

Abstract
Traumatic brain injury (TBI) results in rapid recruitment of leukocytes into the injured brain. Monocytes constitute a significant proportion of the initial infiltrate and have the potential to propagate secondary brain injury or generate an environment of repair and regeneration. Monocytes are a diverse population of cells (classical, intermediate and non-classical), with distinct functions, however, the recruitment order of these subpopulations to the injured brain is largely remains unknown. Thus, we examined which monocyte subpopulations are required for the generation of early inflammatory infiltrate within the injured brain, and whether their depletion attenuates secondary injury or neurocognitive outcome. Global monocyte depletion correlated with significant improvements in brain edema, motor coordination, and working memory, and abrogated neutrophil infiltration into the injured brain. However, targeted depletion of classical monocytes alone had no effect on neutrophil recruitment to the site of injury, implicating the non-classical monocyte in this process. In contrast, mice that have markedly reduced numbers of non-classical monocytes (CX3CR1−/−) exhibited a significant reduction in neutrophil infiltration into the brain after TBI as compared to control mice. Our data suggest a critical role for non-classical monocytes in the pathology of TBI in mice, including important clinical outcomes associated with mortality in this injury process.
Introduction
Traumatic brain injury is a growing and under-recognized public health threat. The Centers for Disease Control estimates nearly 2.5 million people sustain a traumatic brain injury (TBI) each year in the United States, resulting in over 300,000 hospitalizations and 50,000 deaths (1). In fact, TBI-related healthcare expenditure nears 80 billion dollars annually (2–4). The impact of TBI is highlighted not only by its high mortality, but also by the significant long-term complications suffered by survivors (5, 6). More recent data has shown that monocytes constitue a larger proportion of the initial inflammatory infiltrate than previously recognized in TBI (7, 8). This has lead us to speculate that infiltrating monocytic cells have a much larger role in the initial evolution of TBI than previously appreciated (9). The early inflammatory response after TBI is characterized by the generation of reactive oxygen species, release of proteolytic enzymes, and directly cytotoxic cell-cell interaction resulting in the formation of brain edema, secondary brain injury, and further recruitment of inflammatory mediators thereby worsening the index injury (10, 11). This has prompted investigation into cell-specific depletion as a potential therapeutic strategy in TBI. In fact, depletion of circulating monocytes with liposome-encapsulated clodronate has been widely described within central nervous system (CNS) injury literature (12–14). Prior investigation has demonstrated that pre-treatment of mice with clodronate liposomes prior to Theiler’s murine encephalomyelitis virus (TMEV) infection resulted in decreased macrophage infiltration into the CNS, reduced demyelination of spinal cords and late onset of paralysis in an experimental model of virus-induced demyelinating disease (12).
In mice, circulating monocytes can be divided into CD115+Ly6C−hiCD62+ CCR2hi classical monocytes, CD115+Ly6C−loCD62−CCR2lo non-classical monocytes, and an intermediate subset that expresses a spectrum of these markers (15). Monopenic mice such as CCR2−/− mice have yielded mixed results in TBI demonstrating both neuro-protection and pathology (16, 17). While classical monocytes infiltrate into the injured brain giving rise to tissue macrophages, the role of non-classical monocytes in the injury process is less clear. Additionally, non-classical monocytes express high levels of the CX3CR1 fractalkine receptor on their cell surface enabling them to migrate towards the CX3CRL1 expressed by neuronal targets after TBI (18, 19). Non-classical monocytes are also known as patrolling monocytes, as they survey vascular endothelium such as the blood-brain barrier, only extravasating under conditions of inflammation or injury (20). Our understanding of the role of these individual monocytic subtypes in TBI is rapidly evolving (16, 21, 22).
The aim of the current study is to determine the role of infiltrating monocytic subtypes in the early inflammatory infiltrate after TBI. Given the relative paucity of data in the literature with regards to the role of non-classical monocytes in TBI, the contributions of this monocytic subtype in the etiology and evolution of TBI may be vastly under-recognized. We hypothesized that non-classical monocytes drive the pathogenesis of the early TBI-induced inflammatory infiltrate and that depletion of non-classical monocytes would attenuate secondary injury and improve neurocognitive outcome.
Materials and Methods
Study design
The sample size of the experiments (n=15) was determined using data from preliminary experiments. In our prior exeriments we identified a statistical difference in TBI vs sham animals of at least p< 0.05 using a non-paired, non parametric two tailed test (Mann Whitney). Exclusion criteria of live animals were established on the basis of if any of the following occurred after surgery: (i) self-inflicted mutilation or severe abrasion at the site of incision, (ii) severe infection, (iii) excessive loss of weight and or lethargic appearance. Data points were excluded as outliers if values were more than two standard deviations away from the means. Treatment groups were age matched littermates and were randomly chosen. Experiments were repeated three times each. Investigators were not blinded to treatment groups.
Mice
All procedures were approved by the Northwestern University Institutional Animal Care and Use committee. Two mouse strains were used, they are C57BL/6 and B6.SJL-Ptprca Pepcb/BoyJ (CD45.1). All mice were purchased from the Jackson Laboratory and housed at a barrier facility at the Center for Comparative Medicine at Northwestern University (Chicago, IL, USA). Twelve to fourteen -week-old male mice (n=15 per experiment) were used for all experiments.
Controlled cortical impact
Mice were anesthetized with 50 mg/kg Ketamine (Ketaset, Fort Dodge, IA) and 2.5 mg/kg Xylazine (Anased, Shenandoah IA) via intraperitoneal injection. After anesthesia induction, a longitudinal 1cm scalp incision was performed on the mice to reveal both the sagittal and coronal sutures of the skull. The area on the skull overlying the left parietal lobe— 2 mm left of the sagittal suture and 2 mm rostral to the coronal suture- was marked as the target area for the procedure. Next, a 5mm in diameter craniectomy was performed leaving the dura intact, and the TBI mice were placed within the stereotaxic operating frame beneath the electromagnetic impacting rod. The impactor was set for severe injury at a velocity of 2.5m/s and an impacting depth of 2mm and the dwell time was 0.1s. Control mice were given ‘sham injury’ comprised of craniectomy alone without TBI.
Immediately following injury or sham injury, all animals had their scalps sealed with VetBond (3M) (Santa Cruz Animal Health, Dallas, TX). All animals received the anesthetic Buprenorphine SR (SR Veterinary Technologies, Windsor, CO) via subcutaneous injection and were allowed to recover in separate cages over a warming pad. Mice were euthanized at 24h post injury via carbon dioxide inhalation and cervical dislocation. Blood, spleen, and brains were harvested for analysis by flow cytometry.
Histopathology and Immunohistochemistry
Animals were sacrificed and perfused using 2% paraformaldehyde in PBS, brains were excised and fixed in 2% paraformaldehyde. Fixed brains were sent to the Northwestern University mouse histology and phenotyping laboratory for further processing. Briefly, paraffin-embedded brain sections (4 μm) were stained with hematoxylin and eosin (H&E), F4/80, Ly6G or CD45. All images were photographed at ×4 using an Olympus BX41 microscope equipped with an Olympus DP21 camera. Slides were used for qualitative and not quantitative purposes; thus they were not scored by a pathologist.
