

Abstract
Vitamin A (all-trans-retinol) is metabolized to the visual chromophore (11-cis-retinal) in the eyes and to all-trans-retinoic acid, a hormone like compound, in most tissues. A key enzyme in retinoid metabolism is lecithin:retinol acyltransferase (LRAT), which catalyzes the esterification of vitamin A. The importance of LRAT is indicated by pathogenic missense and nonsense mutations, which cause devastating blinding diseases. Retinoid-based chromophore replacement therapy has been proposed as treatment for these types of blindness based on studies in LRAT null mice. Here, we analyzed the structural and biochemical basis for retinal pathology caused by mutations in the human LRAT gene. Most LRAT missense mutations associated with retinal degeneration are localized within the catalytic domain, whereas E14L substitution is localized in an N-terminal α-helix, which has been implicated in interaction with the phospholipid bilayer. To elucidate the biochemical consequences of this mutation, we determined LRAT(E14L)’s enzymatic properties, protein stability, and impact on ocular retinoid metabolism. Bicistronic expression of LRAT(E14L) and enhanced green fluorescence protein revealed instability and accelerated proteosomal degradation of this mutant isoform. Surprisingly, instability of LRAT(E14L) did not abrogate the production of the visual chromophore in a cell-based assay. Instead, expression of LRAT(E14L) led to a rapid increase in cellular levels of retinoic acid upon retinoid supplementation. Thus, our study unveils the potential role of retinoic acid in the pathology of a degenerative retinal disease with important implications for the use of retinoid-based therapeutics in affected patients.
Graphical Abstract
Perception of light is mediated by the light-induced change in the geometric configuration of the visual chromophore (11-cis-retinal) bound to rhodopsin or cone opsins, subsequently triggering a cascade of G-protein-mediated signaling events.1,2 To sustain continuous vision and preserve the health of photoreceptor cells, 11-cis-retinal needs to be regenerated. In vertebrates, thermodynamically unfavorable reisomerization of all-trans-retinal back to its 11-cis configuration occurs via a metabolic pathway known as the retinoid (visual) cycle.3 The critical role of this process for the health of photoreceptors and retinal pigmented epithelium (RPE) cells is accentuated by numerous retinal degenerative diseases caused by mutations in enzymes involved in the visual cycle.4 Among them, Leber congenital amaurosis (LCA) is the most severe. LCA is characterized by the early onset and fast progression that contribute to the exceptional burden of this blinding disease.5 This autosomal recessive ocular disease is associated with mutations in at least 14 genes that encode proteins important for vision6 including key enzymes of the visual cycle: RPE-specific protein with a molecular mass of 65 kDa (RPE65),7–9 retinol dehydrogenase 12 (RDH12),10,11 and lecithin:retinol acyltransferase (LRAT).12
The functional significance of LRAT in ocular retinoid metabolism arises from its ability to selectively convert vitamin A (all-trans-retinol) into retinyl esters by transferring an acyl moiety from the sn-1 position of phosphatidylcholine.13–15 This enzymatic activity is essential for the effective uptake of vitamin A from the systemic circulation into the RPE to build a retinoid storage pool within the cells.16 More importantly, LRAT provides a direct substrate for RPE65-dependent production of 11-cis-retinol.17,18 Consequently, LRAT-deficiency abolishes formation of visual pigments and leads to progressive retinal degeneration.16,19
Currently, there are 13 identified genetic mutations in LRAT that cause LCA or milder retinitis pigmentosa (summarized in ref 21). Six of them result in reading frame shifts and premature termination of translation.12,22–26 The remaining 7 are single amino acid substitutions clustered within the catalytic domain that may directly affect enzyme activity.12,23,27,28 The only exception is E14L, which is located in the N-terminal helix (Figure 1), a region of LRAT that was shown not to be required for vitamin A esterification both in vitro14,29 and in tissue culture experiments.30 Nonetheless, a patient affected by this mutation suffers from a severe form of LCA associated with atrophy of RPE cells.27 This surprising finding suggests an alternative to the visual chromophore depletion mechanism of pathogenesis. Yet, the retinoid-based visual chromophore replacement therapy aimed to preserve vision in patients affected by LRAT mutations was developed based on the studies in LRAT null mice completely lacking the visual chromophore.31,32 Thus, the proper interpretation of ongoing clinical trials as well as the future clinical application of retinoid-based drugs may depend on better understanding pathogenesis related to noninactivating LRAT mutations.
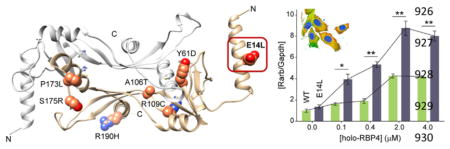
Figure 1.

Position of LCA-associated missense mutations of LRAT within a model of the human enzyme. (A) Side chains of residues substituted in patients diagnosed with retinal degeneration are shown as spheres. A yellow background represents the plane of a phospholipid membrane. Thus, the protein is oriented such that transmembrane helices (TM I and TM II) are perpendicular, whereas N-terminal helices stay parallel to the phospholipid membrane surface (top view). All of the mutations are clustered within the catalytic domains with the exception of E14L, which affects the residue in the N-terminal portion of the enzyme (indicated by the red rectangle). (B) Sequence alignment of the N-termini of human and mouse LRAT. Glutamic acid residue substituted with leucine is marked in green. (C) α-Helical model of N-terminal section of LRAT. Two distinct sides of the α-helix, polar and hydrophobic, are indicated. The color scheme represents electrostatic charge of the side chains (red, negative; blue, positive). (D) Effect of E14L mutation on the orientation of the N-terminal helix with respect to the phospholipid bilayer. Elimination of the negative charge changes in the middle of the polar side of the amphiphilic helix alters the mode of interaction of this part of LRAT with the lipid membrane, allowing it to partition into the hydrophobic core of the membrane. Calculation of the modes and free energies of the orientation of the peptides with phospholipids was performed using PPM server.20
In an attempt to determine the molecular mechanism responsible for E14L-induced progressive retinal degeneration, we analyzed the biochemical characteristics of the mutated enzyme. We investigated the effect of E14L substitution on LRAT’s function in intracellular vitamin A uptake, production of visual chromophore, and retinoid homeostasis. Our study reveals the potential contribution of altered retinoid homeostasis to retinal pathology and indicates diversity in the mechanisms that lead to retinal dystrophies caused by mutations in LRAT. Thus, our findings may have significant implications for choosing the most appropriate therapeutic strategy in patients affected by nondeactivating LRAT mutations.
MATERIAL AND METHODS
Generation of the LRAT Homology Model
A homology model for human LRAT was generated based on the crystal structure of the HRASLS3/LRAT chimeric enzyme (PDB accession 4Q95)15 using SWISS-MODEL server.33,34 Initial model coordinates were examined with COOT35 to optimize the stereochemistry and inter-residue contacts. The model was then energy-minimized in UCSF Chimera.36 The secondary structure of the N-terminus was calculated based on the amino acid sequence with help of PSIPRED server.37 Images of the LRAT homology model were generated in UCSF Chimera.38
Mutagenesis and Stable Transduction of NIH3T3 Cell Lines
NIH3T3 and Phoenix-Eco retroviral producer cell lines as well as mouse LRAT cDNA were purchased from American Type Culture Collection (ATCC). To construct retroviral expression vectors, EcoRI and NotI restriction sites were introduced at the ends of the coding sequence of LRAT’s cDNA by PCR by using the following primers: forward, GAGGTGAATTCAGCTACTCAGGGATGAAGAAC-CCAATGCTGGAAGC; reverse, ACTGACGCGGCCGCA-TGAAGCTAGCCAGACATCATCCACAAGC. The modified LRAT cDNA was cloned into the pMX-IG or pMX-IP retroviral vector provided by Dr. T. Kitamura from the University of Tokyo.39 These vectors allowed for insertion of LRAT cDNA into a multicloning site located upstream of the internal ribosomal entry site (IRES) and enhanced green fluorescence protein (EGFP) or puromycin resistance gene, respectively. Thus, expression of the protein of interest and the EGFP or the antibiotic selection gene occurred from the same mRNA. The LRAT(E14L) mutation was introduced by PCR amplification of the entire plasmid by using Phusion high-fidelity polymerase (New England Biolabs). The constructs were sequenced to confirm that only the desired mutation was introduced into the cDNA and integrity of the IRES site was not compromised.
NIH3T3-LRAT and NIH3T3-LRAT(E14L) stable cell lines were generated by transduction of NIH3T3 cells with retrovirus resulting from transfection of Phoenix-Eco cells with pMXs-IG containing LRAT’s cDNA according to the previously published protocol.40,41 Prior to performing functional experiments, the cells were sorted by a FACSAria cell sorter (BD Biosciences) to select for transduced cells and ensure comparable EGFP fluorescence intensity profiles of NIH3T3-LRAT and NIH3T3-LRAT(E14L).41 The cell lines were cultured in growth medium (GM) composed of Dulbecco’s modified Eagle’s medium, pH 7.2, with 4 mM L-glutamine, 4500 mg/L glucose, and 110 mg/L sodium pyruvate, supplemented with 10% heat-inactivated fetal bovine serum, 100 units/mL penicillin, and 100 units/mL streptomycin. Cells were maintained at 37 °C in 5% CO2.
