

Abstract
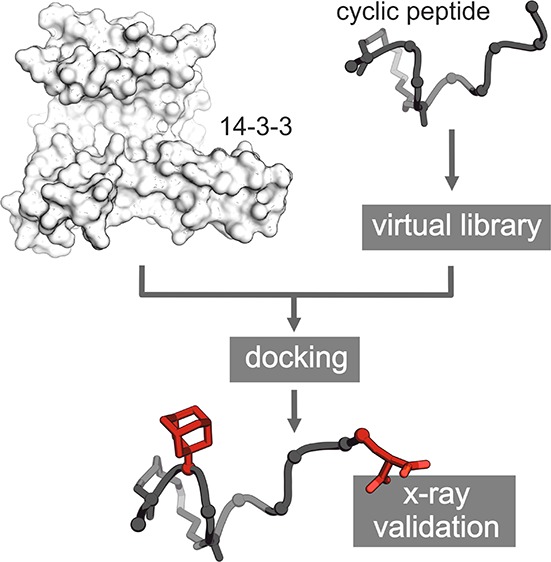
Macrocyclic peptides can interfere with challenging biomolecular targets including protein–protein interactions. Whereas there are various approaches that facilitate the identification of peptide-derived ligands, their evolution into higher affinity binders remains a major hurdle. We report a virtual screen based on molecular docking that allows the affinity maturation of macrocyclic peptides taking non-natural amino acids into consideration. These macrocycles bear large and flexible substituents that usually complicate the use of docking approaches. A virtual library containing more than 1400 structures was screened against the target focusing on docking poses with the core structure resembling a known bioactive conformation. Based on this screen, a macrocyclic peptide 22 involving two non-natural amino acids was evolved showing increased target affinity and biological activity. Predicted binding modes were verified by X-ray crystallography. The presented workflow allows the screening of large macrocyclic peptides with diverse modifications thereby expanding the accessible chemical space and reducing synthetic efforts.
Introduction
Macrocyclic scaffolds are a common structural element among natural products, and they are considered promising candidates for the development of chemical probes and novel therapeutics.1,2 This is mainly due to their ability to bind protein surfaces even if those lack distinct binding pockets. The presence of such pockets is often required for high affinity binding of classic small molecules.3−5 Protein–protein interactions (PPIs) tend to involve flat surfaces rendering their inhibition extremely challenging. Among macrocycles, peptide-derived structures proved to be particularly valuable starting points for the generation of PPI inhibitors.2 Often, the design process starts by macrocyclization of known peptide binding epitopes,2 or by the screening of macrocyclic peptide libraries (e.g., via phage or mRNA display)6,7 resulting in structures that often exhibit good affinity for their target. However, in most cases an evolution toward higher affinity ligands is required to efficiently block PPIs and/or to compensate for affinity losses during the optimization process toward increased bioavailability.2 For that reason, affinity maturation constitutes a bottleneck preventing straightforward use of bioactive macrocyclic peptides. Ideally, the consideration of numerous modifications including natural and non-natural amino acids at all positions of the peptide sequence would be desired.
Given the efforts associated with the chemical synthesis and evaluation of large peptide libraries8 and the complexity of biological screening platforms as well as their restriction to a limited number and type of non-natural building blocks,6,7 computational screening approaches provide an appealing alternative. In the case of small molecules and short peptide ligands, virtual screening based on molecular docking has proven particularly useful.9−13 For macrocyclic scaffolds with relatively small substituents, benchmark studies were able to reproduce known binding modes indicating that docking could also be applied.14−16 However, it is not clear if these approaches allow exhaustive virtual screening and the prediction of novel interactions as molecular docking encounters severe difficulties in scoring new binding modes for extended scaffolds.12,13 In particular, the consideration of macrocycles with large and flexible substituents, as they are often found in peptide-derived PPI inhibitors,2 can be expected to be extremely challenging. For this reason, computationally more demanding methods such as molecular dynamics simulations have been used for protein–peptide docking,17−19 however, at the cost of a dramatically reduced throughput.20−23 An additional challenge in the affinity maturation of macrocyclic ligands occurs when the starting point already shows good target affinity.10 In this case, a precise prediction and scoring of the binding mode is of utmost importance to allow the identification of improved ligands.10 The availability of fast and reliable docking approaches for modified peptide ligands would accelerate the time-consuming optimization process and is highly desired.
Herein, we describe a computational approach based on molecular docking that allows the affinity maturation of macrocyclic peptide ligands. Originally derived from linear bacterial peptide sequence 1 (ESp),24 macrocyclic peptide 2 (βss12)24 served as a starting point. Based on the crystal structure of 2 in complex with its target protein 14–3–3, a virtual library of macrocycles was generated, which was screened against the target. Subsequently, binding poses that resemble the backbone conformation of the starting peptide in its bound state were scored. This selection process allowed the identification of a macrocyclic peptide with two non-natural amino acids, which exhibits increased target affinity as well as increased potency in cell-based assays. Most importantly, predicted side chain binding modes were verified by X-ray crystallography.
Results
Virtual Peptide Library
Aiming for a computational approach that allows the handling of large and flexible structures, we choose macrocyclic peptide 2 as model system.24,25 This peptide binds 14–3–3 proteins which form a family of closely related adaptor proteins that regulates a wide range of cellular processes via numerous PPIs. Peptide 2 involves an 11-mer irregularly structured peptide sequence (dark gray, Figure 1), which is constrained by a hydrophobic side chain-to-side chain cross-link in analogy to so-called hydrocarbon-stapled peptides.26,27 It was originally derived from the natural peptide epitope 1.24 Macrocycle 2 binds to the amphipathic groove of 14–3–3, which recognizes certain phosphorylated proteins and some nonphosphorylated peptide epitopes. Among the synthetic 14–3–3 binders, macrocyclic peptide 2 is the highest affinity ligand (Kd ≈ 0.1 μM).24,28−34 Its relatively high affinity, molecular weight (M = 1280 g mol–1) and flexibility render peptide 2 a challenging starting point for any in silico affinity optimization. The crystal structure of 2 (PDB ID 4n84)24 in complex with isoform zeta of 14–3–3 (14–3–3ζ) served as starting point for our work. All following calculations and biophysical as well as biological experiments were performed with 14–3–3ζ.
