

Abstract
Environmental conditions modulate cell cycle progression in many cell types. A key component of the eukaryotic cell cycle is the protein kinase Wee1, which inhibits the cyclin-dependent kinase Cdk1 in yeast through human cells. In the fission yeast Schizosaccharomyces pombe, the protein kinase Cdr1 is a mitotic inducer that promotes mitotic entry by phosphorylating and inhibiting Wee1. Cdr1 and Wee1 both localize to punctate structures, termed nodes, on the medial cortex, but it has been unknown whether node localization can be altered by physiological signals. Here we investigated how environmental conditions regulate Cdr1 signaling for cell division. Osmotic stress induced hyperphosphorylation of the mitotic inducer Cdr1 for several hours, and cells delayed division for the same time period. This stress-induced hyperphosphorylation required both Cdr1 autophosphorylation and the stress-activated protein kinase Sty1. During osmotic stress, Cdr1 exited cortical nodes and localized in the cytoplasm. Using a series of truncation mutants, we mapped a C-terminal domain that is necessary and sufficient for Cdr1 node localization and found that Sty1 directly phosphorylates this domain in vitro. Sty1 was not required for Cdr1 exit from nodes, indicating the existence of additional regulatory signals. Both Cdr1 phosphorylation and node localization returned to basal levels when cells adapted to osmotic conditions and resumed cell cycle progression. In summary, we identified a mechanism that prevents Cdr1 colocalization with its inhibitory target Wee1 during osmotic stress. Dynamic regulation of protein localization to cortical nodes might represent a strategy to modulate entry into mitosis under differing environmental conditions.
Keywords: cell cycle, mitosis, protein kinase, protein phosphorylation, Schizosaccharomyces pombe, yeast, Cdr1, Wee1, osmotic stress
Introduction
Events of the cell cycle occur in an ordered progression that ensures proper cell division even in the face of a rapidly changing environment. Checkpoints ensure the order of cell cycle events and also modify the timing of cell cycle progression according to intracellular or extracellular cues. For example, environmental changes can alter cell cycle timing to ensure faithful division under all conditions. A commonly encountered environmental shock for many cell types is hyperosmotic stress. Upon hyperosmotic stress, a wide range of cell types, including yeast and human cells, delay entry into mitosis by triggering a G2/M checkpoint (1–3). At the molecular level, entry into mitosis at G2/M is regulated by the phosphorylation status of the cyclin-dependent kinase Cdk1. The inhibitory protein kinase Wee1 phosphorylates and inhibits Cdk1 to prevent mitotic entry during interphase. Prior to mitotic entry, the protein phosphatase Cdc25 reverses this inhibitory phosphorylation to activate Cdk1 and promote entry into mitosis. Because the potential for mitotic entry reflects the balanced activities of Wee1 versus Cdc25, pathways that target either regulator can alter cell cycle timing.
The signaling pathways that link osmotic stress to mitotic entry through Cdk1 have been examined in many different cell types. In human cells, osmotic stress triggers a G2/M checkpoint by activating the p38 stress-activated protein kinase (SAPK)2 pathway (2–7). This checkpoint prevents cells from entering error-prone mitosis before they adapt to the new osmotic environment (2). SAPK prevents mitotic entry by inhibiting Cdc25 (4). This inhibition may be catalyzed by the p38 substrate MAPKAP kinase 2, which directly phosphorylates Cdc25 (8). In fission yeast cells, a similar pathway has been proposed to act downstream of the p38-related SAPK Sty1 during osmotic stress (9, 10). Activated Sty1 phosphorylates the protein kinase Srk1, which is related to MAPKAP kinase 2. Srk1 then phosphorylates Cdc25 to inhibit its nuclear localization (11–13). Thus, SAPK can regulate mitotic entry through Cdc25 in fission yeast, but the possibility that osmotic stress also regulates Wee1 pathways has not been examined.
A different mechanism has been proposed in budding yeast, where SAPK pathways prevent mitotic entry during osmotic stress by acting through Wee1. Activation of the p38-related SAPK Hog1 leads to stabilization of Swe1 (budding yeast Wee1), resulting in G2/M arrest (1, 14). In this pathway, activated Hog1 phosphorylates the checkpoint kinase Hsl1, a known regulator of Swe1 (14). This Hog1–Hsl1–Swe1 pathway has been proposed to act through Hsl7, which interacts with both Hsl1 and Swe1. An opposing model has questioned the role of Hsl7 and instead proposed that Swe1 stabilization is driven by feedback from Cdk1 but not Hsl7 (15). These studies indicate that SAPK can act through Wee1 to prevent mitotic entry during osmotic stress, but the molecular mechanisms remain unclear.
A similar connection between SAPK and Wee1 signaling in fission yeast has not been examined. Two Hsl1-like protein kinases, Cdr1 and Cdr2, act to inhibit Wee1 in fission yeast cells (16–18). Cdr1 directly phosphorylates and inhibits the kinase domain of Wee1 (19–21). Cdr2 assembles a series of membrane-bound multiprotein structures, termed “nodes,” at the cell middle (22). Cdr2 then recruits both Cdr1 and Wee1 to nodes, meaning that Cdr1 overlaps with its inhibitory target Wee1 at nodes (23, 24). Here we focused on Cdr1 because it acts directly on Wee1. We hypothesized that Cdr1 might be a target of stress-activated signaling pathways to link environmental changes with cell cycle progression. By screening a range of conditions, we identified osmotic stress as an environmental cue that induces hyperphosphorylation and relocalization of Cdr1 involving the SAPK Sty1. This mechanism likely contributes to the delay in cell division we observed when fission yeast cells were exposed to osmotic stress.
Results
Osmotic stress induces hyperphosphorylation of Cdr1 and mitotic delay
We sought to identify mechanisms that might regulate the protein kinase Cdr1 according to different environmental and growth conditions. Cdr1 controls the timing of mitotic entry and has been reported to autophosphorylate in vitro (19–21). To investigate Cdr1 phosphorylation in fission yeast cells, we integrated a 5×FLAG epitope tag at the carboxyl terminus of endogenous Cdr1; this tag included a nine-glycine linker and did not interfere with Cdr1 function, as tested by cell length at division. In SDS-PAGE and Western blotting, immunoprecipitated Cdr1 migrated as a smeared band. This band collapsed into a single, faster-migrating species upon treatment with λ phosphatase (Fig. 1A). This confirms that Cdr1 is a phosphoprotein in cells. We next screened Cdr1 phosphorylation by SDS-PAGE band shift in a panel of environmental conditions that change cell cycle progression and/or cell size at division. The migration of Cdr1-FLAG shifted dramatically by 15-min exposure to multiple conditions, particularly osmotic stress, low glucose, and heat stress (Fig. 1B).
Figure 1.

