

Summary
The human leukocyte antigen class I gene HLA‐B27 is the strongest risk factor for ankylosing spondylitis (AS), a chronic inflammatory arthritic disorder. More recently, the Endoplasmic Reticulum Aminopeptidase (ERAP) 1 and 2 genes have been identified by genome wide association studies (GWAS) as additional susceptibility factors. In the ER, these aminopeptidases trim the peptides to a length suitable to fit into the groove of the major histocompatibility complex (MHC) class I molecules. It is noteworthy that an epistatic interaction between HLA‐B27 and ERAP1, but not between HLA‐B27 and ERAP2, has been highlighted. However, these observations suggest a paramount centrality for the HLA‐B27 peptide repertoire that determines the natural B27 immunological function, i.e. the T cell antigen presentation and, as a by‐product, elicits HLA‐B27 aberrant behaviours: (i) the misfolding leading to ER stress responses and autophagy and (ii) the surface expression of homodimers acting as ligands for innate immune receptors. In this context, it has been observed that the HLA‐B27 carriers, besides being prone to autoimmunity, display a far better surveillance to some viral infections. This review focuses on the ambivalent role of HLA‐B27 in autoimmunity and viral protection correlating its functions to the quantitative and qualitative effects of ERAP1 and ERAP2 polymorphisms on their enzymatic activity.
Keywords: ankylosing spondylitis, autoimmunity, endoplasmic reticulum aminopeptidases, HLA‐B27
Introduction
A group of human disorders classified as autoimmune and/or autoinflammatory share a similar immunopathogenic ground in which the principal risk factor is a ‘tagging’ human leukocyte antigen (HLA) class I gene. In addition, a prominent role has been attributed to Endoplasmic Reticulum Aminopeptidase (ERAP)1 and, in some cases, ERAP2 genes. We are referring to immunological disorders such as Behçet's disease (BD), psoriasis (Ps), birdshot chorioretinopathy (BSCR) and ankylosing spondylitis (AS), each associated by a variable relative risk (odds ratio) with the HLA‐B51, HLA‐C*0602, HLA‐A29 or HLA‐B27 genes, respectively 1, 2. Besides peculiar clinical hallmarks due to distinct polygenic backgrounds, such diseases usually affect body sites that undergo physical stress and are located at either external barriers (oral mucosa, skin, gut, eye) or at interior sites (joints, enthesis, cardiac valves, blood vessel walls) 1. The endogenous and/or exogenous aetiological triggers are usually unknown. However, the co‐occurring association with both single HLA class I genes and endoplasmic reticulum (ER) aminopeptidases involved in the final cut of HLA peptides points unequivocally at antigen processing and imbalanced peptide repertoire as putative unifying pathogenic key events. This concept is strengthened further by the fact that the association of ERAP1 in BD, Ps and AS reveals an epistatic gene–gene interaction with HLA‐B51, HLA‐C*0602 and HLA‐B27, respectively 3, 4, 5. This review will discuss how the classical and non‐classical HLA‐B27 functions could be influenced by the peptide repertoire that, in turn, is finely shaped by ERAP1 and 2 allelic variants and how the different settings can affect AS and the superior anti‐viral immunity (Fig. 1).
Figure 1.
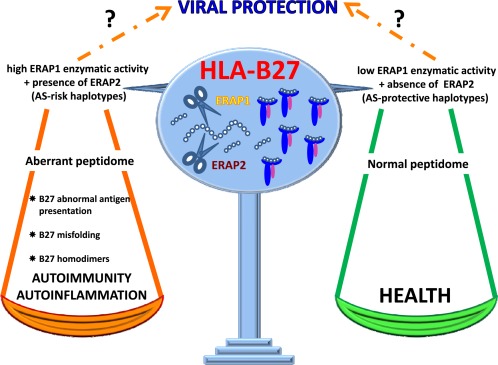
Cartoon illustrating the interaction between human leukocyte antigen (HLA)‐B27 and Endoplasmic Reticulum Aminopeptidase 1 and 2 in ankylosing spondylitis and antiviral defence. HLA‐B27 carriage, in association with high peptide‐trimming activity ERAP1 haplotypes and the concomitant expression of ERAP2 predisposes to AS. This genetic ground would affect the B27 peptidome impairing HLA‐B27 functionality. It is currently unknown whether the so‐called ‘aberrant’ peptidome contains additional pathogenic epitopes and/or loses peptides relevant for the stability of B27 complex or other functions. Indeed, an aberrant peptidome could impact on antigen presentation as well as on the rate of B27 misfolding thus activating ER stress, unfolded protein response or autophagy. In addition, the formation of B27 homodimers can activate cells expressing killer‐cell immunoglobulin receptor (KIR)3DL2 and leukocyte immunoglobulin‐like receptors (LILRB2) innate immune receptors. Conversely, the HLA‐B27 co‐inherited with ERAP1 haplotypes of low enzymatic activity, and in the absence of ERAP2, would display a proper and non‐disease prone B27 peptidome. With regard to viral infections, it is still unknown whether the protective behaviour of HLA‐B27 correlates with a high or low activity of ERAP1, and/or with the presence or absence of ERAP2.
HLA‐B27, a molecule with two faces: the involvement in spondyloarthritis
HLA‐B27 went into the spotlight during early 1970s, when a strong association with AS and other related immune‐mediated conditions [reactive arthritis (ReA), psoriatic arthritis, enteropathic arthritis and acute anterior uveitis] was established 6, 7. During the next 40 years, although rapid progress in many research fields enabled acquisition of a deep knowledge of the biochemical and functional properties of HLA‐B27, its exact role in AS onset has remained elusive.
The normal immunological function of HLA class I molecules is generally to present endogenous peptides of microbial or self‐origin to CD8+ T lymphocytes. These are generally peptides of 8–10 aa in length, which in the case of HLA‐B27 are stabilized through two principal anchors: the first is almost exclusively an Arg at position 2 (P2), whereas the second, at the peptide carboxy‐terminus (pΩ), can admit several different amino acids 8. Accordingly, one of the first and more popular theories to explain the B27 involvement in AS was the so‐called arthritogenic peptide hypothesis. This implies the activation of an autoreactive HLA‐B27‐restricted cytotoxic CD8+ T cell response primed by cross‐reactive microbial antigen(s), thus breaking self‐tolerance and perpetuating an autoimmune process leading to tissue damage 9. In accordance with this hypothesis, HLA‐B27‐restricted CD8+ T cells reactive against both self‐peptides and enterobacterial antigens were found in the synovial fluid of patients with AS and ReA 10. Moreover, in HLA‐B27‐positive AS patients, our group has described autoreactive B27‐mediated cytotoxic T lymphocyte (CTL) responses triggered by a self‐peptide from the vasoactive intestinal peptide receptor type I (VIPR) and cross‐reacting with a viral epitope from latent membrane protein 2 (LMP2) of Epstein–Barr virus (EBV) 11. However, this theory has not received further support, for several reasons. Unlike another related disease, namely ReA 12, AS is not usually preceded by microbial infections, even though the intestinal microbiota has been evoked recently to have an inciting role on B27‐restricted T cell responses 13. Furthermore, the HLA‐B27 transgenic rat model seems to disprove the possibility that HLA‐B27 is uniquely responsible for disease because of its classical antigen‐presenting functions, given that the lack of CD8+ T cells does not prevent spondyloarthritis in this context 14.
