

Abstract
Objective
Strategies improving skeletal muscle mitochondrial capacity are commonly paralleled by improvements in (metabolic) health. We and others previously identified microRNAs regulating mitochondrial oxidative capacity, but data in skeletal muscle are limited. Therefore, the present study aimed to identify novel microRNAs regulating skeletal muscle mitochondrial metabolism.
Methods and results
We conducted an unbiased, hypothesis-free microRNA silencing screen in C2C12 myoblasts, using >700 specific microRNA inhibitors, and investigated a broad panel of mitochondrial markers. After subsequent validation in differentiated C2C12 myotubes, and exclusion of microRNAs without a human homologue or with an adverse effect on mitochondrial metabolism, 19 candidate microRNAs remained. Human clinical relevance of these microRNAs was investigated by measuring their expression in human skeletal muscle of subject groups displaying large variation in skeletal muscle mitochondrial capacity.
Conclusion
The results show that that microRNA-320a, microRNA-196b-3p, microRNA-150-5p, and microRNA-34c-3p are tightly related to in vivo skeletal muscle mitochondrial function in humans and identify these microRNAs as targets for improving mitochondrial metabolism.
Keywords: Skeletal muscle metabolism, Mitochondria, MicroRNA, Screening, Type 2 diabetes, Oxidative capacity
Graphical abstract
Highlights
-
•
A microRNA screen identifies microRNAs modulating muscle mitochondrial metabolism.
-
•
Identified microRNAs correlate with in vivo muscle mitochondrial function in humans.
-
•
MicroRNAs are potential targets to improve skeletal muscle mitochondrial metabolism.
1. Introduction
Mitochondria are the primary regulators of cellular metabolism. These organelles efficiently generate useable energy in the form of ATP from different substrates via oxidative phosphorylation. As such, mitochondria play a critical role in many energy-requiring cellular processes such as apoptosis, autophagy, metabolic pathways, substrate oxidation, Ca2+ homeostasis, and aging [1], [2]. Indeed, decreased mitochondrial function also plays an important role in numerous pathologies such as cardiomyopathy, cancer, neuro-muscular degeneration, Alzheimer's disease, and type 2 diabetes mellitus (T2DM) [3], [4], [5]. A diminished mitochondrial oxidative capacity in skeletal muscle tissue has frequently been reported in humans with T2DM and insulin resistance [6], [7], [8]. More importantly, interventions that improve muscle mitochondrial oxidative capacity, such as exercise training, caloric restriction, and resveratrol supplementation have been shown to result in an improved metabolic health [9], [10], [11]. However, increasing physical activity and reducing caloric intake, the most potent strategies to improve mitochondrial oxidative capacity, are not easily implemented by a mostly overweight or obese population. This increases the need for additional strategies to improve mitochondrial function.
MicroRNAs (miRNAs) are small non-coding RNA strands of approximately 20–22 nucleotides that provide a fascinating mechanism in coordinating complex programs of gene expression. miRNAs regulate the stability and translation of conventional messenger RNAs by base pairing with protein-coding transcripts [12]. As such, miRNAs add an additional level of regulating gene expression and protein output [13], [14], [15], [16] and have been demonstrated to regulate numerous processes including cell function, development, and even aging [13], [17]. An abnormal expression of miRNAs may also contribute to pathophysiological phenotypes such as diabetes, cardiovascular disease, cancer, and neurological disorders [18]. Intriguingly, miRNAs have been discovered that are located inside mitochondria (mitomiRs) [19], and evidence for an active role of miRNAs in the regulation of multiple aspects of mitochondrial function is accumulating [20], [21], [22]. In this context, miRNAs have been reported to be involved in the regulation of mitochondrial biogenesis, energy metabolism, and electron transport chain subunits [23], [24], [25].
In addition, we recently showed that miRNA-199a and miRNA-214, two miRNAs that were markedly up regulated during myocardial hypoxia and elevated in cardiac biopsies from human heart failure patients, actively repress PPARδ, a well-known transcription factor in the regulation of mitochondrial metabolism [26], [27]. Interestingly, in vivo silencing of these miRNAs in an animal model with specific antagomirs (RNA-like oligonucleotides reverse complement to mature miRNAs) resulted in a restoration of PPARδ levels, a normalization of mitochondrial fatty acid oxidation and even a rescue of cardiac failure [27]. However, although some reports exist, little is known about the role of miRNAs in the regulation of mitochondrial metabolism in skeletal muscle, a pivotal tissue in maintaining metabolic health.
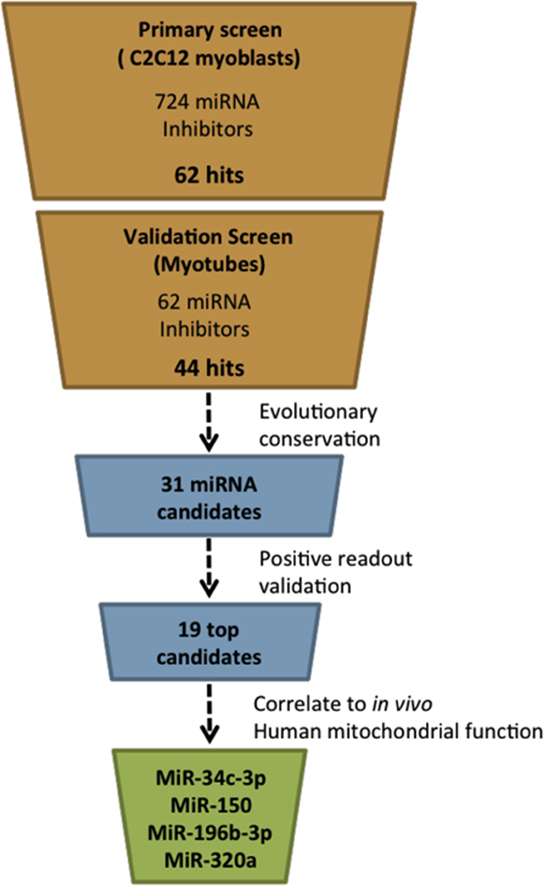
We previously demonstrated the efficacy of a high-throughput screening method to identify compounds targeting different aspects of mitochondrial function [28]. In the present study, we performed a similar unbiased, hypothesis-free screening approach in C2C12 myoblasts, using specific miRNA inhibitors. Top candidates were validated in fully differentiated C2C12 myotubes, leading to the identification of 19 specific, fully conserved miRNAs as positive modulators of mitochondrial metabolism when silenced. To render results relevant to humans, we next quantified the expression of these miRNAs in skeletal muscle biopsies of endurance-trained athletes, lean and obese sedentary subjects, and type 2 diabetic patients and found that the expression of 4 of our validated miRNAs showed a strong relationship with in vivo mitochondrial function in humans.
2. Results
2.1. Optimization of C2C12 myoblast cell culture, transfection conditions and primary screen read-outs
For the primary screening of 724 miRNA inhibitors, we used the C2C12 myoblast cell line and first sought to ensure that the myoblasts would favor mitochondrial oxidative phosphorylation over glycolysis for their energy production. Thus, we compared a high glucose growth medium (HG) with a low glucose medium supplemented with oleic acid (LGO) and subsequently measured redox potential using the AlamarBlue® dye, which is sensitive to the NAD+/NADH ratio, and extra-cellular acidification rate (ECAR), a marker for lactate production. Both redox potential and ECAR were substantially decreased when using LGO medium, by 33% and 57% respectively (Figure 1A), indicating that the LGO medium induced a metabolic shift from glycolysis to oxidative phosphorylation. It should be mentioned that Alamar blue is also used as a cell proliferation assay [29]. However, since C2C12 myoblasts are a fast proliferating cell line, we are confident that the observed decrease is due to a change in redox potential and not due to low proliferation rates. Additionally, cellular ATP levels were similar between the two media types, indicating that the LGO medium is not detrimental for cell viability (Figure 1A). Therefore, the LGO medium was used for all screening purposes in C2C12 myoblasts.
Figure 1.