Hyperphosphorylation of Cdr1 during osmotic stress response. A, Cdr1 is a phosphoprotein in vivo. Immunoprecipitated Cdr1-FLAG was treated with λ phosphatase or mock-treated and then analyzed by SDS-PAGE and Western blotting. B, modification of Cdr1 by different environmental stresses. Cells were exposed to the indicated conditions for 15 min, and then whole-cell extracts were analyzed by SDS-PAGE and Western blotting. C, the change in Cdr1 mobility is due to phosphorylation. Cells were exposed to EMM4S alone or to EMM4S + 1 m KCl for 15 min. Immunoprecipitated (IP) Cdr1 was treated with λ phosphatase and analyzed by SDS-PAGE and Western blotting. Note that slower-migrating bands collapse into a single band when dephosphorylated. D, hyperphosphorylation of Cdr1 during sorbitol treatment. Cells were exposed to the indicated condition for 15 min, and then whole-cell extracts were analyzed by SDS-PAGE and Western blotting. E, time course of asynchronous cells exposed to 1 m KCl. Whole-cell extracts were blotted with anti-FLAG. F, timing of septation delay by osmotic stress of wild-type cells. Cell cycle progression was synchronized by centrifugal elutriation, and then cells were released into YE4S or YE4S + 1 m KCl. Percent septation was used to monitor cell cycle progression; n > 100 cells for each time point. G, timing of septation delay by osmotic stress in cdc25–22 cdr1-FLAG cells, which were arrested in G2 phase by incubation at 37 °C and then released into synchronized cell cycle progression by switching to 25 °C in YE4S or YE4S + 1 m KCl. H, change in Cdr1 phosphorylation during mitotic arrest induced by osmotic stress. Shown is a Western blot of samples from G; Cdr1 mobility was analyzed by SDS-PAGE at the indicated time points. I, timing of septation delay by osmotic stress of cdr1Δ cells, analyzed using centrifugal elutriation as in F.
We investigated osmotic stress in more detail because this condition induced a strong band shift in Cdr1-FLAG. In addition, osmotic stress has been shown to alter cell cycle progression in budding yeast (1). To test whether this shift was due to phosphorylation, we immunoprecipitated Cdr1-FLAG from cells treated with 1 m KCl and from an untreated control. Incubation with λ phosphatase collapsed both bands to the faster-migrating, unphosphorylated state (Fig. 1C). Thus, the KCl-induced band shift in Cdr1 is due to phosphorylation. We refer to this state as hyperphosphorylated because it exceeds the level of phosphorylation observed for Cdr1 under basal growth conditions. This hyperphosphorylation is due to osmotic stress and not salt stress because we observed a similar band shift for cells treated with sorbitol (Fig. 1D).
The osmotic stress response has two phases: an initial, transient phase that alters many signaling and transcriptional pathways, followed by a sustained phase when cells have adapted to growth in the new osmotic environment (25). Our initial experiments revealed hyperphosphorylation 15 min after osmotic stress. To examine the kinetics of this regulation, we collected samples at time intervals following osmotic stress for analysis by SDS-PAGE and Western blotting. We found that Cdr1 was hyperphosphorylated within 15 min of osmotic stress but returns to a basal phosphorylated state within several hours. The species of Cdr1 that migrates fastest by SDS-PAGE, and thus represent the less phosphorylated form, reappeared after 240 min under osmotic stress (Fig. 1E).
To determine how these kinetics relate to cell growth and division, we used centrifugal elutriation to generate a population of small wild-type cells that were synchronized in the cell cycle. The culture was then split, and released into medium containing KCl or into control medium. In control medium, the first cell cycle culminated with a septation peak at 80 min, followed by a second septation peak at 220 min (Fig. 1F). In contrast, the cells released into KCl medium delayed septation until they exhibited a broad peak between 240–320 min (Fig. 1F). Thus, the hyperphosphorylation of Cdr1 and the delay in septation that are induced by osmotic stress follow similar kinetics.
Similar results were obtained using a different method to synchronize cell cycle progression. Specifically, we performed block-and-release experiments with the temperature-sensitive mutant cdc25–22, which arrests cells in G2 phase at high temperature. Following arrest for 4 h, cdc25–22 cells were shifted to the permissive temperature and split into medium containing KCl or control medium. Similar to our elutriation experiment, cells released into KCl medium delayed septation compared with cells released into control medium (Fig. 1G). We directly monitored phosphorylation of Cdr1 in this assay (Fig. 1H). Cdr1 was hyperphosphorylated when cells were released into KCl medium and remained hyperphosphorylated during the delay in septation. Cdr1 returned to the basal phosphorylated state before proceeding to septation. We conclude that Cdr1 hyperphosphorylation correlates with delayed entry into mitosis during osmotic stress, and reversal of Cdr1 hyperphosphorylation correlates with entry into cell division.
We tested whether Cdr1 is required for delayed septation in KCl by performing elutriation experiments on cdr1Δ mutants. Upon release of cdr1Δ mutant cells into medium lacking KCl, the initial septation peak was slightly delayed compared with wild-type cells (Fig. 1I), consistent with loss of the mitotic inducer Cdr1. When released into medium containing KCl, cdr1Δ mutant cells exhibited a long delay in septation similar to wild-type cells (Fig. 1I). This result could reflect additional mechanisms that act redundantly with Cdr1 to delay cell division during osmotic stress. Alternatively, hyperphosphorylation could inhibit Cdr1 function during osmotic stress, and such an inhibitory mechanism would not be perturbed in cells lacking Cdr1. We explored this possibility next by examining the consequences of Cdr1 hyperphosphorylation in cells.
Cdr1 exits cortical nodes when hyperphosphorylated
To determine how hyperphosphorylation impacts Cdr1 behavior in cells, we tested Cdr1 localization during osmotic stress. We used genomic integration to tag the C terminus of Cdr1 with GFPγ, a variant of GFP with improved brightness and photostability (26, 27). cdr1-GFPγ cells divided at a smaller size than wild-type cells, indicating that this tag increases Cdr1 activity. This effect is similar to previous results with other fluorescent tags on Cdr1 (23, 24). During interphase, Cdr1 localizes to a series of membrane-bound cortical nodes. These nodes are organized by the related protein kinase Cdr2, which is required for Cdr1 node localization and acts upstream of Cdr1 in genetic pathways (23, 24). We found that Cdr1-GFPγ remained at cortical nodes when cells were switched to control medium lacking KCl (Fig. 2). In contrast, Cdr1-GFPγ was absent from the cell cortex within 15 min of osmotic stress, induced by switching to medium containing 1 m KCl (Fig. 2). The loss of Cdr1-GFPγ from nodes was seen in virtually all cells during osmotic stress (Fig. 2). Cdr1-GFPγ began returning to nodes 120 min after switching to medium containing 1 m KCl and fully relocalized to nodes after 240 min. This timing mirrors the hyperphosphorylation of Cdr1, as well as the delay in cell division, upon osmotic stress.
Figure 2.

Cdr1 exits nodes during osmotic stress. A, inverted contrast single focal plane images of Cdr1-GFPγ grown in EMM4S to mid-log phase and then split into medium containing 1 m KCl or into control medium (EMM4S). Insets are enlarged images of the medial cortex; yellow boxes indicate the enlarged area. Scale bar = 5 μm. B, quantification of cells containing Cdr1 localization to nodes (n > 100 cells for each time point; error bars represent standard deviation).
