

Abstract
CD22, a sialic-acid binding immunoglobulin type-lectin (Siglec) family member, is an inhibitory co-receptor of the B-cell receptor (BCR) with established roles in health and disease. The restricted expression pattern of CD22 on B-cells and most B-cell lymphomas has made CD22 a therapeutic target for B-cell-mediated diseases. Models to better understand how in vivo targeting of CD22 translates to human disease are needed. Here, we report development of a transgenic mouse expressing human CD22 (hCD22) in B-cells and assess its ability to functionally substitute for murine CD22 (mCD22) for regulation of BCR signaling, antibody responses, homing, and tolerance. Expression of hCD22 on transgenic murine B-cells is comparable to expression on human primary B-cells, and co-localizes with mCD22 on the cell surface. Murine B-cells expressing only hCD22 have identical calcium (Ca2+) flux responses in response to anti-IgM as mCD22-expressing WT B-cells. Furthermore, hCD22 transgenic mice on a mCD22−/− background have restored levels of marginal zone B-cells and antibody responses compared to deficiencies observed in CD22−/− mice. Consistent with these observations, hCD22 transgenic mice develop normal humoral responses in a peanut allergy oral sensitization model. Homing of B-cells to Peyer’s patches (PP) was partially rescued by expression of hCD22 compared to CD22−/− B-cells, although not to WT levels. Notably, Siglec-engaging antigenic liposomes (STALs) formulated with a hCD22 ligand were shown to prevent B-cell activation, increase cell death, and induce tolerance in vivo. This hCD22 transgenic mouse will be a valuable model for investigating the function of hCD22 and pre-clinical studies targeting hCD22.
Introduction
Sialic acid-binding immunoglobulin(Ig)-like lectins (Siglecs) are primarily expressed on immune cells and recognize sialic acid-containing glycan ligands(1–3). There are 15 members of the Siglec family in humans and 9 in mice, however, only four members are highly conserved between mice and man: sialoadhesin (Siglec-1), CD22 (Siglec-2), myelin associated glycoprotein (MAG; Siglec-3), and Siglec-15. The ability of CD22 to regulate B-cell function has been studied in great detail. Consistent with CD22 being one of the conserved members of the Siglec family, murine (mCD22) and human (hCD22) share 60% homology in amino acid sequence and both have a restricted expression pattern on B-cells. However, an even higher homology between mCD22 and hCD22 is observed within its N-terminal ligand binding domain as well as C-terminal cytoplasmic tail, both of which are critically important for the ability of CD22 to regulate B-cell receptor (BCR) signaling (4). Close proximity of CD22 to the BCR is required for CD22 to become phosphorylated through the actions of the Src kinase, Lyn. Phosphorylation occurs within the multiple immunoreceptor tyrosine-based inhibitory motifs (ITIMs) of CD22 and these phosphorylated motifs become docking sites for Src homology (SH2) domain containing proteins, including phosphatases such as SHP-1. Recruitment of such phosphatases to the BCR, through CD22, can strongly blunt BCR signaling under the appropriate conditions(5–9).
The physiological circumstances in which CD22 is in close proximity to the BCR has been the topic of numerous studies, with many lines of evidence suggesting that its sialic acid-containing glycan ligands on either the same cell (cis) or on another cell (trans) can modulate the function of CD22 as an inhibitor of B-cell activation by sequestering it away or enforcing ligation to the B-cell receptor (10–15). The ligands for CD22 are α2-6-linked sialic acids, which are abundantly found on the cell surface of lymphocytes. B-cells lacking α2-6-linked sialic acids from ST6Gal1−/− mice(16) or knock-in mice having a mutant version of CD22 not capable of binding its glycan ligands(5), both have increased association of CD22 with the BCR and, consequently, blunted BCR signaling, demonstrating that interactions between CD22 and cis glycan ligands on the same B-cell keep it sequestered away from the BCR(17). Moreover, a detailed high resolution microscopy study has validated these claims(18). Interestingly, B-cells from CD22−/− mice do generate a modest degree of hyper-responsiveness to BCR signaling following stimulation with anti-IgM(5, 19–22); this modest increase shows that there may be a small amount of co-localization between CD22 and BCR that occurs in a non-ligand dependent manner in wild-type mice. It is worth noting, however, that this hyper-responsive effect on BCR stimulation is restricted to anti-IgM stimulation since neither monomeric nor more multimeric engagement of the BCR results in hyper-responsiveness in CD22−/− B-cells, with hypo-responsiveness even being observed in certain cases (10, 23). In line with a view that CD22 does not simply set a threshold for BCR signaling, CD22−/− mice generate moderately impaired T-independent and T-dependent antibody responses, which has been attributed to a shorter B-cell lifespan and propensity to undergo apoptosis after BCR ligation, at least in vitro(20–22, 24–26).
On the other hand, trans glycan ligands of CD22 have been shown to play several clear roles in regulating the activity of CD22 in a BCR-dependent and -independent manner. When presented on the same cell surface as the cognate antigen for the BCR, CD22 glycan ligands drive co-localization of CD22 and the BCR, resulting in strong Lyn-dependent inhibition of BCR signaling(10). Such effects could play an important role in preventing autoantibody responses to cell surface autoantigens and alloantigens. CD22 has also been shown to be involved in homing of B-cells to several different compartments, which appears to be mediated through interactions with trans glycan ligands on high endothelial venules (HEVs) in these tissues. Originally shown in the bone marrow (BM), HEVs stain brightly with CD22-Fc and long-term homing assays have revealed a role for CD22 in recirculating to the BM(27). More recently, HEVs in Peyer’s patches (PP) and, to a lesser extent the mesenteric lymph nodes, were shown to mediate CD22-dependent homing of B-cells to these locations(28). While these are roles for trans ligands, cis ligands also have the potential playing a role in these events since cis ligands ‘mask’ the ability of CD22 to interact with trans ligands. Altered masking of CD22, such as the ‘unmasking’ of CD22 in the germinal center (GC) that occurs through subtle changes in glycan ligands on GC B-cells, has the potential to fine tune the activity of B-cells in different ways(29). In this regard, it is noteworthy that defects in memory B-cell formation were recently been reported in CD22−/− mice(30).
Given the restricted expression pattern of CD22 on B-cells and its ability to modulate B-cell function, CD22 has garnered significant attention as a therapeutic target. Oncology and systemic lupus erythematosus (SLE) have been two areas of particular interest involving CD22 as a therapeutic target. The majority of B-cell lymphomas express high levels of CD22 and variety of anti-CD22 antibody drug conjugates and bispecific antibodies aimed at destroying B-cell lymphomas are currently in pre-clinical and clinical trials(31). An unconjugated anti-CD22 monoclonal antibody (Epratuzumab) has also shown clinical efficacy in multiple clinical trials involving B-cell lymphomas (non-Hodgkin, acute lymphoblastic leukemia, and diffuse large B-cell lymphoma)(32–36). Epratuzumab showed positive results in early Phase II clinical trials(37–40), but failed to meet the primary endpoint in two larger Phase III trials(41). Despite the fact that Epratuzumab did not have a robust signal in Phase III trials, the mechanism by which it modulates B-cell function continues to be a topic of interest, with a recent study showing that this antibody can augment responses to TLR7. This is an agreement with a previous study that described CD22−/− B-cells as having hyper-responsiveness to a variety of TLR stimulations(42).
The demonstration that CD22 ligand and antigen presented on the same surface drive co-localization of CD22 and BCR has been exploited in the development of Siglec-engaging tolerance-inducing antigenic liposomes (STALs)(10–12, 43). Enforcing ligation of CD22 and the BCR has been shown to strongly inhibit basal BCR signaling, resulting in induction of antigen-specific B-cell apoptosis, which results in deletion of the antigen-specific B-cells from the B-cell repertoire and giving rise to immunological tolerance to the antigen of interest(10, 43). In this way, STALs have been shown to successfully induce tolerance to a biotherapeutic (FVIII) and in allergy (major peanut allergen, Ara h 2)(43, 44). Accordingly, exploiting the ability of CD22 to inhibit BCR signaling represents an attractive therapeutic strategy for guiding antibody responses and it is noteworthy that engaging hCD22 on human primary B-cells has been shown to generate a similar pro-apoptotic pathway in vitro, although the potential of hCD22 to induce antigen-specific B-cell tolerance in vivo has not been tested.
A mouse model to investigate the role of hCD22 in health and disease will be crucial as a pre-clinical model for emerging therapeutics and to understand basic aspects of CD22 B-cell biology. This is particularly important since mCD22 and hCD22 have subtle differences in the glycan ligands they prefer(45), and for this reason ligands with differential affinity and selectivity have been developed for mCD22 and hCD22(12, 46). Human knock-in (Huki) hCD22 mice were previously generated by replacing the mouse Cd22 gene with the cDNA encoding human Cd22. Although the expression of hCD22 in the Huki mouse B-cells was much lower than found in human B-cells, certain functional aspects of hCD22 were recovered in these mice in comparison to CD22−/− mice, such as antibody responses and the endocytotic properties of CD22. However, other important biological features of CD22 were not recapitulated, such as: restoration of B-cell maturation, homing to the BM, and regulation of BCR signaling. It was concluded that the failure of hCD22 to fully functionally substitute for mCD22 on murine B-cells may have been due to either: (1) differences in the ability of the ITIM motifs to recruit SHP-1, (2) subtle differences in interactions of the glycan ligands between mouse and human, or (3) the 10–30× lower expression levels of hCD22 on the surface compared to primary human B-cells(47).
Here we have developed a new transgenic mouse model in which hCD22 is expressed at high levels on B-cells in absence of mCD22. We find that hCD22 regulates BCR signaling in a manner comparable with mCD22. Specifically, compared to CD22−/− mice, mice that only express hCD22 generate antibody responses to TI and TD antigens to the same extent as WT mice, have a normal marginal zone B-cell compartment, and develop antibody responses to an oral antigen to the same degree as WT mice. Furthermore, we find that homing to PP were only partially rescued in hCD22+, while STALs targeting hCD22L in the transgenic mice were able to induce robust immunological tolerance. These results demonstrate that BCR-dependent functions of CD22 are recapitulated by hCD22 in murine B-cells, providing this as an attractive model for pre-clinical studies examining strategies to modulate B-cell functional through hCD22.
Materials and Methods
Animal Studies
The Scripps Research Institute Institutional Animal Care and Use Committee approved all experimental procedures involving mice. CD22−/− were obtained from L. Nitschke (University of Erlangen, Erlangen, Germany). The Scripps Research Institute rodent breeding colony provided wild-type (WT) C57BL/6J mice. Human CD22 transgenic mice were generated by inserting the hCD22 cDNA into the ROSA-26 locus. These mice were then crossed with MB1-cre knock-in mice(48) to drive expression of the hCD22 transgene selectively in B-cells since a STOP(flx/flx) sequence was present upstream of hCD22. Following the generation of mice that expressed both hCD22 and mCD22, these mice were further bred with mCD22−/− mice to generate mCD22−/−hCD22+ transgenic mice. Integration of the hCD22 transgene was verified by PCR of tail snips as well as through expression of hCD22 on B-cells by flow cytometry. All hCD22+ mice used in these studies contain a single copy of hCD22 and are referred to as hCD22+ throughout the text.