Optimizing cell culture and transfection conditions in C2C12 myoblasts. (A) Redox potential, ATP level and extra-cellular acidification rate (ECAR) measured after 24 h of incubation in different media conditions. A reduction in glucose concentration and supplementation with oleic acid shifts cells towards a more oxidative metabolism (n = 4). (B) miRNA-206 luciferase reporter assay 24 h post-transfection with varying concentrations of miRNA-206 targeting locked nucleic acids (LNA). 150 nM of LNA results in the highest level of miRNA-206 silencing (n = 4). (C) Luciferase reporter assay for heat shock protein 60 (HSP60) transcription factor activity, corrected for renilla luciferase activity, after 24 h of doxycycline (15 μg/mL) treatment (n = 8). (D) Luciferase reporter assay for cytochrome C oxidase subunit 4 (COX4) transcription factor activity, corrected for renilla luciferase activity, after 24 h of resveratrol (1 μM) treatment (n = 8). Statistical significance was assessed using one-way ANOVA with Dunnett's correction for multiple testing, and is indicated with *, **, and *** representing p < 0.05, p < 0.01, and p < 0.001, respectively. Graphs represent mean ± SEM.
Next, we used a luciferase reporter plasmid with a binding site for the muscle-specific miRNA-206, a miRNA involved in muscle development and function [30], to determine the optimal transfection conditions for miRNA silencing using locked nucleic acids (LNAs; short antisense oligonucleotides that are able to bind and target specific miRNAs). Transfection of C2C12 myoblasts with increasing concentrations of anti-miRNA-206 LNA resulted in a dose-dependent silencing of the targeted miRNA, with an optimal inhibitory concentration of 150 nM (Figure 1B). Furthermore, using a similar approach, we confirmed via miRNA qPCR that transfection with 150 nM of anti-miRNA-2016 LNA resulted in a robust and optimal miRNA silencing in C2C12 myoblasts (Supplemental Figure 1). Therefore, this LNA concentration was subsequently used in the miRNA silencing screen in C2C12 myoblasts.
To identify miRNAs able to regulate mitochondrial metabolism in skeletal muscle, we included 5 mitochondrial readouts. ATP level, redox potential and mitochondrial membrane potential (ΔΨm) were already optimized in a previous study [28]. Here, we added luciferase reporters for heat shock protein 60 (HSP60), a marker of mitochondrial unfolded protein response (UPRmt) [31], and for cytochrome c oxidase subunit 4 (COX4), a mitochondrial respiratory chain subunit, used here as a marker of mitochondrial biogenesis. Doxycycline, a tetracycline antibiotic that inhibits mitochondrial protein synthesis and thereby induces UPRmt [31], was used as a positive control for the HSP60 luciferase assay. The natural compound resveratrol, known to induce mitochondrial biogenesis [11], [32], [33], was used as a positive control for the COX4 luciferase assay. Twenty-four hours incubation with these two positive controls increased the luciferase activity of HSP60 and COX4 reporters by 1.9 and 2.8-fold, respectively, underscoring the sensitivity of these luciferase reporter assays (Figure 1C and D).
2.2. A miRNA silencing screen in C2C12 myoblasts
We transfected C2C12 myoblasts with a miRNA inhibitor library (miRCURY LNA™), containing 724 individual mouse miRNA inhibitors (Supplemental data file 1). The 5 mitochondrial readouts were measured 24-hours post-transfection. Good discriminative power was achieved for ATP level, redox potential and ΔΨm, with Z-factors of 0.76, 0.52, and 0.64, respectively (Figure 2A). The luciferase reporter assays had Z-scores below 0, indicating a lower discriminative power, i.e. these assays could result in more false positive and negative readouts. There was no significant correlation between the different readouts, demonstrating that these assays were non-redundant and complementary to each other (Figure 2B and C and Ref. [28]).
Figure 2.

Screening of 724 miRNAs in C2C12 myoblasts. (A–C) Screening of 724 miRNA inhibitors, using 5 functional readouts representing different aspects of mitochondrial function and oxidative cell culture conditions, i.e. low glucose concentration (1 g/L) and 1% oleic acid. Cells were plated at 2000 cells per well with the density being nearly confluent (90%) during the day of the assays. (A) Bandwidth separation of the cell-based assays. Plots represent the cumulative density of Z-scores of the raw values for all the points of the screening, including a positive control (benzethonium chloride). The ATP level assay had the best separation capacity with narrow peaks of the positive control and screening values on the density plot and a Z factor close to 1. (B–C) Correlations between 2 luciferase based assays show a poor level of relationship between the other parameters. (B) Pearson's ρ correlation coefficients and their corresponding regression r2 value were calculated for the normalized ratios of every parameter. (C) Spearman's ρ correlation coefficient was calculated for the correlation between rank orders of every parameter.
miRNAs were considered hits when they induced signal changes superior to 2 standard deviations of the negative control (negative control A ± 2σNegative control A), while also deviating from a normal distribution, i.e. when the observed quantile deviates from the theoretical quantile (Figure 3A and B). After applying this two-step approach, the selection requirements were met by 62 miRNAs, reflecting an average hit rate of 1.7% per assay. Hits were visualized using hierarchical clustering to show the different profiles of the hits, i.e. the combination of all mitochondrial parameters (Figure 3C).
Figure 3.

Hit determination of miRNA screen in C2C12 myoblasts. (A) Graphs represent the normalized ratio of all the readouts, calculated as described in the method section. The black line indicates the limit of miRNA inhibitors that induced a change superior to Negative control A ± 2σNegative control A, and the red line the limit of miRNA inhibitors that induced a change superior to Negative control A ± 3σNegative control A. (B) Graphs represent quantile–quantile (QQ) plots, plotting the theoretical quantiles for each assay over the normalized ratio. miRNA inhibitors were considered hits when they induced changes of at least A ± 2σNegative control A and deviated from the normal distribution. The gray points correspond to the compounds inducing a change inferior to Negative control A ± 2σNegative control A, the black points to a change comprised between the Negative control A ± 2σNegative control A and A ± 3σNegative control A, and the red points to a change superior to and A ± 3σNegative control A. (C) Hierarchical clustering of the 5 parameters for the 62 hits. miRNA inhibitors were considered hits when they induced changes of at least A ± 2σNegative control A and deviated from the normal distribution.
2.3. Validation of identified miRNA hits in C2C12 myotubes
In order to validate the identified miRNAs, we next performed a secondary screen with the 62 miRNA hits in fully differentiated C2C12 myotubes, a more advanced muscle cell model that resembles the post-mitotic adult skeletal muscle more closely. The smaller scale of this secondary screen allowed us to include additional functional mitochondrial readouts, i.e. basal and maximal cellular oxygen consumption rates, and additional time points (24 and 48-hours post-transfection). miRNAs were considered as hits when inducing signal changes superior to negative control A ± 2σNegative control A (Figure 4A).
Figure 4.

Hit determination and hierarchical clustering of miRNA validation in C2C12 myotubes. (A) A subset of all readouts is depicted; graphs represent the normalized ratio of all the readouts, calculated as described in the methods section. The black line indicates the limit of miRNA inhibitors that induced a change superior to negative control A ± 2σNegative control A, and the red line the limit of miRNA inhibitors that induced a change superior to Negative control A ± 3σNegative control A. (B) Hierarchical clustering of the 5 parameters for the 44 hits. miRNA inhibitors were considered hits when they induced changes of at least A ± 2σNegative control A.
This secondary screen in myotubes confirmed 44 miRNAs as hits (Figure 4), i.e. miRNAs that, when silenced, induced a significant change in any of the mitochondrial read-outs at any time point post-transfection. Hierarchical clustering was used to identify groups of miRNAs that have similar effects on the different parameters (Figure 4B). Interestingly, 15 of the hits belonged to only 4 miRNA families; i.e. the miRNA-476 family (miRNAs 466f, 466 g, 466i, 466j, 467f, and 669o), the miRNA-17 family (miRNAs 18a-3p, 20a, 93-3p, and 106b-3p), the miRNA-34 family (miRNAs 34a-3p, 34b, and 34c-3p) and the miRNA-154 family (miRNAs 381 and 382). All members of the miRNA-17 and miRNA-476 families, except for miRNA-669o, induced HSP60 promoter activity, suggesting that the loss of these miRNAs resulted in the activation of UPRmt. Members of the miRNA-154 family were clustered together closely in Figure 4 and induced HSP60 promoter activity, albeit only 48 h post-transfection (Figure 4B).
Finally, we carefully assessed all the 44 miRNA hits and excluded those that were not evolutionary conserved in humans. Additionally, hits without a significant positive effect on any of the mitochondrial read-outs in the validation tests were also excluded. The resulting set of 19 miRNAs was subjected to further analyses.
