia_4500BP and South_Africa_2000BP
Gene flow related to Ethiopia_4500BP into the ancestors of YRI more than MSL
Gene flow most closely related to MSL into the common ancestors of the archaic Neanderthals and Denisovans (we exclude this as implausible)
The fourth deviating statistic f4(Archaic, South_Africa_2000BP; Ethiopia_4500BP, MSL) (Z = 3.068) suggests that Ethiopia_4500BP and MSL are not a clade with respect to South_Africa_2000BP. This weakens the case for gene flow into Yoruba alone as a sufficient explanation, since the Yoruba do not enter into this statistic. Instead, either excess basal ancestry in the Mende and gene flow between South_Africa_2000BP and Ethiopia_4500BP could explain this particular statistic.
Admixture models with gene flow events
We proceeded by testing models with one gene flow event positing that YRI have mixture related to either South_Africa_2000BP or Ethiopia_4500BP, or that MSL have ancestry from a basal lineage (Fig. S4C; Fig. S4D; Fig. S4E). We found that neither of these fit the data, with between 4 and 13 f4-statistic outliers with |Z| > 3.
Testing admixture graphs with two admixture events, we found that a model where both YRI and MSL have ancestry from a basal African lineage had its single outlier in an f4 statistic f4(Archaic, Ethiopia_4500BP; Ethiopia_4500BP, YRI) that is more negative in the data than the model (Fig. S4F). This f4-statistic has Ethiopia_4500BP appearing twice, and can thus be rearranged to be an f3 statistic f3(Archaic, YRI; Ethiopia_4500BP) that is not positive enough (Z = 3.157), and can thus be interpreted as the model underrepresenting the external drift in the Ethiopia_4500BP genome. We do not consider this outlier to compromise the model, as the processing of the Ethiopia_4500BP (Mota) genome that we use is pseudo-haploid (single random sequence read), and thus there is no real information about the external drift of the Mota lineage. This outlier may thus reflect a difficulty in modeling external drift for pseudo-haploid samples.
In addition, we found that an admixture graph where both Ethiopia_4500BP and YRI are mixed between MSL- and South_Africa_2000BP lineages does not fit the data (Fig. S4G), with 3 outliers, and the worst being Z = −4.835. However, a model where the YRI has ~2% ancestry from a population that is mixed between South_Africa_2000BP and Ethiopia_4500BP fits the data with 2 outliers that are not too surprising after correcting for multiple hypothesis testing (Fig. S4H). These outliers are f4(Archaic, South_Africa_2000BP; Ethiopia_4500BP, MSL) (Z = 3.068) which was also significant for the tree model without admixture, and f4(Archaic, South_Africa_2000BP; Ethiopia_4500BP, YRI) (Z = 3.018). Both these outliers could be consistent with the presence of basal African ancestry in YRI and MSL, or unmodeled gene flow between the ancestors of South_Africa_2000BP and Ethiopia_4500BP.
We thereby conclude that the most parsimonious admixture graph models identified here posit either the presence of basal African ancestry in Mende and Yoruba (Fig. 3D; Fig. S4F), or alternatively admixture from a source related to both South_Africa_2000BP and Ethiopia_4500BP in the Yoruba but not the Mende (with evidence also for gene flow between South_Africa_2000BP and Ethiopia) (Fig. 3F; Fig. S4G).
Automated grafting of populations onto a skeleton admixture graph
Using the admixture graph model in which the Yoruba and Mende both carry ancestry from a basal western African population, we automatically added additional populations to each possible node in the graph. We evaluated the fit in terms of the number and deviation of outlying f4-statistics, as well as whether the added branches had zero drift length. We show the results of this procedure in Table S6, with the key to the node labels used shown in Fig. S5A. We highlight topologies that we consider optimal fits in Fig. S5B–5D.
We find that the Malawi_Hora_8100BP can be best fitted as mixed between the lineage leading to South_Africa_2000BP, and the lineage related to Ethiopia_4500BP that also is fitted as forming part of the ancestry of YRI and MSL (Fig. S5B). Similarly, the Mbuti can be best fitted as mixed between the lineage related to Ethiopia_4500BP that also is fitted as forming part of the ancestry of YRI and MSL, and secondly a lineage that diverged prior to South_Africa_2000BPbut after the basal West African lineage (Fig. S5C). Non-Africans (Japanese) are fitted as having part Archaic ancestry (Green et al., 2010), with the remainder of their ancestry again being derived from the lineage related to Ethiopia_4500BP that also is fitted as forming part of the ancestry of YRI and MSL (Fig. S5D). This analysis is consistent with the possibility that the same human lineage contributed ancestry both to the source of non-Africans and many African populations today.
Support for a single out-of-Africa founding population
Simple tree models suggest that non-African variation represented by Sardinian, English, Han Chinese and Japanese falls within the variation of African populations. To test whether non-Africans are indeed consistent with being descended from a homogeneous population that separated earlier from the ancestors of a subset of African populations – beyond the known effects of archaic admixture in non-Africans – we used African populations with little or no known West Eurasian mixture (South_Africa_2000BP, Mbuti, Biaka, Mende, Ethiopia_4500BP, Dinka) and tested whether they are consistent with being an unrooted clade with respect to a diverse set of non-Africans (Orcadian, Onge, Mixe, Motala_Mesolithic, Japanese, Anatolia_Neolithic) using qpWave (Patterson et al., 2012; Reich et al., 2012). We found that this model was consistent with the data (P = 0.53) (transition SNPs excluded to a final set of 110,507 transversion SNPs). Even when we add New Guinean highlanders to the set of non-Africans, the single-source model for the out-of-Africa founders is not rejected (P = 0.11).
