

Summary
Progenitor cells differentiate into specialized cell types through coordinated expression of lineage-specific genes and modification of complex chromatin configurations. We demonstrate that a histone deacetylase (Hdac3) organizes heterochromatin at the nuclear lamina during cardiac progenitor lineage restriction. Specification of cardiomyocytes is associated with reorganization of peripheral heterochromatin and, independent of deacetylase activity, Hdac3 tethers peripheral heterochromatin containing lineage-relevant genes to the nuclear lamina. Deletion of Hdac3 in cardiac progenitor cells releases genomic regions from the nuclear periphery, leading to precocious cardiac gene expression and differentiation into cardiomyocytes; in contrast, restricting Hdac3 to the nuclear periphery rescues myogenesis in progenitors otherwise lacking Hdac3. Our results suggest that availability of genomic regions for activation by lineage-specific factors is regulated in part through dynamic chromatin-nuclear lamina interactions and that competence of a progenitor cell to respond to differentiation signals may depend upon coordinated movement of responding gene loci away from the nuclear periphery.
eTOC
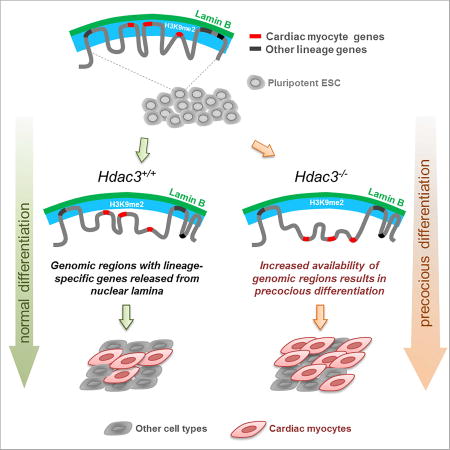
Nuclear architecture provides a distinct layer of gene regulation during development, coordinating cell fate determination through changes in chromatin accessibility that are mediated by chromatin-nuclear lamina interactions
Introduction
Heart development requires precise formation and differentiation of several progenitor populations, including those that can differentiate into cardiac myocytes (CMs), smooth muscle cells and endothelium (Kattman et al., 2006; Moretti et al., 2006; Wu et al., 2006). Specification of these precursor cells to various cell types requires coordinated regulation of numerous lineage-specific gene programs, and the mechanisms governing these processes are of intense interest to developmental biologists and those studying regeneration.
The dynamic ability of progenitor cells to differentiate in response to inductive signals represents a longstanding concept in developmental and stem cell biology known as “competence” (Waddington, 1940). The mechanistic underpinnings of cellular competence have never been thoroughly defined, and it remains unclear why common developmental morphogens have widely different effects depending on the characteristics of the progenitor cell upon which they act. In part, this could be due to concentrations, gradients and combinations of inducing factors, but a growing body of evidence suggests an additional level of gene regulation involving spatial positioning of genomic loci within the nucleus through chromatin-nuclear lamina interactions. The nuclear lamina supports nuclear shape and stiffness, and is a hub for interactions of nuclear proteins and chromatin (Osmanagic-Myers et al., 2015). Chromatin-lamina interactions are cell-type specific and dynamic during differentiation (Kohwi et al., 2013; Meister et al., 2010; Peric-Hupkes et al., 2010; Robson et al., 2016). Chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-seq; Shah et al., 2013), DNA adenine methyltransferase identification (DamID; (Guelen et al., 2008; van Steensel et al., 2001), and fluorescent in situ hybridization (FISH) have defined lamina-associated domains (LADs) - blocks of chromatin associated with the inner nuclear lamina. Consistent with historical electron micrographs, LADs are most commonly heterochromatic and marked by repressive histone modifications (Reddy et al., 2008).
Studies have focused on identifying proteins that tether chromatin to the lamina (Gonzalez-Sandoval et al., 2015; Poleshko et al., 2013; Solovei et al., 2013) as well as defining the critical epigenetic histone modifications and enzymes involved in regulation of nuclear architecture (Gonzalez-Sandoval et al., 2015; Guelen et al., 2008; Harr et al., 2015; Kind et al., 2015; Kind et al., 2013; Therizols et al., 2014; Towbin et al., 2012). One such factor is Hdac3, a histone deacetylase (HDAC) that interacts at the inner nuclear membrane with multiple proteins, including Lamina-associated polypeptide 2 (Lap2β; Somech et al., 2005; Zullo et al., 2012). Hdac3 is required for proper embryogenesis and heart development (Bhaskara et al., 2008; Lewandowski et al., 2015). In addition to the canonical function of HDACs during cardiogenesis (Haberland et al., 2009), emerging data suggest that HDACs have important deacetylase-independent roles (Sun et al., 2013; You et al., 2013). The specific function of Hdac3 in cardiac progenitor specification remains unknown.
Here, we define a role for Hdac3 in organizing heterochromatin at the nuclear periphery during cardiac progenitor cell lineage specification. Deletion of Hdac3 in cardiac progenitor cells (CPCs) induces precocious differentiation into CMs. Surprisingly, Hdac3 deacetylase activity is dispensable for repression of myocyte differentiation, suggesting a non-enzymatic role for Hdac3 in gene regulation. We demonstrate that cardiomyocyte specification is associated with a reorganization of peripheral heterochromatin. Further, we show that H3K9me2 is restricted to a layer of peripheral heterochromatin and has a high degree of overlap with LADs. In combination with high-resolution 3D FISH, we demonstrate that myocyte specification is associated with relocalization of cardiac genes away from the H3K9me2-marked peripheral chromatin layer towards the nuclear interior. Finally, we show that Hdac3 tethers regions containing loci relevant for myocyte determination to the nuclear periphery, and this catalytic-independent function of Hdac3 is required for myogenesis. Together, our data demonstrate that nuclear lamina-chromatin interactions influence cardiac progenitor cell differentiation. We propose that organogenesis is achieved through dynamic spatial reorganization of chromatin, including coordinated sequestration and/or release of genomic regions harboring key developmental genes from the nuclear lamina. Our model predicts that the ability of a progenitor cell to respond to an inductive signal will depend upon the availability of target gene loci for activation by classic developmental transcription factors and that availability is dynamically regulated through chromatin-nuclear lamina interactions during mammalian development.
Results
Hdac3 represses cardiac progenitor differentiation into cardiomyocytes
Cardiac development was modeled in vitro by aggregating mouse embryonic stem cells (ESCs) into embryoid bodies (EBs) and differentiating them into multipotent Flk1+PDGFRα+ progenitors that give rise to CM, endothelial and smooth muscle lineages (Kattman et al., 2011). In our protocol, we observe peak numbers of Flk1+PDGFRα+ CPCs at day 5 of differentiation and Tnnt2+ beating CMs by day 8. To assess the role of Hdac3 in CPCs during cardiogenesis, we overexpressed Hdac3 at day 5 of the in vitro cardiac differentiation protocol and measured, by flow cytometry, a decrease in the number of Tnnt2+ CMs at day 8 compared to control cultures treated with virus encoding GFP alone (Figures 1A and B). We observed statistically significant increases in the numbers of αSMA+ smooth muscle cells and Pecam1+ endothelial cells (Figures 1A and B), suggesting that overexpression of Hdac3 affects lineage determination of CPCs.
Figure 1. Hdac3 represses differentiation of cardiac progenitor into cardiomyocytes.

(A) Schema of cardiac differentiation of mouse ESCs into multipotent progenitors that give rise to indicated differentiated cell types. (B) Percentage of each cell type measured by flow cytometry at day 8. Cells overexpressing Hdac3 (vehicle+Hdac3-Flag) at day 5 of differentiation show a decrease in Tnnt2+ CMs; tamoxifen-mediated Hdac3 deletion (Tamoxifen+GFP) increases the number of Tnnt2+ CMs. (C) Immunoblot of total Hdac3 and loading control (β-actin) following transduction of CMV-creERT; Hdac3f/f ESCs with GFP control or indicated Hdac3 construct with vehicle or tamoxifen treatment. (D) GO analysis of gene expression data from day 8 tamoxifen-mediated Hdac3 deletion compared to control cultures identifies cardiac-specific GO terms associated with Hdac3 loss-of-function. (E) Gene expression (qRT-PCR) of candidate genes chosen from GO terms in (D) relative to Gapdh. (F) Schema of tamoxifen-mediated Hdac3 deletion at day 7 of differentiation. (G) Tamoxifen-mediated Hdac3 deletion starting at day 7 of cardiac differentiation shows no difference in percentage of each cell type measured by flow cytometry at day 8 compared to vehicle. (H) Schema of tamoxifen-mediated Hdac3 deletion and transduction of wildtype or mutant constructs at day 5 of differentiation. (I) Percentage of Tnnt2+ CMs measured by flow cytometry at day 8 (cells treated at day 5 with tamoxifen and transduced with wild-type Hdac3 or Hdac3Y298H mutant). Data represent mean ± SEM, n≥3 replicates for (B, E, G, I). (B, I) analyzed by one-way ANOVA, with a Tukey post-hoc test. (E, G) analyzed by two-tailed Student’s t-test; statistical comparisons are to vehicle. * p< 0.05. See Fig. S1, Table S1.
Next, we performed Hdac3 loss-of-function experiments utilizing ESCs generated from CMV-creERT; Hdac3f/f mice, in which a critical exon of Hdac3 is targeted for Cre recombinase excision. Addition of tamoxifen at day 5 of differentiation effectively reduces Hdac3 protein (Figure 1C); at day 8, we observed significantly more Tnnt2+ CMs compared to vehicle-treated cells (Figure 1B), independent of the presence or absence of the control virus (Figure S1A and B). Increase in Tnnt2+ cells was not due to a change in cell proliferation as measured by phospho-histone H3+ (Figure S1C). Consistent with these observations, in vivo deletion of Hdac3 in either second heart field Islet1+ cells (Islet1Cre/+; Hdac3f/f) or first heart field Nkx2-5+ progenitors (Nkx2-5Cre/+; Hdac3f/f) also results in a small but significant increase in the proportion of CMs in embryonic day 12.5 (E12.5) hearts as determined by flow cytometry (Figures S1D and E).
We assessed the effect of Hdac3 deletion on gene expression of day 8 CMV-creERT; Hdac3f/f EBs that had been treated with tamoxifen or vehicle on day 5 of the differentiation protocol. Gene Ontology (GO) analysis of the top 300 upregulated genes (defined by fold change) in Hdac3-deleted cells yields a number of GO terms associated with cardiac muscle development (Figure 1D, Table S1). We confirmed the upregulation of cardinal myocyte genes by qRT-PCR (Figure 1E). These data suggest that Hdac3 functions as a repressor of a cardiomyocyte gene program.
We observed that Hdac3 can influence cardiac myocyte differentiation only during a limited time frame when multipotent cardiac progenitors are present. Deletion of Hdac3 through addition of tamoxifen 48 hours past peak expression of Flk1+PDGFRα+ (day 7) results in no significant changes in the numbers of cardiomyocyte, smooth muscle or endothelial cells, nor is there any alteration of myocyte gene expression compared to vehicle-treated samples (Figures 1F, 1G, S1F and S1G). This is consistent with in vivo studies demonstrating that loss of Hdac3 in mature CMs is relatively well tolerated despite altered expression of genes regulating metabolic programs (Montgomery et al., 2008; Sun et al., 2011). Thus, promotion of cardiomyogenesis by Hdac3 loss appears to be restricted to the CPC stage.
The role of Hdac3 in cardiac differentiation is independent of its catalytic activity
To determine whether Hdac3 catalytic activity is required for regulation of CPC differentiation, we introduced various Hdac3 constructs in CMV-creERT; Hdac3f/f ESCs (Figure 1H). Deletion of Hdac3 in day 5 EBs promotes cardiomyocyte differentiation, and as expected, concomitant expression of exogenous wild-type Hdac3 restores differentiation to wild-type proportions (Figure 1I). Exogenous expression of a deacetylase-dead Hdac3 mutant (Hdac3 Y298H; Figure 1C) rescues differentiation to the same degree as wild-type Hdac3 (Figure 1I). While Hdac3 Y298H lacks enzymatic activity, it interacts with known binding partners (Lahm et al., 2007; Sun et al., 2013).
We next examined the requirement for Hdac3 catalytic activity during cardiac development. Mice harboring mutations in the deacetylase-activating domains of both NCoR and SMRT (NS-DADm) lack detectable Hdac3 enzymatic activity in vivo (You et al., 2013); however, these mice exhibit no increase in embryonic mortality (You et al., 2013) or ventricular hypoplasia, and display normal cardiac development and morphology through gestation (Figure S1H). This is in striking contrast to the phenotype of Hdac3−/−mutants that are not viable beyond E18.5 and exhibit structural heart disease (Lewandowski et al., 2015). Analysis of cardiomyocyte-specific gene expression reveals no significant change in myocyte gene signature between wild-type and NS-DADm mice (Figure S1I). Together, our in vitro and in vivo studies demonstrate that Hdac3 has a deacetylase-independent function in cardiac development and progenitor differentiation.