The HLA‐B27 family consists of more than 160 alleles (https://www.ebi.ac.uk/cgi-bin/ipd/imgt/hla/allele.cgi), whose ancestral subtype is the HLA‐B*2705 that is distributed ubiquitously worldwide and is strongly AS‐associated 15. Most B27 alleles are rare, so that their relation to AS is unknown. Nevertheless, at least two alleles, the HLA‐B*2706 and HLA‐B*2709, are not risk factors for AS 15, 16, 17. These alleles have limited polymorphic positions compared to the AS‐associated HLA‐B*2705 allele. In particular, the B*2709 differs from the B*2705 only for a His instead of an Asp at position 116, whereas the B*2706, an Asian allele, displays two substitutions (Asp114His, Tyr116Asp) compared to the B*2704 allele, which is strongly AS‐associated in the same geographic area 16. These polymorphisms are clustered mainly around the pockets of the peptide‐binding cleft, and influence the peptide repertoire. Therefore, based on the arthritogenic peptide theory, the disease‐inducing peptides should be exclusive ligands of the AS‐associated alleles and not of B*2706 and B*2709 alleles. In contrast with this assumption, recent biochemical studies from Purcell's group, analysing a large peptide data set from the AS‐associated and non‐AS‐associated B27 alleles expressed by transfected C1R cells, failed to identify qualitative changes in their peptide repertoire 18. Rather, quantitative differences have been found which justify the interest towards a panel of 26 peptides eluted in lower abundance from the non‐AS‐associated alleles 19. In addition, another study in which structure, peptide specificity, folding and stability of either AS‐associated or non‐AS‐associated subtypes on C1R cells were correlated with the constitutive peptidomes, reached similar results. Indeed, very few peptides emerged as connected strictly with the disease 20. Unfortunately, these studies are limited by experimental requirements that allow preferential analysis of certain cell types. Therefore, little is known so far about the peptide repertoire of either AS‐associated or non‐AS‐associated subtypes in the target tissues, where the differences could be more informative.
Alternative areas of intense investigation have considered aberrant and potentially pathogenic biochemical features of HLA‐B27 that set this allotype apart from other HLA class I molecules. First, the HLA‐B27 displays an altered folding rate during the assembly into the ER 9, 21, 22. This misfolding is a consequence of the particularly slow HLA‐B27 maturation rate that triggers the endoplasmic reticulum (ER)‐associated degradation (ERAD) of the heavy chains 22. However, the accumulation of misfolded heavy chains, aggregates or even dimeric structures, which do not transit further along the secretory pathway, generates ER stress and the unfolded protein response (UPR) 22, 23, 24. In turn, the UPR promotes cytokine dysregulation and activates the interleukin (IL)‐23/IL‐17 axis 25. This mechanism has been well documented in transgenic rat model for spondyloarthritis but not in humans, and its pathological implication in AS remains controversial 25, 26. In this regard, the occurrence of B27 misfolding in gut of AS patients does not seem to activate UPR but rather autophagy, that would be the leading mechanism modulating the intestinal production of IL‐23 in the disease 27. Another peculiarity of the HLA‐B27 is related to its expression on the cell surface as a non‐canonical form made by β2m‐free homodimers 9, 21, 28. The formation of homodimers arises from endosomal recycling compartments and is caused by the impaired and highly reactive cysteine 67 (Cys67) located into the B pocket of the peptide groove 29. The pathogenic effects of these structures would depend upon their engagement by both leukocyte immunoglobulin‐like receptors (LILR)B2 and killer‐cell immunoglobulin receptor (KIR)3DL2 innate immune receptors expressed by natural killer (NK) and T cells, thus triggering an inflammatory cascade 30. In patients with spondyloarthropathies, more CD4+ T cells expressing KIR3DL2 have been found and the binding with B27 dimers would licence pathogenic T helper type 17 (Th17) cell polarization 31, 32.
If misfolding as well as homodimer formation can be the cause of AS pathogenesis, then the disease‐associated B27 subtypes should differ from the non‐disease associated HLA‐B*2706 and 09 alleles in this aspect. Several investigations have addressed this issue, and the results were not always consistent 8, 33, 34, 35. One study failed to establish a total correspondence between being a disease‐associated B27 subtype and a lower folding efficiency, as the AS‐associated B*2707 allele behaved as the non‐AS‐associated B*2706 and B*2709 alleles 36. Moreover, the two structurally close but differently AS‐predisposing B*2705 and B*2709 alleles have been shown to have similar intracellular trafficking and propensity to form oligomers 33. However, more recently, it has been described a greater tendency of AS‐associated B*2702, B*2705, B*2707 alleles to accumulate in ‘dynamic’ intracellular vesicles as misfolded/aggregate proteins in comparison with the non‐associated B*2706 allele 37. This behaviour has been correlated with the B27 protein level.
One important question is whether a differential amount of cell surface free heavy chain (FHC) and/or homodimers, recognized by KIR3DL2 on NK and CD4+ T cells, discriminates some B27 alleles from others, justifying their distinct AS‐association. So far, this matter has not been explored in an in vivo setting in which cells from AS patients are compared with the controls expressing the different B27 alleles. However, no significant difference in cell surface expression of FHC among several B27 subtypes expressed on C1R cells has been reported 38. Moreover, one report highlighted that B*2706, unlike B*2709, expressed the highest surface level of FHC, probably reflecting the poor tapasin dependence during the assembly process which produces more dissociation‐prone heterotrimeric complexes 39. More recently, Cauli and co‐workers, using transfected cells, have shown that a higher cell surface FHC expression of B*2705 versus B*2709 could contribute to the differential disease‐association 40.
The formation of B27 dimers and oligomers is a complex process in which a series of unpaired Cys residues (positions 67, 308 and 325) appear to play important roles, but the conserved Cys residues at 101 and 164 could also be relevant 22, 30, 41, 42. These Cys residues are shared by all B27 alleles; therefore, other residues must influence their reactivity and accessibility to the oxidizing environment of the ER for a time sufficient to allow the formation of aberrant disulphide bonds. A pivotal role has been ascribed to residues 114 and 116 located in the F pocket of the binding groove, which notoriously influence the repertoire of bound peptides but also chaperone association, assembly process, maturation rate and, lastly, heavy chain dimerization 43, 44. Interestingly, these residues distinguish the AS‐associated from the non‐AS‐associated alleles 9, 16. Of note, Tyr116 in B*2706, much more than His116 in B*2709, appears to impact positively on the assembly kinetics, thus reducing dimer formation 44. These data can also be interpreted on the basis of recent biophysical and computational analyses. Indeed, several studies have described an enhanced degree of flexibility and disorder of B*2705 and B*2704 peptide‐binding cleft in comparison to that of B*2706 and B*2709 alleles 45, 46, 47. This would influence the tapasin dependence, the folding dynamics and the stability of HLA–peptide complexes overall 46.