Figure 1.
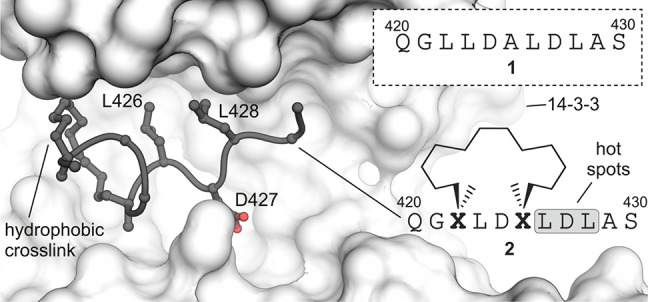
Sequence of linear peptide 1 and crystal structure of cyclic peptide 2 (dark gray) bound to 14–3–3ζ (light gray, PDB ID 4n84). Cross-link and hotspot residues (L426, D427, and L428) are shown explicitly. Peptide sequence of 2 and chemical structure of cross-link are shown (Residues are numbered in accordance to PDB ID 4n84).
Initially, the contribution of all side chains of peptide 2 to 14–3–3ζ binding was assessed employing an alanine scan (Figure S1). This revealed three hotspot residues (L426, D427, and L428, Figure 1) at which alanine substitution results in dramatically reduced affinity (>10-fold, Figure S1). Preceding tests reveal that even minor changes at a hotspot position result in severe affinity reduction (Figure S2). We concluded that variation of these residues is unlikely to result in derivatives with increased binding affinity and focused on the remaining six amino acids (Figure 1). A virtual amino acid library was assembled (Table S2) containing all proteinogenic amino acids (except for glycine and proline) and 223 nonproteinogenic ones. These amino acids were selected based on two criteria: (i) commercial availability of suitably protected analogs allowing a direct use in solid-phase peptide synthesis (SPPS) and (ii) coverage of a diverse chemical space preventing the accumulation of closely related derivatives (Figure S3). Incorporation of all amino acids into the six positions of interest results in a virtual library of 1446 peptides.
Molecular Docking of Virtual Library
Given the successful utilization of molecular docking and scoring approaches for the virtual screening of small molecules and the achievements with respect to small peptide ligands,9,12,35,36 we decided to peruse this strategy. Macrocycle 2 bears large and flexible substituents, features that affect the validity of docking poses and scoring. To reduce the number of invalid docking poses, we considered the implementation of a checkpoint after library docking that would allow focusing on the most relevant docking poses for subsequent scoring. Knowing the importance of the three hotspot residues for binding, we reasoned that high affinity binders will most likely adopt a conformation that positions those residues in analogy to 14–3–3-bound 2. For that reason, pose filtering was applied, ensuring that only docking poses were considered for which the central hotspot amino acids (426–428) resemble the conformation of peptide 2 in its bound state (allowed RMSD ≤ 2.0 Å). These considerations resulted in the following workflow in which the crystal structure of peptide 2 in complex with 14–3–3ζ (PDB ID 4n84) served as starting point (for details see Supporting Information). First, the coordinates of 2 were extracted, and each of the six selected residues was iteratively substituted by each member of the virtual amino acid library (241 building blocks). This resulted in a database of 1446 macrocyclic peptides each of them bearing a single residue variation compared to 2. Second, docking parameters were adjusted to ensure that the conformation of 2 bound to 14–3–3 is reproduced upon docking. These parameters were then used to dock each peptide into the amphipathic groove of 14–3–3ζ using AutoDock Vina35 to sample the conformational space. Thereafter, above-mentioned pose filtering was applied followed by a final rescoring step. For rescoring of docking poses of small peptides, it was shown to be beneficial to consider a combination of knowledge-based and empirical scoring functions since they provide complementary hit lists.12,37 In this respect, Astex Statistical Potential38 (ASP, knowledge-based) and ChemScore39 (empirical) have been described to be suitable for the scoring of peptide conformations12 and were thus selected for rescoring.40 For each peptide, the remaining poses (after pose filtering) were rescored, and only the highest scoring one was considered for the final ranking. For each of the six amino acid positions, the top five ranking peptides per scoring function were visually inspected (in total 60 complexes) to select one peptide per scoring function (#ChemScore, ◊ASP) and position for experimental validation (in total 12 peptides 10–21, Figure 2 and Figure S4).
Figure 2.
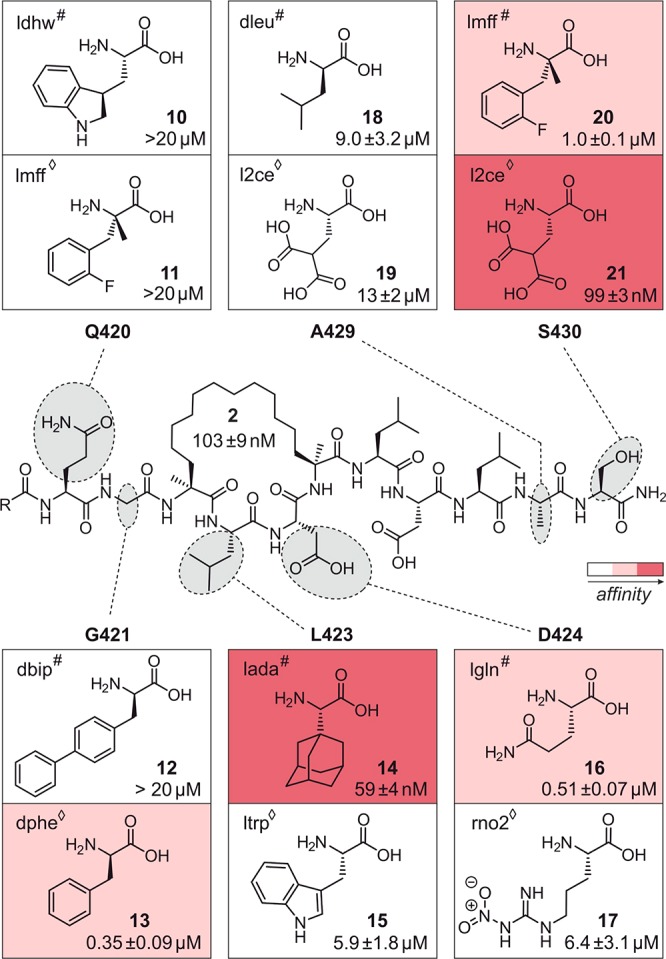
Chemical structure of 2 with varied residues highlighted in gray. Selected 12 variations are shown (two per position: one obtained from ChemScore (#) and the other one from ASP (◊) hit list). Experimentally determined dissociation constants (Kd) for corresponding peptides 10–21 with 14–3–3ζ are given (for details see Figure S7). Variations are color coded in accordance to their affinity for 14–3–3ζ (Kd rages: dark red, <0.1 μM; light red, 0.1–1 μM; white, >1 μM).