2.4. miRNA-34c-3p, miRNA-150-5p, miRNA-196b and miRNA-320a correlate to in vivo mitochondrial function in human skeletal muscle
In order to translate our results to human skeletal muscle, we first aimed to identify human subjects with different mitochondrial capacity and metabolic health. Thus, we selected (from previous studies) four subject groups known to be associated with different mitochondrial abundance and oxidative capacity, including type 2 diabetic patients (T2DM), obese non-diabetic subjects (O), lean sedentary individuals (LS) and endurance-trained athletes (A) [34], [35], [36], [37]. In these subjects, we measured two markers of in vivo mitochondrial function, i.e. maximal aerobic capacity (VO2max) and phosphocreatine (PCr) recovery rate [34], [35], [36], [37].
VO2max was significantly different between all groups, with the exception of T2DM vs. obese, non-diabetic subjects (Table 1). The athlete group had a 2.44-fold (p < 0.001), 2.15-fold (p < 0.001) and 1.44-fold (p < 0.001) higher VO2max compared to T2DM, obese and lean sedentary subjects respectively, while lean sedentary subjects displayed a 1.7-fold (p < 0.001) higher VO2max compared to T2DM subjects. In addition, athletes had a 1.39-fold (p < 0.05) and 1.73-fold (p < 0.001) shorter PCr recovery time compared to obese and T2DM subjects, respectively (Table 1). Finally, T2DM had a 1.25-fold (p < 0.05) and 1.33-fold (p < 0.05) longer PCr recovery time compared to obese and lean sedentary subjects, respectively. Together these results show that, as anticipated, mitochondrial muscle capacity was poorest in T2DM and greatest in athletes.
Table 1.
Subject characteristics. Values presented are mean ± SEM (n = 12 per group). Statistical differences depicted represent changes between the different groups. Significance was assessed using one-way ANOVA with Tukey's correction for multiple testing, and is indicated with #, $, * and § representing changes compared to T2DM, obese, lean sedentary and athletic subjects respectively, and with 1, 2, and 3 symbols representing p < 0.05, p < 0.01, and p < 0.001 respectively.
| T2DM# | Obese$ | Lean untrained∗ | Athletes§ | |
|---|---|---|---|---|
| Subjects | 12 | 12 | 12 | 12 |
| Age (yrs) | 58.64 ± 1.22∗∗∗,§§§ | 57 ± 2.29∗∗∗,§§§ | 21.91 ± 0.72###,$$$ | 25.08 ± 1.25###,$$$ |
| BMI (kg/m2) | 32.37 ± 1.11∗∗∗,§§§ | 30.83 ± 1.14∗∗∗,§§§ | 21.84 ± 0.57###,$$$ | 20.98 ± 0.44###,$$$ |
| Body fat (%) | 34.41 ± 1.76∗∗∗,§§§ | 34.31 ± 2.17∗∗∗,§§§ | 19.0 ± 1.03###,$$$,§ | 12.76 ± 0.60###,$$$,∗ |
| VO2max (ml/min/kg) | 24.55 ± 1.24∗∗∗,§§§ | 27.91 ± 1.36∗∗∗,§§§ | 41.70 ± 0.50###,$$$,§§§ | 59.97 ± 1.20###,$$$,∗∗∗ |
| PCR recovery (s) | 26.95 ± 1.79∗,§§§ | 21.61 ± 1.35§ | 20.22 ± 1.44# | 15.58 ± 1.56###,$ |
| FPG (mmol/L) | 7.68 ± 0.21 | 5.56 ± 0.09### | 5.00 ± 0.09### | 5.13 ± 0.08### |
We next determined the expression of the 19 identified candidate miRNAs in skeletal muscle biopsies of these subjects and found that 8 were differently expressed between groups (Table 2). Thus, miRNA-196b-3p was expressed lowest in the athletes compared to all other groups, and was 1.70-fold (p < 0.01), 1.57-fold (p < 0.05) and 1.55-fold (p < 0.05) higher in T2DM, obese and lean sedentary subjects respectively (Figure 5A). Furthermore, miRNA-320a was 1.32-fold (p < 0.05) lower in athletic compared to T2DM subjects. Next, miRNA-150-5p was 1.68-fold (p < 0.05) higher expressed in athletes compared to the obese/overweight subjects (Figure 5A). In addition, miRNA-106b-3p was expressed 1.37-fold (p < 0.05) lower in athletes compared to obese subjects, miRNA-19a-3p expression was 1.89-fold (p < 0.05) lower in athletes compared to obese subjects, and miRNA-21-3p expression was 1.77-fold (p < 0.01) and 1.65-fold (p < 0.05) lower in athletes compared to T2DM and obese subjects respectively. Furthermore, miRNA-30a-3p was expressed 1.38-fold (p < 0.01) and 1.35-fold (p < 0.05) higher in athletes and lean sedentary compared to obese subjects respectively, and lastly, Let-7f-5p expression was 1.31-fold (p < 0.05) lower in athletes compared to T2DM subjects (Supplemental Figure 2).
Table 2.
miRNA expression per group. Values presented are average fold change ± SEM (n = 12 per group), MicroRNA expression levels were quantified using the 2-ΔΔCt method, normalized against housekeeping microRNA-423, U6 and SNORD48 gene expression and expressed relatively to microRNA expression of a lean subject. Statistical differences depicted represent relative changes between the different groups. Significance was assessed using non-parametric one-way ANOVA with Sidak's correction for multiple testing. Significance is indicated with #, $, * and § representing changes compared to T2DM, obese, lean sedentary and athletic subjects respectively, and with 1 and 2 symbols representing p < 0.05 and p < 0.01 respectively. miRNAs marked with n/d were non-detectable.
| miRNA | T2DM# | Obese$ | Lean untrained∗ | Athletes§ |
|---|---|---|---|---|
| hsa-miR-106b-3p | 1.52 ± 0.17 | 1.45 ± 0.08§ | 1.33 ± 0.10 | 1.06 ± 0.08$ |
| hsa-miR-130b-5p | 1.09 ± 0.21 | 0.91 ± 0.13 | 1.03 ± 0.15 | 0.79 ± 0.08 |
| hsa-miR-196a-3p | n/d | n/d | n/d | n/d |
| hsa-miR-19a-3p | 1.90 ± 0.39 | 1.75 ± 0.24§ | 1.21 ± 0.13 | 0.93 ± 0.10$ |
| hsa-miR-150-5p | 0.85 ± 0.08 | 0.71 ± 0.04§ | 0.90 ± 0.05 | 1.14 ± 0.07$ |
| hsa-miR-196b-3p | 1.67 ± 0.13§§ | 1.55 ± 0.18§ | 1.66 ± 0.17§ | 0.99 ± 0.09††,$,∗ |
| hsa-miR-381-3p | 3.50 ± 1.17 | 2.67 ± 0.49 | 1.84 ± 0.24 | 1.16 ± 0.25 |
| hsa-miR-204-5p | 2.18 ± 0.43 | 1.38 ± 0.20 | 1.33 ± 0.18 | 0,94 ± 0,06 |
| hsa-miR-20a-5p | 1.28 ± 0.26 | 1.13 ± 0.16 | 0.79 ± 0.08 | 0.62 ± 0.06 |
| hsa-miR-21-3p | 6.98 ± 1.56§§ | 5.16 ± 0.48§ | 3.57 ± 0.45 | 3.13 ± 0.45##,$ |
| hsa-miR-296-5p | 0.92 ± 0.08 | 0.91 ± 0.10 | 0.79 ± 0.05 | 0.77 ± 0.06 |
| hsa-miR-302b-5p | n/d | n/d | n/d | n/d |
| hsa-miR-30a-3p | 0.64 ± 0.04 | 0.56 ± 0.02∗,§§ | 0.76 ± 0.03$ | 0.78 ± 0.03$$ |
| hsa-miR-320a | 1.10 ± 0.10$ | 0.94 ± 0.05 | 0.90 ± 0.04 | 0.76 ± 0.05# |
| hsa-miR-34b-5p | 2.26 ± 0.30 | 2.19 ± 0.48 | 1.32 ± 0.19 | 1.61 ± 0.27 |
| hsa-miR-34c-3p | 1.23 ± 0.13 | 1.07 ± 0.19 | 0.90 ± 0.10 | 1.14 ± 0.17 |
| hsa-miR-376b-3p | 3.15 ± 0.29 | 3.37 ± 0.27 | 2.52 ± 0.36 | 2.69 ± 0.50 |
| hsa-miR-382-5p | 2.02 ± 0.37 | 1.63 ± 0.17 | 1.25 ± 0.13 | 1.24 ± 0.16 |
| hsa-let-7f-5p | 1.14 ± 0.07$ | 0.97 ± 0.03 | 0.96 ± 0.05 | 0.87 ± 0.03# |
Figure 5.