HLA‐B27, a molecule with two faces: protection from viral infections
Being a carrier of HLA‐B27 certainly represents a risk condition for the development of autoimmune rheumatic diseases, but there are also benefits concerning a superior protection against a variety of viruses 48, 49. Together with few other HLA class I molecules of the B locus, the HLA‐B27 is associated with long‐term non‐progression to AIDS in patients, called ‘elite controllers’, which maintain a low viral load and remain asymptomatic for longer 50. Furthermore, recent studies have revealed in HLA‐B27 subjects a high rate of spontaneous clearance of hepatitis C virus 48, 51. The reasons for this are not completely understood, although virological and immunological explanations have been anticipated. First, during HIV and hepatitis C virus (HCV) infections, viral escape from HLA‐B27‐restricted cytotoxic T cells targeting immunodominant epitopes is undoubtedly a difficult process. It usually requires multiple compensatory mutations to counterbalance structural and functional constraints having a high cost for the viral fitness 48, 52, 53. Secondly, a number of immunological benefits of the virus‐specific, HLA‐B27‐restricted CD8+ T cells have been described pertaining to broader polyfunctionality and higher functional avidity 54. Furthermore, special thymic selection inducing a larger B27‐driven CD8+ T cell precursor repertoire, preferential usage of certain T cell receptor (TCR) clonotypes associated with higher cross‐reactive and, finally, better capacity of evasion from regulatory T cell (Treg)‐mediated suppression have also been documented 55, 56, 57, 58. Moreover, rapid and efficient processing of the proper immunodominant epitopes would contribute to these successful B27‐restricted T cell responses 59.
Recently, in HLA‐B*2705 subjects, mainly patients with AS, our group has described the capacity to elicit a vigorous HLA‐B27‐restricted CD8+ T cell response against an EBV epitope from EBNA3A (RPPIFIRRL) which was already known as immunodominant in another restriction context, namely the HLA‐B7 molecules 60, 61. This presentation is somewhat intriguing, as the peptide is a suboptimal B27 ligand and is expected to shift into the peptide‐binding cleft to permit the fitting of N‐terminal Arg into the B pocket, while leaving the A pocket empty 60. Of note, almost 70% of B*2705 individuals possess such ‘unexpected’ CD8+ T lymphocytes that share a common TCR β‐chain repertoire. Interestingly, no reactivity has been found in B*2709 healthy donors and, accordingly, the non‐AS‐associated B*2709 allele appears unable to present such suboptimal epitope. This is a further evidence supporting the different plasticity of B*2705 versus the B*2709 antigen‐presenting groove 49. Overall, this finding allows us to speculate that for some HLA‐B27 alleles, possibly those associated with AS, the real pool of bound ligands is larger than anticipated on the basis of biochemical data. This would enhance the ability to mediate anti‐viral protection while increasing the risk of autoimmunity.
The ERAP1 and ERAP2 aminopeptidases: functional role and allelic variants
ERAP1 and ERAP2 are endoplasmic reticulum‐resident aminopeptidases trimming peptides to an optimal length for binding with major histocompatibility complex (MHC) class I molecules 62, 63. They belong to the M1 family of zinc‐metallopeptidases and share 49% of sequence identity. In humans, ERAP1 and ERAP2 genes are located on chromosome 5q15 in opposite orientation and, conceivably, share regulatory elements 62.
ERAP1, besides its role in antigen processing, exerts several other biological functions promoting innate immune responses, or even regulating angiogenesis and hypertension/blood pressure 64. ERAP1 is a highly dynamic molecule switching from a lower‐activity open conformation to a higher‐activity closed conformation 65. This conformational transition is induced by the substrate upon binding to a regulatory site neighbouring the catalytic domain 63. Through a mechanism named ‘molecular ruler’, whereby the enzyme itself acts as a peptide‐length template, ERAP1 trims peptides of 9–16 residues very efficiently while sparing shorter peptides 66, 67, 68. The substrate specificity of ERAP1 is dictated by the N‐ and C‐terminal residues of the peptide as well as by the internal sequence 67, 69, 70, 71. ERAP1 shows preferences for hydrophobic residues, while basic and acidic amino acids are poor substrates; finally, Pro is never hydrolyzed 69, 70. The trimming activity of ERAP2 is complementary to that of ERAP1 for both N‐terminal substrate specificity and peptide length. Indeed, ERAP2 cleaves positively charged residues preferentially and its activity is maximal on octameric substrates and lower on longer peptides 63, 72, 73, 74. Therefore, the two aminopeptidases would operate in a concerted manner, ensuring an efficient generation and/or destruction of MHC class I epitopes for a proper functioning and regulation of the adaptive immunity.
ERAP1 and ERAP2 have been shown to form heterodimeric complexes having an allosteric effect on ERAP1 that acquires an enhanced in vitro trimming activity due to a higher substrate binding affinity 75, 76. However, less than 30% of each enzyme is engaged in the heterodimers 75. Recently, it has also been shown that the ERAP1/ERAP2 dimer could work as a peptide editor by trimming ‘on MHC I’ substrates until the correct length enabling the MHC groove to reach a closed conformation 77.
ERAP1 is a highly polymorphic gene. The most common protein variants, reported as ERAP1 allotypes, are encoded by haplotypes created by missense variant combinations of SNPs harboured in an ancestral haplotype found in humans as well as in primates 78. The most investigated ERAP1 allotypes (from 10 to 13) 78, 79 are distinguished by enzymatic functions with both qualitative (substrate preferences) and quantitative (high, intermediate and low activity variants) effects 5, 64, 80, 81, 82. Interestingly, some non‐synonymous SNPs influence the gene expression level of ERAP1 83.
ERAP2, instead, displays poor polymorphism. In the worldwide population, evolution under balancing selection has maintained two main ERAP2 haplotypes: one expressing the protein and the other ERAP2‐deficient, because the G allelic variant of SNP rs2248374 induces a truncated form that goes through non‐sense‐mediated decay 84. The two haplotypes are almost equally frequent in the different ethnic groups, so that 25% of individuals, being homozygous for the second haplotype, do not express ERAP2. Moreover, a non‐synonymous SNP (rs2549782) encoding for the amino acid substitution N392K affects both enzymatic activity and substrate specificity 72. This functional variant is in strong linkage with SNP rs2248374. Apart from specific ethnic peculiarities, the N392 allelic variant is almost absent in the human populations because of its co‐inheritance with the rs2248374 null‐allele 84, 85.
HLA‐B27 and ERAP1/2 as players in ankylosing spondylitis
In 2007, a genome wide association study (GWAS) revealed the association between five ERAP1 SNPs and an increased risk to develop AS 86. In particular, two of these SNPs, rs30187 (Arg528Lys) and rs27044 (Glu730Gln), reached high statistical significance for AS, as the minor alleles were robustly more frequent in AS patients than controls. Afterwards, several studies replicated these genetic associations, imputed ERAP1 haplotypes and found further associations with other SNPs mapping in the coding, UTR or intronic regions 2, 78, 87. In 2011, it was proved that the association of ERAP1 with AS occurred exclusively in HLA‐B27‐positive patients, pointing at a gene–gene epistatic interaction 5. Hence, the obvious effort has been to understand the impact of AS‐risk ERAP1 polymorphisms on the B27 peptidome and, consequently, on the putative B27 pathogenic functions.
Apart from a few exceptions, a higher enzymatic activity marks individual ERAP1 polymorphisms or the entire haplotypes associated with increased risk of AS (Table 1) 2, 5, 78, 79, 80. Several studies agree to indicate the high trimming Met349/Lys528/Asp575/Arg725/Gln730 haplotype as being associated most strongly with AS risk, while the low trimming Val349/Arg528/Asn575/Gln725/Glu730 haplotype as the most protective 2, 78, 88. In contrast, a report has suggested that rare hyperactive or hypoactive allotype pair combinations found in AS patients could explain the involvement of ERAP1 in the disease 79. However, such study suffered from low statistical power due to the small cohorts analysed.