Experimental Affinities and Structural Validation
Fluorescently labeled versions of all 12 peptides were synthesized applying standard protocols for SPPS and macrocycle formation.24,41 Affinities toward 14–3–3ζ (aa 1–230) were determined using a fluorescence polarization (FP) assay. As reference, the dissociation constant (Kd) of 2 was determined (Kd = 103 nM). Among the 12 peptides (Figure 2), seven show relatively low affinities for 14–3–3ζ (Kd > 1 μM, white) with four of them still being in the low micromolar range (6–13 μM). Five peptides exhibit affinities in the submicromolar range (Kd < 1 μM, light and dark red) rendering them better binders than the wildtype epitope 1. Most strikingly, among those five peptides, there are two that show lower Kd values than starting point 2. In one case, leucine at position 423 (L423) is substituted by l-(1-adamantyl)glycine (lada, peptide 14, Kd = 59 nM), while in the other one, serine at position 430 (S430) is substituted by l-γ-carboxyglutamic acid (l2ce, peptide 21, Kd = 99 nM). Whereas the non-natural amino acid l2ce in 21 carries a malonic acid side chain predicted to engage a positively charged cavity on 14–3–3ζ (Figure S5), lada in 14 bears a bulky and hydrophobic adamantyl moiety presumably interacting with a hydrophobic patch of 14–3–3ζ (Figure S6).
Having identified two variations with increased affinity, we explored the consequences of incorporating both non-natural amino acids simultaneously into the sequence resulting in peptide 22 (AdCe). For a fluorescently labeled version of 22, 14–3–3 affinity was determined using FP. Notably, both variations appear to act additively, resulting in further increased affinity (Kd = 38 ± 3 nM, Figure S7), which is a 2.7-fold increase compared to starting peptide 2 (103 ± 9 nM). To obtain molecular details of the 22–14–3–3ζ interaction and to evaluate the accuracy of our predicted binding modes, we aimed for a crystal structure of the corresponding complex. Cocrystallization of 22 and 14–3–3ζ (aa 1–230) provided crystals of space group P212121 and allowed the determination of a crystal structure with 2.3 Å resolution (PDB ID 5jm4, Table S3). Each asymmetric unit contains two 14–3–3ζ proteins, with each harboring one molecule of 22 in its hydrophobic groove (Figure S8). The corresponding 2Fo–Fc density map (Figures S9) allows the identification of the entire peptide except for the side chain of N-terminal amino acid Q420, which is not resolved. Superimposition of 22 (red) and 2 (gray) in complex with 14–3–3ζ (Figure 3a) reveals good structural overlay (RMSD backbone: 0.67 Å) verifying analogous binding modes. Most importantly, comparison of the 22 crystal structure with docking predictions for lada at position 423 (Figure 3b) and l2ce at position 430 (Figure 3c) show excellent superimposition. As predicted, the adamantyl moiety of lada engages a hydrophobic patch of the 14–3–3ζ groove, and the malonate side chain of l2ce interacts with two arginines of 14–3–3ζ (Figure S10) that are not addressed by 2. This accuracy of the molecular docking is remarkable, considering that the C-terminal part of the peptide backbone is relatively flexible. Interestingly, lada at position 423 originates from the ChemScore (#) hit list, while l2ce at position 430 was suggested after ASP (◊) rescoring (Figure 2). This underlines the usefulness of considering knowledge-based as well as empirical scoring functions for rescoring.
Figure 3.
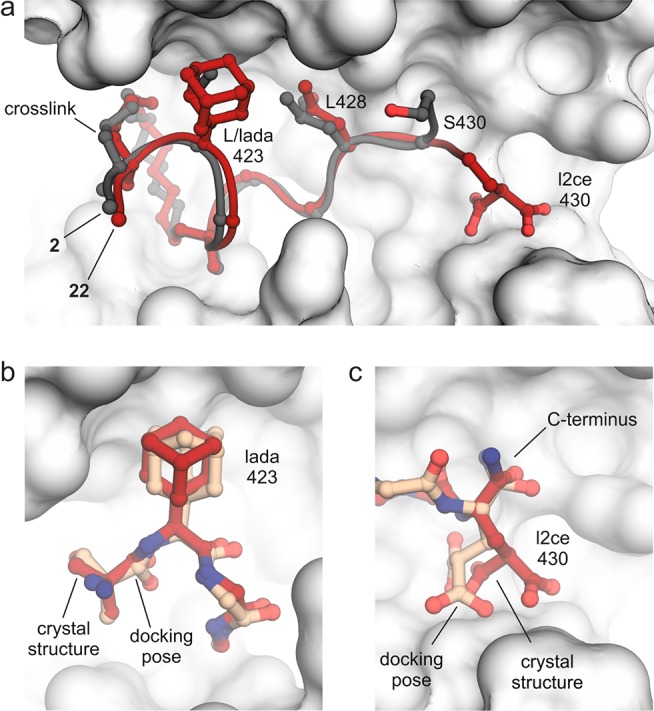
(a) Overlay of crystal structures of 2 (gray, PDB ID 4n84) and 22 (red, PDB ID 5jm4) when bound to 14–3–3ζ (light gray surface representation). Peptide backbones are shown as ribbons. Varied side chains (423 and 430), hotspot residue L428, and cross-link are shown explicitly. (b,c) Superimposition of 22 crystal structure (red, PDB ID 5jm4) with predicted structure (orange, pose with highest score) of lada- and l2ce-modified peptide, respectively. Amino acid of interest and backbone of neighboring amino acids are shown explicitly.