Immunization and blood collection
Whole blood (50 μl) was collected from mice via a retro-orbital bleed to obtain the serum after centrifugation (17,000 g, 1 minute). Serum was aliquoted and stored at −20°C. Liposomes were delivered via the lateral tail vein (I.V.) in a volume of 200μl/mouse. For studies involving a challenge with non-liposomal antigen, mice were injected with either 200μl of OVA-Alum consisting of 100 μg total OVA or 100 μg of NP-Ficoll delivered intraperitoneally (I.P.).
Peanut Allergy Model
Oral sensitization and challenge of mice with crude peanut extract was performed similar to a previously published protocol(44), with minor modifications to account for carrying the model out in C57Bl/6J mice. Specifically, mice were sensitized with 5 mg of crude peanut extract mixed with 15 μg of cholera toxin via gavage for four consecutive weeks. One week after the final sensitization, mice were bled via a retro-orbital bleed, followed by challenge with 1.5 mg of crude peanut extract via I.P. injection and rectal temperature was measured with RET-3 probe (Physitemp Instrument) at 15, 30, 45, and 60 minutes following the challenge.
Flow Cytometry
An LSR II or CantoII flow cytometer (BD Biosciences) was used with up to eight or ten colors, respectively. Briefly, a single cell suspension was obtained and Fc receptors blocked with purified anti-mouse CD16/32 antibody (BioLegend) in FACs Buffer (BSA, BD Bioscience) for 10min on ice. Following the Fc block, the primary antibodies were added in at a concentration directed by the manufacturer or fluorescent liposomes (40μM) for 1hr on ice, then washed 3× with FACs buffer to remove excess antibodies. These cells were stored on ice until run on a flow cytometer. Annexin V was used as directed by the manufacturer (ThermoFisher). Data were analyzed using FlowJo.
Microscopy
Purified B-cells (20 × 106/ml) were incubated at 37°C for 30 min. Cells (1 × 106; 100 μl) were plated onto poly-L-lysine coverslips (BD Biosciences). After 5 min, the medium was gently removed, and chilled 3% paraformaldehyde was added for 5 min at 4°C. Cells were washed twice with PBS and blocked with 5% normal goat serum in PBS for 30 min at room temperature. Slides were probed with FITC-labeled anti-mouse CD22 (1:200), and biotin-labeled anti–human CD22 (20 μg/ml final) in 1% normal goat serum overnight at 4°C. The following day, slides were washed with PBS and stained for 2 hr with anti-FITC AF488 and streptavidin-AF555. Following washing of the slides PBS, slides briefly incubated with 1 μg/ml Hoescht and mounted in anti-fade medium. Images were acquired on a Zeiss confocal microscope at 63× magnification using oil emersion. Quantification of overlap between the signals representing mCD22 and hCD22 was carried out on 7 individual B-cells using ImageJ software and the Coloc2 plug in.
Calcium Flux
Splenocytes were resuspended at 15 × 106 cells/ml in RPMI medium containing 1% FCS, 10 mM HEPES, 1 mM MgCl2, 1 mM EGTA, and penicillin-streptomycin 1% and 1 μM Indo-1 (Invitrogen). Cells were incubated in a 37°C incubator for 30 minutes. Following incubation, a 5-fold volume of the same buffer (without Indo-1) was added and the cells were centrifuged (300 g, 5 minutes). PBMCs were stained with anti-mouse B220 and anti-mouse CD5, for 30 min and washed 2×. Cells were stored on ice in calcium flux running buffer (HBSS, 1% FCS, 1mM CaCl2) and an aliquot (0.5 ml; 1 × 106 cells) was warmed (37°C, 5 minutes) prior to initiating calcium flux measurements. During flow cytometry acquisition and analysis B220+CD5− cells were gated to establish the B-cell population. Cells were stimulated with liposomes or anti-IgM F(ab)’2 fragments (Jackson ImmunoResearch) and Indo-1 fluorescence (violet vs. blue) was monitored by flow cytometry (500–1000 events/s) for 3–5 minutes at 37°C. Stimulation always took place 10 seconds after starting acquisition so that background could be established. For experiments involving GFP+ vs GFP− mice, the GFP is expressed in all tissues and is not cell type specific, the splenocytes were mixed at a 1:1 ratio prior to loading with Indo-1. Data were analyzed using FlowJo using the kinetics functions.
ELISAs
Maxisorp plates were coated (O/N, 4°C) with the relevant protein (100μl/well, 10μg/ml) in PBS. NP4-7-BSA in PBS (Biosearch Technologies) was used to look for anti-NP antibodies. The following day, plates were washed 6× with PBS-T (0.1% Tween 20) and blocked (2 hour, RT) with PBS with 0.1% Tween 20 and 1% BSA. Serum was initially diluted between 10- and 100-fold followed by 2- to 3-fold serial dilution 8 times on the ELISA plate. Plates were incubated (2 hour, RT) with serum (100μl/well), washed 6× with PBS-T, and incubated (1 hour, RT) with the appropriate HRP-conjugated secondary antibodies (1:2000; Santa Cruz Biotechnology Inc.). Following 6 washes in PBS-T, plates were developed (15 minutes, RT) in 75μl/well of TMB substrate (ThermoFisher) and quenched with 75μl/well of 2N H2SO4. Absorbance was measured at 450 nm, and the titer was calculated as the dilution of serum that produced the half-maximal absorbance.
Short-term homing assays
Splenocytes were isolated from wild type, mCD22−/−, or hCD22−/− mice, and stained with CFSE (1μM), CTV (5μM), or both. The staining was switched in every experiment to rule out an effect of the dye. Recipient wild type mice were injected via the tail vein with equal numbers of donor splenocytes that were pre-mixed just prior to injection. After 1.5 hour, T and B lymphocytes from spleen, peripheral lymph nodes (inguinal, axillary, brachial), mesenteric LNs, and Peyer’s patches were isolated and stained with anti-CD3, anti CD4, anti-CD19 and anti-IgD. Propidium iodide was used as a live/dead staining and counting beads were added to calculate total numbers of cells. The homing of live CD19+ IgD+ B-cells, and live CD3+ CD4+ T-cells from each donor was evaluated. The efficiency of B-cell homing to each organ is presented as a relative localization ratio (RLR):the number of B-cells found in each organ is divided by the number of B-cell injected (input), then normalized to the number of T-cells found in the organ divided by the number of T-cells injected (input).
In Vitro B-cell STAL assay
Hy10 hen egg lysozyme (HEL)-specific (49) B-cells from hCD22+ or CD22−/− mice were purified using magnetic bead sort (Miltenyi) with a purity >93% B-cells and cultured at 1×105 B-cells in U-bottom well for 24hrs under the following conditions: unstimulated, stimulated (40 μM Duck Egg Lysozyme (DEL) Liposomes) or DEL+hCD22L (40 μM DEL STALs). B-cells were then stained as described above and analyzed by flow cytometry.
Protein-lipid conjugation
A protocol for the preparation protein-lipid conjugation is described in detail elsewhere(43)
High affinity murine and human CD22 ligands
The synthesis of the high affinity mCD22L, described as 6′BPA-Neu5Gc, and high affinity hCD22L, described as 6′MBP-5F-Neu5Ac, were prepared as described previously(12, 46).
Liposomes
A protocol for the preparation of liposomes is described in detail elsewhere(43). Briefly, all liposomes used in these studies were extruded through a 100 nm filter and passed through a CL-4B column following extrusion. Liposomal composition consisted of 0.03% antigen (OVA), 1.5% CD22L, and 0.1% AF647, all of which were linked to PEG-DSPE according to previous reports (43, 50, 51). Primary human B-cells were isolated from healthy donor blood (The Scripps Research Institute normal blood donor service (NBDS).
Statistics
Statistical significance was determined using an unpaired 2-tailed Student’s t test. P < 0.05 was considered significant.
Results
Human CD22 Expression on B-cells and Ligand Binding
With the goal of developing a transgenic mouse that expresses human CD22 (hCD22) in a B-cell specific manner, the hCD22 transgene was inserted into the ROSA-26 locus with a STOP(flx/flx) cassette upstream of the hCD22 gene(52). To drive expression of the hCD22 transgene exclusively on B-cells, we took advantage of mice that express Cre recombinase under the expression of the B-cell specific mb1 gene. Mb1 encodes the Ig-α signaling subunit of the B-cell receptor and is exclusively expressed in B-cells starting at the pro-B-cell stage in the bone marrow(48). Cre recombinase knocked into the MB1 locus drives expression of Cre in a B-cell specific manner in over 99% of mature B-cells. Accordingly, conditional hCD22-expressing mice were crossed with MB1-Cre mice to drive expression of hCD22 in all B-cells. Initially, this generated mice that expressed both mCD22 and hCD22, which allowed us to determine if the localization pattern of the hCD22 is comparable to mCD22 on the surface of B-cells. B-cells purified from these mCD22+hCD22+ chimeric mouse were analyzed by confocal microscopy for mCD22 and hCD22. Nearly perfect co-localization of mCD22 and hCD22 was observed for single B-cells as well as at the cell-cell interface in cases where two B-cells were in contact (Figure 1A). To quantify the overlap in the single cells, we calculated the Manders’ overlap coefficient (MOC) in 7 individual cells and found that the MOC for hCD22 with mCD22 was 0.86 +/− 0.08, while the MOC for mCD22 with hCD22 was 0.80 +/− 0.11. Given that previous evidence has shown that CD22 localization on individual B-cells (53) and at the site of cell contact(54) is regulated by cis and trans glycan ligands of CD22, respectively, these results demonstrate that the glycan ligands expressed on murine B-cells are capable of correctly regulating the membrane organization of hCD22.
Figure 1. Human CD22 Expression on B-cells its binding to glycan ligands.