Tissue preparation and flow cytometry
Blood was collected into EDTA-containing tubes either via facial vein bleed (from live animals) or via cardiac puncture (from euthanized animals). Whole blood was stained with fluorochrome-conjugated antibodies (see fluorochrome list in Table 1) and erythrocytes were then lysed using BD FACS lysing solution (BD Biosciences, San Jose, CA). Spleen was digested using mixture of Collagenase D and DNase I (Roche, Basel, Switzerland) in HBSS at 37°C for 30 min, and filtered through 40 μm nylon mesh (Falcon). Erythrocytes remaining in the single cell suspension were lysed using BD Pharm Lyse (BD Biosciences) and cells were counted using Countess automated cell counter (Invitrogen, Carlsbad CA); dead cells were discriminated using trypan blue. For brain analysis, mice were perfused through the left ventricle with 20 mL of ice cold PBS. The brains were then excised and place in ice cold HBSS until time to process. The brains were weighed, cut into small pieces and placed into C-tubes (Miltenyi) containing digestion buffer (2.5 mg/mL Liberase TL (Roche, Basel, Switzerland), and 1 mg/mL of DNase I in HBSS). The C-tubes were placed on a MACS dissociator and run on the M_Brain_3 protocol, after which they were placed in an incubator for 30 minutes at 37°C with shaking at 200 rpm. After incubation, the C-tubes were placed back on the MACS dissociator and run on the same protocol as before. The cells released were then passed through a 40 μm nylon mesh with a cell masher, the cells were washed with 100ml of wash buffer (1% BSA in HBSS) per brain sample, microglia and infiltrating cells were isolated using a 30/70 percoll gradient. The cells collected from the interphase of the gradient were washed with HBSS and counted using Countess automated cell counter (Invitrogen); dead cells were discriminated using trypan blue. Cells were stained with live/dead Aqua (Invitrogen) viability dye, incubated with Fc-Block (BD Bioscience) and stained with fluorochrome-conjugated antibodies (see fluorochrome list in Table 1). Data were acquired on BD LSR II flow cytometer (BD Biosciences, San Jose, CA). Compensation and analysis of the flow cytometry data were performed using Flowjo software (TreeStar, Ashland, OR). “Fluorescence minus one” controls were used when necessary to set up gates (Supplemental Figure 1).
Table 1.
Fluorochrome List.
| Antibody | Fluorochrome | Company and Clone |
|---|---|---|
| BLOOD | ||
| CD45 | BD Horizon V500 | 30-F11/BD Bioscience |
| B220 | PECF594 | RA3-6B2/BD Bioscience |
| NK1.1 | Alexa Flour 700 | PK136/BD Bioscience |
| CD11b | e-Flour 450 | M1/70/eBiosciences |
| Ly6G | Percp-Cy 5.5 | 1A8/BD Bioscience |
| Ly6C | APC-Cy7 | AL-21/BD Bioscience |
| CD115 | PE | AFS98/BD Bioscience |
| CD62L | PE-Cy7 | MEL-14/BD Bioscience |
| CD43 | APC | S7/BD Bioscience |
| BRAIN | ||
| Live | ||
| CD45 | APC-Cy7 | 30-F11/BD Bioscience |
| CD64 | APC | X54-5/7.1/BD Bioscience |
| CD11b | eFlour 450 | M1/70/eBiosciences |
| CD11c | PE-Cy7 | HL3/BD Bioscience |
| Ly6G | Alexa Flour 700 | 1A8/BD Bioscience |
| B220 | PECF594 | RA3-6B2/BD Bioscience |
| Siglec F | PECF594 | E50-2440/BD Bioscience |
| CD4 | PECF594 | RM4-5/BD Bioscience |
| CD8 | PECF594 | 53-6.7/BD Bioscience |
| NK1.1 | PECF594 | PK136/BD Bioscience |
List of antibody conjugated fluorochromes used to label blood and brain cells for flowcytometry.
Shielded Bone Marrow Chimeras
Bone marrow was aseptically harvested from tibias and femurs, from 8-week-old B6 CD45.1 donor mice, erythrocytes were lysed (BD Pharm Lyse buffer; BD Biosciences) and the cells were counted using a Countess automated cell counter. 8 weeks old B6.CD45.2 mice received a single 1000-cGy γ-irradiation dose using a Cs-137-based Gammacell 40 irradiator (Best Theratronics, Ottawa, ON, Canada). The mice heads were shielded with a lead bar so as to deliver the irradiation to the body only. 6 hours after shielded irradiation, busulfan (30 mg/kg) was administered to completely ablate the bone marrow of the recipient mice. Donor bone marrow (CD45.1) was transplanted 12 hours later. Shielded bone marrow chimeras were maintained on trimethoprim/sulfamethoxazole (40 mg/5 mg, respectively; Hi-Tech Pharmacal/Akorn, Amityville, NY, USA) diluted in sterile water for 1 week prior to irradiation and for 4 weeks after. Eight weeks after irradiation, 95% of the circulating monocytes were of donor origin.
Monocyte Depletion— Clodronate and anti-CCR2 mAb
All mice weighed between 25g–30g. For pan depletion of monocytes, 24 hours prior to TBI or sham injury, C57BL/6 male mice underwent monocyte or sham depletion via intravenous injection of liposome encapsulated clodronate or liposome encapsulated PBS as control (Liposoma, Amsterdam). Liposomes were administered according to the recommended dose of 10uL/1g of mouse. For selective depletion of Ly6Chi monocytes, C57BL/6 mice were given intravenous injections of an anti-CCR2 monoclonal antibody (clone: MC21) 24hrs prior to TBI at a dose of 25ug/mouse. The antibody was generated and characterized by Mack et al. as described in (23) and gifted to our lab by Steffen Jung (19). The isotype control rat IgG2b (Biolegend) was administered at the same dose. Peripheral blood was assessed before injection to confirm the presence of monocytes. 24 hours post injury the animals were euthanized, blood was collected via cardiac puncture and brains were collected and processed as described above. Leukocyte subsets were identified and counted via flow cytometry as described above. Data was analyzed with the statistical software program PRISM and are reported as the mean ± SEM.
Behavioral Phenotyping and Analysis
Immediate working memory performance was assessed on mice following TBI and sham injured animals by recording spontaneous alternation behavior in a Y-maze. This arena has three arms that radiate from a central triangular area. The arms are spaced 120 degrees apart and are of identical dimensions. Each of the three arms was 60 cm in length, 3.5 cm wide at the bottom and 14 cm wide at the top. Each mouse was placed within the “home” arm and positioned with its nose towards the center of the maze and then released to choose one of the other arms. The mouse is observed for 500 seconds while it freely traverses the maze. The order of arm entries is recorded and analyzed for spontaneous alternation (SA). An alternation is scored for each set of three consecutive choices where no repeated entries occur. An alternation score (# alternations/# of possible alternations * 100) of 50% indicates a random selection. Young, healthy C57BL6 mice typically exhibit scores of 75–80% alternation and aging mice approach 50% alternation (24). The Y maze data are collected by LimeLight software, selected measures such as alternations were exported and analyzed using Microsoft excel and GraphPad Prism software.
Motor coordination and acquisition of skilled behavior in mice was assessed using the TSE RotaRod System. A machine comprising a rotating drum, with individual lanes for each animal. Animal falls from the drum are detected by light-beam sensors, and each lane is timed independently. Motor speed for drum rotation is controlled electronically. Mice were trained at a constant speed for four trials of 60 seconds each per day, for three days. After training at constant speed (12 rpm, 60 seconds, 4 times per day) for three days, the speed of the rod was accelerated from 4 to 40 rpm. Four acceleration trials were performed with 20-min intertrial intervals and the time until they dropped from the rod was recorded. The Rod software was used to define, store and run speed profiles, and all data were exported as an excel file and analyzed using the GraphPad Prism software.