Inhibition of PPI
Knowing that 22 exhibits higher affinity for 14–3–3ζ than 2 (2.7-fold) and wild-type epitope 1 (20-fold), we were interested if this results in more efficient competition with PPIs and increased bioactivity. For that purpose, the competition with phosphorylated binding partners of 14–3–3 was considered. Superimposition of 22 (red) and representative phosphorylated peptides (blue) in complex with 14–3–3 shows substantial overlap (Figure 4a), suggesting competitive binding. To test this hypothesis, we performed FP competition experiments employing a Raf-derived phosphorylated peptide ligand (Figure S12) of 14–3–3ζ as tracer and nonlabeled peptide 1, 2, or 22 as competitor. These measurements (Figure 3b) clearly show efficient competition of 22 (red) with the phosphorylated peptide (half maximal inhibitory concentration: IC50 = 0.8 μM). In line with their lower affinity, 2 (dark gray, IC50 = 1.2 μM) and 1 (light gray, IC50 = 3.3 μM) exhibit reduced competition.
Figure 4.
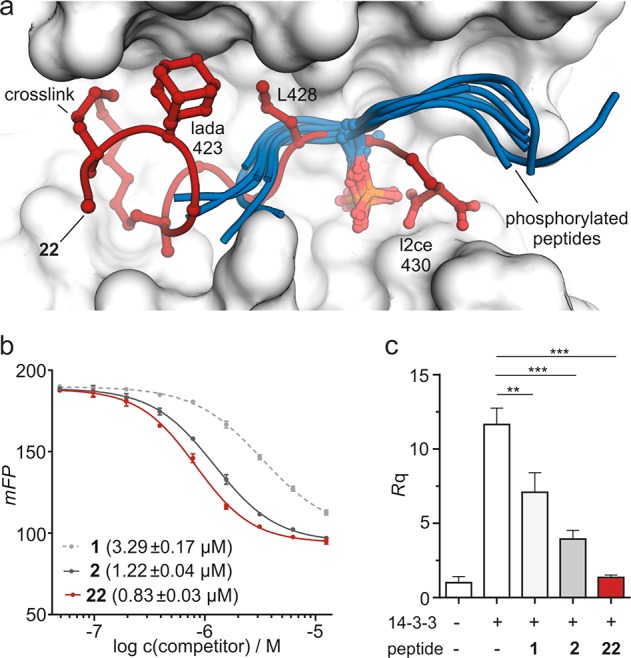
(a) Superimposition of 22 (red, PDB ID 5jm4) and selected phosphorylated peptides (blue, PDB IDs iqja, 1ywt, 2bn5, 2btp, 2c74, 2npm, 2v7d, 3e6y, and 3nkx) bound to 14–3–3. Peptide backbones are shown as ribbons and important side chains explicitly. (b) FP competition using labeled Raf-peptide as tracer (10 nM with 2 μM 14–3–3) and nonlabeled versions of 1, 2, and 22 (including their IC50 values). (c) Inhibition of MMP1 transcription in a cell-based assay after pathway activation with 14–3–3ζ (c = 200 nM). Cells were treated for 24 h in the absence and presence of 1, 2, or 22 (20 μM). Expression levels of mRNA were measured by quantitative real time PCR (for details, see Supporting Information).
Finally, it was tested if improved target affinity also results in enhanced activity in cell-based assays. Since fluorescence microscopy indicates very low cellular uptake of these peptides (Figure S13), we aimed at the inhibition of an extracellular 14–3–3ζ PPI. It is known that 14–3–3 proteins bind to the extracellular domain of transmembrane receptor aminopeptidase N (APN).42,43 This interaction presumably occurs between phosphorylated epitopes on APN and the amphipathic groove of 14–3–3 proteins.33,44 It is known that APN–14–3–3 complex formation induces an intracellular signaling cascade, triggering the expression of a subset of matrix metalloproteinases (MMPs),42,43 and that the inhibition of extracellular 14–3–3 can reduce MMP transcription levels.33 MMPs are important factors in the modulation of the extracellular matrix, and their upregulation is associated with pathological processes such as rheumatoid arthritis or cancer metastasis.45−47 For that reason, MMP inhibition is considered an attractive therapeutic approach. To investigate the potency of 22 in MMP inhibition, we used U87 glioblastoma cells showing robust upregulation of MMP1 mRNA levels (Rq = 11.7, Figure 4c) upon treatment with 14–3–3ζ (c = 200 nM). After incubation with a nonlabeled version of 22 (c = 20 μM), 14–3–3ζ-dependent increase of MMP1 mRNA was drastically reduced (8.6-fold reduction, relative to untreated cells) restoring the levels of nonstimulated cells. Concentration-dependent treatments provide an IC50 value of 8.6 μM for 22 (Figure S14), which exceeds the potency of any synthetic competitive inhibitor reported so far.24,28−34 Compared to 22 (8.6-fold reduction), the nonlabeled versions of 1 and 2 show significantly reduced inhibition of MMP1 transcription (1.6- and 3.0-fold reduction, respectively).