Date of admixture between expanding agriculturalists and previously established foragers
We estimated the date of admixture between expanding agriculturalists related to western Africans (probably Bantu-speakers) and the previously established foragers using the ALDER software (Loh et al., 2012), which uses the rate of decay per generation of the linkage disequilibrium that is created in admixed populations and that can be detected using signed linkage disequilibrium weighted by allele frequency differences between populations taken as proxies for the ancestral populations (Moorjani et al., 2011). To maximize statistical power, we used the full 1240 k data merged with 1000 genomes phase 3 genotype data. We then estimated weighted linkage disequilibrium in 99 unrelated LWK individuals (Luhya from Webuye, Kenya) a Bantu-speaking group, using 85 unrelated West African MSL individuals (Mende from Sierra Leone) and 4 ancient eastern African hunter-gatherer individuals (Ethiopia_4500BP, Tanzania_Pemba_1400BP, Tanzania_Zanzibar_1400BP, Kenya_400BP). The analysis used a total of 1,070,197 SNPs. We obtained significant evidence for one-reference weighted LD decay for both putative source populations (Z ~ 6), as well as for the two-reference weighted LD decay (Z = 4.89, P = 10−5). The estimated date of mixture was 16.8 generations ago, with a standard error of 3.4. Assuming a generation time of 30 years, this suggests that admixture occurred on average 380 to 760 years ago (95% confidence interval). We note that a previous study (Pickrell et al., 2014) did not obtain high-confidence support for West African related admixture in the Luhya, and we hypothesize that our clear demonstration of this is due to both the availability of a more accurate ancestral population in the form of the ancient eastern Africans, as well as to the increased leverage from the large samples size 1000 Genomes data.
Evidence for selective sweeps in the ancestry of present-day San
We performed a genome-wide scan for large genomic regions with excessive allele frequency between present-day San (Khomani and Ju_hoan_North) and the two ancient South_Africa_2000BP, with the Mbuti as a second outgroup. Previous statistics such as the Locus-Specific Branch length (LSBL) (Shriver et al., 2004) and the derivative Population Branch Statistic (PBS) (Yi et al., 2010) also estimate a branch length in a three-population phylogeny, but use FST as their base, which can be undefined when used for loci with fixed differences. Thus, we computed the statistic f3 = (pSan−pSouth_Africa_2000BP)(pSan−pMbuti) for windows of 500 kb, separated by 10 kb. We only retained windows with at least 50 SNPs, resulting in a total of l = 262,047 autosomal loci. We approximated the neutral genome-wide average μf and its standard deviation σf by subsampling 546 of the windows, requiring that these were separated by at least 5 million base pairs (Mb) and thus approximately independent. We then standardized the distribution of the test statistic fw in each window as Z(fw) = (fw − μf)/σf. We show the most deviating windows in Table 2.
Evidence for polygenic selection
Investigating evidence for polygenic selection in African populations is complicated by the fact that most information on the genomic basis of human phenotypic variation has been based on analysis of highly differentiated populations such as Europeans. In the absence of rich phenotypic information about sub-Saharan African populations, we used gene ontology (GO) information, which draws on information from a wide array of studies on humans and nonhuman model organisms. We focused on 208 GO categories that contained at least 50 genes each, which allows us to compute genome-wide block jackknife standard errors using the entire genic regions. We focus on f3-statistics that measure the length of one of the branches in a hypothetical three-way tree-like population history (assuming no admixture). The three populations we focused on were the ancient South_Africa_2000BP (n = 2 pseudohaploid draft genomes), the present-day San (n = 6 complete genomes), and the Mbuti (n = 4 complete genomes). The availability of the two ancient South_Africa_2000BP genomes can in principle inform us about selection in the last ~2000 years since these individuals lived, or starting further back in time in case they are not from a direct ancestral population of the present-day San (our data suggest that they are more closely related to the Khomani San than to the Ju’/hoan). We thus computed the statistic f3(Mbuti, South_Africa_2000BP; San) which is proportional to allele frequency differentiation in the present-day San compared to the other two populations.
We find that the “RESPONSE_TO_RADIATION” GO category is an outlier that shows the greatest degree of differentiation in this analysis. However, this could also be due to genes in this category constantly being under rapid evolution or having other differences compared to other categories. To test this, we computed the statistic f3(San, South_Africa_2000BP; Mbuti), and found no strong signal in the response to radiation category. Instead, the category with the strongest evidence of differentiation in the Mbuti lineage since the divergence from the San groups is “REGULATION_OF_GROWTH”, suggesting the possibility of relatively recent evolution of the shorter stature of present-day rainforest hunter-gatherer populations.
DATA AVAILABILITY
Raw sequence data (bam files) from the 15 newly reported ancient individuals is available from the European Nucleotide Archive under accession no. PRJEB21878. The newly reported SNP genotyping data is available to researchers who send a signed letter to D.R. containing the following text: “(a) I will not distribute the data outside my collaboration; (b) I will not post the data publicly; (c) I will make no attempt to connect the genetic data to personal identifiers for the samples; (d) I will use the data only for studies of population history; (e) I will not use the data for any selection studies; (f) I will not use the data for medical or disease-related analyses; (g) I will not use the data for commercial purposes.”
Supplementary Material
Figure S1. Archaeological context and dating of newly reported human remains from Malawi. Related to Figure 1. A) Locations of the three sites in Malawi. B) Plan of the Hora rock shelter showing locations of the original Hora 1 (male) and Hora 2 (female) burials relative to new excavations from 2016. C) Plan of Fingira Cave showing locations of human remains recovered in 1966 (green dots) and 2016 (green stars). D) Plan of Chencherere II rock shelter redrawn from Crader (1984), showing approximate locations where human remains were recovered in 1972 (red and green stars), and the approximate locations of the remains sampled from Nguludi for this study (blue stars). E) Ultrafiltered collagen FTIR spectra for three individuals from Malawi (MW2 = UCIAMS-186346, MW3 = UCIAMS-18647, MW4 = UCIAMS-186348) compared to well-preserved standards and samples (Modern Carabou, BU4, RP01, POR1). These samples show the typical features for well-preserved collagen. The top of the figure shows the FTIR spectra for a single XAD purified sample (MW1 = PSUAMS-1734) compared against a well preserved standard (Beauford Whale). These data, along with the stable isotopes and C:N ratios, indicate that these samples are well preserved and that the AMS 14C dates are reliable.
Figure S2. Ancient individuals and African population structure. Related to Figure 1. A) full legend and PC1-PC2 scatter plot (same analysis as Figure 1) with all present-day populations indicated with separate symbols. B) PC1-PC2 computed only with southern African populations (and west African Yoruba). C) Symmetry statistics confirming that ancient individuals from the western Cape region in South Africa are more closely related to southern San such as the Khomani rather than more northern San populations such as the Ju_hoan_North.
