

Abstract
The assembly mechanism of the Mn/Fe ligand-binding oxidases (R2lox), a family of proteins that are homologous to the nonheme diiron carboxylate enzymes, has been investigated using time-resolved techniques. Multiple heterobimetallic intermediates are observed through optical and magnetic resonance spectroscopies that exhibit unique spectral features, including visible absorption bands and exceptionally broad EPR signatures. On the basis of comparison to known diiron species and model compounds, the spectra have been attributed to (μ-peroxo)-MnIII/FeIII and high-valent Mn/Fe species. Global spectral analysis coupled with isotopic substitution and kinetic modeling reveals elementary rate constants for the assembly of Mn/Fe R2lox under aerobic conditions. A complete reaction mechanism for cofactor maturation that is consistent with experimental data has been developed. These results suggest that the Mn/Fe cofactor can perform direct C–H bond abstraction, demonstrating the potential for potent chemical reactivity that remains unexplored.
TOC image
INTRODUCTION
Nonheme diiron carboxylate proteins are responsible for a wide range of critical biological transformations, including DNA synthesis,1 fatty acid desaturation,2 hydrocarbon oxidation,3–6 cell signaling,7 antibiotic biosynthesis,8,9 and post-translational modifications of tRNA,10,11 among many others. In recent years, homologs of these proteins have been found to possess noncanonical metal cofactors, including the dimanganese class Ib ribonucleotide reductases (RNRs),12,13 the heterobimetallic Mn/Fe class Ic RNRs,14 and the Mn/Fe ligand-binding oxidases (R2lox).15,16 These systems are of particular interest because of their prevalence in extremophiles and pathogenic bacteria, including Chlamydia trachomatis (Ct) and Mycobacterium tuberculosis (Mt).15 While both CtR2 (R2c) and R2lox exhibit site-selective assembly and perform O2 activation, R2lox contains a bis(carboxylato) motif, with one carboxylate moiety donated by an exogenous fatty acid, and secondary sphere residues that resemble those of the bacterial multicomponent monooxygenases (BMMs) rather than those of RNRs.3,15–18 Consistent with this similarity in outer coordination environment, aerobic assembly of R2lox induces formation of a novel tyrosine-valine ether crosslink within the protein scaffold. This two-electron oxidation process, which formally oxidizes an aliphatic C–H bond, is analogous to reactions performed by the BMMs. By contrast, CtR2 executes reversible, single-electron transfer between the Mn/Fe site and a cysteine thiyl radical species in the active site of the R1 subunit of RNR. This divergent chemistry alludes to the potential for Mn/Fe cofactors to act as surrogates for the ubiquitous diiron proteins.
To probe potential reactivity and begin to understand the molecular basis underlying the selectivity of the R2lox Mn/Fe system, we have undertaken time-resolved spectroscopic investigations of cofactor assembly, O2 binding, and crosslink formation. Distinct intermediates have been observed, and preliminary assignments of these species have been made on the basis of spectral properties and comparison to known compounds. The spectroscopic results have been coupled with kinetic analyses and global fitting routines to resolve rate constants for each elementary step. Taken together, these findings represent an initial glimpse into the mechanism of activation in this unusual heterobimetallic system.
MATERIALS AND METHODS
Expression and Purification of GkR2loxI
Geobacillus kaustophilus (Gk) R2loxI (accession number WP_011232245.1) was heterologously expressed and purified using methods similar to those previously reported.16 Sequence-confirmed plasmids encoding the Gk R2loxI gene along with an ampicillin-resistance gene were transformed into E. coli BL21-DE3* competent cells (New England Biolabs). A 300 mL starter growth of Terrific Broth (TB) media (ForMedium, Norfolk, United Kingdom) containing 50 mg/L carbenicillin (GoldBio Technologies) was inoculated using cell stocks and shaken at 200 rpm for 14–16 hours at 37°C. The starter growth was then divided into 1 L flasks of TB containing 50 mg/mL carbenicillin and grown to an OD of 0.9–1. Cells were shaken with 0.5 mM EDTA for approximately 20 min at 18°C; protein expression was then induced with 0.5 mM IPTG (GoldBio Technologies) and shaken at 200 rpm for 14–16 hours at 18°C.
To harvest the growths, the cells were pelleted via multiple rounds of centrifugation at 5800 × g (Avanti J–E centrifuge, JLA-10.500 rotor). The resultant cell pellets were washed twice with Buffer A (25 mM HEPES, 20 mM imidazole, 300 mM NaCl, 0.5 mM EDTA, at pH 7.0) and stored at −80°C until needed. Pellets were resuspended in Buffer A and lysed using 0.1 mg/g lysate DNase (GoldBio Technologies) + 100 μM PMSF (GoldBio Technologies) dissolved in ethanol (Decon Laboratories). To fully homogenize the cell contents, the lysate was subjected to sonication on ice using 15-second pulses at an amplitude of 20 dB (Q700, Qsonica) followed by a 105-second resting period, for 8 cycles. The lysate was centrifuged at 39,000 × g to remove cellular debris. The resultant supernatant was then heated to 60°C using a water bath, resulting in the formation of a white, cloudy precipitant. These solids were removed with a second round of centrifugation at 39,000 × g. All centrifugation steps were carried out at 4°C.
Supernatant from the final centrifugation was applied to a Ni-nitrilotriacetic acid (NTA) agarose (McLab) gravity flow column that had been equilibrated with Buffer A. The beads were then washed extensively with Buffer B (Buffer A + 40 mM imidazole) and Buffer B’ (Buffer B without EDTA). Purified protein was eluted with Buffer C (25 mM HEPES, 250 mM imidazole, 300 mM NaCl, at pH 7.0) and exchanged into Buffer D (25 mM HEPES, 50 mM NaCl, pH 7.0) using an Amicon Stirred Cell equipped with a 3 kDa membrane filter (EMD Millipore, Billerica, MA). After complete buffer exchange, purified protein was concentrated down to 18–50 mg/mL, divided into 100–300 μL aliquots, flash-frozen in liquid nitrogen, and stored at −80°C until use. Protein purity was verified using SDS-PAGE (Figure S1) and concentrations were obtained using the previously reported extinction coefficient of 47,757 M−1cm−1 at 280 nm.17 We note that cell lysis and purification were performed on the same day for optimized apo-protein yields.
UV-Vis Reconstitution Protocol
Frozen apo-R2lox aliquots were thawed at 4°C and diluted into Buffer E (100 mM HEPES, 50 mM NaCl, pH 7.0) in a semi-micro Spectrosil cuvette (Starna Cells, 10 mm pathlength). To reconstitute R2lox with the appropriate metal cofactor, protein was brought to a final concentration of 100 μM in 700–750 μL by the addition of a 25 μL aliquot of a mixture of aqueous MnII and FeII to achieve a final ratio of 1:1:1 MnII:FeII:apoprotein. Metal stocks were made by weighing out MnCl2·4H2O (Amresco) and (NH4)2Fe(SO4)2·6H2O (Sigma-Aldrich) separately and dissolving in deionized water to make 0.1 M stocks, followed by further dilution as necessary to allow for 25 μL volume; fresh stocks were made each day. All solutions were air-saturated. Metal addition was performed via rapid hand-mixing, resulting in a dead time of approximately 1 min. Absorption spectra from 900–240 nm were collected on a Shimazdu UV-2600 spectrophotometer using a fast scan speed, giving one scan per minute. Cofactor assembly was monitored continuously for 1–12 hours. Experiments to assess possible isotope effects were performed using Buffer E prepared with D2O (Sigma-Aldrich, 99.9%). For all experiments in D2O, apo-protein was thoroughly buffer-exchanged into deuterated Buffer E using centrifugal membrane concentrators (Centricons, MWCO 3 kDa) and incubated at 4°C for 16–20 hours to allow for complete exchange of protons prior to metallation. Care was taken to avoid concentrating the protein above 1 mM, as apo-R2lox stability was observed to be lower in deuterated buffers relative to protiated buffers. Metal solutions were also prepared in D2O. For both protiated and deuterated experiments, aliquots for UV-Vis and stopped-flow absorption (SF-Abs) experiments were taken from the same protein stock. UV-Vis data were collected in parallel with each SF-Abs experiment, with representative traces shown. Difference absorption spectra were generated by subtracting the t = 60 min trace from earlier traces. Reconstitution experiments spanning a longer time period showed an increasing background after ~1 hr (Figure S2), likely reflecting precipitation of FeIII from solution over time.