Hdac3 associates with the nuclear lamina
Hdac3 has been identified in a LAD-regulating complex with cKrox/ThPOK (Zbtb7b, Zinc finger and BTB domain containing 7B), and Lap2β, an inner nuclear membrane protein (Somech et al., 2005; Zullo et al., 2012; Figure 2A). To test whether Hdac3 prevents precocious cardiac progenitor differentiation by tethering LADs at the nuclear periphery, we first confirmed the ability of Hdac3 to interact with Lap2β (Figure 2B). We deleted a 38 amino acid domain of Hdac3 (Hdac3Δ33–70) that mediates interaction with Lap2β, abrogating Hdac3-Lap2β interaction in co-immunoprecipitation assays and proximity ligation assays (Figures 2B and S2A). Hdac3Δ33–70 does retain interaction with NCoR, an obligate Hdac3 co-factor (Sun et al., 2013) (Figure S2B). We asked whether loss of Zbtb7b would affect embryoid body differentiation. shRNA knockdown of Zbtb7b increased ESC differentiation into CMs to a similar degree as Hdac3 deletion (Figure S2C) and support a non-enzymatic LAD tethering function for Hdac3 in myogenesis.
Figure 2. Reorganization of lamina-bound chromatin during cardiogenesis.

(A) Model of Hdac3 interaction with components of the nuclear periphery; ONM: outer nuclear membrane; INM: inner nuclear membrane. (B) Immunoblots of co-IP and input lysates from 293T cells transfected with indicated constructs; IP: antibody used for immunoprecipitation; IB: antibody used for immunoblot. (C) Representative genome tracks of LaminB1 DamID (Peric-Hupkes et al., 2010) and input-normalized Lamin B ChIP-seq from ESCs. Area in gray box is magnified in lower set of tracks. Black bars indicate regions defined as LADs by EDD. (D) Overlap of genes defined in LADs using LaminB ChIP-seq and LaminB1 DamID (Peric-Hupkes et al., 2010). (E) Comparison of normalized log2 gene expression (Wamstad et al., 2012) of genes in LADs and non-LAD regions in ESCs and CMs (median expression with Tukey confidence intervals). (F) Stacked bar graph: number of genes that acquire, remain, or lose LAD residence in CMs compared to ESCs. (G) LaminB ChIP-seq tracks from ESC and CM show the cardiac gene Ace2 losing residence in a LAD in CM compared to ESC; black bars: LADs. (H) Change in Lamin B contact frequency between ESCs and CMs for myocyte signature genes compared to gene set signatures from pluripotent, endoderm, and endothelial cells, and a random gene set; n=number of genes in each set; p: significance of fold change differences between each gene set compared to the myocyte genes analyzed by one-way ANOVA Kruskal-Wallis Test. See Fig. S2, Tables S2, S3.
Dynamic spatial genome reorganization underlies cardiogenesis
Coordinated organization of LAD architecture may simultaneously regulate numerous genes in a differentiation pathway or lineage. Previous work has demonstrated that LADs reorganize during cellular differentiation (Kohwi et al., 2013; Meister et al., 2010; Peric-Hupkes et al., 2010). Therefore, in order to define the scope of chromatin reorganization at the nuclear periphery during cardiogenesis, we performed Lamin B ChIP-seq from ESCs (Figures S2D). Replicates (Jaccard indices: 0.71–0.94, Figure S2E) were merged and LADs were defined using Enriched Domain Detector (EDD; Lund et al., 2014)). We observe ~43% LAD occupancy genome-wide (Figure S2F and S2G) and our LADs strongly correlate to previously published ESC DamID LADs (Jaccard indices: 0.63–0.71, Figures 2C and 2D; (Peric-Hupkes et al., 2010). Likewise, 78% of the genes residing in our LADs were also observed within DamID LADs (Figure 2D and Table S2).
We assessed the differences in ESCs and ESC-derived CMs LADs. In CMs, LADs encompass ~45% of the genome (Figures S2F and S2G). Using RNA sequencing (RNA-seq) data from ESCs and ESC-derived CMs (Wamstad et al., 2012), we determined that genes residing within LADs had reduced expression compared to genes residing outside of LADs (Figure 2E), consistent with previous reports (Guelen et al., 2008). Most genes in ESC LADs reside within CM LADs (Figure 2F, gray box); however, KEGG pathway analysis of genes that lose LAD residence upon ESC to CM differentiation (Figures 2F and 2G) revealed multiple cardiac-related categories (Table S2). Pathway analysis of genes that gain residence in LADs was dominated by non-cardiac categories.
We next determined the probability that genic regions are in contact with the nuclear lamina (Peric-Hupkes et al., 2010); such calculations provide a method for analyzing ChIP-seq data generated from cells that likely reflect heterogeneity in nuclear lamina-bound regions, as reported in single-cell studies (Kind et al., 2015). We calculated a length-normalized probability of Lamin B occupancy for all genes in CMs compared to ESCs. We then compared gene signatures of CMs, pluripotent ESCs, endoderm, and endothelial cells based on publicly available expression data and relevant gene ontology categories (Table S3; CMs: GSE47948; pluripotent ESCs: GSE36114; E8.5 endoderm GSE40823; endothelial cells: GSE52564). As a group, genes in the cardiac myocyte signature show a greater cumulative Lamin B fold change than genes associated with other cell lineages (Figure 2H). To provide an unbiased comparison, we also compared the change in Lamin B occupancy for the myocyte signature genes with that of an equivalent number of genes sampled randomly 500 times; in this analysis, we observed an overall significant difference with p<0.0001 (1-way ANOVA, Figure 2H; see Methods). The myocyte signature includes key genes associated with cardiogenesis such as Titin (Ttn), Actin alpha cardiac muscle 1 (Actc1), and Myocyte enhancer factor 2C (Mef2c), and we observed that 90% (499/552) have lower Lamin B occupancy in CMs compared to ESCs.
H3K9me2 preferentially marks chromatin at the nuclear periphery
We postulated that the reorganization of heterochromatin during differentiation may involve specific histone modifications on peripheral heterochromatin. Dimethylation of lysine 9 on histone H3 (H3K9me2) marks megabase regions of heterochromatin, silent gene deserts, and subtelomeric regions (Black and Whetstine, 2011; Rosenfeld et al., 2009; Wu et al., 2005). H3K9me2 domains have been posited to correlate with binding of Lamin B (Guelen et al., 2008; Wu et al., 2005), though there are conflicting results in the literature (Filion and van Steensel, 2010; Wen et al., 2009). To explore this putative association, we assayed multiple commercially available antibodies, and confirmed specificity of two (Active Motif 39239; Abcam ab1220) for synthetic H3K9me2 peptides but not H3K9me1 or H3K9me3 peptides (Figures S3A and S3B, Table S4). Neither antibody recognized H3K27me2, which has an almost identical amino acid signature as H3K9me2 (ARKSA and ARKST, respectively; Figures S3A and S3B). Abcam ab1220 specifically recognized H3K9me2-bound chromatin in ChIP experiments, while Active Motif 39239 yielded only non-specific signal (Figure S3C). Conversely, Abcam ab1220 was non-specific in immunofluorescence (IF) assays (Figure S3D), while Active Motif 39239 yielded specific signal in IF assays (Figures S3D, S3E). H3K9me2 IF could be specifically abrogated by a H3K9me2 peptide, but not by closely related peptides (Figure S3E).
We compared H3K9me2-marked chromatin with other modified histones found in heterochromatin. H3K9me2 IF shows staining specific to chromatin adjacent to the nuclear lamina in murine ESCs, ESC-derived CMs and freshly isolated E12.5 CMs (Figure 3A). We measured the thickness of the H3K9me2-marked chromatin at more than 30 locations in each cell type, and determined that these cell types had slightly different average thicknesses of peripheral heterochromatin as marked by H3K9me2 (Figure 3B; 0.3µm average H3K9me2 layer thickness in ESCs and ESC-derived CMs versus 0.2µm in freshly isolated E12.5 CMs; one-way ANOVA: p<0.01 compared to ESCs or ESC-derived CMs). H3K9me2 staining (Figures 3A and 3C) was in stark contrast to repressive mark H3K9me3, which was not enriched at the nuclear periphery (Figure 3D). Hdac3 co-localizes with H3K9me2 at the nuclear periphery in undifferentiated ESCs (Figure S3F). Periphery-specific staining was not observed for H3K27me2, H3K27me3, H4K20me2, or H4K20me3 (Figure 3E). Thus, H3K9me2 specifically marks a layer of heterochromatin at the nuclear periphery.
Figure 3. H3K9me2 marks peripheral heterochromatin.

(A) H3K9me2 and Lamin B (Lmnb) immunostaining of indicated cell types. Areas in dotted boxes highlighted in bottom row. (B) Depth of H3K9me2-marked layer of chromatin at nuclear lamina in indicated cell types. 30 locations measured in each cell type, plot shows median distance with Tukey confidence intervals. Analyzed by one-way ANOVA Kruskal-Wallis Test. (C, D) H3K9me2 (C) or H3K9me3 (D) alongside LaminB IF and 3D reconstruction of Z-stack of images in pluripotent ESCs; scale bars: 2.5 µm. (E) IF of skeletal myoblasts with indicated repressive chromatin marks and LaminB. See Fig. S3, Table S4.
To further evaluate the relationship of LADs and H3K9me2-marked chromatin, we performed H3K9me2 ChIP-seq from ESCs and ESC-derived CMs (Figures 4A and Figure S3C). We found H3K9me2 enrichment in large segments in ESCs and ESC-derived CMs (Figures 4A and S4A). We used EDD to identify H3K9me2 domains, which encompassed ~49% of the genome in ESCs and ~52% of the genome in CMs (Figures S4A and S4B). H3K9me2 occupancy is highly correlated with Lamin B occupancy in both ESCs and CMs (10kb bins, Pearson’s r=0.84 for ESCs, Figures 4B and S4C). Consistently, the majority of genes residing within H3K9me2 domains also reside within ESC and CM LADs (Figure 4C and Table S2). Genes within H3K9me2 domains were expressed at lower levels compared to those outside of H3K9me2 domains (Figure 4D), further suggesting that H3K9me2 domains are similar to LADs. We also compared our H3K9me2 ChIP-seq data with ENCODE ChIP-seq datasets (GSM1000089 and GSM1000147), and found that H3K9me2 occupancy was different from H3K9me3 and H3K27me3 (Figure S4D), consistent with our IF images. As with LADs, the majority of genes that reside within ESC H3K9me2 domains reside within CM H3K9me2 domains (Figure 4E). Many non-myocyte genes gain residence in CM H3K9me2 domains (Table S2) and some cardinal myocyte genes that reside in ESC H3K9me2 domains lose residence in CMs (Figure 4F). Taken together, our imaging data and molecular approaches demonstrate that H3K9me2-marked chromatin closely resembles LAD chromatin.
Figure 4. H3K9me2-marked chromatin mirrors lamina bound chromatin.

(A) Representative H3K9me2 and LaminB ChIP-seq tracks from ESCs (Chr 3). Black bars represent LADs. Area in gray box is magnified below top set of tracks. (B) Correlation of LaminB and H3K9me2 occupancy in the genome in 10 kb bins; Pearson’s correlation r = 0.84. (C) Overlap of genes in LADs and H3K9me2 domains. (D) Distribution of normalized log2 gene expression (Wamstad et al., 2012) of genes in H3K9me2 domains and non-H3K9me2 regions in ESC and CMs; median expression with Tukey confidence intervals; significance determined with one-way ANOVA Kruskal-Wallis Test. (E) Number of genes that acquire, remain, or lose residence in H3K9me2 domains in CMs compared to ESCs. (F) H3K9me2 ChIP-seq tracks in ESC and CMs, highlighting Ttn losing residence in a H3K9me2 domain (black bar) in CM compared to ESC. See Fig. S4, Table S2.
Cardiac genes are released from the nuclear periphery during cardiac differentiation
Next, we used FISH to visualize the spatial localization of candidate gene regions that lose Lamin B and/or H3K9me2 occupancy upon myocyte differentiation as suggested by our ChIP-seq experiments. Based on our observation that the thickness of H3K9me2-marked chromatin at the nuclear periphery varies in ESCs and CMs (Figures 3A, 3B, and 5A), we measured precisely the distance of each FISH signal from the nuclear lamina to determine whether it was within the layer of H3K9me2-bound chromatin in that cell type (Figure 5B). Given the limitations with DAPI signal segmentation or 2D FISH approaches, we performed 3D image reconstruction from confocal Z-stacks to accurately determine spatial positioning of loci within or outside the nuclear peripheral heterochromatin layer (Figures 5B–D). Confocal Z-stack images were taken with 40 nm intervals between Z-planes, with an average of 150 stacks per cell providing up to 50 nm resolution between the FISH probe and nuclear lamina signal.