Figure S3. ADMIXTURE clustering analysis of K = 2 to K = 7 clusters. Related to Figure 1. Each bar shows inferred cluster membership in each individual for a given number of clusters. This analysis used 431,134 SNPs that were not in a CpG dinucleotide context. We also show an inset with cluster membership inferred for damage-restricted ancient DNA sequences (PMD-score > 3) in a parallel run with only 111,208 transversion SNPs.
Figure S4. Admixture models of African population history. Related to Figure 3. A) A tree-like model. Tests of all possible triplet topologies predicted to be symmetric by the tree obtained for major African population lineages in the maximum likelihood model in Fig. 3A. B–H) Under each graph we show the outlier f4 statistic with the greatest Z-score between model and empirical values. When the same group appears twice in an f4 statistic (e.g. f4(Archaic, South_Africa_HGs; South_Africa_HGs, YRI), this corresponds to an f3 statistic. Graphs F and H are the most parsimonious fits with only two admixture events.
Figure S5. Automatically adding additional populations to a skeleton graph. Related to Figure 3. A) Skeleton graph invoking widespread admixture from a hypothesized ancestral northeast African population. B–D) Best fitting insertion points for Malawi_Hora_8100BP shotgun sequence data (B), Mbuti (C), and Japanese non-Africans (D), in the skeleton graph displayed in (A). For details see Table S6.
Table S1. Related to Table 1. Details of sequence libraries prepared from ancient samples.
Table S2. Related to Table 1. Details of direct AMS radiocarbon bone dates.
Table S3. Related to Figure 2. Details of ancestry proportions inferred using qpAdm.
Table S4. Related to Table 1. Y-chromosome haplogroup informative polymorphisms inferred for male individuals with sufficient data.
Table S5. Related to Figure 1. Z-scores (computed for D-statistics) in complete genomes from present-day and ancient Africans.
Table S6. Related to Figure 3. Results of systematically inserting additional populations into all possible edges of the skeleton graph in Fig. S6A.
Table S7. Related to Table 1. MtDNA haplogroup informative polymorphisms.
Genome-wide analysis of 16 African individuals who lived up to 8,100 years ago
Forager populations related to southern African San once widespread in eastern Africa
Comparison of ancient and modern Africans reveal recent genomic adaptations
Evidence for a divergent human lineage contributing to west Africans
Acknowledgments
Permission to analyze the remains from Kenya and Tanzania was granted by the National Museums of Kenya; the Antiquities Division of the Ministry of Natural Resources and Tourism, Tanzania; and the Zanzibar Department of Museums and Antiquities. We thank I. Lazaridis, M. Lipson, I. Mathieson and S. Tishkoff for discussions, and I. Kucukkalipci and K. Majander for laboratory support. P.S. was supported by the Wenner-Gren Foundation and the Swedish Research Council (VR grant 2014-453). J.K. and A.M. were supported by the DFG grant KR 4015/1-1 and the Max Planck Society. K.Si. was supported by NSF grant BCS-1613577. M.H., A.H., M.M. and S.P. were supported by the Max Planck Society. A.G.M and J.P. are supported by the National Research Foundation of South Africa. R.R was supported by the South African Medical Research Council. N.B. was supported by ERC starting grant SEALINKS (206148), and R.P was supported by ERC starting grant ADNABIOARC (263441). M.G.T. was supported by Wellcome Trust Senior Investigator Award (grant number 100719/Z/12/Z). D.R. was supported by NIH grant GM100233, by NSF HOMINID BCS-1032255, and is a Howard Hughes Medical Institute investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions
Conceptualization, P.S., J.C.T., M.E.P., V.M.H., R.R., A.G.M., N.B., R.P., J.K., and D.R.; Formal Analysis, P.S., S.M., A.P., and I.O.; Investigation, P.S., A.M., K.S., M.H., T.S., N.R., A.H., M.F., E.H., M.M., K.S., J.C.-R.; Resources, J.C.T., M.E.P., C.C., A.C., E.G.-C., A.O.G., K.M.G., I.T.H., G.H., R.H., M.H., S.L., A.Z.P.M., J.P., C.S., M.G.T., R.T., M.W., V.M.H., A.G.M., N.B.; Data Curation, P.S., M.H., N.R., S.M., A.P., I.O., M.F., E.H., M.M., K.S., D.J.K. and D.R.; Writing, P.S. and D.R.; Supervision, V.H., M.M., S.P., N.N., N.B., R.P., J.K. and D.R.
References
- Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Research. 2009;19:1655–1664. doi: 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behar D, Van Oven M, Rosset S, Metspalu M, Loogvali EL, Silva N, Kivisild T, Torroni A, Villems R. A “Copernican” reassessment of the human mitochondrial DNA tree from its root. Am J Hum Genet. 2012;90:675–684. doi: 10.1016/j.ajhg.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs AW, Heyn P. Preparation of next-generation sequencing libraries from damaged DNA. Ancient DNA: Methods and Protocols. 2012:143–154. doi: 10.1007/978-1-61779-516-9_18. [DOI] [PubMed] [Google Scholar]
- Briggs AW, Stenzel U, Meyer M, Krause J, Kircher M, Pääbo S. Removal of deaminated cytosines and detection of in vivo methylation in ancient DNA. Nucleic acids research. 2010;38:e87–e87. doi: 10.1093/nar/gkp1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronk Ramsey C. Bayesian analysis of radiocarbon dates. Radiocarbon. 2009;51:337–360. [Google Scholar]
- Brown TA, Nelson DE, Vogel JS, Southon JR. Improved collagen extraction by modified Longin method. Radiocarbon. 1988;30:171–177. [Google Scholar]
- Busby GBJ, Band G, Si Le Q, Jallow M, Bougama E, Mangano VD, Amenga-Etego LN, Enimil A, Apinjoh T, Ndila CM, et al. Admixture into and within sub-Saharan Africa. eLife. 2016;5:e15266. doi: 10.7554/eLife.15266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell MC, Ranciaro A, Froment A, Hirbo J, Omar S, Bodo JM, Nyambo T, Lema G, Zinshteyn D, Drayna D. Evolution of functionally diverse alleles associated with PTC bitter taste sensitivity in Africa. Molecular biology and evolution. 2011 doi: 10.1093/molbev/msr293. msr293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cann RL, Stoneking M, Wilson AC. Mitochondrial DNA and human evolution. Nature. 1987;325:31–36. doi: 10.1038/325031a0. [DOI] [PubMed] [Google Scholar]