Brain Water Content Analysis
Immediately following sacrifice, the brain was removed and placed in pre-weighed tubes. The tubes were weighed immediately following removal to obtain wet weight (WW). The tissue was then dried at 56°C for 3 days and weighed to obtain dry weight (DW). Water content was calculated as a ratio of wet weight minus dry weight divided by wet weight (WW−DW)/WW.
Statistical Analysis
Calculations and further data and statistical analysis were performed using Microsoft excel and GraphPad Prism Software (GraphPad Software, San Diego, CA). Due to the difference in the variation between the the clodronate treated and the PBS treated animals that underwent TBI, statistical comparisons were made using the non-paired, non-parametric Mann-Whitney test. Data are reported as the mean ± SEM, unless indicated otherwise.
Data Availability
The data that support the findings in this manuscript are available from the corresponding author upon request.
Results
Murine model of Traumatic brain injury
Preliminary experiments were performed to asses the severity of injury in 12–14 week old male C57BL/6 mice that underwent controlled cortical impact (CCI). Our model induced a severe brain injury at 24-hours post injury as seen by both gross examination as well as histologic section. The histopathologic defects were quite striking with substantial loss from the ipsilateral cortex as well as distortion of deep brain structures in both the ipsilateral and contralateral hemispheres (not shown).We also use immunohistochemistry to show evidence of distribution of infiltrating cells within the brain parenchyma after injury. IHC stained sections from brain-injured mice at 24 hours post-TBI yielded a large volume loss of cortex (Ctx) at the site of impact, as well as infiltration of peripheral immune cells as depicted by CD45+ and Ly6G+ and increased intensity of F4/80 staining within the ipsilateral hemisphere (Fig 1A and 1B). IHC stained coronal sections of sham-injured mouse brains (craniotomy only) showed normal neuroanatomy and no cellular infiltration (Fig 1C and 1D). Although immunohistochemistry is useful for assessing morphology and proliferation, it has a number of drawbacks limiting its use. It requires preparation of fixed tissue which is susceptible to artifact, and it only permits semi quantitative analysis from localized regions of the CNS. Therefore, for the simultaneous characterization and quantification of whole populations of myeloid cells within the CNS we employ flow cytometry (FC) which is a more reliable and reproducible method (25, 26).
Figure 1.
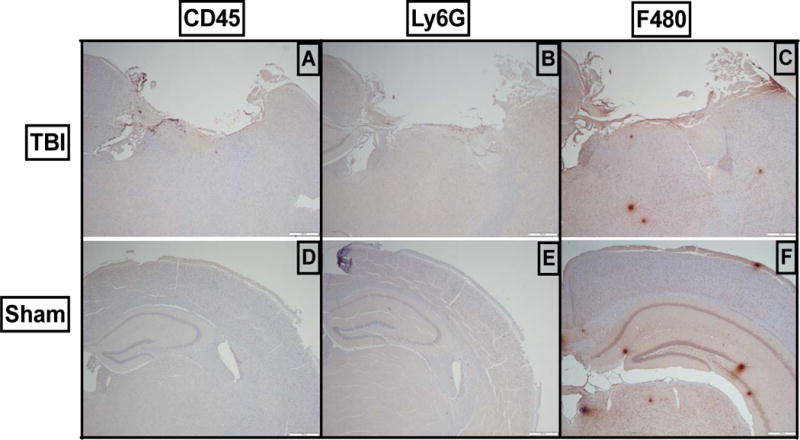
Severe TBI results in disruption of the structural architecture of the brain as well as infiltration of peripheral immune cells into the injured brain. Controlled cortical impact was performed at a velocity of 2.5m/s, a depth of 2mm and a dwell time of 0.2 seconds. Representative sections demonstrate (A, D) infiltration of CD45hi cells into the injured brain as compared to sham-injured animals. (B, E) Infiltration of Ly6G+ cells into the brain after injury compared to sham-injured animals. (C, F) Infiltration of F4/80+ cells into the brain after injury compared to sham-injured animals. All images are at 4× magnification.
Validation of gating strategy using bone marrow chimeras
Flow cytometry was used to characterize and quantify both the inflammatory infiltrate as well as the resident innate immune cells of the brain, microglia, in sham-injured and TBI mice. In order to interrogate our gating strategy and ensure we had adequately differentiated bone marrow derived monocytes and macrophages from resident microglia, we created bone marrow chimeric mice allowing definitive distinction between the two embryologically distinct populations. Eight weeks after bone marrow transplant, all mice had CD45.1 cells in the circulation (Fig 2A) while all brain cells remained CD45.2 (Fig 2B). Twenty-four hours after the induction of TBI, we observed two distinct populations of CD45+ cells within the injured brain—CD45hi and CD45lo. The CD45hi cells were exclusively CD45.1 indicating that this population had infiltrated from the periphery. Similarly, the CD45lo cells were exclusively CD45.2 indicating that these were the brain resident immune cells—microglia. Upon further analysis, we observed that the CD45hi cells was comprised mainly of neutrophils, CD64+ macrophages, and CD64− monocytes with a limited number of T-cells and B-cells that infiltrated from the periphery (Figure 2B). Conversely the CD45lo cells were entirely composed of brain resident microglia (Fig 2C). This study provided solid rationale for the development of our future gating strategies allowing us to phenotypically demarcate peripheral infiltrating cells after acute TBI from the brain resident microglia.
Figure 2.
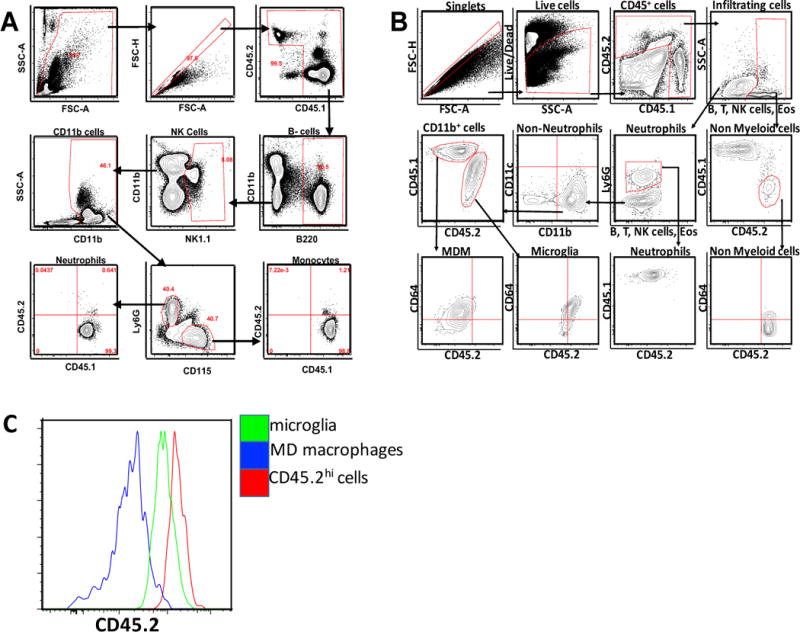
Gating strategy for cells from brains of shielded CD45.1+CD45.2 mixed chimera mice: A) PBMCs from blood of chimeric mice 8 weeks after adoptive transfer, showing that all monocytes and neutrophils are CD45.1. (B) cells from brain of chimeric mice 24h post TBI, showing that resident microglia can be differentiated from infiltrating monocyte derived macrophages. Cells gated on singlets, then live cells then CD45.1 and CD45.2 populations. Arrow indicated directionality of gating. C) Histogram showing the CD45.2 expression between microglia and other peripheral cells compared to monocyte derived cells in a chimeric mouse.
