

Abstract
Antibiotic resistance is a natural feature of diverse microbial ecosystems. Although recent studies of the antibiotic resistome have highlighted barriers to the horizontal transfer of antibiotic resistance genes between habitats, the rapid global spread of genes that confer resistance to carbapenem, colistin and quinolone antibiotics illustrates the dire clinical and societal consequences of such events. Over time, the study of antibiotic resistance has grown from focusing on single pathogenic organisms in axenic culture to studying antibiotic resistance in pathogenic, commensal and environmental bacteria at the level of microbial communities. As the study of antibiotic resistance advances, it is important to incorporate this comprehensive approach to better inform global antibiotic resistance surveillance and antibiotic development. It is increasingly becoming apparent that although not all resistance genes are likely to geographically and phylogenetically disseminate, the threat presented by those that are is serious and warrants an interdisciplinary research focus. In this Review, we highlight seminal work in the resistome field, discuss recent advances in the studies of resistomes, and propose a resistome paradigm that can pave the way for the improved proactive identification and mitigation of emerging antibiotic resistance threats.
Antibiotics have revolutionized modern medicine and have saved millions of lives since their discovery at the beginning of the twentieth century. These crucial drugs are invaluable owing to their ability to kill or inhibit the growth of bacteria without damaging host cells and tissues1,2 (BOX 1). Aside from directly curing deadly infectious diseases, antibiotics have enabled physicians to pursue new treatments such as surgery, organ transplantation and cancer chemotherapy, which were previously infeasible or unsafe owing to the high collateral risk of infection3. Antibiotics remain one of the most commonly prescribed classes of drug, with more than 70 billion clinical doses administered globally in 2010, which represents a 36% increase in antibiotic consumption relative to 2000 (REF.4). Although the first antibiotics were synthetic compounds, most antibiotics are natural products or derivatives thereof5. Many antibiotics were recovered from cultured soil microorganisms, particularly those belonging to the Streptomyces genus3. As might be expected given their origins as natural products of bacterial secondary metabolism, many antibiotics can be detected at low levels in pristine environmental samples and have been found in samples that pre-date human antibiotic use6,7.
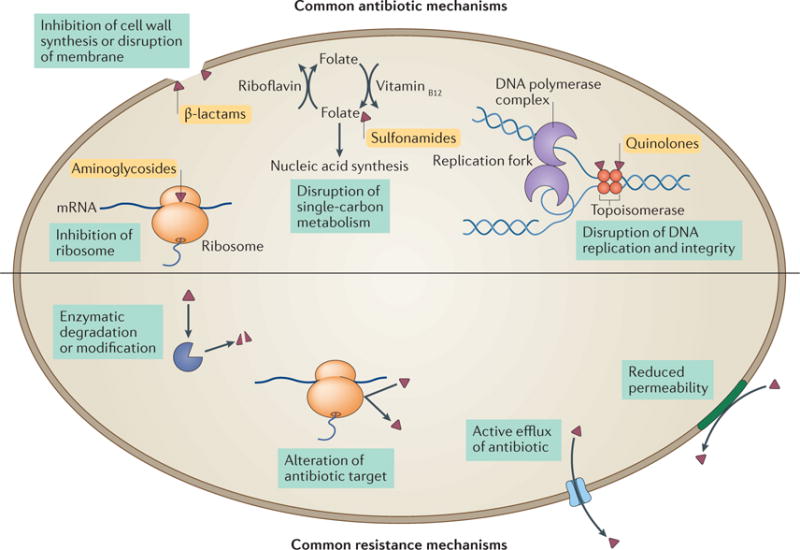
Box 1. Common mechanisms of antibiotic action and antibiotic resistance.
Antibiotic targets are generally conserved across the bacterial domain of life, and are absent from or sufficiently different in eukaryotes. Although hundreds of such targets exist in theory, our current antibiotic arsenal generally attacks the bacterial ribosome (inhibiting protein synthesis; for example, aminoglycosides); cell wall synthesis and lipid membrane integrity (compromising membrane integrity or lysing bacterial cells; for example, β-lactams); single-carbon metabolic pathways (inducing metabolic disruption; for example, sulfonamides); and DNA maintenance (interfering with bacterial replication and genomic integrity; for example, quinolones)2,17,103 (see the figure, top).
Bacteria typically resort to one of a few common strategies to resist antibiotics104 (see the figure, bottom). One resistance mechanism involves the inactivation of the antibiotic through enzymatic degradation or modification of the antibiotic scaffold, which renders it ineffective. Examples of this mechanism include chloramphenicol acetyltransferases105, tetracycline-inactivating enzymes such as TetX39,106 and, most notably, the widespread β-lactamases107,108. A second strategy for resistance is protection, alteration or overexpression of the antibiotic target. This approach is used by vancomycin-resistant enterococci, which enzymatically modify peptidoglycan, thus decreasing the affinity of vancomycin for its target109, and by methicillin-resistant Staphylococcus aureus (MRSA), which expresses a redundant methicillin-insensitive variant of the native penicillin-binding protein110. Two additional resistance mechanisms rely on keeping the antibiotic out of the bacterial cell, through either efflux or altering the permeability of the cell membrane. By expressing either a generalist multidrug efflux pump111–114 or an antibiotic-specific exporter, such as tetracycline efflux pumps115, bacteria keep the intracellular concentration of the antibiotic below inhibitory levels. Alternatively, some bacteria reduce their cell membrane or cell wall permeability to prevent antibiotics from entering the cell by decreasing porin expression or expressing a more selective porin variant116,117. In some cases, bacteria will use several complementary mechanisms to achieve high levels of resistance. For example, high-level carbapenem resistance in clinical isolates of Enterobacter cloacae that lack any known carbapenemase can be achieved through a combination of porin mutations that decrease carbapenem uptake and increased expression of a chromosomal non-carbapenemase β-lactamase118.
As a result of the ubiquity of antibiotic production by environmental microorganisms, defence mechanisms against these antibiotics and thus resistance (BOX 1) also seem to be ancient. Sequencing of ancient environmental DNA preserved in 30,000-year-old permafrost sediments has detected genes that confer resistance to several classes of antibiotic8, and the direct screening of bacteria cultured from a cave that had no anthropogenic contact for the past four million years revealed extensive resistance against 14 antibiotics9. Similarly, the host-associated microbiome of previously uncontacted indigenous people in South America was found to encode several antibiotic resistance genes10. This indicates that resistance to antibiotics far pre-dates the appropriation of these small molecules for human use.
The collection of resistance genes in a given environment, which is termed the ‘resistome’ (REFS11,12), encompasses both intrinsic and acquired resistance genes, as well as proto-resistance genes and silent or cryptic resistance genes. It is thought that many clinically relevant antibiotic resistance genes have their evolutionary origins in environmental microorganisms, which we define for the purposes of this article to comprise microorganisms that are not generally associated with humans. This hypothesis was pioneered by Benveniste and Davies13, who, in 1973, noted biochemically identical resistance-conferring kanamycin acetyltransferases in environmental Streptomyces spp. and in clinical isolates of pathogenic Escherichia coli. Further evidence was provided by a recent functional screen of soil metagenomes, which revealed the presence of environmental antibiotic resistance genes that have >99% nucleotide identity to resistance genes in pathogenic isolates14. Synteny of these genes with other resistance genes and mobility elements suggests that they are likely candidates for past or future dissemination by horizontal gene transfer (FIG. 1).
Figure 1. The synteny of antibiotic resistance genes provides historical context and foreshadows future.
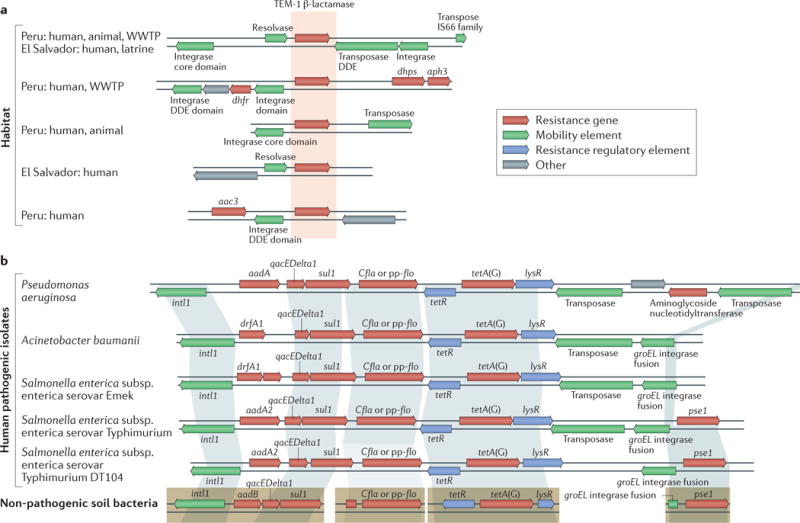
The genetic context of antibiotic resistance genes can provide important evidence regarding the likelihood of past or future horizontal gene transfer. a | A single β-lactamase (TEM-1) was found in 25 different genetic contexts in a recent cross-habitat resistome study, which provided a historical record of past mobility. Five contexts are shown here. Habitats in which a gene was discovered are indicated on the left (‘human’: human-associated microbial community; ‘animal’: animal-associated microbial community; ‘WWTP’: wastewater treatment plant; ‘latrine’: composting latrine). b | Four resistance determinants that were discovered in functional selections of soil metagenomic libraries (bottom) have high identity with resistance genes from clinical pathogenic isolates. Shading indicates >99% nucleotide identity, which suggests recent horizontal transfer. Resistance genes in pathogens are syntenic with mobility elements and other resistance genes, which suggests that they may be present on a mobile multidrug resistance cassette and are therefore likely to undergo horizontal gene transfer. DDE, Asp-Asp-Glu. Part a is adapted with permission from REF.66, Macmillan Publishers Limited. Part b is adapted with permission from REF.14, AAAS.
The ancient origins of antibiotics and antibiotic resistance make it unsurprising that large-scale clinical and agricultural use of these compounds has selected for increasing resistance across the bacterial domain of life. Robust evidence for increasing environmental resistance exists in decades-old soil samples, in which increasing numbers of antibiotic resistance genes correlate with the industrial-scale production and use of these drugs15. The resulting deterioration of the efficacy of our antibacterial armamentarium has come with substantial costs that are expected to increase throughout the next decades. Worldwide, it has been estimated that at least 700,000 people die annually as a result of resistant infections, a figure that is predicted to increase to 10 million annual deaths by the year 2050 if the current trajectory is not altered. Throughout the next few decades, the cumulative global cost of antibiotic resistance is predicted to exceed US$100 trillion16.