Discussion and Conclusions
In conclusion, we report a fast molecular docking approach that allows the affinity maturation of medium-sized modified peptides considering natural and non-natural amino acid variations. The combination of virtual library screening and peptide-adapted molecular docking resulted in a short hit list enriched with 14–3–3ζ binders allowing to lower experimental validation efforts dramatically. We utilized a fast molecular docking approach by focusing only on generated binding poses that locate the three hot spot residues at their original positions. As a result, the bound conformation of the core structure is preserved while keeping the termini of the peptide flexible. For the first time, virtual screening of a large macrocyclic peptide library was successfully applied while omitting the use of time-consuming computational approaches. Notably, the 12 single residue variations (selected from a database of 1446 peptides) contain five hits with submicromolar affinities, which can be useful in the future to improve, e.g., physicochemical properties. The combination of the two highest affinity variations, a hydrophobic and a hydrophilic non-natural amino acid, allowed the design of a peptide with ca. 3-fold increased affinity for 14–3–3ζ. Most importantly, predicted binding modes of these novel side chains were verified by X-ray crystallography underlining the validity of the docking results. The finally evolved macrocyclic PPI inhibitor shows increased potency in PPI inhibition and in a cell-based assay. Our computational workflow allows the use of very large databases of non-natural amino acids, e.g., created by in silico methods. Taking into account the broad chemical space accessible with such libraries, it is an appealing strategy for the development of novel peptide-based inhibitors.
Experimental Section
General
For detailed information about experimental procedures, see the Supporting Information. Peptide synthesis was performed on solid support following standard Fmoc-based protocols with macrocyclization and double-bond reduction following previously published procedures.41 Peptide purity was determined by RP-HPLC via peak integration at λ = 210 nm. All peptides exhibit a purity ≥95%. Expression and purification of 14–3–3ζ (aa 1–230) was performed according to established protocols.24
Structure and Library Preparation for Docking
Coordinates from protein–ligand complex were retrieved from PDB entry 4n84. Water molecules and ions were removed. AutoDock-Tools (ADT) 1.5.6 was used to add polar hydrogens and charges.48 For peptides, rotatable bonds were assigned (all substituents including the two peptide sequences were kept flexible except for amide bonds). Two-dimensional structures of the 18 natural (Gly and Pro are excluded) and 223 nonproteinogenic amino acids were manually created using ChemDraw 14.0 and subsequently converted to 3D structures in protonation states under neutral condition using Maestro 9.3.5.49 The peptide library was created using in-house Python scripts by replacing single amino acids of 2 with each amino acid in the amino acid library.
Docking Engine, Scoring Function, and Docking Experiments
AutoDock Vina 1.1.2 was used as a docking engine.35 The center of the grid box was set to 10/13/10, and the box size was set to 30 Å in each dimension. Docking parameters were chosen as follows: exhaustiveness = 12, weight_gauss1 = 0.7, weight_repulsion = 0.5, weight_hydrophobic = −0.15, weight_hydrogen = −0.6. (other parameters default). During pose filtering, all poses were excluded in which the functional groups of residues L426, D427, and L428 exhibit an rmsd > 2 Å when compared to analog residues in 2 bound to 14–3–3 (PDB ID 4n84). For rescoring, the remaining poses were scored with ChemScore39 and the Astex statistical potential (ASP),38 respectively, using the simplex minimization option as implemented in Gold 5.2.2.4.40 For each peptide, only the highest scoring pose was considered for the final ranking. The top five ranking peptides per scoring function and position were visually inspected (in total 60 complexes) to select one peptide per scoring function and position for experimental validation (in total 12 peptides). For selected peptides and their predicted binding modes, see Supplementary Figure S4.
Fluorescence Polarization Assays
A 0.1 mM solution of the corresponding FITC-labeled peptide in DMSO was diluted with FP buffer (10 mM HEPES, 150 mM NaCl, 0.1% Tween-20, pH 7.4) to 40 nM. 14−3–3ζ (aa 1–230) was diluted with FP buffer in a 2.5-fold dilution series (80 μM–0.5 nM) in a 384-well plate. To 15 μL of the protein solution, 5 μL of the 40 nM peptide stock was added (final peptide concentration, 10 nM; final protein concentrations, 60 μM–0.4 nM). After 1 h, fluorescence polarization was measured (λ(ex) = 485 nm; λ(em) = 525 nm). The dissociation constant (Kd) was determined from the binding curve with GraphPad from Prism. For competition experiments, N-terminally acetylated peptides were diluted 1:1 in a 384-well plate (10 μL, 100 μM–1 nM). 10 μL of a mixture (1:1) of 14–3–3ζ (aa 1–230) and TAMRA-labeled cRaf peptide was added (final concentrations: acetylated peptides = 50 μM–0.5 nM; 14–3–3ζ (aa 1–230) = 800 nM; TAMRA-labeled cRaf peptide = 100 nM). After 1 h, fluorescence polarization was measured (λ(ex) = 530 nm; λ(em) = 585 nm). The half maximal inhibitory concentration (IC50) was determined from the binding curve with GraphPad from Prism.
X-ray Crystallography and Structure Determination
14–3–3ζ (aa 1–230) was prepared in 50 mM HEPES (pH 7.5), 100 mM NaCl, and 2 mM MgCl2. For complexation, 22 was dissolved in DMSO (11 mM) and mixed with the protein in a molar ratio of 1:2 (protein/peptide). The complex was incubated overnight at 4 °C (final protein concentration: 22 mg/mL) and set up for crystallization using NeXtal Screens (Qiagen). Crystals grew within 4 weeks in the following condition: 1.36 M sodium citrate and 15% (v/v) glycerol and showed a diffraction to 2.34 Å. After molecular replacement, the space group was determined to be P212121. Data was collected using PXII beamline for protein crystallography at the Paul Scherrer Institute Swiss Light Source (SLS). Crystallographic analysis was performed using the XDS software package. Molecular replacement was carried out with the CCP4 package, and model building was performed with COOT (Supplementary Table 3). Crystal structure was deposited in the Protein Data Bank (PDB: 5jm4).
Cell Permeability Assay
HeLa cells were grown as a monolayer in 10 cm tissue culture dishes and cultured in DMEM supplemented with 10% fetal calf serum, and nonessential amino acids (at 37 °C in an atmosphere of 5% CO2). For experiments, cells were removed from flasks by treatment with trypsin-EDTA, and 5000 cells were plated in each well of a 96-well microplate and cultured for 24 h. Then, peptides were added at a final concentration of 20 μM with 1% DMSO to the medium and incubated for 4 h. Cells were washed three times with PBS, fixed with 4% paraformaldehyde, and washed another three times with PBS. For nuclear staining, a 3 μM 4′,6-diamidino-2-phenylindole (DAPI) solution in PBS was prepared and left on the cells for at least 5 min. After additional washing steps, the cells were left in PBS, and the distribution of FITC-labeled peptides was analyzed via fluorescence microscopy using a 20× air objective (Axiovert 40 CFL, Zeiss).