Clodronate depletion of monocytes inhibits neutrophil infiltration into the injured brain
Several studies have shown that neutrophils are the first cell subsets to infiltrate into the brain after TBI (7, 27), therefore we used neutrophil infiltration as a readout of acute inflammation in our CCI model. We next set out to determine which infiltrating cells subsets predominate the early infiltrate into the brain at 24 hours post TBI as well as what effect selective depletion of these subsets had on the overall cellular infiltrate. Global monocyte depletion was induced via intravenous injection of liposome-encapsulated clodronate. Clodronate-induced monocyte depletion in the peripheral blood did not effect circulating neutrophil counts (Figure 3A–D). Expectedly, given that infiltrating monocytes differentiate into macrophages within the injured brain after TBI, there was a reduction in CD64+ macrophages within the brain after TBI (Fig 3G–H, p ≤ 0.03) compared to PBS-liposome treated animals. Surprisingly however, animals pre-treated with clodronate prior to TBI exhibited significant absence of neutrophilic infiltrate into the brain (p ≤ 0.05) after TBI as compared to animals that were sham depleted (Fig 3E–F) despite normal levels of circulating neutrophils.
Figure 3.
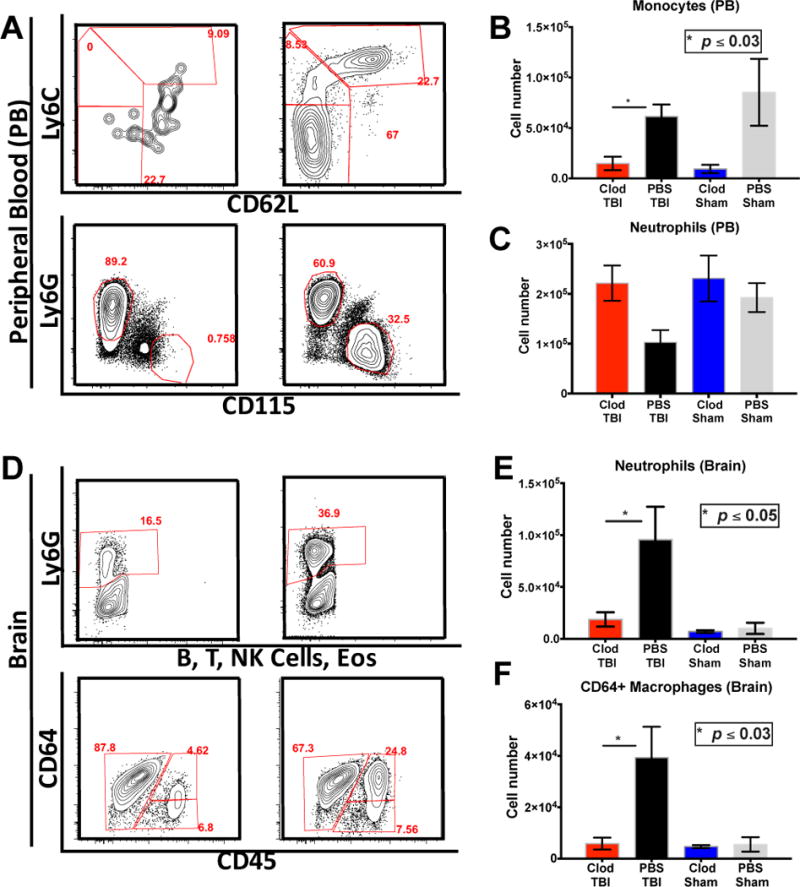
Monocyte depletion inhibits neutrophil recruitment after TBI: (A–C) intravenous administration of one dose of clodronate does not affect neutrophil population whereas all monocyte subsets are depleted in circulation P ≤ 0.05. (D–F) Monocyte depletion abrogates neutrophil infiltration into the brain as well as monocyte derived macrophages. P ≤ 0.05 Mann Whitney test and n=15
Monocyte depletion improves functional outcomes
It is well known that the early neutrophil response contributes to the formation of early and sustained brain edema (10, 11). Therefore, we measured brain edema in TBI and sham-injured mice 30 days post TBI. We found that the clodronate treated mice had significantly lower water content in the brain as compared to the PBS liposome treated mice (Fig 4D, p ≤ 0.03). Consequently we investigated behavioral deficits in these animals to determine wether there was any rescue in behavioral phenotype. We used the Y-maze test of spontaneous alteration (SA) to test working memory—a system for temporarily storing and managing the information required to carry out complex cognitive sasks such as learning, reasoning, and comprehension. SA is a natural, hippocampal-dependent behavior of rodents in which they tend not to repeat exploration of a region that has no reward (24). This behavior relies on working memory and does not require any rewards or punishments. Clodronate depleted animals had improved alternation scores as measured by the Y-maze spatial alternation test (Fig 4C, p ≤ 0.05) compared to sham depleted animals. In addition to improved working memory, the clodronate depleted mice also demonstrated better motor coordination and skill acquisition as determined by rotarod testing. Clodronate treated mice spent longer times on the rod and mainitained their balance better at higher speeds than sham depleted mice (Fig 4A–B, p ≤ 0.02).
Figure 4.
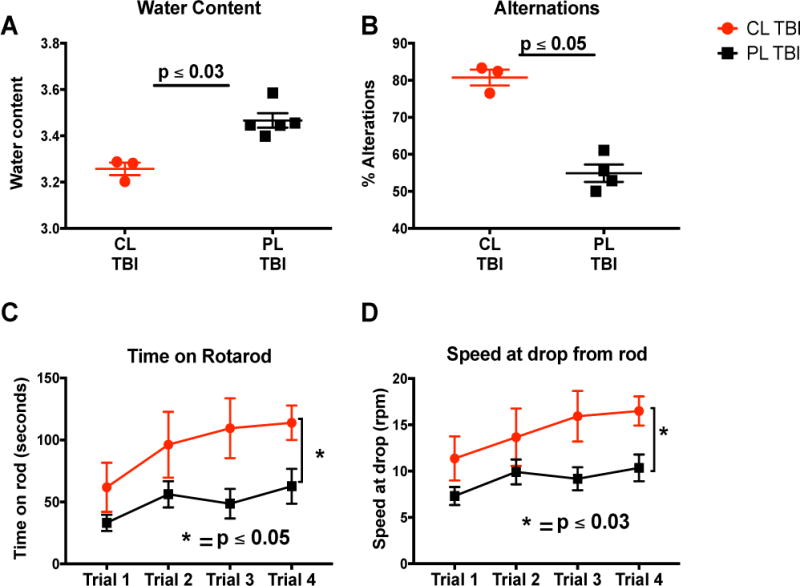
Monocyte depletion improves functional outcome one month after TBI: (A–B) intravenous administration of clodronate improves the (A)time spent on the rotarod P ≤ 0.05and (B) the speed P ≤ 0.03. (C) Monocyte depletion improves spatial alternations in mice after TBI compared to control treatment P ≤ 0.05. (D) Monocyte depletion improves edema in mice brain via abrogation of neutrophil infiltration after TBI P ≤ 0.03. Mice n= 8. Mann whitney test.
