

SUMMARY
Liver cancer has the second highest worldwide cancer mortality rate and has limited therapeutic options. We analyzed 363 hepatocellular carcinoma (HCC) cases by whole exome sequencing and DNA copy number analyses, and 196 HCC also by DNA methylation, RNA, miRNA, and proteomic expression. DNA sequencing and mutation analysis identified significantly mutated genes including LZTR1, EEF1A1, SF3B1, and SMARCA4. Significant alterations by mutation or down-regulation by hypermethylation in genes likely to result in HCC metabolic reprogramming (ALB, APOB, and CPS1) were observed. Integrative molecular HCC subtyping incorporating unsupervised clustering of five data platforms identified three subtypes, one of which was associated with poorer prognosis in three HCC cohorts. Integrated analyses enabled development of a p53 target gene expression signature correlating with poor survival. Potential therapeutic targets for which inhibitors exist include WNT signaling, MDM4, MET, VEGFA, MCL1, IDH1, TERT, and immune checkpoint proteins CTLA-4, PD-1, and PD-L1.
Keywords: Liver cancer, hepatocellular carcinoma (HCC), TERT, TP53, CTNNB1, CDKN2A, promoter hypermethylation, IDH1/2, miR-122, immune checkpoint proteins, HCC subtyping
eTOC
Multiplex molecular profiling of human hepatocellular carcinoma patients provides insight into subtype characteristics and points toward key pathways to target therapeutically.
INTRODUCTION
Liver cancer is the second most common cause of death from cancer worldwide, with 700,000 annual deaths recorded globally in recent years (Ferlay et al., 2015). Hepatocellular carcinoma (HCC), the predominant form of liver cancer, has several known risk factors including chronic hepatitis B virus (HBV) and hepatitis C virus (HCV) infections, alcohol abuse, autoimmune hepatitis, diabetes mellitus, obesity, and several metabolic diseases. In developed nations, there has been a rise in HCC incidence partly attributed to HCV, obesity and diabetes (Yang and Roberts, 2010). The liver injury induced by these risk factors produces a progressive inflammatory milieu that results in a cycle of necrosis and regeneration and the development of chromosomal instability (Karagozian et al., 2014). Genetic and epigenetic alterations that progressively accumulate in a background of increased reactive oxygen species, inflammatory cytokines and fibrosis likely lead to the initiation of HCC (Dhanasekaran et al., 2016). Initiation and progression of HCC is considered a multi-step process but the precise molecular events that underlie HCC formation remain only partially understood (Zucman-Rossi et al., 2015).
Recent studies have explored HCC genomic alterations and have identified frequently mutated genes, including TERT promoter, TP53 and CTNNB1 (β-catenin) (Schulze et al., 2015; Totoki et al., 2014). Despite many potential therapeutic targets, sorafenib, a multikinase inhibitor, is currently the only drug approved for advanced HCC management (Llovet et al., 2008). More than ten drugs have failed to meet clinical end points in phase III trials, indicating a need for new drug discovery for HCC (Llovet and Hernandez-Gea, 2014).
As part of The Cancer Genome Atlas (TCGA) network we have performed the first large scale multi-platform analysis of HCC, including evaluation of somatic mutations and DNA copy number in 363 patients, and examination of DNA methylation, mRNA expression, microRNA (miRNA) expression and protein expression in 196 patients to understand the molecular landscape of HCCs (Table S1A–C). The integrated analyses of multiple data platforms in conjunction with clinical data (Table S1A,B) has facilitated biological insights, identification of therapeutic targets, and the characterization of robust subclasses with prognostic implications that may influence HCC clinical management.
RESULTS
Somatic Mutations
Whole exome sequencing was performed on 363 HCC cases for a mean coverage of 95% of targeted bases with a minimum of 20-fold coverage. In total, 12,136 genes had non-silent mutations, and 26 genes were determined to be significantly mutated genes (SMGs) by the MutSigCV algorithm (Lawrence et al., 2014) (Figure 1, Table S2A–B, Supplemental Methods). Of these 26 genes, 18 were reported as SMGs in at least one previous HCC genome sequencing study (Table S2B). These included the tumor suppressor genes TP53 (31%), AXIN1 (8%) and RB1 (4%) that were inactivated by mutation, the WNT pathway oncogene CTNNB1 (27%), and the chromatin remodeling genes ARID1A (7%), ARID2 (5%) and BAP1 (5%) (Figure 1, Table S2A–B). NFE2L2 and its interactor KEAP1, important in cellular anti-oxidant defenses, were significantly mutated in 3% and 5% of HCC, respectively. Albumin (ALB) and APOB mutations were observed in 13% and 10% of tumors, consistent with previous HCC sequencing studies (Figure 1, Table S2B) (Fujimoto et al., 2016; Schulze et al., 2015). ALB and APOB RNA expression were decreased in HCC relative to normal tissues. HCC stratified by low ALB and APOB expression were associated by Gene Set Enrichment Analysis (GSEA) with increased cell cycle progression, ribosome biogenesis and nucleotide synthesis, and reduced oxidative phosphorylation (data not shown). Because ALB expression accounts for 20% of cellular mRNA (Uhlen et al., 2015) and APOB consumes large amounts of cellular energy by facilitating VLDL secretion (Egusa et al., 1985), there may be selection for ALB or APOB inactivating mutations to divert energy into cancer-relevant metabolic pathways (Fernandez-Banet et al., 2014).
Figure 1. The genomic landscape of liver hepatocellular carcinoma and mutational signatures.
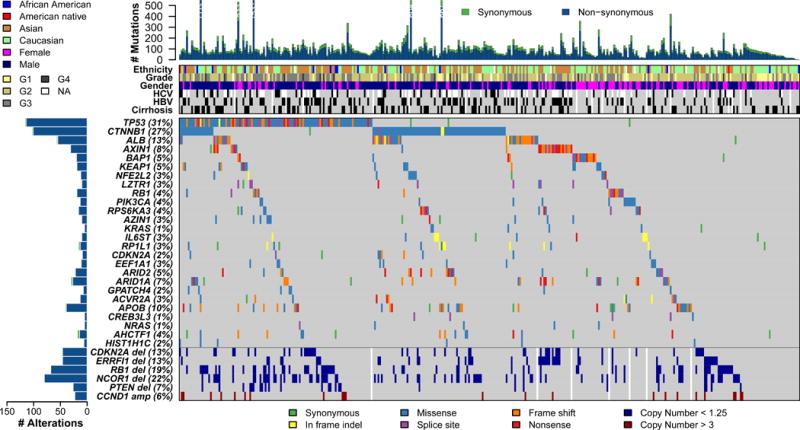
Top panel shows individual tumor mutation rates while the middle panel details ethnicity, tumor grade, age, gender, hepatitis C virus (HCV) and hepatitis B virus (HBV) infection status, and cirrhosis for 363 HCC. Bottom panel shows genes with statistically significant levels of mutation (MutSig suite, false discovery rate, 0.1) and mutation types are indicated in the legend at the bottom. The bottom six rows display significant DNA copy number alterations in likely cancer driver genes.
Among the 26 MutSigCV-identified SMGs were 8 genes not previously considered candidate HCC drivers (Table S2B). LZTR1, encoding an adaptor of CUL3-containing E3 ligase complexes, was mutated in 10 of 377 HCC (3%). Eight LZTR1 mutations were inactivating splice site mutations at codon 217, a mutation observed in adrenocortical and pancreatic cancers (Witkiewicz et al., 2015). LZTR1 germline mutations have been associated with inherited segmental schwannomatosis and somatic LTZR1 mutations are identified as driver mutations in glioblastoma (Frattini et al., 2013; Piotrowski et al., 2014). The translation elongation factor gene EEF1A1 was significantly mutated in 10 tumors and five tumors contained S432I/S mutations, a codon mutation observed in HCC and other cancers (Ahn et al., 2014). Other genes identified as significantly mutated by MutSigCV included AZIN1, RP1L1, GPATCH4, CREB3L3, AHCTF1, and HIST1H1. None of these six genes have been reported as drivers in HCC or other cancers.
In addition to algorithmically curated SMGs, we manually curated two genes with MutSigCV q values close to 0.1 as likely driver genes due to recurrent mutations. SF3B1, a splicing factor gene, was mutated in 10 patients, with mutations in codons N626 and K666 occurring twice each in our HCC tumor set and 11 and 21 times, respectively, across other tumor studies (Cerami et al., 2012). SF3B1 mutations have been reported as likely driver mutations in hematopoietic malignancies (Bonnal et al., 2012). SMARCA4, encoding a chromatin modifier of the SWI/SNF family, was mutated in 11 HCC patient tumors. Mutations at codons 1160 and 1192 occurred twice and were observed at this codon in 6 and 14 other non-HCC tumors, respectively (Cerami et al., 2012). Mutations in SMARCA4 have been observed in some cancer types, including 4 of 36 HCC (Endo et al., 2013).
TERT promoter mutations were the most common somatic mutation, found in 87 of 196 (44%) HCCs analyzed in the TERT promoter region (Figure S1A, Table S3). Two independent TERT promoter mutations (chr5, 1,295,228 G>A (C228T) and 1,295,250 G>A (C250T) were found, consistent with activating mutations previously reported (Horn et al., 2013). Further analysis revealed a germline TERT promoter mutation (C228T) in the blood and tumor of an HBV-positive 29-year-old Asian male with no recorded family history of HCC. Germline TERT mutations (1,295,161 T>G at the transcription start site) were associated with familial melanoma (Horn et al., 2013), but germline mutation at position C228T has not been reported.
Patients with a TERT promoter mutation were older (p=0.0006), predominantly male (p=0.006), more likely to be HCV positive (p=0.04) and less likely to be HBV positive (p=0.02) than patients without the mutation. Molecular correlates of TERT promoter mutation included a strong co-occurrence with CDKN2A silencing by promoter hypermethylation (p = 8.1 × 10−5) (Figure S1A). The CDKN2A gene encodes the tumor suppressor p16INK4A, and downregulation of p16INK4A expression in conjunction with enhanced TERT expression has been shown to be essential for epithelial cell immortalization, a cancer hallmark (Kiyono et al., 1998). TERT RNA was significantly upregulated in the HCC cohort overall (p<0.001) but TERT promoter mutation did not significantly correlate with increased TERT RNA expression.
Mutational Signatures
We performed mutational signature analysis on the core set of 196 HCC applying a Bayesian variant of the non-negative matrix factorization (NMF) algorithm (Tan and Fevotte, 2013) to mutation counts of single nucleotide variants (SNVs) stratified by 96-trinucleotide contexts. This analysis identified three independent mutational signatures (“A”,”B” and “C”, Supplemental Methods) of which two correspond to reported mutation signatures (Alexandrov et al., 2013) (see also http://cancer.sanger.ac.uk/cosmic/signatures). To further identify samples with a significant enrichment of each mutational process we performed a hierarchical clustering of normalized signature activity (Figure S1B). Nine samples significantly associated with the plant-derived carcinogen aristolochic acid (AA) signature had a predominance of A:T-to-T:A transversions at [C|T]AG tri-nucleotide motifs and these samples had a significant enrichment of splice-site mutations (P=10−6 by Wilcoxon rank-sum test) due to overlap of the motif with the canonical splice acceptor site. Seven samples were significantly correlated with mutational signature B (Sig B), associated with Aflatoxin B1 exposure, characterized by an excess of G:C-to-T:A transversions. Aflatoxin B1 exposure is a risk factor for HCC, associated with hotspot mutation R249S. Recurrent TP53-R249S mutant samples had significant enrichment of AFB1 signature activity in comparison to other TP53 mutants (p=0.005 by Wilcoxon rank-sum) or WT samples (p=0.0001) (Figure S1C). HBV-positive samples had much higher AFB1 activity than HBV negative HCC (P=0.005 by Wilcoxon rank-sum), indicating a likely synergistic interaction between Aflatoxin B1 exposure and HBV.
Copy Number Changes
Somatic copy number alterations (SCNA) were determined by profiling HCC on Affymetrix SNP 6.0 arrays and analysis by GISTIC 2.0. Overall patterns of broad and focal alterations across the entire cohort were similar to earlier reports (Guichard et al., 2012; Totoki et al., 2014) (Figure S2A). Most frequent chromosomal arm alterations included copy number gains in 1q and 8q and copy number losses in 8p and 17p (Figure S2A, B). GISTIC 2.0 analysis of all tumors identified 28 significantly reoccurring focal amplifications including those containing well characterized driver oncogenes such as CCND1 and FGF19 (11q13.3), MYC (8q24.21), MET (7q31.2), VEGFA (6p21.1), and MCL1 (1q21.3). Moreover, TERT (5p15.33) was amplified in 10% of HCC. Among 36 deletion events, 13q14.2 (RB1) and 9p21.3 (CDKN2A) were prominent (Figure S2B). Also seen was a 1p36.23 focal deletion peak that includes the tumor suppressor ERRFI1, recently described in gliomas and HCC (Park et al., 2015). We noted a 17p11.2 focal deletion that contained the tumor suppressor NCOR1, which functions as a suppressor of beta catenin expression (Song and Gelmann, 2008).
Methylation Profiling
Comparison of genome-scale DNA methylation profiles in normal tissue and HCC revealed significant amounts of both hypo- and hypermethylation in the tumors. Unsupervised clustering of HCC using CpG sites that showed cancer-specific DNA hypermethylation identified four hypermethylation clusters (Figure 2A). Two clusters (3 and 4) exhibited elevated hypermethylation. Cluster 3 in particular contained all of the tumors with IDH1/2 mutations and exhibited a distinct DNA hypermethylation profile (Figure 2A), consistent with previous data that IDH1/2 mutations are gain of function lesions that increase levels of cellular D-2-hydroxyglutarate that regulate genomic methylation rates (Lu et al., 2012). Cluster 4 HCC were disproportionately enriched for CDKN2A epigenetic silencing, TERT promoter mutations and CTNNB1 mutations (Figure 2A). Asian ethnicity and HBV infection was significantly associated with Cluster 1, while HCV infection was significantly associated with Cluster 4.
Figure 2. Liver cancers show distinct gene hypermethylation patterns.
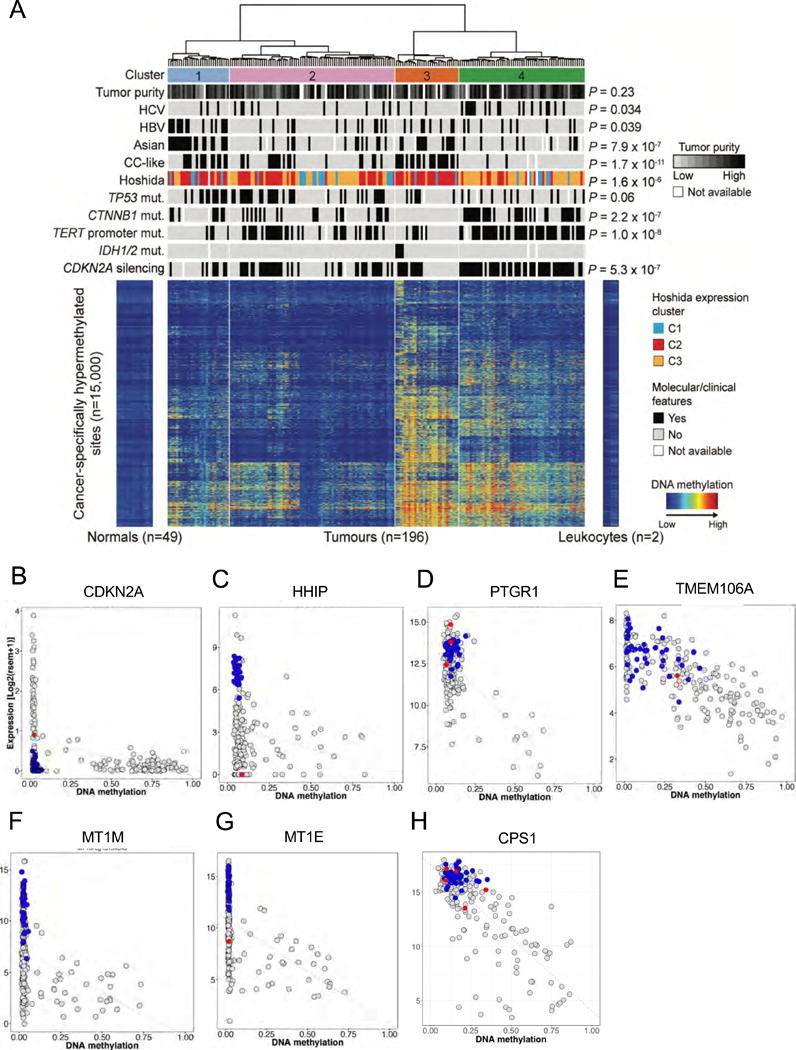
(A) Unsupervised clustering analysis of gene hypermethylation in HCC relative to normal tissue reveals four distinct subgroups. Roughly 15,000 CpG sites showing significant hypermethylation in 196 HCC were analyzed and are shown in heat map format with normal tissues and tumors organized in columns according to cluster designation. Intensity of methylation for each CpG site is indicated by row. Above the heat map the four distinct hypermethylation clusters are shown, and below are bars indicating the distribution of clinical and molecular attributes of the individual tumors by cluster. To the right, P values indicate significant non-random distributions for each attribute. (B–I) Scatter plots of representative CpG sites in gene promoters shown to be frequently hypermethylated in HCC, where gene RNA expression (y axis) is plotted against relative promoter site hypermethylation (x axis). Gray dots are results from tumor samples, blue dots normal tissues, and red dots tumors with mutations in the gene. (B) CDKN2A (cg13601799). (C) HHIP (cg23109129). (D) PTGR1 (cg13831329). (E) TMEM106A (cg21211480). (F) MT1M (cg15134649). (G) MT1E (cg02512505). (H) CPS1 (cg21967368).
Two approaches (see Supplementary Methods) were used to identify those genes with high levels of tumor-specific hypermethylation in conjunction with reduced RNA expression (Table S4A). Seven representative genes frequently hypermethylated in our HCC sample set are shown in Figure 2B–H. These genes displayed significantly reduced RNA expression correlated with high frequency promoter hypermethylation. CDKN2A epigenetic silencing was found in 53% (102/191) of samples whereas CDKN2A mutations were observed in 4% of HCC, indicating that DNA methylation is the predominant mechanism for CDKN2A inactivation (Figure 2B, Table S4A) in HCC. Other highly hypermethylated and downregulated genes included HHIP, a suppressor of hedgehog signaling, a pathway important in hepatocarcinogenesis (Zheng et al., 2013) (Figure 2C, Table S4A), prostaglandin reductase 1 (PTGR1), shown to inhibit lung cancer growth (Zhao et al., 2010) (Figure 2D, Table S4A), and TMEM106A, encoding a pro-apoptotic protein downregulated in gastric cancer (Xu et al., 2014) (Figure 2E, Table S4A). Members of the metallothionein family, MT1M and MT1E, have been implicated as tumor suppressors in HCC and other cancers (Mao et al., 2012) (Figure 2F,G, Table S4A).
Hypermethylation-mediated downregulation of CPS1 (carbamoyl phosphate synthase I), a liver-specific rate-limiting enzyme of the urea cycle reported as a HCC-hypermethylated gene (Liu et al., 2011), may favor glutamine usage in HCC by CAD (carbamoyl phosphate synthase II), which initiates the de novo pyrimidine synthesis pathway, thus favoring cell division (Figure 2H, Table S4A). Consistent with this hypothesis, mean CAD RNA levels were 2.8-fold increased in HCC relative to normal liver tissues (p = 6.7 × 10−34), while mean CPS1 RNA levels were 2.1-fold reduced in HCC compared to normal liver tissue.
Of the 298 genes exhibiting significant HCC-specific hypermethylation, 81 have been reported to be hypermethylated and another 28 have been reported to be downregulated (methylation status unknown) in HCC or other cancers relative to normal tissues (Table S4A). Gene Set Enrichment Analyses (GSEA) of these 298 hypermethylated genes had an enrichment for pathways related to differentiation, stem cell maintenance and targets of the Polycomb repressive complex, a phenomenon previously reported (Widschwendter et al., 2007) (Table S4B).