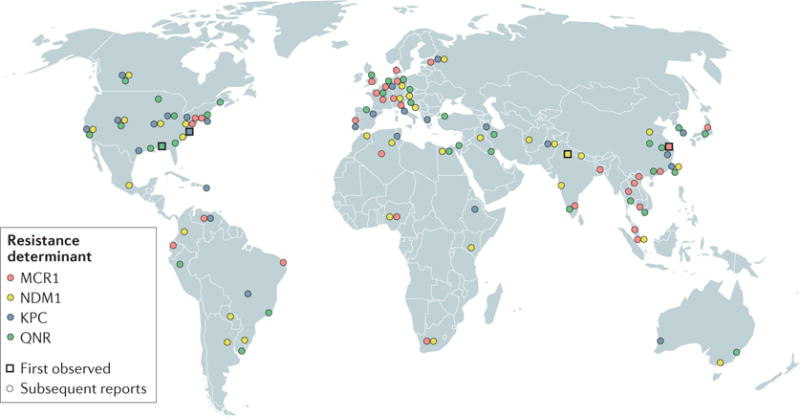
The recent spread of mcr1, kpc, blaNDM1, qnr and other plasmid-borne resistance determinants (BOX 2) emphasizes two themes. The first is that in the antibiotic ‘arms race’ against bacteria, humanity is rarely ahead. This was exemplified by the first widely used antibiotics, the sulfonamides, against which resistance was observed only 6 years following their mass introduction17. More recently, resistance to tigecycline was identified even before its approval for human use, let alone its introduction into the clinic18. Therefore, it is either a sign of hubris or a lack of understanding of evolutionary principles when new antibiotics are advertised as being resistance-proof. The second theme has been our relatively myopic (albeit understandable) focus on resistance in human pathogens. It is a tautology that antibiotics are most effective at treating human pathogens. They would not be antibiotics if they failed to effectively treat pathogens, but our emphasis on a few pathogen ‘trees’ initially resulted in the field of antibiotics being blind to the ‘forest’ of environmental bacteria. The targets of most antibiotics are both highly conserved and essential across bacterial taxa, and have therefore made excellent mediators of intra-kingdom communication and competition among bacteria throughout biological history19. When our view is expanded to include the vast diversity of non-pathogenic bacteria, it is not surprising to see that all genera of the bacterial kingdom have evolved resistance mechanisms that protect these vital processes.
Box 2. Global spread of plasmid-borne resistance.
In the past several decades, various new antibiotic resistance threats have emerged and spread worldwide. In particular, the emergence and spread of plasmid-borne resistance genes (see the figure and Supplementary information S1 (table) for specific reports of plasmid-borne resistance) — which are likely to undergo horizontal gene transfer — have jeopardized many last-line antibiotic therapies, including the cephalosporin and carbapenem families of β-lactam antibiotics, quinolones, such as ciprofloxacin, and an antibiotic of last resort, colistin5. Cephalosporins have been compromised by resistance owing to extended spectrum β-lactamases (ESBLs), which were first reported in 1979 (REF.119), and subsequently spread phylogenetically across the Enterobacteriaceae and geographically across the globe120. Consequently, many infections that were previously treated by drugs such as cefotaxime must now be treated with the carbapenem class of antibiotics. However, the spread of carbapenemases121 — particularly New Delhi metallo-β-lactamase 1 (NDM1)122–124 and Klebsiella pneumoniae carbapenemases (KPCs)125 since their respective discoveries in 2006 and 2001 — has also rendered the carbapenem class of antibiotics vulnerable126 (see the figure).
A similar development has been observed for the quinolones, which were originally touted to be at low risk of displaying the type of transferable resistance that has beleaguered the β-lactam antibiotics. Indeed, whereas chromosomal resistance to this class of antibiotic can readily develop through mutations in DNA gyrase, transferable resistance was slow to develop following the clinical introduction of these antibiotics in the 1960s. Nonetheless, plasmid-mediated resistance through the quinolone resistance (QNR) proteins, first described in 1998, has emerged from quinolone overuse and has subsequently spread worldwide (see the figure). In the search for the origins of QNR resistance determinants, a PCR screen of Gram-negative organisms found homologues in the aquatic bacterium Shewanella algae that have almost 99% amino acid identity127. Shewanella species that lack these homologues were found to have a fourfold to eightfold greater sensitivity to quinolone antibiotics than those that expressed these homologues, which indicates the ability of the qnr-related genes to confer some antibiotic resistance even in their native contexts, in which they probably bind to DNA for some other purpose2,127. The hypothesized horizontal transfer of qnr homologues from environmental to pathogenic bacteria corroborates hypotheses that propose that many resistance genes may have been co-opted by pathogens from the environmental resistome13,14,127,128. Quinolone efficacy has been further reduced by the emergence of the plasmid-borne bifunctional aminoglycoside and quinolone resistance gene aac(6′)-Ib-cr in Shanghai in 2003 (REF.129). This gene, which differs from that encoding a classical aminoglycoside transferase by only two base pairs, is notable in its ability to modify fluoroquinolones and aminoglycosides through acetylation. Alarmingly, aac(6′)-Ib-cr is often colocalized with other resistance genes, such as ESBLs130, on multidrug resistance plasmids.
The spread of the aforementioned resistance mechanisms leaves only a few classes of antibiotic approved for the treatment of infection by carbapenem-resistant members of the Enterobacteriaceae. One of these, the polymyxin class of peptide antibiotics, was first used in the 1960s but was abandoned in favour of other antibiotics owing to its toxicity. Recently, the use of the polymyxin colistin has been revived owing to its efficacy against several multidrug-resistant Gram-negative pathogens131. However, transferable resistance has now begun to threaten colistin. In 2015, a colistin-resistant Escherichia coli isolate from a pig farm in Shanghai was found to contain mcr1 — a plasmid-borne gene that encodes a phosphoethanolamine transferase50. This finding, and the subsequent discovery of mcr1 on plasmids worldwide51–59 (see the figure), foreshadows the fall of colistin as a last-resort antibiotic that lacks transferable plasmid-encoded resistance. With the high levels of antibiotic prescription in medicine and unregulated antibiotic use in agriculture around the world it is likely that it is only a matter of time before completely pan-resistant plasmids are found that combine the above resistance mechanisms. Indeed, the coexistence of NDM1 and MCR1 in members of the Enterobacteriaceae has already been observed55,132, and a recent report documented the co-transfer of mcr1 and ndm5 on a mobile hybrid IncX3–X4 plasmid133.
In this Review, we examine these themes across three eras of antibiotic resistance research. The first era, which coincides with the golden age of antibiotics, was characterized by a focus on resistance in pathogens in the clinic, and by the ready supply of new antibiotics and the diverse antibiotic mechanisms of action. In this era, drawing inspiration from Koch, the research priority was on single resistance genes and enzymes primarily from pathogenic organisms in axenic culture. In the second era of antibiotic resistance studies, we became less restricted by Koch’s postulates and shifted our attention to resistomes. The antibiotic resistance of both benign and pathogenic bacteria was studied at the microbial community level, and metagenomic approaches were incorporated to discover resistance determinants from diverse habitats. As we enter the next era of antibiotic resistance research, we should acknowledge the ubiquity of antibiotic resistance, and take steps to improve the development of new antibiotic therapies and the surveillance of emerging resistance threats, focusing on resistance genes that are the most likely candidates for horizontal transfer to pathogens. In the remainder of this Review, we provide brief overviews of these three eras of resistome studies, and we outline how distinct philosophies from each of these eras could be integrated to improve the translational effect of resistome research going forwards.
The golden age of antibiotics
Both Sir Alexander Fleming and Gerhard Domagk, who discovered penicillin and prontosil, respectively, warned of the danger of bacterial resistance developing following improper use of their antibiotics20,21. One of the first reports of antibiotic resistance was the finding that a cell-free extract from a coliform bacterium that was unaffected by penicillin was able to inactivate penicillin, which indicated enzymatic degradation of the drug22. In the following decades, the extensive use of penicillin in hospitals led to the development of widespread resistance in Staphylococcus aureus and resulted in the 1953 penicillin-resistant S. aureus phage-type 80/81 pandemic, which represented the world’s first superbug until the release of methicillin ended the threat23. Soon after, the emergence of multidrug-resistant strains of Shigella spp., following apparent horizontal gene transfer from faecal strains of E. coli, was reported. Remarkably, there was evidence that such transfers occurred in hospitalized patients during the course of their antibiotic treatment24.
The first era of resistance studies was concomitant with the golden age of antibiotics. During this period, all of the major modern classes of antibiotic were discovered. Within years, many were rendered obsolete in at least some organisms owing to the acquisition and spread of resistance17. For a period of time, novel or previously known mechanisms of resistance that arose in new pathogens were commonly dealt with by developing and releasing a new antibiotic. Indeed, resistant bacteria were even welcomed by Domagk as an opportunity to increase the antibiotic market share20. During this era, resistance genes and bacteria were largely identified one gene and one strain at a time using culture-based methods. This low-throughput approach, combined with incompletely exhausted reserves of natural antibiotics, disguised the otherwise apparent threat that antibiotic-resistant bacteria were destined to become.
High-throughput resistance discovery
The second era of resistome studies has been characterized by a broader ecological view of resistance, which was enabled by several breakthrough technologies. In the past few decades, the development of the new ecological resistome philosophy that recognizes the environment as the plausible original source of resistance genes and as the reservoir of resistance gene diversity has been driven by the technical advances in molecular biology and nucleic acid sequencing.