Quantitative Real Time PCR Analysis
U87 glioblastoma cells were cultivated in DMEM (+10% FCS) at 37 °C at 5% CO2. Cells were plated for 24 h, and medium was changed (DMEM + 1% FCS). After another 24 h, cells were treated with 200 nM 14–3–3ζ, the corresponding peptides in DMEM + 1% FCS and 0.5% DMSO. Untreated and 14–3–3ζ-treated controls were cultivated under the same conditions with 0.5% DMSO. After 24 h of incubation, total RNA was isolated (Quick-RNA MicroPrep Kit, Zymo Research) and reverse transcribed into cDNA (Quanti Tect Reverse Transcription Kit, Qiagen). Next, cDNA was used for quantitative real time PCR (SensiMix SYBR Low-ROX Kit, Bioline) in the Applied Biosystems 7500 Fast Real Time PCR machine (Thermo Fisher Scientific). For relative quantitation, 2−ΔΔCT method was used with the reference gene GAPDH.
Acknowledgments
We are grateful for support from AstraZeneca, Bayer CropScience, Bayer HealthCare, Boehringer Ingelheim, Merck KGaA, and the Max-Planck-Society. O.K. is funded by the German Federal Ministry for Education and Research (Medizinische Chemie in Dortmund, Grant No. BMBF 1316053). T.N.G. thanks the Deutsche Forschungsgemeinschaft (DFG; Emmy Noether program GR3592/2-1) and the European Reasearch Council (ERC; ERC starting grant, no. 678623). This work was supported by the Collaborative Research Centre 1093, funded by the Deutsche Forschungsgemeinschaft (DFG). We appreciate IT support by C. Schwittek (MPI Dortmund).
Glossary
Abbreviations Used
- ACN
acetonitrile
- ADT
AutoDock-Tools
- APN
aminopeptidase N, CD13
- ASP
Astex Statistical Potential
- cRAF
RAF proto-oncogene serine/threonine-protein kinase
- DAP
4′,6-diamidino-2-phenylindole
- DIPEA
N,N-diisopropylethylamine
- DMEM
Dulbecco’s modified Eagle’s medium
- EDT
1,2-ethanedithiol
- FCS
fetal calf serum
- FITC
fluorescein isothiocyanate
- FP
fluorescence polarization
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- IPTG
isopropyl-β-d-1-thiogalactopyranoside
- Kd
dissociation constant (M)
- MMP
matrix metalloproteinase
- NTA
nickelnitrilotriacetic acid
- PEG2
8-amino-3,6-dioxaoctanoyl
- PPI
protein–protein interaction
- PyBop
(benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate
- SASA
solvent accessible surface area
- SEC
size exclusion chromatography
- SLS
swiss light source
- SPPS
solid phase peptide synthesis
- TAMRA
5-carboxytetramethylrhodamine
- TCEP
tris(2-carboxyethyl)phosphine
- TEV
tobacco etch virus
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.7b01221.
Computational setup, peptide synthesis and characterization, protein expression and purification, fluorescence polarization assays, X-ray crystallography and structure determination, cell permeability assay, quantitative real time PCR analysis (PDF)
Highest scored binding mode of 10−21 (ZIP)
Molecular formula strings of peptides (CSV)
Accession Codes
Crystal structure of 22 in complex with 14–3–3 has been released as PDB ID 5jm4.
Author Present Address
§ Department of Cell and Molecular Biology, Uppsala University, BMC Box 596, 751 24 Uppsala, Sweden.
Author Contributions
# These authors contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Villar E. A.; Beglov D.; Chennamadhavuni S.; Porco J. A. Jr.; Kozakov D.; Vajda S.; Whitty A. How proteins bind macrocycles. Nat. Chem. Biol. 2014, 10 (9), 723–731. 10.1038/nchembio.1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelay-Gimeno M.; Glas A.; Koch O.; Grossmann T. N. Structure-based design of inhibitors of protein-protein interactions: mimicking peptide binding epitopes. Angew. Chem., Int. Ed. 2015, 54 (31), 8896–8927. 10.1002/anie.201412070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperandio O.; Reynes C. H.; Camproux A.-C.; Villoutreix B. O. Rationalizing the chemical space of protein-protein interaction inhibitors. Drug Discovery Today 2010, 15 (5–6), 220–229. 10.1016/j.drudis.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Nevola L.; Giralt E. Modulating protein-protein interactions: the potential of peptides. Chem. Commun. 2015, 51 (16), 3302–3315. 10.1039/C4CC08565E. [DOI] [PubMed] [Google Scholar]
- Milroy L. G.; Grossmann T. N.; Hennig S.; Brunsveld L.; Ottmann C. Modulators of protein-protein interactions. Chem. Rev. 2014, 114 (9), 4695–4748. 10.1021/cr400698c. [DOI] [PubMed] [Google Scholar]
- Passioura T.; Katoh T.; Goto Y.; Suga H. Selection-based discovery of druglike macrocyclic peptides. Annu. Rev. Biochem. 2014, 83, 727–752. 10.1146/annurev-biochem-060713-035456. [DOI] [PubMed] [Google Scholar]
- Heinis C.; Winter G. Encoded libraries of chemically modified peptides. Curr. Opin. Chem. Biol. 2015, 26, 89–98. 10.1016/j.cbpa.2015.02.008. [DOI] [PubMed] [Google Scholar]
- Rezaei Araghi R.; Ryan J. A.; Letai A.; Keating A. E. Rapid Optimization of Mcl-1 inhibitors using stapled peptide libraries including non-natural side chains. ACS Chem. Biol. 2016, 11 (5), 1238–1244. 10.1021/acschembio.5b01002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falchi F.; Caporuscio F.; Recanatini M. Structure-based design of small-molecule protein-protein interaction modulators: the story so far. Future Med. Chem. 2014, 6 (3), 343–357. 10.4155/fmc.13.204. [DOI] [PubMed] [Google Scholar]