HBV and HCV Infection
Chronic HBV and HCV infection are major viral risk factors for HCC. In the core TCGA dataset, 44 of 196 (22.4%) patients displayed clinical and molecular evidence of HBV infection. HBV infection was significantly associated with Asian ethnicity, younger age at initial diagnosis, and male gender (Table S5A). HBV+ HCCs were significantly more likely to be mutated in TP53 and significantly less likely to harbor TERT promoter mutations than HBV− HCCs.
Most (37/44, 84%) HBV-infected HCCs exhibited evidence of HBV DNA integration into the host genome by analysis of RNA sequence reads for HBV-chromosomal gene fusion transcripts. Such integrated viral genomes raise the possibility of cis-activation or inactivation of cancer regulatory genes, believed to be an occasional source of driver mutations in HCC. RNA fusion-based HBV integration sites identified by two methods are shown in Table S5B. Roughly 50% of HBV integration sites were within genes, though only two genes had recurrent mutations: MLL4, a histone methyltransferase that regulates proliferation and reported as a frequent HBV integration site (Saigo et al., 2008) and TERT. The five tumors with MLL4 insertions and one of the two TERT insertions displayed the highest levels of MLL4 and TERT RNA expression among all HCC, suggesting an HBV cis-activating event (Table S5C). Among the non-recurring HBV insertional events associated with very high levels of RNA transcription, potential driver events were observed. These include known oncogenes CCND1, CCNE1, and GLI2 (a sonic hedgehog transcription factor). Thus, the effect of HBV on transcriptional levels of key oncogenes demonstrated potential driver events affecting a number of patients.
In our HCC samples, 35 of 196 (17.9%) patients exhibited serological and/or molecular markers of HCV infection, by presence of HCV antibody or HCV RNA as determined by commercial HCV RNA testing or by sequence analysis. HCV infection was significantly higher in white and black patients than Asian patients and in patients with cirrhosis (Figure 1). HCV+ tumors displayed significantly increased frequency of CDKN2A promoter silencing (p = 0.0061) and TERT promoter mutation (p = 0.014).
Multi-Platform Integrative Molecular Subtyping
Unsupervised clustering of data from five platforms (DNA copy number, DNA methylation, mRNA expression, miRNA expression and RPPA) gave a collection of discordant subgroupings specific to each data platform. To reconcile these disparate data types we used a joint multivariate regression approach (see Supplemental Methods) to simultaneously cluster data from the five platforms. This comprehensive approach resolved three major subtypes (Figure 3A and S3A, B). The majority of individual platform cluster solutions concentrated preferentially in one or another of the 3 integrated iCluster solutions with p <0.0001 (Figure S3A, S3C) lending confidence that the aggregate solution captured the main features of each platform. The association of the three iClusters with demographic, pathologic and molecular features of the HCC patients strengthened the clinical relevance of the subtypes defined by the iCluster procedure.
Figure 3. Multiplatform clustering analysis identified three integrated molecular subtypes of liver cancer.
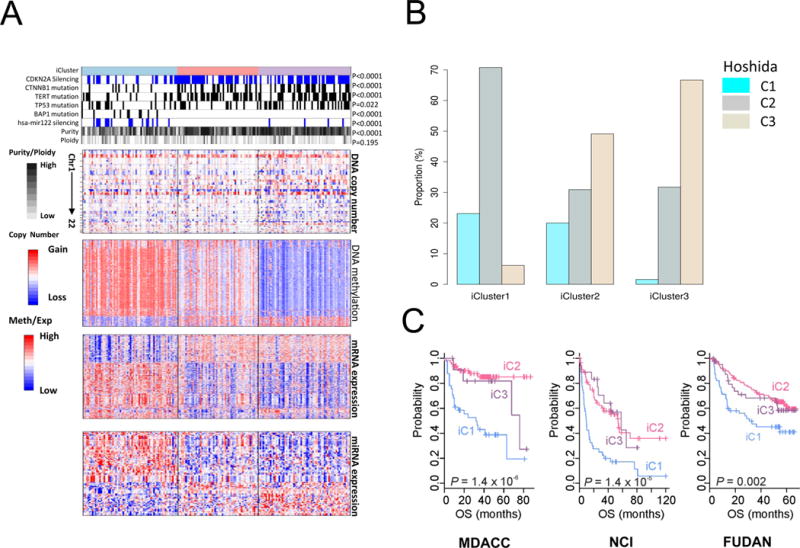
(A) Heat maps organized by iCluster groupings for DNA copy number, DNA methylation status, mRNA expression, and miRNA expression, and correlated with selected molecular features (top tracks). Tumors are in columns, grouped by the iCluster membership. (B) Relative proportions in each iCluster of Hoshida et al. (2009) subtypes defined by RNA expression profiling of a separate HCC cohort. (C) Patient survival outcome fitting three external clinically annotated HCC patient cohort sets of RNA expresson data to the TCGA iClusters (NCI, Lee et al. 2006; Fudan, Roessler et al. 2010; MDACC, Sohn et al. 2016). See also Figure S3.
The first integrated cluster, iClust 1 (n=65), was characterized by clinical associations with younger age, Asian ethnicity, female gender and normal body weight (Table S6, Figure S3B). These tumors exhibited features such as higher tumor grade and presence of macrovascular invasion, and the lowest fraction of differentiated samples by Hoshida classification (Hoshida et al., 2009) (Table S6, Figure 3B, S3B). Molecular correlations with iClust1 included a low frequency of CDKN2A silencing (32%) as compared to iClust2 and iClust3 (69% and 63%, respectively, low frequency of CTNNB1 mutation (12% in iClust1 vs 38% and 43% in iClust2 and iClust3, respectively), low frequency of TERT promoter mutation and low TERT expression (Figure 3A, Table S6). iClust1 tumors exhibited specific changes in miRNA expression, including high expression of miR-181a (a lipid metabolism regulator) and epigenetic silencing of miR-122 (Figure S3D). This subclass was associated with over-expression of proliferation marker genes such as MYBL2, PLK1, and MKI67 (Figure S3D).
In contrast, iClust2 (n=55) and iClust3 (n=63) exhibited a high frequency of CDKN2A silencing by DNA hypermethylation, high frequency of TERT promoter mutation, CTNNB1 mutation, and enrichment for HNF1A mutation. Correlation with clinical variables revealed association of iClust2 with low-grade tumor (P=0.0006) and less microvascular invasion (P=0.01) (Table S6, Figure S3B). iClust3 was characterized by a higher degree of chromosomal instability with distinct 17p loss, high frequency of TP53 mutation and hypomethylation of multiple CpG sites.
To compare the iCluster classification to previous molecular subclasses we assigned each of our patients to one of the three mRNA expression-based subclasses from Hoshida and collaborators (Hoshida et al., 2009), using prediction signatures developed from their expression data. We found correspondence between the iClusters and Hoshida subclasses (C1–C3) (Table S6). iClust1 consisted predominantly of Hoshida C2 patients whereas iClust3 consisted predominantly of Hoshida C3 (Figure 3B).
We further tested the clinical relevance of the iCluster groupings by constructing a subclass prediction model based on the 200 most variably expressed genes compared across the 3 iClusters (Supplemental Methods). We then tested the predictor on three published data sets of three external clinically annotated HCC patient cohorts, with long term follow-up (Lee et al., 2006; Roessler et al., 2010; Sohn et al., 2015). Among all three external cohorts iClust1 had significantly worse prognosis than iClust2 and iClust3 (Figure 3C). There was no difference in overall survival between the three clusters in the TCGA cohort (P=0.561) possibly due to the relatively short follow-up times in this data set (median follow-up 18 months) (Table S1A). Nonetheless, robust replication of poor survival in iCluster 1 in three independent data sets suggests it is a reliable clinical predictor of outcome.
IDH1/2 Mutations AND miR-122 Expression
Analysis of the mutation data revealed two mutations in IDH1 (R132C, R132G) and two mutations in IDH2 (R172K, R172S), in four different tumors. These specific IDH1/2 mutations, seen in multiple human cancers, result in a neomorphic isocitrate dehydrogenase that produces an oncometabolite believed to alter cellular epigenetic programs and block normal differentiation (Lu et al., 2012). IDH1/2 mutations are more frequent in intrahepatic cholangiocarcinomas (CCA) than in HCC, hence the possibility that these tumors actually represented mixed HCC-CCA was considered. We carefully reviewed the histopathology of these tumors and all of them exhibited features of HCC and not of mixed tumor or cholangiocarcinoma.
When the Bayesian compound covariate predictor (BCCP) algorithm (Radmacher et al., 2002) was applied to the mRNA expression data, 11 samples with gene expression patterns similar to the IDH1/2-mutated samples were identified; however, these samples did not have IDH1/2 mutations (Figure 4A). When compared with other molecular subtypes of HCC, the IDH mutant and IDH-like samples exhibited the highest similarity to an hepatic stem cell-like subtype (Lee et al., 2006). These samples were all classified with the poor prognosis iCluster 1 subclass and exhibited similarity to non-differentiated RNA clustering phenotypes (Hoshida C2) (Hoshida et al., 2009), cholangiocarcinoma-like (CCL-HCC) (Woo et al., 2010), silencing of the Hippo pathway (Hippo) (Sohn et al., 2015), and had high Risk Scores based on a gene expression signature of 65 genes (RS65) (Borger et al., 2012) (Figure 4A), suggesting that HCC with the IDH-like gene expression signature represent a poor prognostic subtype of HCC. The IDH-like gene expression signature was present in similar proportions in the followup TCGA extended HCC cohort, and in four other published HCC cohorts with extended follow-up data (Figure 4B). It was associated with significantly worse survival (Figure 4C) in the aforementioned three external cohorts with survival data (see above and also Figure 3C).
Figure 4. HCC with IDH1/2 mutations and with IDH-like gene expression share miRNA and RNA expression profiles and worse clinical outcomes.
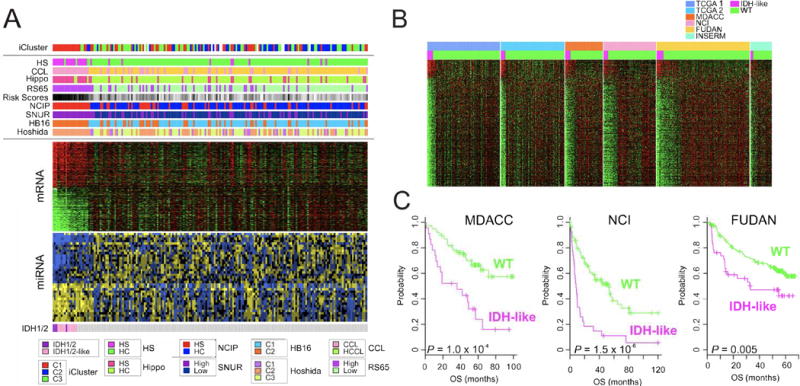
(A) Integrated analysis of IDH1/2 mutations (bottom), mRNA and miRNA expression data (middle), and iCluster and molecular subtypes of HCC (top). HS, hepatic stem cell subtype; CCL, cholangiocarcinoma-like subtype; Hippo, Hippo pathway subtype; RS65, 65-gene risk score subtype; NCIP, National Cancer Institute proliferation subtype; SNUR, Seoul National University recurrence subtype; HB16, 16-gene hepatoblastoma subtype; Hoshida, HCC RNA expression subtype profiling category. (B) Comparison of mRNA expression profiles of two TCGA HCC cohorts and four other HCC cohorts showing subsets of tumors with IDH-like gene expression. (C) Clinical significance of IDH-like subtype in HCC. Patients in three external cohorts were stratified according to IDH-like gene expression signature. See Figure S4.
Genes and microRNAs that were differentially expressed in IDH-mutant and IDH-like tumors were also identified (Figure S4A). Intriguingly, miR-122, which is liver-specific and the most abundant miRNA in liver (Figure S4B), was significantly downregulated in some of the IDH-mutant and IDH-like tumors by promoter DNA hypermethylation (Figure S4C). miR-122 dysregulation has been observed in HCC studies and has been associated with poor survival (Coulouarn et al., 2009). miR-122 regulates the expression of multiple genes including PKM2 (Figure S4D), and is implicated in metabolism as well as HCC progression (Liu et al., 2014). The four IDH-mutant samples had a distinct DNA hypermethylation profile, as seen in other cancer types, while the IDH-like samples lacked the characteristic DNA hypermethylation profile.
P53 Signature
Mutations involving TP53 were found in 31% of patients. We used an alternate method to determine p53 functional status by assessment of p53 transcriptional target expression (p53 signature). The degree of p53 target gene upregulation is used as a surrogate for p53 functionality (See Supplemental Methods “TP53 Signature”). Tumors were stratified based on p53 target gene expression (Figure 5A). While only one HCC with high p53 target expression had a TP53 mutation, 11 out of 48 (23%) samples in the low p53 expression quartile were TP53 wildtype. Thus, many HCCs without TP53 mutations appear to have inactive p53, consistent with the existence of non-mutational p53 inactivating mechanisms (Soussi, 2014). We examined specific inhibitors of p53 function and found that MDM4, a p53 inhibitory protein, was significantly increased in copy number and expression in low signature WT TP53 HCCs relative to other HCCs (p=3.6 × 10−4 and p=5.4 × 10−4, respectively) (Figure 5A–C), providing one possible mechanism for low p53 signatures in non-TP53 mutated HCCs.
Figure 5. P53-induced gene target expression signature for improved clustering of HCC molecular and biological attributes.
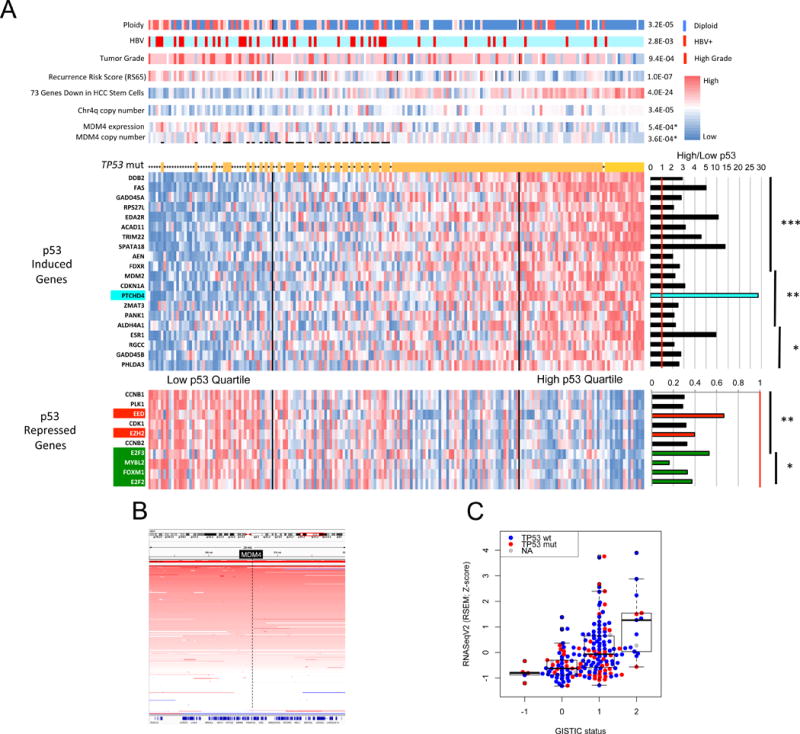
(A) Clustering of 191 HCC by composite expression of known p53 target genes. An expression heat map of 20 p53-induced target genes is shown above that of 10 p53-repressed target genes. To the right are shown mean expression ratios of top quartile p53 target genes relative to bottom quartile. Asterisks indicate level of significance. ***P<1E-10, **P<1E-07, *P<1E-04. Above the p53 target heat map asterisks indicate tumors with a TP53 mutation. Top bars show molecular and clinical attributes and correlation (p values) with high and low p53 target gene expression. MDM4 copy number and expression are significantly increased in those HCC with wildtype TP53 and with low p53 target expression relative to all other HCC (p values with asterisks). (B) Frequent copy number amplification of MDM4 gene in HCC. A segment of chromosome 1 centered on the MDM4 locus (in black box) is shown. The intensity of red bars corresponds to degree of copy number gain. Each horizontal line corresponds to a single tumor. (C) MDM4 copy number gain and amplification correlates with increased RNA expression. RNA expression for each tumor is represented by a red dot (mutant TP53) or blue dot (WT TP53) according to MDM4 copy number (−1 = deletion, 0 = diploid, 1 = copy number gain, 2 = amplification).
Tumors having low p53 target expression exhibited significant associations with increased copy number instability (including high frequency chromosome 4q loss (Rashid et al., 1999)), higher pathological grade, reduced expression of mature hepatocyte marker genes, and increased risk of tumor recurrence (Figure 5A). HCC within the lowest quartile p53 expression displayed a significantly reduced overall survival relative to their high p53 signature counterparts (P = 0.0018) (Figure S5A). Of three external HCC cohorts tested, two showed significantly reduced overall survival of the low p53 signature patients (Figure S5B–D).
Among the p53-regulated HCC target genes PTCHD4 had a 28-fold increased expression in the high relative to the low p53 expression quartiles (Figure 5A). PTCHD4 suppresses sonic hedgehog (SHH) signaling in colorectal cancers (Chung et al., 2014) and SHH signaling is important in liver regeneration. SHH pathway gene expression was significantly upregulated in low p53 signature tumors by GSEA analysis. Another p53-repressed target gene, EZH2, was significantly upregulated in low p53 signature HCC (Figure 5A). EZH2 encodes a histone methyltransferase that epigenetically regulates stem cell maintenance (Volkel et al., 2015) and its enhanced expression in low p53 signature HCC coincides with increased stem/progenitor gene expression (Figure 5A). The low p53 signature HCC had increased expression of the p53-repressed cell cycle positive regulatory genes CCNB1/2, E2F2/3, and FOXM1. We hypothesize that p53 regulates HCC phenotypes in part through the sonic hedgehog pathway via upregulated PTCHD4, the Polycomb repressive complex 2 via downregulated EZH2, and downregulation of S/G2/M promoting cell cycle genes.
Other Signaling Pathways
While most gene and pathway alterations were evenly distributed with respect to iCluster classification, some mutations, such as TERT and CTNNB1, were underrepresented in iCluster1 (Figure 6, Figure S6A,B). As described in previous HCC genomics studies, WNT pathway members were frequently mutated or subject to copy number alterations. Overall, 44% of HCC displayed gene alterations in the WNT signaling axis. Other key pathways included cell cycle regulatory pathways driven by mutations and copy number changes in RB1, CCND1, CDKN2A and RTK/PI-3 kinase signaling driven by PTEN, PIK3CA, MET, and VEGFA copy number/mutational changes. Chromatin modifiers such as BAP1, ARID1A, and ARID2 were significantly mutated genes.
Figure 6. Integrated molecular comparison of somatic alterations in signaling pathways across iCluster groups.
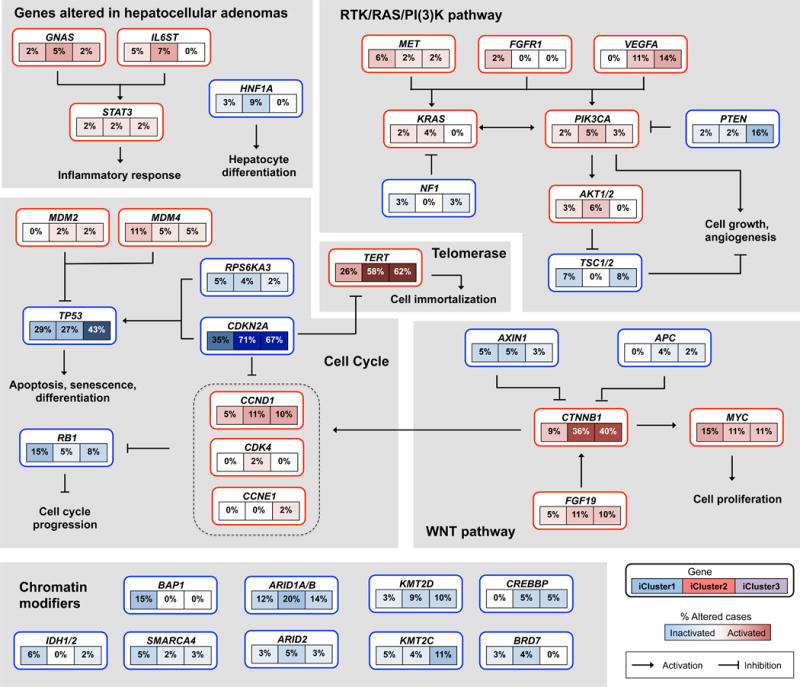
Each gene box includes 3 percentages representing the frequency of activation or inactivation in iCluster 1, 2 and 3 based on the core 196 sample HCC dataset. All somatic changes are tallied together in calculating the percentages of altered cases within each of the iCluster sample groups. Somatic alterations include mutations and copy-number changes (homozygous deletion and high-level amplifications), as well as epigenetic silencing of CDKN2A. Missense mutations are only counted if they have known oncogenic function, have been reported in COSMIC, or occur at known mutational hotspots. Genes are grouped by signaling pathways, with edges showing pairwise molecular interactions.