Cortical nodes are multiprotein structures assembled by Cdr2, which then recruits additional proteins, including Cdr1. Two models could explain the change in Cdr1 localization during osmotic stress. First, nodes could disassemble, causing all node proteins, including Cdr1, to change localization. Alternatively, nodes could remain intact during osmotic stress, with specific proteins such as Cdr1 changing localization through targeted regulation. To distinguish between these models, we tested the localization of Cdr2 during osmotic stress as well as its role in Cdr1 hyperphosphorylation. In contrast to Cdr1, Cdr2-mEGFP remained localized to cortical nodes during osmotic stress (Fig. 3, A and B), indicating that nodes remain at the plasma membrane and that Cdr1 exits these structures. Further, Cdr1 was still hyperphosphorylated during osmotic stress in both cdr2Δ and kinase-dead cdr2(E177A) mutant cells (Fig. 3C), meaning that Cdr2 is not required for Cdr1 hyperphosphorylation. Additionally, because nodes are absent in cdr2Δ cells, this result raises the possibility that node localization is not required for Cdr1 hyperphosphorylation during osmotic stress.
Figure 3.

Cdr2 nodes remain intact during osmotic stress. A, single focal plane inverted images of a time course with or without exposure to 1 m KCl. Scale bar = 5 μm. DIC, differential interference microscopy. B, quantification of cells containing Cdr2 nodes (n > 100 cells for each time point, error bars represent standard deviation). C, phosphorylation of Cdr1 is independent of Cdr2 nodes and Cdr2 kinase activity. cdr2(E177A) is a catalytically inactive mutation. Shown is a Western blot of whole-cell extracts after 15-min exposure to 1 m KCl. Note the presence of hyperphosphorylated Cdr1 in Cdr2 mutants.
Identification of the node localization domain in Cdr1
Our results led us to ask how Cdr1 localizes to nodes and whether node localization is important for Cdr1 function in cell size control. Cdr1 is a 593-amino acid protein that contains an N-terminal protein kinase domain, followed by a short domain termed the essential region (ER) and an uncharacterized C terminus. The ER domain is necessary for Cdr1 kinase activity (18, 21, 28). Based on overexpression experiments, previous studies concluded that the C terminus is not required for Cdr1 function in vivo (18, 28). To gain insight into the requirements for endogenous Cdr1 localization and function, we integrated a series of truncation mutants tagged with GFPγ into the cdr1+ locus under control of the endogenous promoter (Fig. 4, A and B).
Figure 4.

Identification of the domain that targets Cdr1 to nodes. A, schematic of Cdr1 and truncations. Kinase, kinase domain. Localization to nodes is indicated for each strain. B, representative inverted contrast images of Cdr1 truncations expressed as GFPγ fusions at the endogenous locus in cdr2+ (left panel) or cdr2Δ (right panel) background. Images are middle and top focal planes from a deconvolved Z series. Black arrows indicate nodes. Scale bar = 5 μm.
Cdr1 was detected at nodes along the sides of cells in middle focal planes and also at the medial cell cortex in the top focal plane of cells. Cdr1(1–460), which contains both the kinase domain and the ER domain, did not localize to nodes, indicating that the C terminus is required for node localization. In contrast, the much smaller Cdr1(460–593) localized to nodes, suggesting that this region contains a node-binding domain. We further truncated this region and identified a 41-amino acid construct Cdr1(460–500) that localized to medial nodes. In contrast, an internal deletion mutant Cdr1(Δ460–482) did not localize to medial nodes but weakly associated with the cell cortex. Together, these results demonstrate that amino acids 460–500 in the C terminus are both necessary and sufficient for Cdr1 localization to medial cortical nodes.
To confirm our conclusion that the Cdr1 C terminus targets cortical nodes, we tested the localization of each construct in cdr2Δ cells (Fig. 4B). Cdr2 forms these cortical nodes and is required for Cdr1 localization to these sites. Thus, constructs that localize to nodes should lose this localization in cdr2Δ mutants. We found that Cdr1(460–593), Cdr1(460–500), and full-length Cdr1, all of which localized to nodes in wild-type cells, failed to localize to medial cortical nodes in cdr2Δ cells. In contrast, cdr2Δ did not affect the localization of Cdr1(1–460) and Cdr1(Δ460–482). We conclude that the Cdr1 C terminus targets medial cortical nodes in a Cdr2-dependent manner.
We used these constructs to test whether node localization is required for Cdr1 function. We integrated untagged versions of each construct and measured cell length at division. Similar to cdr1Δ, the mutant cdr1(460–593) lacking the kinase domain was elongated at division, consistent with loss of the mitotic inducer function. The mutants cdr1(1–460) and cdr1(Δ460–482), which contain the kinase and ER domains but do not localize to nodes, were also elongated at division, indicating a loss-of-function phenotype (Fig. 5A). Formally, these 23 amino acids could contribute to Cdr1 function beyond localization, but we consider it most likely that this small truncation mutant exhibits the loss-of-function phenotype because of localization defects. We also note that several mutants, notably cdr1(1–460), cdr1(460–593), and cdr1(460–500), appear to be highly vacuolated for unknown reasons (Fig. 4B). By careful examination of these different mutants expressed under the endogenous control system, we can make two important conclusions. First, the kinase domain and ER domain are not sufficient for the function of Cdr1. Second, node localization appears to be essential for Cdr1 function in cell size control.
Figure 5.

Node localization is required for Cdr1 function. A, measurement of cell size at division for the indicated cdr1 alleles (n > 50 cells/strain, values are mean ± S.D.). B, hyperphosphorylation of the indicated Cdr1 mutants upon osmotic stress.
We also tested whether key Cdr1 truncation mutants can be hyperphosphorylated upon osmotic stress. We found that Cdr1(1–460) and Cdr1(Δ460–482), both of which do not localize to nodes, can still be phosphorylated upon osmotic stress (Fig. 5B). The phosphorylation level of these mutants appears to be reduced compared with full-length Cdr1, potentially because of an increased level of protein that remains unphosphorylated (or basally phosphorylated). This result, combined with hyperphosphorylation of full-length Cdr1 in cdr2Δ cells, indicates that node localization is not required for hyperphosphorylation of Cdr1 during osmotic stress.
Cdr1 catalytic activity is required for hyperphosphorylation
Cdr1 has been shown to autophosphorylate in vitro, and we confirmed that the kinase-dead mutant Cdr1(K41A) (20, 21) migrates as a collapsed band by SDS-PAGE (Fig. 6A). To test whether Cdr1 autophosphorylation contributes to its hyperphosphorylation during osmotic stress, we monitored Cdr1(K41A)-FLAG expressed at the endogenous locus. Unlike wild-type Cdr1-FLAG, the kinase-dead mutant Cdr1(K41A)-FLAG was not hyperphosphorylated during exposure to 1 m KCl (Fig. 6A). We next tested whether Cdr1 kinase activity is required for exit from nodes during osmotic stress. Cdr1(K41A)-GFPγ remained at nodes upon osmotic stress, in stark contrast to the wild-type protein (Fig. 6, B and C). We conclude that Cdr1 kinase activity is required for its own hyperphosphorylation and exit from nodes during osmotic stress.
Figure 6.

