

SUMMARY
Eukaryotic gene transcription is regulated at many steps, including RNA Polymerase II (RNAPII) recruitment, transcription initiation, promoter-proximal RNAPII pause release, and transcription termination; however, mechanisms regulating transcription during productive elongation remain poorly understood. Enhancers, which activate gene transcription, themselves undergo RNAPII-mediated transcription, but our understanding of enhancer transcription and enhancer RNAs (eRNAs) remain incomplete. Here we show that transcription at intragenic enhancers interferes with and attenuates host gene transcription during productive elongation. While the extent of attenuation correlates positively with nascent eRNA expression, the act of intragenic enhancer-transcription alone, but not eRNAs, explains the attenuation. Through CRISPR/Cas9-mediated deletions, we demonstrate a physiological role for intragenic enhancer-mediated transcription attenuation in cell-fate determination. We propose that intragenic enhancers not only enhance transcription of one or more genes from a distance but also fine-tune transcription of their host gene through transcription interference, facilitating differential utilization of the same regulatory element for disparate functions.
eTOC Blurb
Cinghu et al. report an unanticipated role for intragenic enhancers in attenuating host gene expression. They show that transcription at intragenic enhancers interferes with and attenuates host gene transcription during productive elongation. Genetic experiments reveal a physiological role for intragenic enhancer-mediated attenuation in cell-fate choice during embryonic stem cell differentiation.
INTRODUCTION
Eukaryotic gene transcription is regulated at many steps (Adelman and Lis, 2012; Jonkers and Lis, 2015; Porrua and Libri, 2015; Proudfoot, 2016). Emerging evidence points to much of the transcription regulation occurring well after RNA Polymerase II (RNAPII) recruitment, through controlled pause and release of promoter-proximal RNAPII during early elongation (Adelman and Lis, 2012; Jonkers and Lis, 2015; Levine, 2011). After its regulated release into productive elongation, RNAPII is generally assumed to processively progress through the gene, terminate, and eventually reinitiate transcription (Adelman and Lis, 2012). However, even after promoter-proximal pause release, RNAPII must still contend with further roadblocks as it transcribes through the length of the gene body (Li et al., 2007; Teves et al., 2014). Certain DNA sequences are more difficult to transcribe than others. Intrinsic pause sites, even on naked DNA, make RNAPII susceptible to transient pausing, which is exacerbated when RNAPII encounters obstacles such as nucleosomes and R-loops, leading to backtracking of RNAPII and transcriptional arrest (Bintu et al., 2012; Kireeva et al., 2005; Li et al., 2007; Teves et al., 2014). Arrested RNAPII is typically reactivated by the prototypic transcription elongation factor TFIIS (Cheung and Cramer, 2011; Kireeva et al., 2005) or targeted for proteolytic degradation (Proudfoot, 2016).
RNAPII pausing has also been observed to occur at intron-exon junctions and near 3′ cleavage/polyadenylation sites, coinciding with splicing factor recruitment (Kwak et al., 2013; Mayer et al., 2015; Nojima et al., 2015). This transient pause event is thought to provide sufficient time for spliceosome assembly and splicing, which would be consistent with exons having the strongest negative effect on elongation rates, delaying transcription through a gene by ~20–30 seconds per exon (Jonkers et al., 2014; Jonkers and Lis, 2015; Martin et al., 2013). Consequently, elongation rates differ greatly among genes (Danko et al., 2013; Jonkers et al., 2014). Although RNAPII elongation rates correlate with gene expression (Danko et al., 2013), our understanding of mechanisms regulating transcription during productive elongation remains incomplete.
Precise spatiotemporal patterns of gene expression during development is regulated through integrated action of many cis-regulatory elements, which include promoters, enhancers, silencers, and insulators. Among this constellation of elements, enhancers, often located at greater distances from their target promoters, and their associated transcription factors (TFs) play a leading role in the activation of transcription (Spitz and Furlong, 2012). A hallmark of enhancers that has been repeatedly demonstrated is that they are relatively insensitive to distance or position relative to their target genes and can activate transcription independent of their orientation (Calo and Wysocka, 2013; Kim and Shiekhattar, 2015; Li et al., 2016; Shlyueva et al., 2014). Several studies have established that many, if not all, functionally active enhancers themselves undergo RNAPII-mediated transcription and produce short enhancer-derived RNAs (eRNAs) (De Santa et al., 2010; Heinz et al., 2015; Kim et al., 2010; Kim and Shiekhattar, 2015; Li et al., 2016; Natoli and Andrau, 2012; Shlyueva et al., 2014). Distinct from mRNAs, eRNAs are generally unstable, less abundant, and rapidly degraded by exosomes (Pefanis et al., 2015). Changes in eRNA expression highly correlate with changes in nearby gene expression; numerous studies have shown that knockdown of eRNAs accompanies a decrease in the expression of corresponding target genes, with several lines of evidence supporting a functional role for eRNAs in perhaps all stages of gene activation (Kim and Shiekhattar, 2015; Li et al., 2016; Schaukowitch et al., 2014). But whether the act of enhancer transcription per se regulates gene expression remains unclear.
Although about half of all annotated enhancers are intragenic (Andersson et al., 2014; Kowalczyk et al., 2012; Shen et al., 2012; Whyte et al., 2013), most, if not all, studies of enhancer function were conducted by cloning the enhancer of interest, even if it is intragenic, either upstream or downstream of transient/transgenic reporters. Consequently, the effects of intragenic enhancer-transcription on host gene expression has not been addressed. Here we report that functionally active intragenic enhancers present yet another obstacle for RNAPII transcribing the host gene. Our studies show that intragenic enhancers, besides activating genes, also attenuate host gene expression. While the extent of attenuation correlates positively with nascent eRNA expression, the act of intragenic enhancer-transcription alone, but not eRNAs, explains the attenuation. Through CRISPR/Cas9-mediated deletions, we demonstrate a functional role for intragenic enhancer-mediated transcription attenuation in cell-fate determination. Our findings suggest that the intragenic enhancer-mediated transcription attenuation could represent a general mechanism to attenuate and fine-tune host gene transcription during productive elongation.
RESULTS
Intragenic Sites of RNAPII Enrichment
To gain insights into regulation of transcription during productive elongation, we examined genome-wide RNAPII ChIP-Seq data in mouse embryonic stem cells (ESCs) (Brookes et al., 2012). We noted intragenic sites of RNAPII enrichment tens of thousands of base pairs downstream of transcription start sites (TSSs) (Figures 1A and S1A)—within genes with a wide range of expression (Figure S1B)—and hypothesized that they might reflect sites regulating transcription during productive elongation. Towards testing this theory, first, we systematically identified intragenic sites of RNAPII enrichment by calculating RNAPII Pausing Index (PI) (Adelman and Lis, 2012), defined as the ratio of RNAPII density at an intragenic site of interest to the median RNAPII density within the ‘gene body’ (Figure 1B). Using a stringent threshold (PI ≥ 10), we identified 1,928 intragenic RNAPII sites (IRSs), located at least 1Kb away from TSSs and transcription end sites (TESs) of all known and predicted genes, which by definition excludes RNAPII pause sites immediately downstream of promoters and near 3′ cleavage/polyadenylation sites (Table S1). For comparison purposes, we also identified 7,530 promoter-proximal RNAPII sites (PRSs), located within 500bp downstream of TSSs (Table S1 and Figure S1C).
Figure 1. Characterization of intragenic RNAPII sites.

(A) ChIP-Seq profiles of RNAPII occupancy at select genes in mouse ESCs(Brookes et al., 2012).
(B) Schematic describing the calculation used to determine promoter-proximal RNAPII sites (PRSs) and intragenic RNAPII sites (IRSs).
(C) Histogram showing distribution of IRSs within genes.
(D) Heatmap representation of nascent RNA expression, as measured using GROSeq( Jonkers et al., 2014) in untreated mouse ESCs, near IRS-containing genes. Each row represents an IRS-containing gene, with genes aligned at their TSSs. Genes are sorted (top to bottom) by increasing distance between TSS and IRS; The distance (d, Kb) between TSS and IRS is indicated by tick marks. Top and bottom panels, data from sense and antisense strands respectively. About 90% of the IRS-containing genes, with IRSs within 200 Kb of their TSSs, are shown.
(E) Genome browser shots of genes Tet2 and Tcfcp2l1 showing GRO-Seq read density in ESCs treated with Triptolide (Trp) or Flavopiridol (FP) for 12.5, 25, and 50 min(Jonkers et al., 2014) and transcription initiation-associated RNA enrichment in mouse ESCs, as measured using Start-Seq(Williams et al., 2015). IRS loci highlighted in yellow.
(F) Heatmap representation of Start-Seq read density at IRS-containing genes. Data representation is similar to that in (D).
See also Figure S1.
IRSs are distributed across gene bodies (Figure 1C), with a median distance of ~40 Kb from TSSs (Figure S1D). After confirming that the RNAPII enrichment at IRSs is consistently observed across other RNAPII data sets in ESCs (Figures S1E and S1F) (Rahl et al., 2010; Shen et al., 2012; Tippmann et al., 2012), we wondered whether IRSs might represent sites of ‘phantom’ RNAPII ChIP enrichment explainable by cross-linking artifacts resulting from their interaction with one or more RNAPII-enriched promoters (Figure S1G). Our analyses of data from global run-on sequencing (GRO-Seq) (Jonkers et al., 2014), which measures nascent RNA from transcriptionally engaged RNAPII, revealed an enrichment for GRO-Seq reads at IRSs (Figure S1H) and a strong positive correlation between RNAPII and GRO-Seq signals (Figure S1I), indicating that RNAPII enrichment at IRSs is unlikely to be ChIP artifact.
We considered two potential explanations for RNAPII enrichment at IRSs: RNAPII pausing during productive elongation and/or transcription initiation. To discriminate between the two possibilities, we analyzed nascent RNA from ESCs treated with Triptolide (Trp), which blocks transcription initiation altogether, or Flavopiridol (FP), which allows transcription initiation but blocks P-TEFb-dependent promoter-proximal RNAPII pause release (Jonkers et al., 2014). Trp effectively blocks transcription initiation, resulting in the clearing of RNAPII from promoters and gene bodies over time (Figures 1D, 1E, and S1J). FP, in contrast, allows RNAPII accumulation at its recruitment sites but blocks the release of paused RNAPII, thus resulting in the clearing of RNAPII from gene bodies, except for at IRSs (Figures 1D, 1E, and S1K). These data suggested that RNAPII enrichment at IRSs is due to intragenic transcription initiation rather than intrinsic RNAPII pausing during productive synthesis. Indeed, our analysis of data from Start-Seq (Williams et al., 2015), which measures transcription initiation-associated RNA, confirmed bidirectional transcription initiation from IRSs (Figure 1E, F).