Figure 5. Cardiac genes are released from the nuclear lamina during cardiac differentiation.

(A–C) Immunostaining (A) and 3D reconstruction (B, C) showing Ttn locus in relation to nuclear lamina and H3K9me2-marked chromatin in ESC. (D) 3D reconstruction showing Ttn locus in relation to nuclear lamina in CM. Bottom panels in (C, D) are higher magnification, slightly rotated images, demonstrating distance of each Ttn allele from lamina (Lmnb). Ttn is located further away from the nuclear periphery in CM (C versus D). (E) Immuno-FISH of indicated loci (red) in each cell type co-stained for LaminB (Lmnb). Number of alleles per cell per locus shown adjacent to immuno-FISH images; 20–30 cells were quantified per condition per locus. Thresholding: see methods. (F) Quantitation of distance (E) performed on an individual locus basis in each cell type as indicated. Scale bars: (A, B) 0.5 µm; (C–E) 5 µm. * p< 0.001. Statistical comparisons analyzed by (E) Fisher’s Exact test; (F) one-way ANOVA with a Tukey post-hoc test, comparisons shown in reference to Kcnc2 loci. See Fig. S5, Table S5.
In ESCs, 3D-reconstructed immuno-FISH images revealed that the Ttn locus was embedded within the layer of H3K9me2 heterochromatin adjacent to the nuclear lamina (Lmnb; Figure 5B and Movie S1). In most ESC-derived Tnnt2+ CMs and in CMs isolated from E12.5 embryos, neither Ttn allele is found within the H3K9me2 layer and are instead centrally located (Figure 5D and Movie S2). To ensure accuracy of the differentiated state, CMs were confirmed by costaining for Tnnt2 (Figures 5E and S5). The peripheral localization of Ttn in ESCs was also seen for Actc1, Mef2c, and Calcitonin/calcitonin-related polypeptide alpha (Calca) loci (Figure 5E). Likewise, in CMs, one or both alleles were observed in the nucleoplasm (Figure 5E, n≥20 cells per condition). The change in distance from the nuclear lamina to each individual locus is statistically significant (one-way ANOVA p < 0.001) and suggests that these loci move from the nuclear periphery to the nucleoplasm during cardiogenesis (Figure 5F and Table S5; gray box in 5F indicates depth of H3K9me2 layer). In contrast to these results, Potassium Channel Voltage Gated KQT-like Subfamily Q Member 1 (Kcnq1) is not peripherally located in ESCs and remains centrally positioned in CMs (Figures 5E and 5F, Table S5), indicating that not all myocyte-relevant genes undergo repositioning from the nuclear periphery to the nucleoplasm during cardiomyocyte differentiation. In addition, Potassium voltage gated channel Shaw-related subfamily member 2 (Kcnc2), expressed primarily in neurons, is associated with the nuclear lamina in ESCs and remains peripherally located in CMs (Figures 5E and 5F, Table S5). These FISH results are consistent with our ChIP-seq observations that cardiac-specific genes showed lower probability of nuclear lamina association compared to genes related to other cell types upon myocyte specification and, together, indicate that a subset of cardiac-specific genes change nuclear position during cardiogenesis.
Loss of Hdac3 results in ectopic locus localization
We tested the hypothesis that Hdac3 is required for tethering chromatin to the nuclear lamina in ESCs. Deletion of Hdac3 in ESCs results in a relocalization of one or both alleles of the cardiac genes Ttn, Actc1, and Calca away from the nuclear periphery as determined by high-resolution FISH (Figure 6A, n ≥ 20 cells per condition). Additional analysis of the distance from the nuclear lamina to each individual locus confirmed a statistically significant difference (one-way ANOVA: p < 0.01) for each of these loci after loss of Hdac3 (Figure 6B, Table S5). Deletion of Hdac3 was confirmed by immunoblot and IF analyses (Figure S6A). Interestingly, neuronal-specific Kcnc2 remained at the periphery following Hdac3 deletion (Figures 6A and 6B, Table S5), suggesting redundancy and/or heterogeneity of complexes required for locus localization at the nuclear lamina and possible cell-type specificity of tethering complexes. We also performed Lamin B ChIP-seq from CMV-creERT; Hdac3f/f ESCs treated with tamoxifen or vehicle. Consistent with the FISH data, we found that, upon loss of Hdac3, more genes lost Lamin B occupancy than gained occupancy. Many neuronal-specific genes were amongst those genes that retained high Lamin B occupancy in the absence of Hdac3 (e.g., Atoh7, Bmyc, Snurf, and 125 olfactory receptor genes; Figure S6B).
Figure 6. Hdac3 is required to retain candidate loci at the nuclear lamina.

(A) Immuno-FISH of indicated loci (red) in CMV-creERT; Hdac3f/f ESCs treated with vehicle or tamoxifen and co-stained for LaminB (Lmnb). Cardiac genes (Ttn, Actc1, Calca) show precocious release from the nuclear periphery upon tamoxifen-mediated Hdac3 deletion. Scoring of number of alleles at the nuclear lamina per cell for each locus is adjacent to immuno-FISH images; 20–30 nuclei were quantified for each condition, for each gene. Thresholding: see methods. (B) Quantitation of distance from the nuclear lamina as observed in immuno-FISH (A) on an individual locus basis in each treatment condition shows significant relocation of cardiac gene loci upon Hdac3 deletion. (C) IF of localization of indicated Hdac3 construct (Flag); Lap2β fusion tethers Hdac3 to the nuclear periphery; Hdac3Δ33-70 loses peripheral localization; right panel of paired image is higher magnification of boxed area in left panel. (D) Immuno-FISH of Actc1 in CMV-creERT; Hdac3f/f ESCs, co-stained for Lmnb, treated with vehicle or tamoxifen, and indicated Hdac3 construct; all Hdac3 constructs rescue locus localization except Hdac3Δ33-70. (E) Scoring number of alleles at the nuclear lamina per cell for Actc1 per condition and construct for (D); 20–30 cells were quantified per condition. Thresholding: see methods. (F) Immuno-FISH (D) quantified on an individual locus basis. Scale bars: 5 µm. * p< 0.01. Statistical comparisons in (A, E) analyzed by Fisher’s Exact test. Statistical comparison in (B) by Student’s t-test, and in (F) by oneway ANOVA with a Tukey post-hoc test; comparison in reference to vehicle treatment. See Fig. S6, Table S5.
To confirm the requirement of Hdac3 for proper locus positioning, we introduced exogenous wild-type or mutant Hdac3 (Figure 6C) while simultaneously deleting endogenous Hdac3 in ESCs, and assayed for repositioning of the Actc1 locus by FISH. Our exogenous Hdac3 constructs include an Hdac3-Lap2β fusion (Hdac3-Lap2β), which localized to the inner nuclear membrane, and the Hdac3Δ33–70 mutant which cannot bind Lap2β but is localized to the nucleus (Figure 6C). Addition of exogenous Hdac3 was sufficient to prevent the nucleoplasmic translocation of Actc1 induced by Hdac3 deletion (Figures 6D–6F, Table S5), and expression of Hdac3-Flag constructs were confirmed by anti-Flag staining (Figure S6C). As with other FISH analyses, we scored the number of cells with 0, 1, or 2 Actc1 alleles at the nuclear lamina (Figure 6E), and whether individual Actc1 loci remained within the peripheral H3K9me2 layer (Figure 6F, gray box). Introduction of the deacetylase-dead Hdac3Y298H mutant phenocopied addition of wild-type Hdac3 (Sun et al., 2013) confirming that the catalytic activity of Hdac3 is dispensable for maintaining the peripheral localization of Actc1 in ESCs (Figures 6D–6F, Table S5). Similarly, introduction of Hdac3-Lap2β was sufficient to prevent the relocalization of the Actc1 loci (Figures 6D–F, Table S5) induced by Hdac3 deletion. Hdac3Δ33–70 mutant was unable to prevent re-localization of the Actc1 loci, indicating that Hdac3 interaction with the nuclear periphery (via Lap2β) is necessary for Hdac3-mediated positioning effects (Figures 6D–6F, Table S5). In total, we scored more than 300 cells with a minimum of 40 individual alleles per condition, which show a non-catalytic function of Hdac3 at the inner nuclear membrane is required for localizing key genomic regions to the nuclear lamina.
Hdac3-mediated sequestration of genomic regions at the nuclear lamina regulates cardiac myocyte differentiation
We examined whether Hdac3 directly represses cardiac differentiation through tethering of myocyte genes to the nuclear lamina. We assessed myogenic differentiation of ESCs after Hdac3 deletion with or without exogenous wild-type and mutant Hdac3 (Figure 7A). Following deletion of endogenous Hdac3 in CMV-creERT; Hdac3f/f EBs at day 5, we observed that myogenesis is markedly enhanced when compared to vehicle-treated, control EBs (Figure 7B). Exogenous expression of the Hdac3-Lap2β fusion or the deacetylase-dead Hdac3Y298H mutant fused to Lap2β was sufficient to restore cardiac differentiation to levels equivalent to EBs transduced with wild-type Hdac3 (Figure 7B). Expression of the Hdac3Δ33–70 mutant (unable to interact with Lap2β), failed to rescue loss of endogenous Hdac3 (Figure 7B). Gene expression analysis from aforementioned day 8 cultures demonstrated that the elevated expression of cardiac-specific genes produced by Hdac3 deficiency was rescued by forms of Hdac3 that are tethered to the nuclear membrane, independent of deacetylase activity (Figure 7C). Consistent with our model, Hdac3Δ33–70 was unable to rescue the loss of Hdac3 (Figures 7B, 7C). Taken together, these data demonstrate that Hdac3 at the nuclear periphery represses cardiogenesis in a deacetylase-independent manner.
Figure 7. Hdac3 requires interaction with the nuclear lamina to repress cardiac differentiation.

(A) Schema of Hdac3 constructs transduced at day 5 of differentiation with simultaneous tamoxifen-mediated Hdac3 deletion. (B) Percentage of Tnnt2+ cardiac myocyte cells on day 8 of differentiation measured by flow cytometry in cells with vehicle or tamoxifen and indicated constructs; all Hdac3 constructs rescue Hdac3 deletion precocious cardiogenesis phenotype, except Hdac3Δ33-70. (C) Gene expression analysis (qRT-PCR) of CM genes and Hdac3 from day 8 cells with vehicle or tamoxifen and indicated constructs; rescue of cardiac gene expression by all constructs, except Hdac3Δ33-70. Data represent mean ± SEM, n≥3 replicates in (B), (C). * p< 0.05, all statistical comparisons are to vehicle+GFP control, analyzed by one-way ANOVA with a Tukey post-hoc test. (D) Differentiation of multipotent CPCs depends on Hdac3 to orchestrate simultaneous release of genomic regions containing lineage-specific loci from the nuclear lamina. Hdac3 tethers chromatin to the lamina. Without Hdac3, cardiac-specific genes lose contact with the nuclear lamina, are less frequently found within the H3K9me2-marked layer of peripheral heterochromatin, and are more likely to be transcriptionally active.
Discussion
We report a role for Hdac3 in mediating the spatial positioning of lineage-specific loci during cardiogenesis. In ESCs, regions of the genome containing key cardiac genes are localized to the nuclear periphery, in an Hdac3-dependent fashion, within H3K9me2-marked heterochromatin. Genomic regions containing cardiac genes release from the nuclear periphery upon differentiation of ESCs into CMs. Hdac3 deletion causes release of cardiac gene regions from the nuclear periphery and leads to precocious myogenesis, and the tethering function of Hdac3 does not require its enzymatic deacetylase activity. Thus, Hdac3-mediated spatial positioning of chromatin is a critical regulator of cardiogenesis (Figure 7D) providing an example of the physiological relevance of peripheral gene localization in organogenesis. We predict that the pathways and players responsible for dynamic tethering of regions of the genome to the nuclear periphery will emerge as a novel system for gene regulation by functioning to control the accessibility of gene promoters and enhancers to canonical activators and repressors of transcription.
Nuclear architecture influences gene regulation and shapes cell identity
It has become increasingly appreciated that progressive lineage restriction requires both activation of lineage-specific gene networks and simultaneous repression of pathways that facilitate alternate fates (Loh et al., 2016). While it is unclear how such a feat of coordinated gene expression is achieved, it is now well-established that nuclear lamina-chromatin interactions correlate with cellular competence. For example, neuronal loci show loss of lamina occupancy as ESCs differentiate into neuronal cell types (Peric-Hupkes et al., 2010), and a similar phenomenon is observed during C. elegans muscle development (Meister et al., 2010). Studies also suggest that lamina-chromatin interactions directly regulate gene programs and cell fate choices. The ability of Drosophila neuroblasts to give rise to various neuronal cell types is prevented when the hunchback genomic locus is transferred from the nucleoplasm to the nuclear periphery and abrogation of hunchback relocalization to the nuclear lamina extends the time window in which neuroblasts can give rise to neurons (Kohwi et al., 2013).