As an alternative to using significantly mutated genes, we employed a computational method to identify signaling pathways that displayed enhanced mutation frequencies across all component genes of that pathway, though each individual gene might not be significantly mutated (Supplemental Methods, Pathway-Associated High Impact Gene Mutations). We tested Reactome pathways for a bias toward evolutionarily conserved nonsynonymous mutations. We identified for each pathway the set of genes that maximized bias toward high Evolutionary Action (EA) mutation scores (a measure of relative evolutionary conservation) compared to the cohort background (q<0.05; Figure S7A–H) (Katsonis and Lichtarge, 2014). Sets that exhibited significant bias after FDR correction, and were more significant than 95% of simulations of similar sized pathways, were considered to be of interest and to point toward cellular functions whose disruption may be advantageous to the tumor (Table S7). Seven of the ten highest-ranked pathway groups contained RAS, RAF, MAPK, PI3K, SOS, and SHC genes and implicated pathways downstream of receptor tyrosine kinases (Figure S7C–H, Table S7). The over-representation of pathways related to receptor tyrosine kinase (RTK) signaling may be related to the sensitivity of HCC to the RTK inhibitor sorafenib.
Immune Phenotyping
Histopathological analyses of our core set of 196 HCCs revealed that 22% displayed high or moderate levels of lymphocyte infiltration. Given the recent success for targeted therapies against immune checkpoint genes such as CTLA-4, PD-1 (PDCD1), and PD-L1 (CD274), we characterized the immune microenvironment in HCC. We first performed unsupervised hierarchical clustering of gene expression using a curated list of sixty-six immune markers that encompass cell surface markers of different immune cell populations (Figure 7A). Expression of the immune markers varied greatly across HCC and tumor adjacent normal tissues. Unsupervised clustering identified six clusters of tumor samples, with the “High 1” and “High 2” clusters exhibiting high expression of the 66 immune markers, including the immune checkpoint genes CTLA4, PDCD1 (PD-1), and CD274 (PD-L1). No significant association was observed with HBV/HCV infection status. Likewise, overall survival was not significantly related to immune clustering.
Figure 7. Characterization of LIHC immune microenvironment using RNA-seq data.
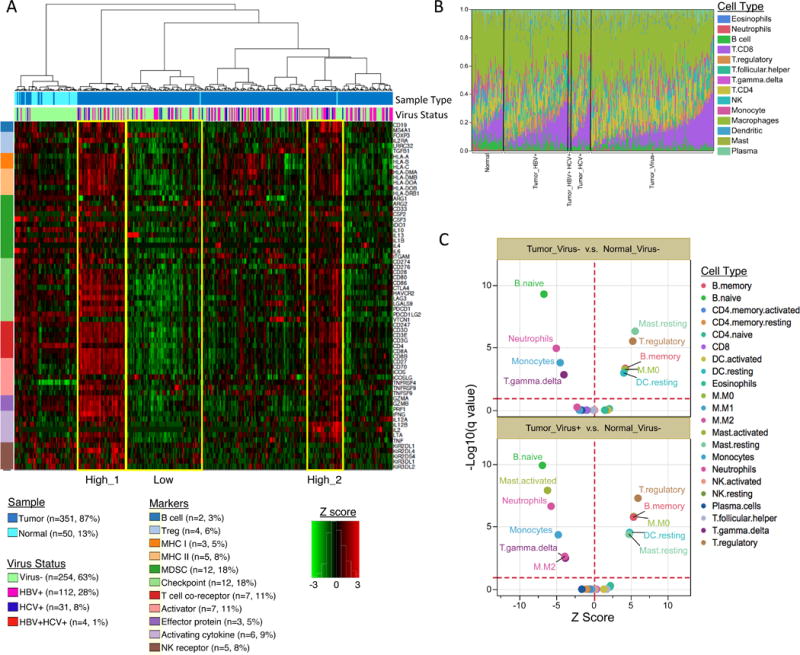
(A) Unsupervised hierarchical clustering of gene expression identifies immune profiles within HCC patients. Sixty-six manually curated immune cell markers were used for clustering. (B) The CIBERSORT-inferred relative fractions of different immune cell types varied across tumor and tumor adjacent normal samples and were not associated with virus status. (C) CIBERSORT cellular composition analysis revealed striking differences in relative compositions of immune cell populations between tumor and tumor-adjacent normal tissues. P values were calculated by Wilcoxon rank-sum test and adjusted for multiple testing (q value). The red dotted lines on the y axis indicate q value of 0.01. The red dotted lines on × axis indicates Z score of 0. The analysis was performed for all CIBERSORT immune cell types but only the significant ones are labeled on the plot.
We further investigated the cellular composition of immune infiltrates in LIHC using the CIBERSORT (Newman et al., 2015) inferred relative fractions of different immune cell types. The immune compositions varied largely across samples (Figure 7B). We observed similar pattern of immune composition between HBV+ and HCV+ tumors (p>0.05), and between HBV/HCV infected and virus negative tumors (p>0.05). Significant differences in immune cellular composition between tumor and tumor-adjacent normal samples were detected, regardless of virus infection status (Figure 7C). In tumors we observed depletion of naïve B cells, activated mast cells (virus+ tumors only), neutrophils, monocytes, gamma delta T cells, and the activated (M2) macrophages (virus+ tumors only), and a significant enrichment of memory B cells, suppressive regulatory T cell (Treg), resting mast cells, resting dendritic cells, and undifferentiated (M0) macrophages (Figure 7C). The ratios of CD8/Treg were significantly decreased in LIHC tumors (p=1.9e-7). These results indicated a transformation of the immune microenvironment in HCC tumor tissues from activating/effector cells to resting/suppressive immune cells.
DISCUSSION
This comprehensive integrated analysis of HCC enhances our understanding of the molecular events relevant to this cancer. The utilization of six distinct data platforms in the current study has facilitated integrated solutions not possible with single platform studies. The robust statistical power provided by a relatively large patient set of 363 HCC enabled us to identify 26 significantly mutated genes through use of the MutSigCV algorithm. Eight of these 26 SMGs had not been identified in previous HCC genomic sequencing studies (Table S2B). Two, LZTR1 and EEF1A1, contained somatic mutations identical to those recurrently observed in other cancers. Two genes, SF3B1 and SMARCA4, exhibited near significance by MutSigCV analysis, and displayed mutations identical to those identified as driver mutations in other cancers (Table S2A,B).
Among the SMGs identified in our HCC dataset were the ALB and APOB genes, key mediators of hepatocyte function in the secretion of blood factors albumin and VLDL. These functions demand a high fraction of hepatocyte transcriptional, translational, and energy resources and thus these processes might be suppressed by the malignant hepatocyte to support cell division requirements. We also noted that a high fraction of HCC exhibited CPS1 hypermethylation accompanied by decreased RNA expression. CPS1 encodes a rate-limiting enzyme for the urea cycle, allowing more efficient removal of ammonia from the body. Reduction of CPS1 could result in shunting of glutamine to initiation of de novo pyrimidine synthesis, consistent with increased CAD and decreased CPS1 expression levels observed in HCC relative to normal hepatocytes. Thus, a key component in the progression of hepatocytes to malignant HCC cells may be metabolic reprogramming through either genetic (ALB, APOB), epigenetic (CPS1) or other mechanisms, converting a cell committed to normal organismal support functions to a cell that supports only its own requirements for growth and division.
These mutation and pathway analyses provide potential directions for future therapeutic efforts. We showed that WNT or p53 signaling or the telomerase promoter are altered in 77% of HCC. WNT pathway small molecule inhibitors are currently in preclinical and clinical development (Pez et al., 2013). Because p53 can be rendered dysfunctional by alterations in upstream regulator function (e.g. MDM2, MDM4), p53 signature analysis may provide a more accurate representation of p53 functional activity and may better predict clinical outcomes than mutation-based studies. A fraction of HCC with WT TP53 have elevated MDM4 expression and currently available MDM4 small molecule inhibitors might be efficacious in these HCC (Jochemsen, 2014). The high frequency of TERT promoter mutations suggests that upregulated TERT expression in HCC might be targeted with telomerase inhibitors currently in development (Ruden and Puri, 2013).
Finally, IDH1/2 mutations were observed in four HCC. The recent development of IDH1 small molecule inhibitors suggests these drugs may be useful in that minority of HCC with IDH mutations (Okoye-Okafor et al., 2015). Although these tumors histopathologically most closely resemble HCCs, they exhibit clinical and genetic features of both cholangiocarcinomas and HCCs, signifying their possible origin from biphenotypic stem cells and suggesting that cholangiocarcinoma and HCC represent two ends of a continuum. Hence, the presence of IDH1/2 mutation in HCC may be associated with a shift towards a biliary phenotype, molecularly, even when the tumors do not resemble mixed tumors by histopathology. The discovery of an expression signature associated with this mutant, found in varying intensity in approximately 10% of the patients in several independent cohorts, supports this view.
Focal HCC amplification events also revealed potential therapeutic targets. Amplification of MET and VEGFA loci indicates that other RTK inhibitors in addition to sorafenib may be effective in HCC. MCL1, frequently amplified in HCC as well as in many other tumor types, encodes an anti-apoptotic protein that induces resistance to several chemotherapeutic agents (Belmar and Fesik, 2015). Numerous small molecule MCL1 inhibitors have been developed and might be tested in corresponding MCL1 amplified HCC patients (Belmar and Fesik, 2015).
Immune phenotyping of HCC by histopathology and gene expression analyses of immune cell markers revealed that a subset of HCC had high levels of immune cell infiltration. The transformation of the immune microenvironment in some HCC from activating/effector cells to resting/suppressive immune cells suggests that therapies targeting the immune checkpoint inhibitors (e.g. CTLA4, PD-1, PD-L1) in HCC might lead to robust responses in those HCC with moderate to high levels of immune cell infiltration (Prieto et al., 2015).
In conclusion, integrated analytic approaches have been applied to multiple data platforms from a large set of clinically annotated HCC to provide a better understanding of molecular targets that may lead to improved therapeutic strategies. The many identified targets indicate that it may be unlikely that one agent can effectively target all or most HCC, and the most effective treatments may entail multiple agents that specifically attack different identified targets.
STAR METHODS TEXT
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, David Wheeler (wheeler@bcm.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Sample Acquisition
The Tissue Source Sites (TSS) contributing biospecimens included in this manuscript include: ABS, Asterand, Inc., Baylor, St. Joseph’s Medical Center Cancer Institute, Christiana Care Health Services, Inc., Emory University, Fox Chase Cancer Center, Hartford Hospital, International Genomics Consortium, ILSbio, LLC., Mayo Clinic, Montefiore Medical Center, Ontario Institute for Cancer Research - Ottawa, Roswell Park Cancer Institute, Saint Mary’s Health Care, St. Joseph - Arizona, University of Calgary Alberta Health Services, University of California San Francisco, University of Florida, University of Michigan, University of Minnesota, University of North Carolina, University of Pittsburgh, and University of Utah.
Approximately 86% of hepatocellular carcinoma cases (consisting of a primary tumor and a germline control) submitted to the BCR and processed passed quality control metrics. Tumor tissue from 184 cases was submitted for reverse phase protein array analysis. The data freeze included 196 cases from LIHC batches 100, 131, 153, 173, 203, 231, 275, 287, 303, 314, 327, 341, 345, and 365.
A descriptive table of clinical features, histological features, and molecular features for the 196 case cohort as well as a patient level summary are shown in Supplemental Table 1A and 1B. A post-freeze set of 167 HCC cases were also examined by exome sequencing and DNA copy number analysis and these are listed in Supplemental Table 1C.
Sample inclusion criteria
Surgical resection of biopsy biospecimens were collected from patients diagnosed with hepatocellular carcinoma (HCC), and had not received prior treatment for their disease (chemotherapy or radiotherapy). Institutional review boards at each tissue source site reviewed protocols and consent documentation and approved submission of cases to TCGA. Cases were staged according to the American Joint Committee on Cancer (AJCC). Each frozen primary tumor specimen had a companion normal tissue specimen (blood or blood components, including DNA extracted at the tissue source site). Adjacent tissue was submitted for some cases. Specimens were shipped overnight using a cryoport that maintained an average temperature of less than −180°C.
Pathology quality control was performed on each tumor and normal tissue (if available) specimen from either a frozen section slide prepared by the BCR or from a frozen section slide prepared by the Tissue Source Site (TSS). Hematoxylin and eosin (H&E) stained sections from each sample were subjected to independent pathology review to confirm that the tumor specimen was histologically consistent with the allowable hepatocellular carcinomas and the adjacent tissue specimen contained no tumor cells. Adjacent tissue with cirrhotic changes was not acceptable as a germline control, but was characterized if accompanied by DNA from a patient-matched blood specimen. The percent tumor nuclei, percent necrosis, and other pathology annotations were also assessed. Tumor samples with ≥60% tumor nuclei and ≤20% or less necrosis were submitted for nucleic acid extraction.
METHOD DETAILS
Sample Processing
RNA and DNA were extracted from tumor and adjacent normal tissue specimens using a modification of the DNA/RNA AllPrep kit (Qiagen). The flow-through from the Qiagen DNA column was processed using a mirVana miRNA Isolation Kit (Ambion). This latter step generated RNA preparations that included RNA <200 nt suitable for miRNA analysis. DNA was extracted from blood using the QiaAmp blood midi kit (Qiagen).
RNA samples were quantified by measuring Abs260 with a UV spectrophotometer and DNA quantified by PicoGreen assay. DNA specimens were resolved by 1% agarose gel electrophoresis to confirm high molecular weight fragments. A custom Sequenom SNP panel or the AmpFISTR Identifiler (Applied Biosystems) was utilized to verify that tumor DNA and germline DNA representing a case were derived from the same patient. Five hundred nanograms of each tumor and normal DNA were sent to Qiagen (Hilden, Germany) for REPLI-g whole genome amplification using a 100 μg reaction scale. RNA was analyzed via the RNA6000 nano assay (Agilent) for determination of an RNA Integrity Number (RIN), and only analytes with RIN ≥7.0 were included in this study. Only cases yielding a minimum of 6.9 μg of tumor DNA, 5.15 μg RNA, and 4.9 μg of germline DNA were included in this study.
Samples with residual tumor tissue were considered for proteomics analysis. When available, a 10 to 20 mg piece of snap-frozen tumor adjacent to the piece used for molecular sequencing and characterization was submitted to MD Anderson for reverse phase protein array analysis.
Contributors: Lisa Iype, Renumathy Dhanasekaran, Tara M. Lichtenberg, Jay Bowen, John A. Demchok, Carmen Helsel, Chad Creighton.
Pathology Review
Standard Review of HCC
Each case had a single digital image of a full scan of an H&E slide available for review. The digital image had a magnification tool that allowed examination of the image at various magnifications. The background liver was not consistently available for review. Each case was reviewed independently by at least 3 liver pathologists, with no clinical or molecular information. Each pathologist has specialty training in liver pathology and extensive experience in diagnostic pathology research. Pathologists had as much time as they needed to review the digital images. The histological data collection sheet had been previously designed and discussed by the participating pathologists. Prior to case review, representative examples of tumor grade and other select histological parameters were circulated in a PowerPoint as a reference guide. Each image was first reviewed to ensure the tumor was consistent with a hepatocellular carcinoma; tumors inconsistent with hepatocellular carcinoma were not further reviewed. After that, the histological data outlined below was collected and submitted through a web based interface. After data submission, the data was reviewed and finalized. For numerical data, the median score was used. For classification data, the majority pathology opinion was used. Tumor grade was scored for both the predominant grade and the least differentiated grade using the following definitions:
Very well differentiated hepatocellular carcinoma: the cytological findings resemble non-neoplastic liver and the H&E differential includes hepatic adenoma, with no more than focal and minimal cytological atypia, and with no architectural atypia.
Well differentiated hepatocellular carcinoma: the tumor shows unequivocal hepatic differentiation on H&E. There is mild but definite cytological atypia and mild architectural atypia.
Moderately differentiated hepatocellular carcinoma: the tumor is clearly cancer based on H&E and the cytological evidence for hepatic differentiation is clear, or hepatic differentiation is strongly suspected from H&E. Moderate cytological and or architectural atypia is present.
Poorly differentiated hepatocellular carcinoma: hepatic differentiation is only suspected or is unclear from the H&E findings. There is marked cytological and or architectural atypia.
Hepatocellular carcinomas have a number of different growth patterns, but most fall into the categories of solid, pseudoacinar, trabecular, or macrotrabecular (trabeculae at least 10 cells in thickness). The predominant pattern was chosen, as well as all other patterns that made up at least 5% of the tumor image.
The tumors were also extensively characterized by their cytological findings. The percent of the tumor with macrovesicular steatosis, glycogen accumulation (clear cell change), hyaline bodies, and Mallory-Dank bodes were estimated to the nearest 10%. Ballooned hepatocytes were scored as none, few, or many. Lymphocytic and neutrophilic intratumoral inflammation was scored separately. Tumors with no or minimal inflammation were scored as 0. Greater degrees of inflammation were scored as mild, moderate, or marked, with marked inflammation defined as tumors with more inflammatory cells than tumor cells. When cholestasis was present, it was scored as mild (less than 5% of tumor area), moderate (6–50% of tumor area), or marked (greater than 50% of tumor area). When scoring intratumoral fibrosis, broad bands of fibrosis that occasionally transverse tumors were not scored and scoring instead focused on capturing “pericellular” or intratumoral patterns of fibrosis. These areas were then scored as none or minimal, mild (intratumoral fibrosis less than 5–25% of surface area), moderate (26 to 50% of surface area), or marked (fibrosis is equal to or greater than the amount of tumor cells)
Tumors were evaluated, on a yes/no basis, for the presence of clearly distinct nodules of HCC with different morphological patterns. The goal was to capture those tumors that have multiple, clearly distinct morphologies on the submitted image. The distinct morphologies are found as separate tumor nodules with clearly delineated borders and this finding is a separate observation from growth pattern.
Finally, tumors were classified into specific subtypes, when possible, using the definitions below. Hepatocellular carcinomas that did not fit into any of these categories were classified as “No Specific Subtype”:
Biphenotypic hepatocellular carcinoma (combined hepatocellular and cholangiocarcinoma). There should be a portion of the tumor that morphologically shows cholangiocarcinoma and a separate component that clearly shows hepatocellular carcinoma.
Cirrhotomimetic hepatocellular carcinoma. This tumor is defined by its growth pattern with tumor nodules that mimic cirrhotic nodules.
Clear cell hepatocellular carcinoma. This subtype was defined as carcinomas with at least 50% clear cell change.
Fibrolamellar carcinoma. This tumor is defined as having large polygonal eosinophilic cells with prominent nucleoli and intratumoral fibrosis. It’s recognized that cases need immunostains to confirm this diagnosis in clinical practice, but the goal was to identify cases with the classic morphological findings.
Granulocyte colony stimulating factor hepatocellular carcinoma. These are moderately to poorly differentiated hepatocellular carcinomas with generally solid growth patterns and striking neutrophilic infiltrates. It is recognized that clinical correlation with the white blood cell count is needed to confirm the diagnosis in clinical practice, but the goal was to identify cases with the classic morphological findings.
Lymphocyte rich hepatocellular carcinoma. This subtype was defined as hepatocellular carcinoma having intratumoral lymphocytes with a density where the lymphocytes are similar or greater in number than tumor cells, and this finding is present in more than 50% of the tumor image
Myxoid hepatocellular carcinoma. This tumor has sinusoids distended by myxoid material. At least 10% of the image should show this finding.