LRAT and LRAT(E14L) cloned into the pMXs-IP vector were used to produce cells expressing both retinoic acid-responsive gene product 6 (STRA6) and LRAT or its mutant (NIH3T3-STRA6-LRAT and NIH3T3-STRA6-LRAT(E14L), respectively). A previously generated NIH3T3-STRA6 stable cell line40,42 was transduced using the same procedure as referenced above. To select the transduced cells, NIH3T3-STRA6-LRAT and NIH3T3-STRA6-LRAT(E14L) were cultured in GM supplemented with puromycin (5 μg/mL).
Immunoblotting of LRAT and RPE65
NIH3T3-LRAT and NIH3T3-LRAT(E14L) cells were plated on 6-well plates at 1 × 106 cells/well, grown for 24 h, and washed with 154 mM NaCl, 5.6 mM Na2HPO4, 1 mM KH2PO4, pH 7.2 (PBS). Cells were detached by scraping and pelleted by centrifugation (1500g, 5 min, 4 °C), resuspended in 200 μL of RIPA lysis buffer (ThermoFisher), and sonicated for 5 s to shear the DNA. Twenty microliters of the cell lysate was mixed with 5 μL of SDS loading buffer (Bio-Rad). Proteins were separated on a 4–20% SDS-PAGE gradient gel (20 μL of each sample) and subsequently transferred onto polyvinylidene fluoride membranes (Bio-Rad). LRAT and β-actin (the control for equal sample loading) were detected by using a primary anti-LRAT monoclonal antibody30 diluted 1:5000, an anti-RPE65 monoclonal antibody (1:5000),43 and an anti-β-actin monoclonal antibody (Sigma-Aldrich) diluted 1:10000, followed by a secondary anti-mouse IgG horse radish peroxidase conjugated antibody (Promega) or chemiluminescent detection reagent (WesternBright, Advansta). Protein bands were visualized using 3,3′,5,5′-tetramethylbenzidine stabilized substrate (Promega). ImageJ software44 was used for the semiquantitative densitometric analysis of the protein bands.
Immunohistochemistry
Localization of LRAT and its E14L mutant was performed by fixing cells with 4% paraformaldehyde in PBS for 10 min. Cells were washed three times with PBST (PBS with 0.1% Triton X-100) and incubated in 1.5% goat serum in PBST for 15 min at room temperature to block nonspecific binding. Cells then were incubated with an anti-LRAT monoclonal antibody30 and a rabbit anti-calreticulin polyclonal antibody (Sigma-Aldrich). Cells were washed in PBST three times and stained with Cy5-conjugated goat anti-mouse IgG (Promega) and Cy3-conjugated goat anti-rabbit IgG (Promega). Cells were mounted in ProLong Gold antifade reagent containing 4′,6-diamidino-2-phenylindole (Molecular Probes) and imaged with a Leica TCS SP2 confocal/multiphoton microscope equipped with a titanium/sapphire laser (Chameleon-XR).
Expression and Purification of RBP4 and CRBP1
Human plasma retinol binding protein (RBP4) was expressed in E. coli, refolded in the presence of all-trans-retinol, and purified according to the detailed protocol published in Golczak et al.40 The final step of the holo-RBP4 purification added to the previous protocol was gel filtration. Fractions containing refolded protein were combined, concentrated to a volume of 5 mL using an Amicon Ultra-4 centrifugal filter with a cutoff 10 kDa (Millipore), and loaded onto a a Superdex 200 (GE Healthcare) gel filtration column equilibrated with 10 mM Tris/HCl buffer, pH 8.0, 150 mM NaCl. Fractions containing holo-RBP4 with an absorbance ratio at 330/280 nm of 0.85 or higher were pooled together, concentrated to 2.2 mg/mL, and stored at −80 °C until further use. To calculate saturation of RBP4 with vitamin A, 0.1 mL of holo protein (10.5 nmol) was extracted with 0.3 mL of hexane. The organic phase was collected and subjected to HPLC-based quantification of all-trans-retinol under the conditions described below in the Retinoid Isomerization Activity Assay section. The amount of vitamin A was calculated to be 9.84 nmol based on a known amount of the synthetic standard. Thus, the saturation of RBP4 with the retinoid ligand was ≈94%. This value corresponded well with a previously reported 0.9 absorbance ratio at 330/280 nm for holo-RBP4 isolated from the serum of healthy human donors45 or human recombinant RBP4 refolded in the presence of vitamin A.46
Human cellular retinol-binding protein 1 (CRBP1) was expressed, purified, and loaded with all-trans-retinol according to the method published in Silvaroli et al.47 without any changes to the established protocol.
Expression of LRAT and Its Mutants in Yeast
cDNA of LRAT and its E14L mutant were amplified by PCR using the following primer pair: forward, GCAGATACTAGTGTTTA-ATTATCAAACAATATCAATAATGAAGAACCCAATGCT-GGAAGCTGC; reverse, CGTCTAGACGCGTTCAG-CCAGACATCATCCACAAGCAGAATGG. For the LRAT N-terminus deletion mutant in which the first 30 amino acids were omitted (del1–30), the forward primer was replaced with GCAGATACTAGTGTTTAATTATCAAACAATATCA-ATAATGGGAGGAGGCACAGGGAAGAACCG. The PCR products were digested with SpeI and MluI restriction enzymes and subcloned into the YepM vector.48 Saccharomyces cerevisiae strain BJ5457 (ATCC) was transfected using Alkali-Cation Yeast Transformation Kit (MP Biomedicals,) and the cells were plated on leucine-deficient selection medium (−Leu) (MP Biomedicals). Colonies of yeast were inoculated into 25 mL of −Leu media that contained 10% glycerol (v/v). The cultures were incubated at 30 °C for 16 h prior to transfer into 2 L of fresh −Leu/glycerol media. Yeasts were grown until OD600 = 1.2–1.4. Then, the cells were harvested by centrifugation (6000g, 15 min), resuspended in 40 mM Tris/HCl, pH 8.0 with 250 mM sucrose, and disrupted by microfluidization at 100 psi (five cycles). The cell homogenate was spun to remove large cellular debris at 12 000g for 20 min. The resulting supernatant was then centrifuged again at 120000g for 1 h to collect microsomal fraction. Expression of LRAT and its mutants in transfected yeast microsomes was confirmed by western blotting. Because the expression level of LRAT and the mutants varied, the concentration of the enzymes was normalized based on the western blot signal by adjusting volume in which collected yeast microsomes were resuspended.
Cellular Retinol Uptake Assay
NIH3T3 cells that express LRAT or the E14L mutant and STRA6 were cultured in 6-well culture plates at a density of 1 × 106 cells/well 16 h prior to performing an experiment. Cells were washed with PBS and serum-free GM that contained 10 μM all-trans-retinol pre-bound to RBP4 delivered in dimethyl sulfoxide. After incubation for 2–24 h, cells were washed with PBS, harvested by centrifugation (1500g, 5 min, 4 °C), and homogenized in 1 mL of PBS/ethanol (v/v). Retinoids were extracted with 4 mL of hexane. The organic phase was collected, dried down in a SpeedVac, and redissolved in 0.3 mL of hexane. Retinoids were analyzed by normal phase HPLC on an Agilent 1100 series HPLC system equipped with a diode array detector using an Agilent-Si column (4.6 × 250 mm, 5 μm). Retinyl esters were separated in a step gradient of ethyl acetate in hexane (1% for 10 min followed by 10% for 20 min) at a flow rate of 2 mL/min, detected at 325 nm, and quantified by correlating peak areas with known quantities of synthetic all-trans-retinyl palmitate.
Retinoid Isomerization Activity Assay
NIH3T3 cell lines that stably express human RPE65 or RPE65 and LRAT or its E14L mutant were seeded in 6-well culture plates at 1 × 106 cells per well in GM. The isomerization reaction was initiated 16 h later by exchange of the GM for one containing 10 μM all-trans-retinol delivered in N,N-dimethylformamide. From this moment, the cells, cell homogenates, and extract were shielded from light. The reaction was carried out for 16 h in a cell culture incubator (37 °C, 5% CO2). The cells and medium were collected, mixed with an equal volume of 4 M KOH in methanol, and incubated at 52 °C for 2.5 h to hydrolyze retinoid esters. Next, an equal volume of hexane was added, and retinoids were extracted by vigorous shaking. Following 15 min centrifugation (4000g, 4 °C) to facilitate phase separation, the hexane layer was collected, dried down, and redissolved in 250 μL of hexane. Extracted retinoids were separated on a normal phase HPLC Agilent-Si column (4.6 × 250 mm, 5 μm) using 10% ethyl acetate in hexane as the mobile phase at an isocratic flow rate of 2.0 mL/min. 11-cis-Retinol was detected at 325 nm and quantified by correlating peak areas with known quantities of a synthetic standard.