Sequencing-based resistance discovery
The development of genomic techniques that enable the culture-independent study of diverse environmental microbial communities has been a crucial driver of the paradigm shift in resistome studies25–28. This is illustrated by the growth of the catalogue of known resistance genes, which has occurred alongside a rapid decrease in sequencing costs. A survey of resistance proteins in the UniProt database29 demonstrates that since 1986 there has been an exponential increase in the number of hypothetical resistance determinants classified as β-lactamases, chloram phenicol acetyltransferases or tetracycline efflux pumps (FIG. 2a). The surfeit of sequencing data exemplifies the second era of resistance studies but in many cases, the lack of functional validation of these genes calls into question whether all of them are resistance determinants. Similarly, the sequence diversity that is captured using cutting-edge techniques may not translate into functional diversity. One technique that has proven useful in the discovery of resistance determinants that are novel, diverse and validated is functional metagenomics (BOX 3), which is a technique that has been developed to mine metagenomes for various phenotypes without a need for culturing30. Functional metagenomics has proven to be essential for the characterization of antibiotic resistomes. Briefly, this technique consists of cloning metagenomic DNA into expression vectors and screening for a phenotype of interest (for example, antibiotic resistance) in a facile host organism. This bypasses the bottleneck that is imposed by bacteria that are difficult to culture and explicitly selects for genes that can spread by horizontal transfer, as only phenotypes that are transferable to a heterologous host are detected. However, antibiotic resistance that results from mutation of the antibiotic target, which is an important contributor to resistance in many organisms, such as Mycobacterium tuberculosis31,32, may not always be detected in functional metagenomic screens, as a single wild-type copy of these genes in the expression host often confers a dominant susceptible phenotype. This is the case for quinolone resistance that results from mutations in the DNA gyrase enzyme31, although the analysis of evolutionary patterns in such genes may enable antibiotic resistance risk estimates to be made33–35. Furthermore, the artificial action of extracting and cloning DNA can lead to the transfer of genes that otherwise might not be mobilizable, which possibly raises concerns in regard to the spread of resistance in cases in which the actual risk may be low, but it may also result in the underestimation of sequences related to mobility elements, which are not explicitly selected for in screens for antibiotic resistance.
Figure 2. Next-generation sequencing and functional metagenomic selection accelerate the cataloguing of known and novel resistance genes.
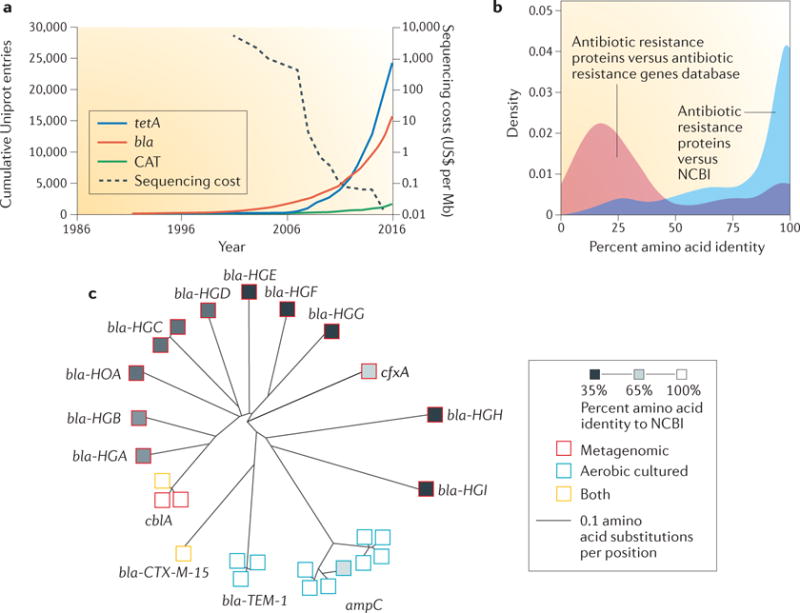
a | The catalogue of resistance genes has increased exponentially in the second era of antibiotic resistome studies owing to improvements in next-generation sequencing. This trend is exemplified by the tetracycline efflux pump-encoding gene tetA, by the bla family encoding β-lactamases and by chloramphenicol acetyl transferase (CAT). Data from the UniProt database29, which was accessed on 12 October 2016; the database was searched using Enzyme Commission numbers (bla: 3.5.2.6; CAT: 2.3.1.28) or a family name (tetA). Data on sequencing costs are from National Human Genome Research Institute webpage28. b | Functional metagenomics is a powerful tool for ascribing antibiotic resistance function to known gene products. Antibiotic resistance proteins identified by functional metagenomic selection of the preterm infant gut microbiome have high amino acid identity to proteins in the National Center for Biotechnology Information (NCBI) database but low amino acid identity to proteins in antibiotic resistance databases. c | In addition, functional metagenomics can reveal entirely sequence-novel resistance determinants. β-Lactamases that were identified by functional metagenomic selection of two human gut resistomes (red squares) encompassed greater phylogenetic diversity than all previously identified classes of β-lactamase (blue squares). Part b is adapted with permission from REF.36, Macmillan Publishers Limited. Part c is adapted with permission from REF.62, AAAS.
Box 3. Functional metagenomic approaches for characterizing resistomes.
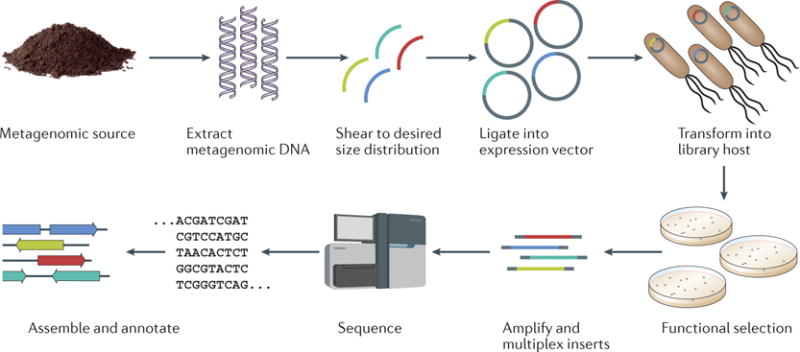
Functional metagenomics is a powerful culture-unbiased and sequence-unbiased tool134. A metagenomic library is created by extracting total metagenomic DNA from a microbial community, shearing the DNA to a target size distribution and cloning these fragments en masse into a screening vector (see the figure). The metagenomic library is then transformed into a heterologous host and selected for a phenotype of interest by, for example, culturing with antibiotics at a concentration that is lethal to the wild-type host27,135. Pairing functional metagenomic selection with next-generation sequencing and better sequence assembly and annotation algorithms14 has simultaneously decreased the cost and increased the throughput of functional metagenomic experiments, and this has facilitated the characterization of resistomes from diverse habitats10,18,36,48,66. Importantly, there is no need to culture members of the target microbial community, and novel resistance genes can be discovered. Furthermore, the genes discovered by functional metagenomics are, by definition, candidates for horizontal transfer, as they must be functional in a heterologous host.
However, there are several limitations of functional metagenomic approaches. For a gene to be identified in a functional metagenomic screen, it must be functional in a heterologous host. Common hosts, such as Escherichia coli, probably present a poor system for the discovery of resistance genes that originated from Gram-positive organisms. Indeed, poor overlap has been reported between clones recovered from the same functional metagenomic library that were subjected to the same screen but in different hosts136,137. In addition, it is generally difficult to screen Gram-positive bacteria-specific antibiotics in E. coli or other common Gram-negative platforms owing to their innate resistance, although in some cases this may be overcome138. Thus, there is a clear need to develop and screen functional libraries in phylogenetically diverse heterologous hosts134,139. Furthermore, functional metagenomic experiments may oversample resistomes, as the method struggles to distinguish resistance genes that pose clinical threats from those that are unlikely to spread beyond their current environment. In addition, genes that confer antibiotic resistance when overexpressed in a heterologous host may not be genuine resistance genes in their native contexts35. To reduce the risk of false-positives, it is important that novel resistance genes are validated using complementary microbiological and biochemical methods, and are cross-checked against existing databases. Finally, the removal of an antibiotic resistance gene from its genomic context often also removes positional information that is pertinent to assessing its risk of dissemination. This can be overcome, in part, by altering insert size, as larger inserts may contain informative regions that flank resistance genes, including mobility elements or plasmid sequences that might indicate the risk of spreading (that is, the mobilome)48,66,140.
The coupling of functional metagenomic selections with high-throughput sequencing and computational methods has facilitated an increase in our ability to catalogue resistomes from diverse habitats. Importantly, functional metagenomics can both ascribe a resistance function to known genes that were not previously identified as resistance determinants and discover resistance genes with novel sequences. For example, a recent analysis of functionally discovered resistance genes found that although many of the gene products had high amino acid identity to known enzymes, most had low identity to known resistance determinants36 (FIG. 2b). Similarly, the utility of functional metagenomics in elucidating entirely new resistance functions is highlighted by its recent application to the discovery of novel rifamycin phosphorylases37, dihydrofolate reductases38 and tetracycline-inactivating enzymes39.
The resistomes of different habitats
Functional metagenomics was first described in 2000 as a method for accessing the diverse biochemical activities encoded in the uncultured soil metagenome30. In an early application of functional metagenomics for the explicit purpose of screening for antibiotic resistance genes, a library that contained 5.4 Gb of metagenomic DNA from a remnant oak savannah site was selected against a panel of aminoglycoside antibiotics, as well as tetracycline and nalidixic acid40. Sequencing of 10 resistant clones revealed that all but one contained novel resistance determinants. Since these initial studies, functional metagenomics has been widely used to screen a diverse range of soil metagenomes, including remote arctic soils41–43, apple orchard soils44, alluvial soils45,46, wetland soils47, agricultural soils38,48 and urban soils49, and has identified novel genes that confer resistance to diverse classes of antibiotic. Agricultural resistomes are of particular interest because the selective pressures of heavy antibiotic use and extensive anthropogenic interaction make this a high-risk habitat for antibiotic resistance selection and dissemination. Indeed, the mcr1 plasmid-mediated colistin resistance determinant was first discovered on a Chinese pig farm50, although it has since been detected in new and banked clinical and environmental samples across the globe51–59 (BOX 2).
In addition to soil habitats, human-associated resistomes have been a recent focus of functional metagenomic investigations. The first human-associated microbial community to be queried for antibiotic resistance by functional metagenomic selection was the oral microbiome. Screening of a small functional library constructed from dental plaque and saliva samples against tetracycline resulted in the discovery of tet(37), a gene that encodes a tetracycline-inactivating enzyme that has low sequence identity to any previously described tetracycline resistance determinant, as well as other resistance genes60. A larger follow-up study revealed resistance against tetracycline, gentamicin and amoxicillin in the oral microbiome61. The first report of a functional metagenomic interrogation of the human gut resistome screened metagenomic DNA from saliva and stool samples from two healthy humans against a panel of 13 antibiotics62. Sequencing of 95 unique inserts, which together conferred resistance to all of the 13 antibiotics that were screened, revealed that they were largely dissimilar to the closest known resistance genes. For example, 10 novel β-lactamases were identified in just two gut microbiomes. Remarkably, these microbiomes encompassed greater phylogenetic β-lactamase diversity than all previously described β-lactamases (FIG. 2c). Additional studies have examined the gut microbiomes of diverse human populations, including healthy adults63, healthy twins and their mothers18,64,65, preterm infants36, uncontacted antibiotic-naive Amerindians10, and families, as well as livestock and pets, from developing countries in Latin America66. Importantly, these studies have made it clear that, although the resident human-associated resistome is enriched after antibiotic therapy36, the gut contains an extensive resistome even in the absence of anthropogenic antibiotic use10,66.