Determination of Apparent Rate Constants
Apparent rate constants were obtained from UV-Vis data using non-linear least-squares curve fitting (Igor Pro v.6.3.7.2) routines. Absorbances at 320 nm as a function of time were fit to double exponential functions. Both fast (kform,1) and slow (kform,2) formation rates were resolved for comparison between H2O and D2O samples. Apparent rate constants for decay were determined using single (620 nm) or double (390 nm and 620 nm in deuterated buffer) exponential functions. Kinetic isotope effects (KIEs) were determined by running experiments in deuterated buffers, assuming complete exchange of all exchangeable protons; reported values for apparent KIEs were calculated from the ratio of kH/kD. At 390 nm, the slow component, k2, in H2O was used for direct comparison to the decay component, k1, in D2O samples.
Stopped-Flow Absorbance Experiments
Single-wavelength stopped-flow (SF) absorbance (Abs) experiments were carried out between 19–23°C in an Applied Photophysics (Surrey, U.K.) SX-20 apparatus with a dead time of 1 ms. Spectra were collected in logarithmic time-spacing over 500 seconds, with a total reaction volume of 0.13–0.14 mL per shot. Solutions of 200 μM apo-R2lox in protiated and deuterated buffers along with metal solutions were prepared as described above and loaded into the stopped-flow instrument with 3 mL plastic syringes (Becton-Dickenson, Franklin Lakes, NJ). Protein and metal solutions were mixed in a 1:1 ratio, resulting in final concentrations of 100 μM. Kinetic traces at wavelengths of interest were obtained across numerous days with multiple protein batches; SF-Abs traces shown represent averages of all data collected. Multi-wavelength diode-array SF-Abs experiments were conducted in the Bollinger/Krebs lab at Pennsylvania State University (PSU, University Park, PA), utilizing a SF-Abs setup described previously.19 Solutions of 300 μM apo-R2lox were mixed with 1 equivalent each MnII and FeII in aerobic buffer. Traces were collected using logarithmic time spacing for a period of 500 seconds, with every 20th trace collected shown. Photodegradation of the sample by the intense white light used in this instrument prevented quantitative analysis of intermediate formation; however, qualitative analysis agrees with single-wavelength data.20
Preparation of 57FeII Solutions
Stock solutions of 57FeII were prepared as previously described.21 Briefly, FeII was prepared via dissolution of solid 57Fe metal (Cambridge Isotope Laboratories, Andover, MA) in 2 equivalents of 1 M H2SO4 that was sparged with nitrogen to remove oxygen. The reaction was shaken at 70 rpm and 60°C in a septum-capped scintillation vial. Upon complete dissolution (approximately 4–5 hours), the FeII stock was diluted using degassed, deionized H2O to generate 10 mM solutions of 57FeII, which were stored at −80°C until use.
Preparation of EPR Samples
Rapid-freeze-quench (RFQ) EPR samples were prepared in the Bollinger/Krebs group at Pennsylvania State University (University Park, PA), utilizing an RFQ apparatus described in previous publications.22 All solutions were saturated with O2. Apo-R2lox at 0.6 mM concentration was prepared in 20% glycerol-containing Buffer E and loaded into one glass syringe, while solutions with the corresponding stoichiometric metal concentrations were loaded into the other syringe. Samples were mixed in a 1:1 volume ratio and rapidly passed through a hose of appropriate length to achieve desired aging time. Reactions were quenched at 0.3 sec, 1 sec, 3 sec, 10 sec, 60 sec, and 10 min via freezing in an isopentane-liquid nitrogen bath at ~113 K with the 60 sec and 10 min timepoints hand-quenched in liquid nitrogen. Samples were frozen in EPR tubes of 4 mm outer diameter and 3 mm inner diameter, 170 mm length (Quartz Scientific, Fairport Harbor, OH), with samples frozen prior to 60 sec requiring manual packing into the EPR tube.
Hand-quenched (HQ) EPR samples were prepared in EPR tubes with 4 mm outer and 3 mm inner diameter, 250 mm length (Quartz Scientific, Fairport Harbor, OH). 150 μL of apo-R2lox was loaded into EPR tubes using a section of PTFE tubing (0.71 mm ID) connected to a needle and syringe. A 25 μL aliquot of metal solutions (giving final ratios of 1:1:1 Mn:Fe:apo-R2lox) was subsequently loaded and hand-mixed within the tube under aerobic conditions, resulting in 175 μL of either 100 μM (H2O and D2O samples) or 300 μM (57Fe samples) protein. An isopropanol-liquid nitrogen bath was maintained at temperatures from 188–198 K for rapid freezing. Samples were frozen at various time points after mixing, ranging from 1–60 min.
EPR Spectroscopy
CW X-band EPR spectra of RFQ samples were collected at 10 K at Pennsylvania State University using a Bruker ESP-300 spectrometer (~9.5 GHz) equipped with an ER/4102 ST resonator (Bruker), a continuous flow helium cryostat (Oxford Instruments), and a temperature controller (ITC 502, Oxford Instruments).23 All other CW X-band EPR spectra from 5–30 K (9.62 GHz) were collected at the Ohio Advanced EPR Facility at Miami University using a Bruker EMX instrument equipped with an Oxford Instruments continuous flow helium cryostat and temperature controller (ESR 900). For time-dependent studies of product formation, the temperature was set to 6.67 K to avoid saturation artifacts.17 Spectra were obtained using a microwave power of 20 mW and a modulation frequency and amplitude of 100 kHz and 10 G, respectively. Spin quantitation was carried out using a 250 μM copper(II) azurin standard measured under non-saturating conditions (200 μW).
Kinetic Model
The kinetic model, with reaction steps given below, was simulated using the KinTek software (v.6.0) with SpectraFit.24,25
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
| (6) |
| (7) |
| (8) |
In the initial steps (Eq. 1–3), we consider divalent metal ion binding to occur sequentially, reversibly, and with low binding affinity based on preliminary anaerobic data (KD ~ 0.1–1 mM; Figure S3), and O2 access to the active site to be diffusion-limited with irreversible binding (kdiff ~ 1010 M−1 s−1). Changes to these parameters did not produce significant changes to the modeled kinetic profiles of intermediates and therefore were considered adequate for these analyses (Figures S4, S5, and Table S1). In addition, a slow reorganization step, (3), is required prior to O2 binding and generation of I1 in order to resolve formation of the first detectable intermediate (Figure S6).
All data fitted to the model were obtained from UV-Vis, SF-Abs, and EPR experiments, where difference spectra were used for the UV-Vis kinetic traces to reduce interference from overlapping product bands. KIEs were determined by comparing fitted rate constants from experiments done in protiated and deuterated buffers. Formation and decay of the first intermediate (I1) were modeled using SF-Abs and UV-Vis data at 620 nm. The second intermediate (I2), was modeled using only the UV-Vis data decaying at 390 nm, as contributions to absorbance from I1 cannot be completely discounted in the SF-Abs data at this wavelength; this is also suggested from the singular value decomposition analysis, discussed below. Quantification of product formation (P) was extracted from EPR intensities at ~374 mT, which is outside the spectral range of MnII, of samples hand-quenched at 1 minute, 3 minutes, 5 minutes, 10 minutes, 20 minutes, and 60 minutes.