Table 1.
Single nucleotide polymorphisms (SNPs) in Endoplasmic Reticulum Aminopeptidase (ERAP)1 and ERAP2 associated with ankylosing spondylitis (AS)
| Amino acid position |
Major/minor allele amino acid (nucleotide) |
AS‐risk allele amino acid (nucleotide) |
Effects | |
|---|---|---|---|---|
| ERAP1 SNPs | ||||
| rs2287987 | 349 | Met (A)/Val (G) | Met (A) |
Trimming activity (substrate‐dependent) |
| rs30187 | 528 | Arg (G)/Lys (A) | Lys (A) |
Expression level, trimming activity, Substrate specificity |
| rs10050860 | 575 | Asp (G)/Asn (A) | Asp (G) | Trimming activity |
| rs17482078 | 725 | Arg (G)/Gln (A) | Arg (G) | Trimming activity |
| rs27044 | 730 | Glu (G) * /Gln (C) | Gln (C) |
Substrate length preference, Trimming activity |
| ERAP2 SNPs | ||||
| rs2549782 | 392 | Asn (T)/Lys (G) | Lys (G) | Trimming activity |
| rs2248374 | null‐allele (G)/expressing allele (A) | expressing allele (A) | Presence/absence | |
*This SNP is also present in an AS‐predisposing haplotype 78.
Seminal work from Lopez de Castro's group has outlined how the B27 peptidome is influenced by ERAP allotypes 89, 90. The effect impacts mainly the P1 residue and, to a lesser extent, the remaining peptide sequence, the peptide length, the amount of specific ligands and the B27 affinity and thermostability of the overall peptide/B27 complexes. It is noteworthy that another study has shown that ERAP1 silencing, as expected, decreased the amount of B27‐bound nonamer peptides and, interestingly, increased the number of longer ligands, especially with extended C‐terminus 91.
A key point which is still the subject of debate is the relationship between the enzymatic activity of ERAP1 variants and the amount of cell surface B27 aberrant FHC/dimeric/oligomeric forms. A correlation between AS‐protective Arg528 and Glu730 ERAP1 variants and a decreased surface expression of B27 FHC on monocytes from AS patients as well as on B27‐expressing cell lines has been reported 92. In contrast, a previous study documented a higher level of FHC induced by the AS‐protective Glu730 ERAP1 variant, while no effect was attributed to Arg528Lys polymorphism 93.
There are also conflicting results on the impact of ERAP1 down‐regulation, which was found to correlate with a lower expression of surface B27 FHC and, consequently, with a lower Th17 expansion 92. Another report showed an opposite effect with a selective increase of FHC for AS‐associated B*2705 and B*2704 subtypes but not for non‐AS‐associated B*2706 and B*2709 alleles on C1R transfectants 93. Similarly, Tran and co‐workers have observed an accumulation of disulphide‐linked HLA‐B27 dimers on U937 monocytic cell lines following ERAP1 knock‐down, while levels of HLA‐B18 and HLA‐B51 were unaffected 94.
Current data do not allow to establish a link between ERAP1 trimming activity, intracellular B27 aberrant forms, cellular stress and disease. Dendritic cells derived from HLA‐B27 patients with AS exhibited ERAP1 over‐expression in comparison with healthy controls, but this did not parallel with an altered amount of overall HLA class I dimers 95. Another study performed in HLA‐B27‐positive and HLA‐B27‐negative AS patients, in the presence of risk or protective ERAP1 variants, did not show significant differences in the expression of ER stress markers nor of proinflammatory cytokines, ruling out the ER stress as cause of disease 96.
A very recent work analysed the HLA‐B27 peptidome from spleen cells of HLA‐B27 transgenic rats in conditions of heterozygous or homozygous deletion of ERAP1 97. Interestingly, the knock‐out genotype of ERAP1 altered approximately one‐third of the B27 peptidome, but was still disease‐permissive 97.
Unlike ERAP1, the association of ERAP2 with AS is independent from HLA‐B27, occurring in both B27‐positive and ‐negative carriers 98, 99. This finding allows speculation of a role for the two aminopeptidases not necessarily converging on the same mechanism. Notably, the ERAP2 null‐variant rs2248374 is strongly protective for AS (Table 1) 99. Hence, ERAP2 could be involved in the AS acting at two levels; that is, coupled or uncoupled with ERAP1. Accordingly, ERAP2 has been shown to influence directly the B*2705 peptidome, destroying some ligands with N‐terminal basic residues and, indirectly, increasing the amount of nonamers through the enhancement of ERAP1 activity 100. The latest work has demonstrated that the effects of ERAP2 on B27 peptidome could change depending on the amount of ERAP1 trimming 101. The net consequence of ERAP2 presence/absence on HLA‐B27 conformations remains to be determined. In one report, the presence of ERAP2 did not influence significantly the expression of folded and unfolded HLA‐B27 molecules, ER stress markers and proinflammatory cytokines in patients versus controls 102. In contrast, another study reported that the loss of ERAP2 induced an increase of FHC B27 level as well as up‐regulation of the UPR pathway 103. Overall, the molecular mechanisms underlying the influence of ERAP2 on AS risk are far from being understood.
HLA‐B27 and ERAP1/2 interplay in the anti‐viral immunity
One of the first studies investigating the role of aminopeptidases in anti‐viral defence reported a genetic association of ERAP2 with natural resistance to HIV 104. The notion that the relevance of ERAP is within the framework of HLA antigen presentation has been clearly supported by an in vivo study performed in a flu‐infected murine model based on HLA‐B27/ERAP–/– or HLA‐B7/ERAP–/– transgenic mice that, unlike humans, express only one ERAP gene (ERAAP) 105. Interestingly, the analysis of cytotoxic T cell responses directed against two influenza nucleoprotein immunodominant peptides restricted for HLA‐B27 (NP) 383–391 and HLA‐B7 (NP) 418–426, a protective allele for AS 106, demonstrated that only the HLA‐B27‐restricted response was ERAP‐dependent and its absence led to the reduction of HLA‐B27 molecules on the cell surface as well as of NP 383–391‐reactive CD8+ T cells 105.
In humans, studies performed in vitro on N‐terminally extended precursors of naturally processed HLA‐B27 antigens from human respiratory syncytial virus (HRSV) have shown that the two aminopeptidases operate in a concerted manner, each using the digestion products of the other as substrate for a further N‐terminal cleavage 107.
Seregin and co‐workers observed, using an in vitro cell‐based antigen presentation system, that the high AS‐risk ERAP1 allotype (Met349/Lys528/Asp575/Arg725/Gln730) compared with the low AS‐risk allotype (Val349/Arg528/Asn575/Gln725/Glu730) influenced antigen presentation by destroying more rapidly the majority of HLA‐B27 peptides, whether from viral, bacterial or self‐origin 108. Hence, the authors speculated that the co‐existence of B27 molecules and ERAP1 allotypes with enhanced enzymatic activity alters the normal presentation of microbial and self‐peptides to the adaptive immune system setting the conditions to autoimmunity 108.
