

Abstract
Catalytic transformation of CO2 to formate is generally realized through bicarbonate hydrogenation in an alkaline environment, while it suffers from a thermodynamic sink due to the considerable thermodynamic stability of the bicarbonate intermediate. Here, we devise a route for the direct catalytic conversion of CO2 over a Schiff-base-modified gold nanocatalyst that is comparable to the fastest known nanocatalysts, with a turnover number (TON) of up to 14,470 over 12 h at 90 °C. Theoretical calculations and spectral analysis results demonstrate that the activation of CO2 can be achieved through a weakly bonded carbamate zwitterion intermediate derived from a simple Lewis base adduct of CO2. However, this can only occur with a hydrogen lacking Lewis base center in a polar solvent. This finding offers a promising avenue for the direct activation of CO2 and is likely to have considerable implications in the fields of CO2 conversion and gold catalytic chemistry.
Bicarbonate reduction is a bottleneck step of the CO2 hydrogenation to formic acid in alkaline solution. Here, the authors show a highly active Schiff-base-modified gold nanocatalyst for the efficient reduction of CO2 to formic acid under relatively mild conditions.
Introduction
Catalytic hydrogenation of CO2 has attracted much attention in recent years, because it not only serves to mitigate the problem of anthropogenic emissions of CO2 but also provides a feasible avenue for carbon recycling and hydrogen energy conversion or storage1–4. Among the various hydrogenation products of CO2, formic acid, which presents as formate in the practical production for the thermodynamic shift of equilibrium forward, is one of the most attractive, owing to its direct employment as a feedstock chemical or hydrogen source for fuel cells5. In addition, the conversion of CO2 to formic acid is believed to be the first and indispensable step in the reduction of CO2 to other chemicals or fuels6, such as methanol, methane, or other hydrocarbons, and a fundamental understanding of this process is thus essential for C1 chemistry7.
The activation of CO2 is often challenging, owing to the high thermodynamic stability of this molecule8,9. Considering that CO2 acts as an electrophile or Lewis acid10, CO2 is generally activated using an electron donor or base. For example, high CO2 conversions have been achieved using homogeneous catalysts containing electron-donating ligands on the metal active sites11,12. However, the catalytic activity in homogeneous catalysis is very sensitive to the ligand used and the ligands are expensive and leachable, which limits their wide-spread application. Without ligand promotion in a heterogeneous catalytic system, CO2 is always activated in a bent conformation by the interaction between the dissolved base with CO2 13. In such a process, CO2 chemically reacts with an aqueous base, such as KOH or NaOH, to give bicarbonates (HCO3 −), which serve as the real precursors for further hydrogenation to formate5,14,15. As such, the production of formate over supported Pd nanoparticles will be boosted when bicarbonate is used instead of gaseous CO2 as the C1 source16. However, such evolution of CO2 to formate through bicarbonate intermediates actually experiences a thermodynamic sink in the reaction, because the bicarbonate species are considerably more stable than the parent CO2 and final formate. As a result, further hydrogenation of bicarbonate is disfavored and requires the enhanced hydrogenation ability of a heterogeneous catalyst. Therefore, the development of a catalyst that can directly activate CO2 for hydrogenation to formate is highly desirable.
Additionally, for the route through bicarbonate, it has been reported that CO2 can also be chemically captured by N-containing organic bases such as amines17. More importantly, such an activation pattern of CO2 is thermally reversible owing to the relatively weak chemical interaction between CO2 and organic bases, which means that the thermodynamic sink caused by bicarbonate in the evolution of CO2 to formate can be avoided if this new pattern of activation can be employed in the process of CO2 conversion18,19. In our previous work, we have developed a new type of Schiff-base-modified gold catalyst, which, unlike conventional catalysts, performs well in the decomposition of formic acid to give H2 20. Taking into account the capability of gold nanocatalysts in various chemo-selective hydrogenation reactions21–24 as well as the reversible behavior of a catalytic process16,25, this kind of catalyst offers great potential in the CO2 transformation to formate by facilitating the activation of CO2 and its subsequent hydrogenation with the help of gold nanoparticles.
Here, we propose an unusual CO2 activation pathway that, through a weakly bonded carbamate zwitterion complex on the Schiff-base-mediated gold catalyst, can effectively avoid the thermodynamic sink of the conventional bicarbonate reactant, and lead to the direct hydrogenation of CO2 to formate.
Results
Theoretical understanding of CO2 activation
Using density functional theory (DFT), we first calculated the chemical interactions between CO2 and N-containing organic bases, including the imine Schiff base and various organic amine bases using the conductor-like screening model (COSMO)26,27. As shown in Table 1, the CO2 molecule can be directly activated by organic bases in polar water solvent to give a carbamate zwitterion intermediate. The free energies for chemisorption of CO2 (∆G 298) are less negative resulting from the weak chemical interaction between CO2 and N-containing bases. Geometrically, the distance between the CO2 molecule and the Lewis base center in this intermediate is short (1.61–1.67 Å), and this results in an increase in the C = O bond length from 1.16 to 1.22 Å and a distortion of the linear O = C = O bond angle to ~137°. This suggests that although the chemical interactions between CO2 and organic bases are relatively weak, the CO2 molecule can still be well-activated through a non-bicarbonate route in a polar solvent environment.
Table 1.
Structural parameters of CO2 bound to N-containing organic bases as zwitterionic Lewis base (LB) adducts
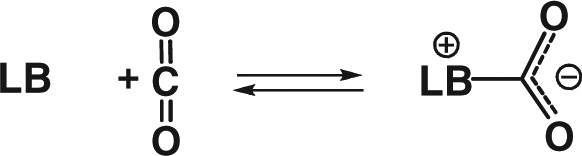
| ||||
|---|---|---|---|---|
| LB | ∆G 298 (Kcal mol−1) | r(N−CO2) (Å) | r(C = O) (Å) | α(O−C−O) (°) |
| Propylamine | −4.40 | 1.61 | 1.22 | 136 |
| Ethylamine | −4.37 | 1.62 | 1.22 | 137 |
| Diethylamine | −2.19 | 1.66 | 1.22 | 137 |
| Triethylamine | 0.83 | 1.66 | 1.22 | 136 |
| N-propylmethanimine | −1.14 | 1.61 | 1.23 | 136 |
B3LYP/def2-TZVP + COSMO with a solvent environment of water. In a non-polar n-hexane environment, the ∆G 298 and changes in the CO2 geometry were negligible
Synthesis and characterization of gold nanocatalysts
Gold nanocatalysts supported on two different alkyl-primary amines-based organic–inorganic hybrid silica materials, namely Au/SiO2-NH2 and Au/SiO2-Schiff, were synthesized according to our previous report using a facile wet-chemistry method20. Briefly, Au/SiO2-Schiff was prepared by aldimine condensation of (3-aminopropyl)triethoxysilane (APTES) with formaldehyde, whereas Au/SiO2-NH2 was obtained by cohydrolysis of APTES and tetraethyl orthosilicate (TEOS), followed by a reduction of the gold precursor with NaBH4. Both samples exhibited a metal loading of ~1.5 wt%, which was determined by inductively coupled plasma mass spectrometry (Supplementary Table 1).