- Chami F. Zanzibar and the Swahili coast from c 30,000 years ago. E&D Vision Pub; 2009. [Google Scholar]
- Chami F, Khator J, Hamis Ali A. The excavation of Mapangani cave, Pemba island, Zanzibar. Studies in the African Past. 2009;9:74–101. [Google Scholar]
- Clark J. Prehistoric origins. The Early History of Malawi Longman; London: 1972. pp. 17–27. [Google Scholar]
- Clark JD. Prehistory in Nyasaland. The Nyasaland Journal. 1956;9:92–119. [Google Scholar]
- Clark JD. The prehistory of southern Africa. Penguin Books; 1959. [Google Scholar]
- Clark JD, Clerk JD. ARCHAEOLOGICAL INVESTIGATION OF A PAINTED ROCK SHELTER AT MWANA WA CHENCHERERE, NORTH OF DEDZA, CENTRAL MALAŴI In July to September, 1972. The Society of Malawi Journal. 1973;26:28–46. [Google Scholar]
- Cole-King P. Kukumba Mbiri Mu Malaŵi: A Summary of Archaeological Research to March 1973. Government Press; 1973. [Google Scholar]
- Coop G, Pickrell JK, Novembre J, Kudaravalli S, Li J, Absher D, Myers RM, Cavalli-Sforza LL, Feldman MW, Pritchard JK. The role of geography in human adaptation. PLoS genetics. 2009;5:e1000500. doi: 10.1371/journal.pgen.1000500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crader DC. Faunal remains from Chencherere II rock shelter, Malawi. The South African Archaeological Bulletin. 1984:37–52. [Google Scholar]
- Crowther A, Prendergast ME, Fuller DQ, Boivin N. Subsistence mosaics, forager-farmer interactions and the transition to food production in eastern Africa. Quaternary International 2017 [Google Scholar]
- Crowther A, Veall MA, Boivin N, Horton M, Kotarba-Morley A, Fuller DQ, Fenn T, Haji O, Matheson CD. Use of Zanzibar copal (Hymenaea verrucosa Gaertn.) as incense at Unguja Ukuu, Tanzania in the 7–8th century CE: chemical insights into trade and Indian Ocean interactions. Journal of Archaeological Science. 2015;53:374–390. [Google Scholar]
- Dabney J, Knapp M, Glocke I, Gansauge MT, Weihmann A, Nickel B, Valdiosera C, García N, Pääbo S, Arsuaga JL. Complete mitochondrial genome sequence of a Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. Proceedings of the National Academy of Sciences. 2013;110:15758–15763. doi: 10.1073/pnas.1314445110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabney J, Meyer M. Length and GC-biases during sequencing library amplification: a comparison of various polymerase-buffer systems with ancient and modern DNA sequencing libraries. Biotechniques. 2012;52:87–94. doi: 10.2144/000113809. [DOI] [PubMed] [Google Scholar]
- DeBusk GH. The distribution of pollen in the surface sediments of Lake Malawi, Africa, and the transport of pollen in large lakes. Review of Palaeobotany and Palynology. 1997;97:123–153. [Google Scholar]
- Fleisher J, LaViolette A. The early Swahili trade village of Tumbe, Pemba Island, Tanzania, AD 600–950. Antiquity. 2013;87:1151–1168. [Google Scholar]
- Fu Q, Hajdinjak M, Moldovan O, Constantin S, Mallick S, Skoglund P, Patterson N, Rohland N, Lazaridis I, Nickel B. An early modern human from Romania with a recent Neanderthal ancestor. Nature. 2015 doi: 10.1038/nature14558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Q, Li H, Moorjani P, Jay F, Slepchenko SM, Bondarev AA, Johnson PLF, Aximu-Petri A, Prufer K, de Filippo C, et al. Genome sequence of a 45,000-year-old modern human from western Siberia. Nature. 2014;514:445–449. doi: 10.1038/nature13810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gansauge MT, Meyer M. Single-stranded DNA library preparation for the sequencing of ancient or damaged DNA. Nature protocols. 2013;8:737–748. doi: 10.1038/nprot.2013.038. [DOI] [PubMed] [Google Scholar]
- Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, Patterson N, Li H, Zhai WW, Fritz MHY, et al. A Draft Sequence of the Neandertal Genome. Science. 2010;328:710–722. doi: 10.1126/science.1188021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronau I, Hubisz MJ, Gulko B, Danko CG, Siepel A. Bayesian inference of ancient human demography from individual genome sequences. Nature genetics. 2011;43:1031–1034. doi: 10.1038/ng.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurdasani D, Carstensen T, Tekola-Ayele F, Pagani L, Tachmazidou I, Hatzikotoulas K, Karthikeyan S, Iles L, Pollard MO, Choudhury A, et al. The African Genome Variation Project shapes medical genetics in Africa. Nature. 2015;517:327–332. doi: 10.1038/nature13997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haak W, Lazaridis I, Patterson N, Rohland N, Mallick S, Llamas B, Brandt G, Nordenfelt S, Harney E, Stewardson K. Massive migration from the steppe is a source for Indo-European languages in Europe. 2015 doi: 10.1038/nature14317. arXiv preprint arXiv:150202783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer MF, Woerner AE, Mendez FL, Watkins JC, Wall JD. Genetic evidence for archaic admixture in Africa. Proceedings of the National Academy of Sciences. 2011;108:15123–15128. doi: 10.1073/pnas.1109300108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helm R, Crowther A, Shipton C, Tengeza A, Fuller D, Boivin N. Exploring agriculture, interaction and trade on the eastern African littoral: preliminary results from Kenya. Azania: Archaeological Research in Africa. 2012;47:39–63. [Google Scholar]
- Helm RM. Conflicting histories: the archaeology of the iron-working, farming communities in the central and southern coast region of Kenya. University of Bristol; 2000. [Google Scholar]
- Henn BM, Gignoux CR, Jobin M, Granka JM, Macpherson JM, Kidd JM, Rodríguez-Botigué L, Ramachandran S, Hon L, Brisbin A, et al. Hunter-gatherer genomic diversity suggests a southern African origin for modern humans. Proceedings of the National Academy of Sciences. 2011;108:5154–5162. doi: 10.1073/pnas.1017511108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg AG, Hua Q, Blackwell PG, Niu M, Buck CE, Guilderson TP, Heaton TJ, Palmer JG, Reimer PJ, Reimer RW. SHCal13 Southern Hemisphere calibration, 0–50,000 years cal BP. Radiocarbon. 2013;55:1889–1903. [Google Scholar]