Intragenic Sites of RNAPII Enrichment Mark Transcriptionally Active Intragenic Enhancers
To determine whether IRSs represent unannotated gene promoters or enhancers, which themselves undergo RNAPII-mediated transcription (De Santa et al., 2010; Kim et al., 2010; Kim and Shiekhattar, 2015; Li et al., 2016), we examined previously defined stereotypical chromatin signatures that distinguish enhancers from promoters (Calo and Wysocka, 2013; Kim and Shiekhattar, 2015; Li et al., 2016; Shlyueva et al., 2014). Unlike annotated and predicted promoters, ~70% of which overlap CpG islands, only ~7% of IRSs overlap CpG islands, which is more in line with ~5% overlap reported for enhancers (Natoli and Andrau, 2012) (Figure S1L). Furthermore, examination of ~4.4M mouse expressed sequence tags (ESTs) revealed enrichment for ends of ESTs at annotated TSSs but not at IRSs (Figure 2A), suggesting that IRSs are less likely to represent unannotated gene promoters.
Figure 2. Intragenic sites of RNAPII enrichment mark transcriptionally active intragenic enhancers.

(A) Density plot showing average expressed sequence tag (EST) enrichment at TSSs, IRSs, and transcription end sites (TESs).
(B) Genome browser shots of genes Tet2 and Tcfcp2l1 showing ChIP-Seq read density profiles of RNAPII, various transcription regulators and chromatin remodelers, and histone modifications in mouse ESCs. Also shown are read density profiles for DNase I hypersensitivity and gene expression (RNA-Seq). IRS loci highlighted in yellow. *Known chromatin interactions.
(C) Relative levels (RPKM) of H3K4me3, H3K4me1 and H3K27ac at IRSs and PRSs in mouse ESCs.
(D) Relative binding levels (RPKM) of master ESC TFs Oct4, Sox2, Nanog, and Prdm14 and other TFs (Klf4, cMyc, Zfx, and E2f1) at IRSs and PRSs in mouse ESCs.
(E) Relative enrichment of TF sequence recognition motifs within IRSs or matched intragenic control regions in comparison to promoters.
(F) Percentage of IRSs involved in chromatin interaction with ≥1 promoter.
(G) Left: Reporter construct used for testing enhancer activity. Right: Relative luciferase activity of IRSs or control regions in mouse ESCs. Error bars represent SEM of three biological replicates.
See also Figure S1.
Further characterization revealed that IRSs exhibit an accessible chromatin architecture—marked by enhanced DNase I hypersensitivity—accompanied by high levels of histone modifications H3K4me1 and H3K27ac, associated with active enhancers, but not H3K4me3, associated with active promoters (Calo and Wysocka, 2013; Creyghton et al., 2010; Ho et al., 2009; Kim and Shiekhattar, 2015; Li et al., 2016; Shlyueva et al., 2014) (Figures 2B, 2C, S1M, and S1N). Using published ChIP-Seq data for various TFs in ESCs (Chen et al., 2008; Ho et al., 2011; Ma et al., 2011; Marson et al., 2008), we found enhanced binding of Oct4, Sox2, Nanog, Prdm14, and Stat3 (TFs generally known to bind enhancers) at IRSs, but not those that typically bind gene promoters (Klf4, Myc, Zfx, and E2f1) (Figures 2B, 2D, and S1O). Consistent with this observation, frequency of known TF binding motifs within IRSs reveals a significant enrichment for motifs targeted by Oct4, Sox2, Nanog, and Stat3 but not Klf4, Myc, TBP, or CTCF (Figure 2E). Additionally, published chromatin interaction maps in ESCs (Schoenfelder et al., 2015; Zhang et al., 2013) reveal 67% of IRSs interacting with one or more promoters (Figure 2F), with 48% of IRSs interacting with their host gene promoter. Altogether, these observations are consistent with previous criteria used to identify active enhancers. We used reporter experiments to confirm that many IRSs, indeed, can enhance gene expression (Figure 2G). While we cannot rule out that some IRSs could be unannotated (alternative) TSSs of coding or long non-coding RNAs, we conclude that IRSs generally represent transcriptionally active intragenic enhancers.
Transcriptionally Active Intragenic Enhancers Attenuate Host Gene Expression
RNAPII engaged in transcription is extremely stably associated with DNA. This intrinsic stability of RNAPII, although obviously advantageous for efficient transcription of long genes, might actually be deleterious whenever RNAPII encounters obstacles such as another stably engaged RNAPII. Indeed, RNAPII transcribing one strand of a DNA fragment can interfere with a convergent RNAPII transcribing the other strand of the same DNA fragment in vitro (Hobson et al., 2012; Prescott and Proudfoot, 2002; Shearwin et al., 2005). Thus, it is conceivable that transcription at intragenic enhancers may interfere with and attenuate host gene transcription during productive elongation. Additionally, eRNAs from intragenic enhancers may be involved in antisense-mediated regulation of sense mRNA via formation of transient double-stranded RNA and/or RNA interference (RNAi). Consistent with these scenarios, the expression of intragenic enhancer-containing genes is significantly lower than that of extragenic enhancer-associated genes (Figure 3A).
Figure 3. Enhancers in intragenic position attenuate host gene expression.

(A) Expression (Brookes et al., 2012) of genes containing intragenic enhancers (IEs; adefined here; bdefined by Whyte et al (Whyte et al., 2013)), genes closest to (≤10 Kb) extragenic enhancers (EEs) (Whyte et al., 2013) and super enhancers (SEs) (Whyte et al., 2013). **P = 3.46e–5; ***P = 7.63e–8 (Wilcoxon-Mann-Whitney U test; two-sided).
(B) Reporter construct containing two reporter genes lacZ (encoding β-galactosidase) and luciferase, fused with a recombinant fragment containing two exons and a single intron, driven by SV40 promoter. Regions of interest were cloned either within the intron (pIntragenic) or downstream of the reporter gene (pExtragenic).
(C, D) Normalized luciferase activity (C) and mRNA levels of the reporter gene (D) in mouse ESCs from pIntragenic and pExtragenic reporter constructs cloned with intragenic enhancers. **P = 0.00598; ***P = 1.93 × 10−7 (Wilcoxon-Mann-Whitney U test; two-sided). Data points represent mean of n = 5 to 15 biological replicates. mRNA data, normalized to Actin.
See also Figure S2 and S3.
To directly test these possibilities, we used a double-reporter construct encoding β-gal and luciferase proteins within a single reading frame but on separate exons, with an intervening intron sequence containing stop codons (Nasim et al., 2002) (Figure S2A). Candidate IRSs (hereinafter referred to as intragenic enhancers) that conform to many if not all previously defined characteristic features of bona fide enhancers (bind one or more TFs; marked by H3K4me1, H3K27ac, and DNase hypersensitivity; undergo active transcription; interact with one or more promoters; enhance reporter expression) were cloned into the double-reporter, either within the intron (pIntragenic) or downstream of the reporter gene (pExtragenic) (Figure 3B), and were subsequently transfected into mouse ESCs. While the cloned intragenic enhancers increased the reporter expression from both intronic and extragenic positions (Figure 3C)—which was expected given that enhancers, regardless of their location, can activate/enhance transcription from their target gene promoters (Calo and Wysocka, 2013; Kim and Shiekhattar, 2015; Li et al., 2016; Shlyueva et al., 2014)—they generated lower luciferase activity from their intragenic position than from their extragenic position (Figures 3C and S2B). To complement these studies, we asked whether transcriptionally active extragenic enhancers, when cloned as intragenic elements, would also behave similarly. Indeed, we observed a similar phenomenon for established extragenic enhancers for Nanog and Prdm14 genes (Figure S2C), suggesting that the relative reduction in the reporter activity is likely due to enhancer location and not due to any intrinsic property of the enhancer.
RNAPII is known to accumulate at splice junctions and has been implicated in co-transcriptional splicing (Jonkers and Lis, 2015; Mayer et al., 2015; Nojima et al., 2015). Although ~90% of intragenic enhancer peaks are at least 100 bp away from splice junctions, we asked if lower luciferase activity in pIntragenic constructs might be due to splicing defects. Because β-gal and luciferase activities were strongly correlated (Figure S2D–F), which is unlikely with splicing inefficiencies because luciferase expression is dependent on the removal of the intron sequence containing three translation stop codons (Figure S2A), we concluded that defective splicing is unlikely to be the cause for lower luciferase activity.
Next, to answer whether muted luciferase activity is due to reduced mRNA levels, we performed RT-qPCR analysis, using primers spanning the β-gal and luciferase exon junctions, and observed lower mRNA levels with the enhancer in the intragenic position than in the extragenic position (Figures 3D, S3A, and S3B). Together, these data suggest that transcriptionally active enhancers in intragenic position, besides enhancing transcription of the host gene and/or other genes, also somehow attenuate host gene expression.
The Extent of Attenuation Correlates Positively with Nascent eRNA expression
To gain insight into mechanisms underlying the observed intragenic enhancer-mediated attenuation in gene expression, we examined nascent RNA expression. Having already ruled out splicing defects as a possible cause, we considered two plausible mechanisms: (1) transcriptional activity at intragenic enhancers interfering with host gene transcription and/or (2) antisense-mediated silencing that involve eRNAs originating from intragenic enhancers. Either way, we reasoned that the higher the intragenic enhancer RNA (ieRNA) expression, especially from the antisense strand, the higher the attenuation on host gene transcription (from the sense strand). To test this theory, we calculated “attenuation coefficient”, for each intragenic enhancer, as the ratio of normalized mRNA expression of the double-reporter gene with the enhancer cloned in extragenic position (pExtragenic) compared to that with the enhancer cloned in intragenic position (pIntragenic) (Figure 3D). As posited, we observed a strong positive correlation between the nascent ieRNA (antisense) expression, measured from native genomic locus, and attenuation coefficient (Figures 4A and S3C), consistent with a role for intragenic enhancer transcription and/or ieRNAs in the negative regulation of host gene expression.
Figure 4. Transcription at intragenic enhancers attenuates host gene expression.

(A) Correlation between attenuation coefficient, calculated based on mRNA data shown in Fig. S3A (summarized as Fig. 3D), and levels of nascent antisense ieRNA expression, assessed from GRO-Seq data in mouse ESCs(Jonkers et al., 2014).
(B) GRO-Seq profiles of nascent RNA at select genes showing relatively higher levels of GRO-Seq signal upstream than downstream of intragenic enhancers (*). GRO-Seq signal (y-axis) above an arbitrary threshold is truncated (pink streak up-top) to highlight the drop-off in the GRO-Seq signal downstream of the intragenic enhancer.
(C) Top: Schematic showing intragenic regions defined as upstream (up) and downstream (down) of intragenic enhancer. Bottom: Distribution of the ratios of GRO-Seq (Jonkers et al., 2014) read density within the ‘up’ region to that within the corresponding ‘down’ region. Only GRO-Seq reads from the sense strand was used.
(D) Correlation between attenuation coefficient, calculated based on data shown in Fig. 3D, and levels of nascent sense ieRNA expression, assessed from GRO-Seq data(Jonkers et al., 2014).
See also Figure S3.