Hyperphosphorylation requires Cdr1 kinase activity. A, α-FLAG Western blot of whole-cell extracts from the indicated strains after KCl treatment. K41A is kinase-dead mutation of Cdr1 (20, 21). B, Cdr1(K41A)-GFPγ does not exit nodes during osmotic stress. Shown are middle focal plane inverted images of a time course with or without 1 m KCl. Insets show enlarged images of Cdr1 present at nodes; yellow boxes indicate the enlarged area. Scale bar = 5 μm. C, quantification of cells with Cdr1(K41A)-GFPγ localization at nodes (n > 100 for each time point, error bars represent standard deviation).
Two models could explain the requirement of Cdr1 kinase activity for regulation under osmotic stress. First, Cdr1 hyperphosphorylation could be independent of its ability to autophosphorylate; for example, through feedback from proteins that act downstream of Cdr1. Alternatively, Cdr1 autophosphorylation could be required for dissociation, and this autophosphorylation could occur either in cis (i.e. intramolecularly) or in trans (i.e. intermolecularly). To distinguish between these different possibilities, we modified Cdr1(K41A)-FLAG cells by introducing a second, catalytically active copy of Cdr1 at the leu1+ locus. This co-expression of active Cdr1 was unable to restore either basal phosphorylation or KCl-induced hyperphosphorylation to kinase-dead Cdr1(K41A)-FLAG (Fig. 7). In addition, co-expression of active Cdr1 did not induce Cdr1(K41A)-GFPγ to exit nodes during osmotic stress (Fig. 7B). These results are inconsistent with both the indirect feedback model and the in trans autophosphorylation model. Rather, these results indicate that Cdr1 autophosphorylates in cis, and this intramolecular modification is required for hyperphosphorylation and node exit during osmotic stress. A small band shift was also observed for kinase-dead Cdr1(K41A)-FLAG during osmotic stress, suggesting that additional kinases might contribute to Cdr1 regulation during this process.
Figure 7.

Intramolecular autophosphorylation by Cdr1. A, Western blots of whole-cell extracts from strains expressing the indicated combinations of wild-type and kinase-dead (K41A) alleles of Cdr1. Strains were treated with 1 m KCl for 15 min. Note that expression of wild-type Cdr1 does not induce hyperphosphorylation of kinase-dead Cdr1(K41A). B, middle focal plan images of Cdr1(K41A)-GFPγ leu1::[6his-2HA-cdr1] in control EMM4S media or in EMM4S + 1 m KCl. Insets are enlarged images of the medial cortex. Scale bar, 5 μm.
Sty1 activation is necessary and sufficient for Cdr1 hyperphosphorylation
Environmental stress commonly elicits a physiological response through SAPK pathways that regulate signaling and transcriptional networks for cellular adaptation to stress. In fission yeast cells, osmotic stress stimulates activation of the SAPK Sty1. During osmotic stress, Sty1 has been shown to prevent mitotic entry by phosphorylating and inhibiting Cdc25, which is also phosphorylated and regulated by the Sty1-associated kinase Srk1 (9–13). We tested the role of these stress-activated kinases in hyperphosphorylation of Cdr1 by Western blotting. We found that Cdr1 hyperphosphorylation during osmotic stress was impaired in sty1Δ mutants cells but not in srk1Δ mutant cells (Fig. 8A). To test whether this effect requires Sty1 kinase activity, we employed the analog-sensitive allele sty1(T97A), which is selectively inhibited by the analog inhibitor 3-BrB-PP1. Inhibition of Sty1 kinase activity by addition of 3-BrB-PP1 to sty1(T97A) cells prevented full hyperphosphorylation of Cdr1 during osmotic stress (Fig. 8B). Sty1 activity is transiently activated by osmotic stress through dual phosphorylation at Thr-171 and Tyr-173 (29). We monitored pThr-171/pTyr-173 activation in our experiments and confirmed that the kinetics of Sty1 activation and inactivation mirrored the timing of Cdr1 hyperphosphorylation (Fig. 8C). We conclude that kinase signaling through Sty1 SAPK is required for full hyperphosphorylation of Cdr1 during osmotic stress.
Figure 8.

Activated SAPK Sty1 is necessary and sufficient for Cdr1 hyperphosphorylation. A, tests for hyperphosphorylation of Cdr1 upon 15-min osmotic stress in srk1Δ and sty1Δ mutants. B, Sty1 kinase activity is necessary for full hyperphosphorylation of Cdr1. Analog-sensitive sty1(T97A) cells were treated with BrB-PP1 inhibitor or with a methanol control as indicated and then exposed to 1 m KCl for 15 min. C, time course of Sty1 activation during 1 m KCl treatment. Activation was monitored by phosphorylation of Thr-171/Tyr-173 in the activation loop; total Sty1 protein was detected with α-FLAG. D, Cdr1 hyperphosphorylation by SISA. Sty1 was activated by removal of 3-BrB-PP1 inhibitor, and whole-cell extracts were analyzed by SDS-PAGE and Western blotting. E, quantification of Cdr1-GFPγ localization in wild-type or analog-sensitive sty1(T97A) mutant cells. Cells were treated as in B (n > 100 cells for each condition; error bars represent standard deviation). F, inverted contrast single focal plane images of Cdr1-GFPγ localization in the indicated strain before or after 30 min of osmotic stress. See E for quantification and controls. Insets are enlarged images of the medial cortex; yellow boxes indicate the enlarged area. Scale bar = 5 μm. G, localization of Cdr1-GFPγ in sty1Δ cells after osmotic stress. Insets are enlarged images of the medial cortex; yellow boxes indicate the enlarged area. Scale bar = 5 μm.
To determine whether Sty1 activation is sufficient for Cdr1 hyperphosphorylation, we employed a recently generated stress-independent Sty1 activation (SISA) strain (30, 31). In SISA cells, the upstream molecular signals that activate Sty1 are engaged, but Sty1 kinase activity can be reversibly inhibited because of the analog-sensitive sty1(T97A) mutation. Thus, Sty1 is maintained in an inactive state by the analog inhibitor 3-BrB-PP1 and then activated by removal of this inhibitor independently of environmental stress. We monitored Cdr1-FLAG phosphorylation in SISA cells grown in the absence of osmotic stress. Upon removal of the inhibitor 3-BrB-PP1, we detected sustained hyperphosphorylation of Cdr1-FLAG (Fig. 8D), showing that Sty1 activation induces Cdr1 hyperphosphorylation independently of osmotic stress. These combined results indicate that Sty1 activation is both necessary and sufficient for Cdr1 hyperphosphorylation.
Next we tested Cdr1-GFPγ localization in analog-sensitive sty1(T97A) mutant cells. Surprisingly, Cdr1-GFPγ still exited nodes during osmotic stress in sty1(T97A) cells treated with the analog inhibitor 3-BrB-PP1 (Fig. 8, E and F). Control experiments confirmed that Cdr1 regulation also was unaffected by addition of 3-BrB-PP1 to wild-type cells, or by the sty1(T97A) mutant in the absence of inhibitor (Fig. 8E). To ensure complete loss of Sty1 signaling, we tested sty1Δ cells and observed exit of Cdr1-GFPγ from nodes during osmotic stress (Fig. 8G). We were unable to quantify this effect because many sty1Δ cells appeared to be dead or dying even after short exposure to osmotic stress. Thus, although Sty1 promotes full hyperphosphorylation of Cdr1 during osmotic stress, it is not required for Cdr1 to exit nodes.