To model experimental data, absorbances and EPR intensities were converted into relative concentration values. Extinction coefficients for conversion of absorbance data were selected based on representative literature values, with ε620, peroxo = 1500 M−1 cm−1 used for formation and decay of I1 at 620 nm, and ε390, IV/IV = 1500 M−1 cm−1 for decay of I2 at 390 nm.26,27 Experimental concentration values were then “assigned” in KinTek to the proposed observable species discussed above, and the model was fit to the data for determination of reaction rate constants. Spin quantitation based on a CuIIAz standard was used to obtain product concentration in the 20-minute EPR sample, which was calculated to be ~40 μM. Concentrations of product at t < 20 min were obtained using the EPR intensity at 374 mT, scaled with respect to the intensity at that field position of the 20-minute sample. However, overlapping contributions from intermediate species in this region obscured attempts to use these values quantitatively; thus, the product intensities were considered only in a relative context. Similarly, relative EPR intensities of the two intermediates were used for comparison to the optical data rather than to obtain absolute concentrations; these data were also not included in the model fitting procedure but instead were used to inform upon the relationship of the different EPR-active species to kinetically resolved intermediates.
A 2D FitSpace analysis of the kinetic model was performed using a chi2 threshold of 0.7143 (Figure S7). Based on the multidimensional analysis of the fitted rate constants, k3 is well-constrained with respect to k4 and moderately constrained with respect to k5. However, the overall rates of product formation (k7) appear relatively insensitive to the final rate constant and remain unbounded, suggesting other processes might reflect rate-limiting steps or additional data is needed to better constrain the global model.
Intermediate Kinetics from EPR
Due to the instability of RFQ samples over time, which were the result of packing defects, formation and decay of intermediates in protiated buffers were monitored from 300 msec to 60 min using a combination of 57Fe and natural abundance (NA) Fe samples, mixed using both RFQ (packed and non-packed) and HQ techniques. These analyses utilized data collected at both PSU and Miami University. The earliest spectra obtained at 300 msec after mixing appear largely to resemble only MnII in both isotopes and are not included in kinetic data (Figure S8). Spectra of packed samples were divided by a factor of 0.6 to account for a ~60% packing efficiency in RFQ technique.28 When comparing intensities directly, spectra collected from 300 μM samples were divided by a factor of 3, normalizing intensities to those of the standard 100 μM samples.
For I1 observed at 30 K, data points for samples mixed and quenched prior to 1 min were obtained using spectra of 57Fe-reconstitutions prepared via RFQ and packed into EPR tubes (Figure S9A), while data points for samples mixed and quenched at and after 1 min were obtained using spectra of NAFe-reconstitutions prepared via HQ techniques (Figure S9B). For the 1 min timepoint, three different samples were available (Figure S10): NAFe HQ, NAFe RFQ (not packed), and 57Fe RFQ, with all of these included in the kinetics to ensure good agreement between techniques. Likewise, two samples at the 5 min timepoint, NAFe HQ and 57Fe HQ, were used. Contributions from excess MnII were removed from NAFe and 57Fe samples by subtraction of MnII-only loaded R2lox and 300 msec RFQ 57Fe sample, respectively. Kinetic data for I1 formation were extracted from the low-field feature centered at ~307 mT. Due to the large splitting in 57Fe samples, an isosbestic point at 308.2 mT was chosen to monitor I1 formation and decay across samples (Figure S11), with excellent agreement when comparing data points extracted from 1 min and 5 min NAFe and 57Fe samples.
For I2, relative intensities monitoring formation of this species were obtained from spectra collected at PSU at 10 K using 300 msec, 1 sec, 3 sec, 10 sec, 47 sec (non-packed), and 10 min samples, all made with 57Fe and RFQ mixing (Figure S12). Spectra monitoring the decay of I2 were collected at Miami University at 6.7 K, using HQ samples prepared with NAFe quenched at 1 min, 3 min, 5 min, 10 min, 20 min, and 60 min (Figure S13). Contributions from MnII were subtracted using the 300 msec RFQ NAFe and 57Fe samples as appropriate for each sample type.
Kinetic data for I2 formation were obtained from the high-field feature at 385 mT; broadening of the high-field features in 57Fe samples was not significant enough to warrant an isosbestic point analysis. I2 decay was monitored at the highest field feature at ~392 mT. This discrepancy in field position was due to the change in parameters between EPR collection at PSU and Miami University. As a result, only relative comparisons between formation and decay of this intermediate can be drawn, with formation and decay traces scaled to unity at their shared ~1 min timepoint.
Singular Value Decomposition
Singular value decomposition (SVD) analyses of the UV-Vis spectra were employed within the KinTek SpectraFit add-on for further examination of the proposed kinetic model. Raw UV-Vis datasets for both H2O and D2O experiments were imported into KinTek for automatic SVD analysis. This global analysis of the multiwavelength data revealed representative spectra and the temporal dependence of each intermediate (Figure S14). Resultant amplitude vectors were in good agreement with the kinetic model given above (Figure S15). The fractional accumulations of each intermediate reproduce kinetic traces obtained from UV-Vis difference spectra. Using the rate constants obtained from fitting the kinetic model to experimental data, the predicted concentration profiles from the SVD analysis were seen to agree with the decomposed spectra. However, the SVD analysis was not used in “fitting” the model.
RESULTS
Assembly of (μ-hydroxo)-MnIII/FeIII product monitored using optical and EPR spectroscopy
The optical signatures of cofactor maturation in R2lox were first characterized using UV-Vis spectroscopy (Figure 1A). Owing to weak divalent metal binding,18 attempts to isolate the metallated MnII/FeII-R2lox protein with anaerobic reconstitutions were unsuccessful (Figure S3). Instead, solutions of apo-R2lox were rapidly mixed with aliquots of aqueous metal ions under aerobic conditions, with full optical spectra recorded each minute following mixing.
Figure 1.
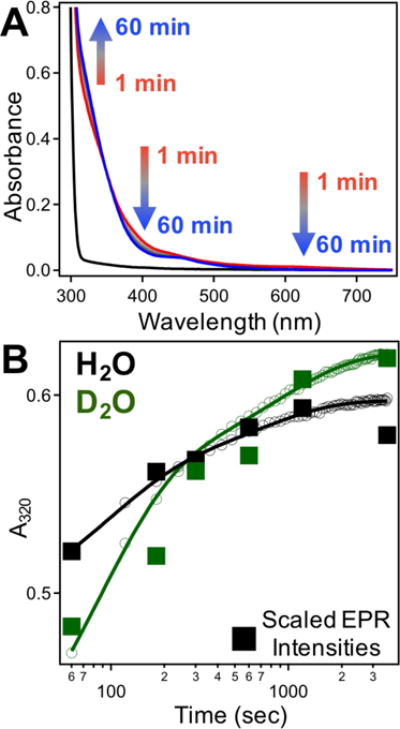
(A) Time-resolved UV-Vis spectra of 100 μM apo-R2lox (black) following addition of 100 μM MnII + 100 μM FeII under aerobic conditions from 1 (red) – 60 (blue) minutes. (B) Kinetic profile of UV-Vis absorption at 320 nm in protiated (black) and deuterated (green) buffers. Scaled EPR intensities at ~374 mT are overlaid (filled squares).
The spectrum of aerobically assembled R2lox features broad ligand-to-metal charge transfer (LMCT) bands from 300–360 nm, similar to those reported for related proteins such as RNR, MMO, and Δ9D.29 The intensity at 320 nm was thus monitored as an indicator for formation of the (μ-hydroxo)–MnIII/FeIII cofactor (“product”). Growth appears to follow biphasic kinetics, with features initially developing on the single-minute timescale (Table 1, Figure 1B).
Table 1.
| I1 | I2 | P | |||
|---|---|---|---|---|---|
| Wavelength | 620 | 390 | 320 | ||
| kform,UV-Vis(min−1) | H2O | n.r.c | n.r. | 0.66 ± 0.06 | 0.09 ± 0.02 |
| D2O | ~2.5d | ~2.5 | 0.68 ± 0.09 | 0.07 ± 0.03 | |
| kform,EPR(min−1) | H2O | 1.95e | 5.52 | 0.38 | |
| D2O | *f | * | 0.16 | ||
| kdecay,UV-Vis(min−1) | H2O | 0.33 ± 0.10 | 0.06 ± 0.01 | –g | |
| D2O | 0.13 ± 0.04 | 0.06 ± 0.02 | – | ||
| kdecay,EPR(min−1) | H2O | 0.39 | 0.08 | – | |
| D2O | 0.17 | 0.06 | – | ||
Values reflect the median values +/− range from 5 (320 nm) and 4 (390 nm and 620 nm) independent experiments.