Our results support these studies and extend our understanding of Hdac3 function beyond that of classic gene regulation. Hdac3 transcriptional repression is known to occur through interactions with cofactor NCoR and SMRT (Li et al., 2000). Hdac3 has been implicated in gene repression at the nuclear periphery (Zullo et al., 2012) where it may also recruit other class I HDACs (Pegoraro et al., 2009). In this study, we demonstrate a critical non-catalytic function for Hdac3 as a LAD tether at the nuclear periphery. Notably, non-catalytic functions of well-studied epigenetic enzymes, including Tet2 (Fuster et al., 2017) and Ezh2 (Kim et al., 2015), have been implicated in progression of atherosclerosis and oncogenesis, respectively. Our observations underscore the importance of understanding the full complement of functions that factors like Hdac3 exert in regulating chromatin biology.
Limiting access to alternate fates
Our data support a model in which chromatin relevant to alternative cell fates may be bound to the lamina scaffold and repressed. We demonstrate that non-cardiac fate gene regions are directed to the nuclear lamina as cardiac cells gain identity. Assessment of DNase hypersensitivity sites in tissues undergoing directed lineage differentiation revealed that chromatin structure and, specifically, access to progenitor-relevant regions of the genome, are progressively altered as cells acquire differentiated identities (Stergachis et al., 2013). During myogenesis in C. elegans, loss of the LAD tether Cec4 causes ectopic activation of intestinal development pathways (Gonzalez-Sandoval et al., 2015). Cardinal pluripotent loci, including Oct4, Klf4, Sox2, and Nanog, gain lamina occupancy as embryonic stem cells differentiate into neuronal cell types (Peric-Hupkes et al., 2010). Thus, it appears that the nuclear lamina provides a flexible scaffold to release regions of the genome required for establishment of cell identity while also limiting access to alternative cell-fate pathways. Cellular plasticity may depend, in part, on regulation of peripheral heterochromatin and reflect LAD stability. Hence, a better understanding of LAD tethers could inform the ability to trans-differentiate one cell type into another by making cells more competent to respond to reprogramming factors.
How is specificity achieved?
It is currently unclear both how tissue-specific chromatin organization is achieved and how LADs themselves are established. LADs are enriched for GAGA motifs (Zullo et al., 2012) and A/T sequences (Meuleman et al., 2013), though it is unlikely that these DNA motifs are sufficient for specificity. Our clear demonstration of H3K9me2 restriction to the nuclear periphery may inform our understanding of LAD establishment and regulation, and ongoing studies will focus on potential “readers” of this histone mark. Organization of peripheral heterochromatin likely involves multiple factors, including cell type-specific proteins embedded within the nuclear envelope that interact (directly or indirectly) with sequence-specific DNA-binding factors, tissue-specific signaling cascades that impact on chromatin and/or the inner nuclear membrane, and non-coding RNAs.
Loss of Hdac3 in cardiac progenitors results in relocalization of genomic regions containing key cardiac genes and activation of those genes, but endothelial and smooth muscle lineage genes remain unaffected. Hdac3 interaction with other peripheral factors, such as the zinc-finger DNA binding protein cKrox (Zbtb7b; Somech et al., 2005; Zullo et al., 2012), may confer specificity for LAD establishment. Proteomic approaches reveal clear differences in the composition of nuclear lamina-associated proteins in different cell types (Korfali et al., 2012), and changing expression of lamin proteins and their interacting partners over the course of differentiation has been shown to impact chromatin localization broadly (Solovei et al., 2013). Additionally, a series of specific nuclear envelope transmembrane muscle proteins, recognize specific loci relevant to muscle fate, facilitate their spatial localization, and impact in vitro differentiation of immortalized cells (Robson et al., 2016). Finally, long non-coding RNAs play a critical role in genome organization at the nuclear lamina (Chen et al., 2016). Characterizing the in vivo role of newly identified tissue-specific proteins and long non-coding RNAs at the nuclear lamina will undoubtedly shed additional light on regulation of peripheral heterochromatin organization.
During normal cardiogenesis, Hdac3 expression is stable; it is unlikely that Hdac3 loss is the endogenous mechanism to trigger cardiac lineage specification. A more likely explanation is that a local signal impacts Hdac3 itself or that some (yet uncharacterized) factor(s) in the Hdac3 tethering complex is sensitive to lineage-specific cues. One hint may come from classic signaling pathway effector molecules, including Erk, Lef, and Smad4, which can localize to the nuclear lamina (Prokocimer et al., 2009). In this regard, combinatorial histone modifications may also serve as an important layer of regulation. For example, histone H3 serine 10 (H3S10) can be phosphorylated by a number of cellular kinases. We observe that H3K9me2 antibodies cannot recognize the K9 dimethyl epitope when the adjacent S10 residue is phosphorylated (Figure S4A, Table S4). This suggests the possibility that recognition of the H3K9me2 modification and LAD organization could be regulated by H3S10 or similar kinases.
Finally, our data suggest that a critical layer of developmental program gene regulation is coordinated by sequestration and/or release of genes at the nuclear periphery. Release of a subset of CM-specific gene regions from the nuclear periphery is required for their activation, but it remains unknown whether release from the periphery is sufficient for expression. We speculate that release from the nuclear periphery equates to “competence” for gene activation, given that during differentiation many gene regions move away from the highly heterochromatic environment of LADs and H3K9me2 enrichment where repression is likely reinforced. We theorize that additional factors, including lineage-specific DNA-binding transcription factors and co-activator complexes, are required for active transcription, and provide an additional level of gene regulation. This is consistent with recent work demonstrating that local chromatin decondensation, but not necessarily transcriptional activation, at the nuclear lamina is sufficient to relocalize chromatin to the nucleoplasm (Therizols et al., 2014). To fully understand the impact that chromatin reorganization at the lamina has on coordinated gene regulation, it will be critical to elucidate 1) how organization impacts competence for transcription, 2) whether transcription precedes, is concomitant with, or follows reorganization of genomic regions from the nuclear lamina, and 3) how lineage-specific transcription factor binding is affected by nuclear organization changes.
Implications for development and disease
Our data support a model in which Hdac3 regulates cardiogenesis by spatially sequestering peripheral heterochromatin. This mechanism may involve controlling whether or not a set of genes is available for activation and repression by conventional regulators. For example, it has been unclear how common inductive signals, such as those in the Bmp, Wnt, or Notch pathways, can have such different effects depending upon the type of progenitor cell receiving the signal; Bmp, for example, might induce a mesenchymal stem cell to form bone while causing a CPC to become muscle. Our data demonstrate that a critical mechanism for lineage restriction is the re-localization of lineage-specific gene-containing regions away from the nuclear periphery, making them competent for activation, and Hdac3 is a factor that contributes to this organizational change by acting as a tether. We propose that different progenitor populations will be characterized by differences in LAD and non-LAD regions, and hence differences in genes that are available for activation within the nucleoplasm. Thus, our work suggests a mechanistic explanation for the concept of cellular competence to respond to a developmental morphogen.
Expanding this work further will likely impact our understanding of human diseases involving defects in the nuclear lamina, collectively known as laminopathies. These include examples of muscular dystrophies, cardiomyopathies and premature aging. Hutchinson-Gilford Progeria syndrome, resulting from the introduction of a cryptic splice site and accumulation of farnesylated Lamin A protein, is characterized by abnormal heterochromatin structure at the periphery and excessive DNA damage (Kubben et al., 2012; Van Bortle and Corces, 2013). Mutations in LMNA or the gene encoding its interacting protein Emerin cause Emery-Dreifuss muscular dystrophy (EMDD), a degenerative muscle disease associated with cardiac abnormalities. Multiple mechanisms by which mutations in LMNA and related genes result in myopathy have been investigated, including abnormal nuclear stiffness and morphology, and abnormal signal transduction (Schreiber and Kennedy, 2013). However, atypical interactions of chromatin with the defective nuclear lamina and subsequent dysregulated gene expression may also contribute to the pathology of the laminopathies. Consistent with this hypothesis, a C. elegans model of EDMD exhibits irregular positioning of heterochromatin (Mattout et al., 2011) and both chromosome positioning and gene expression are altered in cells expressing a LMNA mutation found in patients with cardiomyopathy (Mewborn et al., 2010). It will be exciting to build on this work to further explore the role of nuclear architecture during normal development and in the pathophysiology of laminopathies and related human disorders.
STAR Methods
Contact for reagent and resource sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by Rajan Jain, jainr@mail.med.upenn.edu.
Experimental Model and Subject Details
Mice
Isl1Cre, Nkx2.5Cre, Hdac3flox, and CMV-creERT mice were maintained on mixed CD1/B6/129 genetic backgrounds separated by 4–8 generations of interbreeding from pure parental strains, see STAR methods for sources. NS-DADm mice were maintained on a C57BL/6 background (You et al., 2013). Mice were genotyped using Cre-specific PCR, and primers designed to distinguish between the control and floxed Hdac3 allele. Littermate embryos were analyzed in all experiments unless otherwise noted. Institutional Animal Care and Use Committee approved all animal protocols.
ESC derivation, differentiation and Hdac3 knockout
Embryonic stem cells (ESCs) were derived from CMV-creERT; Hdac3f/f mice as previously described (Jain et al., 2015). Blastocysts were collected at E3.5 and cultured on STO feeder cells in standard ESC media + leukemia inhibitory factor (LIF) + 50µM MEK1 inhibitor (Cell Signaling #9900) for 7 days until the blastocyst hatches and forms colony. Individual colonies were subcultured for about 1 week when MEK1 inhibitor is removed and cells passaged as a normal ESC line. Cardiac differentiation from these cells was adapted from published protocols (Kattman et al., 2011). Briefly, ESCs were cultured and maintained on a feeder layer of mitotically inactivated MEFs in DMEM with 15% FBS (Fisher Scientific #SH3007003) and ESGRO LIF. Differentiation through hanging droplets method was initiated following ESC dissociation and suspension at 5 × 104 cells/ml in DMEM with 10% FBS (Atlanta Biologicals #S11550) without LIF in 20 µl drops. Two days after droplets formation, embryoid bodies (EBs) were transferred in suspension onto poly-HEMA coated dishes. After another two days, EBs were plated on gelatin coated dishes in cardiac differentiation media (StemPro-34 SF medium, Invitrogen #10639-011) supplemented with 5ng/ml VEGF (R&D systems), 10ng/ml bFGF (R&D systems), 12.5ng/ml FGF10 (R&D systems), 2.5µM XAV939 (Cayman Chemical #13596), 1mM Ascorbic Acid (Sigma #A4403) and 2mM Glutamax (Invitrogen #35050-061). Beating cells were visible within 24–48 hours. Hdac3 knockout in CMV-creERT; Hdac3f/f ESCs were performed using 4-Hydroxytamoxifen (Sigma #T176) dissolved in 100% ethanol. Cells were treated with 1 µg/ml tamoxifen for 24 hour to delete Hdac3.
ESCs and CMs that were used for ChIP-seq experiments utilized CCE cells (derived from 129/Sv mouse strain). The protocol was slightly modified: mESCs were cultured in serum-free, feeder-free 2i media (50% DMD-F12 (Invitrogen); 50% Neurobasal medium (Invitrogen); 0.5 × N2 supplement (Invitrogen); 0.5 × B27 supplement (Invitrogen); 0.05% BSA (Invitrogen); L-Glutamine (Invitrogen); Penicilin/Streptomycin (Invitrogen) and 150 uM β-mercaptoethanol (Sigma); 20 ng/ml LIF (eBioscience), 3 µM CHIR (Stemgent) and 1 µM PD325901 (Stemgent)). ESCs were differentiated as EBs to the cardiomyocyte lineage. EBs were generated at day 0 of differentiation in SF-D media (75% IMDM (Invitrogen); 25% Ham’s F12 (Invitrogen); 0.5 × N2 supplement (Invitrogen); 0.5 × B27 supplement (Invitrogen); 0.05% BSA (Invitrogen); L-Glutamine (Invitrogen); Penicillin/Streptomycin (Invitrogen) and 150 µM beta-mercaptoethanol (Sigma) with ascorbic acid (0.5 mM) and monothioglycerol (0.4 mM)). On day 2, EBs were collected, dissociated, and reaggregated in SF-D media with VEGF (5 ng/ml, R&D Systems), Activin A (8 ng/ml, R&D Systems), and BMP4 (1 ng/ml, R&D Systems) to induce cardiac mesoderm formation. At day 4 EBs were collected, washed (with IMDM), and cultured in StemPro (Invitrogen) with VEGF (5 ng/ml, R&D Systems), bFGF (10 ng/ml, R&D Systems), and XAV (10 µM, Stemgent) to induce cardiomyocyte differentiation. Subsequent media changes were done in the same media without XAV, and cells were collected on day 10 of differentiation.