Limitations of resistome studies
Although these studies are extremely valuable in cataloguing the genetic diversity of the global resistome, they have been largely descriptive or focused on collecting data, and thus require substantial downstream mechanistic validation. It is important to emphasize that just because a gene confers resistance when it is overexpressed in a heterologous host, it may not be a bona fide resistance gene in the original host. Indeed, nuanced definitions of antibiotic resistance should consider the function of resistance determinants in their native contexts and the risk of transmission of resistance determinants between bacterial species35. There is extensive evidence that resistome composition is strongly correlated with the phylogenetic structure of a microbial community, and that the synteny of antibiotic resistance genes with mobility elements is observed less frequently in environmental meta genomes (for example, those from soil) than in sequenced pathogen genomes48,67 (FIG. 1b). Nonetheless, the discovery of identical resistance genes in multiple genomic and environmental contexts suggests historical transfer and/or dissemination events. For example, a recent functional metagenomic study identified an identical TEM-1 β-lactamase in 25 different habitats and genetic backgrounds66 (FIG. 1a). This research, when viewed in light of recent emerging resistance threats (BOX 2), emphasizes that although not all resistance genes are threats for widespread dissemination, we should pay close attention to the ones that are. A focus of future studies on antibiotic resistomes should be the quantification of risk of horizontal gene transfer to pathogens based on ‘metadata’ associated with a given resistance gene (for example, synteny with mobility elements, whether it is plasmid-borne or chromosomal, and whether it is found in a close relative of a human pathogen) to model the risk of transmission34,35.
Next-generation approaches to resistance
Strategies to combat antibiotic resistance will require substantial changes both to the antibiotic pipeline and stewardship practices, and to antibiotic resistance surveillance. The ubiquity of antibiotic resistance suggests that the drug development pipeline must include proactive screens for existing environmental resistance threats to new drugs and should explore alternative strategies for combating resistance outside of the traditional antibiotic pipeline. In addition, it is the responsibility of the wider medical and scientific communities to apply and combine the findings from the first two eras of resistome studies in efforts to surveil potential antibiotic resistance hotspots for emerging resistance threats before they spread.
Next-generation therapeutics
A key aspect that requires investigation in the next generation of antibiotic resistance studies is the proactive development of therapeutic strategies to counteract emerging antibiotic resistance. One side of this approach involves having a greater focus on resistance potential when developing new and improved antibiotics. Current antibiotic discovery and development pipelines only take into account resistance mechanisms that are already prevalent in the clinic. In retrospect, we know that this strategy is doomed to fail, as resistance has been observed for every antibiotic that has been implemented for human use17. Going forwards, we should improve on the current reactive strategies by screening promising lead compounds for resistance against diverse functional metagenomic libraries. This will identify existing mechanisms of resistance that are likely to disseminate to the clinical setting following the introduction of the antibiotic for clinical use (FIG. 3).
Figure 3. The integration of next-generation sequencing and screening technologies with drug development and resistance surveillance.
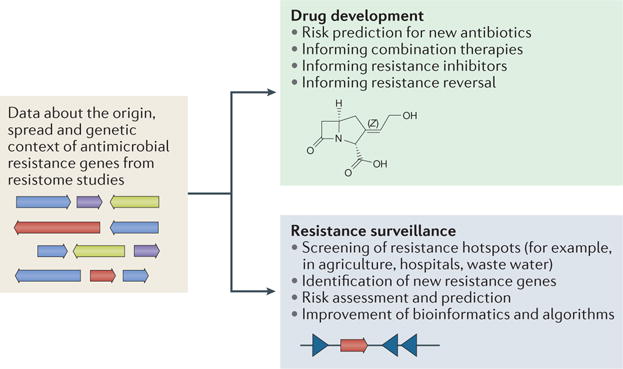
The wide application of functional metagenomic selections and next-generation sequencing will steadily increase our knowledge of functionally annotated antibiotic resistance genes and their genetic context. This will enable more intelligent antibiotic resistance surveillance and drug design by accounting for specific mechanisms of resistance and the risk of resistance spread.
In the near future, our understanding of the resistome will be sufficiently mature to enable the development of strategies that actively combat resistance mechanisms themselves in contrast to current strategies that side-step resistance through the expensive development of new antibiotics that immediately select for new mechanisms of resistance. This new approach, in our opinion, provides a higher likelihood of success than searching for a fabled ‘silver bullet’ antibiotic to which resistance is unlikely to develop. One such strategy is the repurposing of existing antibiotics in synergistic combinations. For example, consider the triple β-lactam combination of meropenem, piperacillin and tazobactam, which synergistically kills methicillin-resistant S. aureus (MRSA) N315 in vitro and in a mouse infection model68. The bacteria were collaterally sensitive to the combination of these three antibiotics, and thus the combination suppressed the evolution of further resistance68. Treatment regimens that induce collateral sensitivity not only enable the elimination of resistant bacteria but also subject resistant bacteria to selective pressure to discard their resistance gene69. Inverting the selective advantage of antibiotic resistance is a promising approach for slowing the rate at which resistance evolves in a population. A recent highlight in the search for selection-inverting compounds involved a screen of nearly 20,000 small molecules for compounds that select against the tetracycline efflux pump TetA. This screen identified two candidates that successfully selected for the loss of the tetA gene70. Perhaps more important are drugs under development and in early-phase testing that target AcrA–AcrB–TolC efflux complexes in Gram-negative bacteria. Unlike TetA proteins, which are tetra cycline-specific, AcrA–AcrB–TolC pumps have broad substrate tolerance and can confer multidrug resistance, particularly in Gram-negative bacteria71.
A final strategy that has already proven to be effective in opposing widespread resistance is the inhibition of antibiotic resistance enzymes72. Small molecules that lack antibiotic activity but enhance the efficacy of antibiotic therapy are broadly termed antibiotic adjuvants73. β-Lactamase inhibitors and TetA efflux selection inversion70,72,74 fall in this category, as do promising examples from recent studies that paired antibiotics with repurposed drugs that are already on the market for other indications, such as the opioid-receptor agonist loperamide, which facilitates tetracycline uptake, and the ADP receptor antagonist ticlopidine, which inhibits teichoic acid biosynthesis3,73,75,76. The continued discovery of antibiotic adjuvants is facilitated and informed by resistome studies that comprehensively describe the collection of possible resistance genes. For example, consider the recently described E. coli-hosted array of more than 40 mechanistically distinct and functionally validated resistance genes77. This platform, which represented resistance to 16 classes of antibiotics, was used to screen a limited small-molecule library for compounds that could enhance the antibiotic activity of gentamicin. Of the 27 compounds that exhibited this activity, eight were found to inhibit aminoglycoside-2″-O-nucleotidyltransferase (ANT(2″)-Ia)-mediated resistance. Two compounds that were selected for further validation exhibited poor antibiotic activity alone and were confirmed to inhibit ANT(2″)-Ia in vitro. By screening drug combinations against a phenotypic resistome array, the authors were able to identify inhibitors of antibiotic resistance genes, thus demonstrating the utility of leveraging resistome data to discover adjuvants that can prolong the efficacy of existing antibiotics77.
Next-generation surveillance
Lastly, it is important to prioritize continued and increased surveillance of the antibiotic resistome, particularly hotspots in which there is a high likelihood of resistance gene evolution and transfer between organisms (FIG. 3). These hotspots include, but are not limited to, agricultural settings in which large quantities of antibiotics and human pathogens, as well as environmental microorganisms, coexist and potentially exchange DNA50; hospitals in which selective pressure is omnipresent owing to the extensive use of antibiotics and antimicrobials, which may co-select for antibiotic resistance24; and wastewater and sewage systems, which directly expose the human gut microbiota to many pharmaceutical residues66. Surveillance requires regular testing of pathogens, commensals and microbial communities for resistance to a broad range of antibiotics. This can occur using culture-dependent or genomic approaches, and necessarily entails the continued development of technologies that will facilitate the identification of antibiotic resistance determinants.
Recent computational and bioinformatic advances have substantially improved our ability to identify antibiotic resistance genes from PCR amplicons and whole-genome sequencing (WGS)27. These databases contain nucleotide or protein sequences for known antibiotic resistance determinants, and can be queried (by BLAST78, for example) to identify the antibiotic resistance genes that are present in a given genome or metagenome. However, these functional predictions are explicitly based only on sequence data and are vulnerable to false-positive predictions, particularly when functional assignments are based on annotations that have not themselves been directly validated biochemically or phenotypically. A key component of our ability to annotate and predict antibiotic resistance genes from genomic data is the continued improvement of antibiotic resistance gene databases through functional validation79–84. In addition, improved probabilistic annotation algorithms, such as profile hidden Markov models, have been shown to perform substantially better than pairwise sequence alignment in terms of precision and recall85,86.
For example, Resfams, a curated database of 166 profile hidden Markov models that were constructed from functionally validated families of antibiotic resistance proteins, correctly predicted 45% more β-lactamases in functionally selected metagenomic contigs than BLAST alignment to antibiotic resistance databases84. In other studies, nucleotide sequences from metagenomic datasets were used to computationally predict qnr genes that might confer resistance to quinolone87,88. Although qnr genes have low similarity to other resistance determinants and relatively high in-family diversity, the authors were able to discover and functionally verify several novel variants87,88. As a result of the development of these tools, we are now able to more confidently identify both transferable and intrinsic resistance determinants from genomes and metagenomes. Going forwards, it is important that these databases are regularly updated with the nucleotide and amino acid sequences of functionally validated, emerging resistance determinants (FIG. 3).