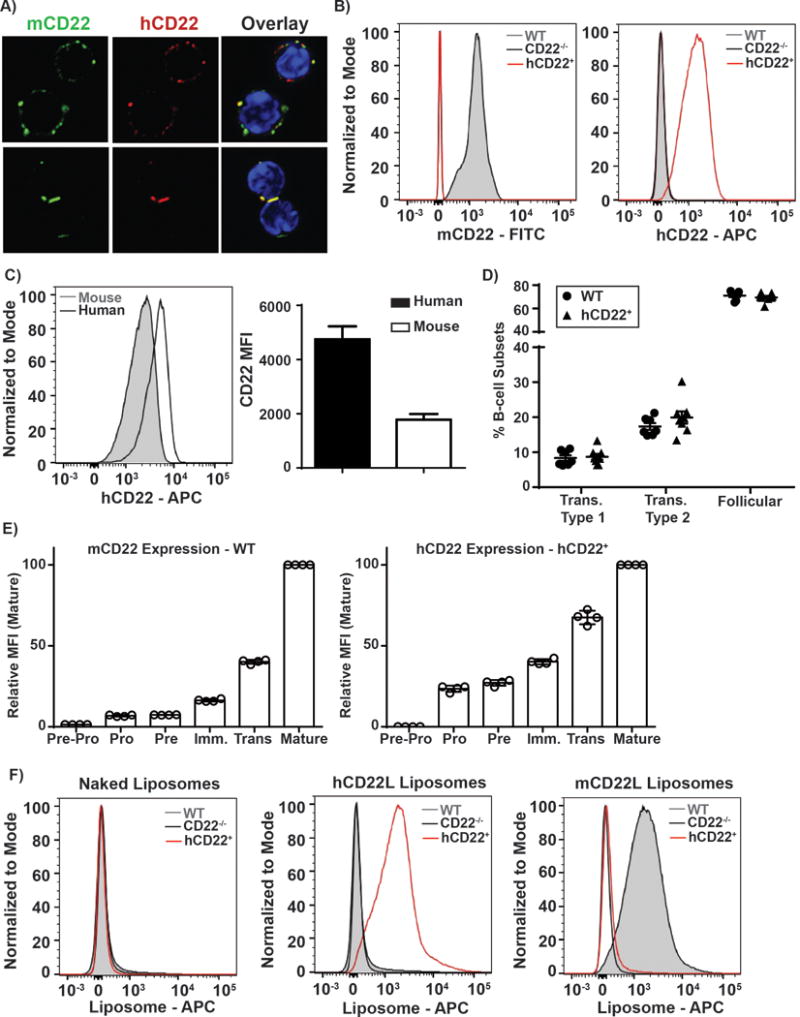
A) B-cells from a mouse expressing both hCD22 and mCD22 were stained with Hoechst, a fluorescent staining dye of DNA and nuclei, mCD22-FITC, and hCD22-PE. Representative images of cells in suspension and cell-cell contact were taken on a Zeiss confocal microscope at 63x magnification. B) WT, hCD22+, and CD22−/− splenocytes were Fc-blocked and stained with B-cells markers: CD19-AF700, B220-BV605, mCD22-FITC, and hCD22-APC. Murine B-cells (CD19+B220+) were analyzed for mCD22 vs. hCD22 markers, representative histograms are presented (left panel, N=6 and middle panel, N=6). C) B-cells from healthy control donors or hCD22+ mice were gated on B-cells, as defined as CD20+CD19+ and murine B-cells as described above were assessed for expression levels of hCD22 (right panel, N=4). D) WT (N=7) and hCD22+ (N=8) splenocytes were stained for B-cell subsets for CD19, B220, mCD22, CD23, CD24, hCD22, and CD21/35. E) WT (N=4) or hCD22+ (N=4) bone marrow cells were stained for mouse or human CD22 expression, respectively, during different stages of B-cell development: B220, CD19, IgM, IgD, CD43, CD24, cKit. F) WT, hCD22+, and CD22−/− splenocytes were Fc-blocked and stained with B-cells markers: CD19-AF700, B220-BV605 and liposomes that incorporated AF647 fluorophore. Murine B-cells (defined by CD19+B220+) were analyzed for AF647 binding to naked liposomes (left panel, N=4), hCD22L decorated liposomes (middle panel, N=4), or mCD22L decorated liposomes (right panel, N=4).
Mice expressing both mCD22 and hCD22 were subsequently bred with mCD22 knock-out (mCD22−/−) mice(20) to generate mice that exclusively express hCD22 on B-cells, which we will from here on out refer to as hCD22+ mice, unless otherwise noted. Flow cytometry analysis of mCD22 and hCD22 expression demonstrated a restricted expression pattern of mCD22 and hCD22 on the B-cells from WT and hCD22+ mice, respectively (Figure 1B). All B-cells in the hCD22+ mice expressed hCD22 at levels comparable to endogenous mCD22, although a direct quantitative comparison cannot be made given that different antibodies were used. Importantly, we were able to directly compare expression levels of hCD22 on these transgenic B-cells to levels on primary human B cells and find that the expression level of hCD22+ on these transgenic B-cells is similar to levels on human B-cells, within ~2.5-fold difference (Figure 1C).
We considered that an inappropriate expression pattern of CD22 on developing B-cells within the bone marrow could be detrimental given that BCR signaling strength is critical at this stage and influences B-cell subset populations in the periphery(24, 30). We find that hCD22+ mice have similar percentages of splenic B-cell subsets compared to WT mice, including: transitional type 1 (CD19+B220+CD24+CD23−CD21/35−), transitional type 2 (CD19+B220+CD24+CD23+CD21/35lo) and follicular B-cells (CD19+B220+CD24−CD23+CD21/35int) (Figure 1D). Consistent with these findings, we find that expression of the hCD22 transgene in the bone marrow follows a similar trend as mCD22 (Figure 1E). Specifically, we find that – similar to mCD22 - pre-pro-B-cells (B220+CD19−IgM−IgD−CD43+CD24−cKit−) lack expression of hCD22, while expression of either hCD22 or mCD22 first becomes detectable at low levels starting at the pro-B-cell stage (B220+CD19+IgM−IgD−CD43+CD24+cKit+), with increasing expression through Pre (B220+CD19+IgM−IgD−CD43loCD24+cKit−), immature (B220+CD19+IgM+IgD−CD43−CD24+cKit−), transitional (B220+CD19+IgMhiIgDloCD43−CD24+cKit−), and mature B-cells (B220+CD19+IgMloIgD+CD43−CD24+cKit−). While the relative expression is not identical at stage of development, this reflects the higher expression of mCD22, in comparison to hCD22, on the mature B-cells. Regardless, the similar trend in hCD22 and mCD22 expression during B-cell development clearly results in no difference in B-cell subsets in the spleen.
Results above showing that hCD22 can be recruited to the site of contact between two B-cells is strong evidence that hCD22 is able to recognize glycan ligands in trans. To more systematically and quantitatively demonstrate this ability, we used fluorescent liposomes bearing high affinity ligands for either mCD22 or hCD22(12, 46). Accordingly, we tested these fluorescent liposomal nanoparticles for binding to WT, hCD22+, or CD22−/− B-cells ex vivo and found that only the hCD22L liposomes bound to hCD22+ B-cell, while only mCD22L liposomes bound to mouse WT B-cells (Figure 1F). CD22−/− B-cells bound to neither liposome. These data establish that B-cells from the hCD22+ mice express functional cell surface hCD22 that is: expressed in the same membrane microdomains as mCD22, expressed at levels on the cell surface that is within a 2-fold range on primary B-cells, and is functionally competent to engage glycan ligands in trans.
BCR signaling and T-Dependent Antibody Responses in hCD22 transgenic mice
Under the appropriate physiological circumstances, CD22 can modulate BCR signaling through recruitment of Src homology (SH2) domain-containing proteins, such as SHP-1. Previous studies have established that B-cells from CD22−/− mice have hyper-responsive Ca2+ flux following stimulation of the BCR by anti-IgM(20). To determine the potential of hCD22 to regulate BCR signaling in response to anti-IgM, we analyzed calcium flux in WT, CD22−/− and hCD22+ B-cells. To do so in quantitative manner, we developed an internally-controlled approach wherein WT GFP+ splenocytes were mixed with splenocytes from either of the three types of mice at a 1:1 ratio, loaded with a Ca2+ flux sensitive dye (Indo-1), and stimulated with anti-IgM followed by analysis by flow cytometry. Ca2+ flux in B-cells (B220+CD5−) from both the GFP+ and GFP− mice were analyzed in parallel to directly compare between the two genotypes of interest. Using this strategy, CD22−/− B-cells reproducibly generate a 50–60% increase in the area under the curve (AUC) compared to WT B-cells (Figure 2A). On the other hand, calcium flux in hCD22+ B-cells was at the same levels of WT B-cells.
Figure 2. Regulation of BCR signaling and T-Dependent Antibody Responses by hCD22.
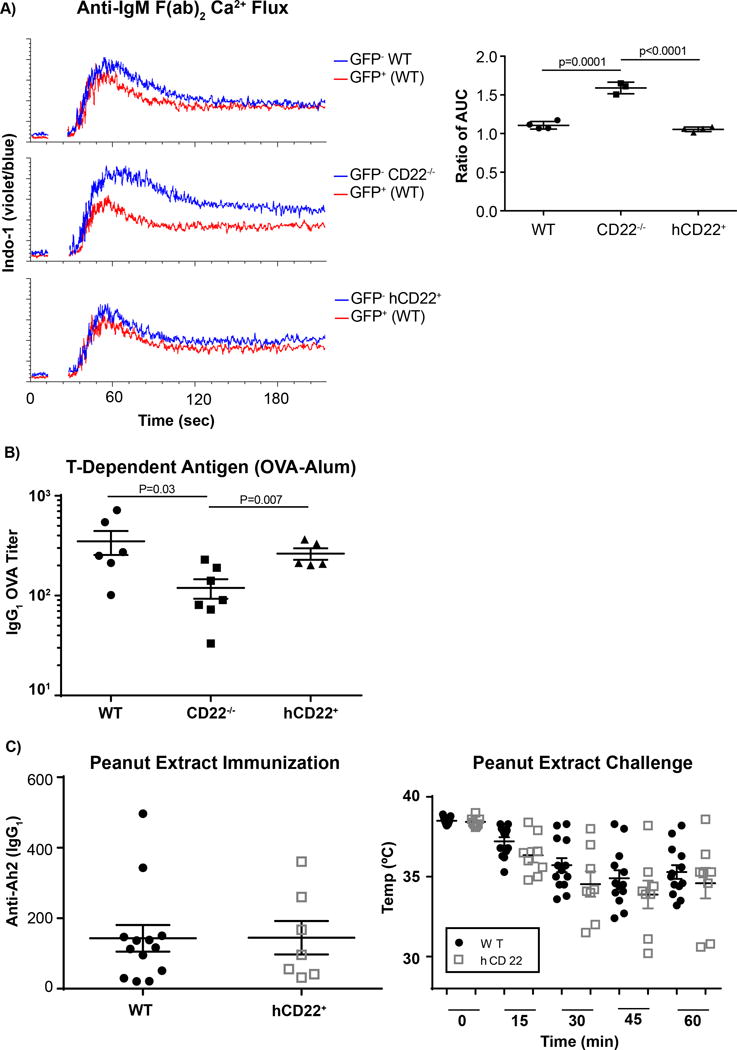
A) Splenocytes from WT-GFP+ mice or WT (top panel, N=4), CD22−/− (middle panel, N=4) or hCD22+ (bottom panel, N=4) were mixed 1:1 and then stained with Indo-1 calcium sensitive dye, after cells were stained for CD19 and CD5. During acquisition, B-cells were defined as CD19+CD5− and monitored for changes the violet versus blue fluorescence of Indo-1. B) WT (N=6), CD22−/− (N=7) or hCD22+ (N=5) mice were injected intraperitoneal with OVA-Alum. Mice were bled 2 weeks after the initial injection and IgG1 titers were assessed for OVA. C) WT (N=13) and hCD22+ (N=7) were orally sensitized with crude peanut extract long with cholera toxin for four consecutive weeks. Following sensitization to peanut, antibody titers to the major peanut allergen (Ara h 2) were determined by ELISA. Mice sensitized to peanut were challenge I.P. with peanut extract and an anaphylactic response, characterized by a decrease in body temperature, was monitored over the first hour following the challenge.