Values reflect data gathered from a single EPR dataset.
Formation of intermediates not resolved on the timescale of UV-Vis experiments in protiated buffer.
Approximate formation of intermediates not consistently observed in deuterated buffer.
Rates of formation are only weakly constrained (Figures S9 and S12).
Fast timepoints not obtained for deuterated samples.
Product decay not applicable.
To investigate the role of protonation on cofactor assembly and product formation, rapid mixing experiments were conducted in deuterated buffers (Figure 1B), though there were no noticeable effects on assembly kinetics. Single-wavelength stopped-flow absorption (SF-Abs) spectroscopy was used to probe assembly on shorter timescales (Figure S16), with product formation again monitored at 320 nm. On this timescale, significant KIEs are evident, with deuterated samples showing absorption increases that are delayed by an apparent factor of ~3 (Figure S16). This observation suggests protonation is involved in at least one of the rate-limiting steps during assembly.
The EPR spectrum of the assembled (μ-hydroxo)-MnIII/FeIII species spans ~60 mT, with 7 peaks centered around g ~ 2, and has been thoroughly characterized using multifrequency pulsed and CW techniques to probe the local electronic structure of the cofactor.17 Using the intensity of the turning point at ~374 mT as an indicator of relative concentration, product assembly kinetics were also monitored using EPR spectroscopy (Figure 1B). Samples were hand-quenched in discrete timepoints following metal addition, as described above, from 1–60 minutes. In this case, isotope effects were observed for samples reconstituted in deuterated buffers, indicating at least one proton-dependent process (Figure S17).
Identification of Mn/Fe intermediates observed during assembly
Difference absorption spectra acquired during R2lox assembly reveal transient optical features with maxima at ~390 nm and ~620 nm that decay as a function of time following metal addition (Figure 2A). These bands decay with apparent first-order rate constants of 0.06 ± 0.01 min−1 and 0.33 ± 0.02 min−1, respectively, in H2O buffer (Figure 2A and Table 1). The effects of isotopic substitution are pronounced only for the decay of the feature at 620 nm, which shows an approximate KIE of 2.5 (Table 1). Deuterated samples also show evidence for development of intermediate features that is not apparent in the protiated experiments (Figures 2B–2C, kform ~ 2.5 ± 0.5 min−1), suggesting the formation step is slowed in D2O. We note that the slight increase in absorbance at 620 nm from t = 1 to t = 2 min in D2O buffer is not always resolved using the hand-mixing technique, while the increase at 390 nm is regularly reproduced. That differential rates of formation and decay are observed at the two wavelengths suggests that these features arise from distinct species. To account for overlapping contributions between intermediate and product absorption spectra, singular value decomposition (SVD) analysis was used to extract model absorption spectra and concentration profiles for each species in both protiated and deuterated buffers (Figures 3, S14). These results are consistent with the presence of at least two intermediates possessing distinct optical spectra that decay with different rates (Figure 3, inset) and the slow appearance of a product-like spectrum.
Figure 2.
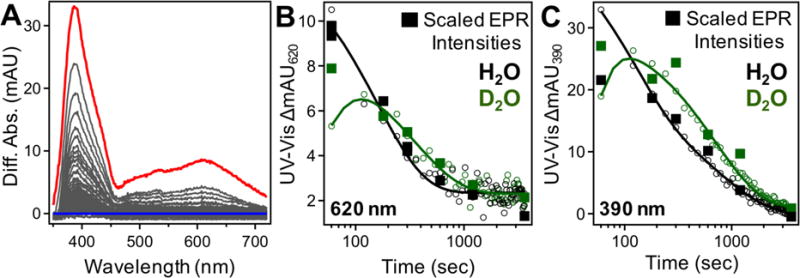
(A) Difference absorption spectra from UV/Vis reconstitution from 1 (red) – 60 (blue) minutes. (B – C) Kinetic profile of UV-Vis absorbance at (B) 620 nm and (C) 390 nm in protiated (black) and deuterated (green) buffers. Relative EPR intensities of intermediates at (B) 30 K and (C) 6.7 K are overlaid (filled squares).
Figure 3.
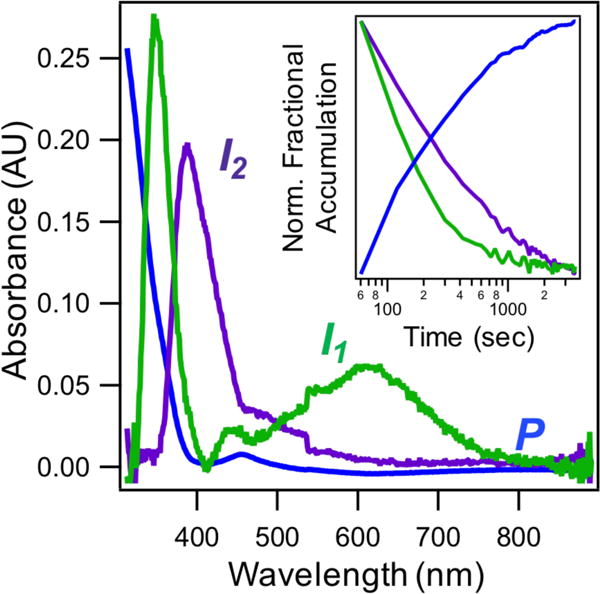
Modeled spectra of UV/Vis intermediates (green and purple traces) and product (blue trace) obtained from SVD analysis. (Inset) SVD-extracted normalized concentration profiles of modeled species.
The formation of these intermediates was also probed using SF-Abs, where an increase in absorption is first observed at 620 nm, with later increases at 390 nm and finally 320 nm (Figure S18); due to significant differences in amplitudes, the subtle temporal differences are best visualized when normalized intensities are compared. The transient species both reach maximum intensities at ~40 sec, with amplitudes on the single mAU scale and modest KIEs (Figure S16). The slight (<1 mAU) increase in SF-Abs intensity at 620 nm after ~250 sec is attributed to increased background scattering from precipitating FeIII. Overlapping temporally from ~60 seconds onwards, scaled UV-Vis intensities show good agreement with SF-Abs traces for both protiated and deuterated (Figure S19) buffers, thereby illustrating the capability for continuous monitoring of this system from ms to hrs.
To further resolve contributions from metal and O2 binding, SF-Abs and UV-Vis experiments were performed with increasing metal concentrations in an attempt to access pseudo-second order kinetic conditions (Figure S20). However, higher amounts of FeII results in increased Fe/Fe cofactor formation and Fe precipitation, complicating analysis.18 Unfortunately, the intense white light required for multiwavelength SF-Abs experiments causes photo-bleaching and sample degradation,20,23 though qualitative analysis of the multichannel SF-Abs results shown in Figure S21 reveals trends similar to those seen in single-wavelength experiments.
As a complementary approach, EPR spectroscopy was also used to study intermediates of R2lox cofactor assembly. Both hand-quenching (HQ) and rapid freeze-quenching (RFQ) techniques were used to trap the O2 activation reaction at time points of interest (Figure 4). At and before 300 ms, only the characteristic MnII EPR signal is present (Figure S8). By 1 sec, a species with a broad EPR spectrum begins to appear (Figure 4B). This spectrum spans ~90 mT at X-band frequencies, which is significantly broader than either MnII (55 mT) or the MnIII/FeIII R2lox product (60 mT). While formation of this intermediate occurs on the timescale of seconds, relative intensities suggest only low accumulation. Decay of this species occurs slowly, with a small amount remaining after 20 minutes (Figures 2C, 4C). EPR samples prepared identically in D2O buffer followed by hand-quenching shows only a negligible effect on the rate of decay, with an approximate KIE of 1 (Figures 2C, S22).
Figure 4.