Sarcomatoid hepatocellular carcinoma. The spindle cell component should make up at least 10% of the tumor image.
Scirrhous hepatocellular carcinoma. Intratumoral fibrosis makes up greater than 50% of the tumor image.
Steatohepatitic hepatocellular carcinoma. This subtype is defined by at least 33% fat, plus ballooned tumor cells that resemble ballooned hepatocytes in steatohepatitis, plus at least mild tumor inflammation. Intratumoral fibrosis may be present but is not required.
The pathology review has limitations imposed by the logistics of this study. One major limit stems from examining a single digital image of a single tumor section, which has risk of sampling effects. This limit is particularly relevant to tumor sub-classification. As one example, fibrolamellar carcinomas can have histological heterogeneity, and the classic findings may not be evident on the scanned slide. As a second example, the requirement for 50% clear cell change to qualify for a clear cell hepatocellular carcinoma is typically applied to the composite percentage of the sections from the entire tumor, and not a single slide. An additional limitation was the inability to consistently collect data on the background, non-neoplastic liver tissues. Finally, diagnostic pathology in clinical practice relies on the combination of morphology and immunohistochemical stains to render the final tumor classification. Immunohistochemical were not available in this study.
Review of IDH1/2 mutated patients
IDH1/2 mutations are frequent in intrahepatic cholangiocarcinomas (CCA) but rare or possibly noexistant in HCC; hence the possibility that these tumors actually represented mixed HCC-CCA or intrahepatic CCAs was considered. First, we reviewed the original pathology report from the tissue source site. The tissue source sites performed the initial pathologic review on the tumor slides and also the surrounding normal liver tissue. They had access to the whole tumor and examined multiple sections before making a diagnosis. They only submitted tissue to the TCGA LIHC project after confirming the diagnosis of HCC. All four of them had been histologically diagnosed as hepatocellular carcinoma and not as mixed HCC-CCA or cholangiocarcinoma. One of the tumors was poorly differentiated; the tissue source site performed albumin in situ hybridization, which was positive, and hence they leaned toward diagnosis of HCC. Subsequently, our TCGA pathology review committee of experienced liver pathologists reviewed submitted images of the H&E slides to independently confirm the diagnosis of HCC. Due to the constraints of the TCGA project process, the pathology review committee did not have access to all slides and blocks from the tumor and were unable to perform additional immunohistochemical analyses of the tumors. Based on the diagnosis of HCC from the tissue source site and its concordance with our independent pathology review we believe that these 4 tumors are likely to be HCC.
Contributors: Michael Torbenson, David Kleiner, Hala Makhlouf, Dhanpat Jain, Sanjay Kakar, Matthew Yeh.
DNA Sequencing and Analysis
Primary DNA Sequencing
Primary DNA exome sequencing was carried out at the Human Genome Sequencing Center at Baylor College of Medicine using approaches standard to TCGA and identical to those described by Totoki et al. (2014). Paired-end DNA sequence libraries were generated following the standard HGSC protocol (https://hgsc.bcm.edu/sites/default/files/documents/Illumina_Barcoded_Paired-End_Capture_Library_Preparation.pdf). Exome capture was performed by pooling 4 samples together into pre-pooled libraries and then capturing with the HGSC VCRome 2.1 capture reagent (42Mb, NimbleGen). Library capture, amplification conditions, and quality control were identical to those described in Totoki et al. (2014). Sequencing was performed on the Illumina HiSeq 2000 platform with one pool perlane following standard protocols identical to those in Totoki et al. (2014). Sequence runs generated between 300–400 successful reads per lane.
Initial sequence analysis was performed by aligning reads to the human genome reference sequence hg19 using the Mercury Pipeline (https://www.hgsc.bcm.edu/software/mercury) exactly as described by Totoki et al. (2014). Once aligned and following base quality recalibration and indel realignment via the Mercury Pipeline, sequence alignment files (BAM files) were checked for contamination by testing the concordance between SNPs in the tumor/normal pairs to the genotypes in the matching SNP Array from the Broad Institute copy number platform. Samples with greater than 5% contamination are annotated and not used for subsequent analyses. Sequence coverage averaged 100× for the cohort, with >90% of target bases covered at 20× or greater in all samples. All BAM files were submitted to CGHub.
Validation Sequencing
Validation sequencing was performed using the Ion Proton platform targeting 3865 amplicons using the AmpliSeq targeted sequencing approach exactly as described by Totoki et al. (2014). Library construction, sequence generation, sequence alignment, and validation criteria were identical to those used by Totoki et al. (2014).
Multi-Center Mutation Calling
Mutations were called by five production or analysis centers within the TCGA Network: Human Genome Sequencing Center (Comprehensive And Reproducible Nucleotide Alterations in Cancer–CARNAC), UCSC (RADIA), BCGSC (Strelka), MD Anderson-Baylor College of Medicine (MuSE), and Broad Institute (MuTect) as described below.
HGSC CARNAC
Mutations were called as described for the HGSC in Totoki et al. (2014).
UCSC RADIA
Single nucleotide somatic mutations were identified by RADIA (RNA AND DNA Integrated Analysis) (Radenbaugh et al., 2014), a method that combines the patient matched normal and tumor DNA whole exome sequencing (DNA-WES) with the tumor RNA sequencing (RNA-Seq) for somatic mutation detection (software available at: https://github.com/aradenbaugh/radia/). The inclusion of the RNA-Seq data in RADIA increases the power to detect somatic mutations, especially at low DNA allelic frequencies. By integrating the DNA and RNA, mutations that would be missed by traditional mutation calling algorithms that only examine the DNA can be rescued back. RADIA classifies somatic mutations into 3 categories depending on the read support from the DNA and RNA: 1) DNA calls – mutations that had high support in the DNA, 2) RNA Confirmation calls – mutations that had high support in both the DNA and RNA, 3) RNA Rescue calls – mutations that had high support in the RNA and weak support in the DNA. Here RADIA identified 32,113 DNA mutations, 6,315 RNA Confirmation mutations, and 741 RNA Rescue mutations.
BCGSC Strelka (Saunders et al., 2012) (v1.0.6) was used to identify somatic single nucleotide variants, and short insertions and deletions from the TCGA LIHC exome dataset. All parameters were set to defaults, with the exception of “isSkipDepthFilters”, which was set to 1 in order to skip depth filtration given the higher coverage in exome datasets. 202 pairs of libraries were analyzed. When a blood sample was available, it served as the matched normal specimen; otherwise, the matched normal tissue was used. The variants were subsequently annotated using SnpEff, and the COSMIC (v61) and dbSNP (v137) databases.
MD Anderson- Baylor College of Medicine. MuSE
We developed a novel approach to mutation calling based on the Markov substitution model for molecular evolution, which models the evolution of the reference allele to the allelic composition of the matched tumor and normal tissue at each genomic locus. To improve overall accuracy, we further adopt a sample-specific error model to identify cutoffs, reflecting the variation in tumor heterogeneity among samples.
Broad Institute
The Firehose pipeline (http://www.broadinstitute.org/cancer/cga/Firehose) performed additional quality control (QC) on the BAM files, mutation calling, small insertion and deletion detection, and annotation of point mutations and indels as follows:
QC on BAM files: The sample cross-individual contamination levels were estimated using the ContEst program (Cibulskis et al., 2011). Tumor normal pairs of samples with contamination less than 4% were used further downstream for analysis.
Somatic mutation Calling and Significantly Mutated Genes: The MuTect algorithm (Cibulskis et al., 2013) was used to detect somatic single nucleotide variants (SSNVs).
Small insertion and deletion detection: The Indelocator algorithm (https://www.broadinstitute.org/cancer/cga/indelocator) was used to detect small indels.
Mutations and indels annotations: Point mutations and indels detected by respective MuTect and Indelocator were annotated using utility named Oncotator (Ramos et al., 2015). Oncotator mapped somatic mutations to respective genes, transcripts, and other relevant features. These annotations correspond to the fields in the Mutation Annotation Format (MAF) files version 2.4: (https://wiki.nci.nih.gov/display/TCGA/Mutation+Annotation+Format+(MAF)+Specification).
Integrating Mutation Calls
Mutation calls from each center were integrated by matched allele aggregation into a multi-center MAF file. The variant and reference coverages for each allele were normalized by direct lookup in the respective BAM files for the samples. Coverages from RNA data were also added for matched samples. Annotation was performed using the CARNAC annotation tools.
The final mutation set validation criteria were:
Accept Tumor validation based on RNA data if greater than two variant alleles observed in RNA and RNA variant allele fraction was greater than 1%.
Accept Normal validation based on RNA data if greater than two variant alleles observed in RNA and RNA variant allele fraction was greater than 0.2%.
Accept Tumor validation based on Proton data if greater than two variant alleles observed in validation sequence and validation variant allele fraction was greater than 1%.
Reject Tumor validation based on Proton data if allele not Accepted by Proton data and the binomial test of allele fraction for validation is significantly less than the allele fraction for the primary sequence.
Accept Normal validation based on Proton data if greater than two variant alleles observed in validation sequence and validation variant allele fraction was greater than 0.2%.
Accept Tumor validation if the allele was called by the Ion Variant Caller in tumor.
Accept Normal validation if the allele was called by the Ion Variant Caller in normal. 8. Final mutation and validation calls were made by integrating the above cases (1–7).
Mutation Significance Analysis
MutSig Suite
MutSig 2CV v3.1 (Lawrence et al., 2014), was applied to the consensus mutation call set filtered by the DNA allelic fraction >= 0.025, to identify 12 significantly mutated genes (Figure 1), including TP53, CTNNB1, ALB, RB1, AXIN1, BAP1, ARID1A, TSC2, IL6ST, APOB, HNF1A, and RPS6KA3 (False Discovery Rate < 0.2). A list of all non-silent gene mutations is shown in Supplemental Table 2A.
Inactivating SMG Analysis
For inactivating SMG analysis the raw MAF file was first filtered using the following filtering strategy; 1) variants were removed if they appeared in a cohort of normal samples, 2) variants were removed if they were observed greater than 2 times in the matched normal sample, had a variant allele fraction less than 0.04, if the gene had greater than 3 variants in the matched sample, or if the base coverage of the normal sample was less than 6. From the filtered data, we compared the rate of inactivating variants (nonsense, frame-shift, splice-site) to all other variation. We report the Chi-squared and Binomial test p-values for the difference in the ratio of inactivating variation in each gene compared with the background rate of the entire cohort (Supplemental Table 2B).
TERT Promoter Sequencing
TERT promoter sequencing was performed by the Sanger sequencing method exactly as described by Totoki et al. (2014). Two amplicons were attempted for each subject and the subject was considered to harbor a TERT promoter mutation if either amplicon generated a positive SNP call. Both automated (via SNPDetector) and manual calling were employed. Cases that failed in amplicon generation are encoded as NA for mutation status of the TERT-promoter. Samples with TERT-promoter status are present in Supplemental Table 3.
Mutation Signature Analysis
The mutation signatures discovery is a process of de-convoluting cancer somatic mutations, stratified by mutation contexts or biologically meaningful subgroups, into a set of characteristic patterns (signatures) and inferring the contributions of discovered signature activity across samples. The common classification of SNVs is based on six base substitutions within the tri-nucleotide sequence context including the bases immediately 5′ and 3′ to each mutated base. Six base substitutions (C>A, C>G, C>T, T>A, T>C, and T>G) with 16 possible combinations of neighboring bases result in 96 possible mutation types. Thus the input data for the mutation signature discovery is given as 96 by M mutation matrix (M= # of sample). Here we applied the Bayesian non-negative matrix factorization algorithm (BayesNMF) (Kasar et al., 2015) to infer the number of mutational signatures and their sample-specific contributions. The mutation count matrix was ingested as an input for the BayesNMF and factored into two matrices, W′ (96 by K) and H′ (K by 2M), approximating X by W′H′. Out of 50 Bayesian NMF runs with a half-normal prior for W′ and H′ seven runs converged to the 2-signature solution, while 43 runs converged to the 3-signature solutions. We used the 3-signature solution (K=3) in downstream analyses (Sig A, Sig B, and Sig C in Supplemental Figure 1a).
To enumerate the number of mutations associated with each mutation signature we performed a scaling transformation, X ~ W′H′ = WH, W = W′U−1 and H= UH′, where U is a K by K diagonal matrix with the element corresponding to the 1-norm of column vectors of W’, resulting in the final signature matrix W and the activity matrix H. Note that the kth column vector of W (wk) represents a normalized mutability of 96 tri-nucleotide mutation contexts in the kth signature and the kth row vector of H (hk) dictates the estimation of mutations associated to the kth signature across samples.
We used cosine similarity to compare our three signatures with thirty signatures (http://cancer.sanger.ac.uk/cosmic/signatures) previously reported. Signature A of this study corresponds most closely with published signature 22 and B with 24. A/22 is associated with exposure to aristolochic acid (AA) and B/24 with exposure to Aflatoxin B1 (AFB1). The etiology of signature C, which corresponds to published signature 5 is unknown.
To identify samples with a significant enrichment of the activity of each mutational process we performed a hierarchical clustering of a normalized signature activity (Supplemental Figure 1B) using the standard R package with a “Euclidean” distance and a “ward.D” linkage option. Nine samples (Red in Supplemental Figure 1a) were tightly clustered with a significantly higher activity of Sig A (aristolochic acid). Twenty-five samples (Blue in Supplemental Figure 1B) were clustered together with the increased activity of Sig B (aflatoxin B1). Interestingly, six of the top seven samples with the highest activity of Sig B were the same as the AFB1-affected samples identified by the independent mutation signature analysis for the 198 TCGA samples.
The enrichment analysis of splice site mutations on nine samples with a high activity of aristolochic acid (AA) signature (Supplemental Figure 1b) and aflatoxin B1 (AFB1) mutations in TP53 R249S mutant samples (Supplemental Figure 1c) was accomplished by two-sided Wilcoxon rank-sum tests.
Contributors: Kyle Covington, Jaegil Kim, Eve Shinbrot, Liu Xi, Amie Radenbaugh, Yu Fan, Wenyi Wang, Katayoon Kasaian, Carrie Cibulskis, Juok Cho.
Identification of Pathogens
Microbial detection in RNA-Seq data
BioBloom Tools – BC Cancer Agency
Our microbial detection pipeline is based on BioBloomTools (BBT, v1.2.4.b1), which is a Bloom filter-based method for rapidly classifying RNA-seq or DNA-seq read sequences (Chu et al., 2014). We generated 43 filters from ‘complete’ NCBI genome reference sequences of bacteria, viruses, fungi and protozoa, using 25-bp k-mers and a false positive rate of 0.02. We ran BBT in paired-end mode with a sliding window to screen FASTQ files from RNA-seq libraries (49-bp PE reads), and whole exome libraries (49-bp PE reads). In a single-pass scan for each library, BBT categorized each read pair as matching the human filter, matching a unique microbial filter, matching more than one filter (multi-match), or matching neither human nor microbe (no-match). For each filter, we then calculated a reads-per-million (RPM) abundance metric as: We applied a threshold of 2 RPM for identifying samples that were positive for hepatitis B.
PathSeq - Broad Institute
The PathSeq algorithm (Kostic et al., 2011) was used to perform computational subtraction of human reads, followed by alignment of residual reads to a combined database of human reference genomes and microbial reference genomes (which includes but is not limited to Hepatitis B virus (HBV), Hepatitis C Virus (HCV) genomes), resulting in the identification of reads mapping to HBV and HCV genomes in RNA sequencing data.
Subjects were classified as HBV-positive by RNA sequencing if at least 1 HBV read in 1 million human reads were present; otherwise, subjects were classified as HBV-negative. In addition, subjects were classified as HCV-positive by RNA sequencing if at least 1 HCV reads in 1 million human reads were present; otherwise, subjects were classified as HCV-negative.
Using PathSeq, human reads were subtracted by first mapping reads to a database of human genomes using BWA (version 0.6.1), Megablast (version 2.2.23), and Blastn (version 2.2.23). Only sequences with perfect or near perfect matches to the human genome were removed in the subtraction process. To identify HBV/HCV reads, the resultant non-human reads were aligned with Megablast to a database of microbial genomes that includes multiple HBV and HCV reference genomes. HBV/HCV reference genomes were obtained from the NCBI nucleotide database (downloaded in June 2012).
Mayo Clinic
To identify viral insertions in these LIHC TCGA DNA-seq and RNA-seq samples, we implemented a workflow with BWA-mem that aligns pair-end reads to viral genomes. An in-house database of viral genomes was built from NCBI RefSeq viral sequences. A set of custom scripts was written to identify reads pairs where one read mapped to the human genome and the second read mapped to a viral genome. The workflow includes the following steps:
Read pairs with at least one read unaligned to the reference genome were extracted from the TCGA GRCh37 aligned BAM files for each sample.
The extracted read pairs were re-aligned to the human genome using BWA-mem. Read pairs where both reads mapped to the human genome were filtered out.
The remaining reads were aligned to the viral genomes in our database using BWA-mem.
Concordant reads that mapped to viral genomes were extracted to compute coverage. Discordant and, if available, softclipped read pairs where only one mate aligned to the human genome were combined and clustered based upon their proximity within the human genome. The cluster cutoff was set to the average insert size of the library. Each cluster was reported as one viral insertion event. The softclipped reads were further used to provide a more precise genomic location of the insertion.
Finally, insertion events with less than 10 supporting reads were filtered out before visually curating the remaining events using IGV.
Consensus virus calls
We deemed a sample positive for Hepatitis B or C if the calls from Broad, BC and Mayo were all above their respective thresholds, or if the clinical data from the tissue source site identified the sample as Hepatitis positive. We chose to maintain the clinical verdict even in cases for which no HBV or HCV was detected by computational methods due to the potential for the virus to have cleared spontaneously or in response to antiviral therapy before the onset of cancer. Using the thresholds determined by each center, 44 tumors and 8 adjacent normals were identified as HBV positive, while 31 tumors and 5 adjacent normals were deemed HCV positive. In every case where an adjacent normal sample was identified as HBV or HCV positive, the matched tumor was also positive.
Viral integration sites inferred from RNA-Seq data
BC Cancer Agency
To detect genomic integration of specific viruses we performed de novo assembly of RNA-seq and DNA-seq sequence data with ABySS v1.3.4 (Simpson et al., 2009), using for each library the reads classified by BBT as human, the virus, multi-match, and no match. We then merged the k-mer assemblies for each library with Trans-ABySS v1.4.8 to generate the working contig set. We re-ran BBT on these contigs, applying only human and specific virus filters, identifying contigs that matched to both filters. We identified any integration breakpoints in such multi-matched contigs by using BLAT v34 to align each contig to the human GRCh37/hg19 reference sequence, and to virus reference sequences. We retained contig alignments in which: a) the aligned human and viral sequences summed to at least 90% of the contig length, and b) the human and viral aligned overlapped by less than 50%. Human breakpoint coordinates were annotated against RefSeq and UCSC (Kuhn et al., 2013) gene annotations (downloaded from the UCSC genome browser on 30-Jun-2013). Breakpoints that had at least 3 spanning mate-pair reads or 5 flanking mate-pair reads were considered potential integration sites.
We identified 27 tumors and 7 adjacent normals as having at least one HBV integration event. In contrast, we detected no HCV integration events. HBV integrated into the human genome in approximately 77% of the samples in which HBV was detectable. In two additional samples, TCGA-CC-A3MA and TCGA-ED-A7PZ, an integration event was detected despite HBV being below threshold. The results are summarized in Supplemental Table 5B.
Broad Institute
An HBV-positive sample was considered integration positive if there were at least 5 spanning read pairs or 10 flanking reads supporting an integration event. In case of HBV-positive, flanking read pairs were defined as having one end of the paired-end read mapped to the HBV genome and its mate pair mapped to the human genome. Spanning reads were defined as having one end of the paired end read spanning the integration junction and its mate pair mapped to either the human or HBV genome. Once HBV reads were obtained, we extracted all mate pairs and used Tophat-2.0.8 (Trapnell et al., 2009) with fusion option enabled to map these paired end reads to a combined database containing the human genome and an HBV genome. Next, spanning reads and flanking reads are identified from the aligned BAM file.
Human genes involved in the integration are identified using the breakpoint coordinates based on RefSeq and UCSC gene annotations (last modified on 30-Jun-2013) from the UCSC genome browser. Similar approach is followed for identification of HCV integration from RNAseq data. These results are summarized in Supplemental Table 5B.