- Jostins L, Xu Y, McCarthy S, Ayub Q, Durbin R, Barrett J, Tyler-Smith C. YFitter: Maximum likelihood assignment of Y chromosome haplogroups from low-coverage sequence data. 2014 arXiv preprint arXiv:14077988. [Google Scholar]
- Juma A. Unguja Ukuu on Zanzibar: An archaeological study of early urbanism 2004 [Google Scholar]
- Kennett DJ, Plog S, George RJ, Culleton BJ, Watson AS, Skoglund P, Rohland N, Mallick S, Stewardson K, Kistler L, et al. Archaeogenomic evidence reveals prehistoric matrilineal dynasty. 2017;8:14115. doi: 10.1038/ncomms14115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M. Analysis of high-throughput ancient DNA sequencing data. In Ancient DNA (Springer) 2012:197–228. doi: 10.1007/978-1-61779-516-9_23. [DOI] [PubMed] [Google Scholar]
- Kircher M, Sawyer S, Meyer M. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic acids research. 2011 doi: 10.1093/nar/gkr771. gkr771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korlević P, Gerber T, Gansauge MT, Hajdinjak M, Nagel S, Ayinuer-Petri A, Meyer M. Reducing microbial and human contamination in DNA extractions from ancient bones and teeth. Bio Techniques. 2015;59:87–93. doi: 10.2144/000114320. [DOI] [PubMed] [Google Scholar]
- Lachance J, Vernot B, Elbers Clara C, Ferwerda B, Froment A, Bodo JM, Lema G, Fu W, Nyambo Thomas B, Rebbeck Timothy R, et al. Evolutionary History and Adaptation from High-Coverage Whole-Genome Sequences of Diverse African Hunter-Gatherers. Cell. 2012;150:457–469. doi: 10.1016/j.cell.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaridis I, Nadel D, Rollefson G, Merrett DC, Rohland N, Mallick S, Fernandes D, Novak M, Gamarra B, Sirak K, et al. Genomic insights into the origin of farming in the ancient Near East. Nature. 2016;536:419–424. doi: 10.1038/nature19310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaridis I, Patterson N, Mittnik A, Renaud G, Mallick S, Kirsanow K, Sudmant PH, Schraiber JG, Castellano S, Lipson M, et al. Ancient human genomes suggest three ancestral populations for present-day Europeans. Nature. 2014;513:409–413. doi: 10.1038/nature13673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorente MG, Jones ER, Eriksson A, Siska V, Arthur KW, Arthur JW, Curtis MC, Stock JT, Coltorti M, Pieruccini P, et al. Ancient Ethiopian genome reveals extensive Eurasian admixture in Eastern Africa. Science. 2015;350:820–822. doi: 10.1126/science.aad2879. [DOI] [PubMed] [Google Scholar]
- Loh PR, Lipson M, Patterson N, Moorjani P, Pickrell JK, Reich D, Berger B. Inference of admixture parameters in human populations using weighted linkage disequilibrium. 2012 doi: 10.1534/genetics.112.147330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse JC, Madsen DB, Culleton BJ, Kennett DJ. Isotope paleoecology of episodic mid-to-late Holocene bison population expansions in the Southern Plains, USA. Quaternary Science Reviews. 2014;102:14–26. [Google Scholar]
- Longin R. New method of collagen extraction for radiocarbon dating. Nature. 1971;230:241–242. doi: 10.1038/230241a0. [DOI] [PubMed] [Google Scholar]
- Mallick S, Li H, Lipson M, Mathieson I, Gymrek M, Racimo F, Zhao M, Chennagiri N, Nordenfelt S, Tandon A, et al. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature. 2016;538:201–206. doi: 10.1038/nature18964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manhire A. A report on the excavations at Faraoskop Rock Shelter in the Graafwater district of the south-western Cape. Southern African Field Archaeology. 1993;2:3–23. [Google Scholar]
- Maricic T, Whitten M, Pääbo S. Multiplexed DNA sequence capture of mitochondrial genomes using PCR products. PloS one. 2010;5:e14004. doi: 10.1371/journal.pone.0014004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall F, Stewart K, Barthelme J. Early domestic stock at Dongodien in northern Kenya. AZANIA: Journal of the British Institute in Eastern Africa. 1984;19:120–127. [Google Scholar]
- Mathieson I, Lazaridis I, Rohland N, Mallick S, Patterson N, Roodenberg SA, Harney E, Stewardson K, Fernandes D, Novak M, et al. Genome-wide patterns of selection in 230 ancient Eurasians. Nature. 2015;528:499–503. doi: 10.1038/nature16152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M, Fu Q, Aximu-Petri A, Glocke I, Nickel B, Arsuaga JL, Martínez I, Gracia A, de Castro JMB, Carbonell E. A mitochondrial genome sequence of a hominin from Sima de los Huesos. Nature. 2014;505:403–406. doi: 10.1038/nature12788. [DOI] [PubMed] [Google Scholar]
- Meyer M, Kircher M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harbor Protocols. 2010;2010 doi: 10.1101/pdb.prot5448. pdb. prot5448. [DOI] [PubMed] [Google Scholar]
- Meyer M, Kircher M, Gansauge MT, Li H, Racimo F, Mallick S, Schraiber JG, Jay F, Prüfer K, de Filippo C, et al. A High-Coverage Genome Sequence from an Archaic Denisovan Individual. Science. 2012;338:222–226. doi: 10.1126/science.1224344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SF. The age of Nachikufan industries in Zambia. The South African Archaeological Bulletin. 1971;26:143–146. [Google Scholar]
- Moorjani P, Patterson N, Hirschhorn JN, Keinan A, Hao L, Atzmon G, Burns E, Ostrer H, Price AL, Reich D. The history of African gene flow into Southern Europeans, Levantines, and Jews. PLoS genetics. 2011;7:e1001373. doi: 10.1371/journal.pgen.1001373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris AG. Isolation and the origin of the Khoisan: Late Pleistocene and Early Holocene human evolution at the southern end of Africa. Human Evolution. 2002;17:231–240. [Google Scholar]
- Morris AG, Heinze A, Chan EK, Smith AB, Hayes VM. First ancient mitochondrial human genome from a prepastoralist southern African. Genome biology and evolution. 2014;6:2647–2653. doi: 10.1093/gbe/evu202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris AG, Ribot I. Morphometric cranial identity of prehistoric Malawians in the light of sub Saharan African diversity. American journal of physical anthropology. 2006;130:10–25. doi: 10.1002/ajpa.20308. [DOI] [PubMed] [Google Scholar]