C2C12 Cell Culture and Transfection
C2C12 skeletal myoblast cells were maintained at 37°C in DMEM supplemented with 10% fetal bovine serum, penicillin and streptomycin. C2C12 cultures were not be allowed to become confluent. C2C12 cells were plated on coverslips and transfected with indicated plasmid DNA using FuGENE6 (Promega #2691) according to manufacturer protocol.
Method Details
Flow cytometry
ES-derived cells and embryonic hearts were fixed, permeabilized and stained for flow cytometry according to standard protocols. Briefly, cells were dissociated in 1mg/ml collagenase solution, fixed in Fixation buffer (eBioscience #00-8222-49), permeabilized in 1x Permeabilization buffer (eBioscience #00-8333-56) and stored in 1% BSA. Embryonic hearts were dissociated in 5mg/ml collagenase at 37°C for 10 minutes with occasional trituration before fixation and permeabilization steps. Cells were stained with primary and secondary antibodies for 1 hour each in Permeabilization buffer, resuspended in Flow Cytometry buffer (eBioscience #00-4222-26) and analyzed on a BD FACSAria II cytometer. Antibodies used were cardiac Troponin T (1:100, Thermo #MS-295-P), Smooth Muscle Actin (1:100, abcam #ab5694), CD31 (1:100, Abcam #ab28364), Phospho-Histone H3 (1:50, Cell Signaling #9701), anti-mouse AlexaFluor 488 (1:200, Invitrogen #A21200) and anti-rabbit AlexaFluor 647 (1:200, Invitrogen #A21244). Flow cytometry data are presented as percent of Troponin T-positive, Smooth Muscle Actin-positive or CD31-positive cells relative to the total sum of cells labeled by one of the three antibodies (i.e. “% cardiac cells”).
RNA isolation and preparation
ES-derived cellular RNA was isolated in Trizol (Invitrogen #15596-026) and RNA was obtained using Qiagen RNeasy spin columns, with on-column DNAse I digestion. Complementary DNA (cDNA) was synthesized according to kit instructions with the Superscript III system (Invitrogen). Quantitative RT-PCR was performed in triplicate using SYBR Green (Applied Biosystems). Gapdh was used as a reference control gene. Quantitative RT-PCR data presented as “Relative gene expression” describe fold change of treatment versus control samples. Quantitative RT-PCR primer sequences are available upon request. Microarray analysis was performed by the University of Pennsylvania Next Generation Sequencing Core using 2-color hybridization technique with a whole genome mouse Agilent array. Two pools each of either vehicle or tamoxifen treated Flk1+PDGFRα+ CMV-creERT; Hdac3flox/flox ESCs were hybridized to the array. DAVID analysis was used to group genes with altered expression in Hdac3-null CPCs versus controls according to functional categories.
Immunoblot analysis
Lysates were run on 4–12% Bis-Tris protein gels (Invitrogen #NP0335) and blots were probed with anti-Hdac3 (1µg/ml, abcam #ab7030), anti-β-actin (1:1000, Cell Signaling #4967), anti-GFP (1:1500, Cell Signaling #2956) or anti-FLAG HRP-conjugated primary antibodies (1:500, Sigma #A8592) according to the instructions of the manufacturer. Anti-rabbit HRP-conjugated secondary antibody (Cell Signaling #7074) was used at 1:2500. Visualization was achieved using ECLPrime (GE Life Sciences #RPN2232) or SuperSignal West Femto Chemiluminescent Substrate (Thermo #34094).
Plasmids, transfection and transduction
Expression plasmids for Hdac3-FLAG and Y298H Hdac3-FLAG were generously provided by Paola Gallinari (Istituto di Ricerche di Biologia Molecolare, Italy). Δ33–70 Hdac3-FLAG was generated by overlap extension PCR according to standard protocols and insert cDNA was fully sequenced. Wild-type and mutant Hdac3 were cloned into AgeI-BsrGI sites of lentivirus FUGW (Addgene #14883) for transduction of ESC-derived cells. Unless otherwise indicated, ES cells were transduced with Hdac3 expression plasmids at day 5 of differentiation (during peak expression of Flk1+Pdgrfα+) and analyzed at day 8. Lap2β was PCR amplified from pEGFP-Lap2β (generously provided by Jan Ellenberg, European Molecular Biology Laboratory, Germany via http://www.euroscarf.de #P30463) and cloned into BsrGI site of FU-Hdac3 or FU-Hdac3 Y298H, creating a fusion protein with Hdac3 or Y298H Hdac3. Lentiviruses were generated in Lenti-X 293T cells (Clontech #632180) according to manufacturer instructions and with the use of standard packaging plasmids (psPAX2, Addgene #12260 and pMD2.G, Addgene #12259). Plasmid transfections were performed with FuGENE 6 (Promega #E2691) according to manufacturer instructions. For confocal imaging C2C12 cells were transfected at 50% confluency and imaged 24hr or 72hr post-transfection. The phenotype rescue experiment was performed in CMV-creERT; Hdac3f/f ESCs by simultaneous addition of tamoxifen for 24 hours, and lentiviral transduction of Hdac3 constructs.
Co-immunoprecipitation
293T cells were transfected with EGFP-Lap2β and/or Hdac3 plasmids for 48 hours and lysates generated as previously described (Zullo et al., 2012). Briefly, cells were collected in hypotonic lysis buffer (420mM NaCl, 20mM HEPES pH 7.9, 0.2mM EDTA, 25% glycerol, supplemented with protease inhibitors), triturated through a 26g syringe and diluted 1:1 in IP lysis buffer (20mM Tris-HCl pH 8, 137mM NaCl, 1% NP-40, 2mM EDTA, supplemented with protease inhibitor). Lysates were precleared twice for one hour each with Protein G Dynabeads (Invitrogen #10003D) and quantified by BCA. 150µg lysates were immunoprecipitated for 4 hours at 4 degrees with Protein G Dynabeads with 2µg mouse IgG (Santa Cruz #sc-2025) or anti-FLAG magnetic beads (Sigma #M8823). Beads were collected, washed 3 times in IP lysis buffer and bead-bound proteins eluted into sample buffer [1x Loading dye (Invitrogen #NP0007), 0.1M DTT, 10% 2-Mercaptoethanol (BioRad #161–0710XTU)]. Samples were boiled for 5 minutes and analyzed by immunoblotting.
Microarray
Microarray services were provided by the University of Pennsylvania Molecular Profiling Facility, including quality control tests of the total RNA samples by Agilent Bioanalyzer and Nanodrop spectrophotometry. All protocols were conducted as described in the Affymetrix WT Plus Reagent Kit Manual and the Affymetrix GeneChip Expression Analysis Technical Manual. Briefly, 250ng of total RNA was converted to first-strand cDNA using reverse transcriptase primed by poly-(T) and random oligomers that incorporated the T7 promoter sequence. Second-strand cDNA synthesis was followed by in vitro transcription with T7 RNA polymerase for linear amplification of each transcript, and the resulting cRNA was converted to cDNA, fragmented, assessed by Bioanalyzer, and biotinylated by terminal transferase end labeling. 5.5 µg of labeled cDNA was added to Affymetrix hybridization cocktails, heated at 99°C for 5 min and hybridized for 16 h at 45°C to Mouse Gene 2.0 ST GeneChips (Affymetrix Inc.) using the GeneChip Hybridization oven 645. The microarrays were then washed at low (6X SSPE) and high (100mM MES, 0.1M NaCl) stringency and stained with streptavidin-phycoerythrin. Fluorescence was amplified by adding biotinylated anti-streptavidin and an additional aliquot of streptavidin-phycoerythrin stain. A GeneChip 3000 7G scanner was used to collect fluorescence signal. Affymetrix Command Console and Expression Console were used to quantitate expression levels for targeted genes; default values provided by Affymetrix were applied to all analysis parameters. Affymetrix “.cel” files were RMA normalized in Partek Genomics Suite (v6.6, Partek, Inc., St. Louis, MO). Principal Component Analysis was used to visualize variation among the samples. Two outliers (Vehicle Replicate 2 and Tamoxifen Replicate 1) were excluded from further analysis. Differential gene expression was evaluated with Significance Analysis of Microarrays (SAMR v2.0, https://github.com/MikeJSeo/SAM).
ChIP and library preparation for sequencing
Murine ESCs and ESC-derived CMs were crosslinked in culture by addition of methanol-free formaldehyde (ThermoFisher; final 1% v/v) and incubated at room temperature for 10 minutes with gentle rotation. Crosslinking was quenched by addition of glycine (final 125mM) and incubated at room temperature for 5 minutes with gentle rotation. Media was discarded and replaced with PBS; cells were scraped and transferred to conical tubes and pelleted by centrifugation (250xg, 5 minutes at room temperature). Supernatant was removed, and pellets were flash frozen on dry ice and stored at −80C.
Six hours before lysing cells, 30uL protein G magnetic beads (per ChIP sample) were washed 3 times in blocking buffer (0.5% BSA in PBS); beads were resuspended in 250uL blocking buffer and 2ug antibody (Lamin B: #sc-6217; H3K9me2: #ab1220) and rotated at 4C for at least 6 hours. Crude nuclei were isolated from frozen crosslinked cells as follows: cell pellet (from 10cm plate) was resuspended in 10mL cold Lysis Buffer 1 (50mM HEPES-KOH pH7.5, 140mM NaCl, 1mM EDTA, 10% Glycerol, 0.5% NP-40, 0.25% Triton X-100, and protease inhibitors), and rotated at 4°C for 10 minutes, followed by centrifugation (250xg, 5 minutes at room temperature). Supernatant was discarded and the pellet was resuspended in 10mL cold Lysis Buffer 2 (10mM Tris-HCl pH 8.0, 200mM NaCl, 1mM EDTA, 0.5mM EGTA, and protease inhibitors), and rotated at room temperature for 10 minutes, followed by centrifugation (250xg, 5 minutes at room temperature). Supernatant was discarded and nuclei were resuspended/lysed in 1mL cold Lysis Buffer 3 (10mM Tris-HCl, pH 8.0, 100mM NaCl, 1mM EDTA, 0.5mM EGTA, 0.1% Na-Deoxycholate, and protease inhibitors) and transferred to pre-chilled 1mL Covaris AFA tubes (Covaris) Samples were sonicated using a Covaris S220 sonicator (high cell chromatin shearing for 15 minutes; Covaris). Lysates were transferred to tubes and Triton X-100 was added (final 1%) followed by centrifugation (top speed, 10 minutes at 4°C in microcentrifuge). Supernatant was transferred to a new tube; protein concentration was measured by Bradford assay. Antibody-conjugated beads were washed 3 times in blocking buffer, resuspended in 50uL blocking buffer and added to 500ug input protein for overnight incubation with rotation at 4°C. 50ug lysate was aliquoted and stored at −20C for input. On day 2, beads were washed 5 times in 1mL RIPA buffer (50mM HEPES-KOH pH 7.5, 500mM LiCl, 1mM EDTA, 1% NP-40, 0.7% Na-Deoxycholate) with 2-minute incubation at room temperature with rotation for each wash. Beads were washed in 1mL final wash buffer (1xTE, 50mM NaCl) for 2 minutes with rotation at room temperature before final resuspension in 210uL elution buffer (50mM Tris-HCl pH 8.0, 10mM EDTA, 1% SDS). To elute, beads were incubated with agitation at 65C for 30 minutes. 200uL eluate was removed to a fresh tube, and all samples (ChIP and reserved inputs) were reverse-crosslinked overnight at 65C with agitation for a minimum of 12 hours, but not more than 18 hours. 200uL 1xTE was added to reverse crosslinked DNA to dilute SDS, and samples were RNaseA treated (final 0.2mg/mL RNase; 37C for 2 hours) and Proteinase K (final 0.2mg/mL Proteinase K; 55C for 2 hours) before phenol:chloroform extraction and resuspension in 10mM Tris-HCl pH 8.0.