LRAT Enzymatic Assay
The acyltransferase activity of LRAT was examined in 20 mM Tris/HCl buffer, pH 7.5, 1 mM DTT, containing 1% bovine serum albumin. Microsomes isolated from yeast expressing LRAT or its mutants served as a source for the enzymatic activity. To eliminate variations resulting from different concentrations of phospholipid added with the microsomes, the reaction mixture was supplemented with 0.5 mM 1,2-diheptanoate-sn-glycero-3-phosphocholine (Avanti Polar Lipids) that was a source of acyl chains. The enzymatic reaction was initiated by the addition of holo-CRBP1 in concentrations ranging between 0.5 and 30 μM. Reaction mixtures were incubated at 30 °C for 3 min and then stopped with 0.3 mL of ethanol. Retinoids were extracted with 0.3 mL of hexane, and their composition was analyzed by HPLC as described above. To calculate the KM values for all-trans-retinol delivered by CRBP1, the initial rate of retinyl ester formation was plotted against holo-CRBP1 concentration and the experimental points were fitted to a Michaelis–Menten model of enzyme kinetics.
Identification of LRAT(E14L) Degradation Pathway
NIH3T3 cells that express LRAT and its E14L mutant were plated in 6-well plates at 1 × 106 cells/well. Six hours later, the following inhibitors of lysosomal or proteasomal protein degradation pathways were added to the GM: chloroquine (200 μM) (Sigma-Aldrich), leupeptin (100 μM) (Sigma-Aldrich), ammonium chloride (10 mM) (Sigma-Aldrich), or bortezomib (25 nM) (Santa Cruz Biotechnology). Cells were grown for 24 h before they were harvested for immunoblotting analysis of LRAT protein level.
QPCR-Based Quantification of Rarβ2 Transcription
NIH3T3-STRA6-LRAT and NIH3T3-STRA6-LRAT(E14L) cells were plated in 6-well plates at 1 × 106 cells/well. Holo-RBP4 was added after 6 h, and the cells were grown for an additional 24 h before they were detached by scraping and pelleted by centrifugation (1500g, 5 min, 4 °C). Cells were washed with PBS twice, resuspended in RLT buffer (Qiagen) supplemented with 10 μL of β-mercaptoethanol per 1 mL, and homogenized by a QIA shredder (Qiagen). Total mRNA isolation was carried out with an RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. RNA concentration and purity were measured with a Nanodrop spectrophotometer (ND-1000, Thermo Scientific). The Applied Biosystems reverse transcription kit (4387406, Applied Biosystems) was used to reverse transcribe up to 2 μg (for a 20 μL reaction) of total RNA to cDNA. RT-qPCR was carried out with TaqMan probes (Applied BioSystems) for the retinoic acid receptor β2 gene, Rarβ2 (Mm01319677_m1). Glyceraldehyde-3-phosphate dehydrogenase, Gapdh (Mm99999915_g1), was used as an endogenous control. All real-time experiments were done with an ABI Step-One Plus RT-qPCR instrument (Applied BioSystems).
Statistical Analysis
Data are represented as the mean ± standard deviation (SD) from at least three independent experiments. For the statistical analysis, results of at least two independent experiments were repeated in triplicates. Significance between the two groups was determined by unpaired Student’s t test. Sigma Plot 11.0 (Systat Software) and Origin 2015 were used to perform statistical analyses.
RESULTS
Location of Substitution Mutations within a Model of Human LRAT Structure and Their Potential Effect on Enzyme Function
Solving the crystal structure of a chimeric enzyme composed of a catalytic subunit of HRASLS3 and LRAT’s specific domain15 allowed us to build a three-dimensional model of human LRAT and search for a plausible mechanism by which pathogenic mutations might affect protein functions. As indicated in Figure 1A, the majority of known substitution mutations causing LCA occur within the catalytic domain in close proximity to the active site. Detailed examination of the LRAT model revealed that these mutations directly affect the orientation of catalytic residues with respect to each other (mutations Y61D and R109C), destabilize the structure of α-helix 4 that contains the catalytic cysteine (mutations A106T, P173L, and S175R), or interfere with the polar end of transmembrane helices (mutation R190H). Importantly, adverse effects of these single amino acid substitutions on LRAT’s activity can be amplified by homodimerization, domain swapping, and membrane localization of the enzyme.
In contrast, the functional effect of the E14L mutation could not be inferred directly from LRAT’s homology model. This substitution occurred in the N-terminal segment (Figure 1B), a conserved portion of the enzyme, whose function remains unknown. Secondary structure prediction performed using the PSIPRED server indicated a strong propensity for the first 20 amino acids of LRAT to fold into an α-helix. Importantly, it contained two discrete sides, polar and hydrophobic, which determined the amphiphilic property of the N-terminus (Figure 1C). The PPM server-based20 calculations of rotational and translational positions of this α-helix with respect to a phospholipid bilayer suggested that the N-terminal peptide has a propensity to interact with the lipid membrane (ΔG = −10.6 and −9.0 kcal/mol for human and mouse peptides, respectively); however, its orientation remained parallel to the membrane surface with tilt angles of 88 ± 10° and 76 ± 7° (Figure 1D), respectively, for the human and mouse peptides. Remarkably, the single amino acid substitution of E14 for L, which occurred at the polar side of the α-helix, lowers the free energy of insertion into a lipid membrane to ΔG = −16.0 and −14.3 kcal/mol for the human and mouse variants of the N-terminal peptide. This sequence modification allowed for the potential formation of a transmembrane segment composed of residues 4–19 that traverses the lipid bilayer at an angle of 36 ± 5° and 37 ± 3° with respect to the membrane normal (Figure 1D). Thus, the E14L mutation altered the mode of interaction between the N-terminus of LRAT and a biological membrane.
E14L Substitution Affects Protein Stability and Intra-cellular Localization
To evaluate consequences of E14L mutation on the stability and enzymatic activity of the protein, we first generated two NIH3T3 cell lines that stably express mouse LRAT or the E14L mutant. We selected the mouse protein over the human variant because there is no antibody that can reliably recognize the human enzyme. Importantly, the sequence of LRAT’s N-terminus is highly conserved (Figure 1B), implying identical biophysical properties of this region in the mouse and human enzymes.
To control for confounding variables that may affect transfection efficiency and thus protein expression levels, we employed a bicistronic retroviral-based system, in which the cDNA of LRAT or its mutant was inserted upstream of the IRES and EGFP sequence.7,39,41 This configuration allowed for expression of both the protein of interest and EGFP from the same mRNA. Thus, by sorting transduced NIH3T3 based on green fluorescence intensity and selecting populations of cells characterized by identical fluorescence profiles, we ensured equivalent mRNA levels for the wild-type (WT) and mutated form of LRAT (Figure 2A).
Figure 2.
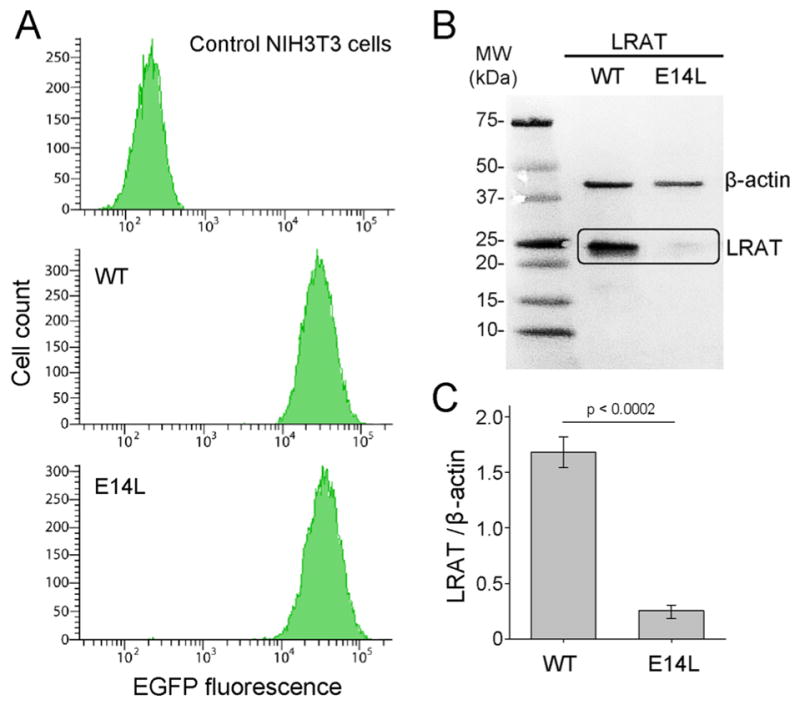
Analysis of mRNA and protein levels of WT LRAT and its E14L mutant. (A) Flow cytometry profiles of EGFP fluorescence in NIH3T3 cells expressing WT LRAT or LRAT(E14L). The pMX-IG retroviral transfer vector contained LRAT-IRES-EGFP cassettes that enabled expression of both LRAT and EGFP from the same mRNA. Thus, a similar distribution of green fluorescence in cells expressing WT LRAT and its E14L variant indicated similar levels of LRAT mRNA. (B) Immunoblot of NIH3T3-LRAT and NIH3T3-LRAT-(E14L) cells. A lower protein level for the mutant enzyme compared to that of WT is visible. (C) Densitometry-based quantification of the relative intensities of bands shown in panel B. Sample loading was normalized based on the signal for β-actin. Error bars correspond to SD obtained from three independent experiments.