CD22−/− mice have been reported to have defective antibody responses in response to both T-Dependent (TD) and T-Independent (TI) antigens(55), which could stem from multiple mechanisms. WT, CD22−/−, or hCD22+ mice were immunized with OVA-Alum - a T-dependent (TD) antigen - and anti-OVA IgG1 titers were assessed 14 days post-immunization. Previously, a modest defect in TD responses in CD22−/− mice was reported(55), which was recapitulated in our immunizations (Figure 2B). hCD22+ mice were able to recover this minor defect to levels that are not statistically different as WT mice. To further assess whether a TD antibody-driven disease can be induced at WT levels in hCD22 mice, WT and hCD22+ mice were orally-sensitized with crude peanut extract along with cholera toxin, which is an established mouse model for food allergies(56, 57). Following four consecutively weekly oral doses of crude peanut extract, antibody titers to one of the major peanut allergens (Ara h 2) were determined by ELISA and found to be at similar levels between the two groups of mice (Figure 2C). Following this sensitization scheme, mice were challenged one week later with peanut extract via I.P. administration, and body temperature was monitored via rectal probe over the first hour post-challenge as a readout of the anaphylactic response. We find no statistical significance between the two groups of mice at any time point, nor the percentage of responders (Figure 2C). These data show that B-cells from our hCD22+ transgenic mice have normal BCR signaling, hCD22+ mice mount normal antibody responses to TD antigens, and hCD22+ can be orally sensitized to antigen to drive an allergic response at the same levels of WT mice.
Recovery of T-Independent Antibody Responses and Marginal Zone B-cells
Marginal zone (MZ) B-cells are strategically positioned in the spleen to respond to multivalent antigens in the blood that are often thought to act as TI antigens (58). Therefore, WT, CD22−/−, and hCD22+ mice were injected with a T-Independent type 2 (TI-2) antigen (NP-Ficoll), and anti-NP titers were analyzed over time. As reported previously(24), CD22−/− mice mounted a blunted TI-2 response compared to WT mice in both IgM and IgG3 isotypes, while hCD22+ mice mounted TI-2 responses comparable to WT mice on days 12 and 18 post-immunization (Figure 3A). BCR signaling strength during B-cell development is thought to play a role in selecting MZ B-cells and it has been previously shown that CD22−/− mice have greatly decreased numbers of MZ B-cells, which may be due to difference in BCR signaling that influences B-cells to favor differentiation into follicular B2 B-cells (59). Accordingly, we examined if the MZ B-cell compartment was normal in hCD22+ mice given that B-cells from these mice had normal Ca2+ flux responses to anti-IgM. Therefore, splenocytes from WT, CD22−/−, and hCD22+ mice were analyzed for the levels of MZ B-cells, as we define as CD19+B220+CD23−CD21/CD35+CD1d+ (24). As reported previously, CD22−/− mice had striking decreases in MZ B-cell compartment, while the hCD22+ transgenic mice had a MZ B-cell compartment that was restored to levels that were not statistically different than WT mice (Figure 3 B, C).
Figure 3. Recovery of T-Independent Antibody Responses and Marginal Zone B-cell in hCD22+ mice.

A) WT, CD22−/− or hCD22+ mice were injected intraperitoneal with NP-Ficoll (N=8/group). Mice were bled once every 6 days and IgM or IgG3 titers were assessed for NP-7. B, C) Splenocytes from WT (left panel, N=6), mCD22−/− (middle panel, N=7), or hCD22+ (right panel, N=7) mice were Fc blocked and stained for MZ B-cells as follows: B220-BV605, CD19-AF700, CD23-Percp-Cy5.5, CD21/35-APC-Cy7, and CD1d-PE. B-cells were gated on as follows B220+CD19+CD23− and assessed for expression of CD1d and CD21/CD35, MZ B-cells were defined as CD1d+CD21/CD35+.
Partial Rescue of Homing to Peyer’s Patches and Mesenteric Lymph Nodes
We have reported previously that CD22−/− B-cells have markedly impaired homing to Peyer’s patches (PP), moderately impaired homing to mesenteric lymph nodes, but normal homing to peripheral lymph nodes and spleen in short-term homing assays (28). The proposed mechanism of action for CD22 in mediating this homing is through differential binding to its endogenous sialic acid-containing glycan ligands on HEVs in these select locations. Because the ligand binding specificity of hCD22 strongly overlaps with that of mCD22 (60), we hypothesized that the deficiency in B-cell homing to PP and mesenteric lymph nodes observed in CD22−/− mice might be restored in hCD22+ mice. To test this hypothesis, an experimental approach was used as described in Figure 4A, wherein we performed a complex mixing and transfer experiment. Splenocytes from WT, CD22−/−, and hCD22+ mice were labeled with a different combination of two fluorescent dyes in order to track the individual cell types. In the example shown in Figure 4A, CD22−/− splenocytes are labeled with CFSE, hCD22+ splenocytes with CTV, and WT splenocytes with both CTV and CFSE (Figure 4A). Moreover, to eliminate the possibility that labeling with different fluorophores biases the results, three independent experiments were conducted where all combinations of genotypes were put into each fluorescent color. Accordingly, splenocytes from all three genotypes were mixed at a 1:1:1 ratio after labelling and transferred into a WT recipient. This transfer system allowed us to monitor the ability of all three genotypes to home to different organs at the same time within the same controlled environment. After an hour and half, organs were removed to assess homing by flow cytometry for CD4+ T-cells (CD4+CD3+) and B-cells (IgD+CD19+) along with the tracing dyes (CFSE, CTV, and dual staining). Based on the fact that T-cells do not express CD22 endogenously or in our transgenic mice, data was normalized to T-cell homing according to the equation described in Figure 4A. We find that hCD22+ B-cells had a statistically significant increase in homing efficiency to both the PP and mesenteric lymph nodes in comparison to CD22−/− B-cells (Figure 4B, C). Organs such as peripheral lymph nodes and spleen previously documented to exhibit CD22-independent homing, had similar relative homing in all three genotypes. Nevertheless, B-cell homing to the PP and mesenteric lymph nodes in hCD22+ mice was not fully restored to the levels observed for WT B-cells. Since the CD22 ligands that mediate homing in these tissues are not fully described, it is possible that there are differences in the recognition by hCD22 and mCD22 that impact this homing function. These results demonstrate that while many of the functions of mCD22 can be restored by hCD22 in mice, not all aspects of CD22 biology are fully recapitulated by hCD22 in mouse.
Figure 4. Partial Rescue of Homing to Peyer’s Patches and Mesenteric Lymph Nodes in hCD22+ B-cells.
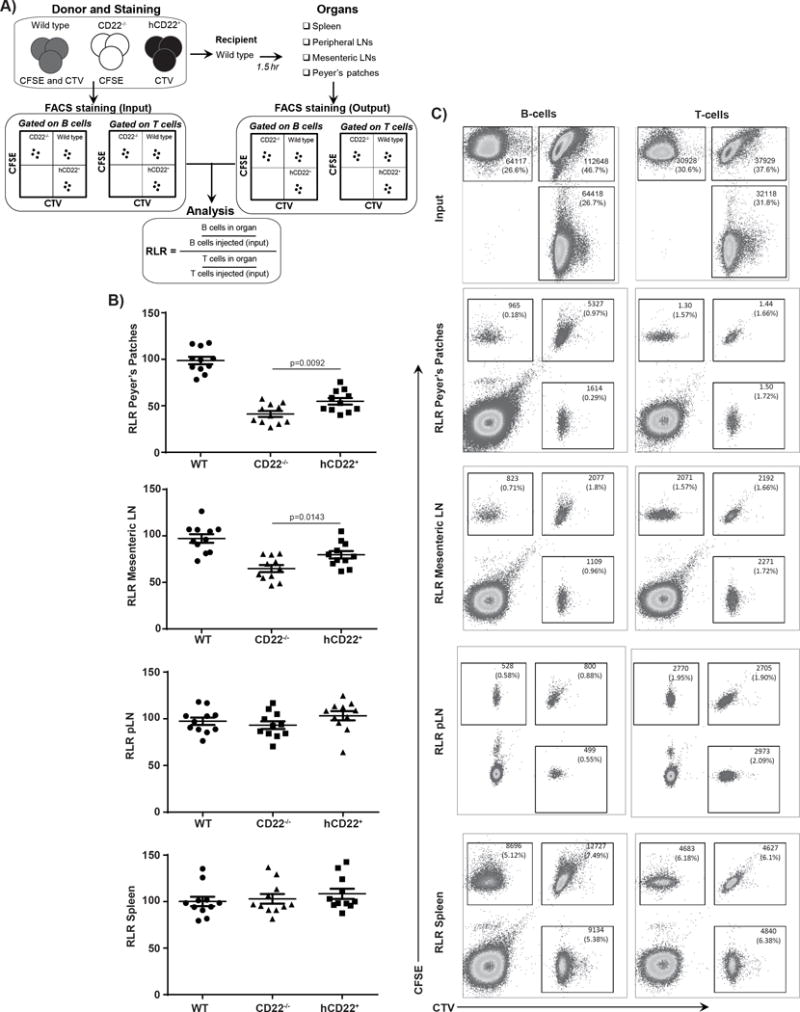
A) WT, CD22−/−, or hCD22+ mice splenocytes were harvested and each was stained with one of the following combinations: CFSE, CTV or CFSE+CTV. The cells were then mixed at a 1:1:1 ratio and injected into a WT recipient host (N=4/experiment, N=3 independent experiments). A small portion was used as an “input” control to measure the ratio of each phenotype injected in at the start. All cells were stained the same before and after harvest, cell were first Fc blocked and then stained with anti-CD3ε, CD4, and CD19, anti-B-cell antibody B220, to classify T-cell and B-cells, respectively, Propidium Iodide was added just before acquisition to determine live/dead cells. 1.5hrs after injection, mice were euthanized and harvested for multiple tissues: spleen, peripheral lymph nodes, mesenteric lymph nodes, and Peyer’s patches. Upon harvesting cells were once again stained for T and B-cells and subsequently gated for live cells and CTV vs CFSE to give relative ratios of T and B-cells. B,C) Relative localization ratio (RLR) is produced from 3 independent experiment (N=4 mice/experiment). Representative pseudo-color plots are shown.