ChIP and input DNA was quantified by Qubit (ThermoFisher) before library preparation using the NEBNext Ultra II DNA library prep kit (NEB). Samples were indexed for multiplex sequencing. Library quality was analyzed by BioAnalyzer (Agilent Genomics) and quantified using qPCR (Kapa Biosystems). Libraries were pooled for multiplex sequencing, requantified, and sequenced on the Illumina NextSeq500 platform (vII; 75bp single end sequencing; Illumina)
ChIP-seq analysis
Following sequencing, fastq files were aligned against the mouse reference genome (mm9) using the STAR aligner (https://github.com/alexdobin/STAR), with default parameters plus options to suppress the matching of spliced reads ‘--outFilterMatchNminOverLread 0.4 --outFilterScoreMinOverLread 0.4’. Duplicate reads were flagged using the MarkDuplicates program from Picard tools. Duplicated and reads matching to more than one genomic location were removed from further analysis using Bamtools (v2.4.1; https://github.com/pezmaster31/bamtools). Replicate ChIP samples were combined using MergeBam program from Picard Tools. Total uniquely mapped reads for merged samples are: ESC Lamin B, 86440728; CM Lamin B, 77143051; ESC H3K9me2, 58181904, CM H3K9me2, 24615479. Four replicates, each, were merged for ESC Lamin B and H3K9me2 analysis, 2 from CCE cells and 2 from vehicle-treated (wild-type) CMV-creERT; Hdac3flox/flox cells. Two replicates were merged for tamoxifen-treated CMV-creERT; Hdac3flox/flox cells (53396419 uniquely mapped reads) and compared to the aforementioned 2 vehicle-treated replicates. DeepTools2 (https://github.com/fidelram/deepTools). bamCoverage and bamCompare tools were used to create coverage tracks (wiggle files) for display on the UCSC genome browser. Spearman rank correlation between individual ChIP and input experiments was performed and visualized using the DeepTools2 multiBamSummary and plotHeatmap tools. Lamin B and H3K9me2 gene occupancy counts were computed by first creating a custom bed file of gene boundaries +/− 5kb using ensembl v67 gene annotations and reads per gene region counted using Rsubread R package. ChIP and input BAM files were down-sampled prior to peak calling using Picard tools DownsampleSam. Lamin B and H3K9me2 domains were called using Enriched Domain Detector (EDD, (Lund et al., 2014) software package (https://github.com/CollasLab/edd) using the parameters ‘-g 10 --bin-size auto --fdr 0.05’. Domains were compared for similarity using the Jaccard statistic as implemented by bedtools suite (v2.26; https://github.com/arq5x/bedtools2/releases) and Jaccard indices heatmaps were created using R. Jaccard indices for replicates of Lamin B ESCs: 0.71 – 0.95; Lamin B CMs: 0.85; H3K9me2 ESCs: 0.82 – 0.94; H3K9me2 CMs: 0.81.
We defined the myocyte signature gene set as myocyte genes that have significantly higher expression in CMs (RPKM > 25) as compared to ESCs (RPKM < 10) based on available RNA-seq data (GSE47948) (441 genes) plus 111 additional genes included in two GO categories: Regulation of Cardiac Muscle Contraction (0055117) and Cardiac Muscle Contraction (0060048). We used publicly available gene sets characteristic of pluripotent ESCs (GEO: GSE36114), E8.5 endoderm (GSE40823), and endothelial cells (GSE52564), and limited the genes in each set to those approximately 550 that were most highly expressed in each cell type.
Immunofluorescence and DNA FISH
C2C12 cells, undifferentiated mouse ESCs, or mouse ESC-derived CMs utilized for IF experiments were grown on glass coverslips, fixed with 4% paraformaldehyde (PFA) for 10 minutes at room temperature (RT), permeabilized with 0.25% Triton X-100 for 10 minutes, blocked in 1% BSA in PBST (8mM Na2HPO4, 150mM NaCl, 2mM KH2PO4, 3mM KCl, 0.05% Tween 20, pH 7.4) and incubated with primary and secondary antibodies for 1 hour each at RT. Samples were counterstained with DAPI solution (Sigma #D9542) for 10 min on RT. All images shown for comparison were taken using a Leica SP8, Leica 3X STED and Zeiss LSM 880 AiryScan confocal microscopes. Primary antibodies used were anti-Lamin B (1:1000, Santa Cruz #sc-6217), anti-Flag M2 (1:400, Sigma #F1804), anti-Hdac3 (1:500, Santa Cruz #sc-17795), anti-H3K9me2 (1:1000, Active Motif #39239), anti-H3K9me2 (1:1000, Abcam #ab1220), anti-H3K9me3 (1:1000, Abcam #ab8898), anti-H3K27me2 (1:1000, Active Motif #39246), anti-H3K27me3 (1:1000, Millipore 07-449), anti-H4K20me2 (1:200, Abcam #ab9052), anti-H4K20me3 (1:1000, Abcam #ab9053), and anti-Troponin T (1:500, Thermo Scientific #MA5-12960). Secondary antibodies used were anti-rabbit AlexaFluor 488 (1:1000, Invitrogen #21206), anti-rabbit AlexaFluor 568 (1:1000, Invitrogen #10042), anti-rabbit AlexaFluor 647 (1:1000, Invitrogen #31573), anti-mouse AlexaFluor 488 (1:1000, Invitrogen #A21202), anti-mouse AlexaFluor 568 (1:1000, Invitrogen #A10037), anti-goat AlexaFluor 488 (1:1000, Invitrogen #A11055), anti-goat AlexaFluor 568 (1:1000, Invitrogen #A11057) and anti-goat AlexaFluor 647 (1:1000, Invitrogen #A21447). To test antibody specificity, blocking peptides were pre-incubated with each antibody: H3K9me2 (Abcam #ab1772), H3K9me3 (Abcam #ab1773) and H3K27me2 (Abcam #ab1781) according to manufacturer’s recommendations.
For DNA FISH cells were grown on gelatin (Millipore #ES-006-B) coated coverslips, fixed with 2% PFA, permeabilized with 0.5% Triton X-100 and stained using standard IF protocols. Next, cells were post fixed with 2% PFA and permeabilized with 0.7% Triton X-100, then hybridized with a labeled DNA probe (Red-dUTP, Abbot Molecular #02N34-040) overnight at 42°C and counterstained with DAPI. DNA FISH probes were labeled using a Nick Translation kit (Abbott Laboratories #32-801300) according to manufacturer’s protocol using BAC DNA clones: Ttn (RP23-310F9), Actc1 (RP23-196J13), Mef2c (RP23-187H18), Calca (RP23-80E16), Kcnq1 (RP23-207G7) and Kcnc2 (RP23-150M6) ordered from ThermoFisher.
Proximity ligation assay
The proximity ligation assay was performed on transfected 293Tx cells. Cells plated on a poly-D-lysine coated coverslip were transfected using FuGENE6 transfection reagent (Promega #E2691) according to manufacturer instructions. Experiments utilized 75 ng of each specified plasmid: Hdac3-Flag, Hdac3-Flag Δ33-70, pCMX-SMRT (provided by Mitchell A. Lazar laboratory, University of Pennsylvania), and pEGFP-Lap2β. Primary antibodies were anti-Flag (Sigma #F1804), anti-GFP (Cell Signaling #2956), and anti-SMRT (mouse ascites provided by Mitchell A. Lazar laboratory). Cells were incubated with antibodies in 3%BSA + 10% goat serum for 90 minutes at room temperature. Staining was performed using the Duolink® In Situ Detection Reagents-Red kit (Sigma-Aldrich) following manufacturer's instructions.
Quantification and statistical analysis
Flow cytometry, qRT-PCR analyses
Data for qRT-PCR and flow cytometry represented as mean ± 1 S.E.M. unless otherwise noted. As indicated in figure legends, for analysis between two samples, a student’s t-test was used, and for analyses among three or more groups, a One-way Analysis of Variance (ANOVA) with a Tukey post-hoc test was used.
Image analysis and quantitation
Confocal 3D images were taken using 0.04 - 0.1 µm step Z-stacks, with a range of 80 to 200 Z-planes per cell. Obtained images were deconvoluted using Huygenes Professional software (Scientific Volume Imaging B.V., The Netherlands). Representative images show a single focal plane. 3D reconstructions were performed using IMARIS 8 software (Bitplane AG, Switzerland). Nuclear lamina, H3K9me2 and Troponin T surfaces were created using the Surfaces tool with automatic settings based on the fluorescent signals from the anti-Lamin B, anti-H3K9me2 and anti-Troponin T antibodies. DNA FISH dots were generated using the Spots tool with a 300nm diameter, created at the intensity mass center of the fluorescent probe signal. Distance from the center of the FISH spot to the edge of the nuclear lamina surface was quantified using the Measurement Points tool. Thickness of the peripheral heterochromatin layer was calculated as the distance from the H3K9me2 surface inner edge to nuclear lamina inner (Lmnb+) edge again using the Measurement Points tool. Average thickness of H3K9me2 in various cell types was quantified by taking 10 random measurements per cell, from 3 independent cells per cell type (total of 30 measurements per cell type). For each experimental condition, 20–30 cells were imaged, and distances from FISH dots to nuclear lamina were quantified. If the distance from FISH dot to nuclear lamina was smaller (or equal) than the thickness of peripheral chromatin in current cell type, then the dot was counted as localized to nuclear periphery. In cases when the FISH signal was imbedded into the nuclear lamina layer, the measurement returned negative distances. P-values were calculated as indicated in the specific figure legends using either a Fisher’s exact test, student’s t-test or One-way Analysis of Variance (ANOVA) with a Tukey post-hoc test.
Data Availability
All ChIP-seq and microarray data are available at NCBI GEO GSE97878.
Supplementary Material
Movie of 3D reconstruction of Titin immuno-FISH in ESCs. Green: Lmnb, Red: Ttn loci, Cyan: H3K9me2.
Movie of 3D reconstruction of Titin immuno-FISH in CMs derived from E12.5 heart. Green: Lmnb, Red: Ttn loci, Cyan: H3K9me2, Yellow: Tnnt2.
Results of microarray analysis of day 8 vehicle- and tamoxifen-treated EB cultures (CMV-creERT; Hdac3f/f); n = 2 samples per condition.
Multiple tables are included in this file: (1) List of genes in LADs and H3K9me2 domains; (2) Genes which either gain or lose localization in LADs and H3K9me2 domains in ESCs and ESC-derived CMs; and (3-6) Gene Ontology analysis of genes that change residence in CM compared to ESC.
This table (1) lists genes included in myocyte signature, pluripotent, endoderm, and endothelial genes; and (2) shows one-way ANOVA analysis with Kruskal-Wallis test of CM genes compared with other lineage genes and 500 random samplings.
Raw intensity values from duplicate peptide array analyses testing specificity of (1) Active Motif 39239 and (2) Abcam ab1220.
Quantitation of the number of cells counted for analyses shown in Figure 3B, Figures 5E–F, Figures 6A–B, and Figures 6D–F. Statistical significance was calculated using a Fisher’s exact test and one-way ANOVA analysis with Kruskal-Wallis test. Also shown on the right side of the table is the quantitation of the scatter plots (number of loci counted) and associated statistical analyses.