- Scott D. E.; Bayly A. R.; Abell C.; Skidmore J. Small molecules, big targets: drug discovery faces the protein-protein interaction challenge. Nat. Rev. Drug Discovery 2016, 15 (8), 533–550. 10.1038/nrd.2016.29. [DOI] [PubMed] [Google Scholar]
- Kruger D. M.; Jessen G.; Gohlke H. How good are state-of-the-art docking tools in predicting ligand binding modes in protein-protein interfaces?. J. Chem. Inf. Model. 2012, 52 (11), 2807–2811. 10.1021/ci3003599. [DOI] [PubMed] [Google Scholar]
- Hauser A. S.; Windshugel B. LEADS-PEP: A benchmark data set for assessment of peptide docking performance. J. Chem. Inf. Model. 2016, 56 (1), 188–200. 10.1021/acs.jcim.5b00234. [DOI] [PubMed] [Google Scholar]
- Rentzsch R.; Renard B. Y. Docking small peptides remains a great challenge: an assessment using AutoDock Vina. Briefings Bioinf. 2015, 16 (6), 1045–1056. 10.1093/bib/bbv008. [DOI] [PubMed] [Google Scholar]
- Alogheli H.; Olanders G.; Schaal W.; Brandt P.; Karlen A. Docking of macrocycles: comparing rigid and flexible docking in glide. J. Chem. Inf. Model. 2017, 57 (2), 190–202. 10.1021/acs.jcim.6b00443. [DOI] [PubMed] [Google Scholar]
- Anighoro A.; de la Vega de Leon A.; Bajorath J. Predicting bioactive conformations and binding modes of macrocycles. J. Comput.-Aided Mol. Des. 2016, 30 (10), 841–849. 10.1007/s10822-016-9973-5. [DOI] [PubMed] [Google Scholar]
- Hochscherf J.; Lindenblatt D.; Steinkruger M.; Yoo E.; Ulucan O.; Herzig S.; Issinger O. G.; Helms V.; Gotz C.; Neundorf I.; Niefind K.; Pietsch M. Development of a high-throughput screening-compatible assay to identify inhibitors of the CK2 alpha/CK2 beta interaction. Anal. Biochem. 2015, 468, 4–14. 10.1016/j.ab.2014.09.003. [DOI] [PubMed] [Google Scholar]
- Antes I. DynaDock: A new molecular dynamics-based algorithm for protein-peptide docking including receptor flexibility. Proteins: Struct., Funct., Genet. 2010, 78 (5), 1084–1104. 10.1002/prot.22629. [DOI] [PubMed] [Google Scholar]
- Dagliyan O.; Proctor E. A.; D’Auria K. M.; Ding F.; Dokholyan N. V. Structural and dynamic determinants of protein-peptide recognition. Structure 2011, 19 (12), 1837–1845. 10.1016/j.str.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciemny M. P.; Debinski A.; Paczkowska M.; Kolinski A.; Kurcinski M.; Kmiecik S. Protein-peptide molecular docking with large-scale conformational changes: the p53-MDM2 interaction. Sci. Rep. 2016, 6, 37532. 10.1038/srep37532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audie J.; Swanson J. Recent work in the development and application of protein-peptide docking. Future Med. Chem. 2012, 4 (12), 1619–1644. 10.4155/fmc.12.99. [DOI] [PubMed] [Google Scholar]
- Allen S. E.; Dokholyan N. V.; Bowers A. A. Dynamic docking of conformationally constrained macrocycles: methods and applications. ACS Chem. Biol. 2016, 11 (1), 10–24. 10.1021/acschembio.5b00663. [DOI] [PubMed] [Google Scholar]
- Lee H.; Heo L.; Lee M. S.; Seok C. GalaxyPepDock: a protein-peptide docking tool based on interaction similarity and energy optimization. Nucleic Acids Res. 2015, 43 (W1), W431–W435. 10.1093/nar/gkv495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedan Y.; Marcu O.; Lyskov S.; Schueler-Furman O. Peptiderive server: derive peptide inhibitors from protein-protein interactions. Nucleic Acids Res. 2016, 44 (W1), W536–W541. 10.1093/nar/gkw385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glas A.; Bier D.; Hahne G.; Rademacher C.; Ottmann C.; Grossmann T. N. Constrained peptides with target-adapted cross-links as inhibitors of a pathogenic protein-protein interaction. Angew. Chem., Int. Ed. 2014, 53 (9), 2489–2493. 10.1002/anie.201310082. [DOI] [PubMed] [Google Scholar]
- Glas A.; Wamhoff E. C.; Krüger D. M.; Rademacher C.; Grossmann T. N. Increased conformational flexibility of a macrocycle–receptor complex contributes to reduced dissociation rates. Chem. - Eur. J. 2017, 10.1002/chem.201702776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafmeister C. E.; Po J.; Verdine G. L. An all-hydrocarbon cross-linking system for enhancing the helicity and metabolic stability of peptides. J. Am. Chem. Soc. 2000, 122 (24), 5891–5892. 10.1021/ja000563a. [DOI] [Google Scholar]
- Cromm P. M.; Spiegel J.; Grossmann T. N. Hydrocarbon stapled peptides as modulators of biological function. ACS Chem. Biol. 2015, 10 (6), 1362–1375. 10.1021/cb501020r. [DOI] [PubMed] [Google Scholar]
- Cromm P. M.; Wallraven K.; Glas A.; Bier D.; Fürstner A.; Ottmann C.; Grossmann T. N. Constraining an irregular peptide secondary structure through ring-closing alkyne metathesis. ChemBioChem 2016, 17 (20), 1915–1919. 10.1002/cbic.201600362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B. C.; Yang H. Z.; Liu Y. C.; Jelinek T.; Zhang L. X.; Ruoslahti E.; Fu H. Isolation of high-affinity peptide antagonists of 14–3-3 proteins by phage display. Biochemistry 1999, 38 (38), 12499–12504. 10.1021/bi991353h. [DOI] [PubMed] [Google Scholar]