Classical monocyte depletion has no effect on neutrophil recruitment in TBI
To further elucidate the specific monocytic subset responsible for our observed neutrophil recruitment into the brain after TBI, we performed a series of experiments that targeted the two major monocyte subsets in mice. In order to determine which subset may be responsible for the observed changes in neutrophil recruitment, we intravenously administered a monoclonal antibody specifically targeting the CCR2 receptor (mAb CCR2) on monocytes. Treatment with mAb CCR2 resulted in near complete depletion of CD115+Ly6C−hiCD62+ CCR2hi classical monocytes (Fig 5A–B p ≤ 0.002) in the blood as compared to mice treated with an isotype control at 24 hours post injury. There was also some decrease in the intermediate monocyte subset in antibody treated mice although it was not significant (Figure 5C). There was no change in the CD115+Ly6C−loCD62−CCR2lo non-classical monocyte population (Fig 5D). In mice treated with the anti-CCR2 antibody, we observed the expected absence of infiltrating monocyte derived CD64+ macrophages within the injured brain after TBI compared to mice treated with isotype control (p ≤ 0.004). However, we continued to observe a high degree of neutrophil infiltration into the injured brain after TBI despite depletion of the CD115+Ly6C−hiCD62+ CCR2hi classical monocyte population.
Figure 5.
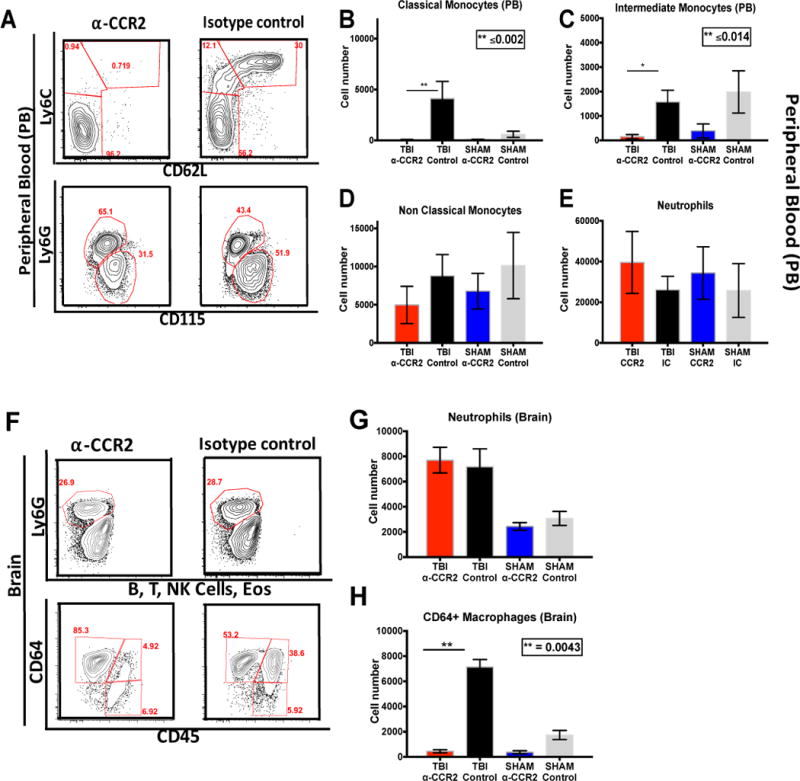
Classical monocytes do not mediate neutrophil infiltration into the brain after TBI: (A–E) intravenous administration of two doses, 25ug each, of anti-CCR2 mAb completely eradicate the circulating classical monocytes P ≤ 0.002, but leaves the non-classical monocyte population unaffected. (F–G) Depletion of classical monocytes does not inhibit neutrophil infiltration into the brain. (H) Depletion of classical monocytes inhibit monocyte derived macrophage recruitment after TBI P ≤ 0.004. All tests are non-paired non parametric Mann-Whitney tests and mice n=15
Non classical monocytes are required for neutrophil recruitment in TBI
These data suggested to us that classical monocytes do not play a critical role in neutrophil recruitment into the injured brain after TBI. Therefore we aimed to determine the role that the CD115+Ly6C−loCD62−CCR2lo non-classical monocytes play in neutrophil recruitment after TBI. To investigate this, we employed transgenic mice that are deficient in CX3CR1. While all blood monocytes express CX3CR1, CX3CR1 deficient mice have markedly fewer non-classical monocytes in circulation. As expected there were fewer CD115+Ly6C−loCD62−CCR2lo monocytes in the blood of CX3CR1 deficient mice with no change in neutrophil numbers compared to heterozygous controls (Fig 6 A–D). However, after TBI, there was a significant decrease in infiltrating neutrophils within the injured brain as compared to heterozygous controls (p ≤ 0.05) (Fig 6 E–F). Interestingly, although there was a decrease in the CD64+ macrophage population it wasn’t significant (Fig 6 H–I). Taken together these data suggest that non-classical monocytes are required for neutrophil infiltration into the injured brain after TBI.
Figure 6.
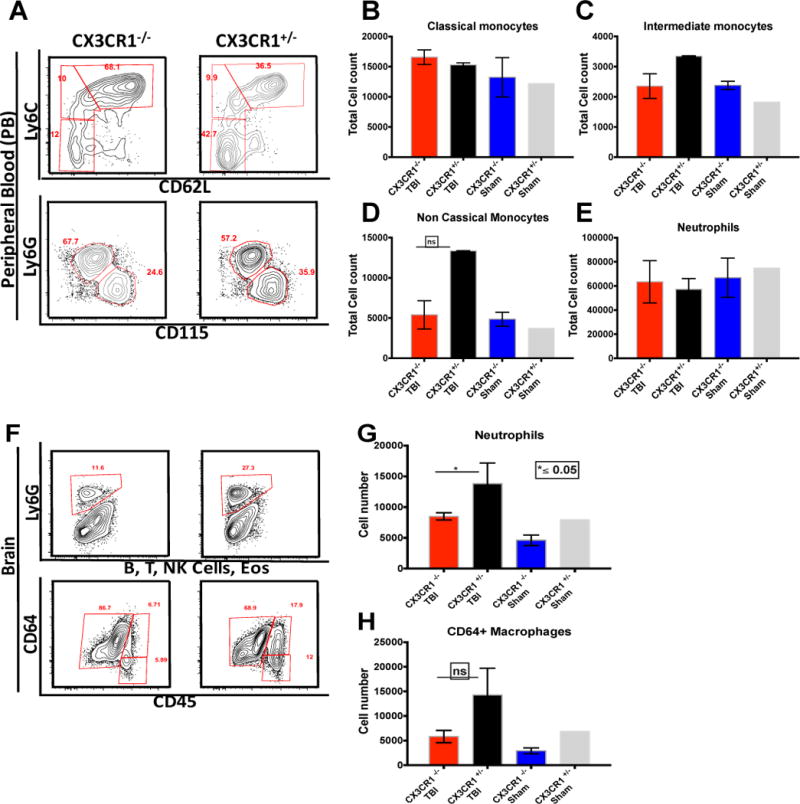
Non-Classical monocytes mediate neutrophil infiltration into the brain after TBI: (A–E) CX3CR1−/− mice have less non-classical monocytes than the CX3CR1GFP/+ mice with no difference in other monocyte subsets or the circulating neutrophils. (F–G) Reduced non-classical monocytes results in inhibition of neutrophil infiltration into the brain P ≤ 0.05 Mann Whitney test. (H) Reduced non-classical monocytes have a dampening effect on the infiltrating monocyte derived macrophage population. N= 15
Discussion
In this study, we demonstrate that depletion of peripheral monocytes results in reduced brain edema, improved working memory, better motor coordination, and improved skill acquisition as compared to nondepleted mice after TBI. Furthermore we show that Ly6Clo non-classical monocytes are required for the recruitment of neutrophils into the injured brain after TBI.