Contributors: Reanne Bowlby, Sara Sadeghi, Karen Mungall Chandra Sekhar Pedamallu, Akinyemi I Ojesina, Matthew Meyerson, Daniel O’Brien, Jean-Pierre Kocher, Betty L. Slagle, Kyle Covington, Lawrence A. Donehower.
Gene Fusion Detection
BCM HGSC
TCGA RNA sequencing data (RNA FastQ files) were downloaded for the 196 patients on this freeze list set from CGHub. deFuse version 0.6.1 (McPherson et al., 2011) with default settings detected a large list of candidate fusion genes. The deFuse results were filtered by removing events identified as “read through” transcription of adjacent genes, requiring coding regions, in-frame (ORF) genes and samples with a defuse confidence score of >80%. Our sample set included 11 tissue adjacent normal (TAN) samples; any fusions that were also identified in the TAN sample set were removed from analysis. To characterize the resultant candidate fusion genes we did the following checks:
Each read spanning a fusion junction was aligned to the reference genome using BLAT in the UCSC Genome Browser to confirm their map locations. The fusions that mapped with 100% identity to each part of the identified fusion (gene1 or gene2) were selected for further analysis. Genes that mapped to multiple locations were discarded.
Each RNA BAM from candidate fusion genes was examined in IGV, to verify the presence of stacked soft clipped reads and changes in coverage at the identified fusion breakpoints. The sequence of each soft clipped read was brought into the UCSC genome browser and mapped using BLAT.
The CBio data portal (http://www.cbioportal.org/) was used to examine copy number data and gene expression data for the gene partners in each fusion identified.
We also utilized copy number data, loading a given patient’s Affymetrix 6.0 .seg files into IGV in tandem with their RNA Seq BAMS, to evaluate the DNA coverage along with soft clipped reads at the identified the mRNA break points. When whole genome sequence BAMS were available for a given patient, we also included those in the evaluation.
MD Anderson
We used the Pipeline for RNAseq Data Analysis (PRADA) to preprocess RNA Seq data and detect gene fusions (Torres-Garcia et al., 2014). PRADA aligns short reads to a composite reference database composed of whole genome sequence (hg19) and transcriptome sequence (Ensembl64). By default, PRADA uses two criteria to select candidate fusions:
a minimum of two discordant read pairs mapping to a candidate gene pair, i.e. two distinct protein coding genes;
a minimum of one junction spanning read mapping to a hypothetical junction constructed from the candidate gene pair.
To construct a hypothetical junction, we used 40 base pairs from either side of two connecting exons, considering the RNAseq read length is 48 base pairs in this data set. All junction spanning reads and discordant reads allowed one mismatch. From these candidate fusions, we filtered out those fusions that had significant sequence similarity (BLASTN, Expect value required to be >0.01). We then calculated the transcriptional allelic fraction (TAF) for each fusion partner. TAF was defined as the fraction of fusion-associated junction spanning reads over all reads that spanned the involving exon boundaries. We required the minimum TAF to be 0.1 for at least one partner gene. Six fusions were included in the final list for their established roles in this cancer type or other cancers despite their lower TAFs. These six fusions included two TCF7L2-VTI1A fusions, three DNAJB1-PRKACA fusions and one FGFR3-TACC3 fusion. Prediction of fusion functional consequence (in-frame, out-of-frame, UTR-CDS, etc.) was performed by PRADA using the Ensembl64 defined gene/transcript model. Only fusions that involved coding regions (in-frame and out-of-frame) were retained for further analysis. More details of the PRADA pipeline were described (http://sourceforge.net/projects/prada/).
We analyzed 196 samples from the freeze list, from which we detected a total of 236 fusions. The number of fusions in each case ranged from 0 to 18. We compared fusions to a list of kinases from Uniprot (http://www.uniprot.org) and cancer genes from the Cancer Gene Census. Out of the 236 fusions 26 involved a kinase gene, and 27 involved a cancer gene. We further aligned the fusions to copy number data. We were able to find a copy number breakpoint for 201 fusions at the vicinity of 500 Kb using the copy number cutoff 0.1 (log ratio). One in-frame SLC12A7-TERT fusion, which had corroborating exon expression pattern and DNA breakpoints near both partner genes.
Mayo Clinic
We converted the TCGA LIHC RNASeq BAM files into FASTQ files and realigned them using the Mayo Analysis Pipeline for RNA Seq (MAP-RSeq) (http://bioinformaticstools.mayo.edu/research/maprseq/). MAP-RSeq uses tophat, a splice-junction aware aligner to map paired-end RNA sequencing reads. Tophat uses bowtie, a memory efficient short read aligner, to quickly map reads to a reference genome and transcriptome, and then uses those alignments to identify known and novel transcript elements within each sample. MAP-RSeq reports 2 fusion events lists, one that displays all possible fusion events detected by tophat-fusion, and a second enriched in confident fusion events using tophat-fusion’s default filtering strategy. Tophat-fusion’s default filtering strategy involves evaluating the number of supporting reads, the genes involved, the mapping uniqueness, and the dissimilarity of the sequence around the fusion breakpoints to detect credible fusion events. The list that includes all possible fusion events was combined with fusions reported by other institutions to establish a consensus set of fusions. The filtered fusions list was used to suggest events for further validation. All fusion events were bioinformatically visualized and curated with IGV and circos plots.
Blueprint Medicines
Gene fusions in the LIHC dataset were discovered using methods previously described (Stransky et al., 2014). Briefly, the RNAseq fastq files were downloaded from CGHub and aligned using the STAR algorithm v2.3.1q (Dobin et al. 2012. doi:10.1093/bioinformatics/bts635) with options described previously. Version hg19 of the human genome, as well as transcriptome and splice junction annotations from the Gencode project v17 were provided to the STAR algorithm as an alignment reference. Next, fusions between any two genes were identified based on the number of chimeric reads (sequencing paired ends mapping to different genes) and split reads (spanning a fusion breakpoint), concordance between the strands of the reads and the genes involved in the putative fusion, and a number of filtering criteria to flag false positive and non-functional fusions. In addition, recurrent kinase fusions observed in a panel of 600 normal samples from TCGA and 1,800 normal samples from the Genotype–Tissue Expression (GTEx) project were also excluded from further analysis. Finally, all recurrent kinase fusions (n≥2) were manually reviewed to identify putative oncogenic drivers with distinctive characteristics of functional kinase fusions. In particular, the following features were required: presence of an intergenic junction between two exons, a predicted in-frame coding sequence and conservation of the complete kinase catalytic domain. Conversely, we excluded false positives from further analysis according to two main criteria: the presence of a homologous or repetitive sequence shared by the two fusion partners causing an alignment artifact, or the very high expression of one or both fusion partners.
Research Center for Advanced Science and Technology (RCAST)
Recent observations of structural rearrangements involving TERT prompted us to specifically investigate TERT mRNA for evidence of fusion transcripts. We extracted all reads that mapped in 5p13.33 (chr5:1–2Mb) from the RNA-seq BAM files. Within this interval we searched for any paired-end reads that mapped more than 100kb apart or mapped to other chromosomes. Anomalous read pairs, which aligned within 10kb of TERT were extracted from RNAseq BAM file, and assembled. TERT-fusion candidates found in four samples were manually checked in Integrative Genomics Viewer (IGV).
Contributors: Eve Shinbrot, Frederick M. Lang, Siyuan Zheng, Roeland G.W. Verhaak, Daniel O’Brien, Jean-Pierre Kocher, Nicolas Stransky, Hiroyuki Aburatani, Yamamoto Shogo.
SNP-Based Copy Number Analysis
DNA from each tumor or germline sample was hybridized to Affymetrix SNP 6.0 arrays using protocols at the Genome Analysis Platform of the Broad Institute as previously described (McCarroll et al., 2008). Briefly, from raw .CEL files, Birdseed was used to infer a preliminary copy number at each probe locus. For each tumour, genome-wide copy number estimates were refined using tangent normalization, in which tumour signal intensities are divided by signal intensities from the linear combination of all normal samples that are most similar to the tumour. This linear combination of normal samples tends to match the noise profile of the tumour better than any set of individual normal samples, thereby reducing the contribution of noise to the final copy-number profile. Individual copy-number estimates then underwent segmentation using Circular Binary Segmentation (Olshen et al., 2004). As part of this process of copy number assessment and segmentation, regions corresponding to germline copy-number alterations were removed by applying filters generated from either the TCGA germline samples from the ovarian cancer analysis or from samples from this collection. Segmented copy number profiles for tumour and matched control DNAs were analyzed using Ziggurat Deconstruction, an algorithm that parsimoniously assigns a length and amplitude to the set of inferred copy-number changes underlying each segmented copy number profile (Mermel et al., 2011). Significant focal copy number alterations were identified from segmented data using GISTIC 2.0 (Mermel et al., 2011). For copy number based clustering, tumours were clustered based on thresholded copy number at reoccurring alteration peaks from GISTIC analysis (all_lesions.conf_99.txt file). Hierarchical clustering was done in R based on Euclidean distance using Ward’s method. Purity, ploidy and whole genome doubling estimates were calculated using the ABSOLUTE algorithm (Carter et al., 2012).
Contributors: Andrew D. Cherniack, Bradley A. Murray, Juliann Shih, Carrie Cibulskis.
DNA Methylation
Assay platform
DNA methylation data were generated using the Illumina Infinium DNA methylation platform (Bibikova et al., 2011), HumanMethylation450 (HM450). The HM450 assay analyzes the DNA methylation status of up to 482,421 CpG and 3,091 non-CpG (CpH) sites throughout the genome. It covers 99% of RefSeq genes with multiple probes per gene and 96% of CpG islands from the UCSC database and their flanking regions. The assay probe sequences and information for each interrogated CpG site on Infinium DNA methylation platform are available from Illumina (www.illumina.com).
The DNA methylation score for each assayed CpG or CpH site is represented as a beta (β) value (β = (M/(M+U)) in which M and U indicate the mean methylated and unmethylated signal intensities for each assayed CpG or CpH, respectively. β-values range from zero to one, with scores of “0” indicating no DNA methylation and scores of “1” indicating complete DNA methylation. A detection P value accompanies each data point and compares the signal intensity difference between the analytical probes and a set of negative control probes on the array. Any data point with a corresponding P value greater than 0.05 is deemed not to be statistically significantly different from background and is thus masked as “NA” in the Level 3 data packages as described below. Further details on the Illumina Infinium DNA methylation assay technology have been described previously (Bibikova et al., 2011).
Sample and data processing
We performed bisulfite conversion on 1μg of genomic DNA from each sample using the EZ-96 DNA Methylation Kit (Zymo Research, Irvine, CA) according to the manufacturer’s instructions. We assessed the amount of bisulfite-converted DNA and completeness of bisulfite conversion using a panel of MethyLight-based quality control (QC) reactions. All the TCGA samples passed our QC tests and entered the Infinium DNA methylation assay pipeline. Bisulfite-converted DNAs were whole-genome-amplified (WGA) and enzymatically fragmented prior to hybridization to BeadChip arrays. BeadArrays were scanned using the Illumina iScan technology to produce IDAT files. Raw IDAT files for each sample were processed with the R/Bioconductor package methylumi. TCGA DNA methylation data packages were then generated using the EGC.tools R package which was developed internally and is publicly available on GitHub (https://github.com/uscepigenomecenter/EGC.tools).
TCGA Data Packages
The data levels and the files contained in each data level package are described below and are present on the TCGA Data Portal website (http://tcga-data.nci.nih.gov/tcga/). Please note that as continuing updates of genomic databases and data archive revisions frequently become available, the data packages on TCGA Data Portal are updated accordingly.
Level 1 data contain raw IDAT files (two per sample) as produced by the iScan system and as mapped by the SDRF. These IDAT files were directly processed by the R/Bioconductor package methylumi. We provided a disease-mapping file (LIHC.mappings.csv) in the AUX directory to facilitate this process. Level 2 data contain background-corrected methylated (M) and unmethylated (U) summary intensities as extracted by the R/Bioconductor package methylumi. Detection P values were computed as the minimum of the two values (one per allele) for the empirical cumulative density function of the negative control probes in the appropriate color channel. Background correction was performed via normal-exponential deconvolution. Multiple-batch archives had the intensities in each of the two channels multiplicatively scaled to match a reference sample (sample with R/G ratio of the normalization control probes closest to 1.0). Level 3 data contain β-value calculations with annotations for HGNC gene symbol, chromosome, and genomic coordinates (UCSC hg19, Feb 2009) for each targeted CpG/CpH site on the array. Probes having a common SNP (Minor Allele Frequency > 0.01, per dbSNP build 135 via the UCSC snp135common track) within 10 bp of the interrogated CpG site or having a 15 bp from the interrogated CpG site which overlapped with a repetitive element (as defined by RepeatMasker and Tandem Repeat Finder Masks contained in the BSgenome.Hsapiens.UCSC.hg19 R package) were masked as “NA” across all samples, and probes with a detection P-value greater than 0.05 in a given sample were masked as “NA” on that array. Probes that were mapped to multiple sites on hg19 were annotated as “NA” for chromosome and 0 for CpG/CpH coordinate.
The following data archives were used for the analyses described in this manuscript.
jhu-usc.edu_LIHC.HumanMethylation450.Level_3.1.13.0
jhu-usc.edu_LIHC.HumanMethylation450.Level_3.2.13.0
jhu-usc.edu_LIHC.HumanMethylation450.Level_3.3.13.0
jhu-usc.edu_LIHC.HumanMethylation450.Level_3.4.13.0
jhu-usc.edu_LIHC.HumanMethylation450.Level_3.5.13.0
jhu-usc.edu_LIHC.HumanMethylation450.Level_3.6.13.0
jhu-usc.edu_LIHC.HumanMethylation450.Level_3.7.13.0
jhu-usc.edu_LIHC.HumanMethylation450.Level_3.8.13.0
jhu-usc.edu_LIHC.HumanMethylation450.Level_3.9.13.0
jhu-usc.edu_LIHC.HumanMethylation450.Level_3.10.13.0
jhu-usc.edu_LIHC.HumanMethylation450.Level_3.11.13.0
jhu-usc.edu_LIHC.HumanMethylation450.Level_3.12.13.0
jhu-usc.edu_LIHC.HumanMethylation450.Level_3.13.13.0
jhu-usc.edu_LIHC.HumanMethylation450.Level_3.14.13.0
Unsupervised clustering analysis
We removed probes which had any “NA”-masked data points and probes that were designed for sequences on X and Y chromosomes.
To capture cancer-specific DNA hypermethylation events, we first selected CpG sites that were not methylated in normal tissues (mean β-value <0.2). To minimize the influence of variable tumour purity levels on a clustering result, we dichotomized the data using a β-value of >0.3 as a threshold for positive DNA methylation. The dichotomization not only ameliorated the effect of tumour sample purity on the clustering, but also removed a great portion of residual batch/platform effects that are mostly reflected in small variations near the two ends of the range of β-values. We also removed CpG sites that were methylated in leukocytes, a major source of contamination present in a tumour sample (mean β-value >0.3). We then performed unsupervised hierarchical clustering on 37,848 CpG sites that were methylated with that threshold in at least 5% of the tumours using a binary distance metric for clustering and Ward’s method for linkage. The cluster assignments were generated by cutting the resulting dendrogram. Figure 2a displays a heatmap of the original β-values for randomly selected 15,000 CpG sites used in the hierarchical clustering. The CpG sites were displayed based on the order of unsupervised hierarchal clustering of the β-values using the Euclidean distance metric and Ward’s linkage method.
To investigate subgroups based on cancer-specific DNA hypomethylation, we first identified CpG sites that were highly methylated in normal tissues (mean β-value >0.8). We dichotomized the data using a β-value of <0.7 as a threshold for loss of DNA methylation. We then performed unsupervised hierarchical clustering based on CpG sites that had hypomethylation in at least 10% of the tumours. We identified three major clusters. The cluster assignments were generated by cutting the dendrogram. To a great extent, these three clusters correlated well with three molecular subtypes defined using iCluster (Supplemental Figure 3c). Of particular interest is approximately one-third of tumours (largely corresponding to iCluster 3) which appear to have an extreme DNA hypomethylation.
Identification of epigenetically silenced genes
We first removed DNA methylation probes overlapping with SNPs, repeats or designed for sequences on X or Y chromosomes or non-CpG sites. The remaining probes were mapped against UCSC Genes using the GenomicFeatures R/Bioconductor package. Probes that were located in a promoter region (defined as the 3 kb region spanning from 1,500 bp upstream to 1,500 bp downstream of the transcription start site) were identified. Level 3 mRNA expression data were log2 transformed (log2 (RSEM+1)) and used to assess the gene expression levels associated with DNA methylation changes. DNA methylation and gene expression data were merged by Entrez Gene IDs. We used two different approaches to identify genes epigenetically silenced in HCC, as described below.
In the first method, we removed the CpG sites that were methylated in normal tissues (mean β-value >0.2). We then dichotomised the DNA methylation data using a β-value of >0.3 as a threshold for positive DNA methylation and eliminated CpG sites methylated in fewer than 5% of the tumor samples. For each probe/gene pair, we applied the following algorithm: 1) organize the tumors as either methylated (β ≥0.3) or unmethylated (β <0.3); 2) compute the mean expression in the methylated and unmethylated groups; 3) compute the standard deviation of the expression in the unmethylated group. We then selected probes for which the mean expression in the methylated group was less than 1.64 standard deviations from the mean expression of the unmethylated group. We labeled each individual tumor sample as epigenetically silenced for a specific probe/gene pair if: a) it belonged to the methylated group and b) the expression of the corresponding gene was lower than the mean of the unmethylated group of samples. If there were multiple probes associated with the same gene, a sample that was identified as epigenetically silenced at more than half the probes for the corresponding gene was also labeled as epigenetically silenced at the gene level. The complete list of 171 genes identified as epigenetically silenced using this method is provided in Supplemental Table 4A.
In the second approach to identify genes silenced by DNA methylation, we applied a previously described method (Noushmehr et al., 2010). Briefly, Student’s t-tests for significant differences in DNA methylation between tumor and adjacent normal tissue were conducted across all CpG loci located in gene promoter regions. Separately, a t-test was used to identify genes that were expressed at significantly different levels between tumor and adjacent normal tissue. The resulting P values were corrected using the Benjamini-Hochberg procedure. We identified 132 genes significantly hypermethylated (FDR-adjusted P < 0.0001 and mean β value difference >0.1) and down-regulated (FDR-adjusted P < 0.0001 and reduced more than twofold) in tumors. For each gene, we selected the DNA methylation probe with the greatest mean expression difference between methylated (β ≥0.3) and unmethylated (β <0.3) groups. We then estimated the frequency of epigenetic silencing for each gene by counting the number of tumors belonging to the methylated group.
CDKN2A (p16INK4A) epigenetic silencing
CDKN2A epigenetic silencing calls were made using the exon level RNA-seq data. CDKN2A DNA methylation status was assessed in each sample based on the probe (cg13601799) located in the p16INK4 promoter CpG island. p16INK4 expression was determined by the log2(RPKM+1) level of its first exon (chr9:21974403-21975038). The epigenetic silencing calls for each sample were made by evaluating a scatter plot showing an inverse association between DNA methylation and expression.
Leukocyte Methylation Signature
The leukocyte methylation signature was calculated as described in Carter et al. (Carter et al., 2012).
Statistics
Statistical analysis and data visualization were carried out using the R/Biocoductor software packages (http://www.bioconductor.org). Cancer-specific DNA methylation was assessed based on unpaired analyses, since matched normal tissues were available for fewer than 25% of the tumour samples.
Contributors: Toshinori Hinoue, Peter W. Laird.
miRNA sequencing
We generated microRNA sequence (miRNA-seq) data for 189 tumor samples and 47 normals using previously described methods (Cancer Genome Atlas, 2012). To identify miRs that were differentially abundant, we ran unpaired two-class SAMseq analyses, with an FDR threshold of 0.05. We assessed potential miRNA targeting for all 189 samples by calculating miR-mRNA Spearman correlations with MatrixEQTL v2.1.1, using gene-level normalized abundance RNAseq (RSEM) data from Firehose (gdac.broadinstitute.org). We calculated correlations with a P-value threshold of 0.05, then filtered the resulting anticorrelations at FDR<0.05. We then extracted miR-gene pairs that corresponded to functional validation publications reported by MiRTarBase v4.5, for stronger (luciferase reporter, qPCR, Western blot) and weaker experimental evidence types.