As indicated by high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) (Fig. 1a, b), the different synthetic procedures gave a similar distribution of small gold nanoparticles, with a size of ~1.5 nm (obtained by image analysis of more than 300 particles). By further careful examination with the aberration-corrected HAADF-STEM technique, gold species were found to be dispersed primarily as sub-nanoclusters with sizes of <2 nm (Supplementary Fig. 1). Meanwhile, single-atom gold species were also observed on both the catalysts, but with only a rather low fraction in the entire gold distribution. 13C CP-MAS NMR spectra suggested that both samples predominantly contained alkylamine groups on the support, but also some unhydrolyzed alkoxysilane groups (Fig. 1c). This was inferred from the chemical shifts at δ = 11, 22, 43, and 58 ppm, as well as the results from quantum mechanics calculations. More interestingly, residual Schiff base (–N = C–) groups were also found in the Au/SiO2-Schiff sample, for which a distinctive resonance peak with a carbon chemical shift at δ = 164 ppm was detected, which was in good agreement with the calculated NMR shift of the imine group (Fig. 1c). The corresponding stretching vibration of this functional group was observed at 1659 cm−1 in the FT-IR spectrum (Supplementary Fig. 2b), and was consistent with our calculated value of 1653 cm−1. Accordingly, as revealed by X-ray photoelectron spectrometry (XPS), an additional N-containing species appeared in the spectrum of the Au/SiO2-Schiff sample, with the lower-binding energy species belonging to the –N = C– group (Supplementary Fig. 3). Moreover, the gold component of both catalysts was found to exhibit predominantly weak electronegativity and to possess a low content of positive gold species, which were ascribed to the electron-donor capability of nitrogen groups toward the small gold clusters and the presence of some of the isolated gold cations on the support, respectively (Supplementary Fig. 4). Thus, we successfully obtained two gold nanocatalysts with or without the modification by Schiff base on the support.
Fig. 1.
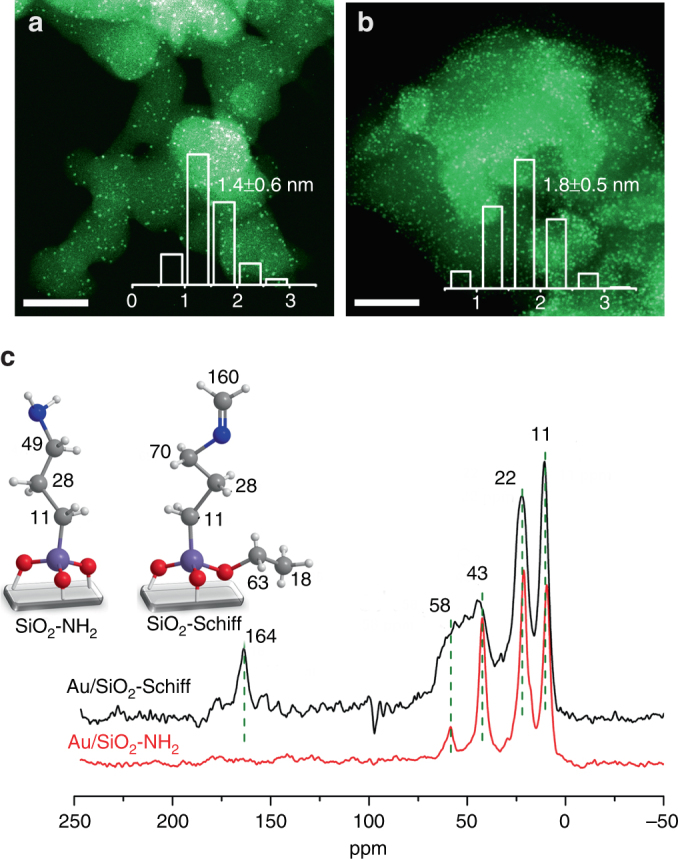
Structural characterization of the two different functionalized gold catalysts. HAADF-STEM images of the prepared Au/SiO2-Schiff a and Au/SiO2-NH2 b catalysts. Scale bar, 50 nm. 13C CP-MAS NMR spectra of Au/SiO2-Schiff and Au/SiO2-NH2 c. The insert shows the results of the quantum mechanics calculation of the NMR shifts
Catalytic hydrogenation of CO2 in liquid phase
The catalytic performance of the as-prepared gold nanocatalysts for CO2 hydrogenation was studied at 90 °C in a H2/CO2 mixture (80 bar) with triethylamine (NEt3) as an additive in the liquid phase. As shown in Table 2, in contrast to the completely inert behavior of Au/SiO2 (entry 1 in Table 2), the CO2 hydrogenation activity of the gold nanocatalysts was greatly enhanced by the presence of an organic base on the support (entries 2 and 3 in Table 2). Moreover, the Schiff-base-modified gold nanocatalyst was found to be considerably more active than its Au/SiO2-NH2 counterpart. Meanwhile, as expected, such an evolution of CO2 only occurred in polar solvents. For example, in water and methanol, the TON reached 9624 and 9806, respectively (entries 4 and 5 in Table 2), whereas in the non-polar solvents of n-hexane (entry 7 in Table 2) and cyclohexane (entry 8 in Table 2), the conversion of CO2 was negligible. The reaction was optimized by experimentally tuning the mixture of methanol and water (Supplementary Table 2), and a TON value as high as 14,470 was achieved in a H2O/methanol mixture containing 20% H2O (vol/vol, entry 2 in Table 2). This TON was comparable with the best result reported in the literature under similar reaction conditions of the heterogeneous conversion of CO2 (Supplementary Table 3).
Table 2.