Induction of B-cell Tolerance by STALs in hCD22+ Mice
Siglec-engaging tolerance-inducing antigenic liposomes (STALs) are liposomal nanoparticles that display both an antigen of interest and a Siglec ligand on their surface, which serve to co-ligate a Siglec with the BCR on the B-cell surface(43). Strong inhibition of BCR signaling is observed towards the antigen on STALs, so much so that even basal BCR signaling can be inhibited(43), resulting in BIM-dependent B-cell apoptosis(10). To determine if hCD22 in our transgenic mice is also capable of inducing this apoptotic mechanism, we bred WT, CD22−/−, and hCD22+ mice onto a Hy10 knock-in background. B-cells from Hy10 mice are specific for Hen Egg Lysozyme (HEL) with the BCR binding to HEL at an extremely high affinity (Ka=4.5×1010 M−1)(61, 62). In contrast, Hy10 BCR binds to duck eggs lysozyme (DEL) at a lower, more physiologically relevant affinity (Ka=1.3×107 M−1). Therefore, splenic B-cells isolated from all 3 genotypes (WT, hCD22+ or CD22−/−) were stimulated with PBS, antigenic DEL liposomes, or DEL STALs displaying either the high affinity mCD22L or hCD22L, and Ca2+ flux responses were monitored by flow cytometry. In line with our previous reports(43, 44), STALs formulated with the mCD22L alone failed to induce Ca2+ flux in WT Hy10 B-cells, while antigenic DEL liposomes showed a robust Ca2+ flux response as expected. Ca2+ flux in WT B-cells were completely abolished by DEL STALs with mCD22L. Similarly, B-cells from Hy10 mice expressing hCD22+ likewise failed to respond to DEL STALs formulated with hCD22L, demonstrating that transgenic expression of hCD22 in B-cells can strongly inhibit the BCR signaling upon co-ligation. Finally, in CD22−/− Hy10 B-cells, Ca2+ responses were strongly induced by all three types of liposomes, confirming that the STAL inhibitory effect on B-cell activation is indeed mediated by CD22L and CD22 interactions (Figure 5A).
Figure 5. Inhibition of B-cell activation and induction of apoptosis by STALs.
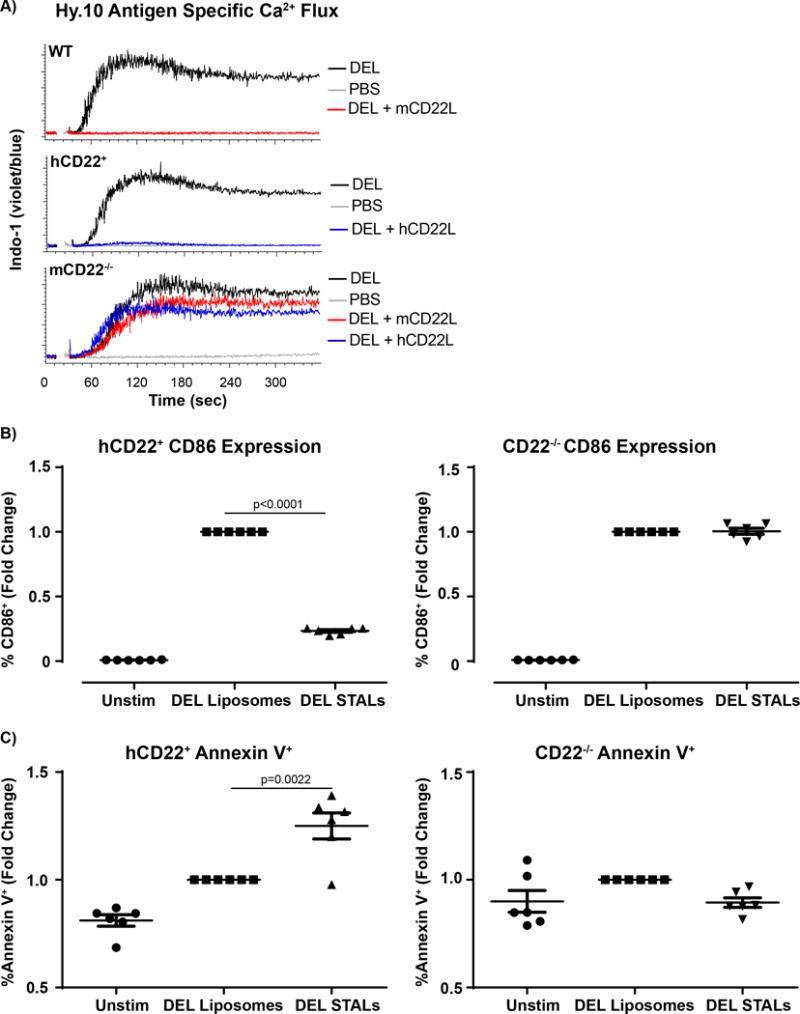
A) Splenocytes from Hy10 mice on a WT (Top panel N=3), hCD22+ (middle panel, N=3) or CD22−/− (bottom panel, N=3) background were stained and gated on as previously described in Figure 2. Splenocytes were stimulated with 5μM liposomes with DEL or DEL+mCD22L or DEL+hCD22L and changes in Indo-1 fluorescence (violet:blue) was assessed by flow cytometry. B,C) Hy10 B-cells from hCD22+ or CD22−/− were purified by negative selection using magnetic bead sort and cultured at 1×105 cells/well for 24hrs under the following conditions: Unstimulated (N=6), Stimulated (DEL Liposomes, N=6) or hCD22L (40μM DEL STALs, N=6). B-cells were Fc blocked and stained with CD19, B220, CD86, Annexin V and Live/Dead to determine B-cell activation and cell death.
In addition to monitoring the initial events of B-cell activation, later events such as upregulation of CD86 and B-cell survival were analyzed at a 24hr time point. hCD22+ or CD22−/− Hy10 B-cells were cultured for 24hr with antigenic DEL liposomes, STALs with hCD22L, or left unstimulated. Compared to antigenic DEL liposomes, CD86 expression in cells incubated with STALs was significantly decreased in hCD22+ B-cells, while CD86 expression in CD22−/− B-cells remained at the same level between STALs and antigenic DEL liposomes (Figure 5B). Cell death, as monitored by upregulated staining with Annexin V, also showed similar trends (Figure 5C). Specifically, STALs significantly increased the percentage of Annexin V+ cells in the hCD22+ mice while CD22−/− B-cells failed to show this effect, demonstrating that apoptosis of hCD22+ B-cells can be induced by STALs through targeting hCD22. Together, these data show that hCD22 can substitute for mCD22 in blocking antigen-specific B-cell responses through co-ligation with the BCR using STALs, resulting in a pro-apoptotic signaling cascade similar to what has been demonstrated previously with mCD22.
In Vivo Tolerance Induction by STALs in hCD22+ mice
We have shown above that B-cells from hCD22+ mice treated with STALs in vitro fail to become activated by antigen and, instead, underwent induced cell death. In WT mice, the consequence of these actions by STALs has been shown previously to result in antigen-specific tolerance in vivo(43), which is defined as a failure to respond to subsequent challenge of antigen. In order to test if STALs can induce tolerance in hCD22+ mice, WT or hCD22+ mice were administered STALs containing OVA and the high affinity mCD22L or hCD22L, respectively (Figure 6A). Appropriate control groups of mice were given PBS or antigenic OVA liposomes. Antibody titers to OVA (anti-OVA IgG1) were determined on day 14 post-immunization. Both WT and hCD22+ mice injected with antigenic OVA liposomes generated significant anti-OVA titers by day 14, while STALs maintained anti-OVA titers at levels that were not statistically different from those in the PBS-injected mice (Figure 6B). On day 15, all mice were challenged with a high dose of antigenic OVA liposomes to assess tolerance induction, and anti-OVA titers were measured 14 days after this challenge (day 29). Mice originally treated on day 0 with PBS generated significant anti-OVA titers, while the mice previously given antigenic OVA liposomes had boosted levels of anti-OVA. In contrast, both WT and hCD22+ originally treated with OVA STALs with mCD22L or hCD22L, respectively, had titers that were significantly blunted compared to the PBS groups (Figure 6B). These data shows that exploiting hCD22 expressed in our hCD22 transgenic mice, with STALs expressing an hCD22L, can induce robust in vivo immunological tolerance.
Figure 6. In Vivo tolerance induction by STALs in hCD22+ mice.
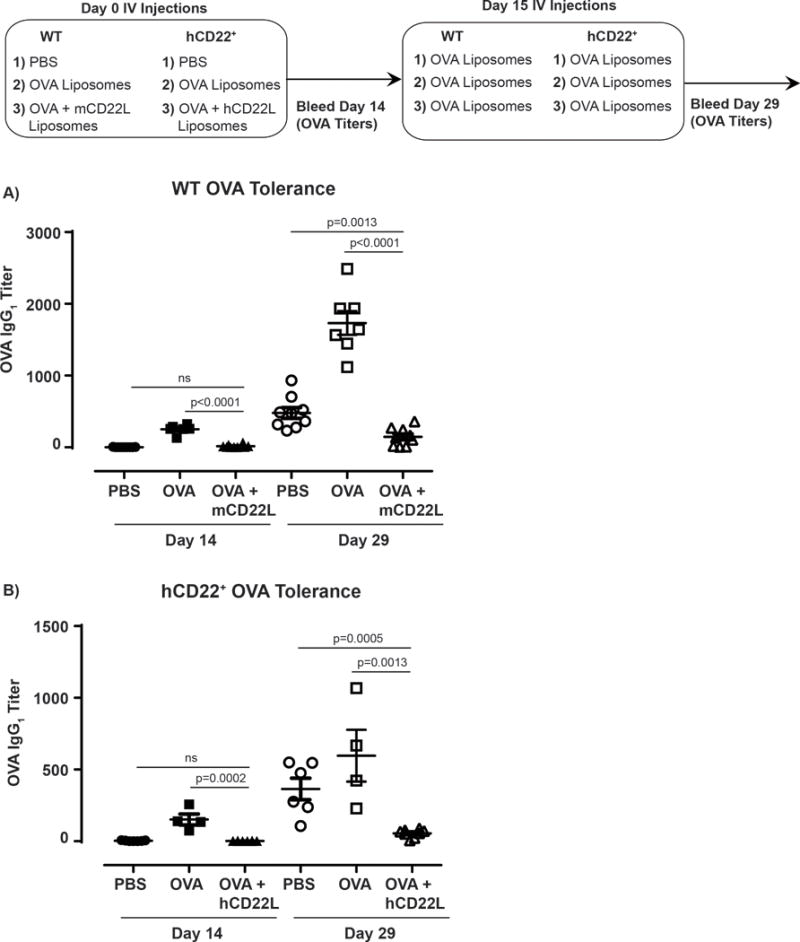
A) Schematic representation of immunization strategy. WT or hCD22+ mice were injected on Day 0 with PBS, OVA Liposomes or OVA STALs (mCD22L or hCD22L, respectively). Mice were bled on day 14 for IgG1 OVA-specific responses and all groups were boosted with OVA Liposomes. Final titers were determined 14 days after boost (day 28) for in vivo tolerization of OVA specific antibody responses. B) WT OVA tolerance summary PBS (N=9), OVA liposomes (N=6), OVA STALS (N=10). hCD22+ OVA tolerance summary PBS (N=7), OVA liposomes (N=4), OVA STALS (N=8). Representative experiment shown, 1 of 2 independent studies.