This table lists qPCR and qRT-PCR primers used in this study.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-β-actin | Cell Signaling | Cat# 4967 |
| Rabbit anti-CD31 | Abcam | Cat# ab28364 |
| Mouse anti-FLAG M2-Peroxidase (HRP) | Sigma-Aldrich | Cat# A8592 |
| Mouse anti-FLAG M2 | Sigma-Aldrich | Cat# F1804 |
| Rabbit anti-GFP (D5.1) | Cell Signaling | Cat# 2956 |
| Rabbit anti-Hdac3 | Abcam | Cat# ab7030 |
| Mouse anti-Hdac3 (B-12) | Santa Cruz | Cat# sc-17795 |
| Rabbit anti-alpha smooth muscle Actin | Abcam | Cat# ab5694 |
| Mouse anti-H3K9me2 | Abcam | Cat# ab1220 |
| Rabbit anti-H3K9me2 | Active Motif | Cat# 39239 |
| Rabbit anti-H3K9me3 | Abcam | Cat# ab8898 |
| Rabbit anti-H3K27me2 | Active Motif | Cat# 39246 |
| Rabbit anti-H3K27me3 | EMD Millipore | Cat# 07-499 |
| Rabbit anti-phospho-Histone H3 (Ser10) | Cell Signaling | Cat# 9701 |
| Rabbit anti-H4K20me2 | Abcam | Cat# ab9052 |
| Rabbit anti-H4K20me3 | Abcam | Cat# ab9053 |
| Rabbit anti-Lamin B1 | Abcam | Cat# ab16048 |
| Goat anti-Lamin B (M-20) | Santa Cruz | Cat# sc-6217 |
| Mouse anti-Troponin T, cardiac isoform Ab-1 | ThermoFisher | Cat# MS-295-P |
| Mouse anti-Cardiac Troponin T (13–11) | ThermoFisher | Cat# MA5-12960 |
| Mouse anti-SMRT (mouse ascites) | M.A. Lazar lab | N/A |
| normal Rabbit IgG | Cell Signaling | Cat# 2729 |
| normal Mouse IgG | Santa Cruz | Cat# sc-3878 |
| normal Goat IgG | Santa Cruz | Cat# sc-2028 |
| Secondary Antibodies | ||
| Chicken anti-mouse AlexaFluor 488 | Invitrogen | Cat# A21200 |
| Goat anti-rabbit AlexaFluor 647 | Invitrogen | Cat# A21244 |
| Donkey anti-Rabbit AlexaFluor 488 | Invitrogen | Cat# A21206 |
| Donkey anti-Rabbit AlexaFluor 568 | Invitrogen | Cat# A10042 |
| Donkey anti-Rabbit AlexaFluor 647 | Invitrogen | Cat# A31573 |
| Donkey anti-Mouse AlexaFluor 488 | Invitrogen | Cat# A21202 |
| Donkey anti-Mouse AlexaFluor 568 | Invitrogen | Cat# A10037 |
| Donkey anti-Goat AlexaFluor 488 | Invitrogen | Cat# A11055 |
| Donkey anti-Goat AlexaFluor 647 | Invitrogen | Cat# A21447 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Red dUTP | Abbott Laboratories | Cat# 02N34-040 |
| Human H3K9me2 peptide | Abcam | Cat# ab1772 |
| Human H3K9me3 peptide | Abcam | Cat# ab1773 |
| Human H3K27me2 peptide | Abcam | Cat# ab1781 |
| Lamin B1 (B-10) Blocking Peptide | Santa Cruz | Cat# sc-6217P |
| Anti-FLAG magnetic beads | Sigma-Aldrich | Cat# M8823 |
| MEK1 inhibitor PD98059 | Cell Signaling | Cat# 9900 |
| 4-Hydroxytamoxifen | Sigma-Aldrich | Cat# T176 |
| Critical Commercial Assays | ||
| Nick Translation kit | Abbott Laboratories | Cat# 32-801300 |
| Duolink | Sigma-Aldrich | Cat# DUO92101 |
| NEBNext Ultra II DNA Library Kit for Illumina | New England Biolabs | Cat# E7645L |
| NextSeq 500/550 High Output v2 kit (75 cycles) | Illumina | FC-404-2005 |
| GeneChip™ WT Plus Reagent Kit | ThermoFisher | Cat# 902280 |
| GeneChip™ Mouse Gene 2.0 ST Array | ThermoFisher | Cat# 902500 |
| Deposited Data | ||
| All genomics data | This paper | GSE97878 |
| Myocyte signature gene set | This paper | GSE47948 |
| Pluripotent ESC gene expression | This paper | GSE36114 |
| E8.5 endoderm cell gene expression | This paper | GSE40823 |
| Endothelial cell gene expression | This paper | GSE52564 |
| H3K27me3 ChIP-seq dataset | ENCODE | GSM1000089 |
| H3K9me3 ChIP-seq dataset | ENCODE | GSM1000147 |
| Experimental Models: Cell Lines | ||
| Mouse: ESC, CCE | Kyoto University | 129/Sv CCE |
| Mouse: ESC, CMV-creERT; Hdac3f/f | This paper | N/A |
| Mouse: C2C12 | ATCC | Cat# CRL-1772 |
| Human: 293T Lenti-X™ cells | Clontech | Cat# 632180 |
| Experimental Models: Organisms/Strains | ||
| Mus Musculus, Mixed, Islet1Cre/+ | S. Evans lab | Now available: Jackson Labs #024242 |
| Mus Musculus, Mixed, Nkx2-5Cre/+ | R.J. Schwartz lab | Now available: Jackson Labs #030047 |
| Mus Musculus, Mixed Hdac3 flox | M.A. Lazar Lab | Now available: Jackson Labs #024119 |
| Mus Musculus, Mixed CMV-creERT | Jackson Labs | Cat# 004682 |
| Mus Musculus, Mixed NS-DADm | M.A. Lazar lab (You et al., 2013) | N/A |
| Oligonucleotides | ||
| Cre-specific genotyping (5’-TGC CAC GAC CAA GTG ACA GC-3’, 5’-CCA GGT TAC GGA TAT AGT TCA TG-3’) | This paper | N/A |
| floxed Hdac3 allele (5’-GCA GTG GTG GTG AAT GGC TT-3’, 5’-CCT GTG TAA CGG GAG CAG AAC TC-3’) | This paper | N/A |
| qPCR primers, see Table S6 | This paper | N/A |
| qRT-PCR primers, see Table S6 | This paper | N/A |
| shRNA oligonucleotides, see Table S6 | This paper | N/A |
| Recombinant DNA | ||
| BAC Mouse RP23-310F9 | ThermoFisher | Cat# RPCI23.C |
| BAC Mouse RP23-196J13 | ThermoFisher | Cat# RPCI23.C |
| BAC Mouse RP23-80E16 | ThermoFisher | Cat# RPCI23.C |
| BAC Mouse RP23-187H18 | ThermoFisher | Cat# RPCI23.C |
| BAC Mouse RP23-207G7 | ThermoFisher | Cat# RPCI23.C |
| BAC Mouse RP23-150M6 | ThermoFisher | Cat# RPCI23.C |
| pEGFP-Lap2β | Euroscarf.de | Cat# P30463 |
| pCMX-SMRT | M.A. Lazar lab | N/A |
| pcDNA3 -Hdac3-FLAG | P. Gallinari lab | N/A |
| pcDNA3 -Hdac3-Y298H–FLAG | P. Gallinari lab | N/A |
| pFUGW-Hdac3-FLAG | This paper | N/A |
| pFUGW-Hdac3-Y298H–FLAG | This paper | N/A |
| pFUGW-Hdac3-FLAG- Lap2β | This paper | N/A |
| pFUGW-Hdac3-Y298H–FLAG- Lap2β | This paper | N/A |
| pFUGW-Δ33-70 Hdac3-FLAG | This paper | N/A |
| psPAX2 | Addgene | Cat# 12260 |
| pMD2.G | Addgene | Cat# 12259 |
| FUGW-GFP | Addgene | Cat# 14883 |
| Software and Algorithms | ||
| IMARIS 8 | Bitplane | http://www.bitplane.com/releasenotes/imaris800.aspx |
| Huygenes Professional | Scientific Volume Imaging | https://svi.nl/HuygensProfessional |
| GraphPad Prism 7 | GraphPad Software, Inc. | https://www.graphpad.com |
| Affymetrix Command Console and Expression Console | ThermoFisher | https://www.thermofisher.com/us/en/home/life-science/microarray-analysis/microarray-analysis-instruments-software-services/microarray-analysis-software/affymetrix-genechip-command-console-software.html |
| Partek Genomics Suite v6.6 | Partek | http://www.partek.com/pgs |
| Significance Analysis of Microarrays v2.0 | Open Source | https://github.com/MikeJSeo/SAM |
| STAR aligner | Open Source | https://github.com/alexdobin/STAR |
| Picard tools | Open Source | http://broadinstitute.github.io/picard/ |
| Bamtools v2.4.1 | Open Source | https://github.com/pezmaster31/bamtools |
| DeepTools2 | Open Source | https://github.com/fidelram/deepTools |
| Enriched Domain Detector | (Lund et al., 2014) | https://github.com/CollasLab/edd |
| Bedtools suite v2.26 | Open Source | https://github.com/arq5x/bedtools2/releases |
Highlights.
Hdac3 retards cardiac progenitor differentiation independent of deacetylase activity
Chromatin-nuclear lamina interactions regulate gene accessibility during development
Gene accessibility may equate to competence of a stem cell to respond to lineage cues
Nuclear architecture provides a distinct layer of gene regulation to impact cell fate
Acknowledgments
This work was supported by NIH RO1 HL131611, R01 HL071546, the Cotswold Foundation, the WW Smith endowed chair, and the Spain Fund for Cardiovascular Research to J.A.E., NIH K08 HL119553, the WW Smith Foundation, the Burroughs Welcome Career Award for Medical Scientists to R.J., NIH 5 T32 GM-007170 and T32 HL007954-13 to M.G., NIH K99/R00 DK099443 to Z.S. and NIH R37 DK43806 to M.A.L. R.J. and J.A.E. received support from the NSF CMMI-1548571. We thank John Tobias for help with transcriptome analysis. We thank Feiyan Liu, Chinmay Trivedi, Jennifer Cohen, Adam Drake and Esteban Toro for their early work on this project, and Casey Brown, Maya Capelson, Shelley Berger and Laurie Boyer for discussions and comments on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
A.P., P.P.S., M.G., C.L.S, J.A.E., R.J. conceived of the project and designed experiments. A.P., M.G., P.P.S., L.J.M, J.L.I., H.A., D.C., Q.W., and J.E.S. performed experiments. Z.S., E.E.M., N.C.D. and M.A.L. contributed reagents. L.L. performed immunostaining on tissue sections. A.B. and M.M. performed bioinformatics analyses. A.P., P.P.S., M.G., L.J.M., C.L.S., J.A.E., R.J. analyzed data and wrote the manuscript. J.A.E. and R.J. supervised the project. All authors discussed results, reviewed and edited the manuscript.
References
- Bhaskara S, Chyla BJ, Amann JM, Knutson SK, Cortez D, Sun Z-W, Hiebert SW. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Molecular cell. 2008;30:61–72. doi: 10.1016/j.molcel.2008.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black JC, Whetstine JR. Chromatin landscape: methylation beyond transcription. Epigenetics. 2011;6:9–15. doi: 10.4161/epi.6.1.13331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CK, Blanco M, Jackson C, Aznauryan E, Ollikainen N, Surka C, Chow A, Cerase A, McDonel P, Guttman M. Xist recruits the X chromosome to the nuclear lamina to enable chromosome-wide silencing. Science. 2016;354:468–472. doi: 10.1126/science.aae0047. [DOI] [PubMed] [Google Scholar]
- Filion GJ, van Steensel B. Reassessing the abundance of H3K9me2 chromatin domains in embryonic stem cells. Nat Genet. 2010;42:4. doi: 10.1038/ng0110-4. author reply 5–6. [DOI] [PubMed] [Google Scholar]
- Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, Wu CL, Sano S, Muralidharan S, Rius C, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017;355:842–847. doi: 10.1126/science.aag1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Sandoval A, Towbin BD, Kalck V, Cabianca DS, Gaidatzis D, Hauer MH, Geng L, Wang L, Yang T, Wang X, et al. Perinuclear Anchoring of H3K9-Methylated Chromatin Stabilizes Induced Cell Fate in C. elegans Embryos. Cell. 2015;163:1333–1347. doi: 10.1016/j.cell.2015.10.066. [DOI] [PubMed] [Google Scholar]
- Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008;453:948–951. doi: 10.1038/nature06947. [DOI] [PubMed] [Google Scholar]
- Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harr JC, Luperchio TR, Wong X, Cohen E, Wheelan SJ, Reddy KL. Directed targeting of chromatin to the nuclear lamina is mediated by chromatin state and A-type lamins. J Cell Biol. 2015;208:33–52. doi: 10.1083/jcb.201405110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain R, Li D, Gupta M, Manderfield LJ, Ifkovits JL, Wang Q, Liu F, Liu Y, Poleshko A, Padmanabhan A, et al. Integration of Bmp and Wnt signaling by Hopx specifies commitment of cardiomyoblasts. Science. 2015;348:aaa6071. doi: 10.1126/science.aaa6071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kattman SJ, Huber TL, Keller GM. Multipotent flk-1+ cardiovascular progenitor cells give rise to the cardiomyocyte, endothelial, and vascular smooth muscle lineages. Developmental cell. 2006;11:723–732. doi: 10.1016/j.devcel.2006.10.002. [DOI] [PubMed] [Google Scholar]
- Kattman SJ, Witty AD, Gagliardi M, Dubois NC, Niapour M, Hotta A, Ellis J, Keller G. Stage-Specific Optimization of Activin/Nodal and BMP Signaling Promotes Cardiac Differentiation of Mouse and Human Pluripotent Stem Cell Lines. Cell stem cell. 2011;8:228–240. doi: 10.1016/j.stem.2010.12.008. [DOI] [PubMed] [Google Scholar]
- Kim KH, Kim W, Howard TP, Vazquez F, Tsherniak A, Wu JN, Wang W, Haswell JR, Walensky LD, Hahn WC, et al. SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2. Nat Med. 2015;21:1491–1496. doi: 10.1038/nm.3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kind J, Pagie L, de Vries SS, Nahidiazar L, Dey SS, Bienko M, Zhan Y, Lajoie B, de Graaf CA, Amendola M, et al. Genome-wide maps of nuclear lamina interactions in single human cells. Cell. 2015;163:134–147. doi: 10.1016/j.cell.2015.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kind J, Pagie L, Ortabozkoyun H, Boyle S, de Vries SS, Janssen H, Amendola M, Nolen LD, Bickmore WA, van Steensel B. Single-cell dynamics of genome-nuclear lamina interactions. Cell. 2013;153:178–192. doi: 10.1016/j.cell.2013.02.028. [DOI] [PubMed] [Google Scholar]