Catalytic performance of the gold nanocatalysts for CO2 hydrogenation
| Entry | Solvent | P(H2)/P(CO2) MPa | Base | Time/h | HCOOH/M | TONa |
|---|---|---|---|---|---|---|
| 1b | H2O/CH3OH | 5.0/3.0 | NEt3 | 12 | – | – |
| 2 | H2O/CH3OH | 5.0/3.0 | NEt3 | 12 | 0.518 | 14,470 |
| 3c | H2O/CH3OH | 5.0/3.0 | NEt3 | 12 | 0.08 | 1026 |
| 4d | H2O | 5.0/3.0 | NEt3 | 12 | 0.689 | 9624 |
| 5d | Methanol | 5.0/3.0 | NEt3 | 12 | 0.702 | 9806 |
| 6d | Ethanol | 5.0/3.0 | NEt3 | 12 | 0.297 | 4148 |
| 7d | n-Hexane | 5.0/3.0 | NEt3 | 12 | 0.025 | 349 |
| 8d | Cyclohexane | 5.0/3.0 | NEt3 | 12 | 0.013 | 181 |
| 9 | H2O/CH3OH | 5.0/0 | KHCO3 | 5 | 0.009 | 251 |
| 10 | H2O/CH3OH | 5.0/3.0 | KHCO3 | 5 | 0.107 | 2989 |
| 11 | H2O/CH3OH | 5.0/0 | NaHCO3 | 5 | 0.007 | 195 |
| 12 | H2O/CH3OH | 5.0/3.0 | NaHCO3 | 5 | 0.063 | 1760 |
| 13e | H2O/CH3OH | 5.0/3.0 | NEt3 | 12 | – | – |
| 14f | H2O/CH3OH | 5.0/3.0 | NEt3 | 12 | 0.021 | 1207 |
Reaction conditions: 5 mg Au/SiO2-Schiff catalyst, 90 °C, 10 mL reagent, 15 mmol base, 600 rpm, H2O/CH3OH (20:80 vol/vol)
aThe TON is calculated by the Supplementary Equation 1. The number of replicates for each experiment was n ≥ 2
bCatalyst was 5 mg Au/SiO2
cCatalyst was 5 mg Au/SiO2-NH2
dCatalyst was 10 mg Au/SiO2-Schiff
eThe single-atom catalyst was prepared by solid-state reduction method
fThe 3.2-nm nanocatalyst was prepared by surface functional group-promoted in situ reduction method
Bicarbonate is a thermodynamically stable form of CO2 in an alkaline environment, and when bicarbonate is used instead of gaseous CO2 as the precursor for hydrogenation, the production of formate is always increased over Pd-based nanocatalysts16,28. However, this is not the case for our Au/SiO2-Schiff catalyst. That is, when bicarbonate was used as the carbon source, the conversion rate decreased inversely, with a TON of only a few hundred (entries 9 and 11 in Table 2). In contrast, when gaseous CO2 was re-introduced into the autoclave, the activity of the catalyst was greatly recovered (entries 10 and 12 in Table 2). This indicated that a non-bicarbonate CO2 hydrogenation route occurred over the Au/SiO2-Schiff nanocatalyst, in which a direct catalytic CO2 hydrogenation was involved29. Notably, to date, no heterogeneous catalyst has been reported for the direct catalytic CO2 hydrogenation to formate.
Moreover, another two Schiff-base-modified gold catalysts, where one had a larger metal particle size (~3.2 nm) and the other contained only the single-atom gold species, were also prepared by the surface-functional group promoted in situ reduction method20 and the milling-mediated solid reduction method, respectively (Supplementary Figs. 5, 6), and were then evaluated at the same reaction conditions. However, the superior activity of the Au/SiO2-Schiff catalyst was greatly reduced or even quenched (entries 13 and 14 in Table 2).This suggests that the hydrogenation of CO2 over gold nanocatalysts exhibits a great size-dependent behavior, with the small sub-nanoclusters being more efficient than the large-sized nanoparticles; however, the single-atom gold species were ineffective as a catalyst in the catalytic hydrogenation of CO2. A similar size dependency of catalytic activity has also been reported previously for the CO oxidation over Au/FeOx materials30.
Discussion
We have successfully achieved the direct catalytic CO2 hydrogenation to formate over amino-functionalized gold nanocatalysts. However, the organic base on the support has a great impact on the catalytic activity, with the Au/SiO2-Schiff catalyst being superior to Au/SiO2-NH2. To gain further insight into the unique behavior of Au/SiO2-Schiff and the discrepancy between the organic bases in terms of CO2 hydrogenation, in situ diffuse reflectance infrared Fourier transform spectroscopy (in situ DRIFTS) was employed to monitor the adsorption and activation of CO2 over these two samples. As shown in Fig. 2a, on exposure of Au/SiO2-NH2 to CO2 in the gas phase, a distinct absorption band originating from CO2 was observed, which was similar to a previous report of CO2 adsorption on amine-grafted SBA-1531. Accordingly, the peaks at 1488 and 1621 cm−1 were ascribed to the deformation vibration of the NH3 + species32, and those at 1431 and 1329 cm−1 were associated to the skeletal symmetric stretching vibrations of COO− with its asymmetric mode at 1568 cm−1 33. These results suggest that an ionic carbamate species forms on the Au/SiO2-NH2 catalyst as a result of proton transfer between two neighboring amine groups. In addition, the low-intensity absorption band at 1690 cm−1 was corresponding to the vibration of a carbonyl group derived from the surface-bound carbamate, and no bicarbonate species was detected31. As for the Au/SiO2-Schiff catalyst, a NH3 + deformation vibration and a COO− skeletal stretching vibration were also detected, as was observed for the Au/SiO2-NH2 catalyst, because of the presence of the alkylamine on the support. In addition, a remarkable absorption peak at 1712 cm−1 was observed for the Au/SiO2-Schiff catalyst, which indicated a new adsorption pattern for CO2 resulting from the interaction between the Schiff base and CO2. More importantly, as seen in Fig. 2b, on evacuation of the vessel containing the CO2-saturated Au/SiO2-Schiff catalyst, the intensity of this new band decreased considerably faster than that of the ionic carbamate species. This suggested that the new species were less stable, and could be removed more easily than the ionic carbamate species. Considering that the formation of the ionic carbamate resulted from the transfer of a proton between two neighboring amines, only primary and secondary amines can capture CO2 as an ionic carbamate31,34. In contrast, the lack of a proton in the Schiff base (-C = N-) prevents it from acting as a proton donor. It can thus act only as a Lewis base for CO2 adsorption, that is, affording a zwitterion adduct on the support35–37. Indeed, we have also simulated the chemisorption of CO2 on the model of a Au55 sub-nanocluster (~1.2 nm) accommodated with an alkyl-imine Schiff base. As shown in Supplementary Fig. 7, gaseous CO2 was found to be captured at the gold/Schiff base interface through a zwitterion intermediate, with an adsorption energy of 0.2–0.4 eV and its oxygen atoms bonded on the low-coordinated sites of the gold nanocluster. The C-O stretch vibrational frequency was calculated to be in the range of 1701–1767 cm−1 for various possible adsorption patterns, which was in agreement with the spectral observations.
Fig. 2.

Spectral analysis of the gold catalysts during CO2 adsorption/hydrogenation. a FTIR spectra of Au/SiO2-NH2 and Au/SiO2-Schiff under a humid CO2 atmosphere. b Time-resolved DRIFTS spectra of the Au/SiO2-Schiff catalyst after wet-CO2 evacuation. In situ DRIFTS spectra of the CO2 hydrogenation reaction over Au/SiO2-Schiff catalyst without c and with d the presence of a surface-bonded carbamate zwitterion
On the other hand, to elucidate the origins of the variation in the Au/SiO2-NH2 catalyst, we compared the thermodynamic stability of the ionic carbamate and carbamate zwitterion species derived from the primary amine groups (Supplementary Fig. 8). The ionic carbamate was found to be thermodynamically more stable than the carbamate zwitterion, with a free energy for the chemisorption of CO2 (∆G 298) of approximately −8.89 kcal mol−1 in water. Correspondingly, this also led to a decrease of the distance between the Lewis base center and CO2 (~1.40 Å). As a result, this may also lead to a thermodynamic sink in CO2 evolution like that occurring through the bicarbonate intermediate path. In contrast, the relatively poor thermal stability of the zwitterion adduct probably offers an important chance to serve as a highly active intermediate for CO2 transformation36. Such an assumption has further been supported by the in situ DRIFTS experiments of CO2 hydrogenation. As shown in Fig. 2c, with only the surface-bonded ionic carbamate on the Au/SiO2-Schiff catalyst by decompression of CO2 in the gas phase, no characterized peak of the formate product was detected under the operational pressure of H2 (10 bar). In sharp contrast, with the presence of the carbamate zwitterion on the catalyst surface, a new absorption peak at 1590 cm−1, which arose from the stretching vibration of the formate species38, was appreciably observed with an increase of the exposure time (Fig. 2d).