Examination of the injured brains in our study confirmed all the classical findings of severe TBI including cerebral edema, contusion, intraparenchymal and extra axial hemorrhage, and loss of brain tissue (28–30). In addition to gross findings, TBI induces significant neuroinflammation as well as a loss of blood brain barrier (BBB) integrity resulting in infiltration of peripheral immune cells into the injured brain (30). Multiple studies have shown that microglia, the resident innate immune cells of the brain, are morphologically and functionally similar to infiltrating monocyte-derived macrophages (31, 32). This is especially true after CNS injury when microglia are activated, making the ability to reliably differentiate between the two cell types problematic (19,27,28). Although advanced flow cytometric techniques have allowed for a greater ability to distinguish between these two similar cell populations, bone marrow chimeric mice have provided the most sensitive model to date (33–35). Therefore, we generated head-shielded, mixed bone marrow chimeric mice based on CD45.1/45.2. Shielding the head of the mice preserves the integrity of the blood brain barrier (BBB) and protects the microglia from damage after irradiation. This enabled us to develop an accurate gating strategy ensuring stringent differentiation between microglia and infiltrating monocyte-derived cells within the injured brain after TBI.
Monocytes and neutrophils are the first immune cell subsets to infiltrate into the brain after TBI and contribute significantly to the pathology of injury (36–39). They secrete inflammatory mediators, propagate continued breakdown of the BBB, and coordinate the evolution of classical neuroinflammation. Recent studies have suggested that monocytes may play a role in neutrophil recruitment to a variety of other inflamed tissues (40, 41). Therefore, we hypothesized that monocytes drive the recruitment of neutrophils into the injured brain after TBI. To test this, global depletion of monocytes with clodronate-encapsulated liposomes was performed. Clodronate-mediated depletion eradicates all monocytic cells in the circulation as well as splenic and hepatic macrophages without affecting other cell types, including microglia (42–44). We observed a conspicuous absence of neutrophilic infiltrate into the injured brain after TBI in clodronate treated mice despite an undisturbed population of neutrophils in circulation (Fig 3E–F). This data suggests that circulating monocytes play a key role in the recruitment of neutrophils into the injured brain after TBI. In addition, we found that mice lacking neutrophil infiltration due to monocyte depletion, demonstrated a significant reduction in brain edema post injury. This is consistent with prior data showing that neutrophil depletion prior to TBI reduced edema formation (45).
In addition to reduced cellular infiltrate and edema, we observed a significant improvement in neurocognitive function in clodronate-depleted animals after TBI (Fig 4 A–C). These findings confirm and advance upon a previous study, which showed improvement in neurocognitive function in mice lacking classical monocytes via CCR2 knockout, but did not observe changes in motor coordination, or asses edema (17). Unlike the CCR2 knockout model, our study utilized clodronate-mediated depletion which resulted in loss of both classical and non-classical monocytes. This lead not only to neurocognitive improvement, but also a reduction in cerebral edema and improvement in motor coordination. This indicated to us that the depletion of non-classical monocyte subset in addition to classical monocytes may be responsible for the observed changes in both neutrophil recruitment as well as the resultant pathology from neutrophil infiltration.
Similar to others, we observed that anti-CCR2 antibody depletes classical monocytes from the circulation and, therefore, infiltration and macrophage differentiation within the injured brain after TBI. However, depletion of classical monocytes alone did not affect neutrophil recruitment into the brain after injury, which remained consistent with levels in animals treated with isotype control antibody (Fig 5 F–G). This finding continued to support our hypothesis that the non-classical component of the monocytic response was responsible for neutrophil recruitment to the site of injury. To better test our theory we aimed to deplete the nonclassicl monocyte population in isolation. While classical monocytes are easily knocked out or depleted with antibodies, there are currently no effective strategies to deplete non-classical Ly6C− monocytes. Therefore, we employed CX3CR1 deficient mice.
Mice with deletions of CX3CR1 have a reduction in both the number and function of their Ly6C− non-classical monocytes (46, 47). This knockout model has been used extensively to study the interplay between CX3CR1+ microglia and the Fractalkine+ neurons in the brain. Microglia utilize fractalkine signaling via CX3CR1 to migrate to their synaptic targets and initiate phagocytosis and synaptic refinement. However, this deficiency does not impair classical monocyte migration and infiltration to the site of injury after TBI (48) (49). In the current study, we too noted no significant difference in monocyte infiltration into the injured brain in CX3CR1−/− mice. However, we did observe a significant reduction in infiltrating neutrophils after TBI in CX3CR1−/− mice as compared to heterozygous controls (p ≤ 0.05). Intriguingly, we have identified a novel role for non-classical monoctyes in the pathology of TBI, the therapeutic implications of which are significant. First, we have shown that targeting a specific monocytic subtype has the ability to fundamentally alter the overall neutrophilic infiltrate within the injured brain, reducing edema and improving neurocognitive and locomotor function. Futhermore, the circulating neutrophil population was unaffected leaving the host with relatively intact peripheral innate immunity. This is of particular importance given the increased susceptibility of the brain injured host to secondary infection (50, 51).
In conclusion, we report that global monocyte depletion abrogates neutrophil infiltration into the injured brain, results in improved working memory, and restores motor coordination and skill acquisition after TBI. Furthermore, our data show a previously unrecognized role for non-classical Ly6Clo monocytes in this process. Although Ly6Chi classical monocytes ultimately differentiate into tissue macrophages within the injured brain, their selective deletion had no effect on neutrophil trafficking and its subsequent generation of cerebral edema (17). These findings provide new insights into the role of monocytic subsets in acute TBI, namely both classical and non-classical subtypes of monocytes are responsible for different aspects of neural toxicity and potential neural repair after CNS injury. The selective depletion of non-classical monocytes represents a potential novel therapeutic strategy for the treatment of TBI, which could be readily translated to human patients. Future studies aimed at identifying each monocytic subtype’s pro-injury and pro-repair gene signature over the time course of injury are currently underway in our laboratory and will undoubtedly allow for the identification of the molecular pathways involved.
Supplementary Material
Acknowledgments
The authors want to thank Salina Dominguez, Alex Schaffer and Fu-Nien Tsai for all technical support.
Sources of Support: This study was supported by NIH grant GM117341 and The American College of Surgeons C. James Carrico Research Fellowship to S.J.S.; NIH grant AR064313 to CMC; NIH grant AR064546, HL134375, AG049665, UH2AR067687 and the United States-Israel Binational Science Foundation (2013247), and the Rheumatology Research Foundation (Agmt 05/06/14) to HRP. HRP was also supported by the Mabel Greene Myers Professor of Medicine.
Footnotes
The authors declare no competing financial interests.
Author contributions: HMM performed the TBI, brain processing and flowcytometry, behavioral phenotyping, data collection and analyses and writing and organization of the manuscript.