- Odner K. Excavations at Narosura, a Stone Bowl Site in the Southern Kenya Highlands. Azania: Archaeological Research in Africa. 1972;7:25–92. [Google Scholar]
- Pagani L, Kivisild T, Tarekegn A, Ekong R, Plaster C, Gallego Romero I, Ayub Q, Mehdi SQ, Thomas Mark G, Luiselli D, et al. Ethiopian Genetic Diversity Reveals Linguistic Stratification and Complex Influences on the Ethiopian Gene Pool. The American Journal of Human Genetics. 2012;91:83–96. doi: 10.1016/j.ajhg.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkington J, Dlamini N. First People: Ancestors of the San. Creda Communications 2015 [Google Scholar]
- Patin E, Lopez M, Grollemund R, Verdu P, Harmant C, Quach H, Laval G, Perry GH, Barreiro LB, Froment A, et al. Dispersals and genetic adaptation of Bantu-speaking populations in Africa and North America. Science. 2017;356:543. doi: 10.1126/science.aal1988. [DOI] [PubMed] [Google Scholar]
- Patterson N, Moorjani P, Luo Y, Mallick S, Rohland N, Zhan Y, Genschoreck T, Webster T, Reich D. Ancient admixture in human history. Genetics. 2012;192:1065–1093. doi: 10.1534/genetics.112.145037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS genetics. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltzer A, Jäger G, Herbig A, Seitz A, Kniep C, Krause J, Nieselt K. EAGER: efficient ancient genome reconstruction. Genome biology. 2016;17:1. doi: 10.1186/s13059-016-0918-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillipson DW. The Early Iron Age in eastern and southern Africa: a critical re-appraisal. AZANIA: Journal of the British Institute in Eastern Africa. 1976;11:1–23. [Google Scholar]
- Pickrell JK, Patterson N, Barbieri C, Berthold F, Gerlach L, Guldemann T, Kure B, Mpoloka SW, Nakagawa H, Naumann C, et al. The genetic prehistory of southern Africa. Nat Commun. 2012;3:1143. doi: 10.1038/ncomms2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, Patterson N, Loh PR, Lipson M, Berger B, Stoneking M, Pakendorf B, Reich D. Ancient west Eurasian ancestry in southern and eastern Africa. Proceedings of the National Academy of Sciences. 2014;111:2632–2637. doi: 10.1073/pnas.1313787111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, Pritchard JK. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS genetics. 2012;8:e1002967. doi: 10.1371/journal.pgen.1002967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plagnol V, Wall JD. Possible ancestral structure in human populations. PLoS Genet. 2006;2:e105. doi: 10.1371/journal.pgen.0020105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleurdeau D, Imalwa E, Détroit F, Lesur J, Veldman A, Bahain JJ, Marais E. “Of Sheep and Men”: Earliest Direct Evidence of Caprine Domestication in Southern Africa at Leopard Cave (Erongo, Namibia) PLOS ONE. 2012;7:e40340. doi: 10.1371/journal.pone.0040340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prendergast ME, Mabulla AZP, Grillo KM, Broderick LG, Seitsonen O, Gidna AO, Gifford-Gonzalez D. Pastoral Neolithic sites on the southern Mbulu Plateau, Tanzania. Azania: Archaeological Research in Africa. 2013;48:498–520. [Google Scholar]
- Prendergast ME, Rouby H, Punnwong P, Marchant R, Crowther A, Kourampas N, Shipton C, Walsh M, Lambeck K, Boivin NL. Continental island formation and the archaeology of defaunation on Zanzibar, eastern Africa. PloS one. 2016;11:e0149565. doi: 10.1371/journal.pone.0149565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prufer K, Racimo F, Patterson N, Jay F, Sankararaman S, Sawyer S, Heinze A, Renaud G, Sudmant PH, de Filippo C, et al. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature. 2014;505:43–49. doi: 10.1038/nature12886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran S, Deshpande O, Roseman CC, Rosenberg NA, Feldman MW, Cavalli-Sforza LL. Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15942–15947. doi: 10.1073/pnas.0507611102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich D, Green RE, Kircher M, Krause J, Patterson N, Durand EY, Viola B, Briggs AW, Stenzel U, Johnson PLF, et al. Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature. 2010;468:1053–1060. doi: 10.1038/nature09710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich D, Patterson N, Campbell D, Tandon A, Mazieres S, Ray N, Parra M, Rojas W, Duque C, Mesa N, et al. Reconstructing Native American population history. Nature. 2012;488:370–374. doi: 10.1038/nature11258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich D, Thangaraj K, Patterson N, Price AL, Singh L. Reconstructing Indian population history. Nature. 2009;461:489–494. doi: 10.1038/nature08365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimer PJ, Bard E, Bayliss A, Beck JW, Blackwell PG, Bronk Ramsey C, Buck CE, Cheng H, Edwards RL, Friedrich M, et al. IntCal13 and Marine13 Radiocarbon Age Calibration Curves 0–50,000 Years cal BP 2013 [Google Scholar]
- Renaud G, Kircher M, Stenzel U, Kelso J. freeIbis: an efficient basecaller with calibrated quality scores for Illumina sequencers. Bioinformatics. 2013 doi: 10.1093/bioinformatics/btt117. btt117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renaud G, Slon V, Duggan AT, Kelso J. Schmutzi: estimation of contamination and endogenous mitochondrial consensus calling for ancient DNA. Genome biology. 2015;16:1. doi: 10.1186/s13059-015-0776-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribot I, Morris AG, Sealy J, Maggs T. Population history and economic change in the last 2000 years in KwaZulu-Natal, RSA. Southern African Humanities. 2010;22:89–112. [Google Scholar]
- Robinson K, Sandelowsky B. The Iron Age of northern Malawi: recent work. AZANIA: Journal of the British Institute in Eastern Africa. 1968;3:107–146. [Google Scholar]
- Robinson KSR. Iron Age of northern Malaŵi: an archaeological reconnaissance. Malaŵi Govt. Ministry of Education and Culture; 1982. [Google Scholar]