In addition, because the evolution of CO2 to formate in our experiments requires the transfer of reactants (both CO2 and H2) from the gas to the liquid phase and then finally to the catalyst surface, the reaction kinetics insights are also of great significance for understanding the catalytic mechanism of CO2 hydrogenation. We therefore further studied the dependence of the hydrogenation rate on the reaction conditions for the Au/SiO2-Schiff catalyst. As shown in Supplementary Table 4, the rate of CO2 hydrogenation was strongly dependent on the reaction temperature. The calculated turnover frequency value increased from 195 to 1950 h−1 as the temperature was increased from 60 to 90 °C. The apparent activation energy (E a) estimated from the Arrhenius plot was ~76 kJ mol−1 (Supplementary Fig. 9), which differed greatly from the previously reported value for the bicarbonate hydrogenation route24. Figure 3 shows the turnover rates for formate production over the Au/SiO2-Schiff catalyst as a function of the H2 pressure [P(H2)] and CO2 pressure [P(CO2)] in the autoclave. The formation rate was linearly dependent on the pressure of H2, although there were two different slopes in the high- and low-pressure ranges. This behavior showed that the diffusion and activation of H2 on a gold surface may be the bottleneck for CO2 hydrogenation at lower P(H2), whereas the elementary step concerned with the split-H attack becomes the kinetic-controlling step when P(H2) is above 30 bar24. In contrast, the rate of CO2 conversion was independent of the CO2 pressure. This was because the polar solvent with added NEt3 in our reaction served as a CO2 reservoir, in which gaseous CO2 was captured as carbamate intermediates. Different organic base additives in liquid may therefore exhibit different effects on the hydrogenation activity of the catalyst. As shown in Supplementary Table 5, NEt3 showed a much higher conversion than the other organic amines studied. Interestingly, N-containing organic bases without a hydrogen on the Lewis base centers, such as NEt3 and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), showed considerably higher CO2 conversions compared with those containing a proton, like primary or secondary amines. This was because Lewis bases such as NEt3 and DBU can capture CO2 through a weakly bonded carbamate zwitterion complex in a polar solvent environment, whereas primary or secondary amines will protonate the carbamate zwitterion to yield an ionic carbamate. As described above, the ionic carbamate was less favorable than the carbamate zwitterion for the transfer of CO2 to the supported Schiff base for further hydrogenation. In addition, the durability of the Au/SiO2-Schiff catalyst was also tested. As shown in Supplementary Table 6, a gradual decrease of the catalytic activity occurred, with a TON value of 5922 at the third catalytic cycle, which was caused by the coalescence of small-sized gold to large particles (Supplementary Fig. 10).
Fig. 3.
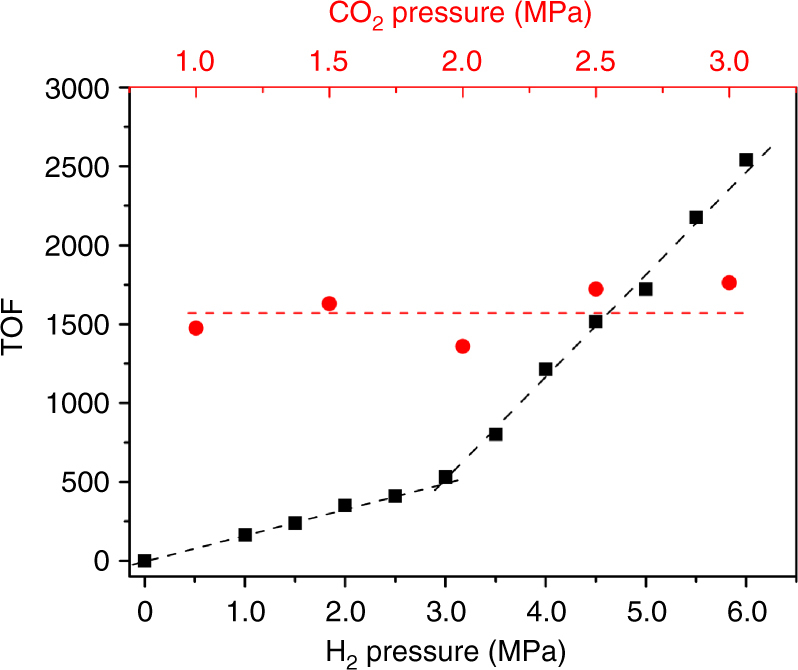
H2 (black square) and CO2 (red circle) pressure-dependent initial reaction rates. Reaction conditions: 5 mg Au/SiO2-Schiff catalyst, 10 mL reagent, 10 mmol NEt3, 600 rpm, H2O/CH3OH (20:80 vol/vol). The solution was pressurized at room temperature with CO2 and completed with H2 to the desired pressure. The system was heated at 90 °C and stirred until a desired formate concentration was reached (0.5–3 h). The initial TOF is calculated by the Supplementary Equation 2
Finally, we performed a mechanistic study of CO2 hydrogenation with DFT calculations based on our previous model of the Au/SiO2-Schiff catalyst under the COSMO solvent effect of water. As shown in Fig. 4, the dissociation of H2 to activated H species occurred on the low-coordinated corner sites of gold nanoclusters, with a barrier of 0.67 eV (TS-1) and a weak exothermic contribution (~0.10 eV). As mentioned above, the CO2 molecule can be facilely captured as a zwitterion intermediate on the interface of Schiff base and gold nanocluster, which can then be hydrogenated by the activated H to an HCO2 intermediate on the gold surface, with a barrier of 1.00 eV (TS-2) and acts as the rate-determining step in the whole of the elementary steps. With the assistance from another H adatom, the HCO2 can easily be further hydrogenated to the cis-HCOOH on the catalyst surface by passing a barrier of 0.58 eV (TS-3), which subsequently desorbs from the gold surface to obtain the desired trans-HCOOH product.
Fig. 4.