miRNA expression levels between subject groups and correlations with in vivo mitochondrial function in human skeletal muscle. (A) Figures (left panels) show correlations for miRNA expression levels plotted against in vivo measures for mitochondrial function, i.e. PCr recovery rate (seconds) and maximal aerobic capacity (VO2max) for all subjects, with the Spearman ρ and p-value given for each miRNA. (B) Relative miRNA expression (middle panels) in type 2 diabetic (T2DM), obese (O), lean sedentary (LS), and Athletic (A) subjects (n = 12). Significance was assessed using one-way ANOVA with the nonparametric Kruskal–Wallis correction for multiple testing, and is indicated with *, **, and *** representing p < 0.05, p < 0.01, and p < 0.001 respectively. (C) Correlations between group means of relative miRNA expression and in vivo mitochondrial function (right panels). Values are means ± SD.
Moreover, we performed a correlation analysis between the expression of these miRNAs and the in vivo measures of mitochondrial capacity, i.e. VO2max and PCr recovery rate. From the 8 miRNAs that were differentially expressed between groups, miRNA-106b-3p, miRNA-19a-3p, miRNA-21-3p, miRNA-30a-3p and Let-7f-5p either did not correlate with in vivo mitochondrial capacity, or were confounded by other parameters such age, BMI or fasting plasma glucose (FPG), as assessed by stepwise regression analysis. On the other hand, miRNA-320a, miRNA-150-5p, miRNA-196b-3p, and miRNA-34c-3p did correlate with in vivo mitochondrial capacity. miRNA-150-5p positively correlated with VO2max (Spearman ρ = 0.54, p < 0.0001) (Figure 5B and C) while miRNA-196b-3p (Spearman ρ = −0.51, p < 0.001) and miRNA-320a (Spearman ρ = −0.42, p < 0.01) both negatively correlated with VO2max (Figure 5B and C). Of note, miRNA-34c-3p positively correlated with PCr recovery rate (Spearman ρ = 0.36, p < 0.05) (Figure 5B and C); however, no differences in expression were observed across the four subject groups (Figure 5A and Supplemental Figure 1). Stepwise regression analysis revealed that neither age, BMI, nor FPG were confounding factors for the observed correlations, further strengthening the association of miRNA-34c-3p, miRNA-150-5p, miRNA-196b-3p, and miRNA-320a expression with in vivo mitochondrial function.
Finally, to further investigate the role of these microRNAs in the direct regulation of mitochondrial function, we quantified – upon silencing of the 4 candidate miRNAs – the expression of 27 genes known to be involved in (the regulation of) various aspects of mitochondrial function such as fatty acid oxidation, mitochondrial dynamics, mitochondrial biogenesis and ETC complex formation. The data, depicted in Figure 6A, show the differential regulation of several of these genes following miRNA silencing. For example, silencing of microRNA-150-5p induced a ∼30% increase in both the expression of PPARGC1a, the well-known regulator of mitochondrial biogenesis and mitochondrial transcription factor A (mtTFA), a gene directly down-stream of PPARGC1a (Figure 6B). Target prediction software revealed that PPARGC1a is a target of microRNA-150-5p, further supporting the direct involvement of this microRNA in the regulation of mitochondrial function (Figure 6C).
Figure 6.

MiRNA-150-5p, miRNA-34c-3p, miRNA-196b-3p, and miRNA-320a alter the expression of genes involved in mitochondrial metabolism in C2C12 myotubes. (A) Hierarchical clustering of the expression of 27 genes involved in different aspects of mitochondrial function, 24 h after individually transfecting LNAs directed against miRNA-150-5p, miRNA-34c-3p, miRNA-196b-3p, and miRNA-320a vs. negative control (NC). Blue and red represent down and up regulation, respectively (B) mRNA levels of miRNA-150-5p predicted target PPARGC1a and its downstream target mtTFA, 24 h post transfection with an anti-miRNA-150-5p. (C) Representation of the binding between the PGC-1α mRNA 3′UTR region and miRNA-150-5p in different species. Mean+/-SD, *p < 0.05; #p = 0.06 (n = 3).
3. Discussion
About a decade ago [12], [13], miRNAs were identified as post-transcriptional regulators of gene expression and protein translation, by interacting with the 3′ untranslated regions (UTR) of mRNAs, thereby modulating the stability and translation of these protein-coding transcripts. Interestingly, several reports have indicated that miRNAs are present in mitochondria and may be important regulators of mitochondrial metabolism. In the current study, we employed an unbiased hypothesis-free screening approach to identify miRNAs involved in the regulation of skeletal muscle mitochondrial metabolism. To this end, we conducted a miRNA silencing screen in C2C12 myoblasts, validated our candidate miRNAs in C2C12 myotubes, and identified 19 candidate miRNAs that are putatively involved in the positive regulation of mitochondrial metabolism in skeletal muscle. Four of these miRNAs (miRNA-34c-3p, miRNA-150-5p, miRNA-196b-3p, and miRNA-320a) were tightly related to in vivo skeletal muscle mitochondrial function in humans, suggesting that these miRNAs are clinically relevant and may be interesting targets for the manipulation of mitochondrial function in humans.
Given the high-throughput nature of our primary screen (>700 miRNA inhibitors, 5 mitochondrial readouts), we used undifferentiated C2C12 myoblasts. The myoblasts were provided with a low glucose media supplemented with oleic acid to ensure that the myoblasts were maximally relying on mitochondrial metabolism to meet their energy demands [28]. This primary silencing screen in C2C12 myoblasts successfully identified 62 miRNA hits (Figure 3) that significantly altered at least one of the mitochondrial readouts when silenced, irrespective of the direction of change.
It has been shown that miRNAs and mitochondria both play important roles in the proliferation of myoblasts as well as in the differentiation into multinucleated myotubes [38], [39], [40]. Furthermore, mitochondrial capacity and oxidative phenotype were shown to increase substantially over the course of the myotube differentiation process [39]. Thus, other aspects than the direct link between miRNAs and mitochondrial metabolism may have interfered with the readouts of our primary silencing screen possibly leading to an over- or underestimation of the outcomes. Therefore, we next sought to confirm our miRNA hits in cultured, differentiated skeletal muscle cells (myotubes) of the mouse C2C12 myoblast cell line which resemble the adult, post-mitotic skeletal muscle more closely. This validation screen in differentiated myotubes confirmed 44 miRNA hits, i.e. miRNAs that – when silenced – had a significant effect on at least one of the readouts on one of the time points (Figure 4). Subsequently, to eliminate hits that are irrelevant for humans, we excluded miRNAs without a human homologue resulting in a list of 19 most promising miRNA candidates. Of these 19 candidates, 4 miRNAs (miRNA-34c-3p, miRNA-150-5p, miRNA-196b-3p, and miRNA-320a) were differentially expressed in subject groups varying in metabolic health and were tightly correlated to in vivo muscle mitochondrial function.
Silencing miRNA-34c-3p induced COX4 promoter activity in C2C12 myoblasts, (Figure 3C), strongly induced HSP60 promoter activity in C2C12 myotubes (Figure 4B), and although its expression did not differ between our subject groups, it did positively correlate with PCr recovery time in human skeletal muscle (Figure 5), indicating that increased miRNA-34c-3p expression relates to a reduced capacity to recover PCr stores. miRNA-34c has been reported to be increased in serum samples of a rat model for mitochondrial toxicity, i.e. rats that received a daily dose of rotenone, a complex 1 inhibitor [41], strongly suggesting its secretion into the circulation as a mitokine [42]. These data combined with our observations collectively show that a reduced miRNA-34c-3p associates with better mitochondrial function in cells, animal models, and humans and indicate that targeting miRNA-34c-3p could be a potential tool to improve skeletal muscle mitochondrial function.
miRNA-320a silencing induced HSP60 promoter activity in both C2C12 myoblasts and myotubes (Figure 3, Figure 4B), suggesting that silencing of this miRNA results in activation of mitochondrial protein quality control. Additionally, its expression was markedly decreased in athletes compared to T2DM subjects and also negatively correlated with VO2max (Figure 5), again demonstrating that decreased miRNA-320a associates with improved mitochondrial function. miRNA-320a has been linked to T2DM in multiple studies [43], a disease repeatedly associated impaired mitochondrial function [6], [44], [45]. A meta-analysis reported increased miRNA-320a concentrations in plasma of T2DM subjects [46], and another study reported increased levels in an insulin resistant subject group compared to controls [47]. Together, this points towards miRNA-320a as another potential target to improve mitochondrial function and metabolic health.
miRNA-196b-3p silencing reduced ATP levels in C2C12 myoblasts (Figure 3C), induced HSP60 promoter activity and slightly increased respiration in C2C12 myotubes (Figure 4B). Although this miRNA has not previously been identified as a regulator for mitochondrial function, its expression was also reduced in athletes compared to all other groups, and negatively correlated with VO2max (Figure 5). This underscores that lower expression levels of this miRNA are linked to an improved in vivo mitochondrial function, possibly via an increased mitochondrial protein quality control.