Immunoblotting analysis of NIH3T3-LRAT revealed robust expression of the WT enzyme. However, the protein level of the E14L mutant was greatly reduced in NIH3T3-LRAT-(E14L) cells (Figure 2B). Densitometry-based quantification of protein levels in relation to a loading marker (β-actin) revealed that the effective concentration of the mutant variant was around 16% of that observed for WT LRAT (Figure 2C).
Because the NIH3T3-LRAT and NIH3T3-LRAT(E14L) cell lines had comparable levels of EGFP transcript, the difference in LRAT protein levels might reffect decreased stability and/or accelerated degradation of the E14L mutant. To distinguish between these two scenarios, we cultured NIH3T3-LRAT-(E14L) cells in the presence of inhibitors of lysosomal or proteasome-dependent proteolysis. Among the tested compounds, only treatment with bortezomib resulted in a significantly increased amount of the mutated enzyme that reached nearly 80% of that observed for WT LRAT (Figure 3A). Next, we examined whether the accumulated LRAT-(E14L) retained proper intracellular localization. Immunohistochemical staining and confocal microscopy of transiently transfected NIH3T3 cells showed that WT LRAT colocalized with the ER marker (calreticulin), which is in agreement with previously published data.30 Importantly, substitution of E14L did not affect ER localization of the enzyme (Figure 3B). From these experiments, we concluded that the E14L mutation in LRAT makes the protein more susceptible to degradation via the proteasomal pathway, which is a major component of ER-associated degradation of misfolded or misassembled membrane proteins.49
Figure 3.
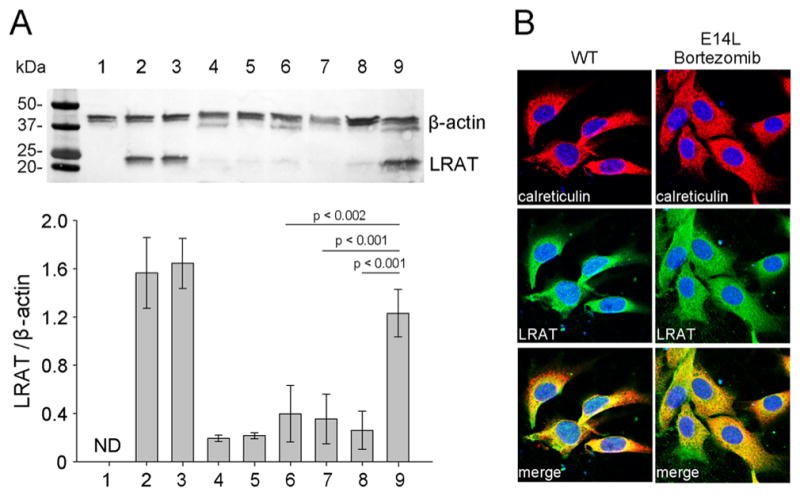
Effect of inhibitors of protein degradation pathways on LRAT(E14L) expression level. (A) NIH3T3-LRAT(E14L) were treated with selected inhibitors 24 h prior to an assessment of the protein expression level by immunoblot. Line 1 shows untransfected NIH3T3 cells; lines 2 and 3 represent untreated or DMSO treated NIH3T3 cells that stably express WT LRAT, and lines 4 and 5 correspond to NIH3T3 cells that express the E14L mutant. Lines 6–9 indicate NIH3T3-LRAT(E14L) cultured in the presence of chloroquine (200 μM), leupeptin (100 μM), ammonium chloride (10 mM), or bortezomib (25 nM), respectively. Relative levels of LRAT expression were normalized based on the intensities of the loading control (β-actin). (B) Subcellular localization of LRAT and its mutant in transfected NIH3T3 cells. To ensure adequate signal, cells were pretreated with 25 nM bortezomib. The ER was visualized by immunohistochemical staining with polyclonal anti-calreticulin primary antibody and Cy3-conjugated secondary antibody (red), whereas LRAT was imaged with monoclonal anti-LRAT antibody and corresponding Cy5-conjugated antibody (green). The merged images show the colocalization of LRAT and the E14L mutant with calreticulin.
Cellular Uptake of All-Trans-Retinol in the Presence of the E14L Mutant
To test whether E14L mutation affected retinoid metabolism, we performed a vitamin A uptake assay. Under physiological conditions, vitamin A is transported in the blood bound to RBP4, and its cellular uptake is mediated by RBP4 receptor, STRA6.50–52 To recapitulate these conditions, WT or E14L LRAT was stably coexpressed in NIH3T3 with STRA6.40,42 The uptake assay was initiated by the addition of all-trans-retinol prebound to RBP4. HPLC-based quantification of retinoids extracted from the cells revealed robust time-dependent accumulation of all-trans-retinyl esters for WT LRAT (Figure 4A). However, in the parallel experiments, the amount of retinyl esters found in cells expressing LRAT(E14L) was at least 4 times lower, indicating impaired vitamin A uptake.
Figure 4.

Uptake of vitamin A and isomerization activity. (A) Time course of retinol uptake in NIH3T3-STRA6-LRAT (●) and NIH3T3-STAR6-LRAT(E14L) (○) cells in the presence of 10 μM holo-RBP4. The retinyl esters were quantified in cellular extracts by HPLC. Values represent the mean ± SD of three independent experiments. (B) Determination of kinetic parameters of all-trans-retinol esterification for WT LRAT (●), LRAT(E14L) (○), and LRAT(del1–30), the N-terminus truncated enzyme (▼). The retinoid substrate was delivered in a prebound form to CRBP1. Microsomes isolated from yeast expressing LRAT and its mutated variants were used as a source of the enzymatic activity. Inset represents immunoblot detection of heterologously expressed enzymes. The proteins were separated on a 4–12% SDS-PAGE gradient gel. (C) UV/vis absorbance spectra for holo-RBP4 (purple) and holo-CRBP1 (orange) show the quality of retinol-binding proteins used in this study.
Intracellular transport of vitamin A depends on cellular retinol-binding proteins.53,54 The most ubiquitous protein from this class that is expressed in the RPE is CRBP1.54 Moreover, accumulated enzyme kinetic data suggest that holo-CRBP1 serves as the preferential substrate for LRAT.55,56 Thus, to verify whether reduced vitamin A uptake might be attributed to impaired interaction of retinol-binding protein with LRAT, we determined kinetic parameters for all-trans-retinol esterification delivered in the form of holo-CRBP1. WT LRAT, LRAT-(E14L), and a deletion mutant lacking the first 30 amino acids were expressed in yeast, and microsomal fractions isolated from these cells served as the source for enzymatic activity. Because the protein level of the E14L mutant was also reduced in a yeast expression system as compared with WT, we scaled up the mutant concentration by adjusting the volume of buffer in which the microsomes were suspended. As shown in Figure 4B, tested LRAT variants revealed comparable enzymatic activity. Analysis of initial reaction rates did not result in statistically significant differences in KM and Vmax values, which were calculated to be 4.0 ± 1.3 μM and 45.0 ± 4.0 pmol/min for WT LRAT; 4.9 ± 1.1 μM and 45.5 ± 3.0 pmol/min for LRAT(E14L); and 5.6 ± 1.8 μM and 48.5 ± 4.4 pmol/min for the del1–30 mutant (Figure 4B,C). Thus, neither E14L nor the entire N-terminus of LRAT is necessary for acquiring substrate from holo-CRBP1.