We identified groups of samples with similar abundance profiles using unsupervised non-negative matrix factorization (NMF) consensus clustering of reads-per-million (RPM) data for the ~300 (25%) most-variant 5p or 3p miRBase v16 mature strands. We chose a 5-cluster solution based on the peaks of the cophenetic and average silhouette width scores.
Contributors: Reanne Bowlby, Gordon Robertson, Denise Brooks.
S8. mRNA Sequencing
Sequencing and quantification
One g of total RNA was converted to mRNA libraries using the lllumina mRNA TruSeq kit (RS-122-2001 or RS-122-2002) following the manufacturer’s directions. Libraries were sequenced 48×7×48bp on the Illumina HiSeq 2000. FASTQ files were generated by CASAVA. RNA reads were aligned to the hg19 genome assembly using MapSplice 0.7.4 (Wang et al., 2010). Gene expression was quantified for the transcript models corresponding to the TCGA GAF2.1 (http://tcga-data.nci.nih.gov/docs/GAF/GAF.hg19.June2011.bundle/outputs/TCGA.hg19.June2011.gaf), using RSEM and normalized within-sample to a fixed upper quartile. For further details on this processing, refer to Description file at the DCC data portal under the V2_MapSpliceRSEM workflow (https://tcga-data.nci.nih.gov/tcgafiles/ftp_auth/distro_ftpusers/anonymous/tumor/lihc/cgcc/unc.edu/illuminahiseq_rnaseqv2/rnaseqv2/unc.edu_LIHC.IlluminaHiSeq_RNASeqV2.mage-tab.1.15.0/DESCRIPTION.txt). FASTQ and BAM files are at CGHUB (https://cghub.ucsc.edu). Quantification of genes, transcripts, exons and junctions can be found at the TCGA Data Portal (https://tcga-data.nci.nih.gov/tcga/).
mRNA expression clustering
Transcription levels quantified by RSEM were filtered to remove genes whose expression was quantified as zero by RSEM in more than 75% of the tumor samples, reducing the set of genes from 20,531 to 15,951. Gene quantifications were subsequently log2 transformed, with zero values set to missing. To identify genes whose expression was variable, the gene set was filtered to remove genes that demonstrated a standard deviation below 2.0 across all tumor samples, resulting in a set of 1,868 genes with high variability in expression. The log2 transformed expression values were then median centered prior to clustering analysis. Cluster analysis was performed using ConsensusClusterPlus (Wilkerson and Hayes, 2010), using agglomerative hierarchical clustering with a 1-Pearson correlation distances and resampling 80% of the samples for 1000 repetitions. The optimal number of clusters was determined using the empirical cumulative distribution function plot.
Contributors: Eric Seiser, Katherine A. Hoadley.
Reverse-Phase Protein Array
RPPA experiments and data processing
Protein was extracted using RPPA lysis buffer (1% Triton X-100, 50 mmol/L Hepes (pH 7.4), 150 mmol/L NaCl, 1.5 mmol/L MgCl2, 1 mmol/L EGTA, 100 mmol/L NaF, 10 mmol/L NaPPi, 10% glycerol, 1 mmol/L phenylmethylsulfonyl fluoride, 1 mmol/L Na3VO4, and aprotinin 10 ug/mL) from human tumors and RPPA was performed as described previously (Hu et al., 2007). Lysis buffer was used to lyse frozen tumors by Precellys homogenization. Tumor lysates were adjusted to 1 μg/μL concentration as assessed by bicinchoninic acid assay (BCA) and boiled with 1% SDS. Tumor lysates were manually serial diluted in two-fold of 5 dilutions with lysis buffer. An Aushon Biosystems 2470 arrayer (Burlington, MA) printed 1,056 samples on nitrocellulose-coated slides (Grace Bio-Labs). Slides were probed with 202 validated primary antibodies followed by corresponding secondary antibodies (Goat anti-Rabbit IgG, Goat anti-Mouse IgG or Rabbit anti-Goat IgG). Signal was captured using a DakoCytomation-catalyzed system and DAB colorimetric reaction. Slides were scanned in CanoScan 9000F. Spot intensities were analyzed and quantified using Microvigene software (VigeneTech Inc., Carlisle, MA), to generate spot signal intensities (Level1data).The software SuperCurveGUI (Hu et al., 2007), available at http://bioinformatics.mdanderson.org/Software/supercurve/, was used to estimate the EC50 values of the proteins in each dilution series (in log2 scale). Briefly, a fitted curve (“supercurve”) was plotted with the signal intensities on the Y-axis and the relative log2 concentration of each protein on the X-axis using the non-parametric, monotone increasing B-spline model. During the process, the raw spot intensity data were adjusted to correct spatial bias before model fitting. A QC metric was returned for each slide to help determine the quality of the slide: if the score is less than 0.8 on a 0–1 scale, the slide was dropped. In most cases, the staining was repeated to obtain a high quality score. If more than one slide was stained for an antibody, the slide with the highest QC score was used for analysis (Level 2 data). Protein measurements were corrected for loading as described (Hu et al., 2007) using median centering across antibodies (level 3 data). In total, 202 antibodies and 184 samples were used. Final selection of antibodies was also driven by the availability of high quality antibodies that consistently pass a strict validation process as previously described (Hennessy et al., 2010). These antibodies are assessed for specificity, quantification and sensitivity (dynamic range) in their application for protein extracts from cultured cells or tumor tissue. Antibodies are labeled as validated and use with caution based on degree of validation by criteria previously described (Hennessy et al., 2010).
Two RPPA arrays were quantitated and processed (including normalization and load controlling) as described previously, using MicroVigene (VigeneTech, Inc., Carlisle, MA) and the R package SuperCurve (version-1.3), available at http://bioinformatics.mdanderson.org/OOMPA (Hu et al., 2007). Raw data (level 1), SuperCurve nonparameteric model fitting on a single array (level 2), and loading corrected data (level 3) were deposited at the DCC.
Data normalization
We performed median centering across all the antibodies for each sample to correct for sample loading differences. Those differences arise because protein concentrations are not uniformly distributed per unit volume. That may be due to several factors, such as differences in protein concentrations of large and small cells, differences in the amount of proteins per cell, or heterogeneity of the cells comprising the samples. By observing the expression levels across many different proteins in a sample, we can estimate differences in the total amount of protein in that sample vs. other samples. Subtracting the median protein expression level forces the median value to become zero, allowing us to compare protein expressions across samples.
Consensus clustering
We performed consensus hierarchical clustering on the RPPA data. 1-Pearson correlation was used as the distance metric and Ward was used as a linkage algorithm. The consensus clustering method clustered the samples and counted how frequently two samples were in the same cluster. The bootstrap resampling analysis identified two robust sample clusters. A total of 184 samples and 202 antibodies were used in the analysis.
Contributors: Rehan Akbani, Shiyun Ling, Zhenlin Ju, Yiling Lu, Gordon Mills.
Integrative Clustering using iCluster
To understand the subgroups formed by integrating various molecular platforms of HCC, we utilized iCluster, which formulates the problem of subgroup discovery as a joint multivariate regression of multiple data types with reference to a set of common latent variables that represent the underlying tumor subtypes (Mo et al., 2013).
Data processing
Five molecular platforms, DNA copy number, DNA methylation, mRNA expression, miRNA expression and RPPA were provided as input to iCluster. Data were pre-processed using the following procedures. Copy number alteration data was derived from CBS segmented data from the Affymetrix SNP6.0 platform, and further reduced to a set of non-redundant regions as described (Mo et al., 2013). For the methylation data (Illumina Infinium 450k arrays), the median absolute deviation was employed to select the top 1000 most variable CpG sites after beta-mixture quantile normalization. Methylation probes with >20% or more missing data and those corresponding to SNP and autosomal chromosomes were removed. For mRNA and miRNA sequence data, lowly expressed genes were excluded based on median-normalized counts, and variance filtering led to 1266 mRNAs and 258 miRNAs for clustering. mRNA and miRNA expression features were log2 transformed, normalized and scaled before using as an input to iCluster.
Model selection
The optimal combination of clusters was determined minimizing a Bayesian Information Criterion (BIC). An ‘elbow’ point was noted at K=3, beyond which the BIC kept increasing and thus the 3-class solution was chosen.
Supplemental Figure 3C shows that the results were highly comparable for individual unsupervised clustering versus integrative clustering, indicating that the iCluster groupings represented the combined information of all platforms and lacked bias to a particular data type.
To compare the resultant iCluster groupings to the molecular subclasses developed by Hoshida (Hoshida et al., 2009), we assigned each of our patients to one of the three Hoshida subclasses using their transcriptional predictors. We found strong concordance between the iClusters and the Hoshida subclasses (see Supplemental Table 6).
Clinical Significance of iClusters
We sought to compare the TCGA iClusters to the subtypes found by Hoshida (Hoshida et al., 2009). To accomplish this, we used gene the expression signatures from Hoshida et al. and used K-mean clustering to group TCGA tissues and assigned membership of tissues according to subtype signature from the original study. Of 619 genes defined by Hoshida et al., expression of 610 genes were available in TCGA mRNA RNA-seq data. TCGA tissues were subgrouped by K-mean cluster (k = 3) and subclasses were assigned according to their expression patterns of subclass signature. These assignments stratified each iCluster grouping to one of 3 Hoshida et al. subclasses (Figure 3B).
To compare the TCGA iClusters to other published studies, we constructed a subtype prediction model using data from the TCGA cohort. For selection of subtype-specific gene sets, multiple 2-class t tests were performed for all possible combinations of the 3 subtypes. Gene expression differences were considered statistically significant if the P value was less than 0.001. Only genes with significant differences in expression in all 2 possible comparisons were considered subtype-specific genes, yielding 1442 significant genes for the iCluster1 subtype, 128 for the iCluster2 subtype, and 329 for the iCluster3 subtype. The top 200 significant genes in iCluster1 and iCluster3 subtypes and 128 genes for the iCluster2 subtype were further selected for development of the prediction model.
To develop a subtype prediction model, we adopted a previously developed model using Bayesian compound covariate predictor algorithms. Briefly, gene expression data for each subtype gene signature (i.e., the 200 significant genes for each subtype, as described above) were used to generate the Bayesian probability of each tissue sample belonging to a particular subtype, generating 3 probability scores for each tumor. Samples in the test cohorts were assigned to 1 of the 3 subtypes according to the highest probability scores.
When the prediction model was applied to the MDACC cohort (n = 100), the iC1 subtype was associated with the worst prognosis and the iC2 and iC3 subtype was associated with the better prognosis (Figure 3C). Consistent with the MDACC, the iC1 subtype was associated with worst prognosis in NCI and Fudan cohorts (see Figure 3C).
Contributors: Arshi Arora, Ronglai Shen, Ju-Seog Lee.
IDH1/2 and IDH1/2-like Mutant Signature
IDH1/2 mutation signature
Four tissues (TCGA-CC-5260, TCGA-DD-A4NA, TCGA-ED-A82E, and TCGA-G3-A25T) had mutations in IDH1 or IDH2 (two mutations in IDH1 (R132C, R132G) and two mutations in IDH2 (R172K, R172S)). Two-sample t-test were carried out to uncover mRNAs differentially expressed between mutant and wildtype HCC tissues and identified 1009 genes (P < 0.0001). Interestingly, several tissues without IDH mutations had highly similar mRNA expression patterns (Figure 4A). When Bayesian compound covariate predictor (BCCP) algorithm (Radmacher et al., 2002) was applied to mRNA expression data to stratify the HCC tissues according to similarity to IDH mutation expression signature, 11 tissues without IDH1/2 mutations were classified into IDH-like subtype (Probability < 2 in range from 0 to 4 in log2 scale).
Stratification of TCGA HCC tissues by known molecular subtypes
To assess concordance between TCGA subtypes and previously identified molecular subtypes, HCC tissues in TCGA cohort were stratified according to molecular signatures from previous studies. Eight tumor-derived prognostic signatures were used to comparison: NCI proliferation (NCIP) signature (Lee et al., 2004), hepatic stem cells (HS) signatures (Lee et al., 2006), Seoul National University recurrence (SNUR) signature (Woo et al., 2008), cholangiocarcinoma-like (CCL) signature (Woo et al., 2010), hepatoblastoma 16 gene (HB16) signature (Cairo et al., 2008), Hippo pathway signature (Sohn et al., 2015), Hoshida signature (Hoshida et al., 2009), and 65-gene risk scores for recurrence (RS65) (Kim et al., 2012). Except for Hoshida signature and RS65 scores, BCCP algorithm was applied to stratify TCGA tumor tissues by using previously defined gene sets and original gene expression data as training set. For stratification according to Hoshida signature, ConsensusClusterPlus package in R (v2.13.2) (Wilkerson and Hayes, 2010) was used to group tissues into three subtypes. RS65 risk scores were was calculated by using recurrence score algorithms as described in a previous study (Kim et al., 2012). Briefly, the risk score for each patient was derived by multiplying the expression level of a gene with its corresponding coefficient (Risk score = sum of Cox coefficient of Gene Gi X expression value of Gene Gi). The risk scores were rescaled 0 to 100 to make 0 as the lowest risk score. Patients were then stratified into two prognostic subtypes (high risk >40). Significance of association between molecular subtypes with IDH1/2 signature was estimated by χ2-test (Figure 4A).
IDH1/2 mutation signature and clinical significance
We next tested the clinical relevance of patients with IDH1/2 mutant and IDH-like HCC by applying IDH mutation expression signature to gene expression data from three independent human HCC cohorts. Gene expression data of 100 HCC tumors generated from a University of Texas MD Anderson Cancer Center (MDACC) (Kim et al., 2012; Sohn et al., 2015) were first used for this analysis. Briefly, a BCCP algorithm was applied to generate probability of IDH mutation signature in each of the human HCC tumors as previously described (Lee et al., 2006; Sohn et al., 2015). When the HCC patients were dichotomized according to IDH1/2 mutation signature probability (Figure 4B), patients with IDH1/2 mutation signature (IDH-like) had significantly worse prognosis than those without IDH1/2 mutation signature (WT) (P = 1.0 × 10−4, Figure 4C), strongly indicating that IDH1/2 mutations or their activation in HCC may dictate clinical outcome and is associated with poor prognosis. The significant association of IDH1/2 mutation signature with worse prognosis was further validated in two independent cohorts (National Cancer Institute (NCI) cohort and Fudan University cohort) (Figure 4C).
miR-122-5p in IDH-like/mut and miR-122 gene targets
We identified miRs that were differentially abundant between IDH-like/IDH-mutant samples and IDH wild type by nonparametric unpaired two-class analysis (Supplemental Figure 4a). Liver-specific miR-122-5p (Supplemental Figure 4B), which is known to be downregulated in HCCs, was strikingly less abundant in the IDH-like/IDH-mutant group (Supplemental Figure 4A). We assessed potential gene targets of this miR through miR-mRNA anticorrelations for n=189 samples (FDR<0.05). The table shows the top 30 significant (FDR<0.05) anticorrelations with miR-122-5p that have been published as validated targets (Supplemental Figure 4D). We noted that miR-122-5p was strongly anticorrelated to PKM2, the M2 isoform of the pyruvate kinase (PK) (rho=−0.62).
Also of note is miR-885-5p with the second largest negative fold change. miR-885-5p is significantly anticorrelated with a number of functionally validated direct targets including CCDC46 (also known as MCM5) (rho=−0.40, FDR=1.2e-06), TP53 (rho=−0.30, FDR=6e-04), CDK2 (rho=−0.29, FDR=0.001) and CTNNB1 (rho=−0.27, FDR=0.003). Alternately, a number of miR-200 family members (miR-200a-5p, 200b-3p and 429) were significantly more abundant in the IDH-like/IDH-mutant group.
Contributors: Lisa Iype, Reanne Bowlby, Toshinori Hinoue, Jae-Jun Shim, Bo Hwa Sohn, Ju-Seog Lee.
p53 Signature
The TCGA HCC tumors with complete exome sequence data, copy number data, and expression data (n=191) were initially stratified by TP53 mutation status. All HCC with TP53 non-synonymous missense, frameshift, nonsense, splice sites, and indels (n = 60) were compared to HCC without TP53 mutations (n = 131) for RNA expression of 20,531 analyzed genes. An unpaired t test was then performed on the expression values for each gene in the two TP53 categories. T test p values for each gene were then ranked from lowest to highest and the gene list cross-indexed with a manually curated list of 155 experimentally validated p53 transcriptional target genes. We identified 30 known p53 target genes that were significantly upregulated (p < 0.005) in WT TP53 HCC compared to MUT TP53 HCC. From these 30 genes we chose 20 p53 target genes that were known to be frequently upregulated in other cancer types with WT TP53 relative to MUT TP53, as described in a previous publication(Parikh et al., 2014). These 20 genes composed the p53 signature.
The HCC were then segregated by p53 signature. To do this, the RNA expression values for each of the 20 target genes were ranked from 1 to 191 across the HCC samples. The expression ranks for all 20 target genes were then summed and the HCC ranked by 20 gene score totals. For many analyses, the HCC quartile with the lowest summed scores (low p53 signature) were compared to the quartile with the highest summed scores (high p53 signature). The ranking of the HCC by signature score and expression of each of the p53 target genes is shown in Figure 5A. We also examined 10 p53 repressed target genes for each of the HCC and the relative expression levels of these 10 genes are also shown in Figure 5a.
For the log-rank survival analyses in Supplemental Figure 5 we utilized the available followup survival data on the TCGA HCC dataset and stratified the HCC by high and low p53 signature quartiles and an intermediate quartile composed of the second and third ranked p53 signature quartiles. The same analysis was performed in three external cohorts: a 242 HCC patient cohort from Fudan, China; a 100 HCC patient cohort from M.D. Anderson Cancer Center; a 113 HCC patient cohort from the National Cancer Institute.
To examine the association of p53 signature status and molecular/clinical correlates we performed unpaired t tests comparing the high and low p53 signature quartile values that had parameters measured by continuous variables (e.g. recurrence risk score, MDM4 expression, MDM4 copy number). For discrete variables we used a chi-square test to compare the values in the high and low p53 signature quartiles (e.g. ploidy, HBV status, tumor grade). P values are shown for individual clinical and molecular parameters at the top of Figure 5A.
Contributor: Lawrence A. Donehower.
Pathway-Associated High Impact Gene Mutations
For all mutations in the HCC cohort, the Evolutionary Action (EA) method1 was applied to predict the functional impact of missense mutations. Nonsense mutations received a heuristic score of maximal EA impact. To identify individual genes with a strong EA mutational bias, we compared the distribution of each gene’s EA scores to that of the cancer as a whole using a one-sided two-sample Kolmogorov Smirnov test. Genes with an FDR-corrected q-value<0.05 were deemed significant single-gene results.
We used the Reactome pathway database (v49) to define groups of functionally related genes. Reactome is hosted by the European Bioinformatics Institute, encompasses 7,498 genes across 1580 pathways, and represents high-quality, manually-curated pathway information. Using all mutated genes that were not significant in single-gene analysis, we identified for each Reactome pathway the set of genes that maximized bias toward high EA mutations using leave-one-out analysis. Sets that exhibited significant bias (q<0.05) after FDR correction and were also more significant than at least 95% of 1,000 size-matched pathway simulations were considered to be of interest. Sets of interest were then ranked by their fold improvement over the threshold set by the simulations (Supplemental Figure 7, Supplemental Table 7).
Contributors: Amanda Koire, Panagiotis Katsonis, Teng-Kuei Hsu, Olivier Lichtarge.
Immune Signature
The normalized RNA-seq gene expression data and the CIBORSORT cellular composition data was downloaded from TCGA Synapse (Syn4976369 and Syn7337221, respectively). The unsupervised hierarchical clustering of gene expression was done using the Next-Generation (Clustered) Heat Maps (NG-CHM, http://bioinformatics.mdanderson.org/chm).
Contributor: Linghua Wang.