Finally, we used in vitro thiophosphate assays to test whether Sty1 directly phosphorylates Cdr1. These assays exploit the ability of analog-sensitive kinase alleles, such as sty1(T97A), to use a bio-orthogonal ATPγS molecule in which a thiophosphate moiety replaces the γ-phosphate. The analog-sensitive kinase thiophosphorylates its direct substrates, which can then be alkylated for detection by a specific anti-thiophosphate ester antibody. Thus, only direct substrates of the analog-sensitive kinase become thiophosphorylated, eliminating concerns about phosphorylation by contaminating kinases in vitro. We immunoprecipitated both Sty1-FLAG and Sty1(T97A)-FLAG that had been activated by stressing cells with KCl. We also purified from bacteria the constructs GST-Cdr1(341–593) and GST alone. Sty1(T97A)-FLAG thiophosphorylated Cdr1(341–593) but not GST alone, and Sty1-FLAG had no effect in this assay (Fig. 9). We conclude that Cdr1 is a direct substrate of Sty1.
Figure 9.
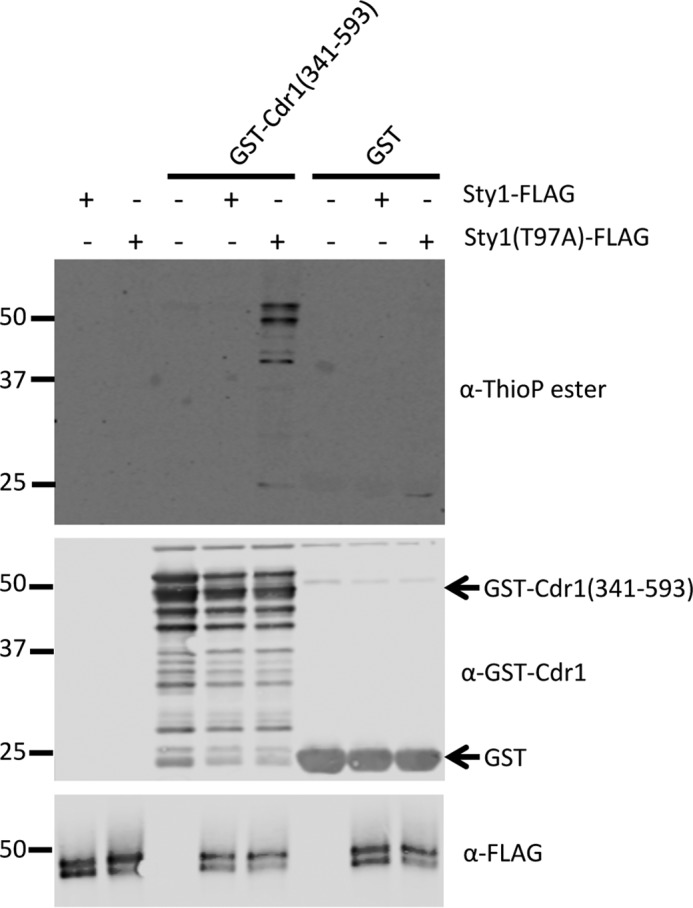
Sty1 directly phosphorylates Cdr1 in vitro. Shown is an in vitro thiophosphate kinase assay using ATPγS. Sty1-FLAG and Sty1(T97A)-FLAG were immunoprecipitated and then added to thiophosphate kinase assays with substrate GST-Cdr1(341–593) or GST alone. Proteins were detected by Western blotting with the indicated antibodies, and phosphorylation was detected using an antibody against thiophosphate ester.
Discussion
Our work has revealed a mechanism linking osmotic stress with regulation of the cell cycle kinase Cdr1. Our results suggest a working model for the factors involved in this process (Fig. 10). In unstressed cells, Cdr1 can phosphorylate and inhibit Wee1 at cortical nodes, where both proteins localize. Osmotic stress induces transient hyperphosphorylation of Cdr1; this modification requires Cdr1 autophosphorylation as well as activated Sty1. During osmotic stress and hyperphosphorylation, Cdr1 exits nodes and is displaced from its inhibitory target Wee1. Thus, hyperphosphorylation amounts to inhibition of Cdr1. This regulatory change is predicted to increase Wee1 activity, resulting in delayed mitotic entry. Consistent with this prediction, we showed that fission yeast cells delay mitotic entry when encountering osmotic stress, similar to budding yeast cells (1, 14, 15).
Figure 10.
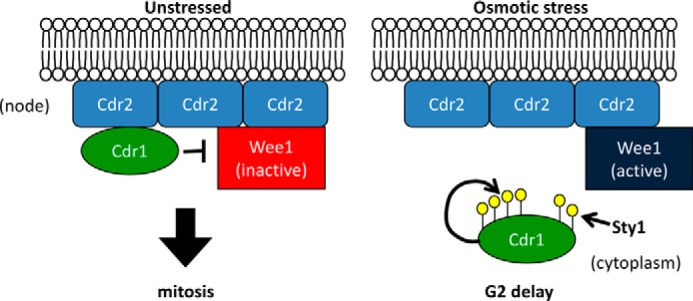
Working model for the regulation of Cdr1 during osmotic stress response. See the text for discussion.
Our work suggests that hyperphosphorylation of Cdr1 contributes to mitotic delay, and a key future goal will be to test the precise role of Cdr1 in this process. cdr1Δ mutants have no defect in the mitotic delay, which is consistent with a mechanism that inhibits Cdr1 function. However, if the mitotic delay during osmotic stress were caused primarily by inhibition of Cdr1, then cdr1 mutant cells should exhibit an equivalent delay under unstressed conditions. Loss of the mitotic inducer Cdr1 is known to delay mitotic entry, leading to increased cell size at division, and we observed delayed division under unstressed conditions, but the delay experienced by wild-type cells under osmotic stress is far more extreme. Thus, the contribution of Cdr1 regulation to mitotic delay during osmotic stress remains open and will be a focus of future work. We anticipate that gain-of-function Cdr1 mutants, which cannot be inhibited, will be needed to clarify how Cdr1 acts within a larger cell cycle regulatory network that delays mitotic entry during osmotic stress.
A critical step in regulation of Cdr1 during osmotic stress in fission yeast cells is a change in protein localization. Our work reveals that fission yeast employs a regulatory mechanism that is different from either of the models proposed in budding yeast. Rather, it appears that environmental stresses can act through Cdr1 in fission yeast by modulating the composition of membrane-bound cortical nodes. This means that nodes are not static structures but, rather, exhibit dynamic properties that can be targeted by physiological signals to alter cell cycle timing.