Influence of the LRAT Mutation on RPE65-Dependent Isomerization Activity
On the basis of studies of Lrat−/− mice, the main cause of retinal degeneration in LRAT inactivating mutations is the absence of retinoids in RPE cells and thus lack of production of visual chromophore.16 Because LRAT-dependent esterification is not a rate-limiting step of the visual cycle,57 partial enzymatic activity of the LRAT mutant might be sufficient to sustain regeneration of 11-cis retinoids. To test this hypothesis, we compared kinetics of 11-cis-retinol production in NIH3T3 cells expressing RPE657 and WT or mutated LRAT. The analysis of retinoid composition revealed robust production of 11-cis-retinol in the presence of LRAT(E14L) reaching 327 ± 11.2 pmol per 1 × 106 cells after 16 h of incubation, which was comparable to the amount detected in cells expressing WT LRAT (398 ± 14.9 pmol per 1 × 106) (Figure 5). However, the initial rate of isomerization was slower as compared to that of WT enzyme (27.4 pmol/h/106 cells for the mutant vs 40.4 pmol/h/106 cells for the WT enzyme). Nevertheless, the significantly lower capacity to esterify vitamin A caused by the instability of the E14L variant did not abolish RPE65-dependent production of 11-cis retinoids in the cell culture assay.
Figure 5.

REP65-dependent production of 11-cis-retinol in the presence of LRAT(E14L). (A) HPLC analysis of retinoid composition extracted from NIH3T3 cells that stably express RPE65 and LRAT or its E14L mutant. Chromatography peaks were identified based on elution time and characteristic UV/vis absorbance spectra. Peak ‘a’ corresponds to 11-cis-retinol (UV/vis spectrum shown in panel B), whereas peak ‘b’ corresponds to 13-cis-retinol. (C) Time course of 11-cis-retinol production in NIH3T3-STRA6-LRAT (●) and NIH3T3-STAR6-LRAT(E14L) (○) cells. (D) Immunoblot analysis of expression of RPE65, LRAT, and its E14L mutant in NIH3T3 stable cell lines used for the 11-cis-retinol production assay.
Correlation between Decreased LRAT Activity and RA Levels
We observed that the E14L mutation did not affect visual chromophore production in a cell culture system, suggesting that retinal degeneration in affected patients is caused by a different pathology. An alternative mechanism may involve the role of LRAT in intracellular retinoid homeostasis. In fact, several lines of evidence indicate that LRAT activity influences retinoic acid levels in vivo.58,59 To test whether the intracellular concentration of retinoic acid is influenced by retinol esterification capacity, we compared the transcriptional response of a retinoic acid target gene in NIH3T3-STRA6-LRAT and NIH3T3-STRA6-LRAT(E14L) cells incubated with holo-RBP4. Unlike other cell types, transcription of Hoxa1 or Cyp26a1 is not retinoid acid-dependent in mouse fibroblasts.60 Thus, Rarβ2 was chosen as a reporter gene to evaluate the retinoid acid status.60 As shown in Figure 6, the mRNA level for Rarβ2 increased upon incubation with holo-RBP in both cell lines. However, the magnitude of this increase was much higher in cells expressing the E14L LRAT variant. Importantly, while cells containing WT LRAT were able to effectively buffer retinoic acid levels for up to 0.4 μM holo-RBP4, conversely, the presence of mutated enzyme led to a rapid increase of retinoic acid at a much lower concentration of holo-RBP4. Effectively, comparable mRNA levels for Rarβ2 were detected at 0.1 μM for the LRAT mutant and 2.0 μM of holo-RBP4 for the WT enzyme. Moreover, the maximum level of Rarβ2 induction was much higher for E14L.
Figure 6.
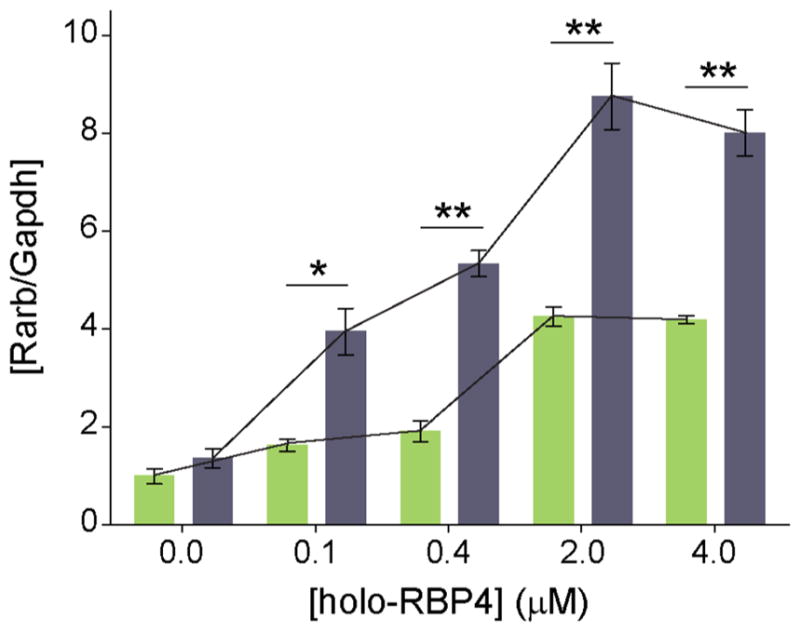
Alteration of RA homeostasis in NIH3T3-STRA6-LRAT-(E14L) cells. Dose-dependence effect of holo-RBP4 on RA regulated induction of Rarβ2 transcript. Total RNA was collected from NIH3T3-STRA6-LRAT (green bars) and NIH3T3-STRA6-LRAT(E14L) (gray bars) cells 24 h after addition of holo-RBP4 and reverse-transcribed to generate cDNA, which was subsequently used for real-time PCR (quantitative mRNA level of Rarβ2 transcription). Each experimental point represents the relative level of Rarβ2 in relation to Gapdh. Experiments were repeated three times using independent RNA samples. The results are representative of three independent biological experiments (mean ± SD). The asterisks depict significance: *p < 0.01, **p < 0.001.
DISCUSSION
The goal of this study was to assess the functional consequences of the noninactivating E14L mutation in the N-terminal part of LRAT that is associated with a severe form of retina and RPE degeneration.27 Alignment of the amino acid sequences of LRAT and highly related HRAS-like suppressors indicates that the extended N-terminus is a unique feature of LRAT.15 However, functional significance of this lipid membrane-interacting extension is unknown. The hypothesis that it might be involved in the recruitment of retinol-binding proteins turned out to be incorrect. The kinetic parameters of vitamin A esterification in the presence of full-length LRAT, its E14L mutant, or LRAT lacking the first 30 amino acids were virtually the same (Figure 4B). Thus, vitamin A esterification by LRAT(E14L) in the cell is not influenced by the inability to access the retinoid substrate bound to CRBP1. Further biochemical characterization of LRAT’s E14L mutant indicated that the mutated enzyme retains its intracellular localization and acyltransferase activity. However, E14L substitution makes the protein vulnerable for accelerated proteasomal degradation that dramatically lowers the concentration of the enzyme in NIH3T3 cells. The mechanism that leads to this instability is not entirely clear. On the basis of the secondary structure prediction, E14 is located in the middle of N-terminal amphiphilic α-helix that interacts with the phospholipid membrane by adopting a lateral orientation with respect to the bilayer normal (Figure 1). Nevertheless, E14L substitution does not per se cause destabilization of the secondary structure. Instead, elimination of a negative charge attributed to the glutamic acid residue transforms the overall character of the N-terminal α-helix from amphiphilic to predominantly hydrophobic. This change might have several implications for the interaction of LRAT with lipid membranes and therefore for the accelerated degradation of the protein. First of all, LRAT belongs to the class of tail-anchored (TA) membrane proteins that possess a single transmembrane α-helix located at the C-terminus.30 Therefore, unlike the majority of polytopic membrane proteins that utilize cotranslational membrane insertion, LRAT depends on the Get3 (guided entry of TA proteins 3)-mediated post-translational pathway for proper insertion into the ER.61 Consequently, TA proteins are synthesized by ribosomes in the cytosol where their canonical C-terminal transmembrane segment is recognized by Get3, which shields it from the aqueous environment until insertion into the ER membrane. Thus, it is important that the N-terminal portion of a TA protein is at least partially soluble. Increased hydrophobicity of LRAT(E14L) N-terminal peptide may cause recognition of this part of the protein as partially unfolded by heat shock proteins and lead to rapid ubiquitination and proteosomal degradation of the mutated enzyme. Alternatively, upon proper insertion of the E14L mutant in the phospholipid membrane, altered interaction of the N-terminus with the lipid bilayer may trigger ER-associated degradation pathways that also include ubiquitination that is essential for retrotranslocation of the polypeptide chain and subsequent degradation.49 One of the putative sites of ubiquitination within LRAT’s N-terminus is the ε-NH2 group of residue K15. However, in silico analysis of the potential ubiquitination sites did not reveal K15 as a primary residue susceptible for this post-translational modification. Moreover, ubiquitination is more prevalent at a lysine that is flanked by negatively charged residues.62 Thus, the exchange of polar glutamic acid for hydrophobic leucine residue at the vicinity of K15 would rather diminish the efficiency of the modification. Nevertheless, we cannot exclude the possibility that the altered sequence of the N-terminal α-helix triggers fusion of ubiquitin to the α-NH2 group of the N-terminal residue, initiating the degradation pathway of mutated LRAT.63
In the past, the pathogenesis of retinal degeneration caused by mutations in key enzymes involved in the retinoid cycle was thought to be metabolic blockage that abolished production of the visual chromophore.64 On the basis of this assumption, a chromophore replacement therapy has been developed that includes systemic administration of 9-cis-retinyl acetate as a pro-drug that allows for bypassing the blockage.32,65 Upon hydrolysis to the corresponding alcohol followed by oxidation, this pro-drug yields 9-cis-retinal that binds to rod and cone opsins as a substitute of 11-cis-retinal.66 This approach has been recently shown to partially restore visual function in LCA patients affected by a subset of RPE65 or LRAT mutations.31 In the context of this clinical trial, our data indicate that for some LCA patients, the mechanism of pathology underlying retinal degeneration might be more complex than previously thought and may include factors such as retinoid toxicity resulting from a metabolic imbalance in vitamin A homeostasis. It is particularly important to consider the metabolic fate of retinoid-based drug in the case of LRAT mutations that result in partial inactivation of the enzyme. LRAT activity not only provides the substrate for RPE65 and is required for cellular uptake of vitamin A16,67 but also plays an important role in maintaining retinoic acid homeostasis.58,59 By esterifying excess vitamin A, LRAT limits the pool of retinol that is available for oxidation to retinal and further retinoic acid. Thus, deficiency in LRAT activity leads to insufficient ability to buffer retinoic acid concentration by the cells in response to systemic administration of vitamin A or a retinoid-based drug such as 9-cis-retinyl acetate. Both all-trans- and 9-cis-retinoic acid are metabolites with profound signaling activity via nuclear retinoic acid receptors and are highly cytotoxic when in excess.68 This mechanism of maintaining retinoid homeostasis is particularly important for RPE cells, which are a key source of retinoic acid for the retina, especially during development.69 Thus, prolonged imbalances or spikes in the levels of intracellular retinoic acid may be additional stressors contributing to RPE atrophy and photoreceptor cell degeneration.