- Kohwi M, Lupton JR, Lai S-L, Miller MR, Doe CQ. Developmentally regulated subnuclear genome reorganization restricts neural progenitor competence in Drosophila. Cell. 2013;152:97–108. doi: 10.1016/j.cell.2012.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korfali N, Wilkie GS, Swanson SK, Srsen V, de Las Heras J, Batrakou DG, Malik P, Zuleger N, Kerr AR, Florens L, et al. The nuclear envelope proteome differs notably between tissues. Nucleus. 2012;3:552–564. doi: 10.4161/nucl.22257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubben N, Adriaens M, Meuleman W, Voncken JW, van Steensel B, Misteli T. Mapping of lamin A- and progerin-interacting genome regions. Chromosoma. 2012;121:447–464. doi: 10.1007/s00412-012-0376-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahm A, Paolini C, Pallaoro M, Nardi MC, Jones P, Neddermann P, Sambucini S, Bottomley MJ, Lo Surdo P, Carfí A, et al. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:17335–17340. doi: 10.1073/pnas.0706487104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandowski SL, Janardhan HP, Trivedi CM. Histone Deacetylase 3 Coordinates Deacetylase-independent Epigenetic Silencing of Transforming Growth Factor-beta1 (TGF-beta1) to Orchestrate Second Heart Field Development. J Biol Chem. 2015;290:27067–27089. doi: 10.1074/jbc.M115.684753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Wang J, Wang J, Nawaz Z, Liu JM, Qin J, Wong J. Both corepressor proteins SMRT and N-CoR exist in large protein complexes containing HDAC3. EMBO J. 2000;19:4342–4350. doi: 10.1093/emboj/19.16.4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh KM, Chen A, Koh PW, Deng TZ, Sinha R, Tsai JM, Barkal AA, Shen KY, Jain R, Morganti RM, et al. Mapping the Pairwise Choices Leading from Pluripotency to Human Bone, Heart, and Other Mesoderm Cell Types. Cell. 2016;166:451–467. doi: 10.1016/j.cell.2016.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund E, Oldenburg AR, Collas P. Enriched domain detector: a program for detection of wide genomic enrichment domains robust against local variations. Nucleic Acids Res. 2014;42:e92. doi: 10.1093/nar/gku324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattout A, Pike BL, Towbin BD, Bank EM, Gonzalez-Sandoval A, Stadler MB, Meister P, Gruenbaum Y, Gasser SM. An EDMD mutation in C. elegans lamin blocks muscle-specific gene relocation and compromises muscle integrity. Current biology : CB. 2011;21:1603–1614. doi: 10.1016/j.cub.2011.08.030. [DOI] [PubMed] [Google Scholar]
- Meister P, Towbin BD, Pike BL, Ponti A, Gasser SM. The spatial dynamics of tissue-specific promoters during C. elegans development. Genes Dev. 2010;24:766–782. doi: 10.1101/gad.559610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuleman W, Peric-Hupkes D, Kind J, Beaudry JB, Pagie L, Kellis M, Reinders M, Wessels L, van Steensel B. Constitutive nuclear lamina-genome interactions are highly conserved and associated with A/T-rich sequence. Genome Res. 2013;23:270–280. doi: 10.1101/gr.141028.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mewborn SK, Puckelwartz MJ, Abuisneineh F, Fahrenbach JP, Zhang Y, MacLeod H, Dellefave L, Pytel P, Selig S, Labno CM, et al. Altered chromosomal positioning, compaction, and gene expression with a lamin A/C gene mutation. PLoS One. 2010;5:e14342. doi: 10.1371/journal.pone.0014342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery RL, Potthoff MJ, Haberland M, Qi X, Matsuzaki S, Humphries KM, Richardson JA, Bassel-Duby R, Olson EN. Maintenance of cardiac energy metabolism by histone deacetylase 3 in mice. The Journal of clinical investigation. 2008;118:3588–3597. doi: 10.1172/JCI35847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti A, Caron L, Nakano A, Lam JT, Bernshausen A, Chen Y, Qyang Y, Bu L, Sasaki M, Martin-Puig S, et al. Multipotent embryonic isl1+ progenitor cells lead to cardiac, smooth muscle, and endothelial cell diversification. Cell. 2006;127:1151–1165. doi: 10.1016/j.cell.2006.10.029. [DOI] [PubMed] [Google Scholar]
- Osmanagic-Myers S, Dechat T, Foisner R. Lamins at the crossroads of mechanosignaling. Genes Dev. 2015;29:225–237. doi: 10.1101/gad.255968.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegoraro G, Kubben N, Wickert U, Gohler H, Hoffmann K, Misteli T. Ageing-related chromatin defects through loss of the NURD complex. Nat Cell Biol. 2009;11:1261–1267. doi: 10.1038/ncb1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peric-Hupkes D, Meuleman W, Pagie L, Bruggeman SWM, Solovei I, Brugman W, Gräf S, Flicek P, Kerkhoven RM, van Lohuizen M, et al. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Molecular cell. 2010;38:603–613. doi: 10.1016/j.molcel.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poleshko A, Mansfield KM, Burlingame CC, Andrake MD, Shah NR, Katz RA. The human protein PRR14 tethers heterochromatin to the nuclear lamina during interphase and mitotic exit. Cell Rep. 2013;5:292–301. doi: 10.1016/j.celrep.2013.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokocimer M, Davidovich M, Nissim-Rafinia M, Wiesel-Motiuk N, Bar DZ, Barkan R, Meshorer E, Gruenbaum Y. Nuclear lamins: key regulators of nuclear structure and activities. J Cell Mol Med. 2009;13:1059–1085. doi: 10.1111/j.1582-4934.2008.00676.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy KL, Zullo JM, Bertolino E, Singh H. Transcriptional repression mediated by repositioning of genes to the nuclear lamina. Nature. 2008;452:243–247. doi: 10.1038/nature06727. [DOI] [PubMed] [Google Scholar]
- Robson MI, de Las Heras JI, Czapiewski R, Le Thanh P, Booth DG, Kelly DA, Webb S, Kerr AR, Schirmer EC. Tissue-Specific Gene Repositioning by Muscle Nuclear Membrane Proteins Enhances Repression of Critical Developmental Genes during Myogenesis. Mol Cell. 2016;62:834–847. doi: 10.1016/j.molcel.2016.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld JA, Wang Z, Schones DE, Zhao K, DeSalle R, Zhang MQ. Determination of enriched histone modifications in non-genic portions of the human genome. BMC Genomics. 2009;10:143. doi: 10.1186/1471-2164-10-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber KH, Kennedy BK. When lamins go bad: nuclear structure and disease. Cell. 2013;152:1365–1375. doi: 10.1016/j.cell.2013.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah PP, Donahue G, Otte GL, Capell BC, Nelson DM, Cao K, Aggarwala V, Cruickshanks HA, Rai TS, McBryan T, et al. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes & development. 2013;27:1787–1799. doi: 10.1101/gad.223834.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovei I, Wang AS, Thanisch K, Schmidt CS, Krebs S, Zwerger M, Cohen TV, Devys D, Foisner R, Peichl L, et al. LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell. 2013;152:584–598. doi: 10.1016/j.cell.2013.01.009. [DOI] [PubMed] [Google Scholar]
- Somech R, Shaklai S, Geller O, Amariglio N, Simon AJ, Rechavi G, Gal-Yam EN. The nuclear-envelope protein and transcriptional repressor LAP2beta interacts with HDAC3 at the nuclear periphery, and induces histone H4 deacetylation. Journal of cell science. 2005;118:4017–4025. doi: 10.1242/jcs.02521. [DOI] [PubMed] [Google Scholar]
- Stergachis AB, Neph S, Reynolds A, Humbert R, Miller B, Paige SL, Vernot B, Cheng JB, Thurman RE, Sandstrom R, et al. Developmental fate and cellular maturity encoded in human regulatory DNA landscapes. Cell. 2013;154:888–903. doi: 10.1016/j.cell.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Feng D, Fang B, Mullican SE, You S-H, Lim H-W, Everett LJ, Nabel CS, Li Y, Selvakumaran V, et al. Deacetylase-Independent Function of HDAC3 in Transcription and Metabolism Requires Nuclear Receptor Corepressor. Molecular cell. 2013;52:769–782. doi: 10.1016/j.molcel.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Singh N, Mullican SE, Everett LJ, Li L, Yuan L, Liu X, Epstein JA, Lazar MA. Diet-induced lethality due to deletion of the Hdac3 gene in heart and skeletal muscle. The Journal of biological chemistry. 2011;286:33301–33309. doi: 10.1074/jbc.M111.277707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therizols P, Illingworth RS, Courilleau C, Boyle S, Wood AJ, Bickmore WA. Chromatin decondensation is sufficient to alter nuclear organization in embryonic stem cells. Science. 2014;346:1238–1242. doi: 10.1126/science.1259587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towbin BD, González-Aguilera C, Sack R, Gaidatzis D, Kalck V, Meister P, Askjaer P, Gasser SM. Step-wise methylation of histone H3K9 positions heterochromatin at the nuclear periphery. Cell. 2012;150:934–947. doi: 10.1016/j.cell.2012.06.051. [DOI] [PubMed] [Google Scholar]
- Van Bortle K, Corces VG. Spinning the web of cell fate. Cell. 2013;152:1213–1217. doi: 10.1016/j.cell.2013.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B, Delrow J, Henikoff S. Chromatin profiling using targeted DNA adenine methyltransferase. Nat Genet. 2001;27:304–308. doi: 10.1038/85871. [DOI] [PubMed] [Google Scholar]
- Waddington CH. Organisers & genes. Cambridge Eng.: The University Press; 1940. [Google Scholar]
- Wamstad JA, Alexander JM, Truty RM, Shrikumar A, Li F, Eilertson KE, Ding H, Wylie JN, Pico AR, Capra JA, et al. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell. 2012;151:206–220. doi: 10.1016/j.cell.2012.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen B, Wu H, Shinkai Y, Irizarry RA, Feinberg AP. Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nat Genet. 2009;41:246–250. doi: 10.1038/ng.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R, Terry AV, Singh PB, Gilbert DM. Differential subnuclear localization and replication timing of histone H3 lysine 9 methylation states. Mol Biol Cell. 2005;16:2872–2881. doi: 10.1091/mbc.E04-11-0997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SM, Fujiwara Y, Cibulsky SM, Clapham DE, Lien C-L, Schultheiss TM, Orkin SH. Developmental origin of a bipotential myocardial and smooth muscle cell precursor in the mammalian heart. Cell. 2006;127:1137–1150. doi: 10.1016/j.cell.2006.10.028. [DOI] [PubMed] [Google Scholar]
- You S-H, Lim H-W, Sun Z, Broache M, Won K-J, Lazar MA. Nuclear receptor co-repressors are required for the histone-deacetylase activity of HDAC3 in vivo. Nature structural & molecular biology. 2013;3:1–7. doi: 10.1038/nsmb.2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zullo JM, Demarco IA, Piqué-Regi R, Gaffney DJ, Epstein CB, Spooner CJ, Luperchio TR, Bernstein BE, Pritchard JK, Reddy KL, et al. DNA sequence-dependent compartmentalization and silencing of chromatin at the nuclear lamina. Cell. 2012;149:1474–1487. doi: 10.1016/j.cell.2012.04.035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie of 3D reconstruction of Titin immuno-FISH in ESCs. Green: Lmnb, Red: Ttn loci, Cyan: H3K9me2.
Movie of 3D reconstruction of Titin immuno-FISH in CMs derived from E12.5 heart. Green: Lmnb, Red: Ttn loci, Cyan: H3K9me2, Yellow: Tnnt2.
Results of microarray analysis of day 8 vehicle- and tamoxifen-treated EB cultures (CMV-creERT; Hdac3f/f); n = 2 samples per condition.
Multiple tables are included in this file: (1) List of genes in LADs and H3K9me2 domains; (2) Genes which either gain or lose localization in LADs and H3K9me2 domains in ESCs and ESC-derived CMs; and (3-6) Gene Ontology analysis of genes that change residence in CM compared to ESC.
This table (1) lists genes included in myocyte signature, pluripotent, endoderm, and endothelial genes; and (2) shows one-way ANOVA analysis with Kruskal-Wallis test of CM genes compared with other lineage genes and 500 random samplings.
Raw intensity values from duplicate peptide array analyses testing specificity of (1) Active Motif 39239 and (2) Abcam ab1220.
Quantitation of the number of cells counted for analyses shown in Figure 3B, Figures 5E–F, Figures 6A–B, and Figures 6D–F. Statistical significance was calculated using a Fisher’s exact test and one-way ANOVA analysis with Kruskal-Wallis test. Also shown on the right side of the table is the quantitation of the scatter plots (number of loci counted) and associated statistical analyses.
This table lists qPCR and qRT-PCR primers used in this study.
Data Availability Statement
All ChIP-seq and microarray data are available at NCBI GEO GSE97878.