Regulated localization of Cdr1 to cortical nodes led us to define the requirements for Cdr1 node localization. We identified a 41-amino acid domain that was both required and sufficient for node localization. This node localization domain resides in the Cdr1 C terminus, a region proposed previously to be dispensable for Cdr1 function based on overexpression studies (18, 28). We showed that this domain is required for node localization and for Cdr1 function. This result strongly suggests that nodes are the sites of Wee1 inhibition by Cdr1. Osmotic stress is likely to regulate the function of this localization domain, so one might predict that sites for hyperphosphorylation reside within this domain. A fragment containing this domain was directly phosphorylated by Sty1 in vitro. However, we also found that Cdr1(Δ460–482), which lacks the localization domain, is still phosphorylated during osmotic stress, although perhaps to a lesser degree. Further, Sty1 was not required for changes in Cdr1 localization during osmotic stress, indicating that it might regulate other aspects of Cdr1 function in cells. Thus, at least some phosphorylation sites are likely to reside outside of the localization domain. Indeed, the Cdr1 protein sequence contains nine minimal motifs for phosphorylation by MAPK/SAPK, such as Sty1 (pSer/pThr-Pro), but only three of these motifs are found within the localization domain. A critical step for the future will be mapping the sites of Cdr1 hyperphosphorylation during osmotic stress. Our model predicts that mutating these sites to prevent phosphorylation will generate a gain-of-function Cdr1 mutant. These sites are likely to be a combination of autophosphorylation sites and Sty1 phosphorylation sites and have the potential to reveal mechanisms for regulating the activity of the novel C-terminal localization domain.
More broadly, cell cycle progression is altered in response to heat, glucose, and nitrogen stress, although many of the mechanisms remain to be defined. We discovered that a range of environmental stresses cause varied modifications to Cdr1. We focused on uncovering the mechanism for regulation during osmotic stress, and it will be interesting to compare and contrast this mechanism with regulation under other conditions. For example, heat stress and low glucose induced clear modification of Cdr1. Heat stress leads to activation of Sty1 (32–34), raising the possibility for a Sty1-dependent mechanism related to osmotic stress. Low glucose was recently connected to Sty1-independent signaling through the Cdr2–Cdr1–Wee1 pathway (35). Beyond these conditions, Cdr1 and Cdr2 were initially identified in genetic screens for defective cell cycle response during nitrogen starvation (36). It will be interesting to compare and contrast the role of Cdr1 regulation under these conditions. Through such future studies, we will be positioned to understand whether cells use different pathways for different stresses or, alternatively, whether cells use the same pathways redundantly under changing environmental conditions.
An open question that remains is why cells from yeast through humans delay mitotic entry during osmotic stress. The answer remains unclear for yeast cells, but human cells are proposed to encounter mutagenic mitosis in the absence of this checkpoint. Specifically, human cells acquire double-strand DNA breaks during osmotic shock, and these breaks require a G2/M checkpoint for proper repair (2). The evolutionary conservation of a mitotic checkpoint under osmotic stress suggests a critical role in cell survival. Our work provides mechanistic insight into a signaling pathway that contributes to this process. Further studies on these pathways will lead to a broader understanding of the connection between environmental conditions and cell division control.
Experimental procedures
Yeast strains and growth
Standard Schizosaccharomyces pombe medium and methods were used (37). The strains used in this study are listed in supplemental Table S1. The plasmids used in this study are listed in supplemental Table S2. PCR-based homologous recombination was used for chromosomal tagging (38). Mutant Cdr1 alleles were created by PCR-amplifying Pcdr1-cdr1-GFPγ::hph-Tcdr1 or Pcdr1-cdr-Tcdr1 for untagged mutants from genomic DNA of JM3733 or JM366, respectively. These constructs were then ligated into a pQE30 plasmid. Mutations were created using site-directed mutagenesis by QuikChange II kit (Stratagene) according to the protocol of the manufacturer. To construct the Cdr1 kinase-dead mutant, lysine at position 41 was converted to alanine and integrated into cdr1Δ::ura4+ (JM2102) mutants at the endogenous locus through 5-fluoroorotic acid (Sigma) counterselection and verified by colony PCR. Site-directed mutagenesis was performed to create internal truncations that were then integrated into the cdr1Δ::ura4+ strain at the endogenous locus through hygromycin selection for GFPγ and verified using 5-fluoroorotic acid. The truncation allele (1–460) was created by chromosomal tagging. The cdr1-GFPγ cells shown in Fig. 2 have the cdc25-degron-DAmP mutation, which elongates cells closer to wild-type length. Cdr1 is present in very low abundance, and the number of nodes scales with cells size (39, 40). Therefore, cell elongation with cdc25-degron-DAmP made node imaging more reliable; similar results were obtained without this mutation.
The SISA strain was always grown on plates containing 5 μm 3-BrB-PP1 or 30 μm 3-BrB-PP1 when first streaking from a glycerol stock. To monitor Cdr1 phosphorylation in a SISA background, cells were grown at 25 °C in YE4S with a final concentration of 5 μm 3-BrB-PP1 to inhibit Sty1 activity. To activate Sty1 independently of stress, the culture was centrifuged at 4400 rpm for 2 min and then divided so that one half of the culture was resuspended in YE4S with 5 μm 3-BrB-PP1 and the other half was washed in a culture volume of YE4S (twice) and resuspended in YE4S lacking inhibitor (31). To test Cdr1 hyperphosphorylation in the sty1(T97A) mutant, sty1(T97A)-9gly-5FLAG cdr1–9gly-5FLAG cells were grown in EMM4S at 25 °C. Cells were treated with either 5 μm 3-BrB-PP1 (dissolved in methanol) or methanol control for 6 h. Cells were then split to either EMM4S + 1 m KCl, or EMM4S control. Samples were then harvested for Western blot analysis.
Immunoprecipitation and Western blotting
For immunoprecipitations, JM3057 was grown in EMM4S until log phase. 100 ml of logarithic phase cells were harvested and then lysed in 200 μl of lysis buffer (20 mm HEPES (pH 7.4), 1 mm EDTA, 300 mm NaCl, 0.2% Triton X-100, 1 mm PMSF, complete EDTA-free protease inhibitor tablets (Roche), 50 mm NaF, 50 mm β-glycerophosphate, and 1 mm sodium orthovanadate) with 200 μl of glass beads using a Mini-beadbeater-16 (Biospec) at 4 °C for 1 min twice. Cell extracts were clarified at 14,000 rpm for 5 min at 4 °C. Lysates were then incubated at 4 °C for 1 h with FLAG M2 magnetic beads (Sigma). Beads were washed vigorously with lysis buffer lacking phosphatase inhibitors (20 mm HEPES (pH 7.4), 1 mm EDTA, and 300 mm NaCl). Immunoprecipitates were treated with 400 units of λ phosphatase or mock-treated (New England Biolabs Inc.) at 30 °C for 30 min while shaking. For λ phosphatase treatment of KCl-stressed cells, samples were treated as above after splitting cells into EMM4S or EMM4S + 1 m KCl for 15 min.
For Western blotting, whole-cell extracts were lysed in 150 μl of sample buffer (65 mm Tris (pH 6.8), 3% SDS, 10% glycerol, 10% 2-mercaptoethanol, 50 mm NaF, 50 mm β-glycerophosphate, and 1 mm sodium orthovanadate) in a Mini-beadbeater-16 for 2 min. Gels were run at 20 mA until the 75-kDa marker was at the bottom of the gel. Blots were probed with anti-FLAG M2 (Sigma). Sty1 activation was observed using phospho-p38 (Thr-180/Tyr-182) (Cell Signaling Technology, 9211S). HA was probed using HA.11 clone 16B12 monoclonal antibody from Covance (MMS-101R).