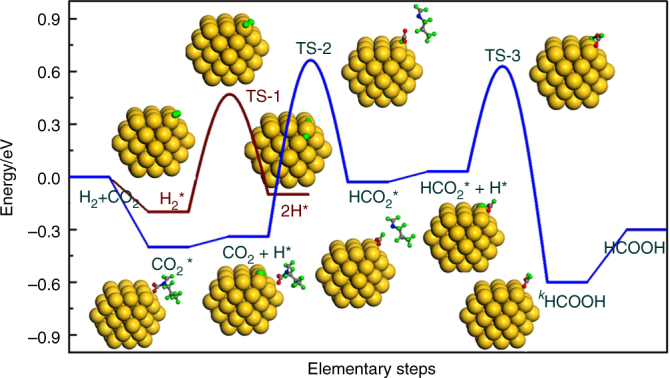
Free energy diagram for CO2 hydrogenation over the Au/SiO2-Schiff catalyst. The geometry with a close-packed Au55 sub-nanocluster (~1.2 nm) accommodated with an alkyl-imine Schiff base under the COSMO solvent effect of water was used to model the real Au/SiO2-Schiff catalyst in water. Energy profile was constructed based on the DFT calculation analysis of each elementary step
From the combination of catalytic performance, spectrum characterization, kinetic analysis, and DFT calculations, we have therefore proposed a possible catalytic process for CO2 hydrogenation over Au/SiO2-Schiff with a NEt3 additive. As shown in Fig. 5, a gaseous CO2 molecule was captured by the NEt3 through a carbamate zwitterion intermediate, which also served as a reservoir for CO2 in the liquid phase. This weakly chemisorbed CO2 could migrate and be transferred to the gold–Schiff base interface but retained its carbamate zwitterion nature. At the same time, the low-coordinated sites of the gold nanoclusters also participated in the activation and dissociation of H2 to the activated H species. Then, the carbamate zwitterion intermediates were hydrogenated by the H species at the gold–Schiff base interface, and the final formate was thus obtained after a two-step hydrogenation and acid–base neutralization in an alkaline environment. Notably, the electron-rich gold surface, caused by the electron donation from nitrogen groups, might also be beneficial for the hydrogenation of CO2, since it could offer a more negative hydride and lead to a higher reactivity of the nucleophilic attack to the carbon center of CO2 39. It was therefore also not surprising that the single-atom gold species was inert in the catalytic hydrogenation of CO2 owing to its single site and cation nature. Single-site gold cationic entities have been previously reported as the only active sites in acetylene hydrochlorination40, which indicates that different gold active sites are involved in the distinguishing catalytic processes.
Fig. 5.
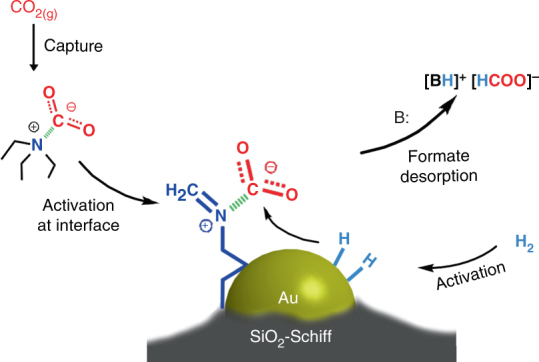
Proposed synergetic mechanism for the hydrogenation of CO2 to formate. The activation of CO2 can be achieved through a weakly bonded carbamate zwitterion intermediate at the gold–Schiff base interface, and then hydrogenated by H species to give formate with the help of gold nanoclusters. The electron-rich gold surface is speculated to be beneficial for the hydrogenation of CO2, since it can offer a more negative hydride and lead to a higher reactivity of nucleophilic attack to the carbon center of CO2
In conclusion, we have delineated a non-bicarbonate route for the direct catalytic hydrogenation of CO2 to formate using a Schiff-base-modified gold nanocatalyst. Theoretical calculations and in situ high-pressure DRIFTS results demonstrated that the CO2 molecule is activated by the formation of a weak carbamate zwitterionic intermediate at the gold–Schiff base interface, which plays a crucial role on the subsequent transformation to formate. Moreover, the catalytic process benefits from the NEt3 additive in a polar solvent, which allows the formation of a reservoir of CO2 by capturing the gaseous CO2 through the same type of zwitterion intermediate. As a result, the Schiff-base-modified gold nanocatalyst exhibits an unusual catalytic performance compared with those of other functionalized or traditional supported gold nanocatalysts, and shows excellent activity toward CO2 hydrogenation to formate. This finding offers a promising approach for the direct hydrogenation of CO2 and is likely to have considerable implications in the field of CO2 conversion chemistry.
Methods
Sample preparation
Preparation of Schiff-base-modified SiO 2 (SiO 2 -Schiff): The SiO2-Schiff powders were prepared by the APTES with formaldehyde. Typically, 10 mL HCHO solution (37%) was added into 0.12 mol L−1 APTES aqueous solutions (500 mL) with stirring at 40 °C for 1 h. The resulting white precipitation was filtered and washed with deionized water, and then transferred into a Teflon-lined autoclave and maintained at 150 °C for 12 h. After the autoclave was cooled down to room temperature naturally, the precipitate was filtered and washed with an excess amount of deionized water. Following drying at 120 °C for 12 h, a stable Schiff-base-functionalized SiO2 support was obtained.
Synthesis of Au/SiO 2 -Schiff: Briefly, 0.5 g SiO2-Schiff was impregnated with an ethanol solution of HAuCl4 (0.095 mM, 400 mL). After stirring at 80 °C for 15 min, 15 mL of NaBH4 (20 mg) aqueous solution was introduced into the mixture to obtain a supported gold catalyst. After stirring for 1 h, the precipitate was filtered and washed with an excess amount of deionized water. Following drying in vacuum at 120 °C for 12 h, the Au/SiO2-Schiff catalyst was obtained.
Preparation of the Au/SiO 2 -Schiff catalyst by a surface functional group-promoted in situ reduction method: Briefly, 0.5 g SiO2-Schiff was impregnated with an aqueous solution of HAuCl4 (0.095 mM, 400 mL). After stirring at 25 °C for 48 h, the mixture was filtered and washed with an excess amount of deionized water. Following drying in vacuum at 120 °C for 12 h, Au/SiO2-Schiff with a diameter of 3.2 nm was obtained.
Preparation of single-atom gold catalyst by the solid reduction method: Briefly, 0.2 g SiO2-Schiff was impregnated with an aqueous solution of HAuCl4 (0.005 mM, 200 mL). After stirring at 25 °C for 10 min, the mixture was filtered and washed with ethanol several times. After drying in vacuum at 25 °C for 4 h, the resulting white powders were mixed with 0.1 g NaBH4 and then ground in an agate mortar for 2 h at room temperature. The color change from white to pale yellow indicated the reduction of the gold precursor. After grinding, the resultant product was washed with deionized water and then dried in a vacuum at 50 °C overnight. This solvent-free approach was beneficial for restraining the crystal nucleation and to yield a single-atom catalyst with low metal loadings (~0.1 wt.%).
Preparation of SiO 2 -NH 2: The primary amine functionalized silica was synthesized by the cohydrolysis of APTS and TEOS. Typically, 4.0 mL of TEOS was mixed with 4.0 mL of APTES solution and 100 mL of ethanol under stirring at room temperature, to which 10 mL of ammonium hydroxide solution (28 wt%) was added to initiate the hydrolysis of the organosilanes. After stirring at room temperature for 5 h, the precipitate was filtered and washed with an excess amount of deionized water. Following drying at 120 °C for 12 h, the SiO2-NH2 was obtained.