Finally, miRNA-150-5p silencing reduced ΔΨm in C2C12 myoblasts (Figure 3C), reduced COX4 and HSP60 promoter activity, and slightly increased basal respiration in C2C12 myotubes (Figure 4B). Furthermore, the expression of PPARGC1a and its downstream target mtTFA increased upon miRNA-150-5p silencing and target prediction software combined with the MitoCarta 2.0 database [48] revealed the potential regulation of >50 mitochondrial-related genes by miRNA-150-5p, including PPARGC1a. Additionally, the expression of miRNA-150-5p positively correlated with VO2max in humans and was also increased in skeletal muscle biopsies of athletes compared to obese subjects, indicating that increased miRNA-150-5p expression levels were associated with increased mitochondrial capacity in human skeletal muscle. In contrast to our findings, miRNA-150-5p was previously reported to be upregulated in T2DM subjects; however, this finding was based on microarray data and its expression was not validated using more quantitative assays, such as RT-qPCR [43]. miRNA-150-5p has been suggested to be enriched in mitochondria of rat hippocampus [49], supporting a role for miRNA-150-5p in mitochondrial metabolism. In addition, increased miRNA-150-5p expression has been reported in human plasma samples post-exercise, matching the increased expression in skeletal muscle of our athlete group and suggesting the secretion of this miRNA into the circulation [50]. Taken together, these results support the notion that miRNA-150-5p plays an important role in the direct regulation of mitochondrial function in muscle.
In conclusion, we here performed an extensive screening of miRNA inhibitors in C2C12 myoblasts and C2C12 myotubes and identified miRNAs that are involved in the regulation of mitochondrial metabolism in skeletal muscle. Of these, we identified miRNA-34c-3p, miRNA-150-5p, miRNA-196b-3p, and miRNA-320a as clinically relevant candidates for the modulation of skeletal muscle mitochondrial metabolism in humans. Since mitochondrial function is linked to metabolic health and disease progression in both animals and humans, these miRNAs may be interesting targets for the treatment of chronic metabolic diseases. Furthermore, since certain miRNAs have been reported to be present in serum, they may potentially convey the presence of mitochondrial stress between different tissues as true mitokines, an area that warrants future investigation.
4. Methods
4.1. miRNA inhibitor library and individual inhibitors
A mouse miRCURY LNA™ miRNA inhibitor library, containing 724 miRNA inhibitors was purchased from Exiqon A/S (Vedbaek, Denmark). miRCURY LNA™ miRNA inhibitor control/negative control A (Exiqon A/S, Vedbaek, Denmark), an aspecific LNA inhibitor, was used as negative control. This negative control (sequence: TAACACGTCTATACGCCCA) does not target any known mature mammalian miRNA of the online miRbase database.
4.2. C2C12 myoblasts culturing and transfection conditions primary screen
C2C12 mouse myoblast cells were obtained from ATCC (Manassas, Virginia, USA) and maintained below 80% confluence in high glucose growth media (Dulbecco's Modified Eagle Medium (DMEM) 4.5 g/L glucose supplemented with 10% FBS, 2% HEPES and 1% Non-essential amino acids). Cells were seeded at 2,000 cells per well in a volume of 50 μl high glucose growth media, in 384-well plates. Twenty-four hours after seeding, the cell media was changed to 40 μL serumless DMEM. Subsequently, we added 10 μl transfection reagent containing the miRNA inhibitors, a renilla reporter plasmid and either a COX4, HSP60 or a negative control luciferase reporter plasmid using lipofectamine 2000, according to the manufacturer's guidelines. The purchased miRNA inhibitor stocks were diluted to a concentration of 12 μM, and the final concentration for transfection (150 nM) was established in optimization experiments using increasing concentrations of a miRNA inhibitor against the muscle-specific miRNA-206.
Three hours post-transfection the cell media was replaced with 50 μL low glucose media (DMEM 1 g/L glucose with 10% FBS, 2% HEPES, and 1% Non-essential amino acids) supplemented with 1% oleic acid. Assays were typically performed 24 h after transfection.
4.3. C2C12 myotubes culture and transfection conditions validation screen
For the validation experiments in differentiated C2C12 myotubes, C2C12 myoblasts were seeded in 96-well plates at 25.000 cells/cm2 in growth media and reached full confluence in 24 h. Subsequently, differentiation was initiated by replacing the growth media for high glucose differentiation media (DMEM 4.5 g/l with 2% Horse serum, 2% HEPES and 1% Non-essential amino acids). The media was changed every other day, and mature myotubes were obtained after 5 days of differentiation.
At day 5 or 6 of differentiation, the cell media was changed to 40 μL serumless DMEM. Subsequently, we added 10 μL transfection reagent containing the miRNA inhibitors, a renilla reporter plasmid, and either a COX4, HSP60, or a negative control luciferase reporter plasmid using lipofectamine 2000, according to the manufacturer's protocol. The final concentration for transfection (25 nM) was established in optimization experiments using increasing concentrations of a miRNA inhibitor against the muscle-specific miRNA-206.
Three hours post-transfection, the cell media was replaced with 50 μL high glucose differentiation media. Assays were always performed at day 7 of differentiation, either 24 or 48 h after transfection.
4.4. Mitochondrial read-outs primary and validation screen
4.4.1. ATP, membrane potential and redox potential
ATP levels were measured using Cell Titer Glo (Promega, Madison, WI, USA), by adding a volume of CellTiterGLO reagent equal to the cell media directly to the cells in culture. Directly afterwards, the plates were wrapped in aluminum foil and shaken for 10 min at room temperature. After another 5 min of stabilization at room temperature, luminescence was recorded using the Tecan infinity 500 (Tecan, Männedorf, Switzerland).
Mitochondrial membrane potential (ΔΨm) was measured using the fluorescent tetramethylrhodamine methyl ester (TMRM) probe (T-668, Invitrogen, Paisley, UK). Briefly, cells were incubated in fresh culture medium and TMRM was added to the culture medium at a final concentration of 100 nM for 45 min at 37 °C before washing with 50 μl of PBS. Finally, plates were read using the Tecan infinity 500 (Tecan, Männedorf, Switzerland) according to manufacturer's protocol.
The Alamar Blue assay (Life Technologies, Paisley, UK) was used as a marker for redox potential. AlamarBlue® reagent was prediluted 2.5 times and 5 μL was added to every well, subsequently the plates were wrapped in aluminum foil and incubated for 3 h at 37 °C with 5% CO2. Finally, plates were read using the Tecan infinity 500 (Tecan, Männedorf, Switzerland) according to manufacturer's protocol.
4.4.2. Luciferase reporter assays
Luciferase reporter assays were performed using the Dual-Glo® Luciferase Assay System (Promega) to assess HSP60 and COX4 promoter activity. Cells for the Dual-Glo luciferase reporter assays were cultured in white, clear bottom plates. On the day of the assay, a volume of reagent equal to the culture medium volume was added to each well and mixed on a plate shaker for 10 min at RT. Firefly luminescence was then measured using the Tecan infinity 500 (Tecan, Männedorf, Switzerland). To measure renilla luciferase activity, a volume of Dual-Glo Stop & Glo reagent was added to the culture media in each well (v/v) and mixed on a plate shaker for 10 min at RT. Unfortunately, 2 luciferase assay plates were lost due to technical malfunction, resulting in a partial lack of data with respect to HSP60 and COX4 promoter activity in the primary silencing screen in C2C12 myoblasts (Figure 3C).
As a negative control, negative control A was transfected on every plate and used for normalization purposes. Furthermore, cell death was induced with by adding 1 μM of the detergent benzethonium chloride as a positive control for reduced ATP levels, redox potential and ΔΨm [51]. Readouts for ATP level, redox potential, TMRM, HSP60, and COX4 assays were obtained using a Tecan Infinite 500 (Tecan, Männedorf, Switzerland). All analyses were performed in duplicate.