Interactive Exploration
To gain greater insight into the development and progression of hepatocellular carcinoma, we have integrated all of the data types produced by TCGA and described in this paper into a single “feature matrix”. From this single heterogeneous dataset, significant pairwise associations have been inferred using statistical analysis and can be visually explored in a genomic context using Regulome Explorer, an interactive web application (http://explorer.cancerregulome.org). In addition to associations that are inferred directly from the TCGA data, additional sources of information and tools are integrated into the visualization for more extensive exploration (e.g., NCBI Gene, miRBase, the UCSC Genome Browser, etc).
Feature Matrix Construction
A feature matrix was constructed using all available clinical, sample, and molecular data for 196 unique patient/tumor samples. The clinical information includes features such as age and tumor size; while the sample information includes features derived from molecular data such as single-platform cluster assignments. The molecular data includes mRNA and microRNA expression levels (Illumina HiSeq data), protein levels (RPPA data), copy number alterations (derived from segmented Affymetrix SNP data as well as GISTIC regions of interest and arm-level values), DNA methylation levels (Illumina Infinium Methylation 450k array), and somatic mutations. For mRNA expression data, gene level RSEM values from RNA-seq were log2 transformed, and filtered to remove low-variability genes (bottom 25% removed, based on interdecile range). For miRNA expression data, the summed and normalized microRNA quantification files were log2 transformed, and filtered to remove low-variability microRNAs (bottom 25% removed, based on interdecile range). For methylation data, probes were filtered to remove the bottom 25% based on interdecile range. For somatic mutations, several binary mutation features indicating the presence or absence of a mutation in each sample were generated. Mutation types considered were synonymous, missense, nonsense and frameshift. Protein domains (InterPro) including any of these mutation types were annotated as such, with nonsense and frameshift annotations being propagated to all subsequent protein domains.
Pairwise Statistical Significance
Statistical association among the diverse data types in this study was evaluated by comparing pairs of features in the feature matrix. Hypothesis testing was performed by testing against null models for absence of association, yielding a p-value. P-values for the association between and among clinical and molecular data types were computed according to the nature of the data levels for each pair: categorical vs. categorical (Chi-square test or Fisher’s exact test in the case of a 2×2 table); categorical vs. continuous (Kruskal-Wallis test) or continuous vs. continuous (probability of a given Spearman correlation value). Ranked data values were used in each case. To account for multiple-testing bias, the p-value was adjusted using the Bonferroni correction.
Exploring significant feature associations
Regulome Explorer allows the user to interactively explore significant associations between various types of features – associations between molecular features (like methylation and gene expression), associations between molecular features and derived numeric features (like RS65 Score), and associations between molecular features and categorical features such as clinical features or clusters derived from prior analysis (like iCluster).
Contributors: Lisa Iype, Sheila M. Reynolds.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification methods and statistical analysis methods for each of the various data platforms and for integrated analyses are described and referenced in their respective Method Detail subsections.
DATA AND SOFTWARE AVAILABILITY
The TCGA HCC (LIHC) clinical data and raw data from the individual platform data (DNA exome sequencing data, RNA expression data, miRNA expression data, DNA methylation data, copy number data, and RPPA proteomics data) are archived in the Genomic Data Commons https://portal.gdc.cancer.gov/legacy-archive/search/f. At this web page select from the Project listing the “TCGA-LIHC”. Results files generated from these archives can be found in at the Synapse archive web site in ID syn2318326 found at the following URL: https://www.synapse.org/#!Synapse:syn2318326/files/.
Access to patient genetic data is controlled by dbGaP. Permission to access is granted through the https://dbgap.ncbi.nlm.nih.gov/aa/wga.cgi?page=login. Software used for the analyses for each of the data platforms and integrated analyses are described and referenced in the individual Method Detail subsections and listed in the Key Resources Table.
KEY RESOURCES TABLE
Supplementary Material
Figure S1 (related to Figure 1). TERT alterations and exomic mutational signatures in HCC. (A) TERT promoter mutations, gene amplification, and fusion RNAs likely to result in increased TERT RNA expression occur in over 50% of the core 196 HCC dataset. These are indicated in the top bar. The second and third bars indicate that CDKN2A RNA expression is frequently reduced by both promoter hypermethylation (second bar) and deep deletion (third bar). Chi-square analysis showed that CDKN2A silencing is highly correlated with TERT alterations (p = 8.1 × 10−5). (B) Hierarchical clustering of normalized signature activity in the core 196 HCC dataset. Hierarchical clustering of the normalized signature activity in TCGA LIHC identified the aristolochic acid (AA) cluster of 9 cases with the enriched Sig.A activity (COSMIC 22) and the aflatoxin B1 (AFB1) cluster of 25 cases affected by the Sig.B (COSMIC 24) activity. Six out of top seven samples with the highest Sig.B activity in the AFB1 cluster were overlapping with the AFB1-affected samples identified by the independent mutation signature analysis (Schulze et el., 2015, Nature Genet. 47:505-511). (C) Boxplot of AFB1-signature associated mutations in TP53-R249S (known AFB1 hotspot site) mutant samples, other TP53 mutant samples, and TP53 wild-type samples reveals a significant enrichment of AFB1 activity in samples harboring TP53-R249S mutations (P=0.0052 vs. other TP53 mutant group and 0.00011 vs. TP53 wild-type group, Wilcoxon rank-sum test).
Figure S2 (related to Figure 1). HCC copy number alterations and their molecular and clinical correlates. (A) In the heat map at the right part of the panel, copy number losses or gains (see legend at right) in individual tumors (vertical axis) are plotted by chromosomal location (horizontal axis). Tumors were hierarchically clustered by significantly reoccurring copy number alterations identified by GISTIC 2.0 analysis of the entire dataset. Vertical sidebars (left) show the cluster designations, patient information, gene mutations and other clinical and molecular correlates. (B) GISTIC 2.0 amplifications and deletions across all HCC. Chromosomal locations of peaks of significantly recurring focal amplifications (red) and deletions (blue) are plotted by false discovery rates. Annotated peaks have an FDR < .2 and encompass 50 or fewer genes. Peaks are annotated with candidate driver oncogenes, tumor suppressors, fragile site genes (green) or by cytoband. The number of genes within each peak is shown next to driver genes or cytobands.
Figure S3 (related to Figure 3). iCluster correlation with molecular, demographic and clinical features of HCC. (A) Prediction signatures for iCluster subtypes of HCC in the TCGA dataset. iCluster subtype-specific gene expression signatures were identified by applying multiple t tests (P < 0.001). Among the significant genes in each subtype, the top 200 genes were selected for development of prediction models (all 128 significant genes were used for the iC2 subtype). Data are presented in matrix format in which each row represents an individual gene and each column represents a tissue sample. Each cell in the matrix represents the expression level of a gene feature in an individual tissue sample. The coloring in the cells reflects relatively high (red) and low (green) expression levels, as indicated in the scale bar (log2 transformed scale). The three transcription-based prediction models were used to classify patients from published studies. (B) Correlation of iCluster groupings of patients with demographic and clinical categories. (C) Each patient was assigned to a molecular cluster category from the indicated individual platform. Colored tiles indicate the cluster membership of the patient within a given molecular platform. To the right are shown p values for the distribution of each platform cluster within the iCluster groupings. (D) Differential expression of selected miRNAs and genes by iCluster analysis.
Figure S4 (related to Figure 4). miR-122-5p, IDH-like/mut vs wild type and miR-122 gene targeting. (A) The top 15 significantly up and down regulated miRNAs in 15 IDH-like/IDH-mutant samples relative to IDH wildtype samples. For each miRNA, the bar plot shows the fold change between the medians and the boxplot shows the log10 abundance between the two groups. (B) miR-122-5p abundance in tumor and normal samples from a range of TCGA tissue types. (C) Scatter plot correlating miRNA-122 expression with hypermethylation of its promoter (representative CpG site cg00481280). Note a fraction of HCC (bottom right) with high methylation and low expression (red dots indicate the five most hypermethylated tumors). (D) The top 30 significant (FDR<0.05) miRNA-mRNA anti-correlations that have been functionally validated as direct targets of miR-122-5p.
Figure S5 (related to Figure 5). HCC with low p53 target expression signatures display poorer prognosis. (A) TCGA HCC (192 patients) in the lowest quartile of p53 target gene expression were compared for overall survival to those in the highest quartile of p53 target gene expression by log-rank survival analyses. (B) An external cohort of 242 HCC patients from Fudan, China were compared for survival of patients with high vs. low p53 signatures as described for panel a. (C) A 100 HCC patient cohort from M.D. Anderson Cancer Center was compared by p53 low and high signature quartiles for overall survival. (D) A 113 HCC patient cohort from the National Cancer Institute was compared by p53 low and high signature quartiles for overall survival.
Figure S6 (related to Figure 6). Stratification of HCC pathways and gene alterations by iCluster categories. (A) Pathway stratification by iCluster categories. Shown are percentages of samples in each subgroup that have at least one alteration in any of the genes associated to each pathway (as shown in Fig. 6). Cells are shaded green in proportion to their percentage of alteration. (B) Driver gene stratification by iCluster categories. The oncoprint summarizes co- occurrence and mutual exclusivity of some of the most relevant somatic alterations across samples and iClusters. “Deep deletions” correspond to cases where more than half of the copies of a gene are deleted from the baseline ploidy. Missense mutations are only included if they have known oncogenic function, have previously been reported in COSMIC, or occur at known mutational hotspots.
Figure S7 (related to Figure 6). Enrichment of functionally important gene mutations within specific HCC signaling pathways. Mutation Action Profiles (MAPs) in HCC. (A) Mutation Action Profiles (MAPs) of two representative genes not under positive selection. Nonsynonymous mutations are binned in deciles by their Action score, with higher scores indicating a larger functional impact. Absolute counts of mutations in each bin are represented by the height of the bin. (B) MAPs of TP53 and CTNNB1, two individual genes under strong positive selection. (C-H) MAPs of selected gene sets under positive selection collectively. Each component gene is represented by a color and plots are labeled with the Reactome pathway associated with the gene set.
Supplemental Table 1A. Clinical and molecular attributes of TCGA HCC
Supplemental Table 1B. Descriptive table of clinical and histological features
Supplemental Table 1C. Extended HCC data set (exome sequencing and copy number data)
Supplemental Table 2A. Somatic gene mutations in 363 TCGA HCC
Supplemental Table 2B. Significantly mutated HCC genes identified across 10 genome sequencing studies (and other studies)
Supplemental Table 3. TERT promoter mutations and their correlates in HCC
Supplemental Table 4A. Hypermethylated genes in HCC identified by two methods
Supplemental Table 4B. GSEA Analysis of hypermethylated genes in HCC
Supplemental Table 5A. HBV Infection status and clinical and molecular parameters
Supplemental Table 5B. RNA-Seq HBV integration events detected at both the BC Cancer Agency (BBT) and Broad Institute (PathSeq)
Supplemental Table 5C. Identification of HBV RNA fusions with likely cancer driver activities in 196 TCGA HCC
Supplemental Table 6. Summary of iCluster molecular and clinical correlates
Supplemental Table 7. Pathways in HCC with enriched frequencies of functionally impactful mutations in pathway component genes
Highlights.
Analysis of hepatocellular carcinomas integrates data of multiple genomic platforms
Mutated genes reveal oncogenic processes altering hepatocyte energy balance
Multiplex analyses suggest a key role for Sonic hedgehog signaling in HCC
IDH mutations point to a HCC subgroup molecularly similar to cholangiocarcinoma
Acknowledgments
We are grateful to the patients who contributed to this study. This work was supported by these grants from the U.S. National Institutes of Health: 5U24CA143799, 5U24CA143835, 5U24CA143840, 5U24CA143843, 5U24CA143845, 5U24CA143848, 5U24CA143858, 5U24CA143866, 5U24CA143867, 5U24CA143882, 5U24CA143883, 5U24CA144025, U54HG003067, U54HG003079, U54HG003273, and P30CA16672.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
The TCGA consortium contributed collectively to this study. Biospecimens were provided by the Tissue Source Sites and processed by the Biospecimen Core Resource. Data generation and analyses were performed by the Genome Sequencing Center, Genome Characterization Centers, and Genome Data Analysis centers. All data were released through the Data Coordinating Center. Project activities were coordinated by the National Cancer Institute and National Human Genome Research Institute Project Teams. Initial guidance in the project design was provided by the Disease Working Group. We acknowledge the following TCGA investigators of the Analysis Working Group, who contributed substantially to the analysis and writing of this manuscript: Project Leaders: David A. Wheeler and Lewis R. Roberts; data coordinator: Chad J. Creighton; manuscript coordinators: Lawrence A. Donehower, Renu Dhanasekaran, and Lisa Iype; analysis coordinator: Ju-Seog Lee; writing team: Lawrence A. Donehower, David A. Wheeler, Lewis R. Roberts., Renu Dhanasekaran, Ju-Seog Lee, Lisa Iype, Ina Felau; DNA sequence analysis: Jaegil Kim, Kyle Covington, David A. Wheeler; mRNA analysis: Katherine A. Hoadley; miRNA analysis: Reanne Bowlby, A. Gordon Robertson; DNA methylation analysis: Toshinori Hinoue and Peter W. Laird; HBV/HCV analysis: Reanne Bowlby, Betty Slagle, Akinyemi Ojesina, Pedamallu Chandra Sekhar; copy-number analysis: Andrew D. Cherniak; pathway analysis: Francisco Sanchez-Vega, Lawrence A. Donehower, Ju-Seog Lee, and Lisa Iype; immunophenotyping: Linghua Wang; clinical data: Renu Dhanasekaran; pathology and clinical expertise: Michael S. Torbenson, Matthew M. Yeh, Sanjay Kakar, Dhanpat Jain, Hala Makhlouf.
There were no identified conflicts of interest by any of the authors.
See accompanying Excel file labeled “LIHC manuscript author list”.
Supplemental Tables: See Excel files labeled “Supplemental Tables-5-8-2017”
References
- Ahn SM, Jang SJ, Shim JH, Kim D, Hong SM, Sung CO, Baek D, Haq F, Ansari AA, Lee SY, et al. Genomic portrait of resectable hepatocellular carcinomas: implications of RB1 and FGF19 aberrations for patient stratification. Hepatology. 2014;60:1972–1982. doi: 10.1002/hep.27198. [DOI] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmar J, Fesik SW. Small molecule Mcl-1 inhibitors for the treatment of cancer. Pharmacol Ther. 2015;145:76–84. doi: 10.1016/j.pharmthera.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98:288–295. doi: 10.1016/j.ygeno.2011.07.007. [DOI] [PubMed] [Google Scholar]
- Bonnal S, Vigevani L, Valcarcel J. The spliceosome as a target of novel antitumour drugs. Nat Rev Drug Discov. 2012;11:847–859. doi: 10.1038/nrd3823. [DOI] [PubMed] [Google Scholar]
- Borger DR, Tanabe KK, Fan KC, Lopez HU, Fantin VR, Straley KS, Schenkein DP, Hezel AF, Ancukiewicz M, Liebman HM, et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist. 2012;17:72–79. doi: 10.1634/theoncologist.2011-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairo S, Armengol C, De Reynies A, Wei Y, Thomas E, Renard CA, Goga A, Balakrishnan A, Semeraro M, Gresh L, et al. Hepatic stem-like phenotype and interplay of Wnt/beta-catenin and Myc signaling in aggressive childhood liver cancer. Cancer Cell. 2008;14:471–484. doi: 10.1016/j.ccr.2008.11.002. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J, Sadeghi S, Raymond A, Jackman SD, Nip KM, Mar R, Mohamadi H, Butterfield YS, Robertson AG, Birol I. BioBloom tools: fast, accurate and memory-efficient host species sequence screening using bloom filters. Bioinformatics. 2014;30:3402–3404. doi: 10.1093/bioinformatics/btu558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung JH, Larsen AR, Chen E, Bunz F. A PTCH1 homolog transcriptionally activated by p53 suppresses Hedgehog signaling. J Biol Chem. 2014;289:33020–33031. doi: 10.1074/jbc.M114.597203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, McKenna A, Fennell T, Banks E, DePristo M, Getz G. ContEst: estimating cross-contamination of human samples in next-generation sequencing data. Bioinformatics. 2011;27:2601–2602. doi: 10.1093/bioinformatics/btr446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulouarn C, Factor VM, Andersen JB, Durkin ME, Thorgeirsson SS. Loss of miR-122 expression in liver cancer correlates with suppression of the hepatic phenotype and gain of metastatic properties. Oncogene. 2009;28:3526–3536. doi: 10.1038/onc.2009.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanasekaran R, Bandoh S, Roberts LR. Molecular pathogenesis of hepatocellular carcinoma and impact of therapeutic advances. F1000Res. 2016;5 doi: 10.12688/f1000research.6946.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egusa G, Beltz WF, Grundy SM, Howard BV. Influence of obesity on the metabolism of apolipoprotein B in humans. J Clin Invest. 1985;76:596–603. doi: 10.1172/JCI112011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo M, Yasui K, Zen Y, Gen Y, Zen K, Tsuji K, Dohi O, Mitsuyoshi H, Tanaka S, Taniwaki M, et al. Alterations of the SWI/SNF chromatin remodelling subunit-BRG1 and BRM in hepatocellular carcinoma. Liver Int. 2013;33:105–117. doi: 10.1111/liv.12005. [DOI] [PubMed] [Google Scholar]
- Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- Fernandez-Banet J, Lee NP, Chan KT, Gao H, Liu X, Sung WK, Tan W, Fan ST, Poon RT, Li S, et al. Decoding complex patterns of genomic rearrangement in hepatocellular carcinoma. Genomics. 2014;103:189–203. doi: 10.1016/j.ygeno.2014.01.003. [DOI] [PubMed] [Google Scholar]
- Frattini V, Trifonov V, Chan JM, Castano A, Lia M, Abate F, Keir ST, Ji AX, Zoppoli P, Niola F, et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet. 2013;45:1141–1149. doi: 10.1038/ng.2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto A, Furuta M, Totoki Y, Tsunoda T, Kato M, Shiraishi Y, Tanaka H, Taniguchi H, Kawakami Y, Ueno M, et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat Genet. 2016 doi: 10.1038/ng.3547. [DOI] [PubMed] [Google Scholar]
- Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, Calderaro J, Bioulac-Sage P, Letexier M, Degos F, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44:694–698. doi: 10.1038/ng.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennessy BT, Lu Y, Gonzalez-Angulo AM, Carey MS, Myhre S, Ju Z, Davies MA, Liu W, Coombes K, Meric-Bernstam F, et al. A Technical Assessment of the Utility of Reverse Phase Protein Arrays for the Study of the Functional Proteome in Non-microdissected Human Breast Cancers. Clin Proteomics. 2010;6:129–151. doi: 10.1007/s12014-010-9055-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K, et al. TERT promoter mutations in familial and sporadic melanoma. Science. 2013;339:959–961. doi: 10.1126/science.1230062. [DOI] [PubMed] [Google Scholar]
- Hoshida Y, Nijman SM, Kobayashi M, Chan JA, Brunet JP, Chiang DY, Villanueva A, Newell P, Ikeda K, Hashimoto M, et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009;69:7385–7392. doi: 10.1158/0008-5472.CAN-09-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, He X, Baggerly KA, Coombes KR, Hennessy BT, Mills GB. Nonparametric quantification of protein lysate arrays. Bioinformatics. 2007;23:1986–1994. doi: 10.1093/bioinformatics/btm283. [DOI] [PubMed] [Google Scholar]