Elutriation
Cells were grown in YE4S medium until log phase at 32 °C. A total of 350 OD cells were subjected to centrifugal elutriation using a Beckman JE5.0 rotor with a 4-ml elutriation chamber. Cells were loaded at 4100 rpm and 30 ml/min. Early G2 cells were eluted by reducing the rotor speed in 50-rpm decrements and fractionated. Fractions containing a homogenous population of small cells were combined into YE4S with/without 1 m KCl. The collected cells were grown at 32 °C, and cultures were sampled every 20 min for imaging and Western blotting. Samples for the septation index were fixed with 70% ethanol and stained with Blankophor. The septation index was calculated (n > 100 for each time point).
Production of GST-Cdr1 antibody
A C-terminal fragment of Cdr1 (341–593) was subcloned into the pGEX6P1 vector and expressed as a GST fusion protein in Escherichia coli strain BL21(DE3). Expression was induced by addition of isopropyl 1-thio-β-d-galactopyranoside, followed by growth for an additional 4 h at 25 °C. Cells were harvested by centrifugation and lysed using a French press in lysis buffer (1× PBS, 300 mm NaCl, 2 mm EDTA, 1 mm DTT, and complete EDTA-free protease inhibitor tablets (Roche)). Triton X-100 was then added to a final concentration of 1%. Extracts were clarified by centrifuging at 15,000 rpm at 4 °C using a SS-34 rotor for 20 min. Lysates were then incubated with glutathione-agarose (Sigma) for 2 h at 4 °C. 20 mm glutathione (pH 8.0) was used to elute purified protein. Following dialysis into PBS, the purified protein was then used to immunize a rabbit for antibody production (Thermo Fisher Scientific). For affinity purification, the same fragment was then coupled to CNBr-activated Sepharose 4B (GE Healthcare, 17-0430-01) according to the protocol of the manufacturer, and affinity purification was performed as described previously (41)
In vitro kinase assay
The C-terminal fragment of Cdr1 (341–593) was purified from E. coli as described above. Sty1-FLAG and analog-sensitive Sty1(T97A)-FLAG were immunoprecipitated from S. pombe after activating Sty1 with 1 m KCl for 10 min. After washing α-FLAG-M2 beads extensively, immunoprecipitated Sty1 was resuspended in 20 mm Tris (pH 8), 100 mm NaCl, 50 mm NaF, 50 mm β-glycerophosphate, and 1 mm sodium orthovanadate. Sty1 constructs and substrates were incubated together in 20 mm Tris (pH 8), 10 mm MgCl2, and 20 μm ATPγS at 30 °C for 30 min. The kinase reaction was stopped by adding 20 mm EDTA and alkylated by adding 2.5 mm p-nitrobenzyl mesylate and incubating at room temperature for 1 h. Sty1-FLAG beads were removed, and the remaining sample (containing substrate) was resuspended in SDS-PAGE sample buffer and boiled. Phosphorylation was detected using an antibody against thiophosphate ester (Abcam, 92570). The Cdr1 fragment and GST were detected with an antibody raised against a GST–Cdr1(341–593) fusion.
Microscopy
Microscopy was performed at room temperature with a Delta Vision imaging system (Applied Precision), an Olympus IX-71 inverted wide-field microscope, a Photometrics CoolSNAP HQ2 camera, and an Insight solid-state illumination unit. For Fig. 4, images were taken as a Z series of the top half of the cell with a 0.4-μm step size and subjected to iterative deconvolution in SoftWorRX (GE/Applied Precision). For all time courses, cells were grown in EMM4S to mid-log phase; the culture was then split and resuspended in EMM4S (control) or EMM4S +1 M KCl. sty1(T97A)-9gly-5FLAG cdr1-GFPγ was grown in EMM4S. Cells were treated as described above to monitor phosphorylation. A sample of each culture was then pelleted by centrifugation and resuspended in either EMM4S or EMM4S + 1 m KCl (with methanol or BrB-PP1) for 30 min. Similarly, sty1Δ cdr1-GFPγ was grown in EMM4S and then split to EMM4S or EMM4S + 1 m KCl, and imaged 30 min after this stress. The quantification was done in triplicate using >100 cells. Cell size measurements were performed in EMM4S at 32 °C (n > 50/strain).
Author contributions
H. E. O. designed, performed, and analyzed all experiments, contributed to the preparation of the figures, and contributed to writing the paper. J. B. M. designed and analyzed all experiments, contributed to the preparation of the figures, and contributed to writing the paper.
Supplementary Material
Acknowledgments
We thank the members of the Moseley laboratory for critical reading of the manuscript and K. Sawin for sharing the SISA yeast strain.
This work was supported by National Institutes of Health Grant R01 GM099774 (to J. B. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Tables S1 and S2.
- SAPK
- stress-activated protein kinase
- ER
- essential region
- SISA
- stress-independent Sty1 activation.
References
- 1. Alexander M. R., Tyers M., Perret M., Craig B. M., Fang K. S., and Gustin M. C. (2001) Regulation of cell cycle progression by Swe1p and Hog1p following hypertonic stress. Mol. Biol. Cell 12, 53–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dmitrieva N. I., Bulavin D. V., Fornace A. J. Jr, and Burg M. B. (2002) Rapid activation of G2/M checkpoint after hypertonic stress in renal inner medullary epithelial (IME) cells is protective and requires p38 kinase. Proc. Natl. Acad. Sci. U.S.A. 99, 184–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Michea L., Ferguson D. R., Peters E. M., Andrews P. M., Kirby M. R., and Burg M. B. (2000) Cell cycle delay and apoptosis are induced by high salt and urea in renal medullary cells. Am. J. Physiol. Renal Physiol. 278, F209–F218 [DOI] [PubMed] [Google Scholar]
- 4. Bulavin D. V., Higashimoto Y., Popoff I. J., Gaarde W. A., Basrur V., Potapova O., Appella E., and Fornace A. J. Jr. (2001) Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature 411, 102–107 [DOI] [PubMed] [Google Scholar]
- 5. Matsusaka T., and Pines J. (2004) Chfr acts with the p38 stress kinases to block entry to mitosis in mammalian cells. J. Cell Biol. 166, 507–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mikhailov A., Shinohara M., and Rieder C. L. (2004) Topoisomerase II and histone deacetylase inhibitors delay the G2/M transition by triggering the p38 MAPK checkpoint pathway. J. Cell Biol. 166, 517–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mikhailov A., Shinohara M., and Rieder C. L. (2005) The p38-mediated stress-activated checkpoint: a rapid response system for delaying progression through antephase and entry into mitosis. Cell Cycle 4, 57–62 [DOI] [PubMed] [Google Scholar]
- 8. Manke I. A., Nguyen A., Lim D., Stewart M. Q., Elia A. E., and Yaffe M. B. (2005) MAPKAP kinase-2 is a cell cycle checkpoint kinase that regulates the G2/M transition and S phase progression in response to UV irradiation. Mol. Cell 17, 37–48 [DOI] [PubMed] [Google Scholar]