Another study has reported that the silencing of ERAP1 as well as the AS‐protective allelic variant Arg528 reduced the presentation of the HIV‐Gag immunodominant HLA‐B27 epitope, KK10 91. Interestingly, the decreased CTL recognition of the cells expressing the Arg528 ERAP1 variant or the minigenes containing KK10 precursors was reversed by the combination Arg528/730Glu, supporting the concept that the global ERAP1 haplotype modulates the fine enzymatic specificity 91.
It is evident from the above that we still have a fragmented view of the role of ERAP1 and 2 in the HLA‐B27‐mediated adaptive immunity. It would be most interesting to assess whether the more effective protection against specific viral infections conferred by the HLA‐B27 comes from a synergic interplay with particular ERAP1 and 2 haplotypes (Fig. 1).
Closing remarks
It is amazing that after so many decades of intense studies, the role of HLA‐B27, the gene associated more strongly with AS pathogenesis, remains uncertain. The new entries, ERAP1 and ERAP2, point strongly at the shaping of the B27 peptidome as a crucial event. Genetic association studies suggest that ERAP1 allotypes with high trimming activity, together with the presence of ERAP2, are strong predisposing factors for AS. However, we still do not know whether or not they contribute directly by destroying the ‘normal peptidome’ or by generating harmful peptides. Alternatively, their activity can impact upon the B27 complex stability and, consequently, on the chance to form dimers or oligomeric structures. A deeper knowledge of the interplay between HLA‐B27 and ERAP1/2 in tuning the antiviral response would certainly help in the understanding of their interconnection in autoimmune diseases.
Disclosure
The authors declare no disclosures.
Acknowledgements
The authors wish to thank Ceschina Foundation and Sapienza University of Rome (Progetti di Ateneo) for their financial support.
References
- 1. McGonagle D, Aydin SZ, Gül A, Mahr A, Direskeneli H. ‘MHC‐I‐opathy’‐unified concept for spondyloarthritis and Behçet disease. Nat Rev Rheumatol 2015; 11:731–40. [DOI] [PubMed] [Google Scholar]
- 2. López de Castro JA, Alvarez‐Navarro C, Brito A, Guasp P, Martín‐Esteban A, Sanz‐Bravo A. Molecular and pathogenic effects of endoplasmic reticulum aminopeptidases ERAP1 and ERAP2 in MHC‐I‐associated inflammatory disorders: towards a unifying view. Mol Immunol 2016; 77:193–204. [DOI] [PubMed] [Google Scholar]
- 3. Kirino Y, Bertsias G, Ishigatsubo Y et al Genome‐wide association analysis identifies new susceptibility loci for Behçet's disease and epistasis between HLA‐B*51 and ERAP1. Nat Genet 2013; 45:202–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Strange A, Capon F, Spencer CC et al A genome‐wide association study identifies new psoriasis susceptibility loci and an interaction between HLA‐C and ERAP1. Nat Genet 2010; 42:985–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Evans DM, Spencer CC, Pointon JJ et al Interaction between ERAP1 and HLA‐B27 in ankylosing spondylitis implicates peptide handling in the mechanism for HLA‐B27 in disease susceptibility. Nat Genet 2011; 43:761–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brewerton DA, Hart FD, Nicholls A, Caffrey M, James DC, Sturrock RD. Ankylosing spondylitis and HL‐A 27. Lancet 1973; 1:904–7. [DOI] [PubMed] [Google Scholar]
- 7. Schlosstein L, Terasaki PI, Bluestone R, Pearson CM. High association of an HL‐A antigen, W27, with ankylosing spondylitis. N Engl J Med 1973; 288:704–6. [DOI] [PubMed] [Google Scholar]
- 8. Marcilla M, López de Castro JA. Peptides: the cornerstone of HLA‐B27 biology and pathogenetic role in spondyloarthritis. Tissue Antigens 2008; 71:495–506. [DOI] [PubMed] [Google Scholar]
- 9. Bowness P. HLA‐B27. Annu Rev Immunol 2015; 33:29–48. [DOI] [PubMed] [Google Scholar]
- 10. Hermann E, Yu DT, Meyer zum Büschenfelde KH, Fleischer B. HLA‐B27‐restricted CD8 T cells derived from synovial fluids of patients with reactive arthritis and ankylosing spondylitis. Lancet 1993; 342:646–50. [DOI] [PubMed] [Google Scholar]
- 11. Fiorillo MT, Maragno M, Butler R, Dupuis ML, Sorrentino R. CD8+ T cell autoreactivity to an HLA‐B27‐restricted self‐epitope correlates with ankylosing spondylitis. J Clin Invest 2000; 106:47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schmitt SK. Reactive arthritis. Infect Dis Clin North Am 2017; 31:265–77. [DOI] [PubMed] [Google Scholar]
- 13. Yang L, Wang L, Wang X, Xian CJ, Lu H. A possible role of intestinal microbiota in the pathogenesis of ankylosing spondylitis. Int J Mol Sci 2016; 17:E2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Taurog JD, Dorris ML, Satumtira N et al Spondylarthritis in HLA‐B27/human beta2‐microglobulin‐transgenic rats is not prevented by lack of CD8. Arthritis Rheum 2009; 60:1977–84. [DOI] [PubMed] [Google Scholar]
- 15. Khan MA. An update on the genetic polymorphism of HLA‐B*27 with 213 alleles encompassing 160 subtypes (and still counting). Curr Rheumatol Rep 2017; 19:9. [DOI] [PubMed] [Google Scholar]
- 16. Reveille JD, Maganti RM. Subtypes of HLA‐B27: history and implications in the pathogenesis of ankylosing spondylitis. Adv Exp Med Biol 2009; 649:159–76. [DOI] [PubMed] [Google Scholar]
- 17. Mathieu A, Cauli A, Fiorillo MT, Sorrentino R. HLA‐B27 and ankylosing spondylitis geographic distribution as the result of a genetic selection induced by malaria endemic? A review supporting the hypothesis. Autoimmun Rev 2008; 7:398–403. [DOI] [PubMed] [Google Scholar]
- 18. Schittenhelm RB, Sian TC, Wilmann PG, Dudek NL, Purcell AW. Revisiting the arthritogenic peptide theory: quantitative not qualitative changes in the peptide repertoire of HLA‐B27 allotypes. Arthritis Rheumatol 2015; 67:702–13. [DOI] [PubMed] [Google Scholar]
- 19. Schittenhelm RB, Sivaneswaran S, Lim Kam Sian TC, Croft NP, Purcell AW. Human leukocyte antigen (HLA) B27 allotype‐specific binding and candidate arthritogenic peptides revealed through heuristic clustering of data‐independent acquisition mass spectrometry (DIA‐MS) data. Mol Cell Proteomics 2016; 15:1867–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. García‐Medel N, Sanz‐Bravo A, Alvarez‐Navarro C et al Peptide handling by HLA‐B27 subtypes influences their biological behavior, association with ankylosing spondylitis and susceptibility to endoplasmic reticulum aminopeptidase 1 (ERAP1). Mol Cell Proteomics 2014; 13:3367–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Antoniou AN, Lenart I, Guiliano DB. Pathogenicity of misfolded and dimeric HLA‐B27 molecules. Int J Rheumatol 2011; 2011:486856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Colbert RA, Tran TM, Layh‐Schmitt G. HLA‐B27 misfolding and ankylosing spondylitis. Mol Immunol 2014; 57:44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Smith JA. The role of the unfolded protein response in axial spondyloarthritis. Clin Rheumatol 2016; 35:1425–31. [DOI] [PubMed] [Google Scholar]