It is tempting to hypothesize that not all LRAT mutations could lead to a similar increase in retinoic acid. In the presence of inactivating mutations, the spike in retinoic acid concentration is acute and occurs shortly after retinoid administration as indicated by studies on Lrat−/− mice.59,70 Because the absence of LRAT activity prevents the formation of vitamin A storage in the liver, an elevated concentration of retinoic acid is transient. In the case of low residual activity of LRAT, such as in the E14L mutant, one may expect that the acute elevation in retinoic acid is followed by a long lasting supply of vitamin A bound to RPB4 that contributes to the persistent imbalance in retinoic acid homeostasis. Thus, an increased concentration of retinoic acid lasts longer. This effect can be particularly prominent in ocular tissues shown to be preferentially supplied with retinoids in a RPB4/STRA6-dependent manner.71 If this scenario holds true, higher toxicity of retinoic acid should be observed in patients affected by E14L substitution as compared to LRATs’ inactivating mutations. Moreover, one can expect that potential adverse effects of visual chromophore replacement therapy will depend on the overall vitamin A status of a patient.
Taking into account the above considerations, our data have important implications for the proper design and assessment of clinical trials aiming to evaluate retinoid-based compounds as well as the future application of such drugs. It has become apparent that functional analysis of LCA-causing LRAT mutations on enzyme activity will help determine whether an LCA patient should be treated with visual chromophore replacement therapy. Therefore, more in-depth studies are needed to evaluate functional consequences of mutations not only in LRAT but also in other enzymes of the retinoic cycle that are associated with progressive retinal degenerative diseases.
Acknowledgments
We thank J. von Lintig from the Department of Pharmacology and J. Lin from the Department of Ophthalmology, CWRU, for fruitful discussions and suggestions that contributed to this manuscript. We also thank P.D. Kiser and K. Palczewski from the Department of Pharmacology, CWRU, for RPE65 and LRAT monoclonal antibodies. This work was supported by grant EY023948 from the National Eye Institute of the National Institutes of Health (NIH) (M.G.). Molecular graphics and analyses were performed with the UCSF Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311).
ABBREVIATIONS
- CRBP1
cellular retinol-binding protein 1
- EGFP
enhanced green fluorescence protein
- ER
endoplasmic reticulum
- HPLC
high-performance liquid chromatography
- IRES
internal ribosomal entry site
- LCA
Leber congenital amaurosis
- LRAT
lecithin:retinol acyltransferase
- Rarβ2
retinoic acid receptor β2 gene
- RBP4
serum retinol-binding protein
- RPE
retinal pigmented epithelium
- RPE65
retinal pigmented epithelium-specific protein with molecular mass 65 kDa
- RDH12
retinol dehydrogenase 12
- TA
tail-anchored
- WT
wild-type
Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.Wald G. The molecular basis of visual excitation. Nature. 1968;219:800–807. doi: 10.1038/219800a0. [DOI] [PubMed] [Google Scholar]
- 2.Hubbard R, Wald G. Cis-trans isomers of vitamin a and retinene in the rhodopsin system. J Gen Physiol. 1952;36:269–315. doi: 10.1085/jgp.36.2.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kiser PD, Golczak M, Palczewski K. Chemistry of the retinoid (visual) cycle. Chem Rev. 2014;114:194–232. doi: 10.1021/cr400107q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Travis GH, Golczak M, Moise AR, Palczewski K. Diseases caused by defects in the visual cycle: Retinoids as potential therapeutic agents. Annu Rev Pharmacol Toxicol. 2007;47:469–512. doi: 10.1146/annurev.pharmtox.47.120505.105225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanein S, Perrault I, Gerber S, Tanguy G, Barbet F, Ducroq D, Calvas P, Dollfus H, Hamel C, Lopponen T, Munier F, Santos L, Shalev S, Zafeiriou D, Dufier JL, Munnich A, Rozet JM, Kaplan J. Leber congenital amaurosis: Comprehensive survey of the genetic heterogeneity, refinement of the clinical definition, and genotype-phenotype correlations as a strategy for molecular diagnosis. Hum Mutat. 2004;23:306–317. doi: 10.1002/humu.20010. [DOI] [PubMed] [Google Scholar]
- 6.Den Hollander AI, Black A, Bennett J, Cremers FP. Lighting a candle in the dark: Advances in genetics and gene therapy of recessive retinal dystrophies. J Clin Invest. 2010;120:3042–3053. doi: 10.1172/JCI42258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bereta G, Kiser PD, Golczak M, Sun W, Heon E, Saperstein DA, Palczewski K. Impact of retinal disease-associated rpe65 mutations on retinoid isomerization. Biochemistry. 2008;47:9856–9865. doi: 10.1021/bi800905v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gu SM, Thompson DA, Srikumari CR, Lorenz B, Finckh U, Nicoletti A, Murthy KR, Rathmann M, Kumaramanickavel G, Denton MJ, Gal A. Mutations in rpe65 cause autosomal recessive childhood-onset severe retinal dystrophy. Nat Genet. 1997;17:194–197. doi: 10.1038/ng1097-194. [DOI] [PubMed] [Google Scholar]
- 9.Morimura H, Fishman GA, Grover SA, Fulton AB, Berson EL, Dryja TP. Mutations in the rpe65 gene in patients with autosomal recessive retinitis pigmentosa or leber congenital amaurosis. Proc Natl Acad Sci U S A. 1998;95:3088–3093. doi: 10.1073/pnas.95.6.3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Janecke AR, Thompson DA, Utermann G, Becker C, Hubner CA, Schmid E, McHenry CL, Nair AR, Ruschendorf F, Heckenlively J, Wissinger B, Nurnberg P, Gal A. Mutations in rdh12 encoding a photoreceptor cell retinol dehydrogenase cause childhood-onset severe retinal dystrophy. Nat Genet. 2004;36:850–854. doi: 10.1038/ng1394. [DOI] [PubMed] [Google Scholar]
- 11.Perrault I, Hanein S, Gerber S, Barbet F, Ducroq D, Dollfus H, Hamel C, Dufier JL, Munnich A, Kaplan J, Rozet JM. Retinal dehydrogenase 12 (rdh12) mutations in leber congenital amaurosis. Am J Hum Genet. 2004;75:639–646. doi: 10.1086/424889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thompson DA, Li Y, McHenry CL, Carlson TJ, Ding X, Sieving PA, Apfelstedt-Sylla E, Gal A. Mutations in the gene encoding lecithin retinol acyltransferase are associated with early-onset severe retinal dystrophy. Nat Genet. 2001;28:123–124. doi: 10.1038/88828. [DOI] [PubMed] [Google Scholar]
- 13.MacDonald PN, Ong DE. Evidence for a lecithin-retinol acyltransferase activity in the rat small intestine. J Biol Chem. 1988;263:12478–12482. [PubMed] [Google Scholar]
- 14.Golczak M, Palczewski K. An acyl-covalent enzyme intermediate of lecithin:retinol acyltransferase. J Biol Chem. 2010;285:29217–29222. doi: 10.1074/jbc.M110.152314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Golczak M, Sears AE, Kiser PD, Palczewski K. Lrat-specific domain facilitates vitamin a metabolism by domain swapping in hrasls3. Nat Chem Biol. 2014;11:26–32. doi: 10.1038/nchembio.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Batten ML, Imanishi Y, Maeda T, Tu DC, Moise AR, Bronson D, Possin D, Van Gelder RN, Baehr W, Palczewski K. Lecithin-retinol acyltransferase is essential for accumulation of all-trans-retinyl esters in the eye and in the liver. J Biol Chem. 2004;279:10422–10432. doi: 10.1074/jbc.M312410200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gollapalli DR, Rando RR. All-trans-retinyl esters are the substrates for isomerization in the vertebrate visual cycle. Biochemistry. 2003;42:5809–5818. doi: 10.1021/bi0341004. [DOI] [PubMed] [Google Scholar]