Preparation of Au/SiO 2 -NH 2: The Au/SiO2-NH2 was prepared by the similar NaBH4 reduction method as mentioned above, except for the use of a primary amine-modified SiO2 as the support.
Unfunctionalized Au/SiO2 was synthesized by heat treating the Au/SiO2-NH2 in an O2 atmosphere at 800 °C for 20 s. Prior to the heat treatment, Au/SiO2-NH2 was first encapsulated by SiO2 layers through the hydrolysis of TEOS in an alkaline environment. For more details about the synthesis and characterization, please refer to the Supplementary Methods.
Materials characterization
The Au loading of the catalysts was measured by inductively coupled plasma atomic emission spectroscopy (ICP-AES) on an IRIS Intrepid II XSP instrument (Thermo Electron Corporation). All the solid-state NMR experiments were performed on a Bruker AvanceIII 600 spectrometer equipped with a 14.1 T wide-bore magnet. The resonance frequencies were 156.4 MHz for 13C.1H → 13C CP/MAS NMR experiments were performed on a 4 mm MAS probe with a spinning rate of 12 kHz.1H → 13C CP/MAS NMR experiments were carried out with a contact time of 5 ms and a recycle delay of 2 s. The chemical shifts were referenced to adamantane with the upfield methine peak at 29.5 ppm. HAADF-STEM images were obtained using a JEOL JEM-2100F at 200 kV. The samples for electron microscopy were prepared by grinding and subsequent dispersion of the powder in ethanol and applying a drop of the very dilute suspension on the carbon-coated grids. XPS was performed on a Kratos Axis Ultra DLA X-ray photoelectron spectrometer equipped with an Al Kα radiation source (1486.6 eV, 15 kV). All binding energies were calibrated with the C1s peak at 284.8 eV for the adventitious carbon. The FTIR spectra were acquired with a spectrometer (BRUKER Equinox 55) equipped with a DLATGS detector and operated at a resolution of 4 cm−1.
In situ DRIFTS experiments
Spectral analysis of the catalysts during CO2 adsorption/hydrogenation was carried out using an Equinox 55 infrared spectrometer (Bruker) equipped with a high-temperature/high-pressure DRIFTS reactor cell and liquid nitrogen-cooled DLATGS detector.
All spectra were obtained with a resolution of 4 cm−1 and an accumulation of 32 scans. The sample cup of the cell was filled with a finely powdered sample. Prior to the CO2 adsorption studies, catalyst samples were pre-treated in situ in a pure He stream (30 mL min−1), heated up to 120 °C at a rate of 10 °C min−1, then kept at the final temperature for 2 h. After pre-treatment, the cell was cooled to the required temperature in He.
Spectra of CO2 absorption were recorded at 80 °C in a pure CO2 stream (10 mL min−1). After adsorption saturation, the system was purged with He (30 mL min−1) to obtain the desorption spectra. The CO2 absorption/desorption spectra in a humid atmosphere were realized by bubbling through water.
For the high-pressure DRIFTS experiments, the free wet-CO2 was carefully released from the vessel until the carbamate zwitterion was decomposed completely from the CO2-saturated Au/SiO2-Schiff catalyst, with only surface-bonded ionic carbamate left behind on the catalyst surface (at this moment, CO2 remained in the gas phase with <1 bar). Then, the vessel was sealed and pressured with 10 bar of H2 to trace the evolution of the surface species on the surface. For comparison, to understand the contribution of the carbamate zwitterion species on CO2 conversion, the vessel was directly pressurized with 10 bar of H2 to monitor the spectral evolution of CO2-satuated Au/SiO2-Schiff (with 1 bar of CO2).
Catalytic activity measurement of CO2 reduction
Preliminary studies with different catalysts were carried out in a magnetically driven Parr autoclave (50 mL) containing a mixture of H2O-MeOH (20/80 v/v, 10 mL) and base (15 mmol). The solutions were purged three times with high purity CO2, and then pressurized up to 30 bar of CO2 and completed up to 80 bar with H2 (P(H2)/P(CO2) = 5/3). The system was heated to 90 °C and stirring was started until the reaction was completed. The reaction mixture was transferred into a centrifuge tube and the solid catalyst was separated by centrifugation. Formate product concentrations were monitored by high-performance liquid chromatography (HPLC, Agilent 1100) on an anion-exclusion column (AminexHPX-87H) using an aqueous H2SO4 solution (5 mM) as the eluent and an ultraviolet detector (λ = 210 nm).
For kinetic measurements, 5 mg of Au/SiO2-Schiff was added into the Parr autoclave containing a mixture of H2O-MeOH (20/80 v/v, 10 mL) and base (10 mmol). The solutions were pressurized up to 10–30 bar with CO2 and then completed to 10–60 bar with H2. The system was heated to the required temperature (60–90 °C) and stirring was started (600 rpm). After reaction for the desired time, the reactor was cooled down and depressurized. The reaction mixture was transferred into a centrifuge tube and the solid catalyst was separated by centrifugation. The formate product concentrations were monitored by the method mentioned above.
Computational methods
The quantum mechanics calculations: The calculation of the NMR shift was carried out using the Gaussian09 quantum chemical package. The geometries were optimized using M062X functional41, and Ahlrichs’s basis sets were used for all atoms at the def2-TZVP level42. The second order Møller–Plesset perturbation method43 and the pcS-2 basis set44 were used for computing the chemical shift, with the first peak at δ = 11 from the experiment used as a reference.
The chemical interactions between CO2 and N-containing organic bases were modeled with NWChem software at the B3LYP/def2-tzvp level45. Different solvent environments were simulated by the COSMO, with the dielectric constant of n-hexane, methanol, and water of 1.89, 32.63, and 78.54, respectively.
The simulation of the chemisorption of CO2 was performed with the program package DMol3 in the Materials Studio of Accelrys Inc46,47. Considering the flexibility of the carbon chain, the geometry with a close-packed Au55 nanocluster (~1.2 nm) accommodated by an alkyl-imine Schiff base in COSMO of water was used to model the practical Au/SiO2-Schiff catalyst in water. The localized double-numerical basis sets with polarization functions (DNP) were used, and the exchange-correlation functional of PBE was employed. The vibrational frequency was corrected by the experimental measurement of gas CO2 at 2340 cm−1. Transition states were obtained by the linear synchronous transit (LST) and quadratic synchronous transit (QST) methods, and were confirmed by the only one imaginary frequency as well as the nudged elastic band (NEB) method.
Data availability
The data that support the findings of this study are available from the corresponding author upon request.