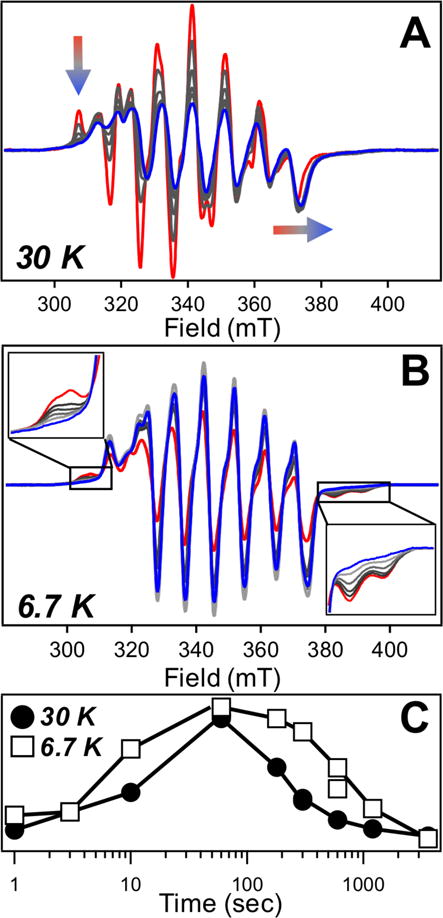
EPR-active intermediates monitored from 1 min (red) – 60 min (blue). (A) Narrow I1 species measured at high temperatures. Transient features indicated on figure. (B) Broad I2 species seen at low temperature with transient features magnified. (C) Scaled kinetic profile of intermediates.
The EPR spectra of RFQ and HQ samples measured at higher temperatures reveal a different signal with 9 well-resolved peaks, including a sharp, low-field peak at 307 mT, and a spectral breadth of ~66 mT (Figure 4A). This species is also present at ~1 sec, though it decays much more rapidly (Figure 4C); essentially all contribution to this signal is gone by 5 min. Moreover, the formation and decay of this species are slowed in deuterated buffer (Figures 2B, S23). The distinct temporal behavior and spectral features suggest the presence of a second intermediate, separate from the aforementioned broad species (Figure S24). However, owing to the very different relaxation properties and spectral overlap with both MnII and product features, direct comparison of the concentrations of each intermediate is not possible at this point in time; only relative amounts are accessible.
Superimposing the relative EPR intensities onto the absorption profiles as a function of time suggests correspondence between the transient species observed optically and those observed via EPR (Figure S25). The slowly-relaxing species with a narrow EPR signature appears to overlap with the absorbance at 620 nm, showing good agreement across both isotopes (Figure 2B). On the other hand, the kinetic profile of the species with the broad EPR spectrum matches that of the transient absorption signal at 390 nm, with a longer lifetime and negligible KIE for the decay process (Figure 2C). That such temporal correlations can be drawn across the measurement methods suggests these two intermediates are related to each other and the assembly process.
EPR characterization of Mn/Fe intermediate species
To further characterize the electronic structures of each intermediate, reconstitution experiments using both 57Fe and 55Mn were carried out. RFQ and HQ-EPR measurements on these samples across different timescales and temperatures confirm that both transient EPR signals derive from spin-coupled, homovalent Mnn+/Fen+ species (Figure 5). In the case of the ~90 mT wide, long-lived intermediate, a representative spectrum has been obtained through scaled subtraction of both MnII and product at t = 3 sec (Figure 5, bottom). Approximately 10 broad peaks are observed approximately centered at g ~ 2, characteristic of coupling to a 55Mn nucleus (I = 5/2). The 55Mn hyperfine coupling and the g-tensor both appear to be anisotropic, with peak splittings of ~220 and ~240 MHz on the low- and high-field sides, respectively, contributing to the spectral breadth.30 Anisotropic 57Fe coupling is also observed, as evidenced by a distinct peak splitting of ~90 MHz on the low-field edge and only slight broadening on the high-field side (Figure 5), suggesting weak-to-moderate exchange coupling between the metal ions. This species relaxes rapidly, showing a Curie-like linear dependence of EPR intensity with the reciprocal of the measurement temperature (Figures S26, S27) that is characteristic of a spin-coupled, S = ½ metallocofactor. Notably, this spectrum somewhat resembles that of chemically reduced MnIII/FeIII CtR2.14
Figure 5.
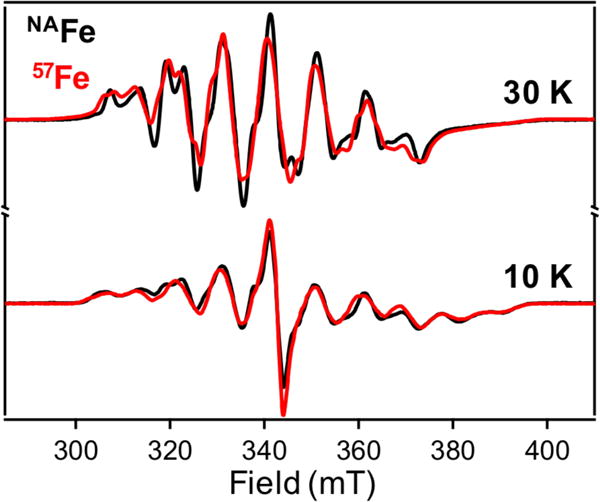
CW X-band EPR spectra of Mn/Fe R2lox assembly intermediates prepared with natural abundance Fe (black) or 57Fe (red). (Top) HQ spectra (1 min) measured at 30 K. (Bottom) RFQ spectra (3 sec) measured at 10 K.
The EPR signal from the short-lived intermediate can also be attributed to a Mn/Fe spin-coupled system, as the multiple bands are indicative of 55Mn coupling. The peak-to-peak splitting between the central bands is ~280 MHz, and additional, near-isotropic splittings of ~50 MHz occur upon introduction of 57Fe. The slow relaxation properties lead to saturation of the EPR signal even at higher temperatures (Figure S28), which, while obscuring attempts to quantitate this species, is suggestive of a well-isolated ground state within the spin-coupled manifold. In both cases, the spectra of product MnIII/FeIII and free MnII overlap with the spectral features of the intermediates. Due to this complication, identifying a unique set of spin Hamiltonian parameters using only a single-frequency CW EPR spectrum for simulation was not feasible. Advanced EPR experiments are underway to identify and further characterize these EPR-active species.
DISCUSSION
Postulated structures of Mn/Fe R2lox assembly intermediates
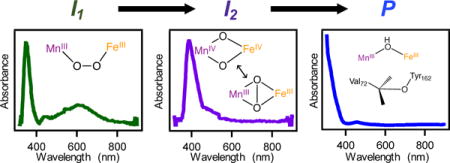
Structures of the observed intermediates have been tentatively assigned on the basis of comparison to model synthetic compounds and related biological systems. The first kinetically resolvable intermediate exhibits optical absorption features in the visible region of the spectrum, reminiscent of the low energy ligand-to-metal charge transfer transitions found in (μ-peroxo)–FeIII/FeIII species.26,29,31–34 Similar intermediates have been isolated in a number of nonheme diiron carboxylate proteins, including MMO, D Δ9D, RNR, CmlI and hDOHH. These species generally feature broad absorption bands centered around 700 nm with extinction coefficients of ~1500 M−1cm−1; often accompanying these low-energy transitions are similarly or more intense bands at higher energies (400–500 nm).9,20,26,35 In addition, a recent model compound containing a (μ-peroxo)–Mn2 III intermediate showed absorption centered at 640 nm.36 Thus, we suggest that the transient species observed during Mn/Fe cofactor assembly with the visible absorption feature around 620 nm is a (μ-peroxo)–MnIII/FeIII intermediate (Figure 6). The rates of formation and decay, along with the kinetic isotope effects, of this putative (μ-peroxo)–MnIII/FeIII intermediate are similar to those seen for the slowly relaxing, EPR-active species measured at 30 K (Figure 2B), suggesting the signals originate from the same intermediate species. Although this state has yet to be observed in any other protein or model compound, the experimental data reported here are consistent with the spectral features expected of such a species.
Figure 6.