- 18.Kiser PD, Zhang J, Badiee M, Li Q, Shi W, Sui X, Golczak M, Tochtrop GP, Palczewski K. Catalytic mechanism of a retinoid isomerase essential for vertebrate vision. Nat Chem Biol. 2015;11:409–415. doi: 10.1038/nchembio.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Batten ML, Imanishi Y, Tu DC, Doan T, Zhu L, Pang J, Glushakova L, Moise AR, Baehr W, Van Gelder RN, Hauswirth WW, Rieke F, Palczewski K. Pharmacological and raav gene therapy rescue of visual functions in a blind mouse model of leber congenital amaurosis. PLoS Med. 2005;2:e333. doi: 10.1371/journal.pmed.0020333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lomize MA, Pogozheva ID, Joo H, Mosberg HI, Lomize AL. Opm database and ppm web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2012;40:D370–376. doi: 10.1093/nar/gkr703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chelstowska S, Widjaja-Adhi MA, Silvaroli JA, Golczak M. Molecular basis for vitamin a uptake and storage in vertebrates. Nutrients. 2016;8:676. doi: 10.3390/nu8110676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Littink KW, van Genderen MM, van Schooneveld MJ, Visser L, Riemslag FC, Keunen JE, Bakker B, Zonneveld MN, den Hollander AI, Cremers FP, van den Born LI. A homozygous frameshift mutation in lrat causes retinitis punctata albescens. Ophthalmology. 2012;119:1899–1906. doi: 10.1016/j.ophtha.2012.02.037. [DOI] [PubMed] [Google Scholar]
- 23.Senechal A, Humbert G, Surget MO, Bazalgette C, Bazalgette C, Arnaud B, Arndt C, Laurent E, Brabet P, Hamel CP. Screening genes of the retinoid metabolism: Novel lrat mutation in leber congenital amaurosis. Am J Ophthalmol. 2006;142:702–704. doi: 10.1016/j.ajo.2006.04.057. [DOI] [PubMed] [Google Scholar]
- 24.Scholl HP, Moore AT, Koenekoop RK, Wen Y, Fishman GA, van den Born LI, Bittner A, Bowles K, Fletcher EC, Collison FT, Dagnelie G, Degli Eposti S, Michaelides M, Saperstein DA, Schuchard RA, Barnes C, Zein W, Zobor D, Birch DG, Mendola JD, Zrenner E. Safety and proof-of-concept study of oral qlt091001 in retinitis pigmentosa due to inherited deficiencies of retinal pigment epithelial 65 protein (rpe65) or lecithin:retinol acyltransferase (lrat) PLoS One. 2015;10:e0143846. doi: 10.1371/journal.pone.0143846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Collin RW, van den Born LI, Klevering BJ, de Castro-Miro M, Littink KW, Arimadyo K, Azam M, Yazar V, Zonneveld MN, Paun CC, Siemiatkowska AM, Strom TM, Hehir-Kwa JY, Kroes HY, de Faber JT, van Schooneveld MJ, Heckenlively JR, Hoyng CB, den Hollander AI, Cremers FP. High-resolution homozygosity mapping is a powerful tool to detect novel mutations causative of autosomal recessive rp in the dutch population. Invest Ophthalmol Visual Sci. 2011;52:2227–2239. doi: 10.1167/iovs.10-6185. [DOI] [PubMed] [Google Scholar]
- 26.Wang X, Wang H, Sun V, Tuan HF, Keser V, Wang K, Ren H, Lopez I, Zaneveld JE, Siddiqui S, Bowles S, Khan A, Salvo J, Jacobson SG, Iannaccone A, Wang F, Birch D, Heckenlively JR, Fishman GA, Traboulsi EI, Li Y, Wheaton D, Koenekoop RK, Chen R. Comprehensive molecular diagnosis of 179 leber congenital amaurosis and juvenile retinitis pigmentosa patients by targeted next generation sequencing. J Med Genet. 2013;50:674–688. doi: 10.1136/jmedgenet-2013-101558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dev Borman A, Ocaka LA, Mackay DS, Ripamonti C, Henderson RH, Moradi P, Hall G, Black GC, Robson AG, Holder GE, Webster AR, Fitzke F, Stockman A, Moore AT. Early onset retinal dystrophy due to mutations in lrat: Molecular analysis and detailed phenotypic study. Invest Ophthalmol Visual Sci. 2012;53:3927–3938. doi: 10.1167/iovs.12-9548. [DOI] [PubMed] [Google Scholar]
- 28.Preising MN, Paunescu K, Friedburg C, Lorenz B. genetic and clinical heterogeneity in lca patients. The end of uniformity. Ophthalmologe. 2007;104:490–498. doi: 10.1007/s00347-007-1533-x. [DOI] [PubMed] [Google Scholar]
- 29.Bok D, Ruiz A, Yaron O, Jahng WJ, Ray A, Xue L, Rando RR. Purification and characterization of a trans-membrane domain-deleted form of lecithin retinol acyltransferase. Biochemistry. 2003;42:6090–6098. doi: 10.1021/bi0342416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moise AR, Golczak M, Imanishi Y, Palczewski K. Topology and membrane association of lecithin: Retinol acyltransferase. J Biol Chem. 2007;282:2081–2090. doi: 10.1074/jbc.M608315200. [DOI] [PubMed] [Google Scholar]
- 31.Koenekoop RK, Sui R, Sallum J, van den Born LI, Ajlan R, Khan A, den Hollander AI, Cremers FP, Mendola JD, Bittner AK, Dagnelie G, Schuchard RA, Saperstein DA. Oral 9-cis retinoid for childhood blindness due to leber congenital amaurosis caused by rpe65 or lrat mutations: An open-label phase 1b trial. Lancet. 2014;384:1513–1520. doi: 10.1016/S0140-6736(14)60153-7. [DOI] [PubMed] [Google Scholar]
- 32.Maeda T, Cideciyan AV, Maeda A, Golczak M, Aleman TS, Jacobson SG, Palczewski K. Loss of cone photoreceptors caused by chromophore depletion is partially prevented by the artificial chromophore pro-drug, 9-cis-retinyl acetate. Hum Mol Genet. 2009;18:2277–2287. doi: 10.1093/hmg/ddp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Gallo Cassarino T, Bertoni M, Bordoli L, Schwede T. Swiss-model: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42:W252–W258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arnold K, Bordoli L, Kopp J, Schwede T. The swiss-model workspace: A web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 35.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 36.Yang Z, Lasker K, Schneidman-Duhovny D, Webb B, Huang CC, Pettersen EF, Goddard TD, Meng EC, Sali A, Ferrin TE. Ucsf chimera, modeller, and imp: An integrated modeling system. J Struct Biol. 2012;179:269–278. doi: 10.1016/j.jsb.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Buchan DW, Minneci F, Nugent TC, Bryson K, Jones DT. Scalable web services for the psipred protein analysis workbench. Nucleic Acids Res. 2013;41:W349–357. doi: 10.1093/nar/gkt381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. Ucsf chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 39.Kitamura T, Koshino Y, Shibata F, Oki T, Nakajima H, Nosaka T, Kumagai H. genomics. Exp Hematol. 2003;31:1007–1014. [PubMed] [Google Scholar]
- 40.Golczak M, Maeda A, Bereta G, Maeda T, Kiser PD, Hunzelmann S, von Lintig J, Blaner WS, Palczewski K. Metabolic basis of visual cycle inhibition by retinoid and nonretinoid compounds in the vertebrate retina. J Biol Chem. 2008;283:9543–9554. doi: 10.1074/jbc.M708982200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Golczak M, Bereta G, Maeda A, Palczewski K. Molecular biology and analytical chemistry methods used to probe the retinoid cycle. Methods Mol Biol. 2010;652:229–245. doi: 10.1007/978-1-60327-325-1_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Isken A, Golczak M, Oberhauser V, Hunzelmann S, Driever W, Imanishi Y, Palczewski K, von Lintig J. Rbp4 disrupts vitamin a uptake homeostasis in a stra6-deficient animal model for matthew-wood syndrome. Cell Metab. 2008;7:258–268. doi: 10.1016/j.cmet.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Golczak M, Kiser PD, Lodowski DT, Maeda A, Palczewski K. Importance of membrane structural integrity for rpe65 retinoid isomerization activity. J Biol Chem. 2010;285:9667–9682. doi: 10.1074/jbc.M109.063941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Collins TJ. Imagej for microscopy. BioTechniques. 2007;43:S25–S30. doi: 10.2144/000112517. [DOI] [PubMed] [Google Scholar]