Transcription at Intragenic Enhancers Interferes with and Attenuates Host Gene Transcription
If the attenuation is due to transcriptional interference (Shearwin et al., 2005), we reasoned that at least some of the elongating RNAPII transcribing the gene likely must have aborted productive transcription and released truncated RNA transcripts. Indeed, examination of nascent RNA expression across the gene length revealed that host gene expression is higher upstream compared to downstream of intragenic enhancers (Figures 4B and 4C). To ensure that the observed decreases in nascent RNA expression downstream of intragenic enhancers are not due to progressive drop-off in processivity as RNAPII traverses through the length of the gene, we repeated this analysis for genes not harboring intragenic enhancers, taking gene center as the reference point. We found that the drop-off in nascent RNA expression is significantly more for genes harboring intragenic enhancers than those do not (P-value = 1.85e–17, Wilcoxon rank-sum test), consistent with premature termination of transcription by RNAPII due to transcriptional interference (Hobson et al., 2012; Prescott and Proudfoot, 2002; Shearwin et al., 2005). We also noted a positive correlation, not as strong, between levels of nascent sense transcripts from intragenic enhancers and attenuation coefficient (Figures 4D and S3D), which is to be expected given that most functional enhancers, like promoters (Core et al., 2008; Preker et al., 2008; Seila et al., 2008), exhibit bidirectional transcription (Figure 1F) (Heinz et al., 2015; Kim and Shiekhattar, 2015; Li et al., 2016). In vitro studies have shown that rear-end collision of RNAPII elongation complexes, transcribing the same DNA strand, results in backtracking (or dislodgement) of trailing (leading, respectively) RNAPII (Saeki and Svejstrup, 2009). Thus, it is conceivable that RNAPII transcribing intragenic enhancers on the sense strand may also interfere with RNAPII transcribing the host gene on the same strand.
To gain insights into the extent of transcription interference, we examined the relationship between the host gene nascent RNA expression and the extent of attenuation (x-axis in Fig. 4C). Our analysis revealed that the higher the host gene nascent RNA expression upstream of the intragenic enhancer, the higher the attenuation (Figure S3E), suggesting that higher rates of transcription initiation from host gene promoter perhaps increases the odds of interference with RNAPIIs transcribing intragenic enhancers. This result, coupled with strong positive correlation between nascent ieRNA expression and attenuation coefficient (Figure 4A), suggests that the extent of transcription interference may range from occasional to frequent depending on many factors including initiation frequencies at the promoter and the enhancer, elongation rates, and processivity.
Role of Intragenic Enhancer RNAs and RNAi machinery in Host Gene Transcription Attenuation
Next, to address whether eRNAs originating from intragenic enhancers might also attenuate host gene expression through antisense-mediated regulation, we took advantage of ESCs deficient in Exosc3, the essential subunit of the RNA exosome complex. Compared to wildtype ESCs, Exosc3-null ESCs exhibit elevated levels of ieRNAs, long non-coding RNAs (lncRNAs), and upstream antisense RNAs, but not mRNAs (Pefanis et al., 2015). Despite the elevated levels of ieRNAs in Exosc3-deficient ESCs (Figure 5A, B), we observed no more attenuation (Figure 5C), suggesting that ieRNAs per se may not be sufficient to explain attenuation. Since RNA exosome-mediated regulation of eRNA levels could occur via two distinct mechanisms, namely via post-transcriptional eRNA degradation or possibly through repression of eRNA synthesis by promoting early transcription termination (Andrulis et al., 2002; Lemay et al., 2014; Pefanis et al., 2015; Preker et al., 2008), to distinguish between the two, we biochemically fractionated chromatin-associated, nucleoplasmic, and cytpoplasmic transcripts in wildtype and Exosc3-null ESCs. Contrary to mRNAs, which are abundant in the cytoplasmic and nucleoplasmic fractions, eRNAs and lncRNAs are abundant in the chromatin-associated and nucleoplasmic fractions (Figure 5D), consistent with their short half-lives due to rapid degradation. Most if not all of the increases in the total eRNA levels in Exosc3-null ESCs is due to increases in the nucleoplasmic and cytoplasmic fractions (Figure 5D, E), suggesting that RNA exosome-mediated regulation of ieRNAs is through post-transcriptional degradation and not through repression of ieRNA synthesis. Consequently, the lack of increased attenuation in Exosc3-deficient ESCs (Figure 5B–C), despite the increase in ieRNA level, suggests ieRNA transcripts are unlikely to be involved in attenuating their cognate host gene expression.
Figure 5. The role of eRNAs from intragenic enhancers in host gene transcription attenuation.

(A) Read density plot showing average expression (RPKM) profile in wildtype (WT) and Exosc3 knockout (KO) mouse ESCs, as measured using RNA-Seq (Pefanis et al., 2015), near transcription start site (TSS), intragenic enhancer (IE), and transcription end site (TES).
(B) RT-qPCR analysis of RNA expression in wildtype and Exosc3 knockdown (KD) mouse ESCs. Data are normalized to Actin. Error bars represent SEM of three biological replicates. ** P < 0.01 (Student’s t-test; two-sided)
(C) Normalized luciferase activity from pIntragenic and pExtragenic reporter constructs, cloned with intragenic enhancers, in control KD and Exosc3 KD mouse ESCs. Data are normalized to control sequence from Ran locus. Error bars represent SEM of three biological replicates.
(D) Proportion of transcripts within chromatin-associated, nucleoplasmic, and cytoplasmic fractions of WT (left) or Exosc3 KO (right) mouse ESCs. Data, normalized to Actin expression in whole cell, represent mean of three biological replicates.
(E) RNA expression in chromatin-associated, nucleoplasmic, or cytoplasmic fractions of WT or Exosc3 KO mouse ESCs. Expression in WT cells (whole cell or various fractions) is set to 1 (black bar). Expression in KO cells (whole cell or various fractions) was calculated relative to that in WT cells (corresponding whole cell or respective fractions). Data are normalized to Actin expression in whole cell. Error bars represent SEM of three biological replicates.
(F) Overlap between genomic sites encoding Ago2-associated RNAs, as determined using CLIP-Seq in mouse ESCs (Leung et al., 2011), and intragenic enhancers.
(G) Normalized luciferase activity from pIntragenic and pExtragenic reporter constructs (Figure 3B), cloned with intragenic enhancers, in wildtype and Dicer1-deficient mouse ESCs. Data are normalized to control sequence from Ran locus. Error bars represent SEM of three biological replicates.
(H) Overlap between intragenic enhancers and Drosha and Dgcr8 binding sites in mouse ESCs (Knuckles et al., 2017).
See also Figure S4.
Next, to determine whether the observed attenuation might involve the endo-siRNA pathway, wherein synthesis of ieRNAs yield siRNAs/miRNAs that can suppress host gene expression posttranscriptionally, we analyzed published Argonaute (Ago2) CLIP-Seq data from ESCs and observed no enrichment for RNAs originating from intragenic enhancers (Figure 5F), suggesting that ieRNAs are not loaded into Ago2-containing RNA-induced silencing complexes. Consistent with this notion, we observed no less attenuation in Dicer knockout ESCs, defective in RNAi, compared to wildtype ESCs (Figure 5G).
Besides their key roles in siRNA/miRNA biogenesis, RNAi factors also promote transcription termination (Skourti-Stathaki et al., 2014) and degradation of nascent transcripts (Knuckles et al., 2017). Antisense transcription, induced by R-loops, may result in the formation of transient double-stranded RNA, which in turn can attract Dicer-Argonaute machinery and promote H3K9me2 deposition and HP1γ recruitment, effectively creating localized patches of heterochromatin (Skourti-Stathaki et al., 2014). The resulting heterochromatin prolongs and reinforces RNAPII pausing at R-loop-associated regions, leading to transcription termination (Skourti-Stathaki et al., 2014). To answer whether premature termination of host gene transcription (Figures 4B and 4C) might involve such a mechanism, we analyzed published datasets and found no evidence of enrichment for R-loops (Chen et al., 2015) or H3K9me2 (Kurimoto et al., 2015) at intragenic enhancers (Figure S4). Nor did we find evidence of enrichment for RNAi factors Drosha and Dgcr8, known to physically associate with chromatin and promote degradation of nascent mRNA (Knuckles et al., 2017), at intragenic enhancers (Figure 5H). Collectively, these data (Figures 4, 5, and S4) suggest that ieRNA transcripts per se are not sufficient to explain transcription interference nor attenuation. Given that nascent ieRNA expression correlates positively with the extent of attenuation (Figure 3A), ruling out ieRNA transcripts leaves the act of transcription at intragenic enhancers itself as a plausible mechanism that can explain the attenuation of host gene expression.
Role for Intragenic Enhancer-Mediated Attenuation in Cell Fate Determination
To determine whether the intragenic enhancer-mediated attenuation has any physiological relevance, we used the CRISPR/Cas9 system to delete candidate intragenic enhancers in ESCs. Removal of intragenic enhancer from highly expressed Tet2 or Ino80 genes, not surprisingly, decreased the host gene expression (Figures S5A and S5B), consistent with its known interaction with host gene promoter (Schoenfelder et al., 2015; Zhang et al., 2013). Because deletion of the enhancer eliminates its attenuation as well as activating functions, we suspected that the loss of any attenuation may have been masked by the presumably dominant enhancer activation function, resulting in net loss in gene expression. In an attempt to uncouple the enhancer and attenuator functions, we focused on candidate intragenic enhancer-containing genes with low-to-moderate expression in ESCs (Figure S1B), with the reasoning that the enhancer’s dominant role at these genes is presumably as an attenuator. As expected, removal of intragenic enhancer from Meis1 or Mapk4, resulted in derepression (~2–3 fold) of respective host genes (Figures 6A and S5C) but not other nearby genes for which the deleted sequence may be an extragenic enhancer (Figures 6D, 6E, and S5D–F).
Figure 6. A role for intragenic enhancer-mediated attenuation in cell-fate determination.

(A) RT-qPCR analysis of mRNA levels of intragenic enhancer-containing genes Meis1 and Mapk4 in corresponding wild-type, enhancer heterozygous (+/−) and enhancer knockout (−/−) mouse ESCs. As a control, an intronic region (showing no signs of an enhancer) from Cdk6 gene was deleted and Cdk6 expression was measured. Data are normalized to Actin. Error bars represent SEM of three biological replicates. *P < 0.01 (Student’s t-test; two-sided).
(B) Morphology and alkaline phosphatase staining of wildtype, Meis1 enhancer (+/−) and Meis1 enhancer (−/−) ESCs.
(C) mRNA levels of pluripotency-associated ESC identity genes (left) and differentiation/developmental genes (right) in wildtype, Meis1 enhancer (+/−) and Meis1 enhancer (−/−) ESCs. Data are normalized to Actin. Error bars represent SEM of three biological replicates. *P < 0.05 (Student’s t-test; two-sided).