- Rohland N, Harney E, Mallick S, Nordenfelt S, Reich D. Partial uracil–DNA–glycosylase treatment for screening of ancient DNA. Philosophical Transactions of the Royal Society of London B: Biological Sciences. 2015;370 doi: 10.1098/rstb.2013.0624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohland N, Hofreiter M. Comparison and optimization of ancient DNA extraction. Biotechniques. 2007;42:343–352. doi: 10.2144/000112383. [DOI] [PubMed] [Google Scholar]
- Russell T, Silva F, Steele J. Modelling the spread of farming in the Bantu-speaking regions of Africa: an archaeology-based phylogeography. PLoS One. 2014;9:e87854. doi: 10.1371/journal.pone.0087854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadr K. Livestock First Reached Southern Africa in Two Separate Events. PloS one. 2015;10:e0134215. doi: 10.1371/journal.pone.0134215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadr K, Smith A, Plug I, Orton J, Mütti B. Herders and foragers on Kasteelberg: interim report of excavations. The South African Archaeological Bulletin. 2003:1999–2002. 27–32. [Google Scholar]
- Sandelowsky B. Ph D dissertation. University of California; Berkeley: 1972. Later Stone Age Assemblages from Malawi and their Technologies. Unpublished. [Google Scholar]
- Scally A, Durbin R. Revising the human mutation rate: implications for understanding human evolution. Nature Reviews Genetics. 2012;13:745–753. doi: 10.1038/nrg3295. [DOI] [PubMed] [Google Scholar]
- Schlebusch C. Issues raised by use of ethnic-group names in genome study. Nature. 2010;464:487–487. doi: 10.1038/464487a. [DOI] [PubMed] [Google Scholar]
- Schlebusch CM, Skoglund P, Sjödin P, Gattepaille LM, Hernandez D, Jay F, Li S, De Jongh M, Singleton A, Blum MGB, et al. Genomic Variation in Seven Khoe-San Groups Reveals Adaptation and Complex African History. Science. 2012;338:374–379. doi: 10.1126/science.1227721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuenemann VJ, Peltzer A, Welte B, van Pelt WP, Molak M, Wang CC, Furtwängler A, Urban C, Reiter E, Nieselt K, et al. Ancient Egyptian mummy genomes suggest an increase of Sub-Saharan African ancestry in post-Roman periods. 2017;8:15694. doi: 10.1038/ncomms15694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sealy J, Patrick M, Morris A, Alder D. Diet and dental caries among later stone age inhabitants of the Cape Province, South Africa. American Journal of Physical Anthropology. 1992;88:123–134. doi: 10.1002/ajpa.1330880202. [DOI] [PubMed] [Google Scholar]
- Shipton C, Crowther A, Kourampas N, Prendergast ME, Horton M, Douka K, Schwenninger JL, Faulkner P, Quintana Morales EM, Langley MC, et al. Reinvestigation of Kuumbi Cave, Zanzibar, reveals Later Stone Age coastal habitation, early Holocene abandonment and Iron Age reoccupation. Azania: Archaeological Research in Africa. 2016;51:197–233. [Google Scholar]
- Shriver MD, Kennedy GC, Parra EJ, Lawson HA, Sonpar V, Huang J, Akey JM, Jones KW. The genomic distribution of population substructure in four populations using 8,525 autosomal SNPs. Human Genomics. 2004;1:274. doi: 10.1186/1479-7364-1-4-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair P, Juma A, Chami F. Excavations at Kuumbi Cave on Zanzibar. 2006;2005 [Google Scholar]
- Skoglund P, Mallick S, Bortolini MC, Chennagiri N, Hünemeier T, Petzl-Erler ML, Salzano FM, Patterson N, Reich D. Genetic evidence for two founding populations of the Americas. Nature. 2015 doi: 10.1038/nature14895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoglund P, Malmström H, Raghavan M, Storå J, Hall P, Willerslev E, Gilbert MTP, Götherström A, Jakobsson M. Origins and genetic legacy of Neolithic farmers and hunter-gatherers in Europe. Science. 2012;336:466–469. doi: 10.1126/science.1216304. [DOI] [PubMed] [Google Scholar]
- Skoglund P, Northoff BH, Shunkov MV, Derevianko AP, Pääbo S, Krause J, Jakobsson M. Separating endogenous ancient DNA from modern day contamination in a Siberian Neandertal. Proceedings of the National Academy of Sciences. 2014a doi: 10.1073/pnas.1318934111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoglund P, Posth C, Sirak K, Spriggs M, Valentin F, Bedford S, Clark GR, Reepmeyer C, Petchey F, Fernandes D, et al. Genomic insights into the peopling of the Southwest Pacific. Nature. 2016;538:510–513. doi: 10.1038/nature19844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoglund P, Sjödin P, Skoglund T, Lascoux M, Jakobsson M. Investigating Population History Using Temporal Genetic Differentiation. Molecular Biology and Evolution. 2014b;31:2516–2527. doi: 10.1093/molbev/msu192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AB. In: Kasteelberg. Guide to Archaeological Sites in the South-western Cape. Smith AB, Mutti B, editors. Cape Town: South African Association of Archaeologists Conference; 1992a. pp. 28–30. [Google Scholar]
- Smith AB. Pastoralism in Africa. Johannesburg: Witwatersrand University Press; 1992b. [Google Scholar]
- Smith BW. Rock art in south-central Africa: a study based on the pictographs of Dedza District, Malawi and Kasam District, Zambia. University of Cambridge; 1995. [Google Scholar]
- Stafford TW, Hare PE, Currie L, Jull AJT, Donahue DJ. Accelerator radiocarbon dating at the molecular level. Journal of Archaeological Science. 1991;18:35–72. [Google Scholar]
- Stuiver M, Polach HA. Discussion: reporting of 14 C data. Radiocarbon. 1977;19:355–363. [Google Scholar]
- Tishkoff SA, Reed FA, Friedlaender FR, Ehret C, Ranciaro A, Froment A, Hirbo JB, Awomoyi AA, Bodo JM, Doumbo O, et al. The Genetic Structure and History of Africans and African Americans. Science. 2009;324:1035–1044. doi: 10.1126/science.1172257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Klinken GJ. Bone collagen quality indicators for palaeodietary and radiocarbon measurements. Journal of Archaeological Science. 1999;26:687–695. [Google Scholar]
- Van Oven M, Kayser M. Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum Mutat. 2009;30:E386–E394. doi: 10.1002/humu.20921. [DOI] [PubMed] [Google Scholar]