Discussion
CD22, a negative regulator of B-cell activation with restricted expression on B-cells, has gained significant interest since a number of anti-CD22 antibodies have been tested in clinical trials for B-cell lymphomas (acute lymphoblastic leukemia, follicular lymphoma, diffuse large B-cell lymphoma) and autoimmunity (systemic lupus erythematosus) (31). Targeting CD22 outside of oncology has proven to be difficult because the mechanism of action remains not fully understood, which may be partially due to an incomplete understanding for roles of CD22 in B-cell biology, as evidenced by recent studies that continue to find new roles for this critical receptor (28–30, 63). Having an animal model that recapitulates the biology of hCD22 in mouse is crucial in future basic and pre-clinical studies. In a previous study with Huki hCD22 knock-in mice, hCD22 expression on B-cells was 10–30 fold lower than human primary B-cells, which likely accounts for hCD22 not robustly substituting for mCD22 in some respects (47). Our strategy, reported here, was to drive transgenic hCD22+ expression in a B-cell-specific manner through the breeding of the well-established MB1-Cre mouse line with the transgenic hCD22 mice that requires expression of Cre to drive the transgene expression. Breeding this mouse model to a mCD22−/− background was not trivial, due to mb1-cre and CD22 residing are on the same chromosome(64), but the choice of this particular Cre-expressing line was worthwhile given that MB1-Cre drives Cre expression in >99% of B-cells, compared to another commonly used B-cell specific Cre mouse - CD19-cre - that is only 85–90% efficient(65). This mb1-cre driven hCD22 expression resulted in no differences in splenic B-cell subsets and, importantly, had a similar B-cell developmental expression pattern in the bone marrow as mCD22 in WT B-cells. Therefore, differences in receptor selection should be minimal. This transgenic hCD22 mouse has enabled us to precisely assess whether hCD22 can functionally substitute for mCD22.
In our hCD22+ mice, hCD22 is expressed at similar levels to that of mCD22 on B-cells, although a direct and quantitative comparison is difficult to make due to a variety of factors. Our hCD22+ mice do have ~2.5-fold lower expression than human peripheral blood B-cells. An interesting possible explanation for a decrease in expression is the size difference between mouse and human B-cells. It has been reported that mouse B-cells have an average diameter of 5–10μm while human B-cells range between 10–20 μm(66). This would decrease the surface area of the cell and limit the number of molecules on the membrane. Another reason for this could simply be a difference in promoter usage, although neither one is definitively proven here. An important factor that controls the ability of CD22 to negatively regulate BCR signaling is localization of CD22 to clathrin-coated pits (67). Notably, this membrane localization is driven by interactions with glycan ligands on their cognate receptors, which are neighboring CD22 receptors(68), with the net effect being that CD22 is largely secluded away from the BCR(18, 53, 69). In this aspect, it is satisfying that hCD22 on our transgenic B-cells high degree of colocalization with mCD22, demonstrating the cis glycan ligands on murine B-cells are sufficient to regulate hCD22 localization. Although the predominant type of sialic acid on murine B-cells is α2-6-linked N-glycolylneuraminic acid (Neu5Gc), while on human B-cells it is α2-6-linked N-acetylneuraminic acid (Neu5Ac), it is noteworthy that hCD22 does not discriminate between these two types of sialic acid, unlike mCD22 that strongly prefers the glycolyl version(29, 70). The ability of hCD22 to recognize sialic acid glycan-containing glycan ligands on mouse B-cells is likely a key factor in its ability to functionally substitute for mCD22 in the various aspects we report herein.
Consistent with the ability of sialic acid-containing glycans expressed on murine B-cells to regulate the membrane organization of hCD22, we have shown that B-cells from hCD22 transgenic mice have normal Ca2+ flux responses to anti-mouse IgM, as compared to hyper-responsiveness of CD22−/− B-cells. Reduced numbers of MZ B-cells in CD22−/− mice were previously speculated to be due to abnormal calcium flux responses(59), although another possible reason is the inability to home to the MZ, since MZ B-cells were shown to have ‘unmasked’ CD22(71). Regardless of the precise mechanism, a restored MZ B-cell population was clearly observed in our hCD22+ mice. This normal MZ B-cell population in hCD22+ mice may help to explain the rescue in a TI-2 antibody response in hCD22+ mice, compared to CD22−/− mice, given that MZ B-cells are thought to be important for generating antibody to highly multivalent antigens, such as TI-2 antigens(24). TD antibody responses were also normal in hCD22+ mice, as compared to modestly impaired responses in CD22−/− mice. Recovery of TD antibody responses could relate to a role for CD22 in regulating B-cell survival post-stimulation, or possibly represent a downstream role for CD22 in B-cell maturation. Indeed, the level of BCR activation is also known to effect class switch recombination and affinity maturation(72). Cumulatively, these results demonstrate that hCD22 expressed on murine B-cells is appropriately regulated by glycan ligands on murine B-cells, leading to WT levels of: BCR stimulation, MZ B-cell compartment, and antibody responses.
Multiple roles for trans ligand interactions were also investigated for hCD22 expressed on B-cells. Recruitment to the site of cell contact between two B-cells revealed that hCD22 was recruited similarly as mCD22. Furthermore, trans ligand interactions with fluorescent liposomes bearing a selective CD22L also revealed appropriate levels of trans binding for hCD22 expressed on murine B-cells, compared to its mCD22 counterpart. Interestingly, in a biological role for trans CD22-ligand interactions, we find a partially rescued phenotype for hCD22+ homing to PPs and mesenteric lymph nodes, which have previously shown to be CD22 dependent(28). While the rescue of homing in comparison to CD22−/− B-cells is encouraging, the incomplete rescue compare to WT B-cells suggest there are still factors we do not completely understand. One of these factors could be a specialized sialic acid-containing glycan on PP HEVs in mouse that has specificity for mCD22 over hCD22. Parsing out which glycan ligands overlap between the species is not a trivial task given the large ensemble of sialic acid-containing glycans that are biosynthetically possible, particularly on the HEVs themselves. Alternatively, the incomplete rescue in homing could potentially stem from an intrinsic role for hCD22 on B-cells that is ligand-independent. These will be important questions to address in future studies, which could reveal important aspects of the role for CD22 in regulating B-cells.
An additional role for hCD22 in responding to trans ligands was investigated using STALs. STALs drive co-localization of CD22 with the BCR on antigen-specific B-cells, and they show potential as a novel approach for inducing immunological tolerance(43, 44). hCD22+ B-cells stimulated with antigenic liposomes formulated with a high affinity and selective hCD22L failed to respond as compared to antigenic liposomes without the hCD22L. The failure of STALs to induce Ca2+ flux response or upregulate CD86 expression at later time points are key pieces of data showing that ITIMs present on the cytoplasmic tail of hCD22 are competent at inhibiting BCR signaling in murine B-cells. Indeed, the sequence homology between the ITIMs in mouse and humans is high(4). Previous studies in mice have demonstrated that STALs induce an apoptotic mechanism, resulting in deletion of the antigen-specific B-cells from the B-cell repertoire and the induction of antigen-specific B-cell tolerance(10). We additionally have shown that STALs induce upregulation of Annexin V on hCD22+ B-cells in vitro, and OVA STALs formulated with a hCD22L indeed drive induced tolerance to OVA in hCD22+ mice to a similar degree as can be achieved by targeting mCD22 in WT mice. Previously, we had shown that STALs could induce a tolerogenic signal in primary naïve and memory human B-cells in vitro(43). Accordingly, these results represent the first studies demonstrating the in vivo potential of STALs targeting hCD22 to induce tolerance. Future studies investigating the ability to induce tolerance through hCD22 in antibody-driven autoimmunity, such as rheumatoid arthritis (RA) or SLE, and allergies should be valuable.
Altogether, we have shown that expression of hCD22 on murine B-cells restores many biological features of mCD22 such as: migration to the site of cell contact, the appropriate regulation of Ca2+ flux responses in response to BCR stimulation, antibody production, homing, and B-cell tolerization. However, not all aspects of homing were fully recovered in these mice as noted in B-cell homing to the PP, revealing important aspects that merits future investigation that could reveal molecular clues as the mechanism and precise glycan ligands used by CD22 to home to this location. This new hCD22-expressing mice should prove to be a valuable tool to assess the mechanisms of action of anti-CD22 monoclonal antibody therapies as well as future optimization of therapies that aim to functionally modulate hCD22 using glycan ligands, such STALs for the induction of B-cell tolerance.
Footnotes
DOD (W81XWH-16-1-0303 to MM, W81XWH-16-1-0302 to MK), NIAID (AI099141 & R01 AI050143 to JCP, and AI47822 to ECB), NIGMS (R01GM37734 to ECB), the Swiss National Sciences Foundation (P2GEP3_162055 to RB), and the NCATS (UL1TR001114 for the NBDS program).