Electronic supplementary material
Acknowledgements
We acknowledge the National Natural Science Foundation of China (21476226, 21676045, 21776269, 21176037), China Ministry of Science and Technology under contact of 2016YFB0600902, the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant XDA09030101, XDB17020400), Dalian Science Foundation for Distinguished Young Scholars (2016RJ04), the Youth Innovation Promotion Association CAS, the Natural Science Foundation of Liaoning Province (201602169), and the Fundamental Research Funds for the Central Universities (DUT15LK29) for financial support. The calculations were performed at Shanghai Supercomputing Center.
Author contributions
Q.L. designed the project and performed the catalyst preparation, characterizations, and catalytic tests. X.Y. performed the DFT calculations and completed the paper. L.L. assisted with the DRIFTS characterization. S.M. assisted with the aberration-corrected HAADF-STEM characterization. Y.L. collaborated with the quantum mechanics calculation of the NMR shifts. Yq.L. and T.Z. participated in beneficial discussions and helped to prepare the manuscript. X.Y., X.W., and Y.H. proposed, planned, designed, and supervised the project. All authors reviewed and commented on the manuscript.
Competing interests
The authors declare no competing financial interests.
Footnotes
Electronic supplementary material
Supplementary Information accompanies this paper at doi:10.1038/s41467-017-01673-3.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Xiaofeng Yang, Email: yangxf2003@dicp.ac.cn.
Xinkui Wang, Email: wangxinkui@dlut.edu.cn.
Yanqiang Huang, Email: yqhuang@dicp.ac.cn.
References
- 1.Schuchmann K, Müller V. Direct and reversible hydrogenation of CO2 to formate by a bacterial carbon dioxide reductase. Science. 2013;342:1382–1385. doi: 10.1126/science.1244758. [DOI] [PubMed] [Google Scholar]
- 2.Wang W, Wang S, Ma X, Gong J. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 2011;40:3703–3727. doi: 10.1039/c1cs15008a. [DOI] [PubMed] [Google Scholar]
- 3.Mellmann D, Sponholz P, Junge H, Beller M. Formic acid as a hydrogen storage material-development of homogeneous catalysts for selective hydrogen release. Chem. Soc. Rev. 2016;45:3954–3988. doi: 10.1039/C5CS00618J. [DOI] [PubMed] [Google Scholar]
- 4.Eppinger J, Huang KW. Formic acid as a hydrogen energy carrier. ACS Energy Lett. 2017;2:188–195. doi: 10.1021/acsenergylett.6b00574. [DOI] [Google Scholar]
- 5.Moret S, et al. Direct synthesis of formic acid from carbon dioxide by hydrogenation in acidic media. Nat. Commun. 2014;5:4017. doi: 10.1038/ncomms5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang X, Shi H, Kwak JH, Szanyi J. Mechanism of CO2 hydrogenation on Pd/Al2O3 catalysts: kinetics and transient DRIFTS-MS studies. ACS Catal. 2015;5:6337–6349. doi: 10.1021/acscatal.5b01464. [DOI] [Google Scholar]
- 7.Porosoff MD, Yan B, Chen JG. Catalytic reduction of CO2 by H2 for synthesis of CO, methanol and hydrocarbons: challenges and opportunities. Energy Environ. Sci. 2016;9:62–73. doi: 10.1039/C5EE02657A. [DOI] [Google Scholar]
- 8.Ostapowicz TG, Schmitz M, Krystof M, Klankermayer J, Leitner W. Carbon dioxide as a C1 building block for the formation of carboxylic acids by formal catalytic hydrocarboxylation. Angew. Chem. Int. Ed. 2013;52:12119–12123. doi: 10.1002/anie.201304529. [DOI] [PubMed] [Google Scholar]
- 9.Kunkel C, Viñes F, Illas F. Transition metal carbides as novel materials for CO2 capture, storage, and activation. Energy Environ. Sci. 2016;9:141–144. doi: 10.1039/C5EE03649F. [DOI] [Google Scholar]
- 10.Kendall JL, Canelas DA, Young JL, DeSimone JM. Polymerizations in supercritical carbon dioxide. Chem. Rev. 1999;99:543–564. doi: 10.1021/cr9700336. [DOI] [PubMed] [Google Scholar]
- 11.Ohnishi YY, Matsunaga T, Nakao Y, Sato H, Sakaki S. Ruthenium (II)-catalyzed hydrogenation of carbon dioxide to formic acid. Theoretical study of real catalyst, ligand effects, and solvation effects. J. Am. Chem. Soc. 2005;127:4021–4032. doi: 10.1021/ja043697n. [DOI] [PubMed] [Google Scholar]
- 12.Himeda Y, Onozawa-Komatsuzaki N, Sugihara H, Kasuga K. Simultaneous tuning of activity and water solubility of complex catalysts by acid-base equilibrium of ligands for conversion of carbon dioxide. Organometallics. 2007;26:702–712. doi: 10.1021/om060899e. [DOI] [Google Scholar]
- 13.Hao C, Wang S, Li M, Kang L, Ma X. Hydrogenation of CO2 to formic acid on supported ruthenium catalysts. Catal. Today. 2011;160:184–190. doi: 10.1016/j.cattod.2010.05.034. [DOI] [Google Scholar]
- 14.Gunasekar GH, Park K, Jung KD, Yoon S. Recent developments in the catalytic hydrogenation of CO2 to formic acid/formate using heterogeneous catalysts. Inorg. Chem. Front. 2016;3:882–895. doi: 10.1039/C5QI00231A. [DOI] [Google Scholar]
- 15.Su J, Lu M, Lin H. High yield production of formate by hydrogenating CO2 derived ammonium carbamate/carbonate at room temperature. Green Chem. 2015;17:2769–2773. doi: 10.1039/C5GC00397K. [DOI] [Google Scholar]
- 16.Bi QY, et al. An aqueous rechargeable formate-based bydrogen battery driven by heterogeneous Pd catalysis. Angew. Chem. Int. Ed. 2014;53:13583–13587. doi: 10.1002/anie.201409500. [DOI] [PubMed] [Google Scholar]
- 17.Werz PD, Kainz J, Rieger B. Thermo- and pH-responsive nanogel particles bearing secondary amine functionalities for reversible carbon dioxide capture and release. Macromolecules. 2015;48:6433–6439. doi: 10.1021/acs.macromol.5b01367. [DOI] [Google Scholar]
- 18.Li YN, et al. In situ hydrogenation of captured CO2 to formate with polyethyleneimine and Rh/monophosphine system. Green Chem. 2013;15:2825–2829. doi: 10.1039/c3gc41265b. [DOI] [Google Scholar]
- 19.Kothandaraman J, Goeppert A, Czaun M, Olah GA, Prakash GS. Conversion of CO2 from air into methanol using a polyamine and a homogeneous ruthenium catalyst. J. Am. Chem. Soc. 2016;138:778–781. doi: 10.1021/jacs.5b12354. [DOI] [PubMed] [Google Scholar]