(D, E) Top: Chromosomal view of Hi-C interaction maps of Meis1 (D) and Mapk4 (E) loci in mouse ESCs(Dixon et al., 2012). Hi-C maps capture all genome-wide interactions, functional as well as structural. Black/gray arrows represent gene structures and the red vertical bar within Meis1 and Mapk4 genes represent intragenic enhancers. Ovals highlight interaction strengths between the regions containing intragenic enhancer and gene promoter(s). Bottom: mRNA levels of genes within Meis1 (or Mapk4) locus in wildtype or Meis1 (Mapk4, respectively) enhancer (−/−) ESCs. Data are normalized to Actin. Error bars represent SEM of three biological replicates.
(F) Depletion of Meis1 in Meis1 enhancer (−/−) ESCs restores the expression of differentiation genes. Data are normalized to Actin. Error bars represent SEM of three biological replicates. *P < 0.05 (Student’s t-test; two-sided).
(G) mRNA levels of early differentiation markers (top row, mesoderm; middle row, endoderm; bottom row, ectoderm) during embryoid body formation of wildtype, Meis1 enhancer (+/−) and Meis1 enhancer (−/−) ESCs. Data are normalized to Actin. Error bars represent SEM of three biological replicates.
See also Figures S5 and S6.
Notably, ESCs lacking Meis1 intragenic enhancer exhibit severe morphological defects, loss of alkaline phosphatase activity, decreased expression of pluripotency-associated genes, and increased expression of several differentiation-associated genes (Figures 6B, C)—all consistent with ESC differentiation. Silencing Meis1 in ESCs lacking Meis1 intragenic enhancer, at least partially, restored the differentiation phenotype (Figures S5G and 6F), suggesting that Meis1 derepression is the cause for the observed phenotype. Indeed, Meis1 overexpression in ESCs recapitulates the ESC differentiation phenotype observed in ESCs lacking Meis1 intragenic enhancer (Figure S5H–J). Furthermore, subjecting ESCs lacking Meis1 enhancer to undergo embryoid body differentiation revealed their propensity to preferentially differentiate towards mesoderm lineage, as evidenced by a dramatic increase in the levels of mesoderm specification genes (Brachyury, Eomes, and Gsc), presumably at the expense of endoderm and ectoderm lineage markers (Figures 6G and S5K). Our findings, consistent with Meis1’s mesoderm-centric essential roles in definitive hematopoiesis and normal cardiac development (Azcoitia et al., 2005; Mahmoud et al., 2013), highlight, at least in the case of Meis1, a physiological role for intragenic enhancer-mediated attenuation in determining cell-fate choice during early embryonic development.
Enhancer strength and attenuation
To gain insights into why intragenic enhancers within low-to-moderately expressed Meis1 and Mapk4 exhibit strong attenuator activity whereas those within highly expressed Tet2 and Ino80 do not, we grouped intragenic enhancers into four bins based on their host gene expression and found that enhancers within lowly expressed genes have significantly lower TF occupancy and enhancer-associated marks (Figure S6A–D), suggesting that they may be relatively weak enhancers, with presumably limited activation potential. Consistent with this observation, a smaller percentage of enhancers within lowly expressed genes are involved in enhancer-promoter interactions (Figure S6E); even those that interact make fewer and weaker contacts (Figure S6E,F), with a vast majority interacting with host gene promoters. Derepression of Meis1 and Mapk4 upon respective intragenic enhancer deletions suggest that any gains in host gene activation from a weak intragenic enhancer is insufficient to overcome losses from enhancer transcription interfering with the host gene transcription; the enhancer, in effect, serves the role of a repressor. In contrast, robust host gene activation by strong intragenic enhancers, as could have been the case for Tet2 and Ino80 (their intragenic enhancers designated as part of a “super-enhancer”), may overwhelm and mask any attenuation by enhancer transcription, but not without enhancer transcription attenuating some of the host gene transcription. In such cases, the net effect would still be gene activation, with enhancer deletion resulting in net loss in host gene expression.
Functional characterization of intragenic enhancer-containing genes
Characterization of genes containing transcriptionally active intragenic enhancers in ESCs revealed an enrichment for ESC identity genes, including those associated with ESC self-renewal, differentiation and early embryonic development (Figure S7A), suggesting a potential role for intragenic enhancers in fine-tuning the transcription of key cell identity genes. To explore this further, we used data from murine bone marrow-derived macrophages (Ostuni et al., 2013) to annotate putative intragenic enhancers (Table S2). As in ESCs, macrophage enhancers are enriched for H3K4me1, H3K27ac, and sequence motifs bound by macrophage-specific master TFs (PU.1, JunB, Stat1), but not ubiquitously expressed TFs (Myc, Zfx and CTCF) (Figures S7B and S7C). Comparison of intragenic enhancer-associated genes in ESCs and macrophages revealed a minimal overlap (Figure S7D, E). We extended our analyses to other mammalian cells (Calabrese et al., 2012; Ghamari et al., 2013; Tippmann et al., 2012) and found that intragenic enhancers are largely cell type-specific (Figure S7F, Tables S3–5), consistent with gene ontology analysis revealing an enrichment for genes associated with cell type-specific biological processes (Figure S7G).
DISCUSSION
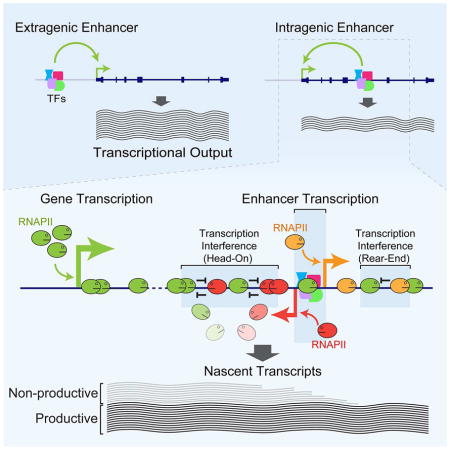
Enhancers are thought to activate or increase transcription from the target gene promoter by facilitating the recruitment of co-activators and other accessory factors to the promoter to potentiate transcription initiation and/or sustain transcription (Calo and Wysocka, 2013; Heinz et al., 2015; Kim and Shiekhattar, 2015; Li et al., 2016; Shlyueva et al., 2014). Recent genomewide studies have established that enhancers themselves recruit RNAPII and undergo RNAPII-mediated transcription to generate short non-coding eRNAs. Several lines of evidence support a functional role for eRNAs in perhaps all stages of gene activation, from facilitating chromatin accessibility, enhancer-promoter loop formation, RNAPII recruitment, promoter-proximal RNAPII pause release to modulating TF-DNA binding (Kim and Shiekhattar, 2015; Li et al., 2016; Schaukowitch et al., 2014). However, whether the act of enhancer transcription, rather than the eRNA transcript itself, has any functional role in regulating target gene expression remains enigmatic. Here we report an unanticipated role for transcriptionally active intragenic enhancers in attenuating host gene expression. Our studies suggest that the act of transcription at intragenic enhancers interferes with and attenuates host gene transcription. Evidence from our analyses of nascent RNA supports premature termination of host gene transcription at or near the intragenic enhancer (Figures 4B and 4C), consistent with enhancer transcription (or other enhancer property) interfering with host gene transcription during productive elongation.
A recent study reported that convergent antisense transcription (CAT), occurring immediately downstream of gene TSSs (~150±50bp), is a characteristic feature of “lower-expressed” genes (with RNAPII density in the gene body used as a proxy for gene expression) and surmised that it may be contributing to gene’s lower expression likely via interference with transcription initiation and/or release of RNAPII from promoter-proximal pausing (Mayer et al., 2015). However, another study investigating CAT within 2 Kb downstream of TSSs observed no correlation between levels of CAT and gene expression (Lavender et al., 2016) and concluded that CAT is not inhibitory. Besides the differences in the cell types, CAT definition, data types and thresholds used for analyses, the discrepancy between these two studies is explainable by the apparent requirement for CAT to traverse past the gene TSS for it to have a negative impact on gene expression (Chen et al., 2016). Given that intragenic enhancer-transcription we studied is located thousands, if not tens of thousands, of base pairs downstream of the host gene promoter (~98% and ~90% of the enhancers are at least 2 and 5 Kb from TSS, respectively) and that enhancer transcription (on the antisense strand) does not seem to extend very far (Figure 1D), it is highly unlikely that enhancer transcription directly interferes with transcription initiation and/or RNAPII pause release at the host gene promoter. It is conceivable, however, that transcription at intragenic enhancers disrupts enhancer-promoter interaction and thus transcription initiation and/or RNAPII pause release at the host gene promoter; but, such a mechanism would have to be exclusive to (or, at least, have a bias toward) intragenic enhancers because the same enhancer has higher activation potential from an extragenic as opposed to an intragenic position (Figure 3). Thus, we infer that any interference by intragenic enhancer transcription (or other enhancer property) on host gene transcription occurs likely during productive elongation, except when the enhancer is near the host gene TSS (within ~200bp), in which case direct interference with transcription initiation and/or RNAPII pause release may also be in play.
While we could not find evidence supporting a role for intragenic enhancer-derived RNAs (ieRNAs) in host gene transcription attenuation, we cannot rule out a role for ieRNAs in antisense-mediated regulation of sense RNA through RNAi or other mechanisms (Faghihi and Wahlestedt, 2009; Pelechano and Steinmetz, 2013; Skourti-Stathaki et al., 2014). While ieRNAs themselves may have additional regulatory functions, our findings suggest that the act of transcription at intragenic enhancers, by itself, can negatively regulate host gene transcription during productive elongation through transcription interference (Figure 7). Although our conclusions are based on candidate intragenic enhancers we investigated, the attenuation function, in theory, can exist for all actively transcribed intragenic enhancers. Further studies are required to investigate, on a genome-wide scale, the net impact of intragenic enhancer-mediated attenuation on host gene expression, which may entail uncoupling the enhancers’ activation and attenuation functions. Nevertheless, our findings suggest that intragenic enhancers serve more regulatory functions than previously appreciated and that intragenic enhancer-mediated transcription attenuation could represent a general mechanism to attenuate and fine-tune host gene transcription, much like a transcriptional rheostat, during productive elongation.
Figure 7. Model for transcription at intragenic enhancers interfering with and attenuating host gene transcription during productive elongation.

RNAPII transcribing intragenic enhancers (shown as red and orange oval faces) interferes with and attenuates the host gene transcription (RNAPII shown as green oval faces) during productive elongation, with the extent of interference ranging from occasional to frequent depending on many factors including initiation frequencies at the promoter and the enhancer, elongation rates, and processivity. Transcription interference may involve one or more events including excessive torsional stress due to positive DNA supercoiling (Chong et al., 2014; Ma et al., 2013), RNAPII stalling/collision, modifications to the chromatin structure/architecture, and antisense-mediated regulation, all eventually leading to premature termination of host gene transcription and non-productive transcripts. The net effect of intragenic enhancer-mediated regulation of host gene expression may range from repression to fine-tuning or activation depending on the difference between gains from enhancer-mediated host gene activation and losses due to enhancer-mediated interference and attenuation of host gene transcription.