- Wu H.; Ge J. Y.; Yao S. Q. Microarray-assisted high-throughput identification of a cell-permeable small-molecule binder of 14–3-3 proteins. Angew. Chem., Int. Ed. 2010, 49 (37), 6528–6532. 10.1002/anie.201003257. [DOI] [PubMed] [Google Scholar]
- Arrendale A.; Kim K.; Choi J. Y.; Li W.; Geahlen R. L.; Borch R. F. Synthesis of a phosphoserine mimetic prodrug with potent 14–3-3 protein inhibitory activity. Chem. Biol. 2012, 19 (6), 764–771. 10.1016/j.chembiol.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bier D.; Rose R.; Bravo-Rodriguez K.; Bartel M.; Ramirez-Anguita J. M.; Dutt S.; Wilch C.; Klarner F. G.; Sanchez-Garcia E.; Schrader T.; Ottmann C. Molecular tweezers modulate 14–3-3 protein-protein interactions. Nat. Chem. 2013, 5 (3), 234–239. 10.1038/nchem.1570. [DOI] [PubMed] [Google Scholar]
- Thiel P.; Roglin L.; Meissner N.; Hennig S.; Kohlbacher O.; Ottmann C. Virtual screening and experimental validation reveal novel small-molecule inhibitors of 14–3-3 protein-protein interactions. Chem. Commun. 2013, 49 (76), 8468–8470. 10.1039/c3cc44612c. [DOI] [PubMed] [Google Scholar]
- Milroy L. G.; Bartel M.; Henen M. A.; Leysen S.; Adriaans J. M. C.; Brunsveld L.; Landrieu I.; Ottmann C. Stabilizer-guided inhibition of protein-protein interactions. Angew. Chem., Int. Ed. 2015, 54 (52), 15720–15724. 10.1002/anie.201507976. [DOI] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. Software news and update AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31 (2), 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch O.; Jager T.; Heller K.; Khandavalli P. C.; Pretzel J.; Becker K.; Flohe L.; Selzer P. M. Identification of M-tuberculosis thioredoxin reductase inhibitors based on high-throughput docking using constraints. J. Med. Chem. 2013, 56 (12), 4849–4859. 10.1021/jm3015734. [DOI] [PubMed] [Google Scholar]
- Kruger D. M.; Evers A. Comparison of structure- and ligand-based virtual screening protocols considering hit list complementarity and enrichment factors. ChemMedChem 2010, 5 (1), 148–158. 10.1002/cmdc.200900314. [DOI] [PubMed] [Google Scholar]
- Mooij W. T. M.; Verdonk M. L. General and targeted statistical potentials for protein-ligand interactions. Proteins: Struct., Funct., Genet. 2005, 61 (2), 272–287. 10.1002/prot.20588. [DOI] [PubMed] [Google Scholar]
- Eldridge M. D.; Murray C. W.; Auton T. R.; Paolini G. V.; Mee R. P. Empirical scoring functions 0.1. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput.-Aided Mol. Des. 1997, 11 (5), 425–445. 10.1023/A:1007996124545. [DOI] [PubMed] [Google Scholar]
- Jones G.; Willett P.; Glen R. C.; Leach A. R.; Taylor R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267 (3), 727–748. 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- Kim Y. W.; Grossmann T. N.; Verdine G. L. Synthesis of all-hydrocarbon stapled alpha-helical peptides via ring-closing olefin metathesis. Nat. Protoc. 2011, 6, 761–771. 10.1038/nprot.2011.324. [DOI] [PubMed] [Google Scholar]
- Ghaffari A.; Li Y. Y.; Kilani R. T.; Ghahary A. 14–3-3 sigma associates with cell surface aminopeptidase N in the regulation of matrix metalloproteinase-1. J. Cell Sci. 2010, 123 (17), 2996–3005. 10.1242/jcs.069484. [DOI] [PubMed] [Google Scholar]
- Nefla M.; Sudre L.; Denat G.; Priam S.; Andre-Leroux G.; Berenbaum F.; Jacques C. The pro-inflammatory cytokine 14–3-3 epsilon is a ligand of CD13 in cartilage. J. Cell Sci. 2015, 128 (17), 3250–3262. 10.1242/jcs.169573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaffari A.; Li Y. Y.; Kilani R. T.; Ghahary A. 14–3-3 sigma associates with cell surface aminopeptidase N in the regulation of matrix metalloproteinase-1. J. Cell Sci. 2010, 123 (17), 2996–3005. 10.1242/jcs.069484. [DOI] [PubMed] [Google Scholar]
- Jiang W. G.; Sanders A. J.; Katoh M.; Ungefroren H.; Gieseler F.; Prince M.; Thompson S. K.; Zollo M.; Spano D.; Dhawan P.; Sliva D.; Subbarayan P. R.; Sarkar M.; Honoki K.; Fujii H.; Georgakilas A. G.; Amedei A.; Niccolai E.; Amin A.; Ashraf S. S.; Ye L.; Helferich W. G.; Yang X.; Boosani C. S.; Guha G.; Ciriolo M. R.; Aquilano K.; Chen S.; Azmi A. S.; Keith W. N.; Bilsland A.; Bhakta D.; Halicka D.; Nowsheen S.; Pantano F.; Santini D. Tissue invasion and metastasis: Molecular, biological and clinical perspectives. Semin. Cancer Biol. 2015, 35, S244–S275. 10.1016/j.semcancer.2015.03.008. [DOI] [PubMed] [Google Scholar]
- Nagase H.; Visse R.; Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 2006, 69 (3), 562–573. 10.1016/j.cardiores.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Konttinen Y. T.; Ainola M.; Valleala H.; Ma J.; Ida H.; Mandelin J.; Kinne R. W.; Santavirta S.; Sorsa T.; Lopez-Otin C.; Takagi M. Analysis of 16 different matrix metalloproteinases (MMP-1 to MMP-20) in the synovial membrane: different profiles in trauma and rheumatoid arthritis. Ann. Rheum. Dis. 1999, 58 (11), 691–697. 10.1136/ard.58.11.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30 (16), 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maestro, version 9.7; Schrodinger LLC: New York, 2014. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.