TBJ performed the blood collection, processing and flowcytometry. She also assisted with the behavioral phenotyping and data collection.
CMC performed data analyses on all of the flowcytometry data and assisted with data interpretation.
SJS, designed the study, assisted with the induction of TBI, oversaw data interpretation and editing of the manuscript.
HRP Assisted with study design, data interpretation and editing of manuscript.
References
- 1.Faul M. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths 2002–2006. Centers for Disease Control and Prevention, National Center for Injury Prevention and Control; Atlanta (GA): 2010. [Google Scholar]
- 2.Corso P, Finkelstein E, Miller T, Fiebelkorn I, Zaloshnja E. Incidence and lifetime costs of injuries in the United States. Injury prevention: journal of the International Society for Child and Adolescent Injury Prevention. 2006;12:212–218. doi: 10.1136/ip.2005.010983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pearson WS, Sugerman DE, McGuire LC, Coronado VG. Emergency department visits for traumatic brain injury in older adults in the United States: 2006–08. The western journal of emergency medicine. 2012;13:289–293. doi: 10.5811/westjem.2012.3.11559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Whitlock JA, Jr, Hamilton BB. Functional outcome after rehabilitation for severe traumatic brain injury. Archives of physical medicine and rehabilitation. 1995;76:1103–1112. doi: 10.1016/s0003-9993(95)80117-0. [DOI] [PubMed] [Google Scholar]
- 5.Schwarzbold M, Diaz A, Martins ET, Rufino A, Amante LN, Thais ME, Quevedo J, Hohl A, Linhares MN, Walz R. Psychiatric disorders and traumatic brain injury. Neuropsychiatric disease and treatment. 2008;4:797–816. doi: 10.2147/ndt.s2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whelan-Goodinson R, Ponsford J, Johnston L, Grant F. Psychiatric disorders following traumatic brain injury: their nature and frequency. The Journal of head trauma rehabilitation. 2009;24:324–332. doi: 10.1097/HTR.0b013e3181a712aa. [DOI] [PubMed] [Google Scholar]
- 7.Jin X, Ishii H, Bai Z, Itokazu T, Yamashita T. Temporal changes in cell marker expression and cellular infiltration in a controlled cortical impact model in adult male C57BL/6 mice. PloS one. 2012;7:e41892. doi: 10.1371/journal.pone.0041892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2009;29:13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trahanas DM, Cuda CM, Perlman H, Schwulst SJ. Differential Activation of Infiltrating Monocyte-Derived Cells After Mild and Severe Traumatic Brain Injury. Shock. 2015;43:255–260. doi: 10.1097/SHK.0000000000000291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holmin S, Soderlund J, Biberfeld P, Mathiesen T. Intracerebral inflammation after human brain contusion. Neurosurgery. 1998;42:291–298. doi: 10.1097/00006123-199802000-00047. discussion 298–299. [DOI] [PubMed] [Google Scholar]
- 11.Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. Journal of leukocyte biology. 2010;87:779–789. doi: 10.1189/jlb.1109766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Christophi GP, Hudson CA, Panos M, Gruber RC, Massa PT. Modulation of macrophage infiltration and inflammatory activity by the phosphatase SHP-1 in virus-induced demyelinating disease. J Virol. 2009;83:522–539. doi: 10.1128/JVI.01210-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Getts DR, Terry RL, Getts MT, Muller M, Rana S, Shrestha B, Radford J, Van Rooijen N, Campbell IL, King NJ. Ly6c+ “inflammatory monocytes” are microglial precursors recruited in a pathogenic manner in West Nile virus encephalitis. The Journal of experimental medicine. 2008;205:2319–2337. doi: 10.1084/jem.20080421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tran EH, Hoekstra K, van Rooijen N, Dijkstra CD, Owens T. Immune invasion of the central nervous system parenchyma and experimental allergic encephalomyelitis, but not leukocyte extravasation from blood, are prevented in macrophage-depleted mice. J Immunol. 1998;161:3767–3775. [PubMed] [Google Scholar]
- 15.Ziegler-Heitbrock L, Ancuta P, Crowe S, Dalod M, Grau V, Hart DN, Leenen PJ, Liu YJ, MacPherson G, Randolph GJ, Scherberich J, Schmitz J, Shortman K, Sozzani S, Strobl H, Zembala M, Austyn JM, Lutz MB. Nomenclature of monocytes and dendritic cells in blood. Blood. 2010;116:e74–80. doi: 10.1182/blood-2010-02-258558. [DOI] [PubMed] [Google Scholar]
- 16.Semple BD, Bye N, Rancan M, Ziebell JM, Morganti-Kossmann MC. Role of CCL2 (MCP-1) in traumatic brain injury (TBI): evidence from severe TBI patients and CCL2−/− mice. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2010;30:769–782. doi: 10.1038/jcbfm.2009.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsieh CL, Niemi EC, Wang SH, Lee CC, Bingham D, Zhang J, Cozen ML, Charo I, Huang EJ, Liu J, Nakamura MC. CCR2 deficiency impairs macrophage infiltration and improves cognitive function after traumatic brain injury. Journal of neurotrauma. 2014;31:1677–1688. doi: 10.1089/neu.2013.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 19.Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, Hume DA, Perlman H, Malissen B, Zelzer E, Jung S. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38:79–91. doi: 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, Sarnacki S, Cumano A, Lauvau G, Geissmann F. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. 2007;317:666–670. doi: 10.1126/science.1142883. [DOI] [PubMed] [Google Scholar]
- 21.Soares HD, Hicks RR, Smith D, McIntosh TK. Inflammatory leukocytic recruitment and diffuse neuronal degeneration are separate pathological processes resulting from traumatic brain injury. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1995;15:8223–8233. doi: 10.1523/JNEUROSCI.15-12-08223.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Corps KN, Roth TL, McGavern DB. Inflammation and neuroprotection in traumatic brain injury. JAMA Neurol. 2015;72:355–362. doi: 10.1001/jamaneurol.2014.3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mack M, Cihak J, Simonis C, Luckow B, Proudfoot AE, Plachy J, Bruhl H, Frink M, Anders HJ, Vielhauer V, Pfirstinger J, Stangassinger M, Schlondorff D. Expression and characterization of the chemokine receptors CCR2 and CCR5 in mice. J Immunol. 2001;166:4697–4704. doi: 10.4049/jimmunol.166.7.4697. [DOI] [PubMed] [Google Scholar]
- 24.Weiss C, Shroff A, Disterhoft JF. Spatial learning and memory in aging C57BL/6 mice. Neurosci Res Commun. 1998;23:77–92. [Google Scholar]
- 25.Collins CE, Young NA, Flaherty DK, Airey DC, Kaas JH. A rapid and reliable method of counting neurons and other cells in brain tissue: a comparison of flow cytometry and manual counting methods. Front Neuroanat. 2010;4:5. doi: 10.3389/neuro.05.005.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nikodemova M, Watters JJ. Efficient isolation of live microglia with preserved phenotypes from adult mouse brain. Journal of neuroinflammation. 2012;9:147. doi: 10.1186/1742-2094-9-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clausen F, Hanell A, Bjork M, Hillered L, Mir AK, Gram H, Marklund N. Neutralization of interleukin-1beta modifies the inflammatory response and improves histological and cognitive outcome following traumatic brain injury in mice. Eur J Neurosci. 2009;30:385–396. doi: 10.1111/j.1460-9568.2009.06820.x. [DOI] [PubMed] [Google Scholar]