4.4.3. Oxygen consumption rate measurements
Oxygen consumption rate (OCR) and extra-cellular acidification rate (ECAR) were measured in C2C12 myotubes using the Seahorse XF96 equipment (Seahorse bioscience Inc., North Billerica, MA, USA). At the day of the assay, the cell media was exchanged by 175 μL XF media according the manufacturer's guidelines and the cells were placed at 37 °C with ambient CO2 concentrations for 45 min. Subsequently, the assay was started and oxygen consumption rate was measured, 3 consecutive times with 3 min intervals, at basal conditions, after an oligomycin injection, and after an FCCP injection, both resulting in a final concentration of 1 μM. After the assay, the cells were washed twice with PBS, aspirated to dryness and stored at −20 °C for protein corrections. Protein concentrations were determined using the Bradford protein assay according to the manufacturer's guidelines.
4.5. Human muscle biopsies
Muscle biopsies were collected from 4 previously conducted studies, all performed at the department of Human Biology and Human Movement Sciences at Maastricht University [34], [35], [36], [37]. Four subject groups were included: 1) overweight/obese type 2 diabetic patients (n = 12); 2) overweight/obese, non-diabetic subjects (n = 12); 3) young, lean sedentary individuals (n = 12) and 4) young, endurance-trained athletes (n = 12). Subjects were selected on the availability of muscle tissue and data for in vivo mitochondrial function (PCr recovery) and maximal aerobic capacity (VO2max), and were subsequently selected in order for the groups to match for age and BMI (i.e. lean sedentary subjects vs. endurance-trained athletes and overweight/obese, non-diabetic subjects vs. type 2 diabetic patients).
All subjects were male, non-smoking, and weight stable for at least 6 months. All type 2 diabetic subjects were diagnosed with type 2 diabetes for at least one year, used metformin, and were allowed to use statins. The overweight/obese, non-diabetic subjects did not use any medication and were excluded if they suffered from uncontrolled hypertension, cardiovascular disease, or liver dysfunction, or if they had first degree relatives with type 2 diabetes mellitus. The overweight/obese type 2 diabetic patients and the overweight/obese, non-diabetic subjects were matched for age and BMI.
Lean sedentary subjects were included if they participated in no more than one hour of exercise per week for the last 2 years and if their VO2max was lower than 45 ml/min/kg. Endurance-trained athletes were included if they participated in endurance training at least 3 times per week for the last 2 years and if their VO2max was above 55 ml/min/kg. Lean, sedentary subjects and endurance-trained athletes were matched for age and BMI.
All biopsies were collected after an overnight fast and in the basal state (i.e. before any intervention). Subjects were instructed to refrain from physical exercise at least 3 days before the biopsy was taken.
4.6. Subject characterization
4.6.1. Body composition
Depending on the study, body composition was either determined via hydrostatic weighing, using the method of Siri [34], or via dual X-ray absorptiometry (DEXA, Discovery A; Hologic, Bedford, MA).
4.6.2. VO2max test
VO2max describes the maximal amount of oxygen that can be utilized by a subject, and this was assessed using an incremental bike-stress-test as described previously [52].
4.6.3. In vivo mitochondrial function
In vivo mitochondrial function was determined as phosphocreatine (PCr) resynthesis rate, assessed by magnetic resonance spectrometry (MRS). All subjects underwent a 31P-MRS measurement which was performed on a 3T whole-body MRI scanner (Achieve 3T-X, Philips Healthcare) as previously described [53]. In short, a 6-cm coil was positioned on the m. vastus lateralis and subjects performed knee-extension exercises inside the MRI on an MR compatible ergometer. The exercise protocol started with 1 min of rest, followed by 4 min of knee extension exercises, followed by a recovery period of 5 min. The initial weight was set at 50–60% of the maximal estimated knee-extension power output. MR settings included: repetition time of 4000 ms with a spectral bandwidth of 1500 Hz and an offset frequency of −1.9 ppm [53].
4.7. Plasma analysis
A fasted blood sample was drawn from the cubital vein and collected in EDTA containing tubes. After centrifugation, the plasma was frozen in liquid nitrogen, followed by long-term storage at −80 °C. Plasma glucose concentrations were determined using enzymatic assays on Cobas Bio Fara and Mira analyzers (glucose: hexokinase method [Roche, Basel, Switzerland]).
4.8. Quantitative PCR in human samples and C2C12 myotubes
4.8.1. miRNA RT-qPCR in human muscle biopsies
In a sterile glass tube, 700 μL of Trizol was combined with 10–15 mg of frozen muscle tissue and homogenized with an Ultra Turrax for 1 min at 17.000 rpm. The Ultra Turrax was cleaned in between samples with chloroform, 100% EtOH and MilliQ. The homogenized sample was transferred to a 2 mL Eppendorf tube and placed on ice. RNA isolation was performed using the miRNeasy mini kit (Qiagen, Germany) according to the manufacturer's guidelines. However, to increase miRNA yield, the final eluate was again added to the column membrane and centrifuged at 10.000 rpm for 1 min; this procedure was repeated twice. RNA concentrations were measured using the Nanodrop (Biocompare, California, US), and quality was assessed using a bioanalyzer (Agilent Technologies, California, US). All samples had RIN values between 7 and 10. Subsequently, RNA concentrations were diluted to a concentration of 5 ng/μL with nuclease free water. The synthetic RNA spike-in UniSp6 was added, and cDNA was synthesized using the Universal cDNA Synthesis kit II (Exiqon, Denmark) according to the manufacturer's protocol. Isolated cDNA samples were analyzed using predesigned 384-well Pick-&-Mix miRNA PCR Panel plates (Exiqon, Denmark) according to the manufacturer's guidelines using a CFX384 Touch Real-time PCR detection system (Bio-Rad, the Netherlands).
4.8.2. RT-qPCR in C2C12 myotubes
Cells were washed with 1 mL cold 1xPBS and harvested in 700 μL Trizol reagent (Invitrogen, Breda, the Netherlands) and stored at −80 °C for later use. RNA was isolated using the RNeasy mini kit (Qiagen, Venlo, the Netherlands). RNA quantity and quality was assessed spectrophotometrically (ND-1000, Nanodrop Technologies, Wilmington, USA) with 6000 Nano chips (Bioanalyzer2100; Agilent, Amstelveen, The Netherlands). cDNA was synthesized using the High-Capacity RNA-to-cDNA Kit (Applied Biosystems). For the PCR, we used SensiMix SYBR Hi-ROX kit (Bioline, London, United Kingdom), and, for each PCR reaction. We used, 5 μL 2× universal master mix, 0.6 μL 10 μM forward and reverse primer, 1.3 μL nuclease-free water and 2.5 μL (20× diluted) cDNA sample per 10 μL reaction, with the following protocol: 50 °C for 2 min, 95 °C for 10 min and 40 cycles of 95 °C for 15 s followed by 60 °C for 1 min. Relative expression was calculated using the 2−ΔΔCt method, comparing the expression of miRNA silenced C2C12 myotubes to C2C12 myotube transfected with Negative control A, using the geometrical mean of 36B4 and B2M as a reference. Heatmaps were generated from the average relative expression of 3 independent experiments, using R (R Foundation for Statistical Computing, Vienna, Austria).
4.9. Data analysis and statistics
For the primary silencing screen, data were analyzed using the cellHTS2 package from R software (http://www.r-project.org/). ATP, redox potential, and TMRM data were normalized per plate using the normalized percentage of inhibition function (normalized ratio), calculated as follows: , where xki is a result point, is the average for all miRNA inhibitor points and is the average of the benzethonium chloride negative control points. Because the HSP60 and COX4 luciferase assays had less discriminative power with the negative control, data were normalized using the robust Z-score method. In short, the mean of all samples was subtracted from the sample measurement and divided by the mean standard deviation of all samples with the following formula: , in which xi is the samples measurement, μx represents the mean of all samples, and sx is the mean of all sample standard deviations. Z scores and Z factors were calculated from these normalized values. Theoretical quantiles were calculated from the normalized values using R software. To correct for variation in luciferase reporter transfection efficiencies, the luciferase reporter assay values were always corrected for total renilla signal, by dividing the firefly luciferase signal by the renilla luciferase signal. Compounds were considered as hits when inducing a signal change superior to negative control A ± 2σNegative control A and when deviating from normal distribution, i.e. when squared distance between observed quantile and theoretical quantile was above a certain threshold. This threshold was arbitrarily set at ≥0.005 for assay readouts. Clustering of the 62 hits was performed on the Hierarchical Clustering module available on the GenePattern website (http://www.broadinstitute.org/cancer/software/genepattern), using Euclidean distance and single linkage as analysis parameters.