- 24. Navid F, Colbert RA. Causes and consequences of endoplasmic reticulum stress in rheumatic disease. Nat Rev Rheumatol 2017; 13:25–40. [DOI] [PubMed] [Google Scholar]
- 25. DeLay ML, Turner MJ, Klenk EI, Smith JA, Sowders DP, Colbert RA. HLA‐B27 misfolding and the unfolded protein response augment interleukin‐23 production and are associated with Th17 activation in transgenic rats. Arthritis Rheum 2009; 60:2633–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zeng L, Lindstrom MJ, Smith JA. Ankylosing spondylitis macrophage production of higher levels of interleukin‐23 in response to lipopolysaccharide without induction of a significant unfolded protein response. Arthritis Rheum 2011; 63:3807–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ciccia F, Accardo‐Palumbo A, Rizzo A et al Evidence that autophagy, but not the unfolded protein response, regulates the expression of IL‐23 in the gut of patients with ankylosing spondylitis and subclinical gut inflammation. Ann Rheum Dis 2014; 73:1566–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rysnik O, McHugh K, van Duivenvoorde L et al Non‐conventional forms of HLA‐B27 are expressed in spondyloarthritis joints and gut tissue. J Autoimmun 2016; 70:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bird LA, Peh CA, Kollnberger S, Elliott T, McMichael AJ, Bowness P. Lymphoblastoid cells express HLA‐B27 homodimers both intracellularly and at the cell surface following endosomal recycling. Eur J Immunol 2003; 33:748–59. [DOI] [PubMed] [Google Scholar]
- 30. Shaw J, Hatano H, Kollnberger S. The biochemistry and immunology of non‐canonical forms of HLA‐B27. Mol Immunol 2014; 57:52–8. [DOI] [PubMed] [Google Scholar]
- 31. Ridley A, Hatano H, Wong‐Baeza I et al Activation‐induced killer cell immunoglobulin‐like receptor 3DL2 binding to HLA‐B27 licenses pathogenic T cell differentiation in spondyloarthritis. Arthritis Rheumatol 2016; 68:901–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jethwa H, Bowness P. The interleukin (IL)‐23/IL‐17 axis in ankylosing spondylitis: new advances and potentials for treatment. Clin Exp Immunol 2016; 183:30–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Giquel B, Carmouse S, Denais C et al Two HLA‐B27 alleles differently associated with spondyloarthritis, B*2709 and B*2705, display similar intracellular trafficking and oligomer formation. Arthritis Rheum 2007; 56:2232–42. [DOI] [PubMed] [Google Scholar]
- 34. Blanco‐Gelaz MA, Suárez‐Alvarez B, Díaz‐Peña R, López‐Larrea C. HLA‐B27 polymorphism at position 116 critically influences the association with TAP/tapasin, intracellular trafficking and conformational homodimers formation. Mol Immunol 2009; 46:1304–11. [DOI] [PubMed] [Google Scholar]
- 35. Rana MK, Luthra‐Guptasarma M. Differences in conformational stability of the two alpha domains of the disease‐associated and non‐disease‐associated subtypes of HLA‐B27. Int J Biol Macromol 2017; 94:233–45. [DOI] [PubMed] [Google Scholar]
- 36. Galocha B, de Castro JA. Folding of HLA‐B27 subtypes is determined by the global effect of polymorphic residues and shows incomplete correspondence to ankylosing spondylitis. Arthritis Rheum 2008; 58:401–12. [DOI] [PubMed] [Google Scholar]
- 37. Jeanty C, Sourisce A, Noteuil A et al HLA‐B27 subtype oligomerization and intracellular accumulation patterns correlate with predisposition to spondyloarthritis. Arthritis Rheumatol 2014; 66:2113–23. [DOI] [PubMed] [Google Scholar]
- 38. Vázquez MN, López de Castro JA. Similar cell surface expression of beta2‐microglobulin‐free heavy chains by HLA‐B27 subtypes differentially associated with ankylosing spondylitis. Arthritis Rheum 2005; 52:3290–9. [DOI] [PubMed] [Google Scholar]
- 39. Goodall JC, Ellis L, Hill Gaston JS. Spondylarthritis‐associated and non‐spondylarthritis‐associated B27 subtypes differ in their dependence upon tapasin for surface expression and their incorporation into the peptide loading complex. Arthritis Rheum 2006; 54:138–47. [DOI] [PubMed] [Google Scholar]
- 40. Cauli A, Shaw J, Giles J et al The arthritis‐associated HLA‐B*27:05 allele forms more cell surface B27 dimer and free heavy chain ligands for KIR3DL2 than HLA‐B*27:09. Rheumatology 2013; 52:1952–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lenart I, Guiliano DB, Burn G et al The MHC Class I heavy chain structurally conserved cysteines 101 and 164 participate in HLA‐B27 dimer formation. Antioxid Redox Signal 2012; 16:33–43. [DOI] [PubMed] [Google Scholar]
- 42. Campbell EC, Antoniou AN, Powis SJ. The multi‐faceted nature of HLA class I dimer molecules. Immunology 2012; 136:380–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fussell H, Nesbeth D, Lenart I et al Novel detection of in vivo HLA‐B27 conformations correlates with ankylosing spondylitis association. Arthritis Rheum 2008; 58:3419–24. [DOI] [PubMed] [Google Scholar]
- 44. Guiliano DB, North H, Panayoitou E et al Polymorphisms in the F pocket of HLA‐B27 subtypes strongly impact on assembly, chaperone interactions and heavy chain misfolding. Arthritis Rheum 2017; 69:610–21. [DOI] [PubMed] [Google Scholar]
- 45. Loll B, Fabian H, Huser H et al Increased conformational flexibility of HLA‐B*27 subtypes associated with ankylosing spondylitis. Arthritis Rheumatol 2016; 68:1172–82. [DOI] [PubMed] [Google Scholar]
- 46. Abualrous ET, Fritzsche S, Hein Z et al F pocket flexibility influences the tapasin dependence of two differentially disease‐associated MHC class I proteins. Eur J Immunol 2015; 45:1248–57. [DOI] [PubMed] [Google Scholar]
- 47. Narzi D, Becker CM, Fiorillo MT, Uchanska‐Ziegler B, Ziegler A, Böckmann RA. Dynamical characterization of two differentially disease associated MHC class I proteins in complex with viral and self‐peptides. J Mol Biol 2012; 415:429–42. [DOI] [PubMed] [Google Scholar]
- 48. Neumann‐Haefelin C. HLA‐B27‐mediated protection in HIV and hepatitis C virus infection and pathogenesis in spondyloarthritis: two sides of the same coin? Curr Opin Rheumatol 2013; 25:426–33. [DOI] [PubMed] [Google Scholar]
- 49. Sorrentino R, Bockmann RA, Fiorillo MT. HLA‐B27 and antigen presentation: at the crossroads between immune defense and autoimmunity. Mol Immunol 2014; 57:22–7. [DOI] [PubMed] [Google Scholar]
- 50. Deeks SG, Walker BD. Human immunodeficiency virus controllers: mechanisms of durable virus control in the absence of antiretroviral therapy. Immunity 2007; 27:406–16. [DOI] [PubMed] [Google Scholar]
- 51. Neumann‐Haefelin C, McKiernan S, Ward S et al Dominant influence of an HLA‐B27 restricted CD8+ T cell response in mediating HCV clearance and evolution. Hepatology 2006; 43:563–72. [DOI] [PubMed] [Google Scholar]