References
- 1.Crocker PR, Paulson JC, Varki A. Siglecs and their roles in the immune system. Nature reviews Immunology. 2007;7:255–266. doi: 10.1038/nri2056. [DOI] [PubMed] [Google Scholar]
- 2.Macauley MS, Crocker PR, Paulson JC. Siglec-mediated regulation of immune cell function in disease. Nature reviews Immunology. 2014;14:653–666. doi: 10.1038/nri3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pillai S, Netravali IA, Cariappa A, Mattoo H. Siglecs and immune regulation. Annual review of immunology. 2012;30:357–392. doi: 10.1146/annurev-immunol-020711-075018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Torres RM, Law CL, Santos-Argumedo L, Kirkham PA, Grabstein K, Parkhouse RM, Clark EA. Identification and characterization of the murine homologue of CD22, a B lymphocyte-restricted adhesion molecule. Journal of immunology. 1992;149:2641–2649. [PubMed] [Google Scholar]
- 5.Muller J, Obermeier I, Wohner M, Brandl C, Mrotzek S, Angermuller S, Maity PC, Reth M, Nitschke L. CD22 ligand-binding and signaling domains reciprocally regulate B-cell Ca2+ signaling. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:12402–12407. doi: 10.1073/pnas.1304888110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen J, McLean PA, Neel BG, Okunade G, Shull GE, Wortis HH. CD22 attenuates calcium signaling by potentiating plasma membrane calcium-ATPase activity. Nature immunology. 2004;5:651–657. doi: 10.1038/ni1072. [DOI] [PubMed] [Google Scholar]
- 7.Doody GM, Justement LB, Delibrias CC, Matthews RJ, Lin J, Thomas ML, Fearon DT. A role in B cell activation for CD22 and the protein tyrosine phosphatase SHP. Science. 1995;269:242–244. doi: 10.1126/science.7618087. [DOI] [PubMed] [Google Scholar]
- 8.Blasioli J, Paust S, Thomas ML. Definition of the sites of interaction between the protein tyrosine phosphatase SHP-1 and CD22. The Journal of biological chemistry. 1999;274:2303–2307. doi: 10.1074/jbc.274.4.2303. [DOI] [PubMed] [Google Scholar]
- 9.Fujimoto M, Bradney AP, Poe JC, Steeber DA, Tedder TF. Modulation of B lymphocyte antigen receptor signal transduction by a CD19/CD22 regulatory loop. Immunity. 1999;11:191–200. doi: 10.1016/s1074-7613(00)80094-1. [DOI] [PubMed] [Google Scholar]
- 10.Macauley MS, Paulson JC. Siglecs induce tolerance to cell surface antigens by BIM-dependent deletion of the antigen-reactive B cells. Journal of immunology. 2014;193:4312–4321. doi: 10.4049/jimmunol.1401723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lanoue A, Batista FD, Stewart M, Neuberger MS. Interaction of CD22 with alpha2,6-linked sialoglycoconjugates: innate recognition of self to dampen B cell autoreactivity? European journal of immunology. 2002;32:348–355. doi: 10.1002/1521-4141(200202)32:2<348::AID-IMMU348>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 12.Duong BH, Tian H, Ota T, Completo G, Han S, Vela JL, Ota M, Kubitz M, Bovin N, Paulson JC, Nemazee D. Decoration of T-independent antigen with ligands for CD22 and Siglec-G can suppress immunity and induce B cell tolerance in vivo. The Journal of experimental medicine. 2010;207:173–187. doi: 10.1084/jem.20091873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Razi N, Varki A. Masking and unmasking of the sialic acid-binding lectin activity of CD22 (Siglec-2) on B lymphocytes. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:7469–7474. doi: 10.1073/pnas.95.13.7469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kelm S, Gerlach J, Brossmer R, Danzer CP, Nitschke L. The ligand-binding domain of CD22 is needed for inhibition of the B cell receptor signal, as demonstrated by a novel human CD22-specific inhibitor compound. The Journal of experimental medicine. 2002;195:1207–1213. doi: 10.1084/jem.20011783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jin L, McLean PA, Neel BG, Wortis HH. Sialic acid binding domains of CD22 are required for negative regulation of B cell receptor signaling. The Journal of experimental medicine. 2002;195:1199–1205. doi: 10.1084/jem.20011796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hennet T, Chui D, Paulson JC, Marth JD. Immune regulation by the ST6Gal sialyltransferase. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:4504–4509. doi: 10.1073/pnas.95.8.4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Collins BE, Blixt O, Bovin NV, Danzer CP, Chui D, Marth JD, Nitschke L, Paulson JC. Constitutively unmasked CD22 on B cells of ST6Gal I knockout mice: novel sialoside probe for murine CD22. Glycobiology. 2002;12:563–571. doi: 10.1093/glycob/cwf067. [DOI] [PubMed] [Google Scholar]
- 18.Gasparrini F, Feest C, Bruckbauer A, Mattila PK, Muller J, Nitschke L, Bray D, Batista FD. Nanoscale organization and dynamics of the siglec CD22 cooperate with the cytoskeleton in restraining BCR signalling. The EMBO journal. 2016;35:258–280. doi: 10.15252/embj.201593027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O’Keefe TL, Williams GT, Davies SL, Neuberger MS. Hyperresponsive B cells in CD22-deficient mice. Science. 1996;274:798–801. doi: 10.1126/science.274.5288.798. [DOI] [PubMed] [Google Scholar]
- 20.Nitschke L, Carsetti R, Ocker B, Kohler G, Lamers MC. CD22 is a negative regulator of B-cell receptor signalling. Current biology: CB. 1997;7:133–143. doi: 10.1016/s0960-9822(06)00057-1. [DOI] [PubMed] [Google Scholar]
- 21.Otipoby KL, Andersson KB, Draves KE, Klaus SJ, Farr AG, Kerner JD, Perlmutter RM, Law CL, Clark EA. CD22 regulates thymus-independent responses and the lifespan of B cells. Nature. 1996;384:634–637. doi: 10.1038/384634a0. [DOI] [PubMed] [Google Scholar]
- 22.Sato S, Miller AS, Inaoki M, Bock CB, Jansen PJ, Tang ML, Tedder TF. CD22 is both a positive and negative regulator of B lymphocyte antigen receptor signal transduction: altered signaling in CD22-deficient mice. Immunity. 1996;5:551–562. doi: 10.1016/s1074-7613(00)80270-8. [DOI] [PubMed] [Google Scholar]
- 23.Horikawa K, Martin SW, Pogue SL, Silver K, Peng K, Takatsu K, Goodnow CC. Enhancement and suppression of signaling by the conserved tail of IgG memory-type B cell antigen receptors. The Journal of experimental medicine. 2007;204:759–769. doi: 10.1084/jem.20061923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Samardzic T, Marinkovic D, Danzer CP, Gerlach J, Nitschke L, Wirth T. Reduction of marginal zone B cells in CD22-deficient mice. European journal of immunology. 2002;32:561–567. doi: 10.1002/1521-4141(200202)32:2<561::AID-IMMU561>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 25.Onodera T, Poe JC, Tedder TF, Tsubata T. CD22 regulates time course of both B cell division and antibody response. Journal of immunology. 2008;180:907–913. doi: 10.4049/jimmunol.180.2.907. [DOI] [PubMed] [Google Scholar]
- 26.Poe JC, Fujimoto Y, Hasegawa M, Haas KM, Miller AS, Sanford IG, Bock CB, Fujimoto M, Tedder TF. CD22 regulates B lymphocyte function in vivo through both ligand-dependent and ligand-independent mechanisms. Nature immunology. 2004;5:1078–1087. doi: 10.1038/ni1121. [DOI] [PubMed] [Google Scholar]
- 27.Nitschke L, Floyd H, Ferguson DJ, Crocker PR. Identification of CD22 ligands on bone marrow sinusoidal endothelium implicated in CD22-dependent homing of recirculating B cells. The Journal of experimental medicine. 1999;189:1513–1518. doi: 10.1084/jem.189.9.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee M, Kiefel H, LaJevic MD, Macauley MS, Kawashima H, O’Hara E, Pan J, Paulson JC, Butcher EC. Transcriptional programs of lymphoid tissue capillary and high endothelium reveal control mechanisms for lymphocyte homing. Nature immunology. 2014;15:982–995. doi: 10.1038/ni.2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Macauley MS, Kawasaki N, Peng W, Wang SH, He Y, Arlian BM, McBride R, Kannagi R, Khoo KH, Paulson JC. Unmasking of CD22 Co-receptor on Germinal Center B-cells Occurs by Alternative Mechanisms in Mouse and Man. The Journal of biological chemistry. 2015;290:30066–30077. doi: 10.1074/jbc.M115.691337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chappell CP, Draves KE, Clark EA. CD22 is required for formation of memory B cell precursors within germinal centers. PloS one. 2017;12:e0174661. doi: 10.1371/journal.pone.0174661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Angata T, Nycholat CM, Macauley MS. Therapeutic Targeting of Siglecs using Antibody- and Glycan-Based Approaches. Trends in pharmacological sciences. 2015;36:645–660. doi: 10.1016/j.tips.2015.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grant BW, Jung SH, Johnson JL, Kostakoglu L, Hsi E, Byrd JC, Jones J, Leonard JP, Martin SE, Cheson BD. A phase 2 trial of extended induction epratuzumab and rituximab for previously untreated follicular lymphoma: CALGB 50701. Cancer. 2013;119:3797–3804. doi: 10.1002/cncr.28299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Furman RR, Coleman M, Leonard JP. Epratuzumab in non-Hodgkin’s lymphomas. Current treatment options in oncology. 2004;5:283–288. doi: 10.1007/s11864-004-0019-1. [DOI] [PubMed] [Google Scholar]
- 34.Leonard JP, Goldenberg DM. Preclinical and clinical evaluation of epratuzumab (anti-CD22 IgG) in B-cell malignancies. Oncogene. 2007;26:3704–3713. doi: 10.1038/sj.onc.1210370. [DOI] [PubMed] [Google Scholar]
- 35.Leonard JP, Coleman M, Ketas JC, Chadburn A, Ely S, Furman RR, Wegener WA, Hansen HJ, Ziccardi H, Eschenberg M, Gayko U, Cesano A, Goldenberg DM. Phase I/II trial of epratuzumab (humanized anti-CD22 antibody) in indolent non-Hodgkin’s lymphoma. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2003;21:3051–3059. doi: 10.1200/JCO.2003.01.082. [DOI] [PubMed] [Google Scholar]
- 36.Goldenberg DM. Epratuzumab in the therapy of oncological and immunological diseases. Expert review of anticancer therapy. 2006;6:1341–1353. doi: 10.1586/14737140.6.10.1341. [DOI] [PubMed] [Google Scholar]
- 37.Strand V, Petri M, Kalunian K, Gordon C, Wallace DJ, Hobbs K, Kelley L, Kilgallen B, Wegener WA, Goldenberg DM. Epratuzumab for patients with moderate to severe flaring SLE: health-related quality of life outcomes and corticosteroid use in the randomized controlled ALLEVIATE trials and extension study SL0006. Rheumatology. 2014;53:502–511. doi: 10.1093/rheumatology/ket378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wallace DJ, Goldenberg DM. Epratuzumab for systemic lupus erythematosus. Lupus. 2013;22:400–405. doi: 10.1177/0961203312469692. [DOI] [PubMed] [Google Scholar]