Despite these advances, the prediction of resistance phenotype on the basis of genotype remains a challenge. WGS-based methods are a promising approach to improve the speed and accuracy of diagnostics89–91. An additional advantage of WGS-based diagnostics is the possibility for the improved identification of antibiotic resistance by target modification, which is often not feasible using existing PCR-based or microarray-based methods91–93. A current highlight of sequencing-based diagnostics is the Mykrobe predictor, which identified antibiotic susceptibility profiles for 12 antibiotics from raw, unassembled Illumina sequencing data from S. aureus and M. tuberculosis isolates, as well as from in silico-simulated heterogeneous populations, with high sensitivity and specificity94. A recently described alternative approach uses draft genome assemblies as inputs and a machine-learning approach to accurately predict resistance profiles for multidrug-resistant Enterobacteriaceae isolates86. Although it is currently cost-prohibitive and time-prohibitive to use WGS as a clinical diagnostic tool, continued reductions in sequencing costs and improvements in computational algorithms may soon make this approach feasible. Faster and more accurate diagnostics will considerably decrease the number of empirical and unsuitable antibiotic prescriptions, thus limiting further selection for resistance (FIG. 3).
Two recent examples from the literature highlight the importance of surveillance efforts as we attempt to prevent the spread of antibiotic resistance. These examples pertain to the current last-resort antibiotics against carbapenem-resistant members of the Enterobacteriaceae: namely, colistin and tigecycline. The first example is the emergence of the mcr1 gene as a transmissible threat to colistin use. The seminal example of a plasmid-borne colistin-resistance gene was found through an active surveillance programme in China that monitored antimicrobial resistance in animals that were reared to be consumed by humans50. Soon after its discovery, several retrospective studies demonstrated that plasmid-borne mcr1 had already spread worldwide and into the clinic95 (BOX 2). In this case, surveillance provided crucial early detection. The second example of the importance of surveillance relates to tigecycline, a semi-synthetic tetracycline antibiotic96. Owing to its increased affinity for the bacterial ribosome compared with other tetracyclines, and to the presence of a glycyl tail, tigecycline is subject to reduced resistance through ribosomal protection or drug efflux97. However, tigecycline is sensitive to the tetracycline-inactivating protein TetX98. Although this protein was originally discovered in Bacteroides fragilis — a usually non-pathogenic human gut commensal — several clinical isolates have recently tested positive for tetX99,100, and tetracycline inactivation has been discovered to be more widespread in environmental metagenomes than was previously thought39. Continued surveillance of this apparent emerging resistance mechanism is prudent, as tigecycline and other next-generation tetracycline derivatives are being designed to exclusively evade resistance through ribosomal protection and efflux101,102. The increased use of next-generation tetracyclines imposes a strong selective pressure on the acquisition and spread of tetracycline-inactivating enzymes from the environmental resistome. Foresight of this event should enable the proactive development of next-generation therapeutic strategies before the widespread dissemination of a resistance threat.
Conclusions
Extensive study of the antibiotic resistome throughout the past few decades has enabled us to begin to understand and address existing antibiotic resistance threats. However, it is evident that antibiotic resistance will continue to evolve and spread despite our best efforts to develop new antibacterial agents. Unless current practice is changed, it will be increasingly difficult to establish and maintain even a transient lead over bacteria in this ‘arms race’. As research into antibiotic resistance expands, it is important to adopt an explicitly proactive approach to antibiotic resistance identification and surveillance, as well as antibiotic therapy development. This proactive approach involves using a combination of functional metagenomics, next-generation sequencing and cutting-edge computational methods to monitor the evolution and dissemination of resistance before a given resistance determinant emerges in a pathogen or in the clinical setting, as well as proactively developing next-generation therapies that target these resistance determinants. The spread of plasmid-borne carbapenem, quinolone and polymyxin resistance in recent years is a sobering reminder of our need to mitigate existing threats and to anticipate emerging resistance mechanisms before they circulate widely in the clinical setting if we wish to alter the current bleak antibiotic resistance trajectory. Recent advances in the field highlight the promise that the next generation of resistome studies hold for characterizing and countering emerging resistance threats.
Supplementary Material
Acknowledgments
This work was supported, in part, by awards to G.D. through the Edward Mallinckrodt Jr. Foundation (Scholar Award), and from the US National Institutes of Health (NIH) Director’s New Innovator Award (http://commonfund.nih.gov/newinno-vator/), the National Institute of Diabetes and Digestive and Kidney Diseases (http://www.niddk.nih.gov/), the National Institute of General Medical Sciences (NIGMS; http://www.nigms.nih.gov/) and the National Institute of Allergy and Infectious Diseases (https://www.niaid.nih.gov/) of the NIH under award numbers DP2DK098089, R01GM099538 and R01AI123394, respectively. T.S.C. received support from a National Institute of Child Health and Development training grant through award number T32 HD049305 (K. H. Moley is named as the Principal Investigator on this grant). A.J.G. received support from a NIGMS training grant through award number T32 GM007067 (J. Skeath is named as the Principal Investigator on this grant). The content of this Review is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Glossary
- Antibiotics
Drugs that inhibit the growth of bacteria or kill them
- Natural products
Small molecules that are naturally produced by living organisms
- Proto-resistance genes
Genes that have the potential to evolve a resistance function
- Cryptic resistance genes
A resistance gene that is embedded in a bacterial chromosome, but that is not obviously associated with antibiotic resistance. The respective gene is usually either not expressed or expressed at low levels
- Metagenomes
The collective genetic material in a given environment
- Synteny
The occurrence of multiple genes in the same genetic locus
- Horizontal gene transfer
The transmission of genetic material between bacterial organisms by transformation, transduction or conjugation, in contrast to vertical transmission through heredity
- Axenic
A term that describes a culture that contains only a single species
- Koch’s postulates
A series of criteria proposed by Robert Koch in 1890 that can be used to establish a causal relationship between a microorganism and a disease
- Collaterally sensitive
A term that describes organisms that develop resistance to one antibiotic and, as a result of the new mutation, are more sensitive to another antibiotic
- PCR
A molecular biology technique that is used to amplify nucleic acids of known sequence
- Whole-genome sequencing
(WGS). The use of next-generation sequencing to determine the complete sequence of an organism’s genome
- Hidden Markov models
Statistical models that are widely used in biological sequence analysis and annotation
- Semi-synthetic
A term that describes a small molecule that is produced by chemical modification of a natural product
- Next-generation sequencing
High-throughput nucleic acid sequencing technologies that have emerged in the past few decades to enable substantial increases in sequencing capacity
Footnotes
Competing interests statement
The authors declare no competing interests.
References
- 1.Walsh CT, Wencewicz TA. Prospects for new antibiotics: a molecule-centered perspective. J Antibiot (Tokyo) 2014;67:7–22. doi: 10.1038/ja.2013.49. [DOI] [PubMed] [Google Scholar]
- 2.Aminov RI. A brief history of the antibiotic era: lessons learned and challenges for the future. Front Microbiol. 2010;1:134. doi: 10.3389/fmicb.2010.00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown ED, Wright GD. Antibacterial drug discovery in the resistance era. Nature. 2016;529:336–343. doi: 10.1038/nature17042. [DOI] [PubMed] [Google Scholar]
- 4.Van Boeckel TP, et al. Global antibiotic consumption 2000 to 2010: an analysis of national pharmaceutical sales data. Lancet Infect Dis. 2014;14:742–750. doi: 10.1016/S1473-3099(14)70780-7. [DOI] [PubMed] [Google Scholar]
- 5.Wright PM, Seiple IB, Myers AG. The evolving role of chemical synthesis in antibacterial drug discovery. Angew Chem Int Ed Engl. 2014;53:8840–8869. doi: 10.1002/anie.201310843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walsh C. Antibiotics: Actions, Origins, Resistance. ASM Press; 2003. [Google Scholar]
- 7.Tyndall J. Observations on the optical deportment of the atmosphere in reference to the phenomena of putrefaction and infection. Br Med J. 1876;1:121–124. doi: 10.1136/bmj.1.787.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.D’Costa VM, et al. Antibiotic resistance is ancient. Nature. 2011;477:457–461. doi: 10.1038/nature10388. This study identifies resistance genes in 30,000-year-old Beringian permafrost sediments, thus providing strong genetic evidence that resistance genes in environmental microorganisms pre-date the anthropogenic use of antibiotics. [DOI] [PubMed] [Google Scholar]
- 9.Bhullar K, et al. Antibiotic resistance is prevalent in an isolated cave microbiome. PLoS ONE. 2012;7:e34953. doi: 10.1371/journal.pone.0034953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clemente JC, et al. The microbiome of uncontacted Amerindians. Sci Adv. 2015;1:e1500183. doi: 10.1126/sciadv.1500183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wright GD. The antibiotic resistome: the nexus of chemical and genetic diversity. Nat Rev Microbiol. 2007;5:175–186. doi: 10.1038/nrmicro1614. [DOI] [PubMed] [Google Scholar]
- 12.D’Costa VM, McGrann KM, Hughes DW, Wright GD. Sampling the antibiotic resistome. Science. 2006;311:374–377. doi: 10.1126/science.1120800. By coining the term resistome, this study cements the concept of microbial communities as reservoirs of antibiotic resistance genes by characterizing the multidrug-resistant phenotypes of numerous soil isolates. [DOI] [PubMed] [Google Scholar]
- 13.Benveniste R, Davies J. Aminoglycoside antibiotic-inactivating enzymes in actinomycetes similar to those present in clinical isolates of antibiotic-resistant bacteria. Proc Natl Acad Sci USA. 1973;70:2276–2280. doi: 10.1073/pnas.70.8.2276. This historic paper provides extensive biochemical evidence supporting the hypothesis that antibiotic resistance genes have their eutionary origins in antibiotic-producing bacteria. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forsberg KJ, et al. The shared antibiotic resistome of soil bacteria and human pathogens. Science. 2012;337:1107–1111. doi: 10.1126/science.1220761. This study developed the PARFuMS pipeline, a high-throughput implementation of functional metagenomics, and applied it to discover the first evidence of the sharing of multidrug resistance gene cassettes between non-pathogenic soil bacteria and human pathogens. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knapp CW, Dolfing J, Ehlert PA, Graham DW. Evidence of increasing antibiotic resistance gene abundances in archived soils since 1940. Environ Sci Technol. 2010;44:580–587. doi: 10.1021/es901221x. [DOI] [PubMed] [Google Scholar]
- 16.O’Neill J. Tackling drug-resistant infections globally: final report and recommendations. amr-review.org. 2016 https://amr-review.org/sites/default/files/160518_Final%20paper_with%20cover.pdf.