While the attenuating function of intragenic enhancer-transcription is perhaps an unintended by-product or unavoidable consequence of enhancer activation, given that enhancers can activate transcription of their target gene promoters independent of their position or location relative to their targets (Calo and Wysocka, 2013; Kim and Shiekhattar, 2015; Li et al., 2016; Shlyueva et al., 2014), evolution could have selected for enhancers to exist outside of protein-coding genes and still have them perform the same function. Our Meis1 studies, which reveal an essential role for intragenic enhancer-mediated attenuation in maintaining ESCs in their pluripotent state, highlight that intragenic sites may have been selected to function as enhancers so that not only can they activate transcription of one or more genes from a distance but also fine-tune transcription of their host gene through transcription interference. Given that the precise level of master TF Oct4 alone can determine three distinct fates of ESCs (Niwa et al., 2000), it is conceivable that refinement of expression of key cell identity genes through intragenic enhancer-mediated attenuation may prevent abrupt changes in their expression levels, which could influence cell fate decisions.
We propose a highly generalizable model wherein transcription at enhancers, weak and strong, interferes with and attenuates host gene expression. The net effect of intragenic enhancer-mediated regulation may range from repression (as in the case of Meis1 and Mapk4) to fine-tuning or activation (as may be the case for Tet2 and Ino80) depending on the difference between gains from enhancer-mediated host gene activation and losses due to enhancer-mediated interference and attenuation of host gene transcription. Under this model, the attenuation function of transcriptionally active intragenic enhancers within highly expressed genes would provide for a fine-tuned enhancement of host gene expression by preventing excessive transcription activation. Such an intrinsic negative feedback mechanism would keep the enhancer and the promoter from activating each other in an exaggerated manner. Intragenic enhancer-mediated attenuation, in addition to providing a potential mechanism to fine-tune transcription of key cell identity genes, may also inhibit expression of genes with inherently “leaky” transcription while keeping them poised for activation. An obvious implication of our finding is that antisense transcription upstream of TSSs (Core et al., 2008; Preker et al., 2008; Seila et al., 2008) may play an active role in attenuating read-through sense transcription, originating from enhancers/genes located immediately upstream, via transcription interference, as previously shown (Nguyen et al., 2014). Such a mechanism would explain why genes with divergent transcription, as a group, have higher expression than those without it (Lavender et al., 2016).
In summary, our studies suggest that transcriptionally active intragenic enhancers not only enhance transcription of one or more genes from a distance but also fine-tune transcription of the host gene through transcription interference, facilitating differential utilization of the same regulatory element for disparate functions.
STAR METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Meis1, rabbit polyclonal | Abcam | Ab124686 |
| Anti-Ran, mouse monoclonal | BD Biosciences | 610341 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 2-Mercaptoethanol | Sigma | M3148 |
| ApaI Enzyme | NEB | R0114S |
| DMEM | Thermo Fisher Scientific | 11965-084 |
| EmbryoMax nucleosides | Millipore | ES-008-D |
| EmeraldAmp GT PCR Master Mix | Takara | RR310A |
| ESGRO Complete PLUS Clonal Grade Medium | Millipore | SF001-500 |
| Fetal Bovine Serum (FBS) | Gemini | 100-125 |
| Gelatin | Sigma | G1890 |
| Inactivated MEFs | Gibco | A24903 |
| LIF | Millipore | ESG1107 |
| Lipofectamine 2000 | Invitrogen | 1168019 |
| Meis1 FlexiTube siRNA | Qiagen | SI01303610 |
| NaCl, 5M | Sigma | S5150 |
| Negative Control siRNA | Qiagen | 1027310 |
| Non-Essential Amino Acids Solution | Thermo Fisher Scientific | 11140050 |
| Protease Inhibitors | Roche | 4693159001 |
| QIAzol Lysis Reagent | Qiagen | 79306 |
| SalI Enzyme | NEB | R3138S |
| Sodium Deoxycholate | Sigma | 30970 |
| SsoFast EvaGreen supermix | Bio-Rad | 1725201 |
| Tris HCL, pH 8.0 | Sigma | T2663 |
| Critical Commercial Assays | ||
| Alkaline Phosphatase Detection Kit | Stemgent | 00-0055 |
| Dual-Light Luciferase & β-Galactosidase Reporter Gene Assay System | Thermo Fisher Scientific | T1003 |
| iScript cDNA Synthesis Kit | Bio-Rad | 1708891 |
| QIAquick Gel Extraction Kit | Qiagen | 28706 |
| Rneasy Mini Kit | Qiagen | 74104 |
| TOPO™ TA Cloning™ Kit | Invitrogen | 450641 |
| Experimental Models: Cell Lines | ||
| Mouse ESCs (E14Tg2a) | ATCC | CRL-1821 |
| Mouse ESCs, Dicer knock-out (KO) | Novus Biologicals | NBP1-96751 |
| Mouse ESCs, Exosc3 knock-out (KO) | Prof. Uttiya Basu | N/A |
| Mouse ESCs, Meis1 intragenic enhancer (+/−) | This paper | N/A |
| Mouse ESCs, Meis1 intragenic enhancer (−/−) | This paper | N/A |
| Mouse ESCs, Mapk4 intragenic enhancer (+/−) | This paper | N/A |
| Mouse ESCs, Mapk4 intragenic enhancer (−/−) | This paper | N/A |
| Mouse ESCs, Cdk6 intronic control region (+/−) | This paper | N/A |
| Mouse ESCs, Cdk6 intronic control region (−/−) | This paper | N/A |
| Mouse ESCs, Tet2 intragenic enhancer (−/−) | This paper | N/A |
| Mouse ESCs, Ino80 intragenic enhancer (−/−) | This paper | N/A |
| Oligonucleotides | ||
| Primers used for PCR amplification of enhancer and control sequences (used in the double-reporter plasmid assay) | This paper | Table S6 |
| Guide RNA sequences used for CRISPR/Cas9-mediated targeted deletions | This paper | Table S7 |
| Screening primers used to test CRISPR/Cas9-mediated targeted deletions | This paper | Table S8 |
| Gene-specific primers used for RT-qPCR analysis | This paper | Table S9 |
| Recombinant DNA | ||
| Plasmid: maxGFP | Lonza | VDF-1012 |
| Plasmid: pExtragenic | This paper | N/A |
| Plasmid: pGL3-Basic | Promega | E1751 |
| Plasmid: pIntragenic | This paper | N/A |
| Plasmid: pRL-TK | Promega | E2231 |
| Plasmid: pTN23 | Prof. I.C. Eperon | N/A |
| Plasmid: pX330-U6-Chimeric_BB-CBh-hSpCas9 | Addgene | 42230 |
| Publicly Available Datasets Used For Analyses | ||
| Mouse ESCs, RNAPII ChIP-Seq | (Brookes et al., 2012) | GEO GSE34520 |
| Mouse ESCs, RNAPII ChIP-Seq | (Rahl et al., 2010) | GEO GSE20485 |
| Mouse ESCs, RNAPII ChIP-Seq | (Shen et al., 2012) | GEO GSE29184 |
| Mouse ESCs, RNAPII ChIP-Seq | (Tippmann et al., 2012) | GEO GSE33252 |
| Mouse ESCs, RNAPII-S2P ChIP-Seq | (Rahl et al., 2010) | GEO GSE20485 |
| Mouse ESCs, RNAPII-S5P ChIP-Seq | (Rahl et al., 2010) | GEO GSE20485 |
| Mouse ESCs, H3K4me1 ChIP-Seq | (Creyghton et al., 2010) | GEO GSE24164 |
| Mouse ESCs, H3K4me3 ChIP-Seq | ENCODE | GEO GSE31039 |
| Mouse ESCs, H3K9me2 ChIP-Seq | (Kurimoto et al., 2015) | GEO GSE60204 |
| Mouse ESCs, H3K27ac ChIP-Seq | ENCODE | GEO GSE31039 |
| Mouse ESCs, H3K27me3 ChIP-Seq | (Ho et al., 2011) | GEO GSE27708 |
| Mouse ESCs, H3K36me3 ChIP-Seq | ENCODE | GEO GSE31039 |
| Mouse ESCs, Brg1 ChIP-Seq | (Ho et al., 2009) | GEO GSE14344 |
| Mouse ESCs, cMyc ChIP-Seq | (Chen et al., 2008) | GEO GSE11431 |
| Mouse ESCs, Dgcr8 ChIP-Seq | (Knuckles et al., 2017) | GEO GSE92257 |
| Mouse ESCs, Drosha ChIP-Seq | (Knuckles et al., 2017) | GEO GSE92257 |
| Mouse ESCs, E2f1 ChIP-Seq | (Chen et al., 2008) | GEO GSE11431 |
| Mouse ESCs, Klf4 ChIP-Seq | (Chen et al., 2008) | GEO GSE11431 |
| Mouse ESCs, Nanog ChIP-Seq | (Marson et al., 2008) | GEO GSE11724 |
| Mouse ESCs, nMyc ChIP-Seq | (Chen et al., 2008) | GEO GSE11431 |
| Mouse ESCs, Oct4 ChIP-Seq | (Marson et al., 2008) | GEO GSE11724 |
| Mouse ESCs, p300 ChIP-Seq | (Creyghton et al., 2010) | GEO GSE24164 |
| Mouse ESCs, Prdm14 ChIP-Seq | (Ma et al., 2011) | GEO GSE42616 |
| Mouse ESCs, Sox2 ChIP-Seq | (Marson et al., 2008) | GEO GSE11724 |
| Mouse ESCs, Stat3 ChIP-Seq | (Ho et al., 2011) | GEO GSE27708 |
| Mouse ESCs, Zfx ChIP-Seq | (Chen et al., 2008) | GEO GSE11431 |
| Mouse ESCs, Dnase-Seq | ENCODE | GEO GSE37074 |
| Mouse ESCs, GRO-Seq | (Jonkers et al., 2014) | GEO GSE48895 |
| Mouse ESCs, Start-Seq | (Williams et al., 2015) | GEO GSE43390 |
| Mouse ESCs, RNA-Seq | (Brookes et al., 2012) | GEO GSE34520 |
| Mouse ESCs, DRIP-RNA-Seq | (Chen et al., 2015) | GEO GSE67581 |
| Mouse ESCs, Hi-C | (Dixon et al., 2012) | GEO GSE35156 |
| Mouse ESCs, Capture Hi-C | (Schoenfelder et al., 2015) | ArrayExpress E-MTAB-2414 |
| Mouse ESCs, RNAPII ChIA-PET | (Zhang et al., 2013) | GEO GSE44067 |
| Mouse ESCs, Ago2 CLIP-Seq | (Leung et al., 2011) | http://rowley.mit.edu/pubs/Ago2_CLIP/Ago2_CLIP.html |
| Mouse Exosc3 WT ESCs, RNA-Seq | (Pefanis et al., 2015) | SRA SRP042355 |
| Mouse Exosc3 KO ESCs, RNA-Seq | (Pefanis et al., 2015) | SRA SRP042355 |
| Mouse BMDM, RNAPII ChIP-Seq | (Ostuni et al., 2013) | GEO GSE38377 |
| Mouse BMDM, JunB ChIP-Seq | (Ostuni et al., 2013) | GEO GSE38377 |
| Mouse BMDM, PU.1 ChIP-Seq | (Ostuni et al., 2013) | GEO GSE38377 |
| Mouse BMDM, Stat6 ChIP-Seq | (Ostuni et al., 2013) | GEO GSE38377 |
| Mouse Terminal Neuron, RNAPII ChIP-Seq | (Tippmann et al., 2012) | GEO GSE33252 |
| Mouse Trophoblast Stem Cell, RNAPII ChIP-Seq | (Calabrese et al., 2012) | GEO GSE39406 |
| Murine Erythroleukemia | (Ghamari et al., 2013) | GEO GSE46849 |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, Dr. Raja Jothi (jothi@nih.gov).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse ES Cell Culture
Mouse ESCs (E14Tg2a, ATCC) were maintained on gelatin-coated plates in the ESGRO complete plus clonal grade medium (Millipore), as previously described (Cinghu et al., 2014; Oldfield et al., 2014). For experiments, ESCs were cultured on gelatin-coated plates in M15 medium: DMEM (Invitrogen) supplemented with 15% FBS, 10μM 2-mercaptoethanol, 0.1 mM nonessential amino acids (Invitrogen), 1× EmbryoMax nucleosides (Millipore), and 1U/ml of ESGRO mLIF (Millipore). Dicer KO mouse ESCs (Novus Biologicals) and Exosc3 KO mouse ESCs, which was a kind gift from U. Basu (University of Columbia) (Pefanis et al., 2015), were grown on inactivated MEFs (Gibco) using the M15 medium. All cells used in the study were routinely tested for mycoplasma contamination.