- 52. McMichael AJ. Triple bypass: complicated paths to HIV escape. J Exp Med 2007; 204:2785–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schneidewind A, Brockman MA, Sidney J et al Structural and functional constraints limit options for cytotoxic T‐lymphocyte escape in the immunodominant HLA‐B27‐restricted epitope in human immunodeficiency virus type 1 capsid. J Virol 2008; 82:5594–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Almeida JR, Price DA, Papagno L et al Superior control of HIV‐1 replication by CD8+ T cells is reflected by their avidity, polyfunctionality, and clonal turnover. J Exp Med 2007; 204:2473–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kosmrlj A, Read EL, Qi Y et al Effects of thymic selection of the T‐cell repertoire on HLA class I‐associated control of HIV infection. Nature 2010; 465:350–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chen H, Ndhlovu ZM, Liu D et al TCR clonotypes modulate the protective effect of HLA class I molecules in HIV‐1 infection. Nat Immunol 2012; 13:691–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ladell K, Hashimoto M, Iglesias MC et al A molecular basis for the control of preimmune escape variants by HIV‐specific CD8+ T cells. Immunity 2013; 38:425–36. [DOI] [PubMed] [Google Scholar]
- 58. Elahi S, Dinges WL, Lejarcegui N et al Protective HIV‐specific CD8+ T cells evade Treg cell suppression. Nat Med 2011; 17:989–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Schmidt J, Iversen AK, Tenzer S et al Rapid antigen processing and presentation of a protective and immunodominant HLA‐B*27‐restricted hepatitis C virus‐specific CD8+ T‐cell epitope. PLOS Pathog 2012; 8:e1003042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tedeschi V, Vitulano C, Cauli A et al The ankylosing spondylitis‐associated HLA‐B*2705 presents a B*0702‐restricted EBV epitope and sustains the clonal amplification of cytotoxic T cells in patients. Mol Med 2016; 22:215–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hill A, Worth A, Elliott T et al Characterization of two Epstein–Barr virus epitopes restricted by HLA‐B7. Eur J Immunol 1995; 25:18–24. [DOI] [PubMed] [Google Scholar]
- 62. Hattori A, Tsujimoto M. Endoplasmic reticulum aminopeptidases: biochemistry, physiology and pathology. J Biochem 2013; 154:219–28. [DOI] [PubMed] [Google Scholar]
- 63. Stratikos E, Stern LJ. Antigenic peptide trimming by ER aminopeptidases – insights from structural studies. Mol Immunol 2013; 55:212–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Alvarez‐Navarro C, López de Castro JA. ERAP1 structure, function and pathogenetic role in ankylosing spondylitis and other MHC‐associated diseases. Mol Immunol 2014; 57:12–21. [DOI] [PubMed] [Google Scholar]
- 65. Kochan G, Krojer T, Harvey D et al Crystal structures of the endoplasmic reticulum aminopeptidase‐1 (ERAP1) reveal the molecular basis for N‐terminal peptide trimming. Proc Natl Acad Sci USA 2011; 108:7745–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. York IA, Chang SC, Saric T et al The ER aminopeptidase ERAP1 enhances or limits antigen presentation by trimming epitopes to 8–9 residues. Nat Immunol 2002; 3:1177–84. [DOI] [PubMed] [Google Scholar]
- 67. Chang SC, Momburg F, Bhutani N, Goldberg AL. The ER aminopeptidase, ERAP1, trims precursors to lengths of MHC class I peptides by a ‘molecular ruler’ mechanism. Proc Natl Acad Sci USA 2005; 102:17107–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Nguyen TT, Chang SC, Evnouchidou I et al Structural basis for antigenic peptide precursor processing by the endoplasmic reticulum aminopeptidase ERAP1. Nat Struct Mol Biol 2011; 18:604–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hearn A, York IA, Rock KL. The specificity of trimming of MHC class I‐presented peptides in the endoplasmic reticulum. J Immunol 2009; 183:5526–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Reeves E, Edwards CJ, Elliott T, James E. Naturally occurring ERAP1 haplotypes encode functionally distinct alleles with fine substrate specificity. J Immunol 2013; 191:35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Evnouchidou I, Momburg F, Papakyriakou A et al The internal sequence of the peptide‐substrate determines its N‐terminus trimming by ERAP1. PLOS ONE 2008; 3:e3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Evnouchidou I, Birtley J, Seregin S et al A common single nucleotide polymorphism in endoplasmic reticulum aminopeptidase 2 induces a specificity switch that leads to altered antigen processing. J Immunol 2012; 189:2383–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Birtley JR, Saridakis E, Stratikos E, Mavridis IM. The crystal structure of human endoplasmic reticulum aminopeptidase 2 reveals the atomic basis for distinct roles in antigen processing. Biochemistry 2012; 51:286–95. [DOI] [PubMed] [Google Scholar]
- 74. Mpakali A, Giastas P, Mathioudakis N, Mavridis IM, Saridakis E, Stratikos E. Structural basis for antigenic peptide recognition and processing by endoplasmic reticulum (ER) aminopeptidase 2. J Biol Chem 2015; 290:26021–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Saveanu L, Carroll O, Lindo V et al Concerted peptide trimming by human ERAP1 and ERAP2 aminopeptidase complexes in the endoplasmic reticulum. Nat Immunol 2005; 6:689–97. [DOI] [PubMed] [Google Scholar]
- 76. Evnouchidou I, Weimershaus M, Saveanu L, van Endert P. ERAP1–ERAP2 dimerization increases peptide‐trimming efficiency. J Immunol 2014; 193:901–8. [DOI] [PubMed] [Google Scholar]
- 77. Chen H, Li L, Weimershaus M, Evnouchidou I, van Endert P, Bouvier M. ERAP1–ERAP2 dimers trim MHC I‐bound precursor peptides; implications for understanding peptide editing. Sci Rep 2016; 6:28902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ombrello MJ, Kastner DL, Remmers EF. Endoplasmic reticulum‐associated amino‐peptidase 1 and rheumatic disease: genetics. Curr Opin Rheumatol 2015; 27:349–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Reeves E, Colebatch‐Bourn A, Elliott T, Edwards CJ, James E. Functionally distinct ERAP1 allotype combinations distinguish individuals with ankylosing spondylitis. Proc Natl Acad Sci USA 2014; 111:17594–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Martín‐Esteban A, Gómez‐Molina P, Sanz‐Bravo A, López de Castro JA. Combined effects of ankylosing spondylitis‐associated ERAP1 polymorphisms outside the catalytic and peptide‐binding sites on the processing of natural HLA‐B27 ligands. J Biol Chem 2014; 289:3978–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Stamogiannos A, Koumantou D, Papakyriakou A, Stratikos E. Effects of polymorphic variation on the mechanism of endoplasmic reticulum aminopeptidase 1. Mol Immunol 2015; 67:426–35. [DOI] [PubMed] [Google Scholar]