- 17.Lewis K. Platforms for antibiotic discovery. Nat Rev Drug Discov. 2013;12:371–387. doi: 10.1038/nrd3975. [DOI] [PubMed] [Google Scholar]
- 18.Moore AM, et al. Pediatric fecal microbiota harbor diverse and novel antibiotic resistance genes. PLoS ONE. 2013;8:e78822. doi: 10.1371/journal.pone.0078822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yim G, Wang HH, Davies J. Antibiotics as signalling molecules. Phil Trans R Soc B. 2007;362:1195–1200. doi: 10.1098/rstb.2007.2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gradmann C. Re-inventing infectious disease: antibiotic resistance and drug development at the Bayer Company 1945–1980. Med Hist. 2016;60:155–180. doi: 10.1017/mdh.2016.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fleming A. Nobel Lectures: Physiology or Medicine 1942–1962. Elsevier; 1964. pp. 83–93. [Google Scholar]
- 22.Abraham EP, Chain E. An enzyme from bacteria able to destroy penicillin 1940. Rev Infect Dis. 1988;10:677–678. [PubMed] [Google Scholar]
- 23.Chambers HF, Deleo FR. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat Rev Microbiol. 2009;7:629–641. doi: 10.1038/nrmicro2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Akiba T, Koyama K, Ishiki Y, Kimura S, Fukushima T. On the mechanism of the development of multiple-drug-resistant clones of Shigella. Jpn J Microbiol. 1960;4:219–227. doi: 10.1111/j.1348-0421.1960.tb00170.x. [DOI] [PubMed] [Google Scholar]
- 25.Rappe MS, Giovannoni SJ. The uncultured microbial majority. Annu Rev Microbiol. 2003;57:369–394. doi: 10.1146/annurev.micro.57.030502.090759. [DOI] [PubMed] [Google Scholar]
- 26.Anantharaman K, et al. Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat Commun. 2016;7:13219. doi: 10.1038/ncomms13219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adu-Oppong B, Gasparrini AJ, Dantas G. Genomic and functional techniques to mine the microbiome for novel antimicrobials and antimicrobial resistance genes. Ann NY Acad Sci. 2017;1388:42–58. doi: 10.1111/nyas.13257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wetterstrand KA. DNA sequencing costs: data from the NHGRI Genome Sequencing Program (GSP) genome.gov. 2016 www.genome.gov/sequencingcostsdata.
- 29.UniProt Consortium. UniProt: a hub for protein information. Nucleic Acids Res. 2015;43:D204–D212. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rondon MR, et al. Cloning the soil metagenome: a strategy for accessing the genetic and functional diversity of uncultured microorganisms. Appl Environ Microbiol. 2000;66:2541–2547. doi: 10.1128/aem.66.6.2541-2547.2000. This is a seminal paper in the development of functional metagenomic techniques. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martinez JL, Baquero F. Mutation frequencies and antibiotic resistance. Antimicrob Agents Chemother. 2000;44:1771–1777. doi: 10.1128/aac.44.7.1771-1777.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pino M, Power P, Gutkind G, Di Conza JA. INQ-1, a chromosome-encoded AmpC β-lactamase from Inquilinus limosus. J Antimicrob Chemother. 2014;69:560–562. doi: 10.1093/jac/dkt378. [DOI] [PubMed] [Google Scholar]
- 33.McGrath M, Gey van Pittius NC, van Helden PD, Warren RM, Warner DF. Mutation rate and the emergence of drug resistance in Mycobacterium tuberculosis. J Antimicrob Chemother. 2014;69:292–302. doi: 10.1093/jac/dkt364. [DOI] [PubMed] [Google Scholar]
- 34.Martinez JL, Baquero F, Andersson DI. Predicting antibiotic resistance. Nat Rev Microbiol. 2007;5:958–965. doi: 10.1038/nrmicro1796. [DOI] [PubMed] [Google Scholar]
- 35.Martinez JL, Coque TM, Baquero F. What is a resistance gene? Ranking risk in resistomes. Nat Rev Microbiol. 2015;13:116–123. doi: 10.1038/nrmicro3399. This article examines the concept of risk in antibiotic resistance and develops criteria for evaluating the level of concern a resistance gene should command based on the clinical importance of the resisted antibiotic, the novelty of the resistance mechanism, the presence of the resistance gene in a pathogen and the vicinity of the resistance gene to mobilization elements, among other factors. [DOI] [PubMed] [Google Scholar]
- 36.Gibson MK, et al. Developmental dynamics of the preterm infant gut microbiota and antibiotic resistome. Nat Microbiol. 2016;1:16024. doi: 10.1038/nmicrobiol.2016.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spanogiannopoulos P, Waglechner N, Koteva K, Wright GD. A rifamycin inactivating phosphotransferase family shared by environmental and pathogenic bacteria. Proc Natl Acad Sci USA. 2014;111:7102–7107. doi: 10.1073/pnas.1402358111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Torres-Cortes G, et al. Characterization of novel antibiotic resistance genes identified by functional metagenomics on soil samples. Environ Microbiol. 2011;13:1101–1114. doi: 10.1111/j.1462-2920.2010.02422.x. [DOI] [PubMed] [Google Scholar]
- 39.Forsberg KJ, Patel S, Wencewicz TA, Dantas G. The tetracycline destructases: a novel family of tetracycline-inactivating enzymes. Chem Biol. 2015;22:888–897. doi: 10.1016/j.chembiol.2015.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Riesenfeld CS, Goodman RM, Handelsman J. Uncultured soil bacteria are a reservoir of new antibiotic resistance genes. Environ Microbiol. 2004;6:981–989. doi: 10.1111/j.1462-2920.2004.00664.x. This is one of the first studies to apply functional metagenomic selections to the study of antibiotic resistance. In this study, screening of soil metagenomes for antibiotic resistance reveals several novel resistance genes and demonstrates the promise of functional metagenomics for the study of resistomes. [DOI] [PubMed] [Google Scholar]
- 41.Allen HK, Moe LA, Rodbumrer J, Gaarder A, Handelsman J. Functional metagenomics reveals diverse β-lactamases in a remote Alaskan soil. ISME J. 2009;3:243–251. doi: 10.1038/ismej.2008.86. [DOI] [PubMed] [Google Scholar]
- 42.Lang KS, et al. Novel florfenicol and chloramphenicol resistance gene discovered in Alaskan soil by using functional metagenomics. Appl Environ Microbiol. 2010;76:5321–5326. doi: 10.1128/AEM.00323-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perron GG, et al. Functional characterization of bacteria isolated from ancient arctic soil exposes diverse resistance mechanisms to modern antibiotics. PLoS ONE. 2015;10:e0069533. doi: 10.1371/journal.pone.0069533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Donato JJ, et al. Metagenomic analysis of apple orchard soil reveals antibiotic resistance genes encoding predicted bifunctional proteins. Appl Environ Microbiol. 2010;76:4396–4401. doi: 10.1128/AEM.01763-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tao W, Lee MH, Wu J, Kim NH, Lee SW. Isolation and characterization of a family VII esterase derived from alluvial soil metagenomic library. J Microbiol. 2011;49:178–185. doi: 10.1007/s12275-011-1102-5. [DOI] [PubMed] [Google Scholar]
- 46.Tao W, et al. Characterization of two metagenome-derived esterases that reactivate chloramphenicol by counteracting chloramphenicol acetyltransferase. J Microbiol Biotechnol. 2011;21:1203–1210. doi: 10.4014/jmb.1107.07034. [DOI] [PubMed] [Google Scholar]
- 47.Jeon JH, et al. Novel metagenome-derived carboxylesterase that hydrolyzes β-lactam antibiotics. Appl Environ Microbiol. 2011;77:7830–7836. doi: 10.1128/AEM.05363-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Forsberg KJ, et al. Bacterial phylogeny structures soil resistomes across habitats. Nature. 2014;509:612–616. doi: 10.1038/nature13377. This study finds that soil resistomes are highly correlated with bacterial phylogeny, and describes a paucity of mobile genetic elements that are syntenic with resistance genes in soil bacteria relative to pathogens, which indicates that the majority of the soil resistome is not poised for facile horizontal acquisition by pathogens. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McGarvey KM, Queitsch K, Fields S. Wide variation in antibiotic resistance proteins identified by functional metagenomic screening of a soil DNA library. Appl Environ Microbiol. 2012;78:1708–1714. doi: 10.1128/AEM.06759-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu YY, et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect Dis. 2016;16:161–168. doi: 10.1016/S1473-3099(15)00424-7. [DOI] [PubMed] [Google Scholar]
- 51.Carnevali C, et al. Occurence of mcr-1 colistin-resistant Salmonella enterica isolates recovered from human and animals in Italy, 2012 to 2015. Antimicrob Agents Chemother. 2016;60:7532–7534. doi: 10.1128/AAC.01803-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ortega-Paredes D, Barba P, Zurita J. Colistin-resistant Escherichia coli clinical isolate harbouring the mcr-1 gene in Ecuador. Epidemiol Infect. 2016;144:2967–2970. doi: 10.1017/S0950268816001369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Teo JQ, et al. mcr-1 in multidrug-resistant blaKPC-2-producing clinical Enterobacteriaceae isolates in Singapore. Antimicrob Agents Chemother. 2016;60:6435–6437. doi: 10.1128/AAC.00804-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fernandes MR, et al. First report of the globally disseminated IncX4 plasmid carrying the mcr-1 gene in a colistin-resistant Escherichia coli sequence type 101 isolate from a human infection in Brazil. Antimicrob Agents Chemother. 2016;60:6415–6417. doi: 10.1128/AAC.01325-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Delgado-Blas JF, Ovejero CM, Abadia-Patino L, Gonzalez-Zorn B. Coexistence of mcr-1 and blaNDM-1 in Escherichia coli from Venezuela. Antimicrob Agents Chemother. 2016;60:6356–6358. doi: 10.1128/AAC.01319-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kline KE, et al. Investigation of first identified mcr-1 gene in an isolate from a U.S. patient — Pennsylvania, 2016. MMWR Morb Mortal Wkly Rep. 2016;65:977–978. doi: 10.15585/mmwr.mm6536e2. [DOI] [PubMed] [Google Scholar]