- 39.Wallace DJ, Kalunian K, Petri MA, Strand V, Houssiau FA, Pike M, Kilgallen B, Bongardt S, Barry A, Kelley L, Gordon C. Efficacy and safety of epratuzumab in patients with moderate/severe active systemic lupus erythematosus: results from EMBLEM, a phase IIb, randomised, double-blind, placebo-controlled, multicentre study. Annals of the rheumatic diseases. 2014;73:183–190. doi: 10.1136/annrheumdis-2012-202760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wallace DJ, Hobbs K, Clowse ME, Petri M, Strand V, Pike M, Merrill JT, Leszczynski P, Neuwelt CM, Jeka S, Houssiau F, Keiserman M, Ordi-Ros J, Bongardt S, Kilgallen B, Galateanu C, Kalunian K, Furie R, Gordon C. Long-Term Safety and Efficacy of Epratuzumab in the Treatment of Moderate-to- Severe Systemic Lupus Erythematosus: Results From an Open-Label Extension Study. Arthritis care & research. 2016;68:534–543. doi: 10.1002/acr.22694. [DOI] [PubMed] [Google Scholar]
- 41.Clowse ME, Wallace DJ, Furie RA, Petri MA, Pike MC, Leszczynski P, Neuwelt CM, Hobbs K, Keiserman M, Duca L, Kalunian KC, Galateanu C, Bongardt S, Stach C, Beaudot C, Kilgallen B, Gordon C, E.I. Group Efficacy and Safety of Epratuzumab in Moderately to Severely Active Systemic Lupus Erythematosus: Results From Two Phase III Randomized, Double-Blind, Placebo-Controlled Trials. Arthritis & rheumatology. 2017;69:362–375. doi: 10.1002/art.39856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Giltiay NV, Shu GL, Shock A, Clark EA. Targeting CD22 with the monoclonal antibody epratuzumab modulates human B-cell maturation and cytokine production in response to Toll-like receptor 7 (TLR7) and B-cell receptor (BCR) signaling. Arthritis research & therapy. 2017;19:91. doi: 10.1186/s13075-017-1284-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Macauley MS, Pfrengle F, Rademacher C, Nycholat CM, Gale AJ, von Drygalski A, Paulson JC. Antigenic liposomes displaying CD22 ligands induce antigen-specific B cell apoptosis. The Journal of clinical investigation. 2013;123:3074–3083. doi: 10.1172/JCI69187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Orgel KA, Duan S, Wright BL, Maleki SJ, Wolf JC, Vickery BP, Burks AW, Paulson JC, Kulis MD, Macauley MS. Exploiting CD22 on antigen-specific B cells to prevent allergy to the major peanut allergen Ara h 2. The Journal of allergy and clinical immunology. 2017;139:366–369 e362. doi: 10.1016/j.jaci.2016.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Powell LD, Sgroi D, Sjoberg ER, Stamenkovic I, Varki A. Natural ligands of the B cell adhesion molecule CD22 beta carry N-linked oligosaccharides with alpha-2,6-linked sialic acids that are required for recognition. The Journal of biological chemistry. 1993;268:7019–7027. [PubMed] [Google Scholar]
- 46.Rillahan CD, Macauley MS, Schwartz E, He Y, McBride R, Arlian BM, Rangarajan J, Fokin VV, Paulson JC. Disubstituted Sialic Acid Ligands Targeting Siglecs CD33 and CD22 Associated with Myeloid Leukaemias and B Cell Lymphomas. Chemical science. 2014;5:2398–2406. doi: 10.1039/C4SC00451E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wohner M, Born S, Nitschke L. Human CD22 cannot fully substitute murine CD22 functions in vivo, as shown in a new knockin mouse model. European journal of immunology. 2012;42:3009–3018. doi: 10.1002/eji.201242629. [DOI] [PubMed] [Google Scholar]
- 48.Hobeika E, Thiemann S, Storch B, Jumaa H, Nielsen PJ, Pelanda R, Reth M. Testing gene function early in the B cell lineage in mb1-cre mice. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:13789–13794. doi: 10.1073/pnas.0605944103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cinamon G, Zachariah MA, Lam OM, Foss FW, Jr, Cyster JG. Follicular shuttling of marginal zone B cells facilitates antigen transport. Nature immunology. 2008;9:54–62. doi: 10.1038/ni1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen WC, Kawasaki N, Nycholat CM, Han S, Pilotte J, Crocker PR, Paulson JC. Antigen delivery to macrophages using liposomal nanoparticles targeting sialoadhesin/CD169. PloS one. 2012;7:e39039. doi: 10.1371/journal.pone.0039039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen WC, Completo GC, Sigal DS, Crocker PR, Saven A, Paulson JC. In vivo targeting of B-cell lymphoma with glycan ligands of CD22. Blood. 2010;115:4778–4786. doi: 10.1182/blood-2009-12-257386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thai TH, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, Murphy A, Frendewey D, Valenzuela D, Kutok JL, Schmidt-Supprian M, Rajewsky N, Yancopoulos G, Rao A, Rajewsky K. Regulation of the germinal center response by microRNA-155. Science. 2007;316:604–608. doi: 10.1126/science.1141229. [DOI] [PubMed] [Google Scholar]
- 53.Collins BE, Smith BA, Bengtson P, Paulson JC. Ablation of CD22 in ligand-deficient mice restores B cell receptor signaling. Nature immunology. 2006;7:199–206. doi: 10.1038/ni1283. [DOI] [PubMed] [Google Scholar]
- 54.Collins BE, Blixt O, DeSieno AR, Bovin N, Marth JD, Paulson JC. Masking of CD22 by cis ligands does not prevent redistribution of CD22 to sites of cell contact. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:6104–6109. doi: 10.1073/pnas.0400851101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jellusova J, Wellmann U, Amann K, Winkler TH, Nitschke L. CD22 × Siglec-G double-deficient mice have massively increased B1 cell numbers and develop systemic autoimmunity. Journal of immunology. 2010;184:3618–3627. doi: 10.4049/jimmunol.0902711. [DOI] [PubMed] [Google Scholar]
- 56.Li XM, Serebrisky D, Lee SY, Huang CK, Bardina L, Schofield BH, Stanley JS, Burks AW, Bannon GA, Sampson HA. A murine model of peanut anaphylaxis: T- and B-cell responses to a major peanut allergen mimic human responses. The Journal of allergy and clinical immunology. 2000;106:150–158. doi: 10.1067/mai.2000.107395. [DOI] [PubMed] [Google Scholar]
- 57.Smit JJ, Willemsen K, Hassing I, Fiechter D, Storm G, van Bloois L, Leusen JH, Pennings M, Zaiss D, Pieters RH. Contribution of classic and alternative effector pathways in peanut-induced anaphylactic responses. PloS one. 2011;6:e28917. doi: 10.1371/journal.pone.0028917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martin F, Kearney JF. Marginal-zone B cells. Nature reviews Immunology. 2002;2:323–335. doi: 10.1038/nri799. [DOI] [PubMed] [Google Scholar]
- 59.Cariappa A, Tang M, Parng C, Nebelitskiy E, Carroll M, Georgopoulos K, Pillai S. The follicular versus marginal zone B lymphocyte cell fate decision is regulated by Aiolos, Btk, and CD21. Immunity. 2001;14:603–615. doi: 10.1016/s1074-7613(01)00135-2. [DOI] [PubMed] [Google Scholar]
- 60.Blixt O, Collins BE, van den Nieuwenhof IM, Crocker PR, Paulson JC. Sialoside specificity of the siglec family assessed using novel multivalent probes: identification of potent inhibitors of myelin-associated glycoprotein. The Journal of biological chemistry. 2003;278:31007–31019. doi: 10.1074/jbc.M304331200. [DOI] [PubMed] [Google Scholar]
- 61.Lavoie TB, Drohan WN, Smith-Gill SJ. Experimental analysis by site-directed mutagenesis of somatic mutation effects on affinity and fine specificity in antibodies specific for lysozyme. Journal of immunology. 1992;148:503–513. [PubMed] [Google Scholar]
- 62.Fischer MB, Goerg S, Shen L, Prodeus AP, Goodnow CC, Kelsoe G, Carroll MC. Dependence of germinal center B cells on expression of CD21/CD35 for survival. Science. 1998;280:582–585. doi: 10.1126/science.280.5363.582. [DOI] [PubMed] [Google Scholar]
- 63.Dorner T, Shock A, Goldenberg DM, Lipsky PE. The mechanistic impact of CD22 engagement with epratuzumab on B cell function: Implications for the treatment of systemic lupus erythematosus. Autoimmunity reviews. 2015;14:1079–1086. doi: 10.1016/j.autrev.2015.07.013. [DOI] [PubMed] [Google Scholar]
- 64.Law CL, Torres RM, Sundberg HA, Parkhouse RM, Brannan CI, Copeland NG, Jenkins NA, Clark EA. Organization of the murine Cd22 locus. Mapping to chromosome 7 and characterization of two alleles. Journal of immunology. 1993;151:175–187. [PubMed] [Google Scholar]
- 65.Rickert RC, Roes J, Rajewsky K. B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nucleic acids research. 1997;25:1317–1318. doi: 10.1093/nar/25.6.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Reth M. Matching cellular dimensions with molecular sizes. Nature immunology. 2013;14:765–767. doi: 10.1038/ni.2621. [DOI] [PubMed] [Google Scholar]
- 67.John B, Herrin BR, Raman C, Wang YN, Bobbitt KR, Brody BA, Justement LB. The B cell coreceptor CD22 associates with AP50, a clathrin-coated pit adapter protein, via tyrosine-dependent interaction. Journal of immunology. 2003;170:3534–3543. doi: 10.4049/jimmunol.170.7.3534. [DOI] [PubMed] [Google Scholar]
- 68.Han S, Collins BE, Bengtson P, Paulson JC. Homomultimeric complexes of CD22 in B cells revealed by protein-glycan cross-linking. Nature chemical biology. 2005;1:93–97. doi: 10.1038/nchembio713. [DOI] [PubMed] [Google Scholar]
- 69.Nitschke L. CD22 and Siglec-G regulate inhibition of B-cell signaling by sialic acid ligand binding and control B-cell tolerance. Glycobiology. 2014;24:807–817. doi: 10.1093/glycob/cwu066. [DOI] [PubMed] [Google Scholar]
- 70.Blixt O, Head S, Mondala T, Scanlan C, Huflejt ME, Alvarez R, Bryan MC, Fazio F, Calarese D, Stevens J, Razi N, Stevens DJ, Skehel JJ, van Die I, Burton DR, Wilson IA, Cummings R, Bovin N, Wong CH, Paulson JC. Printed covalent glycan array for ligand profiling of diverse glycan binding proteins. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:17033–17038. doi: 10.1073/pnas.0407902101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Danzer CP, Collins BE, Blixt O, Paulson JC, Nitschke L. Transitional and marginal zone B cells have a high proportion of unmasked CD22: implications for BCR signaling. International immunology. 2003;15:1137–1147. doi: 10.1093/intimm/dxg114. [DOI] [PubMed] [Google Scholar]
- 72.Niiro H, Clark EA. Regulation of B-cell fate by antigen-receptor signals. Nature reviews Immunology. 2002;2:945–956. doi: 10.1038/nri955. [DOI] [PubMed] [Google Scholar]