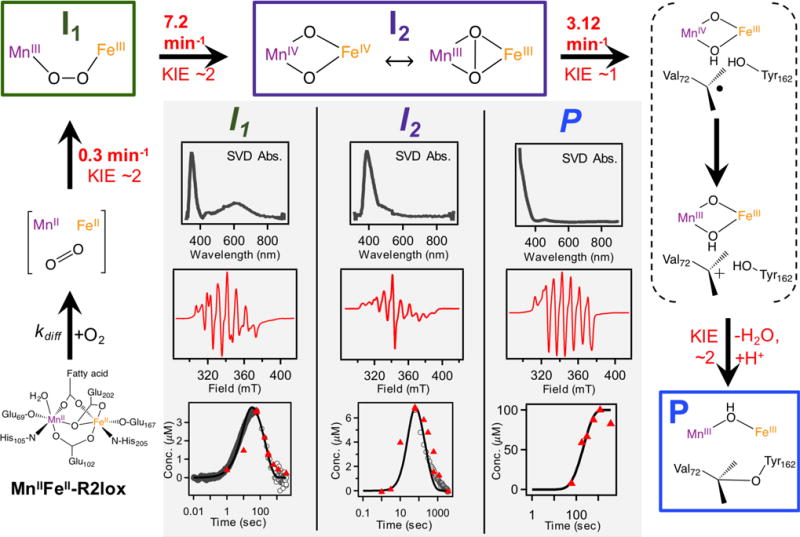
Proposed mechanism for Mn/Fe R2lox assembly. Modeled rate constants from this global kinetic model are inherently distinct from apparent rate constants obtained from fitting experimental data to simple exponential functions (Table 1). Product KIE obtained from isotope exchange EPR data. (Inset) Optical spectra obtained from SVD analysis (top) and representative EPR spectra (middle) of each intermediate as well as product. (Bottom) Relative concentrations obtained from experimental data overlaid with model simulations.
The second intermediate features an intense absorption band at 390 nm, evident in both the difference optical spectra and the SVD analysis, with kinetics and isotope effects that align with those of the broad, EPR-active intermediate (Figure 2C). The optical signatures of this species are remarkably similar to those seen in the MnIV/FeIV state generated upon O2 activation of the heterobimetallic cofactor in CtR2.27 On this basis, the identity of the second intermediate observed during aerobic Mn/Fe R2lox assembly has been tentatively assigned to be a high-valent Mn/Fe intermediate (Figure 6). We note that the EPR spectral features in the high-valent intermediate of R2lox are distinct from those seen in the MnIV/FeIV state of CtR2, which has recently been found to have a “diamond-core” structure. That different EPR spectra are seen for the two species suggests a structure other than the canonical diamond-core, further highlighting important distinctions between CtR2 and R2lox.37
A proposed mechanism for Mn/Fe R2lox assembly
Based on these results, an updated reaction scheme is proposed for cofactor maturation in Mn/Fe R2lox (Figure 6). This scheme is similar to those seen for the standard nonheme diiron carboxylate proteins.38 The first transient species, a purported (μ-peroxo)–MnIII/FeIII state (I1), forms upon binding of O2 to the metal centers. At this point, the molecular structure of this possible (μ-peroxo) species cannot be determined; for simplicity, this intermediate is shown as the cis-μ-1,2-peroxo (μ-η−1: η −1) species, though other arrangements and conformations cannot be excluded. This species then decays into the high-valent “Mn/Fe” species (I2), which is shown in Figure 6 with either a “diamond-core” MnIV/FeIV structure or a possible MnIII/FeIII species with a μ2-peroxide ligand. Owing to the large g- and A-tensor anisotropy, the EPR spectra would be consistent with a MnIII state; however, the UV/Vis signature of this intermediate is very similar to previously published MnIV/FeIV species. We therefore refrain from making an absolute assignment on the identity of I2 beyond a high-valent species and are actively pursuing further characterization experiments. Direct oxidation of either the valine or tyrosine residue via H-atom abstraction or PCET, respectively, is proposed to occur from the high-valent I2 state, generating either a valyl or tyrosyl radical and a (μ-oxo, μ-hydroxo)-MnIV/FeIII state not unlike that seen in CtR2.14,39 A valyl radical is expected to rapidly reduce the metal centers, forming a carbocation that undergoes nucleophilic attack from the nearby tyrosine residue to produce the protein crosslink. On the other hand, a tyrosyl radical could precede valine oxidation, though ultimately the same series of events would be observed. At this stage, the final trivalent oxidation state in each metal has been generated, albeit with a (μ-oxo, μ-hydroxo)–MnIII/FeIII state. Protonation with loss of water forms the product resting state, which has been extensively characterized crystallographically and spectroscopically.16,17,40
The initial O2 adduct shown in Figure 6 prior to I1 formation is similar to the “O” state in sMMO.41 Protonation and dissociation of the bridging Glu202 residue from the metal centers is necessary for oxygen binding, which may reflect a conformationally-gated step in the mechanism not unlike those seen for related non-heme diiron proteins. Formation of I1 occurs relatively slowly at a rate of 0.3 min−1, with a modest KIE of ~2, likely due to proton movement associated with the conformational change. Decay of I1 into I2 also features a KIE of ~2 and proceeds more rapidly (7.2 min−1), preventing significant accumulation of I1. The subsequent decay of I2 is observed to occur with a rate of 3.12 min−1 in both protiated and deuterated buffers. As mentioned above, this high-valent state is suggested to be responsible for C–H bond activation, which ultimately forms the tyrosine-valine crosslink. The previously proposed mechanism for R2lox assembly based on computational modeling had implicated formation of a tyrosyl radical prior to valine oxidation.16 However, multiwavelength SF-Abs data lack the spectroscopic signatures characteristic of tyrosyl radical formation (Figure S21),42,43 though rapid decay of such a species may also preclude intermediate accumulation. Moreover, a significant KIE would be expected for generation of a tyrosyl radical, yet decay of I2 appears to be isotope independent. These results prompted an alternative hypothesis in which a valyl rather than a tyrosyl radical is proposed, taking into consideration that both molecules have similar bond dissociation free energies.44 Future efforts aimed at selective isotopic labeling will further probe this hypothesis. Formation of the (μ-oxo, μ-hydroxo)–MnIV/FeIII species accompanies valyl radical generation and represents a relatively unstable species that we are unable to resolve in any of our experimental studies, largely because such a species is EPR silent. This mixed-valent species likely decays rapidly into a MnIII/FeIII state that is expected to closely resemble the product. Final protonation and water loss occurs with a KIE of ~2 (Figure S17). The different product KIEs observed in EPR and via optical absorption profiles at 320 nm are attributed to differences in selectivity of the measurement; overlapping contributions from multiple intermediates in the near-UV region of the spectrum may obscure clear identification of a given species, while the EPR measurements select for the (μ-hydroxo)-MnIII/FeIII product.
CONCLUSIONS
We have characterized the assembly of the Mn/Fe R2lox cofactor using time-resolved optical and EPR spectroscopy and developed a kinetic model for O2 activation that is consistent with the experimental data. During assembly, the Mn/Fe cofactor progresses through two distinct, identifiable intermediates, as evidenced by distinct optical and EPR spectra along with differential rates of formation, decay, and kinetic isotope effects. The first, with moderately intense features in the visible region of the optical spectrum, has been assigned to a (μ-peroxo)–MnIII/FeIII intermediate, potentially the first heterobimetallic peroxo species reported to date. This decays into a state with optical bands characteristic of a high-valent Mn/Fe species, which performs C–H bond activation. This observation highlights the unique chemical potential of this heterobimetallic cofactor for performing challenging oxidation reactions, with implications for in vivo functionality.
On a more fundamental level, the natural occurrence of these heterobimetallic cofactors is particularly challenging to understand in the context of the Irving-Williams series of divalent metal stabilities, which asserts that FeII should bind more tightly to a protein than MnII.45 In fact, in the absence of MnII, both R2lox and CtR2 will assemble with Fe/Fe cofactors, with formation of the tyrosine-valine crosslink also seen in Fe/Fe R2lox.18,40,46 However, we have previously shown that in vitro combination of apo-R2lox, MnII, and FeII results in selective generation of the heterobimetallic species.18 While our initial emphasis has been placed on characterizing assembly and maturation of the Mn/Fe cofactor, analogous work is also being pursued on the Fe/Fe system to investigate the factors contributing to this selective assembly process. In conclusion, these studies lay an important foundation for further spectroscopic characterization of the intermediates identified here and shed light on possible roles that these heterobimetallic Mn/Fe ligand-binding oxidases may play in host organisms.