- Jochemsen AG. Reactivation of p53 as therapeutic intervention for malignant melanoma. Curr Opin Oncol. 2014;26:114–119. doi: 10.1097/CCO.0000000000000033. [DOI] [PubMed] [Google Scholar]
- Karagozian R, Derdak Z, Baffy G. Obesity-associated mechanisms of hepatocarcinogenesis. Metabolism. 2014;63:607–617. doi: 10.1016/j.metabol.2014.01.011. [DOI] [PubMed] [Google Scholar]
- Kasar S, Kim J, Improgo R, Tiao G, Polak P, Haradhvala N, Lawrence MS, Kiezun A, Fernandes SM, Bahl S, et al. Whole-genome sequencing reveals activation-induced cytidine deaminase signatures during indolent chronic lymphocytic leukaemia evolution. Nat Commun. 2015;6:8866. doi: 10.1038/ncomms9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsonis P, Lichtarge O. A formal perturbation equation between genotype and phenotype determines the Evolutionary Action of protein-coding variations on fitness. Genome Res. 2014;24:2050–2058. doi: 10.1101/gr.176214.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SM, Leem SH, Chu IS, Park YY, Kim SC, Kim SB, Park ES, Lim JY, Heo J, Kim YJ, et al. Sixty-five gene-based risk score classifier predicts overall survival in hepatocellular carcinoma. Hepatology. 2012;55:1443–1452. doi: 10.1002/hep.24813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyono T, Foster SA, Koop JI, McDougall JK, Galloway DA, Klingelhutz AJ. Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature. 1998;396:84–88. doi: 10.1038/23962. [DOI] [PubMed] [Google Scholar]
- Kostic AD, Ojesina AI, Pedamallu CS, Jung J, Verhaak RG, Getz G, Meyerson M. PathSeq: software to identify or discover microbes by deep sequencing of human tissue. Nat Biotechnol. 2011;29:393–396. doi: 10.1038/nbt.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn RM, Haussler D, Kent WJ. The UCSC genome browser and associated tools. Brief Bioinform. 2013;14:144–161. doi: 10.1093/bib/bbs038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Chu IS, Heo J, Calvisi DF, Sun Z, Roskams T, Durnez A, Demetris AJ, Thorgeirsson SS. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology. 2004;40:667–676. doi: 10.1002/hep.20375. [DOI] [PubMed] [Google Scholar]
- Lee JS, Heo J, Libbrecht L, Chu IS, Kaposi-Novak P, Calvisi DF, Mikaelyan A, Roberts LR, Demetris AJ, Sun Z, et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat Med. 2006;12:410–416. doi: 10.1038/nm1377. [DOI] [PubMed] [Google Scholar]
- Liu AM, Xu Z, Shek FH, Wong KF, Lee NP, Poon RT, Chen J, Luk JM. miR-122 targets pyruvate kinase M2 and affects metabolism of hepatocellular carcinoma. PLoS One. 2014;9:e86872. doi: 10.1371/journal.pone.0086872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Dong H, Robertson K, Liu C. DNA methylation suppresses expression of the urea cycle enzyme carbamoyl phosphate synthetase 1 (CPS1) in human hepatocellular carcinoma. Am J Pathol. 2011;178:652–661. doi: 10.1016/j.ajpath.2010.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llovet JM, Hernandez-Gea V. Hepatocellular carcinoma: reasons for phase III failure and novel perspectives on trial design. Clin Cancer Res. 2014;20:2072–2079. doi: 10.1158/1078-0432.CCR-13-0547. [DOI] [PubMed] [Google Scholar]
- Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao J, Yu H, Wang C, Sun L, Jiang W, Zhang P, Xiao Q, Han D, Saiyin H, Zhu J, et al. Metallothionein MT1M is a tumor suppressor of human hepatocellular carcinomas. Carcinogenesis. 2012;33:2568–2577. doi: 10.1093/carcin/bgs287. [DOI] [PubMed] [Google Scholar]
- McCarroll SA, Kuruvilla FG, Korn JM, Cawley S, Nemesh J, Wysoker A, Shapero MH, de Bakker PI, Maller JB, Kirby A, et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat Genet. 2008;40:1166–1174. doi: 10.1038/ng.238. [DOI] [PubMed] [Google Scholar]
- McPherson A, Hormozdiari F, Zayed A, Giuliany R, Ha G, Sun MG, Griffith M, Heravi Moussavi A, Senz J, Melnyk N, et al. deFuse: an algorithm for gene fusion discovery in tumor RNA-Seq data. PLoS Comput Biol. 2011;7:e1001138. doi: 10.1371/journal.pcbi.1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo Q, Wang S, Seshan VE, Olshen AB, Schultz N, Sander C, Powers RS, Ladanyi M, Shen R. Pattern discovery and cancer gene identification in integrated cancer genomic data. Proc Natl Acad Sci U S A. 2013;110:4245–4250. doi: 10.1073/pnas.1208949110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okoye-Okafor UC, Bartholdy B, Cartier J, Gao EN, Pietrak B, Rendina AR, Rominger C, Quinn C, Smallwood A, Wiggall KJ, et al. New IDH1 mutant inhibitors for treatment of acute myeloid leukemia. Nat Chem Biol. 2015;11:878–886. doi: 10.1038/nchembio.1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olshen AB, Venkatraman ES, Lucito R, Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–572. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- Parikh N, Hilsenbeck S, Creighton CJ, Dayaram T, Shuck R, Shinbrot E, Xi L, Gibbs RA, Wheeler DA, Donehower LA. Effects of TP53 mutational status on gene expression patterns across 10 human cancer types. J Pathol. 2014;232:522–533. doi: 10.1002/path.4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E, Kim N, Ficarro SB, Zhang Y, Lee BI, Cho A, Kim K, Park AK, Park WY, Murray B, et al. Structure and mechanism of activity-based inhibition of the EGF receptor by Mig6. Nat Struct Mol Biol. 2015;22:703–711. doi: 10.1038/nsmb.3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pez F, Lopez A, Kim M, Wands JR, Caron de Fromentel C, Merle P. Wnt signaling and hepatocarcinogenesis: molecular targets for the development of innovative anticancer drugs. J Hepatol. 2013;59:1107–1117. doi: 10.1016/j.jhep.2013.07.001. [DOI] [PubMed] [Google Scholar]
- Piotrowski A, Xie J, Liu YF, Poplawski AB, Gomes AR, Madanecki P, Fu C, Crowley MR, Crossman DK, Armstrong L, et al. Germline loss-of-function mutations in LZTR1 predispose to an inherited disorder of multiple schwannomas. Nat Genet. 2014;46:182–187. doi: 10.1038/ng.2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto J, Melero I, Sangro B. Immunological landscape and immunotherapy of hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2015;12:681–700. doi: 10.1038/nrgastro.2015.173. [DOI] [PubMed] [Google Scholar]
- Radenbaugh AJ, Ma S, Ewing A, Stuart JM, Collisson EA, Zhu J, Haussler D. RADIA: RNA and DNA integrated analysis for somatic mutation detection. PLoS One. 2014;9:e111516. doi: 10.1371/journal.pone.0111516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radmacher MD, McShane LM, Simon R. A paradigm for class prediction using gene expression profiles. J Comput Biol. 2002;9:505–511. doi: 10.1089/106652702760138592. [DOI] [PubMed] [Google Scholar]
- Ramos AH, Lichtenstein L, Gupta M, Lawrence MS, Pugh TJ, Saksena G, Meyerson M, Getz G. Oncotator: cancer variant annotation tool. Hum Mutat. 2015;36:E2423–2429. doi: 10.1002/humu.22771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid A, Wang JS, Qian GS, Lu BX, Hamilton SR, Groopman JD. Genetic alterations in hepatocellular carcinomas: association between loss of chromosome 4q and p53 gene mutations. Br J Cancer. 1999;80:59–66. doi: 10.1038/sj.bjc.6690321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler S, Jia HL, Budhu A, Forgues M, Ye QH, Lee JS, Thorgeirsson SS, Sun Z, Tang ZY, Qin LX, et al. A unique metastasis gene signature enables prediction of tumor relapse in early-stage hepatocellular carcinoma patients. Cancer Res. 2010;70:10202–10212. doi: 10.1158/0008-5472.CAN-10-2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruden M, Puri N. Novel anticancer therapeutics targeting telomerase. Cancer Treat Rev. 2013;39:444–456. doi: 10.1016/j.ctrv.2012.06.007. [DOI] [PubMed] [Google Scholar]
- Saigo K, Yoshida K, Ikeda R, Sakamoto Y, Murakami Y, Urashima T, Asano T, Kenmochi T, Inoue I. Integration of hepatitis B virus DNA into the myeloid/lymphoid or mixed-lineage leukemia (MLL4) gene and rearrangements of MLL4 in human hepatocellular carcinoma. Hum Mutat. 2008;29:703–708. doi: 10.1002/humu.20701. [DOI] [PubMed] [Google Scholar]
- Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28:1811–1817. doi: 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- Schulze K, Imbeaud S, Letouze E, Alexandrov LB, Calderaro J, Rebouissou S, Couchy G, Meiller C, Shinde J, Soysouvanh F, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet. 2015;47:505–511. doi: 10.1038/ng.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I. ABySS: a parallel assembler for short read sequence data. Genome Res. 2009;19:1117–1123. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn BH, Shim JJ, Kim SB, Jang KY, Kim SM, Kim JH, Hwang JE, Jang HJ, Lee HS, Kim SC, et al. Inactivation of Hippo pathway is significantly associated with poor prognosis in hepatocellular carcinoma. Clin Cancer Res. 2015 doi: 10.1158/1078-0432.CCR-15-1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song LN, Gelmann EP. Silencing mediator for retinoid and thyroid hormone receptor and nuclear receptor corepressor attenuate transcriptional activation by the beta-catenin-TCF4 complex. J Biol Chem. 2008;283:25988–25999. doi: 10.1074/jbc.M800325200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soussi T. The TP53 gene network in a postgenomic era. Hum Mutat. 2014;35:641–642. doi: 10.1002/humu.22562. [DOI] [PubMed] [Google Scholar]
- Stransky N, Cerami E, Schalm S, Kim JL, Lengauer C. The landscape of kinase fusions in cancer. Nat Commun. 2014;5:4846. doi: 10.1038/ncomms5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan VY, Fevotte C. Automatic relevance determination in nonnegative matrix factorization with the beta-divergence. IEEE Trans Pattern Anal Mach Intell. 2013;35:1592–1605. doi: 10.1109/TPAMI.2012.240. [DOI] [PubMed] [Google Scholar]
- Torres-Garcia W, Zheng S, Sivachenko A, Vegesna R, Wang Q, Yao R, Berger MF, Weinstein JN, Getz G, Verhaak RG. PRADA: pipeline for RNA sequencing data analysis. Bioinformatics. 2014;30:2224–2226. doi: 10.1093/bioinformatics/btu169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totoki Y, Tatsuno K, Covington KR, Ueda H, Creighton CJ, Kato M, Tsuji S, Donehower LA, Slagle BL, Nakamura H, et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat Genet. 2014;46:1267–1273. doi: 10.1038/ng.3126. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- Volkel P, Dupret B, Le Bourhis X, Angrand PO. Diverse involvement of EZH2 in cancer epigenetics. Am J Transl Res. 2015;7:175–193. [PMC free article] [PubMed] [Google Scholar]
- Wang K, Singh D, Zeng Z, Coleman SJ, Huang Y, Savich GL, He X, Mieczkowski P, Grimm SA, Perou CM, et al. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 2010;38:e178. doi: 10.1093/nar/gkq622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, Weisenberger DJ, Campan M, Young J, Jacobs I, et al. Epigenetic stem cell signature in cancer. Nat Genet. 2007;39:157–158. doi: 10.1038/ng1941. [DOI] [PubMed] [Google Scholar]
- Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010;26:1572–1573. doi: 10.1093/bioinformatics/btq170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, Mollaee M, Wagner KU, Koduru P, Yopp A, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun. 2015;6:6744. doi: 10.1038/ncomms7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo HG, Lee JH, Yoon JH, Kim CY, Lee HS, Jang JJ, Yi NJ, Suh KS, Lee KU, Park ES, et al. Identification of a cholangiocarcinoma-like gene expression trait in hepatocellular carcinoma. Cancer Res. 2010;70:3034–3041. doi: 10.1158/0008-5472.CAN-09-2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo HG, Park ES, Cheon JH, Kim JH, Lee JS, Park BJ, Kim W, Park SC, Chung YJ, Kim BG, et al. Gene expression-based recurrence prediction of hepatitis B virus-related human hepatocellular carcinoma. Clin Cancer Res. 2008;14:2056–2064. doi: 10.1158/1078-0432.CCR-07-1473. [DOI] [PubMed] [Google Scholar]
- Xu D, Qu L, Hu J, Li G, Lv P, Ma D, Guo M, Chen Y. Transmembrane protein 106A is silenced by promoter region hypermethylation and suppresses gastric cancer growth by inducing apoptosis. J Cell Mol Med. 2014;18:1655–1666. doi: 10.1111/jcmm.12352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JD, Roberts LR. Hepatocellular carcinoma: A global view. Nat Rev Gastroenterol Hepatol. 2010;7:448–458. doi: 10.1038/nrgastro.2010.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Weng CC, Tong M, Wei J, Tai HH. Restoration of leukotriene B(4)-12-hydroxydehydrogenase/15- oxo-prostaglandin 13-reductase (LTBDH/PGR) expression inhibits lung cancer growth in vitro and in vivo. Lung Cancer. 2010;68:161–169. doi: 10.1016/j.lungcan.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Zeng W, Gai X, Xu Q, Li C, Liang Z, Tuo H, Liu Q. Role of the Hedgehog pathway in hepatocellular carcinoma (review) Oncol Rep. 2013;30:2020–2026. doi: 10.3892/or.2013.2690. [DOI] [PubMed] [Google Scholar]
- Zucman-Rossi J, Villanueva A, Nault JC, Llovet JM. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology. 2015;149:1226–1239.e1224. doi: 10.1053/j.gastro.2015.05.061. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 (related to Figure 1). TERT alterations and exomic mutational signatures in HCC. (A) TERT promoter mutations, gene amplification, and fusion RNAs likely to result in increased TERT RNA expression occur in over 50% of the core 196 HCC dataset. These are indicated in the top bar. The second and third bars indicate that CDKN2A RNA expression is frequently reduced by both promoter hypermethylation (second bar) and deep deletion (third bar). Chi-square analysis showed that CDKN2A silencing is highly correlated with TERT alterations (p = 8.1 × 10−5). (B) Hierarchical clustering of normalized signature activity in the core 196 HCC dataset. Hierarchical clustering of the normalized signature activity in TCGA LIHC identified the aristolochic acid (AA) cluster of 9 cases with the enriched Sig.A activity (COSMIC 22) and the aflatoxin B1 (AFB1) cluster of 25 cases affected by the Sig.B (COSMIC 24) activity. Six out of top seven samples with the highest Sig.B activity in the AFB1 cluster were overlapping with the AFB1-affected samples identified by the independent mutation signature analysis (Schulze et el., 2015, Nature Genet. 47:505-511). (C) Boxplot of AFB1-signature associated mutations in TP53-R249S (known AFB1 hotspot site) mutant samples, other TP53 mutant samples, and TP53 wild-type samples reveals a significant enrichment of AFB1 activity in samples harboring TP53-R249S mutations (P=0.0052 vs. other TP53 mutant group and 0.00011 vs. TP53 wild-type group, Wilcoxon rank-sum test).
Figure S2 (related to Figure 1). HCC copy number alterations and their molecular and clinical correlates. (A) In the heat map at the right part of the panel, copy number losses or gains (see legend at right) in individual tumors (vertical axis) are plotted by chromosomal location (horizontal axis). Tumors were hierarchically clustered by significantly reoccurring copy number alterations identified by GISTIC 2.0 analysis of the entire dataset. Vertical sidebars (left) show the cluster designations, patient information, gene mutations and other clinical and molecular correlates. (B) GISTIC 2.0 amplifications and deletions across all HCC. Chromosomal locations of peaks of significantly recurring focal amplifications (red) and deletions (blue) are plotted by false discovery rates. Annotated peaks have an FDR < .2 and encompass 50 or fewer genes. Peaks are annotated with candidate driver oncogenes, tumor suppressors, fragile site genes (green) or by cytoband. The number of genes within each peak is shown next to driver genes or cytobands.
Figure S3 (related to Figure 3). iCluster correlation with molecular, demographic and clinical features of HCC. (A) Prediction signatures for iCluster subtypes of HCC in the TCGA dataset. iCluster subtype-specific gene expression signatures were identified by applying multiple t tests (P < 0.001). Among the significant genes in each subtype, the top 200 genes were selected for development of prediction models (all 128 significant genes were used for the iC2 subtype). Data are presented in matrix format in which each row represents an individual gene and each column represents a tissue sample. Each cell in the matrix represents the expression level of a gene feature in an individual tissue sample. The coloring in the cells reflects relatively high (red) and low (green) expression levels, as indicated in the scale bar (log2 transformed scale). The three transcription-based prediction models were used to classify patients from published studies. (B) Correlation of iCluster groupings of patients with demographic and clinical categories. (C) Each patient was assigned to a molecular cluster category from the indicated individual platform. Colored tiles indicate the cluster membership of the patient within a given molecular platform. To the right are shown p values for the distribution of each platform cluster within the iCluster groupings. (D) Differential expression of selected miRNAs and genes by iCluster analysis.
Figure S4 (related to Figure 4). miR-122-5p, IDH-like/mut vs wild type and miR-122 gene targeting. (A) The top 15 significantly up and down regulated miRNAs in 15 IDH-like/IDH-mutant samples relative to IDH wildtype samples. For each miRNA, the bar plot shows the fold change between the medians and the boxplot shows the log10 abundance between the two groups. (B) miR-122-5p abundance in tumor and normal samples from a range of TCGA tissue types. (C) Scatter plot correlating miRNA-122 expression with hypermethylation of its promoter (representative CpG site cg00481280). Note a fraction of HCC (bottom right) with high methylation and low expression (red dots indicate the five most hypermethylated tumors). (D) The top 30 significant (FDR<0.05) miRNA-mRNA anti-correlations that have been functionally validated as direct targets of miR-122-5p.
Figure S5 (related to Figure 5). HCC with low p53 target expression signatures display poorer prognosis. (A) TCGA HCC (192 patients) in the lowest quartile of p53 target gene expression were compared for overall survival to those in the highest quartile of p53 target gene expression by log-rank survival analyses. (B) An external cohort of 242 HCC patients from Fudan, China were compared for survival of patients with high vs. low p53 signatures as described for panel a. (C) A 100 HCC patient cohort from M.D. Anderson Cancer Center was compared by p53 low and high signature quartiles for overall survival. (D) A 113 HCC patient cohort from the National Cancer Institute was compared by p53 low and high signature quartiles for overall survival.
Figure S6 (related to Figure 6). Stratification of HCC pathways and gene alterations by iCluster categories. (A) Pathway stratification by iCluster categories. Shown are percentages of samples in each subgroup that have at least one alteration in any of the genes associated to each pathway (as shown in Fig. 6). Cells are shaded green in proportion to their percentage of alteration. (B) Driver gene stratification by iCluster categories. The oncoprint summarizes co- occurrence and mutual exclusivity of some of the most relevant somatic alterations across samples and iClusters. “Deep deletions” correspond to cases where more than half of the copies of a gene are deleted from the baseline ploidy. Missense mutations are only included if they have known oncogenic function, have previously been reported in COSMIC, or occur at known mutational hotspots.
Figure S7 (related to Figure 6). Enrichment of functionally important gene mutations within specific HCC signaling pathways. Mutation Action Profiles (MAPs) in HCC. (A) Mutation Action Profiles (MAPs) of two representative genes not under positive selection. Nonsynonymous mutations are binned in deciles by their Action score, with higher scores indicating a larger functional impact. Absolute counts of mutations in each bin are represented by the height of the bin. (B) MAPs of TP53 and CTNNB1, two individual genes under strong positive selection. (C-H) MAPs of selected gene sets under positive selection collectively. Each component gene is represented by a color and plots are labeled with the Reactome pathway associated with the gene set.
Supplemental Table 1A. Clinical and molecular attributes of TCGA HCC
Supplemental Table 1B. Descriptive table of clinical and histological features
Supplemental Table 1C. Extended HCC data set (exome sequencing and copy number data)
Supplemental Table 2A. Somatic gene mutations in 363 TCGA HCC
Supplemental Table 2B. Significantly mutated HCC genes identified across 10 genome sequencing studies (and other studies)
Supplemental Table 3. TERT promoter mutations and their correlates in HCC
Supplemental Table 4A. Hypermethylated genes in HCC identified by two methods
Supplemental Table 4B. GSEA Analysis of hypermethylated genes in HCC
Supplemental Table 5A. HBV Infection status and clinical and molecular parameters
Supplemental Table 5B. RNA-Seq HBV integration events detected at both the BC Cancer Agency (BBT) and Broad Institute (PathSeq)
Supplemental Table 5C. Identification of HBV RNA fusions with likely cancer driver activities in 196 TCGA HCC
Supplemental Table 6. Summary of iCluster molecular and clinical correlates
Supplemental Table 7. Pathways in HCC with enriched frequencies of functionally impactful mutations in pathway component genes