For the validation experiments in C2C12 myotubes, all miRNA inhibitors were corrected over the average signal of Negative Control A for each plate, resulting in a normalized ratio. Compounds were considered as hits when inducing a signal change superior to negative control A ± 2σNegative control A. Furthermore, regarding the oxygen consumption rates, the average oxygen consumption rate was calculated from 3 repeated measures for basal and maximal respiration, and each well was corrected for total protein.
Relative miRNA expression levels were calculated using the 2−ΔΔCt method and were expressed relative to one of the lean sedentary subjects. Furthermore, occasional outliers were removed using the ROUT method [54]. Threshold values were equalized between all PCR runs. Furthermore, to correct for inter plate variation, we calculated the average of all inter plate calibrator Cq values of each plate. Subsequently, the calibration factor was calculated for each plate by determining the difference between the plate average and overall average. Finally, each plate was calibrated by subtracting the calibration factor from all plate Cq values.
Human miRNA expression data was correlated to in vivo read out parameters for mitochondrial function. Additionally, to determine that no confounding factors such as age and BMI were driving these correlations, stepwise regression analysis was performed. All correlation and stepwise regression analysis were performed using SPSS. Furthermore, because PCR data are generally not normally distributed, differences in miRNA expression between subject groups were assessed using one-way ANOVA with the nonparametric Kruskal–Wallis correction for multiple testing. Subject characteristics were analyzed using one-way ANOVA with Tukey's correction for multiple testing.
4.10. Study approval
Skeletal muscle biopsies were collected from 4 previously conducted studies, all performed at the department of Human Biology and Human Movement Sciences at Maastricht University [33], [34], [35], [36]. The institutional medical ethics committee approved the aforementioned studies (clinical trial reg. no. NCT00943059 [33], NTR2002 [34], [35] and NCT01298375 [36]), and all participants gave their written informed consent prior to inclusion in the study.
Author contributions
D.D. designed and performed the experiments, analyzed data, and wrote the manuscript. A.H. and P.A. designed and performed the experiments and analyzed data. J.J. and X.W. assisted during the experiments. L.J.d.W. contributed to the initial aspects of study design. P.S., J.A., and J.H. contributed to the design of the study, analyzed and interpreted the data and reviewed and edited the manuscript. All authors reviewed and approved the final version of the manuscript.
Acknowledgments
The work of J.H. is supported by a Vidi (Grant 917.14.358) for innovative research from the Netherlands Organization for Scientific Research (NWO) and a Senior Fellowship from the Dutch Diabetes Research Foundation (grant number 2013.82.1639). The research of J.A. is supported by grants from the École Polytechnique Fédérale de Lausanne, the Swiss National Science Foundation (31003A-140780), the AgingX program of the Swiss Initiative for Systems Biology (51RTP0-151019), and the NIH (R01AG043930). L.D.W. was supported by grant 311549 from the European Research Council (ERC) and a VICI award (918-156-47) from NWO. The authors thank the biomolecular screening facility (BSF) of the École Polytechnique Fédérale de Lausanne for providing technical support in the high-throughput screening aspects.
Footnotes
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molmet.2017.08.007.
Conflict of interest
The authors report no conflicts of interest relevant to this article.
Appendix A. Supplementary data
The following are the supplementary data related to this article:
References
- 1.Weber T.A., Reichert A.S. Impaired quality control of mitochondria: aging from a new perspective. Experimental Gerontology. 2010;45:503–511. doi: 10.1016/j.exger.2010.03.018. [DOI] [PubMed] [Google Scholar]
- 2.Pan S., Ryu S.Y., Sheu S.S. Distinctive characteristics and functions of multiple mitochondrial Ca2+ influx mechanisms. Science China Life Sciences. 2011;54:763–769. doi: 10.1007/s11427-011-4203-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dominic E.A., Ramezani A., Anker S.D., Verma M., Mehta N., Rao M. Mitochondrial cytopathies and cardiovascular disease. Heart. 2014;100:611–618. doi: 10.1136/heartjnl-2013-304657. [DOI] [PubMed] [Google Scholar]
- 4.Johannsen D.L., Ravussin E. The role of mitochondria in health and disease. Current Opinion in Pharmacology. 2009;9:780–786. doi: 10.1016/j.coph.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin M.T., Beal M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 6.Kelley D.E., He J., Menshikova E.V., Ritov V.B. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes. 2002;51:2944–2950. doi: 10.2337/diabetes.51.10.2944. [DOI] [PubMed] [Google Scholar]
- 7.Hoeks J., Schrauwen P. Muscle mitochondria and insulin resistance: a human perspective. Trends in Endocrinology & Metabolism. 2012;23:444–450. doi: 10.1016/j.tem.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 8.Petersen K.F., Dufour S., Befroy D., Garcia R., Shulman G.I. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. New England Journal of Medicine. 2004;350:664–671. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Civitarese A.E., Carling S., Heilbronn L.K., Hulver M.H., Ukropcova B., Deutsch W.A. Calorie restriction increases muscle mitochondrial biogenesis in healthy humans. PLoS Medicine. 2007;4:e76. doi: 10.1371/journal.pmed.0040076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meex R.C., Schrauwen-Hinderling V.B., Moonen-Kornips E., Schaart G., Mensink M., Phielix E. Restoration of muscle mitochondrial function and metabolic flexibility in type 2 diabetes by exercise training is paralleled by increased myocellular fat storage and improved insulin sensitivity. Diabetes. 2010;59:572–579. doi: 10.2337/db09-1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Timmers S., Konings E., Bilet L., Houtkooper R.H., van de Weijer T., Goossens G.H. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metabolism. 2011;14:612–622. doi: 10.1016/j.cmet.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bartel D.P. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 14.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 15.Lytle J.R., Yario T.A., Steitz J.A. Target mRNAs are repressed as efficiently by microRNA-binding sites in the 5′ UTR as in the 3′ UTR. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:9667–9672. doi: 10.1073/pnas.0703820104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Farh K.K., Grimson A., Jan C., Lewis B.P., Johnston W.K., Lim L.P. The widespread impact of mammalian MicroRNAs on mRNA repression and evolution. Science. 2005;310:1817–1821. doi: 10.1126/science.1121158. [DOI] [PubMed] [Google Scholar]
- 17.Smith-Vikos T., Slack F.J. MicroRNAs and their roles in aging. Journal of Cell Science. 2012;125:7–17. doi: 10.1242/jcs.099200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ali A.S., Ali S., Ahmad A., Bao B., Philip P.A., Sarkar F.H. Expression of microRNAs: potential molecular link between obesity, diabetes and cancer. Obesity Reviews. 2011;12:1050–1062. doi: 10.1111/j.1467-789X.2011.00906.x. [DOI] [PubMed] [Google Scholar]
- 19.Sripada L., Tomar D., Singh R. Mitochondria: one of the destinations of miRNAs. Mitochondrion. 2012;12:593–599. doi: 10.1016/j.mito.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 20.Bian Z., Li L.M., Tang R., Hou D.X., Chen X., Zhang C.Y. Identification of mouse liver mitochondria-associated miRNAs and their potential biological functions. Cell Research. 2010;20:1076–1078. doi: 10.1038/cr.2010.119. [DOI] [PubMed] [Google Scholar]
- 21.Li P., Jiao J., Gao G., Prabhakar B.S. Control of mitochondrial activity by miRNAs. Journal of Cellular Biochemistry. 2012;113:1104–1110. doi: 10.1002/jcb.24004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barrey E., Saint-Auret G., Bonnamy B., Damas D., Boyer O., Gidrol X. Pre-microRNA and mature microRNA in human mitochondria. PLoS One. 2011;6 doi: 10.1371/journal.pone.0020220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu Y., Zhao C., Sun X., Liu Z., Zhang J. MicroRNA-761 regulates mitochondrial biogenesis in mouse skeletal muscle in response to exercise. Biochemical and Biophysical Research Communications. 2015;467:103–108. doi: 10.1016/j.bbrc.2015.09.113. [DOI] [PubMed] [Google Scholar]