Supplementary Material
Acknowledgments
We gratefully acknowledge Drs. J. Martin Bollinger, Jr. & Carsten Krebs for assistance with RFQ & multiwavelength SF-Abs experiments; Drs. Martin Högbom and Julia J. Griese for helpful discussions & GkR2loxI plasmid; Dr. Rob McCarrick at the Ohio Advanced EPR Facility at Miami University for assisting with variable temperature EPR experiments; and Dr. Marina Bahktina at the Biophysical Interaction and Characterization Facility at OSU for assistance with single wavelength SF-Abs experiments. H.S.S. thanks the Department of Chemistry and Biochemistry at The Ohio State University for funding; N.E.T. acknowledges the SOLAR Award, the URO Research Scholar Award, and a Pelotonia Undergraduate Fellowship; P.T.M. acknowledges an NIH CMBP training grant (T32-GM086252); E.J.B. thanks the N.I.H. for a National Research Service Award (GM116353-01).
Footnotes
Supporting Information.
The following file is available free of charge.
Supplemental spectra, stopped-flow absorption kinetic traces, table of fitted rate constants, diagrams of full kinetic schemes, global spectral analyses for both isotopes, and SDS-PAGE of purified protein (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Notes
The authors declare no competing financial interests.
References
- 1.Huang M, Parker MJ, Stubbe J. Choosing the right metal: case studies of class I ribonucleotide reductases. J Biol Chem. 2014;289:28104–28111. doi: 10.1074/jbc.R114.596684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fox BG, Lyle KS, Rogge CE. Reactions of the diiron enzyme stearoyl-acyl carrier protein desaturase. Acc Chem Res. 2004;37:421–429. doi: 10.1021/ar030186h. [DOI] [PubMed] [Google Scholar]
- 3.Sazinsky MH, Lippard SJ. Correlating structure with function in bacterial multicomponent monooxygenases and related diiron proteins. Acc Chem Res. 2006;39:558–66. doi: 10.1021/ar030204v. [DOI] [PubMed] [Google Scholar]
- 4.Fox BG, Hendrich MP, Surerus KK, Andersson KK, Froland WA, Lipscomb JD, Munck E. Mössbauer, EPR, and ENDOR studies of the hydroxylase and reductase components of methane monooxygenase from Methylosinus trichosporium OB3b. J Am Chem Soc. 1993;115:3688–3701. [Google Scholar]
- 5.Lippard SJ. Hydroxylation of C–H bonds at carboxylate-bridged diiron centres. Philos Trans R Soc Lond Math Phys Eng Sci. 2005;363:861–877. doi: 10.1098/rsta.2004.1532. [DOI] [PubMed] [Google Scholar]
- 6.Bailey LJ, Fox BG. Crystallographic and catalytic studies of the peroxide-shunt reaction in a diiron hydroxylase. Biochemistry. 2009;48:8932–8939. doi: 10.1021/bi901150a. [DOI] [PubMed] [Google Scholar]
- 7.Jasniewski AJ, Engstrom LM, Vu VV, Park MH, Que L. X-ray absorption spectroscopic characterization of the diferric-peroxo intermediate of human deoxyhypusine hydroxylase in the presence of its substrate eIF5a. JBIC J Biol Inorg Chem. 2016;21:605–618. doi: 10.1007/s00775-016-1373-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Komor AJ, Rivard BS, Fan R, Guo Y, Que L, Lipscomb JD. Mechanism for six-electron aryl-N-oxygenation by the non-heme diiron enzyme CmlI. J Am Chem Soc. 2016;138:7411–7421. doi: 10.1021/jacs.6b03341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Korboukh VK, Li N, Barr EW, Bollinger JM, Jr, Krebs C. A long-lived, substrate-hydroxylating peroxodiiron(III/III) intermediate in the amine oxygenase, AurF, from Streptomyces thioluteus. J Am Chem Soc. 2009;131:13608–13609. doi: 10.1021/ja9064969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Subedi BP, Corder AL, Zhang S, Foss FW, Pierce BS. Steady-state kinetics and spectroscopic characterization of enzyme–tRNA interactions for the non-heme diiron tRNA-monooxygenase, MiaE. Biochemistry. 2015;54:363–376. doi: 10.1021/bi5012207. [DOI] [PubMed] [Google Scholar]
- 11.Mathevon C, Pierrel F, Oddou JL, Garcia-Serres R, Blondin G, Latour JM, Ménage S, Gambarelli S, Fontecave M, Atta M. tRNA-modifying MiaE protein from Salmonella typhimurium is a nonheme diiron monooxygenase. Proc Natl Acad Sci. 2007;104:13295–13300. doi: 10.1073/pnas.0704338104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cox N, Ogata H, Stolle P, Reijerse E, Auling G, Lubitz W. A tyrosyl–dimanganese coupled spin system is the native metalloradical cofactor of the R2f subunit of the ribonucleotide reductase of Corynebacterium ammoniagenes. J Am Chem Soc. 2010;132:11197–11213. doi: 10.1021/ja1036995. [DOI] [PubMed] [Google Scholar]
- 13.Cotruvo JA, Stubbe J. An active dimanganese(III)-tyrosyl radical cofactor in Escherichia coli class Ib ribonucleotide reductase. Biochemistry. 2010;49:1297–1309. doi: 10.1021/bi902106n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang W, Yun D, Saleh L, Barr, Eric R, Xing Gang, W Z, Hoffart Lee, B J, Maslak Monique-Anne, P L, Krebs C, Bollinger JM., Jr A manganese(IV)/iron(III) cofactor in Chlamydia trachomatis ribonucleotide reductase. Science. 2007;316:1188–1192. doi: 10.1126/science.1141179. [DOI] [PubMed] [Google Scholar]
- 15.Andersson CS, Högbom M. A Mycobacterium tuberculosis ligand-binding Mn/Fe protein reveals a new cofactor in a remodeled R2-protein scaffold. Proc Natl Acad Sci. 2009;106:5633–5638. doi: 10.1073/pnas.0812971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Griese JJ, Roos K, Cox N, Shafaat HS, Branca RMM, Lehtio J, Graslund A, Lubitz W, Siegbahn PEM, Hogbom M. Direct observation of structurally encoded metal discrimination and ether bond formation in a heterodinuclear metalloprotein. Proc Natl Acad Sci. 2013;110:17189–17194. doi: 10.1073/pnas.1304368110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shafaat HS, Griese JJ, Pantazis DA, Roos K, Andersson CS, Popović-Bijelić A, Gräslund A, Siegbahn PEM, Neese F, Lubitz W, Högbom M, Cox N. Electronic structural flexibility of heterobimetallic Mn/Fe cofactors: R2lox and R2c proteins. J Am Chem Soc. 2014;136:13399–13409. doi: 10.1021/ja507435t. [DOI] [PubMed] [Google Scholar]
- 18.Kutin Y, Srinivas V, Fritz M, Kositzki R, Shafaat HS, Birrell J, Bill E, Haumann M, Lubitz W, Högbom M, Griese JJ, Cox N. Divergent assembly mechanisms of the manganese/iron cofactors in R2lox and R2c proteins. J Inorg Biochem. 2016;162:164–177. doi: 10.1016/j.jinorgbio.2016.04.019. [DOI] [PubMed] [Google Scholar]
- 19.Price JC, Barr EW, Tirupati B, Bollinger JM, Jr, Krebs C. The first direct characterization of a high-valent iron intermediate in the reaction of an α-ketoglutarate-dependent dioxygenase: a high-spin Fe(IV) complex in taurine/α-ketoglutarate dioxygenase (TauD) from Escherichia coli. Biochemistry. 2003;42:7497–7508. doi: 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- 20.Pandelia ME, Li N, Nørgaard H, Warui DM, Rajakovich LJ, Chang W, Booker SJ, Krebs C, Bollinger JM., Jr Substrate-triggered addition of dioxygen to the diferrous cofactor of aldehyde-deformylating oxygenase to form a diferric-peroxide intermediate. J Am Chem Soc. 2013;135:15801–15812. doi: 10.1021/ja405047b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ravi N, Bollinger JM, Jr, Huynh BH, Stubbe J, Edmondson DE. Mechanism of assembly of the tyrosyl radical-diiron (III) cofactor of E. coli ribonucleotide reductase: 1. Mössbauer characterization of the diferric radical precursor. J Am Chem Soc. 1994;116:8007–8014. [Google Scholar]