- 28.Guterman A, Smith RW. Neurological sequelae of boxing. Sports Med. 1987;4:194–210. doi: 10.2165/00007256-198704030-00004. [DOI] [PubMed] [Google Scholar]
- 29.Blennow K, Hardy J, Zetterberg H. The neuropathology and neurobiology of traumatic brain injury. Neuron. 2012;76:886–899. doi: 10.1016/j.neuron.2012.11.021. [DOI] [PubMed] [Google Scholar]
- 30.Lagraoui M, Latoche JR, Cartwright NG, Sukumar G, Dalgard CL, Schaefer BC. Controlled cortical impact and craniotomy induce strikingly similar profiles of inflammatory gene expression, but with distinct kinetics. Frontiers in neurology. 2012;3:155. doi: 10.3389/fneur.2012.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nayak D, Roth TL, McGavern DB. Microglia development and function. Annu Rev Immunol. 2014;32:367–402. doi: 10.1146/annurev-immunol-032713-120240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, Mulinyawe SB, Bohlen CJ, Adil A, Tucker A, Weissman IL, Chang EF, Li G, Grant GA, Hayden Gephart MG, Barres BA. New tools for studying microglia in the mouse and human CNS. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:E1738–1746. doi: 10.1073/pnas.1525528113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ford AL, Goodsall AL, Hickey WF, Sedgwick JD. Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein-reactive CD4+ T cells compared. J Immunol. 1995;154:4309–4321. [PubMed] [Google Scholar]
- 34.Lassmann H, Schmied M, Vass K, Hickey WF. Bone marrow derived elements and resident microglia in brain inflammation. Glia. 1993;7:19–24. doi: 10.1002/glia.440070106. [DOI] [PubMed] [Google Scholar]
- 35.Diserbo M, Agin A, Lamproglou I, Mauris J, Staali F, Multon E, Amourette C. Blood-brain barrier permeability after gamma whole-body irradiation: an in vivo microdialysis study. Can J Physiol Pharmacol. 2002;80:670–678. doi: 10.1139/y02-070. [DOI] [PubMed] [Google Scholar]
- 36.Price CJ, Menon DK, Peters AM, Ballinger JR, Barber RW, Balan KK, Lynch A, Xuereb JH, Fryer T, Guadagno JV, Warburton EA. Cerebral neutrophil recruitment, histology, and outcome in acute ischemic stroke: an imaging-based study. Stroke; a journal of cerebral circulation. 2004;35:1659–1664. doi: 10.1161/01.STR.0000130592.71028.92. [DOI] [PubMed] [Google Scholar]
- 37.Buck BH, Liebeskind DS, Saver JL, Bang OY, Yun SW, Starkman S, Ali LK, Kim D, Villablanca JP, Salamon N, Razinia T, Ovbiagele B. Early neutrophilia is associated with volume of ischemic tissue in acute stroke. Stroke; a journal of cerebral circulation. 2008;39:355–360. doi: 10.1161/STROKEAHA.107.490128. [DOI] [PubMed] [Google Scholar]
- 38.Carlos TM, Clark RS, Franicola-Higgins D, Schiding JK, Kochanek PM. Expression of endothelial adhesion molecules and recruitment of neutrophils after traumatic brain injury in rats. Journal of leukocyte biology. 1997;61:279–285. doi: 10.1002/jlb.61.3.279. [DOI] [PubMed] [Google Scholar]
- 39.Zhuang J, Shackford SR, Schmoker JD, Anderson ML. The association of leukocytes with secondary brain injury. J Trauma. 1993;35:415–422. doi: 10.1097/00005373-199309000-00014. [DOI] [PubMed] [Google Scholar]
- 40.Carlin LM, Stamatiades EG, Auffray C, Hanna RN, Glover L, Vizcay-Barrena G, Hedrick CC, Cook HT, Diebold S, Geissmann F. Nr4a1-dependent Ly6C(low) monocytes monitor endothelial cells and orchestrate their disposal. Cell. 2013;153:362–375. doi: 10.1016/j.cell.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Misharin AV, Cuda CM, Saber R, Turner JD, Gierut AK, Haines GK, 3rd, Berdnikovs S, Filer A, Clark AR, Buckley CD, Mutlu GM, Budinger GR, Perlman H. Nonclassical Ly6C(−) monocytes drive the development of inflammatory arthritis in mice. Cell reports. 2014;9:591–604. doi: 10.1016/j.celrep.2014.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sunderkotter C, Nikolic T, Dillon MJ, Van Rooijen N, Stehling M, Drevets DA, Leenen PJ. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J Immunol. 2004;172:4410–4417. doi: 10.4049/jimmunol.172.7.4410. [DOI] [PubMed] [Google Scholar]
- 43.Huitinga I, van Rooijen N, de Groot CJ, Uitdehaag BM, Dijkstra CD. Suppression of experimental allergic encephalomyelitis in Lewis rats after elimination of macrophages. The Journal of experimental medicine. 1990;172:1025–1033. doi: 10.1084/jem.172.4.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Polfliet MM, Goede PH, van Kesteren-Hendrikx EM, van Rooijen N, Dijkstra CD, van den Berg TK. A method for the selective depletion of perivascular and meningeal macrophages in the central nervous system. Journal of neuroimmunology. 2001;116:188–195. doi: 10.1016/s0165-5728(01)00282-x. [DOI] [PubMed] [Google Scholar]
- 45.Kenne E, Erlandsson A, Lindbom L, Hillered L, Clausen F. Neutrophil depletion reduces edema formation and tissue loss following traumatic brain injury in mice. Journal of neuroinflammation. 2012;9:17. doi: 10.1186/1742-2094-9-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hanna RN, Carlin LM, Hubbeling HG, Nackiewicz D, Green AM, Punt JA, Geissmann F, Hedrick CC. The transcription factor NR4A1 (Nur77) controls bone marrow differentiation and the survival of Ly6C- monocytes. Nature immunology. 2011;12:778–785. doi: 10.1038/ni.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Landsman L, Bar-On L, Zernecke A, Kim KW, Krauthgamer R, Shagdarsuren E, Lira SA, Weissman IL, Weber C, Jung S. CX3CR1 is required for monocyte homeostasis and atherogenesis by promoting cell survival. Blood. 2009;113:963–972. doi: 10.1182/blood-2008-07-170787. [DOI] [PubMed] [Google Scholar]
- 48.Gyoneva S, Kim D, Katsumoto A, Kokiko-Cochran ON, Lamb BT, Ransohoff RM. Ccr2 deletion dissociates cavity size and tau pathology after mild traumatic brain injury. Journal of neuroinflammation. 2015;12:228. doi: 10.1186/s12974-015-0443-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM. Control of microglial neurotoxicity by the fractalkine receptor. Nature neuroscience. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- 50.Lucin KM, V, Sanders M, Popovich PG. Stress hormones collaborate to induce lymphocyte apoptosis after high level spinal cord injury. Journal of neurochemistry. 2009;110:1409–1421. doi: 10.1111/j.1471-4159.2009.06232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Woiciechowsky C, Asadullah K, Nestler D, Eberhardt B, Platzer C, Schoning B, Glockner F, Lanksch WR, Volk HD, Docke WD. Sympathetic activation triggers systemic interleukin-10 release in immunodepression induced by brain injury. Nat Med. 1998;4:808–813. doi: 10.1038/nm0798-808. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings in this manuscript are available from the corresponding author upon request.