- 57.Wong SC, et al. Colistin-resistant Enterobacteriaceae carrying the mcr-1 gene among patients in Hong Kong. Emerg Infect Dis. 2016;22:1667–1669. doi: 10.3201/eid2209.160091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brauer A, et al. Plasmid with colistin resistance gene mcr-1 in ESBL-producing Escherichia coli strains isolated from pig slurry in Estonia. Antimicrob Agents Chemother. 2016;60:6933–6936. doi: 10.1128/AAC.00443-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.von Wintersdorff CJ, et al. Detection of the plasmid-mediated colistin-resistance gene mcr-1 in faecal metagenomes of Dutch travellers. J Antimicrob Chemother. 2016;71:3416–3419. doi: 10.1093/jac/dkw328. [DOI] [PubMed] [Google Scholar]
- 60.Diaz-Torres ML, et al. Novel tetracycline resistance determinant from the oral metagenome. Antimicrob Agents Chemother. 2003;47:1430–1432. doi: 10.1128/AAC.47.4.1430-1432.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Diaz-Torres ML, et al. Determining the antibiotic resistance potential of the indigenous oral microbiota of humans using a metagenomic approach. FEMS Microbiol Lett. 2006;258:257–262. doi: 10.1111/j.1574-6968.2006.00221.x. [DOI] [PubMed] [Google Scholar]
- 62.Sommer MO, Dantas G, Church GM. Functional characterization of the antibiotic resistance reservoir in the human microflora. Science. 2009;325:1128–1131. doi: 10.1126/science.1176950. This study is the first to apply functional metagenomic selections for antibiotic resistance genes harboured by the human gut microbiome. The authors find that the human gut hosts a diverse resistome that had previously been severely undersampled by culture-dependent methods. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cheng G, et al. Functional screening of antibiotic resistance genes from human gut microbiota reveals a novel gene fusion. FEMS Microbiol Lett. 2012;336:11–16. doi: 10.1111/j.1574-6968.2012.02647.x. [DOI] [PubMed] [Google Scholar]
- 64.Moore AM, et al. Gut resistome development in healthy twin pairs in the first year of life. Microbiome. 2015;3:27. doi: 10.1186/s40168-015-0090-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fouhy F, et al. Identification of aminoglycoside and β-lactam resistance genes from within an infant gut functional metagenomic library. PLoS ONE. 2014;9:e108016. doi: 10.1371/journal.pone.0108016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pehrsson EC, et al. Interconnected microbiomes and resistomes in low-income human habitats. Nature. 2016;533:212–216. doi: 10.1038/nature17672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pal C, Bengtsson-Palme J, Kristiansson E, Larsson DG. The structure and diversity of human, animal and environmental resistomes. Microbiome. 2016;4:54. doi: 10.1186/s40168-016-0199-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gonzales PR, et al. Synergistic, collaterally sensitive β-lactam combinations suppress resistance in MRSA. Nat Chem Biol. 2015;11:855–861. doi: 10.1038/nchembio.1911. This study reports on the ability of a combination of three β-lactam antibiotics to kill several clinical methicillin-resistant S. aureus strains. Notably, the authors find that although resistance could evolve to each individual antibiotic, the evolution of resistance was suppressed when the antibiotics were used in combination owing to the reciprocal collateral sensitivity of each drug. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Baym M, Stone LK, Kishony R. Multidrug evolutionary strategies to reverse antibiotic resistance. Science. 2016;351:aad3292. doi: 10.1126/science.aad3292. This review explores strategies to combat the emergence of antibiotic resistance, including strategies to directly inhibit antibiotic resistance enzymes and induce selection inversion through drug combination synergy and collateral sensitivity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stone LK, et al. Compounds that select against the tetracycline-resistance efflux pump. Nat Chem Biol. 2016;12:902–904. doi: 10.1038/nchembio.2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lomovskaya O, Zgurskaya HI, Totrov M, Watkins WJ. Waltzing transporters and ‘the dance macabre’ between humans and bacteria. Nat Rev Drug Discov. 2007;6:56–65. doi: 10.1038/nrd2200. [DOI] [PubMed] [Google Scholar]
- 72.Drawz SM, Papp-Wallace KM, Bonomo RA. New β-lactamase inhibitors: a therapeutic renaissance in an MDR world. Antimicrob Agents Chemother. 2014;58:1835–1846. doi: 10.1128/AAC.00826-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wright GD. Antibiotic adjuvants: rescuing antibiotics from resistance. Trends Microbiol. 2016;24:862–871. doi: 10.1016/j.tim.2016.06.009. [DOI] [PubMed] [Google Scholar]
- 74.Palmer AC, Angelino E, Kishony R. Chemical decay of an antibiotic inverts selection for resistance. Nat Chem Biol. 2010;6:105–107. doi: 10.1038/nchembio.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ejim L, et al. Combinations of antibiotics and nonantibiotic drugs enhance antimicrobial efficacy. Nat Chem Biol. 2011;7:348–350. doi: 10.1038/nchembio.559. [DOI] [PubMed] [Google Scholar]
- 76.Brown D. Antibiotic resistance breakers: can repurposed drugs fill the antibiotic discovery void? Nat Rev Drug Discov. 2015;14:821–832. doi: 10.1038/nrd4675. [DOI] [PubMed] [Google Scholar]
- 77.Cox G, et al. A common platform for antibiotic dereplication and adjuvant discovery. Cell Chem Biol. 2017;24:98–109. doi: 10.1016/j.chembiol.2016.11.011. [DOI] [PubMed] [Google Scholar]
- 78.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 79.Wallace JC, Port JA, Smith MN, Faustman EM. FARME DB: a functional antibiotic resistance element database. Database (Oxford) 2017;2017:baw165. doi: 10.1093/database/baw165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu B, Pop M. ARDB — Antibiotic Resistance Genes Database. Nucleic Acids Res. 2009;37:D443–D447. doi: 10.1093/nar/gkn656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McArthur AG, et al. The comprehensive antibiotic resistance database. Antimicrob Agents Chemother. 2013;57:3348–3357. doi: 10.1128/AAC.00419-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xavier BB, et al. Consolidating and exploring antibiotic resistance gene data resources. J Clin Microbiol. 2016;54:851–859. doi: 10.1128/JCM.02717-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zankari E, et al. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother. 2012;67:2640–2644. doi: 10.1093/jac/dks261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gibson MK, Forsberg KJ, Dantas G. Improved annotation of antibiotic resistance determinants reveals microbial resistomes cluster by ecology. ISME J. 2015;9:207–216. doi: 10.1038/ismej.2014.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Eddy SR. A new generation of homology search tools based on probabilistic inference. Genome Inform. 2009;23:205–211. [PubMed] [Google Scholar]
- 86.Pesesky MW, et al. Evaluation of machine learning and rules-based approaches for predicting antimicrobial resistance profiles in Gram-negative Bacilli from whole genome sequence data. Front Microbiol. 2016;7:1887. doi: 10.3389/fmicb.2016.01887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Boulund F, Johnning A, Pereira MB, Larsson DG, Kristiansson E. A novel method to discover fluoroquinolone antibiotic resistance (qnr) genes in fragmented nucleotide sequences. BMC Genomics. 2012;13:695. doi: 10.1186/1471-2164-13-695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Flach CF, Boulund F, Kristiansson E, Larsson DJ. Functional verification of computationally predicted qnr genes. Ann Clin Microbiol Antimicrob. 2013;12:34. doi: 10.1186/1476-0711-12-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bertelli C, Greub G. Rapid bacterial genome sequencing: methods and applications in clinical microbiology. Clin Microbiol Infect. 2013;19:803–813. doi: 10.1111/1469-0691.12217. [DOI] [PubMed] [Google Scholar]
- 90.Didelot X, Bowden R, Wilson DJ, Peto TE, Crook DW. Transforming clinical microbiology with bacterial genome sequencing. Nat Rev Genet. 2012;13:601–612. doi: 10.1038/nrg3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zumla A, et al. Rapid point of care diagnostic tests for viral and bacterial respiratory tract infections — needs, advances, and future prospects. Lancet Infect Dis. 2014;14:1123–1135. doi: 10.1016/S1473-3099(14)70827-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kothari A, Morgan M, Haake DA. Emerging technologies for rapid identification of bloodstream pathogens. Clin Infect Dis. 2014;59:272–278. doi: 10.1093/cid/ciu292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pulido MR, Garcia-Quintanilla M, Martin-Pena R, Cisneros JM, McConnell MJ. Progress on the development of rapid methods for antimicrobial susceptibility testing. J Antimicrob Chemother. 2013;68:2710–2717. doi: 10.1093/jac/dkt253. [DOI] [PubMed] [Google Scholar]
- 94.Bradley P, et al. Rapid antibiotic-resistance predictions from genome sequence data for Staphylococcus aureus and Mycobacterium tuberculosis. Nat Commun. 2015;6:10063. doi: 10.1038/ncomms10063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Du H, Chen L, Tang YW, Kreiswirth BN. Emergence of the mcr-1 colistin resistance gene in carbapenem-resistant Enterobacteriaceae. Lancet Infect Dis. 2016;16:287–288. doi: 10.1016/S1473-3099(16)00056-6. [DOI] [PubMed] [Google Scholar]
- 96.Kasbekar N. Tigecycline: a new glycylcycline antimicrobial agent. Am J Health Syst Pharm. 2006;63:1235–1243. doi: 10.2146/ajhp050487. [DOI] [PubMed] [Google Scholar]
- 97.Sun Y, et al. The emergence of clinical resistance to tigecycline. Int J Antimicrob Agents. 2013;41:110–116. doi: 10.1016/j.ijantimicag.2012.09.005. [DOI] [PubMed] [Google Scholar]
- 98.Moore IF, Hughes DW, Wright GD. Tigecycline is modified by the flavin-dependent monooxygenase TetX. Biochemistry. 2005;44:11829–11835. doi: 10.1021/bi0506066. [DOI] [PubMed] [Google Scholar]