- Veeramah KR, Wegmann D, Woerner A, Mendez FL, Watkins JC, Destro-Bisol G, Soodyall H, Louie L, Hammer MF. An early divergence of KhoeSan ancestors from those of other modern humans is supported by an ABC-based analysis of autosomal resequencing data. Molecular biology and evolution. 2012;29:617–630. doi: 10.1093/molbev/msr212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Nielsen R. Estimating population divergence time and phylogeny from single nucleotide polymorphisms data with outgroup ascertainment bias. Molecular Ecology. 2012;21:974–986. doi: 10.1111/j.1365-294X.2011.05413.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissensteiner H, Pacher D, Kloss-Brandstätter A, Forer L, Specht G, Bandelt HJ, Kronenberg F, Salas A, Schönherr S. HaploGrep 2: mitochondrial haplogroup classification in the era of high-throughput sequencing. Nucleic acids research. 2016;44:W58–W63. doi: 10.1093/nar/gkw233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi X, Liang Y, Huerta-Sanchez E, Jin X, Cuo ZXP, Pool JE, Xu X, Jiang H, Vinckenbosch N, Korneliussen TS, et al. Sequencing of 50 Human Exomes Reveals Adaptation to High Altitude. Science. 2010;329:75–78. doi: 10.1126/science.1190371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubieta LF. Learning through practise: Cheŵa women’s roles and the use of rock art in passing on cultural knowledge. Journal of Anthropological Archaeology. 2016;43:13–28. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Archaeological context and dating of newly reported human remains from Malawi. Related to Figure 1. A) Locations of the three sites in Malawi. B) Plan of the Hora rock shelter showing locations of the original Hora 1 (male) and Hora 2 (female) burials relative to new excavations from 2016. C) Plan of Fingira Cave showing locations of human remains recovered in 1966 (green dots) and 2016 (green stars). D) Plan of Chencherere II rock shelter redrawn from Crader (1984), showing approximate locations where human remains were recovered in 1972 (red and green stars), and the approximate locations of the remains sampled from Nguludi for this study (blue stars). E) Ultrafiltered collagen FTIR spectra for three individuals from Malawi (MW2 = UCIAMS-186346, MW3 = UCIAMS-18647, MW4 = UCIAMS-186348) compared to well-preserved standards and samples (Modern Carabou, BU4, RP01, POR1). These samples show the typical features for well-preserved collagen. The top of the figure shows the FTIR spectra for a single XAD purified sample (MW1 = PSUAMS-1734) compared against a well preserved standard (Beauford Whale). These data, along with the stable isotopes and C:N ratios, indicate that these samples are well preserved and that the AMS 14C dates are reliable.
Figure S2. Ancient individuals and African population structure. Related to Figure 1. A) full legend and PC1-PC2 scatter plot (same analysis as Figure 1) with all present-day populations indicated with separate symbols. B) PC1-PC2 computed only with southern African populations (and west African Yoruba). C) Symmetry statistics confirming that ancient individuals from the western Cape region in South Africa are more closely related to southern San such as the Khomani rather than more northern San populations such as the Ju_hoan_North.
Figure S3. ADMIXTURE clustering analysis of K = 2 to K = 7 clusters. Related to Figure 1. Each bar shows inferred cluster membership in each individual for a given number of clusters. This analysis used 431,134 SNPs that were not in a CpG dinucleotide context. We also show an inset with cluster membership inferred for damage-restricted ancient DNA sequences (PMD-score > 3) in a parallel run with only 111,208 transversion SNPs.
Figure S4. Admixture models of African population history. Related to Figure 3. A) A tree-like model. Tests of all possible triplet topologies predicted to be symmetric by the tree obtained for major African population lineages in the maximum likelihood model in Fig. 3A. B–H) Under each graph we show the outlier f4 statistic with the greatest Z-score between model and empirical values. When the same group appears twice in an f4 statistic (e.g. f4(Archaic, South_Africa_HGs; South_Africa_HGs, YRI), this corresponds to an f3 statistic. Graphs F and H are the most parsimonious fits with only two admixture events.
Figure S5. Automatically adding additional populations to a skeleton graph. Related to Figure 3. A) Skeleton graph invoking widespread admixture from a hypothesized ancestral northeast African population. B–D) Best fitting insertion points for Malawi_Hora_8100BP shotgun sequence data (B), Mbuti (C), and Japanese non-Africans (D), in the skeleton graph displayed in (A). For details see Table S6.
Table S1. Related to Table 1. Details of sequence libraries prepared from ancient samples.
Table S2. Related to Table 1. Details of direct AMS radiocarbon bone dates.
Table S3. Related to Figure 2. Details of ancestry proportions inferred using qpAdm.
Table S4. Related to Table 1. Y-chromosome haplogroup informative polymorphisms inferred for male individuals with sufficient data.
Table S5. Related to Figure 1. Z-scores (computed for D-statistics) in complete genomes from present-day and ancient Africans.
Table S6. Related to Figure 3. Results of systematically inserting additional populations into all possible edges of the skeleton graph in Fig. S6A.
Table S7. Related to Table 1. MtDNA haplogroup informative polymorphisms.
Data Availability Statement
Raw sequence data (bam files) from the 15 newly reported ancient individuals is available from the European Nucleotide Archive under accession no. PRJEB21878. The newly reported SNP genotyping data is available to researchers who send a signed letter to D.R. containing the following text: “(a) I will not distribute the data outside my collaboration; (b) I will not post the data publicly; (c) I will make no attempt to connect the genetic data to personal identifiers for the samples; (d) I will use the data only for studies of population history; (e) I will not use the data for any selection studies; (f) I will not use the data for medical or disease-related analyses; (g) I will not use the data for commercial purposes.”