- 22.Bollinger JM, Jr, Hangtong W, Ravi N, Hanh Huynh B, Edmondson D, Stubbe J. In: [20] Use of rapid kinetics methods to study the assembly of the diferric-tyrosyl radical cofactor of E coli ribonucleotide reductase. Enzymology B-M, editor. Academic Press; 1995. pp. 278–303. [DOI] [PubMed] [Google Scholar]
- 23.Rajakovich LJ, Nørgaard H, Warui DM, Chang W, Li N, Booker SJ, Krebs C, Bollinger JM, Jr, Pandelia ME. Rapid reduction of the diferric-peroxyhemiacetal intermediate in aldehyde-deformylating oxygenase by a cyanobacterial ferredoxin: evidence for a free-radical mechanism. J Am Chem Soc. 2015;137:11695–11709. doi: 10.1021/jacs.5b06345. [DOI] [PubMed] [Google Scholar]
- 24.Johnson KA, Simpson ZB, Blom T. Global Kinetic Explorer: A new computer program for dynamic simulation and fitting of kinetic data. Anal Biochem. 2009;387:20–29. doi: 10.1016/j.ab.2008.12.024. [DOI] [PubMed] [Google Scholar]
- 25.Johnson KA, Simpson ZB, Blom T. FitSpace Explorer: An algorithm to evaluate multidimensional parameter space in fitting kinetic data. Anal Biochem. 2009;387:30–41. doi: 10.1016/j.ab.2008.12.025. [DOI] [PubMed] [Google Scholar]
- 26.Trehoux A, Mahy JP, Avenier F. A growing family of O2 activating dinuclear iron enzymes with key catalytic diiron(III)-peroxo intermediates: biological systems and chemical models. Coord Chem Rev. 2016;322:142–158. [Google Scholar]
- 27.Jiang W, Hoffart LM, Krebs C, Bollinger JM., Jr A manganese(IV)/iron(IV) intermediate in assembly of the manganese(IV)/iron(III) cofactor of Chlamydia trachomatis ribonucleotide reductase. Biochemistry. 2007;46:8709–8716. doi: 10.1021/bi700906g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bollinger JM., Jr Personal communication 2016 Jun; [Google Scholar]
- 29.Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee SK, Lehnert N, Neese F, Skulan AJ, Yang YS, Zhou J. Geometric and electronic structure/function correlations in non-heme iron enzymes. Chem Rev. 2000;100:235–350. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- 30.Teutloff C, Schäfer KO, Sinnecker S, Barynin V, Bittl R, Wieghardt K, Lendzian F, Lubitz W. High-field EPR of MnIIIMnIV and MnIIMnIII states of dimanganese catalase and related model systems. Magn Reson Chem. 2005;43:S51–S64. doi: 10.1002/mrc.1685. [DOI] [PubMed] [Google Scholar]
- 31.Liu KE, Wang D, Huynh BH, Edmondson DE, Salifoglou A, Lippard SJ. Spectroscopic detection of intermediates in the reaction of dioxygen with the reduced methane monooxygenase/hydroxylase from Methylococcus capsulatus (Bath) J Am Chem Soc. 1994;116:7465–7466. [Google Scholar]
- 32.Liu KE, Valentine AM, Wang D, Huynh BH, Edmondson DE, Salifoglou A, Lippard SJ. Kinetic and spectroscopic characterization of intermediates and component interactions in reactions of methane monooxygenase from Methylococcus capsulatus (Bath) J Am Chem Soc. 1995;117:10174–10185. [Google Scholar]
- 33.Lee SK, Lipscomb JD. Oxygen activation catalyzed by methane monooxygenase hydroxylase component: proton delivery during the O–O bond cleavage steps. Biochemistry. 1999;38:4423–4432. doi: 10.1021/bi982712w. [DOI] [PubMed] [Google Scholar]
- 34.Broadwater JA, Ai J, Loehr TM, Sanders-Loehr, Fox BG. Peroxodiferric intermediate of stearoyl-acyl carrier protein δ9 desaturase: oxidase reactivity during single turnover and implications for the mechanism of desaturation. Biochemistry. 1998;37:14664–14671. doi: 10.1021/bi981839i. [DOI] [PubMed] [Google Scholar]
- 35.Makris TM, Vu VV, Meier KK, Komor AJ, Rivard BS, Münck E, Que L, Lipscomb JD. An unusual peroxo intermediate of the arylamine oxygenase of the chloramphenicol biosynthetic pathway. J Am Chem Soc. 2015;137:1608–1617. doi: 10.1021/ja511649n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coggins MK, Sun X, Kwak Y, Solomon EI, Rybak-Akimova E, Kovacs JA. Characterization of metastable intermediates formed in the reaction between a Mn(II) complex and dioxygen, including a crystallographic structure of a binuclear Mn(III)–peroxo species. J Am Chem Soc. 2013;135:5631–5640. doi: 10.1021/ja311166u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang W, Yun D, Saleh L, Bollinger JM, Jr, Krebs C. Formation and function of the manganese(IV)/iron(III) cofactor in Chlamydia trachomatis ribonucleotide reductase. Biochemistry. 2008;47:13736–13744. doi: 10.1021/bi8017625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Krebs C, Bollinger JM, Jr, Booker SJ. Cyanobacterial alkane biosynthesis further expands the catalytic repertoire of the ferritin-like “di-iron-carboxylate” proteins. Curr Opin Chem Biol. 2011;15:291–303. doi: 10.1016/j.cbpa.2011.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kwak Y, Jiang W, Dassama LMK, Park K, Bell CB, Liu LV, Wong SD, Saito M, Kobayashi Y, Kitao S, Seto M, Yoda Y, Alp EE, Zhao J, Bollinger JM, Jr, Krebs C, Solomon EI. Geometric and electronic structure of the Mn(IV)Fe(III) cofactor in class Ic ribonucleotide reductase: correlation to the class Ia binuclear non-heme iron enzyme. J Am Chem Soc. 2013;135:17573–17584. doi: 10.1021/ja409510d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Griese JJ, Kositzki R, Schrapers P, Branca RMM, Nordström A, Lehtiö J, Haumann M, Högbom M. Structural basis for oxygen activation at a heterodinuclear manganese/iron cofactor. J Biol Chem. 2015;290:25254–25272. doi: 10.1074/jbc.M115.675223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Banerjee R, Meier KK, Münck E, Lipscomb JD. Intermediate P* from soluble methane monooxygenase contains a diferrous cluster. Biochemistry. 2013;52:4331–4342. doi: 10.1021/bi400182y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bollinger JM, Jr, Edmondson DE, Huynh BH, Filley J, Norton, Stubbe J. Mechanism of assembly of the tyrosyl radical-dinuclear iron cluster cofactor of ribonucleotide reductase. Science. 1991;253:292–298. doi: 10.1126/science.1650033. [DOI] [PubMed] [Google Scholar]
- 43.Larsson A, Sjöberg BM. Identification of the stable free radical tyrosine residue in ribonucleotide reductase. EMBO J. 1986;5:2037–2040. doi: 10.1002/j.1460-2075.1986.tb04461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Warren JJ, Tronic TA, Mayer JM. Thermochemistry of proton-coupled electron transfer reagents and its implications. Chem Rev. 2010;110:6961–7001. doi: 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Irving H, Williams R. The stability of transition-metal complexes. J Chem Soc Resumed. 1953:3192–3210. [Google Scholar]
- 46.Högbom M, Stenmark P, Voevodskaya N, McClarty G, Gräslund A, Nordlund P. The radical site in Chlamydial ribonucleotide reductase defines a new R2 subclass. Science. 2004;305:245–248. doi: 10.1126/science.1098419. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.