METHOD DETAILS
Transient Transfection
For transfections, ESCs were cultured in M15 medium and transfected with 50nM siRNA or 100ng plasmid DNA using Lipofectamine 2000 (Invitrogen) as per manufacturer protocol. Genes-pecific siRNAs used: Meis1 (Qiagen) and non-targeting negative control (Qiagen).
Double-Reporter Plasmids and Assay
The double-reporter construct was based upon pTN23 (Nasim et al., 2002) plasmid, which was a kind gift from I.C. Eperon (University of Leicester). The construct encodes β-gal and luciferase proteins within a single reading frame but on separate exons, with an intervening intron sequence containing stop codons. Using site directed mutagenesis, multiple point mutations were introduced in the intronic and extragenic (downstream of the reporter gene) regions to facilitate the cloning of DNA segments of interest (intragenic enhancer, extragenic enhancer, or control sequences). Newly created multiple cloning site (MCS) contains restriction sites for restriction enzymes Sal1, Xma1/Sma1, Apa1 and Cla1. Enhancer and control DNA sequences of interest were PCR amplified using mouse genomic DNA as a template, digested with SalI and ApaI (NEB), and cloned into the intronic or extragenic MCS. See Table S6 for the list of primers used for PCR amplification. Mouse ESCs were transfected with 200ng double-reporter plasmids along with 50ng of maxGFP internal control plasmids using lipofectamine 2000 transfection reagent (Invitrogen). Forty-eight hours after transfection, cells were lysed using the lysis buffer from the Dual-Light Luciferase & β-Galactosidase Reporter Gene Assay System (Thermo Fisher). Firefly luciferase, β-gal and GFP activities were measured using the Synergy 2 Microplate reader (BioTek). Normalized luciferase or β-galactosidase (β-gal) activity was calculated as the ratio of the luminescence signal divided by the maxGFP fluorescence signal. To measure reporter activities in Exosc3-depleted ESCs, the double-reporter plasmids were co-transfected with Exosc3 or non-targeting negative control siRNAs (Qiagen), at 50nM concentration, using lipofectamine 2000 (Invitrogen). Forty-eight hours after transfection, the reporter assay was performed as described above.
CRISPR/Cas9-Mediated Targeted Deletion
Genomic regions of interest (~1100 bp) were deleted in mouse ESCs by co-transfecting two Cas9 containing plasmids (pX330), each carrying a unique guide RNA (gRNA) to direct a double strand break (DSB) at a specific genomic locus. gRNAs were designed with the assistance of the CRISPR Design Tool (crispr.mit.edu) to minimize off-target effects and cloned into the pX330 plasmid, then sequenced verified prior to transfection. See Table S6 for the list of guide RNA sequences. After transfection using lipofectamine 2000 (Invitrogen), ESCs were seeded at low density to allow for selection of individual colonies. Colonies were individually expanded and split for future culture or genomic DNA isolation. 100ng of genomic DNA from these colonies or mouse genomic DNA (control) was screened in a PCR reaction (EmeraldAmp, Takara) with primer pairs annealing to the region outside the DSB sites. See Table S6 for the list of screening primers used to confirm targeted deletions. PCR reactions were separated on 1% agarose gels. ESC DNA gave one of three results: one band migrating the same as control DNA (wildtype/WT), one band migrating faster than control DNA (null), or two bands (heterozygote/het), one corresponding to the wild-type allele and one corresponding to the null allele. Bands were excised from the gel and purified using the QIAquick Kit (Qiagen), cloned using the TOPO TA cloning kit (Invitrogen), and sequenced (Genewiz) to confirm the status of the deletion. Additionally, genotypes were confirmed by screening with a PCR reaction containing a primer pair annealing to the genomic region inside the location of the two DSBs. Wildtype and heterozygotes gave a single product matching mouse genomic DNA, while nulls gave no PCR product. See Tables S7 and S8 for the list of guide RNA sequences and screening primers used.
Quantitative RT-PCR
Quantitative RT-PCR analysis was performed in biological triplicates or quintuplicates. Total RNAs were prepared from cells with the RNeasy Mini kit (Qiagen). Subcellular RNA fractions were prepared as described previously (Bhatt et al 2012). Briefly, cytoplasmic and nucleoplasmic RNA was purified using Qiagen RNeasy columns and chromatin RNA was isolated using QiAzol (Qiagen), followed by further purification with RNeasy columns. All samples were eluted into 100 ul RNAse-free water. cDNAs were generated using the iScript kit (Bio-Rad) according to the manufacturer’s instructions. For each biological replicate, quantitative PCR reactions were performed in technical triplicates using the SsoFast EvaGreen supermix (Bio-Rad) on the Bio-Rad CFX-384 or CFX-96 Real-Time PCR System, and the data normalized to Actin. Data from individual control/WT samples were normalized to 1, and data from individual mutant/KO/KD samples were normalized to the corresponding control/WT sample. Data from biological replicates are plotted as mean +/− S.E.M. See Table S6 for the list of gene-specific primers used. In the case of double-reporter RNA analysis, primers spanning the Lacz-Luc exon-exon junction were used to quantify the reporter expression, which was normalized to maxGFP expression to control for transfection efficiency. See Table S6 for primers spanning the Lacz-Luc exon-exon junction and GFP primers to control for transfection. The attenuation coefficient, for each enhancer, is calculated as the ratio of normalized mRNA expression of the double-reporter gene in pExtragenic compared to that in pIntragenic.
Alkaline Phosphatase (AP) Staining
AP staining was performed using the Alkaline Phosphatase Detection Kit (Stemgent), according to the manufacturer’s instructions.
Embryoid Body Differentiation
Wildtype, Meis1-enhancer heterozygote (+/−) and Meis1-enhancer null (−/−) mouse ESCs were grown on non-adhering plates, using the M15 media without the LIF component, and allowed to form cell aggregates in suspension culture for 10 days. Embryoid bodies (EBs) were collected on days 0, 2, 4, 6, 8, and 10, and RNA was isolated as described below. Quantitative RT-PCR reactions were performed, as described below, to assess the expression of various pluripotency- and differentiation-associated genes.
Western Blot
Cell pellets, lysed in RIPA buffer (25 mM Tris HCl, pH 7.4, 150 mM NaCl, 1% Nonidet P-40, 1% sodium deoxycholate) with protease inhibitors, were sonicated using Bioruptor (Diagenode) for three cycles (30 s on and 50 s off). The lysate was boiled with SDS/PAGE sample buffer, loaded onto a NuPAGE gel, and transferred to 0.22μM PVDF membranes. Each membrane was treated with appropriate primary and secondary antibodies. The membrane was then incubated with a horseradish peroxidase- conjugated secondary antibody and developed with enhanced chemilluminescence PLUS reagent (Amersham). Loading was normalized based on Ran.
Identification of Intragenic Sites of RNAPII Enrichment
Intragenic sites of RNAPII enrichment were determined based on RefSeq gene annotations. “Gene body” is defined as the region beginning 1 Kb downstream of transcription start site (TSS) to transcription end site (TES). For every RefSeq-annotated gene, a 1 Kb sliding window, that slides by 10 bp each time, was used to record the number of RNAPII ChIP-Seq reads mapping to 1 Kb regions within the gene body; “median gene body RNAPII signal” was then calculated as the median of the recorded RNAPII read counts across all such 1 Kb windows within the “gene body”. The 1 Kb sliding window begins its slide from 1 Kb downstream of TSS, with the window centered at 1 Kb downtream of TSS, and ends at TES, with the window centered at TES. For every 1 Kb region within the entire length of the gene, RNAPII Pausing Index (PI) (Adelman and Lis, 2012), defined as the relative ratio of RNAPII density within that 1 Kb region to median “gene body” RNAPII density, is computed. Regions (1Kb in length) with PI ≥ 10 were defined as intragenic sites of RNAPII enrichment, with the mid-points defined as peaks. Those peaks that fall anywhere between TSS and 500 bp downstream of TSS were defined as promoter-proximal RNAPII sites (PRSs), provided that they are not within the “gene body” of any known/predicted genes (UCSC known genes). Those peaks that fall near TES (within D bp upstream of TES, where D = max[1000, min(2000, 5% of the gene length)]) were discarded as cleavage/polyadenylation-related RNAPII accumulation near 3′ end of the genes (Core et al., 2008; Kwak et al., 2013; Nojima et al., 2015). Those peaks that fall anywhere between D bp downstream of TSS and D bp upstream of TES were designated as intragenic RNAPII sites (IRSs), provided that they are at least 1 Kb away from TSSs of all known/predicted genes (UCSC known genes). Peaks that fall within the region between 500 and D bp downstream of TSS were left unclassified and discarded. Consequently, IRSs were defined only for genes that are at least 2 Kb in length. These definitions resulted in 7,530 PRSs and 1,928 IRSs in ESCs.