- 9. Millar J. B., Buck V., and Wilkinson M. G. (1995) Pyp1 and Pyp2 PTPases dephosphorylate an osmosensing MAP kinase controlling cell size at division in fission yeast. Genes Dev. 9, 2117–2130 [DOI] [PubMed] [Google Scholar]
- 10. Shiozaki K., and Russell P. (1995) Cell-cycle control linked to extracellular environment by MAP kinase pathway in fission yeast. Nature 378, 739–743 [DOI] [PubMed] [Google Scholar]
- 11. López-Avilés S., Grande M., González M., Helgesen A. L., Alemany V., Sanchez-Piris M., Bachs O., Millar J. B., and Aligue R. (2005) Inactivation of the Cdc25 phosphatase by the stress-activated Srk1 kinase in fission yeast. Mol. Cell 17, 49–59 [DOI] [PubMed] [Google Scholar]
- 12. López-Avilés S., Lambea E., Moldón A., Grande M., Fajardo A., Rodríguez-Gabriel M. A., Hidalgo E., and Aligue R. (2008) Activation of Srk1 by the mitogen-activated protein kinase Sty1/Spc1 precedes its dissociation from the kinase and signals its degradation. Mol. Biol. Cell 19, 1670–1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Smith D. A., Toone W. M., Chen D., Bahler J., Jones N., Morgan B. A., and Quinn J. (2002) The Srk1 protein kinase is a target for the Sty1 stress-activated MAPK in fission yeast. J. Biol. Chem. 277, 33411–33421 [DOI] [PubMed] [Google Scholar]
- 14. Clotet J., Escoté X., Adrover M. A., Yaakov G., Garí E., Aldea M., de Nadal E., and Posas F. (2006) Phosphorylation of Hsl1 by Hog1 leads to a G2 arrest essential for cell survival at high osmolarity. EMBO J. 25, 2338–2346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. King K., Kang H., Jin M., and Lew D. J. (2013) Feedback control of Swe1p degradation in the yeast morphogenesis checkpoint. Mol. Biol. Cell 24, 914–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Breeding C. S., Hudson J., Balasubramanian M. K., Hemmingsen S. M., Young P. G., and Gould K. L. (1998) The cdr2(+) gene encodes a regulator of G2/M progression and cytokinesis in Schizosaccharomyces pombe. Mol. Biol. Cell 9, 3399–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kanoh J., and Russell P. (1998) The protein kinase Cdr2, related to Nim1/Cdr1 mitotic inducer, regulates the onset of mitosis in fission yeast. Mol. Biol. Cell 9, 3321–3334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Russell P., and Nurse P. (1987) The mitotic inducer nim1+ functions in a regulatory network of protein kinase homologs controlling the initiation of mitosis. Cell 49, 569–576 [DOI] [PubMed] [Google Scholar]
- 19. Coleman T. R., Tang Z., and Dunphy W. G. (1993) Negative regulation of the wee1 protein kinase by direct action of the nim1/cdr1 mitotic inducer. Cell 72, 919–929 [DOI] [PubMed] [Google Scholar]
- 20. Parker L. L., Walter S. A., Young P. G., and Piwnica-Worms H. (1993) Phosphorylation and inactivation of the mitotic inhibitor Wee1 by the nim1/cdr1 kinase. Nature 363, 736–738 [DOI] [PubMed] [Google Scholar]
- 21. Wu L., and Russell P. (1993) Nim1 kinase promotes mitosis by inactivating Wee1 tyrosine kinase. Nature 363, 738–741 [DOI] [PubMed] [Google Scholar]
- 22. Morrell J. L., Nichols C. B., and Gould K. L. (2004) The GIN4 family kinase, Cdr2p, acts independently of septins in fission yeast. J. Cell Sci. 117, 5293–5302 [DOI] [PubMed] [Google Scholar]
- 23. Martin S. G., and Berthelot-Grosjean M. (2009) Polar gradients of the DYRK-family kinase Pom1 couple cell length with the cell cycle. Nature 459, 852–856 [DOI] [PubMed] [Google Scholar]
- 24. Moseley J. B., Mayeux A., Paoletti A., and Nurse P. (2009) A spatial gradient coordinates cell size and mitotic entry in fission yeast. Nature 459, 857–860 [DOI] [PubMed] [Google Scholar]
- 25. Hohmann S., Krantz M., and Nordlander B. (2007) Yeast osmoregulation. Methods Enzymol. 428, 29–45 [DOI] [PubMed] [Google Scholar]
- 26. Zhang C., and Konopka J. B. (2010) A photostable green fluorescent protein variant for analysis of protein localization in Candida albicans. Eukaryot. Cell 9, 224–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee S., Lim W. A., and Thorn K. S. (2013) Improved blue, green, and red fluorescent protein tagging vectors for S. cerevisiae. PLoS ONE 8, e67902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wu L., and Russell P. (1997) Nif1, a novel mitotic inhibitor in Schizosaccharomyces pombe. EMBO J. 16, 1342–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pérez P., and Cansado J. (2010) Cell integrity signaling and response to stress in fission yeast. Curr. Protein Pept. Sci. 11, 680–692 [DOI] [PubMed] [Google Scholar]
- 30. Mutavchiev D. R., Leda M., and Sawin K. E. (2016) Remodeling of the fission yeast Cdc42 cell-polarity module via the Sty1 p38 stress-activated protein kinase pathway. Curr. Biol. 26, 2921–2928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zuin A., Carmona M., Morales-Ivorra I., Gabrielli N., Vivancos A. P., Ayté J., and Hidalgo E. (2010) Lifespan extension by calorie restriction relies on the Sty1 MAP kinase stress pathway. EMBO J. 29, 981–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Degols G., Shiozaki K., and Russell P. (1996) Activation and regulation of the Spc1 stress-activated protein kinase in Schizosaccharomyces pombe. Mol. Cell. Biol. 16, 2870–2877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nguyen A. N., and Shiozaki K. (1999) Heat-shock-induced activation of stress MAP kinase is regulated by threonine- and tyrosine-specific phosphatases. Genes Dev. 13, 1653–1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shiozaki K., Shiozaki M., and Russell P. (1998) Heat stress activates fission yeast Spc1/StyI MAPK by a MEKK-independent mechanism. Mol. Biol. Cell 9, 1339–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kelkar M., and Martin S. G. (2015) PKA antagonizes CLASP-dependent microtubule stabilization to re-localize Pom1 and buffer cell size upon glucose limitation. Nat. Commun. 6, 8445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Young P. G., and Fantes P. A. (1987) Schizosaccharomyces pombe mutants affected in their division response to starvation. J. Cell Sci. 88, 295–304 [DOI] [PubMed] [Google Scholar]
- 37. Moreno S., Klar A., and Nurse P. (1991) Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 194, 795–823 [DOI] [PubMed] [Google Scholar]
- 38. Bähler J., Wu J. Q., Longtine M. S., Shah N. G., McKenzie A. 3rd, Steever A. B., Wach A., Philippsen P., and Pringle J. R. (1998) Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast 14, 943–951 [DOI] [PubMed] [Google Scholar]
- 39. Deng L., and Moseley J. B. (2013) Compartmentalized nodes control mitotic entry signaling in fission yeast. Mol. Biol. Cell 24, 1872–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pan K. Z., Saunders T. E., Flor-Parra I., Howard M., and Chang F. (2014) Cortical regulation of cell size by a sizer cdr2p. eLife 3, e02040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Greenfield E. A. (2014) Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.