- 99.Deng M, et al. Molecular epidemiology and mechanisms of tigecycline resistance in clinical isolates of Acinetobacter baumannii from a Chinese university hospital. Antimicrob Agents Chemother. 2014;58:297–303. doi: 10.1128/AAC.01727-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Leski TA, et al. Multidrug-resistant tet(X)-containing hospital isolates in Sierra Leone. Int J Antimicrob Agents. 2013;42:83–86. doi: 10.1016/j.ijantimicag.2013.04.014. [DOI] [PubMed] [Google Scholar]
- 101.Sutcliffe JA, O’Brien W, Fyfe C, Grossman TH. Antibacterial activity of eravacycline (TP-434), a novel fluorocycline, against hospital and community pathogens. Antimicrob Agents Chemother. 2013;57:5548–5558. doi: 10.1128/AAC.01288-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Macone AB, et al. In vitro and in vivo antibacterial activities of omadacycline, a novel aminomethylcycline. Antimicrob Agents Chemother. 2014;58:1127–1135. doi: 10.1128/AAC.01242-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Davies J, Davies D. Origins and evolution of antibiotic resistance. Microbiol Mol Biol Rev. 2010;74:417–433. doi: 10.1128/MMBR.00016-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Walsh C. Molecular mechanisms that confer antibacterial drug resistance. Nature. 2000;406:775–781. doi: 10.1038/35021219. [DOI] [PubMed] [Google Scholar]
- 105.Shaw WV, et al. Primary structure of a chloramphenicol acetyltransferase specified by R plasmids. Nature. 1979;282:870–872. doi: 10.1038/282870a0. [DOI] [PubMed] [Google Scholar]
- 106.Yang W, et al. TetX is a flavin-dependent monooxygenase conferring resistance to tetracycline antibiotics. J Biol Chem. 2004;279:52346–52352. doi: 10.1074/jbc.M409573200. [DOI] [PubMed] [Google Scholar]
- 107.Neu HC. Effect of β-lactamase location in Escherichia coli on penicillin synergy. Appl Microbiol. 1969;17:783–786. doi: 10.1128/am.17.6.783-786.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bush K. Bench-to-bedside review: the role of β-lactamases in antibiotic-resistant Gram-negative infections. Crit Care. 2010;14:224. doi: 10.1186/cc8892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Courvalin P. Vancomycin resistance in gram-positive cocci. Clin Infect Dis. 2006;42(Suppl 1):S25–S34. doi: 10.1086/491711. [DOI] [PubMed] [Google Scholar]
- 110.Katayama Y, Ito T, Hiramatsu K. A new class of genetic element, Staphylococcus cassette chromosome mec, encodes methicillin resistance in Staphylococcus aureus. Antimicrob Agents Chemother. 2000;44:1549–1555. doi: 10.1128/aac.44.6.1549-1555.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ogawa W, Onishi M, Ni R, Tsuchiya T, Kuroda T. Functional study of the novel multidrug efflux pump KexD from Klebsiella pneumoniae. Gene. 2012;498:177–182. doi: 10.1016/j.gene.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 112.Ruggerone P, Murakami S, Pos KM, Vargiu AV. RND efflux pumps: structural information translated into function and inhibition mechanisms. Curr Top Med Chem. 2013;13:3079–3100. doi: 10.2174/15680266113136660220. [DOI] [PubMed] [Google Scholar]
- 113.Hinchliffe P, Symmons MF, Hughes C, Koronakis V. Structure and operation of bacterial tripartite pumps. Annu Rev Microbiol. 2013;67:221–242. doi: 10.1146/annurev-micro-092412-155718. [DOI] [PubMed] [Google Scholar]
- 114.Piddock LJ. Clinically relevant chromosomally encoded multidrug resistance efflux pumps in bacteria. Clin Microbiol Rev. 2006;19:382–402. doi: 10.1128/CMR.19.2.382-402.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Roberts MC. Tetracycline resistance determinants: mechanisms of action, regulation of expression, genetic mobility, and distribution. FEMS Microbiol Rev. 1996;19:1–24. doi: 10.1111/j.1574-6976.1996.tb00251.x. [DOI] [PubMed] [Google Scholar]
- 116.Tamber S, Hancock RE. On the mechanism of solute uptake in Pseudomonas. Front Biosci. 2003;8:s472–s483. doi: 10.2741/1075. [DOI] [PubMed] [Google Scholar]
- 117.Poulou A, et al. Outbreak caused by an ertapenem-resistant, CTX-M-15-producing Klebsiella pneumoniae sequence type 101 clone carrying an OmpK36 porin variant. J Clin Microbiol. 2013;51:3176–3182. doi: 10.1128/JCM.01244-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Babouee Flury B, et al. Association of novel nonsynonymous single nucleotide polymorphisms in ampD with cephalosporin resistance and phylogenetic variations in ampC, ampR, ompF, and ompC in Enterobacter cloacae isolates that are highly resistant to carbapenems. Antimicrob Agents Chemother. 2016;60:2383–2390. doi: 10.1128/AAC.02835-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sanders CC, Sanders WE. Emergence of resistance to cefamandole: possible role of cefoxitin-inducible β-lactamases. Antimicrob Agents Chemother. 1979;15:792–797. doi: 10.1128/aac.15.6.792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Perez F, Endimiani A, Hujer KM, Bonomo RA. The continuing challenge of ESBLs. Curr Opin Pharmacol. 2007;7:459–469. doi: 10.1016/j.coph.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Potter RF, D’Souza AW, Dantas G. The rapid spread of carbapenem-resistant Enterobacteriaceae. Drug Resist Updat. 2016;29:30–46. doi: 10.1016/j.drup.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yong D, et al. Characterization of a new metallo-β-lactamase gene, blaNDM-1, and a novel erythromycin esterase gene carried on a unique genetic structure in Klebsiella pneumoniae sequence type 14 from India. Antimicrob Agents Chemother. 2009;53:5046–5054. doi: 10.1128/AAC.00774-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Castanheira M, et al. Early dissemination of NDM-1-and OXA-181-producing Enterobacteriaceae in Indian hospitals: report from the SENTRY Antimicrobial Surveillance Program, 2006–2007. Antimicrob Agents Chemother. 2011;55:1274–1278. doi: 10.1128/AAC.01497-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kumarasamy KK, et al. Emergence of a new antibiotic resistance mechanism in India, Pakistan, and the UK: a molecular, biological, and epidemiological study. Lancet Infect Dis. 2010;10:597–602. doi: 10.1016/S1473-3099(10)70143-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yigit H, et al. Novel carbapenem-hydrolyzing β-lactamase, KPC-1, from a carbapenem-resistant strain of Klebsiella pneumoniae. Antimicrob Agents Chemother. 2001;45:1151–1161. doi: 10.1128/AAC.45.4.1151-1161.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Pesesky MW, et al. KPC and NDM-1 genes in related Enterobacteriaceae strains and plasmids from Pakistan and the United States. Emerg Infect Dis. 2015;21:1034–1037. doi: 10.3201/eid2106.141504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Poirel L, Rodriguez-Martinez JM, Mammeri H, Liard A, Nordmann P. Origin of plasmid-mediated quinolone resistance determinant QnrA. Antimicrob Agents Chemother. 2005;49:3523–3525. doi: 10.1128/AAC.49.8.3523-3525.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Allen HK, et al. Call of the wild: antibiotic resistance genes in natural environments. Nat Rev Microbiol. 2010;8:251–259. doi: 10.1038/nrmicro2312. [DOI] [PubMed] [Google Scholar]
- 129.Robicsek A, et al. Fluoroquinolone-modifying enzyme: a new adaptation of a common aminoglycoside acetyltransferase. Nat Med. 2006;12:83–88. doi: 10.1038/nm1347. [DOI] [PubMed] [Google Scholar]
- 130.Jacoby GA, Strahilevitz J, Hooper DC. Plasmid-mediated quinolone resistance. Microbiol Spectr. 2014 doi: 10.1128/microbiolspec.PLAS-0006-2013. http://dx.doi.org/10.1128/microbiolspec.PLAS-0006-2013. [DOI] [PMC free article] [PubMed]
- 131.Li J, et al. Colistin: the re-emerging antibiotic for multidrug-resistant Gram-negative bacterial infections. Lancet Infect Dis. 2006;6:589–601. doi: 10.1016/S1473-3099(06)70580-1. [DOI] [PubMed] [Google Scholar]
- 132.Zheng B, et al. Coexistence of MCR-1 and NDM-1 in clinical Escherichia coli isolates. Clin Infect Dis. 2016;63:1393–1395. doi: 10.1093/cid/ciw553. [DOI] [PubMed] [Google Scholar]
- 133.Sun J, et al. Co-transfer of blaNDM-5 and mcr-1 by an IncX3-X4 hybrid plasmid in Escherichia coli. Nat Microbiol. 2016;1:16176. doi: 10.1038/nmicrobiol.2016.176. This study describes, for the first time, the co-transfer of resistance genes against two last-resort antibiotics — namely, colistin and carbapenems — by a plasmid in E. coli. The authors demonstrate through sequence analysis that this plasmid probably originated through the recombination of IncX3 and IncX4 plasmids. [DOI] [PubMed] [Google Scholar]
- 134.Lam KN, Cheng J, Engel K, Neufeld JD, Charles TC. Current and future resources for functional metagenomics. Front Microbiol. 2015;6:1196. doi: 10.3389/fmicb.2015.01196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Pehrsson EC, Forsberg KJ, Gibson MK, Ahmadi S, Dantas G. Novel resistance functions uncovered using functional metagenomic investigations of resistance reservoirs. Front Microbiol. 2013;4:145. doi: 10.3389/fmicb.2013.00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Craig JW, Chang FY, Kim JH, Obiajulu SC, Brady SF. Expanding small-molecule functional metagenomics through parallel screening of broad-host-range cosmid environmental DNA libraries in diverse proteobacteria. Appl Environ Microbiol. 2010;76:1633–1641. doi: 10.1128/AEM.02169-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Martinez A, et al. Genetically modified bacterial strains and novel bacterial artificial chromosome shuttle vectors for constructing environmental libraries and detecting heterologous natural products in multiple expression hosts. Appl Environ Microbiol. 2004;70:2452–2463. doi: 10.1128/AEM.70.4.2452-2463.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Stokes JM, et al. Cold stress makes Escherichia coli susceptible to glycopeptide antibiotics by altering outer membrane integrity. Cell Chem Biol. 2016;23:267–277. doi: 10.1016/j.chembiol.2015.12.011. [DOI] [PubMed] [Google Scholar]
- 139.Liebl W, et al. Alternative hosts for functional (meta) genome analysis. Appl Microbiol Biotechnol. 2014;98:8099–8109. doi: 10.1007/s00253-014-5961-7. [DOI] [PubMed] [Google Scholar]
- 140.Siefert JL. Defining the mobilome. Methods Mol Biol. 2009;532:13–27. doi: 10.1007/978-1-60327-853-9_2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.