RNA-Seq, GRO-Seq, Start-Seq, and DRIP-RNA-Seq Data Analysis
Sequence reads from RNA-Seq experiments were aligned to the mouse reference genome (mm9 assembly) and UCSC annotated transcripts using Tophat (Kim et al., 2013) version v2.0.4. Gene expression was calculated using Cufflinks version v2.0.2, and represented as reads/fragments per kilobase of transcript per million mapped reads (RPKM/FPKM). See KEY RESOURCES TABLE for the list of publicly available RNA-Seq datasets used for analysis. For GRO-Seq, Start-Seq, and DRIP-RNA-Seq analysis, mapped reads from previously published spike-in control-normalized GRO-Seq (GSE48895) (Jonkers et al., 2014), Start-Seq (GSE43390) (Williams et al., 2015), and DRIP-RNA-Seq (GSE67581) (Chen et al., 2015) data were used.
ChIP-Seq Data Analysis
Sequence reads generated from ChIP-Seq experiments, by various groups, were processed uniformly by aligning to the mouse reference genome (mm9 assembly) using Bowtie (Langmead et al., 2009) version 0.12.8. See KEY RESOURCES TABLE for the list of publicly available ChIP-Seq datasets used for analysis. Only reads that mapped to unique genomic regions with at most two mismatches were retained for follow up analysis. For visualization and generation of screenshots from the UCSC Genome Browser, each ChIP-Seq data set was first normalized by the total number of reads and then to one million reads. ChIP-Seq read density plots were generated by calculating the number of reads within +/−5 kb upstream and downstream of sites of interest in 100 bp windows, and normalized to reads per base/kilobase per million reads (RPM/RPKM) and plotted as histograms. Data for heatmaps were generated similarly.
Chromatin Interaction Map Analysis
Chromatin interaction between IRSs and promoters were determined based on published Capture Hi-C (E-MTAB-2414) (Schoenfelder et al., 2015) and RNAPII ChIA-PET (GSE44067) (Zhang et al., 2013) data sets in ESCs. An IRS is considered to interact with a promoter if the 1 Kb region centered around the IRS center and 1 Kb region centered around transcription start site (promoter) has been observed to interact in the Capture Hi-C and/or and RNAPII ChIA-PET interaction maps.
Ago2 CLIP-Seq Data Analysis
To assess whether RNA synthesized from intragenic enhancers might be involved in RNAi-mediated gene silencing, we compared intragenic enhancers (IRSs) to the list of published genomic sites encoding Ago2-associated RNAs in ESCs (Leung et al., 2011), as determined using Ago2-CLIP-Seq. RNA from an intragenic enhancer is considered to be loaded into Ago2-containing RNA-induced silencing complexes if one or more genomic sites encoding Ago2-associated RNAs are located within the 1000 nt region of the intragenic enhancer.
Expressed Sequence Tag (EST) Analysis
ESTs are short sub-sequences of cDNA sequences. Mouse ESTs (source: dbEST; URL: http://www.ncbi.nlm.nih.gov/dbEST/) aligned to the mouse reference genome (mm9 assembly), obtained from UCSC Genome Browser, were used for analysis. About 4.37 million mapped strand-specific mouse ESTs were used to generate EST density plots by calculating the number of EST starts within +/−5 kb upstream and downstream of sites of interest (TSS/TES/intragenic enhancer) in 100 bp windows, and normalized to tags per kilobase per million tags and plotted as histograms.
Sequence Analysis
TRAP (Thomas-Chollier et al., 2011) was used to search for known TF motifs, obtained from Jaspar (Portales-Casamar et al., 2010) and TRANSFAC (Qian et al., 2006) databases, within DNA sequences spanning 500 nucleotides around the centers of IRSs or matched intragenic control sequences (defined as the DNA sequences spanning 500 nucleotides around the gene mid-point). Statistical significance for enrichment of sequence motifs within IRSs or intragenic control regions were calculated in reference to sequences from promoter regions. Benjamini-Hochberg method was used for multiple-testing correction. CpG island annotations were downloaded from the UCSC genome browser.
Functional Enrichment Analysis
Intragenic enhancer-containing genes in ESCs were analyzed in relation to the rank-ordered list of all mouse genes (Cinghu et al., 2014) that are likely to be associated with ESC maintenance vs. ESC differentiation.
QUANTIFICATION AND STATISTICAL ANALYSIS
See Methods Details for details of quantification and statistical analysis.
Supplementary Material
List of IRSs and PRSs in mouse ESCs (see enclosed Excel table).
List of IRSs and PRSs in mouse bone marrow-derived macrophages (see enclosed Excel table).
List of IRSs and PRSs in mouse terminal neurons (see enclosed Excel table).
List of IRSs and PRSs in mouse trophoblast stem cells (see enclosed Excel table).
List of IRSs and PRSs in mouse murine erythroleukemia cells (see enclosed Excel table).
Primers used for PCR amplification of enhancer and control sequences, CRISPR/Cas9-mediated targeted deletion experiments, and RT-qPCR analysis.
HIGHLIGHTS.
Intragenic enhancers, besides activating genes, also attenuate host gene expression
Transcription at intragenic enhancers interferes with host gene transcription
The act of enhancer transcription alone, but not the eRNA, explains the attenuation
Intragenic enhancer-mediated attenuation determines cell fate choice
Acknowledgments
We thank T.K. Archer, T. Henriques, L. Ho, G. Hu, D. Fargo, T.A. Kunkel, S. Peddada, P.A. Wade for useful discussions and comments on the manuscript. This work was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences (R.J.: 1ZIAES102625).
Footnotes
AUTHOR CONTRIBUTIONS
R.J. conceived the study. S.C., P.Y., and R.J. designed the study. S.C. performed all the experiments. P.Y. performed all the bioinformatics analyses. J.P.K. generated CRISPR clones. D.K. contributed to bioinformatics analyses. A.E.C. contributed to cloning. S.C., P.Y., J.P.K., D.K., A.E.C., A.J.O., K.A., and R.J. analyzed the data. R.J. wrote the manuscript with input from all authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adelman K, Lis JT. Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat Rev Genet. 2012;13:720–731. doi: 10.1038/nrg3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, Chen Y, Zhao X, Schmidl C, Suzuki T, et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507:455–461. doi: 10.1038/nature12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrulis ED, Werner J, Nazarian A, Erdjument-Bromage H, Tempst P, Lis JT. The RNA processing exosome is linked to elongating RNA polymerase II in Drosophila. Nature. 2002;420:837–841. doi: 10.1038/nature01181. [DOI] [PubMed] [Google Scholar]
- Azcoitia V, Aracil M, Martinez AC, Torres M. The homeodomain protein Meis1 is essential for definitive hematopoiesis and vascular patterning in the mouse embryo. Dev Biol. 2005;280:307–320. doi: 10.1016/j.ydbio.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Bintu L, Ishibashi T, Dangkulwanich M, Wu YY, Lubkowska L, Kashlev M, Bustamante C. Nucleosomal elements that control the topography of the barrier to transcription. Cell. 2012;151:738–749. doi: 10.1016/j.cell.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes E, de Santiago I, Hebenstreit D, Morris KJ, Carroll T, Xie SQ, Stock JK, Heidemann M, Eick D, Nozaki N, et al. Polycomb associates genome-wide with a specific RNA polymerase II variant, and regulates metabolic genes in ESCs. Cell Stem Cell. 2012;10:157–170. doi: 10.1016/j.stem.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese JM, Sun W, Song L, Mugford JW, Williams L, Yee D, Starmer J, Mieczkowski P, Crawford GE, Magnuson T. Site-specific silencing of regulatory elements as a mechanism of X inactivation. Cell. 2012;151:951–963. doi: 10.1016/j.cell.2012.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo E, Wysocka J. Modification of enhancer chromatin: what, how, and why? Mol Cell. 2013;49:825–837. doi: 10.1016/j.molcel.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PB, Chen HV, Acharya D, Rando OJ, Fazzio TG. R loops regulate promoter-proximal chromatin architecture and cellular differentiation. Nat Struct Mol Biol. 2015;22:999–1007. doi: 10.1038/nsmb.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- Chen Y, Pai AA, Herudek J, Lubas M, Meola N, Jarvelin AI, Andersson R, Pelechano V, Steinmetz LM, Jensen TH, et al. Principles for RNA metabolism and alternative transcription initiation within closely spaced promoters. Nat Genet. 2016;48:984–994. doi: 10.1038/ng.3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung AC, Cramer P. Structural basis of RNA polymerase II backtracking, arrest and reactivation. Nature. 2011;471:249–253. doi: 10.1038/nature09785. [DOI] [PubMed] [Google Scholar]
- Chong S, Chen C, Ge H, Xie XS. Mechanism of transcriptional bursting in bacteria. Cell. 2014;158:314–326. doi: 10.1016/j.cell.2014.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinghu S, Yellaboina S, Freudenberg JM, Ghosh S, Zheng X, Oldfield AJ, Lackford BL, Zaykin DV, Hu G, Jothi R. Integrative framework for identification of key cell identity genes uncovers determinants of ES cell identity and homeostasis. Proc Natl Acad Sci U S A. 2014;111:E1581–1590. doi: 10.1073/pnas.1318598111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Waterfall JJ, Lis JT. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008;322:1845–1848. doi: 10.1126/science.1162228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danko CG, Hah N, Luo X, Martins AL, Core L, Lis JT, Siepel A, Kraus WL. Signaling pathways differentially affect RNA polymerase II initiation, pausing, and elongation rate in cells. Mol Cell. 2013;50:212–222. doi: 10.1016/j.molcel.2013.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Santa F, Barozzi I, Mietton F, Ghisletti S, Polletti S, Tusi BK, Muller H, Ragoussis J, Wei CL, Natoli G. A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol. 2010;8:e1000384. doi: 10.1371/journal.pbio.1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faghihi MA, Wahlestedt C. Regulatory roles of natural antisense transcripts. Nat Rev Mol Cell Biol. 2009;10:637–643. doi: 10.1038/nrm2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghamari A, van de Corput MP, Thongjuea S, van Cappellen WA, van Ijcken W, van Haren J, Soler E, Eick D, Lenhard B, Grosveld FG. In vivo live imaging of RNA polymerase II transcription factories in primary cells. Genes Dev. 2013;27:767–777. doi: 10.1101/gad.216200.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Romanoski CE, Benner C, Glass CK. The selection and function of cell type-specific enhancers. Nat Rev Mol Cell Biol. 2015;16:144–154. doi: 10.1038/nrm3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho L, Jothi R, Ronan JL, Cui K, Zhao K, Crabtree GR. An embryonic stem cell chromatin remodeling complex, esBAF, is an essential component of the core pluripotency transcriptional network. Proc Natl Acad Sci U S A. 2009;106:5187–5191. doi: 10.1073/pnas.0812888106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho L, Miller EL, Ronan JL, Ho WQ, Jothi R, Crabtree GR. esBAF facilitates pluripotency by conditioning the genome for LIF/STAT3 signalling and by regulating polycomb function. Nat Cell Biol. 2011;13:903–913. doi: 10.1038/ncb2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobson DJ, Wei W, Steinmetz LM, Svejstrup JQ. RNA polymerase II collision interrupts convergent transcription. Mol Cell. 2012;48:365–374. doi: 10.1016/j.molcel.2012.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers I, Kwak H, Lis JT. Genome-wide dynamics of Pol II elongation and its interplay with promoter proximal pausing, chromatin, and exons. Elife. 2014;3:e02407. doi: 10.7554/eLife.02407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers I, Lis JT. Getting up to speed with transcription elongation by RNA polymerase II. Nat Rev Mol Cell Biol. 2015;16:167–177. doi: 10.1038/nrm3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–187. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TK, Shiekhattar R. Architectural and Functional Commonalities between Enhancers and Promoters. Cell. 2015;162:948–959. doi: 10.1016/j.cell.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kireeva ML, Hancock B, Cremona GH, Walter W, Studitsky VM, Kashlev M. Nature of the nucleosomal barrier to RNA polymerase II. Mol Cell. 2005;18:97–108. doi: 10.1016/j.molcel.2005.02.027. [DOI] [PubMed] [Google Scholar]