- 82. Evnouchidou I, Kamal RP, Seregin SS et al Cutting Edge: coding single nucleotide polymorphisms of endoplasmic reticulum aminopeptidase 1 can affect antigenic peptide generation in vitro by influencing basic enzymatic properties of the enzyme. J Immunol 2011; 186:1909–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Costantino F, Talpin A, Evnouchidou I et al ERAP1 gene expression is influenced by nonsynonymous polymorphisms associated with predisposition to spondyloarthritis. Arthritis Rheumatol 2015; 67:1525–34. [DOI] [PubMed] [Google Scholar]
- 84. Andrés AM, Dennis MY, Kretzschmar WW et al Balancing selection maintains a form of ERAP2 that undergoes nonsense‐mediated decay and affects antigen presentation. PLOS Genet 2010; 6:e1001157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Vanhille DL, Hill LD, Hilliard DD et al A novel ERAP2 haplotype structure in a Chilean population: implications for ERAP2 protein expression and preeclampsia risk. Mol Genet Genomic Med 2013; 1:98–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Burton PR, Clayton DG, Cardon LR et al Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat Genet 2007; 39:1329–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Reeves E, Elliott T, James E, Edwards CJ. ERAP1 in the pathogenesis of ankylosing spondylitis. Immunol Res 2014; 60:257–69. [DOI] [PubMed] [Google Scholar]
- 88. Roberts AR, Appleton LH, Cortes A et al ERAP1 association with ankylosing spondylitis is attributable to common genotypes rather than rare haplotype combinations. Proc Natl Acad Sci USA 2017; 114:558–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. García‐Medel N, Sanz‐Bravo A, Van Nguyen D et al Functional interaction of the ankylosing spondylitis‐associated endoplasmic reticulum aminopeptidase 1 polymorphism and HLA‐B27 in vivo . Mol Cell Proteomics 2012; 11:1416–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Sanz‐Bravo A, Campos J, Mazariegos MS, López de Castro JA. Dominant role of the ERAP1 polymorphism R528K in shaping the HLA‐B27 peptidome through differential processing determined by multiple peptide residues. Arthritis Rheumatol 2015; 67:692–701. [DOI] [PubMed] [Google Scholar]
- 91. Chen L, Fischer R, Peng Y et al Critical role of endoplasmic reticulum aminopeptidase 1 in determining the length and sequence of peptides bound and presented by HLA‐B27. Arthritis Rheumatol 2014; 66:284–94. [DOI] [PubMed] [Google Scholar]
- 92. Chen L, Ridley A, Hammitzsch A et al Silencing or inhibition of endoplasmic reticulum aminopeptidase 1 (ERAP1) suppresses free heavy chain expression and Th17 responses in ankylosing spondylitis. Ann Rheum Dis 2016; 75:916–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Haroon N, Tsui FW, Uchanska‐Ziegler B, Ziegler A, Inman RD. Endoplasmic reticulum aminopeptidase 1 (ERAP1) exhibits functionally significant interaction with HLA‐B27 and relates to subtype specificity in ankylosing spondylitis. Ann Rheum Dis 2012; 71:589–95. [DOI] [PubMed] [Google Scholar]
- 94. Tran TM, Hong S, Edwan JH, Colbert RA. ERAP1 reduces accumulation of aberrant and disulfide‐linked forms of HLA‐B27 on the cell surface. Mol Immunol 2016; 74:10–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Campbell EC, Fettke F, Bhat S, Morley KD, Powis SJ. Expression of MHC class I dimers and ERAP1 in an ankylosing spondylitis patient cohort. Immunology 2011; 133:379–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Kenna TJ, Lau MC, Keith P et al Disease‐associated polymorphisms in ERAP1 do not alter endoplasmic reticulum stress in patients with ankylosing spondylitis. Genes Immun 2015; 16:35–42. [DOI] [PubMed] [Google Scholar]
- 97. Barnea E, Melamed Kadosh D, Haimovich Y et al The human leukocyte antigen (HLA)‐B27 peptidome in vivo, in spondyloarthritis‐susceptible HLA‐B27 transgenic rats and the effect of ERAP1 deletion. Mol Cell Proteomics 2017; 16:642–662s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Cortes A, Hadler J, Pointon JP et al Identification of multiple risk variants for ankylosing spondylitis through high‐density genotyping of immune‐related loci. Nat Genet 2013; 45:730–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Robinson PC, Costello ME, Leo P et al ERAP2 is associated with ankylosing spondylitis in HLA‐B27‐positive and HLA‐B27‐negative patients. Ann Rheum Dis 2015; 74:1627–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Martín‐Esteban A, Guasp P, Barnea E, Admon A, López de Castro JA. Functional interaction of the ankylosing spondylitis‐associated endoplasmic reticulum aminopeptidase 2 with the HLA‐B*27 peptidome in human cells. Arthritis Rheumatol 2016; 68:2466–75. [DOI] [PubMed] [Google Scholar]
- 101. Martín‐Esteban A, Sanz‐Bravo A, Guasp P, Barnea E, Admon A, López de Castro JA. Separate effects of the ankylosing spondylitis associated ERAP1 and ERAP2 aminopeptidases determine the influence of their combined phenotype on the HLA‐B*27 peptidome. J Autoimmun 2017; 79:28–38. [DOI] [PubMed] [Google Scholar]
- 102. Robinson PC, Lau E, Keith P et al ERAP2 functional knockout in humans does not alter surface heavy chains or HLA‐B27, inflammatory cytokines or endoplasmic reticulum stress markers. Ann Rheum Dis 2015; 74:2092–5. [DOI] [PubMed] [Google Scholar]
- 103. Zhang Z, Ciccia F, Zeng F et al Brief report: functional interaction of endoplasmic reticulum aminopeptidase 2 and HLA‐B27 activates the unfolded protein response. Arthritis Rheumatol 2017; 69:1009–15. [DOI] [PubMed] [Google Scholar]
- 104. Cagliani R, Riva S, Biasin M et al Genetic diversity at endoplasmic reticulum aminopeptidases is maintained by balancing selection and is associated with natural resistance to HIV‐1 infection. Hum Mol Genet 2010; 19:4705–14. [DOI] [PubMed] [Google Scholar]
- 105. Akram A, Lin A, Gracey E, Streutker CJ, Inman RD. HLA‐B27, but not HLA‐B7, immunodominance to influenza is ERAP dependent. J Immunol 2014; 192:5520–8. [DOI] [PubMed] [Google Scholar]
- 106. Cortes A, Pulit SL, Leo PJ et al Major histocompatibility complex associations of ankylosing spondylitis are complex and involve further epistasis with ERAP1. Nat Commun 2015; 6:7146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Lorente E, Barriga A, Johnstone C, Mir C, Jiménez M, López D. Concerted in vitro trimming of viral HLA‐B27‐restricted ligands by human ERAP1 and ERAP2 aminopeptidases. PLoS One 2013; 8:e79596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Seregin SS, Rastall DP, Evnouchidou I et al Endoplasmic reticulum aminopeptidase‐1 alleles associated with increased risk of ankylosing spondylitis reduce HLA‐B27 mediated presentation of multiple antigens. Autoimmunity 2013; 46:497–508. [DOI] [PMC free article] [PubMed] [Google Scholar]