- 45.Kanai M, Raz A, Goodman DS. Retinol-binding protein: The transport protein for vitamin a in human plasma. J Clin Invest. 1968;47:2025–2044. doi: 10.1172/JCI105889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xie Y, Lashuel HA, Miroy GJ, Dikler S, Kelly JW. Recombinant human retinol-binding protein refolding, native disulfide formation, and characterization. Protein Expression Purif. 1998;14:31–37. doi: 10.1006/prep.1998.0944. [DOI] [PubMed] [Google Scholar]
- 47.Silvaroli JA, Arne JM, Chelstowska S, Kiser PD, Banerjee S, Golczak M. Ligand binding induces conformational changes in human cellular retinol-binding protein 1 (crbp1) revealed by atomic resolution crystal structures. J Biol Chem. 2016;291:8528–8540. doi: 10.1074/jbc.M116.714535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Figler RA, Omote H, Nakamoto RK, Al-Shawi MK. Use of chemical chaperones in the yeast saccharomyces cerevisiae to enhance heterologous membrane protein expression: High-yield expression and purification of human p-glycoprotein. Arch Biochem Biophys. 2000;376:34–46. doi: 10.1006/abbi.2000.1712. [DOI] [PubMed] [Google Scholar]
- 49.MacGurn JA, Hsu PC, Emr SD. Ubiquitin and membrane protein turnover: From cradle to grave. Annu Rev Biochem. 2012;81:231–259. doi: 10.1146/annurev-biochem-060210-093619. [DOI] [PubMed] [Google Scholar]
- 50.Quadro L, Blaner WS, Salchow DJ, Vogel S, Piantedosi R, Gouras P, Freeman S, Cosma MP, Colantuoni V, Gottesman ME. Impaired retinal function and vitamin a availability in mice lacking retinol-binding protein. EMBO J. 1999;18:4633–4644. doi: 10.1093/emboj/18.17.4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kawaguchi R, Yu J, Honda J, Hu J, Whitelegge J, Ping P, Wiita P, Bok D, Sun H. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin a. Science. 2007;315:820–825. doi: 10.1126/science.1136244. [DOI] [PubMed] [Google Scholar]
- 52.Chen Y, Clarke OB, Kim J, Stowe S, Kim YK, Assur Z, Cavalier M, Godoy-Ruiz R, von Alpen DC, Manzini C, Blaner WS, Frank J, Quadro L, Weber DJ, Shapiro L, Hendrickson WA, Mancia F. Structure of the stra6 receptor for retinol uptake. Science. 2016;353:aad8266–12. doi: 10.1126/science.aad8266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ghyselinck NB, Bavik C, Sapin V, Mark M, Bonnier D, Hindelang C, Dierich A, Nilsson CB, Hakansson H, Sauvant P, Azais-Braesco V, Frasson M, Picaud S, Chambon P. Cellular retinol-binding protein i is essential for vitamin a homeostasis. EMBO J. 1999;18:4903–4914. doi: 10.1093/emboj/18.18.4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saari JC, Nawrot M, Garwin GG, Kennedy MJ, Hurley JB, Ghyselinck NB, Chambon P. Analysis of the visual cycle in cellular retinol-binding protein type i (crbpi) knockout mice. Invest Ophthalmol Vis Sci. 2002;43:1730–1735. [PubMed] [Google Scholar]
- 55.Levin MS. Cellular retinol-binding proteins are determinants of retinol uptake and metabolism in stably transfected caco-2 cells. J Biol Chem. 1993;268:8267–8276. [PubMed] [Google Scholar]
- 56.Ong DE, MacDonald PN, Gubitosi AM. Esterification of retinol in rat liver. Possible participation by cellular retinol-binding protein and cellular retinol-binding protein ii. J Biol Chem. 1988;263:5789–5796. [PubMed] [Google Scholar]
- 57.Rattner A, Smallwood PM, Nathans J. Identification and characterization of all-trans-retinol dehydrogenase from photoreceptor outer segments, the visual cycle enzyme that reduces all-trans-retinal to all-trans-retinol. J Biol Chem. 2000;275:11034–11043. doi: 10.1074/jbc.275.15.11034. [DOI] [PubMed] [Google Scholar]
- 58.Isken A, Holzschuh J, Lampert JM, Fischer L, Oberhauser V, Palczewski K, von Lintig J. Sequestration of retinyl esters is essential for retinoid signaling in the zebrafish embryo. J Biol Chem. 2007;282:1144–1151. doi: 10.1074/jbc.M609109200. [DOI] [PubMed] [Google Scholar]
- 59.Liu L, Tang XH, Gudas LJ. Homeostasis of retinol in lecithin: Retinol acyltransferase gene knockout mice fed a high retinol diet. Biochem Pharmacol. 2008;75:2316–2324. doi: 10.1016/j.bcp.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kashyap V, Gudas LJ. Epigenetic regulatory mechanisms distinguish retinoic acid-mediated transcriptional responses in stem cells and fibroblasts. J Biol Chem. 2010;285:14534–14548. doi: 10.1074/jbc.M110.115345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hegde RS, Keenan RJ. Tail-anchored membrane protein insertion into the endoplasmic reticulum. Nat Rev Mol Cell Biol. 2011;12:787–798. doi: 10.1038/nrm3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hicke L. Protein regulation by monoubiquitin. Nat Rev Mol Cell Biol. 2001;2:195–201. doi: 10.1038/35056583. [DOI] [PubMed] [Google Scholar]
- 63.Ciechanover A, Ben-Saadon R. N-terminal ubiquitination: More protein substrates join in. Trends Cell Biol. 2004;14:103–106. doi: 10.1016/j.tcb.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 64.Kiser PD, Palczewski K. Retinoids and retinal diseases. Annu Rev Vis Sci. 2016;2:197–234. doi: 10.1146/annurev-vision-111815-114407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Maeda T, Maeda A, Matosky M, Okano K, Roos S, Tang J, Palczewski K. Evaluation of potential therapies for a mouse model of human age-related macular degeneration caused by delayed all-trans-retinal clearance. Invest Ophthalmol Visual Sci. 2009;50:4917–4925. doi: 10.1167/iovs.09-3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Van Hooser JP, Liang Y, Maeda T, Kuksa V, Jang GF, He YG, Rieke F, Fong HK, Detwiler PB, Palczewski K. Recovery of visual functions in a mouse model of leber congenital amaurosis. J Biol Chem. 2002;277:19173–19182. doi: 10.1074/jbc.M112384200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moiseyev G, Crouch RK, Goletz P, Oatis J, Jr, Redmond TM, Ma JX. Retinyl esters are the substrate for isomerohydrolase. Biochemistry. 2003;42:2229–2238. doi: 10.1021/bi026911y. [DOI] [PubMed] [Google Scholar]
- 68.McCaffery PJ, Adams J, Maden M, Rosa-Molinar E. Too much of a good thing: Retinoic acid as an endogenous regulator of neural differentiation and exogenous teratogen. Eur J Neurosci. 2003;18:457–472. doi: 10.1046/j.1460-9568.2003.02765.x. [DOI] [PubMed] [Google Scholar]
- 69.Mey J, McCaffery P, Klemeit M. Sources and sink of retinoic acid in the embryonic chick retina: Distribution of aldehyde dehydrogenase activities, crabp-i, and sites of retinoic acid inactivation. Dev Brain Res. 2001;127:135–148. doi: 10.1016/s0165-3806(01)00127-4. [DOI] [PubMed] [Google Scholar]
- 70.O’Byrne SM, Wongsiriroj N, Libien J, Vogel S, Goldberg IJ, Baehr W, Palczewski K, Blaner WS. Retinoid absorption and storage is impaired in mice lacking lecithin:Retinol acyltransferase (lrat) J Biol Chem. 2005;280:35647–35657. doi: 10.1074/jbc.M507924200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Amengual J, Zhang N, Kemerer M, Maeda T, Palczewski K, Von Lintig J. Stra6 is critical for cellular vitamin a uptake and homeostasis. Hum Mol Genet. 2014;23:5402–5417. doi: 10.1093/hmg/ddu258. [DOI] [PMC free article] [PubMed] [Google Scholar]