- 20.Liu Q, et al. A Schiff base modified gold catalyst for green and efficient H2 production from formic acid. Energy Environ. Sci. 2015;8:3204–3207. doi: 10.1039/C5EE02506K. [DOI] [Google Scholar]
- 21.Grirrane A, Corma A, García H. Gold-catalyzed synthesis of aromatic azo compounds from anilines and nitroaromatics. Science. 2008;322:1661–1664. doi: 10.1126/science.1166401. [DOI] [PubMed] [Google Scholar]
- 22.Haruta M, Daté M. Advances in the catalysis of Au nanoparticles. Appl. Catal. A Gen. 2001;222:427–437. doi: 10.1016/S0926-860X(01)00847-X. [DOI] [Google Scholar]
- 23.Preti D, Resta C, Squarcialupi S, Fachinetti G. Carbon dioxide hydrogenation to formic acid by using a heterogeneous gold catalyst. Angew. Chem. Int. Ed. 2011;50:12551–12554. doi: 10.1002/anie.201105481. [DOI] [PubMed] [Google Scholar]
- 24.Filonenko GA, Vrijburg WL, Hensen EJ, Pidko EA. On the activity of supported Au catalysts in the liquid phase hydrogenation of CO2 to formates. J. Catal. 2016;343:97–105. doi: 10.1016/j.jcat.2015.10.002. [DOI] [Google Scholar]
- 25.Hull JF, et al. Reversible hydrogen storage using CO2 and a proton-switchable iridium catalyst in aqueous media under mild temperatures and pressures. Nat. Chem. 2012;4:383–388. doi: 10.1038/nchem.1295. [DOI] [PubMed] [Google Scholar]
- 26.Andzelm J, Kölmel C, Klamt A. Incorporation of solvent effects into density functional calculations of molecular energies and geometries. J. Chem. Phys. 1995;103:9312–9320. doi: 10.1063/1.469990. [DOI] [Google Scholar]
- 27.York DM, Karplus M. A smooth solvation potential based on the conductor-like screening model. J. Phys. Chem. A. 1999;103:11060–11079. doi: 10.1021/jp992097l. [DOI] [Google Scholar]
- 28.Shao X, et al. Pd@ C3N4 nanocatalyst for highly efficient hydrogen storage system based on potassium bicarbonate/formate. AIChE J. 2016;62:2410–2418. doi: 10.1002/aic.15218. [DOI] [Google Scholar]
- 29.Wang WH, Himeda Y, Muckerman JT, Manbeck GF, Fujita E. CO2 hydrogenation to formate and methanol as an alternative to photo- and electrochemical CO2 reduction. Chem. Rev. 2015;115:12936–12973. doi: 10.1021/acs.chemrev.5b00197. [DOI] [PubMed] [Google Scholar]
- 30.Herzing AA, Kiely CJ, Carley AF, Landon P, Hutchings GJ. Identification of active gold nanoclusters on iron oxide supports for CO oxidation. Science. 2008;321:1331–1335. doi: 10.1126/science.1159639. [DOI] [PubMed] [Google Scholar]
- 31.Danon A, Stair PC, Weitz E. FTIR study of CO2 adsorption on amine-grafted SBA-15: elucidation of adsorbed species. J. Phys. Chem. C. 2011;115:11540–11549. doi: 10.1021/jp200914v. [DOI] [Google Scholar]
- 32.Bossa JB, Theulé P, Duvernay F, Borget F, Chiavassa T. Astron. Astrophys. 2008. Carbamic acid and carbamate formation in NH3:CO2 ices-UV irradiation versus thermal processes; pp. 719–724. [Google Scholar]
- 33.Foo GS, et al. Elucidation of surface species through in situ FTIR spectroscopy of carbon dioxide adsorption on amine-grafted SBA-15. ChemSusChem. 2017;10:266–276. doi: 10.1002/cssc.201600809. [DOI] [PubMed] [Google Scholar]
- 34.McCann N, et al. Kinetics and mechanism of carbamate formation from CO2(aq), carbonate species, and monoethanolamine in aqueous solution. J. Phys. Chem. A. 2009;113:5022–5029. doi: 10.1021/jp810564z. [DOI] [PubMed] [Google Scholar]
- 35.Buß F, Mehlmann P, Mück-Lichtenfeld C, Bergander K, Dielmann F. Reversible carbon dioxide binding by simple lewis base adducts with electron-rich phosphines. J. Am. Chem. Soc. 2016;138:1840–1843. doi: 10.1021/jacs.5b13116. [DOI] [PubMed] [Google Scholar]
- 36.Wang YB, Wang YM, Zhang WZ, Lu XB. Fast CO2 sequestration, activation, and catalytic transformation using N-heterocyclic olefins. J. Am. Chem. Soc. 2013;135:11996–12003. doi: 10.1021/ja405114e. [DOI] [PubMed] [Google Scholar]
- 37.Villiers C, Dognon JP, Pollet R, Thuéry P, Ephritikhine M. An isolated CO2 adduct of a nitrogen base: crystal and electronic structures. Angew. Chem. Int. Ed. 2010;49:3465–3468. doi: 10.1002/anie.201001035. [DOI] [PubMed] [Google Scholar]
- 38.Jiang K, Xu K, Zou S, Cai WB. B-doped Pd catalyst: boosting room-temperature hydrogen production from formic acid-formate solutions. J. Am. Chem. Soc. 2014;136:4861–4864. doi: 10.1021/ja5008917. [DOI] [PubMed] [Google Scholar]
- 39.Lv XY, Lu G, Wang ZQ, Xu ZN, Guo GC. Computational evidence for Lewis base-promoted CO2 hydrogenation to formic acid on gold surface. ACS Catal. 2017;7:4519–4526. doi: 10.1021/acscatal.7b00277. [DOI] [Google Scholar]
- 40.Malta G, et al. Identification of single-site gold catalysis in acetylene hydrochlorination. Science. 2017;355:1399–1403. doi: 10.1126/science.aal3439. [DOI] [PubMed] [Google Scholar]
- 41.Zhao Y, Truhlar DG. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008;120:215–241. doi: 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- 42.Weigend F, Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005;7:3297–3305. doi: 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- 43.Head-Gordon M, Pople JA, Frisch MJ. MP2 energy evaluation by direct methods. Chem. Phys. Lett. 1988;153:503–506. doi: 10.1016/0009-2614(88)85250-3. [DOI] [Google Scholar]
- 44.Jensen F. Basis set convergence of nuclear magnetic shielding constants calculated by density functional methods. J. Chem. Theory Comput. 2008;4:719–727. doi: 10.1021/ct800013z. [DOI] [PubMed] [Google Scholar]
- 45.Valiev M, et al. NWChem: a comprehensive and scalable open-source solution for large scale molecular simulations. Comput. Phys. Commun. 2010;181:1477–1489. doi: 10.1016/j.cpc.2010.04.018. [DOI] [Google Scholar]
- 46.Delley B. An all-electron numerical method for solving the local density functional for polyatomic molecules. J. Chem. Phys. 1990;92:508–517. doi: 10.1063/1.458452. [DOI] [Google Scholar]
- 47.Delley B. From molecules to solids with the Dmol3 approach. J. Chem. Phys. 2000;113:7756–7764. doi: 10.1063/1.1316015. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon request.