- 24.Shen L., Chen L., Zhang S., Du J., Bai L., Zhang Y. MicroRNA-27b regulates mitochondria biogenesis in myocytes. PLoS One. 2016;11 doi: 10.1371/journal.pone.0148532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mohamed J.S., Hajira A., Pardo P.S., Boriek A.M. MicroRNA-149 inhibits PARP-2 and promotes mitochondrial biogenesis via SIRT-1/PGC-1alpha network in skeletal muscle. Diabetes. 2014;63:1546–1559. doi: 10.2337/db13-1364. [DOI] [PubMed] [Google Scholar]
- 26.Burkart E.M., Sambandam N., Han X., Gross R.W., Courtois M., Gierasch C.M. Nuclear receptors PPARbeta/delta and PPARalpha direct distinct metabolic regulatory programs in the mouse heart. Journal of Clinical Investigation. 2007;117:3930–3939. doi: 10.1172/JCI32578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.el Azzouzi H., Leptidis S., Dirkx E., Hoeks J., van Bree B., Brand K. The hypoxia-inducible microRNA cluster miR-199a approximately 214 targets myocardial PPARdelta and impairs mitochondrial fatty acid oxidation. Cell Metabolism. 2013;18:341–354. doi: 10.1016/j.cmet.2013.08.009. [DOI] [PubMed] [Google Scholar]
- 28.Andreux P.A., Mouchiroud L., Wang X., Jovaisaite V., Mottis A., Bichet S. A method to identify and validate mitochondrial modulators using mammalian cells and the worm C. elegans. Scientific Reports. 2014;4:5285. doi: 10.1038/srep05285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rampersad S.N. Multiple applications of Alamar Blue as an indicator of metabolic function and cellular health in cell viability bioassays. Sensors (Basel) 2012;12:12347–12360. doi: 10.3390/s120912347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Townley-Tilson W.H., Callis T.E., Wang D. MicroRNAs 1, 133, and 206: critical factors of skeletal and cardiac muscle development, function, and disease. International Journal of Biochemistry & Cell Biology. 2010;42:1252–1255. doi: 10.1016/j.biocel.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Houtkooper R.H., Mouchiroud L., Ryu D., Moullan N., Katsyuba E., Knott G. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013;497:451–457. doi: 10.1038/nature12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lagouge M., Argmann C., Gerhart-Hines Z., Meziane H., Lerin C., Daussin F. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 33.Baur J.A., Pearson K.J., Price N.L., Jamieson H.A., Lerin C., Kalra A. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van de Weijer T., Phielix E., Bilet L., Williams E.G., Ropelle E.R., Bierwagen A. Evidence for a direct effect of the NAD+ precursor Acipimox on muscle mitochondrial function in humans. Diabetes. 2015;64:1193–1201. doi: 10.2337/db14-0667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Phielix E., Meex R., Moonen-Kornips E., Hesselink M.K., Schrauwen P. Exercise training increases mitochondrial content and ex vivo mitochondrial function similarly in patients with type 2 diabetes and in control individuals. Diabetologia. 2010;53:1714–1721. doi: 10.1007/s00125-010-1764-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Phielix E., Meex R., Ouwens D.M., Sparks L., Hoeks J., Schaart G. High oxidative capacity due to chronic exercise training attenuates lipid-induced insulin resistance. Diabetes. 2012;61:2472–2478. doi: 10.2337/db11-1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vosselman M.J., Hoeks J., Brans B., Pallubinsky H., Nascimento E.B., van der Lans A.A. Low brown adipose tissue activity in endurance-trained compared with lean sedentary men. International Journal of Obesity (London) 2015;39:1696–1702. doi: 10.1038/ijo.2015.130. [DOI] [PubMed] [Google Scholar]
- 38.Rochard P., Rodier A., Casas F., Cassar-Malek I., Marchal-Victorion S., Daury L. Mitochondrial activity is involved in the regulation of myoblast differentiation through myogenin expression and activity of myogenic factors. Journal of Biological Chemistry. 2000;275:2733–2744. doi: 10.1074/jbc.275.4.2733. [DOI] [PubMed] [Google Scholar]
- 39.Remels A.H., Langen R.C., Schrauwen P., Schaart G., Schols A.M., Gosker H.R. Regulation of mitochondrial biogenesis during myogenesis. Molecular and Cellular Endocrinology. 2010;315:113–120. doi: 10.1016/j.mce.2009.09.029. [DOI] [PubMed] [Google Scholar]
- 40.Duguez S., Sabido O., Freyssenet D. Mitochondrial-dependent regulation of myoblast proliferation. Experimental Cell Research. 2004;299:27–35. doi: 10.1016/j.yexcr.2004.05.017. [DOI] [PubMed] [Google Scholar]
- 41.Baumgart B.R., Gray K.L., Woicke J., Bunch R.T., Sanderson T.P., Van Vleet T.R. MicroRNA as biomarkers of mitochondrial toxicity. Toxicology and Applied Pharmacology. 2015;312:26–33. doi: 10.1016/j.taap.2015.10.007. [DOI] [PubMed] [Google Scholar]
- 42.Kim K.H., Jeong Y.T., Oh H., Kim S.H., Cho J.M., Kim Y.N. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nature Medicine. 2013;19:83–92. doi: 10.1038/nm.3014. [DOI] [PubMed] [Google Scholar]
- 43.Karolina D.S., Tavintharan S., Armugam A., Sepramaniam S., Pek S.L., Wong M.T. Circulating miRNA profiles in patients with metabolic syndrome. Journal of Clinical Endocrinology & Metabolism. 2012;97:E2271–E2276. doi: 10.1210/jc.2012-1996. [DOI] [PubMed] [Google Scholar]
- 44.Schrauwen-Hinderling V.B., Kooi M.E., Hesselink M.K., Jeneson J.A., Backes W.H., van Echteld C.J. Impaired in vivo mitochondrial function but similar intramyocellular lipid content in patients with type 2 diabetes mellitus and BMI-matched control subjects. Diabetologia. 2007;50:113–120. doi: 10.1007/s00125-006-0475-1. [DOI] [PubMed] [Google Scholar]
- 45.Phielix E., Schrauwen-Hinderling V.B., Mensink M., Lenaers E., Meex R., Hoeks J. Lower intrinsic ADP-stimulated mitochondrial respiration underlies in vivo mitochondrial dysfunction in muscle of male type 2 diabetic patients. Diabetes. 2008;57:2943–2949. doi: 10.2337/db08-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Villard A., Marchand L., Thivolet C., Rome S. Diagnostic value of cell-free circulating MicroRNAs for obesity and type 2 diabetes: a meta-analysis. Journal of Molecular Biomarkers & Diagnosis. 2015;6 doi: 10.4172/2155-9929.1000251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Flowers E., Aouizerat B.E., Abbasi F., Lamendola C., Grove K.M., Fukuoka Y. Circulating microRNA-320a and microRNA-486 predict thiazolidinedione response: moving towards precision health for diabetes prevention. Metabolism. 2015;64:1051–1059. doi: 10.1016/j.metabol.2015.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Calvo S.E., Clauser K.R., Mootha V.K. MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Research. 2016;44:D1251–D1257. doi: 10.1093/nar/gkv1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang W.X., Visavadiya N.P., Pandya J.D., Nelson P.T., Sullivan P.G., Springer J.E. Mitochondria-associated microRNAs in rat hippocampus following traumatic brain injury. Experimental Neurology. 2015;265:84–93. doi: 10.1016/j.expneurol.2014.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.de Gonzalo-Calvo D., Davalos A., Montero A., Garcia-Gonzalez A., Tyshkovska I., Gonzalez-Medina A. Circulating inflammatory miRNA signature in response to different doses of aerobic exercise. Journal of Applied Physiology (1985) 2015;119:124–134. doi: 10.1152/japplphysiol.00077.2015. [DOI] [PubMed] [Google Scholar]
- 51.Enomoto R., Suzuki C., Ohno M., Ohasi T., Futagami R., Ishikawa K. Cationic surfactants induce apoptosis in normal and cancer cells. Annals of the New York Academy of Sciences. 2007;1095:1–6. doi: 10.1196/annals.1397.001. [DOI] [PubMed] [Google Scholar]
- 52.Kuipers H., Verstappen F.T., Keizer H.A., Geurten P., van Kranenburg G. Variability of aerobic performance in the laboratory and its physiologic correlates. International Journal of Sports Medicine. 1985;6:197–201. doi: 10.1055/s-2008-1025839. [DOI] [PubMed] [Google Scholar]
- 53.Lindeboom L., Nabuurs C.I., Hoeks J., Brouwers B., Phielix E., Kooi M.E. Long-echo time MR spectroscopy for skeletal muscle acetylcarnitine detection. Journal of Clinical Investigation. 2014;124:4915–4925. doi: 10.1172/JCI74830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Motulsky H.J., Brown R.E. Detecting outliers when fitting data with nonlinear regression – a new method based on robust nonlinear regression and the false discovery rate. BMC Bioinformatics. 2006;7:123. doi: 10.1186/1471-2105-7-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.