- Knuckles P, Carl SH, Musheev M, Niehrs C, Wenger A, Buhler M. RNA fate determination through cotranscriptional adenosine methylation and microprocessor binding. Nat Struct Mol Biol. 2017 doi: 10.1038/nsmb.3419. [DOI] [PubMed] [Google Scholar]
- Kowalczyk MS, Hughes JR, Garrick D, Lynch MD, Sharpe JA, Sloane-Stanley JA, McGowan SJ, De Gobbi M, Hosseini M, Vernimmen D, et al. Intragenic enhancers act as alternative promoters. Mol Cell. 2012;45:447–458. doi: 10.1016/j.molcel.2011.12.021. [DOI] [PubMed] [Google Scholar]
- Kurimoto K, Yabuta Y, Hayashi K, Ohta H, Kiyonari H, Mitani T, Moritoki Y, Kohri K, Kimura H, Yamamoto T, et al. Quantitative Dynamics of Chromatin Remodeling during Germ Cell Specification from Mouse Embryonic Stem Cells. Cell Stem Cell. 2015;16:517–532. doi: 10.1016/j.stem.2015.03.002. [DOI] [PubMed] [Google Scholar]
- Kwak H, Fuda NJ, Core LJ, Lis JT. Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science. 2013;339:950–953. doi: 10.1126/science.1229386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavender CA, Cannady KR, Hoffman JA, Trotter KW, Gilchrist DA, Bennett BD, Burkholder AB, Burd CJ, Fargo DC, Archer TK. Downstream Antisense Transcription Predicts Genomic Features That Define the Specific Chromatin Environment at Mammalian Promoters. PLoS Genet. 2016;12:e1006224. doi: 10.1371/journal.pgen.1006224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemay JF, Larochelle M, Marguerat S, Atkinson S, Bahler J, Bachand F. The RNA exosome promotes transcription termination of backtracked RNA polymerase II. Nat Struct Mol Biol. 2014;21:919–926. doi: 10.1038/nsmb.2893. [DOI] [PubMed] [Google Scholar]
- Leung AK, Young AG, Bhutkar A, Zheng GX, Bosson AD, Nielsen CB, Sharp PA. Genome-wide identification of Ago2 binding sites from mouse embryonic stem cells with and without mature microRNAs. Nat Struct Mol Biol. 2011;18:237–244. doi: 10.1038/nsmb.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine M. Paused RNA polymerase II as a developmental checkpoint. Cell. 2011;145:502–511. doi: 10.1016/j.cell.2011.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- Li W, Notani D, Rosenfeld MG. Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat Rev Genet. 2016;17:207–223. doi: 10.1038/nrg.2016.4. [DOI] [PubMed] [Google Scholar]
- Ma J, Bai L, Wang MD. Transcription under torsion. Science. 2013;340:1580–1583. doi: 10.1126/science.1235441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, Swigut T, Valouev A, Rada-Iglesias A, Wysocka J. Sequence-specific regulator Prdm14 safeguards mouse ESCs from entering extraembryonic endoderm fates. Nat Struct Mol Biol. 2011;18:120–127. doi: 10.1038/nsmb.2000. [DOI] [PubMed] [Google Scholar]
- Mahmoud AI, Kocabas F, Muralidhar SA, Kimura W, Koura AS, Thet S, Porrello ER, Sadek HA. Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature. 2013;497:249–253. doi: 10.1038/nature12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marson A, Levine SS, Cole MF, Frampton GM, Brambrink T, Johnstone S, Guenther MG, Johnston WK, Wernig M, Newman J, et al. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell. 2008;134:521–533. doi: 10.1016/j.cell.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin RM, Rino J, Carvalho C, Kirchhausen T, Carmo-Fonseca M. Live-cell visualization of pre-mRNA splicing with single-molecule sensitivity. Cell Rep. 2013;4:1144–1155. doi: 10.1016/j.celrep.2013.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer A, di Iulio J, Maleri S, Eser U, Vierstra J, Reynolds A, Sandstrom R, Stamatoyannopoulos JA, Churchman LS. Native elongating transcript sequencing reveals human transcriptional activity at nucleotide resolution. Cell. 2015;161:541–554. doi: 10.1016/j.cell.2015.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasim MT, Chowdhury HM, Eperon IC. A double reporter assay for detecting changes in the ratio of spliced and unspliced mRNA in mammalian cells. Nucleic Acids Res. 2002;30:e109. doi: 10.1093/nar/gnf108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natoli G, Andrau JC. Noncoding transcription at enhancers: general principles and functional models. Annu Rev Genet. 2012;46:1–19. doi: 10.1146/annurev-genet-110711-155459. [DOI] [PubMed] [Google Scholar]
- Nguyen T, Fischl H, Howe FS, Woloszczuk R, Serra Barros A, Xu Z, Brown D, Murray SC, Haenni S, Halstead JM, et al. Transcription mediated insulation and interference direct gene cluster expression switches. Elife. 2014;3:e03635. doi: 10.7554/eLife.03635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa H, Miyazaki J, Smith AG. Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat Genet. 2000;24:372–376. doi: 10.1038/74199. [DOI] [PubMed] [Google Scholar]
- Nojima T, Gomes T, Grosso AR, Kimura H, Dye MJ, Dhir S, Carmo-Fonseca M, Proudfoot NJ. Mammalian NET-Seq Reveals Genome-wide Nascent Transcription Coupled to RNA Processing. Cell. 2015;161:526–540. doi: 10.1016/j.cell.2015.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldfield AJ, Yang P, Conway AE, Cinghu S, Freudenberg JM, Yellaboina S, Jothi R. Histone-fold domain protein NF-Y promotes chromatin accessibility for cell type-specific master transcription factors. Mol Cell. 2014;55:708–722. doi: 10.1016/j.molcel.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostuni R, Piccolo V, Barozzi I, Polletti S, Termanini A, Bonifacio S, Curina A, Prosperini E, Ghisletti S, Natoli G. Latent enhancers activated by stimulation in differentiated cells. Cell. 2013;152:157–171. doi: 10.1016/j.cell.2012.12.018. [DOI] [PubMed] [Google Scholar]
- Pefanis E, Wang J, Rothschild G, Lim J, Kazadi D, Sun J, Federation A, Chao J, Elliott O, Liu ZP, et al. RNA exosome-regulated long non-coding RNA transcription controls super-enhancer activity. Cell. 2015;161:774–789. doi: 10.1016/j.cell.2015.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelechano V, Steinmetz LM. Gene regulation by antisense transcription. Nat Rev Genet. 2013;14:880–893. doi: 10.1038/nrg3594. [DOI] [PubMed] [Google Scholar]
- Porrua O, Libri D. Transcription termination and the control of the transcriptome: why, where and how to stop. Nat Rev Mol Cell Biol. 2015;16:190–202. doi: 10.1038/nrm3943. [DOI] [PubMed] [Google Scholar]
- Portales-Casamar E, Thongjuea S, Kwon AT, Arenillas D, Zhao X, Valen E, Yusuf D, Lenhard B, Wasserman WW, Sandelin A. JASPAR 2010: the greatly expanded open-access database of transcription factor binding profiles. Nucleic Acids Res. 2010;38:D105–110. doi: 10.1093/nar/gkp950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preker P, Nielsen J, Kammler S, Lykke-Andersen S, Christensen MS, Mapendano CK, Schierup MH, Jensen TH. RNA exosome depletion reveals transcription upstream of active human promoters. Science. 2008;322:1851–1854. doi: 10.1126/science.1164096. [DOI] [PubMed] [Google Scholar]
- Prescott EM, Proudfoot NJ. Transcriptional collision between convergent genes in budding yeast. Proc Natl Acad Sci U S A. 2002;99:8796–8801. doi: 10.1073/pnas.132270899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proudfoot NJ. Transcriptional termination in mammals: Stopping the RNA polymerase II juggernaut. Science. 2016;352:aad9926. doi: 10.1126/science.aad9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Z, Cai YD, Li Y. Automatic transcription factor classifier based on functional domain composition. Biochemical and biophysical research communications. 2006;347:141–144. doi: 10.1016/j.bbrc.2006.06.060. [DOI] [PubMed] [Google Scholar]
- Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA, Young RA. c-Myc regulates transcriptional pause release. Cell. 2010;141:432–445. doi: 10.1016/j.cell.2010.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeki H, Svejstrup JQ. Stability, flexibility, and dynamic interactions of colliding RNA polymerase II elongation complexes. Mol Cell. 2009;35:191–205. doi: 10.1016/j.molcel.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaukowitch K, Joo JY, Liu X, Watts JK, Martinez C, Kim TK. Enhancer RNA facilitates NELF release from immediate early genes. Mol Cell. 2014;56:29–42. doi: 10.1016/j.molcel.2014.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenfelder S, Furlan-Magaril M, Mifsud B, Tavares-Cadete F, Sugar R, Javierre BM, Nagano T, Katsman Y, Sakthidevi M, Wingett SW, et al. The pluripotent regulatory circuitry connecting promoters to their long-range interacting elements. Genome Res. 2015;25:582–597. doi: 10.1101/gr.185272.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seila AC, Calabrese JM, Levine SS, Yeo GW, Rahl PB, Flynn RA, Young RA, Sharp PA. Divergent transcription from active promoters. Science. 2008;322:1849–1851. doi: 10.1126/science.1162253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearwin KE, Callen BP, Egan JB. Transcriptional interference--a crash course. Trends Genet. 2005;21:339–345. doi: 10.1016/j.tig.2005.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, et al. A map of the cis-regulatory sequences in the mouse genome. Nature. 2012;488:116–120. doi: 10.1038/nature11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlyueva D, Stampfel G, Stark A. Transcriptional enhancers: from properties to genome-wide predictions. Nat Rev Genet. 2014;15:272–286. doi: 10.1038/nrg3682. [DOI] [PubMed] [Google Scholar]
- Skourti-Stathaki K, Kamieniarz-Gdula K, Proudfoot NJ. R-loops induce repressive chromatin marks over mammalian gene terminators. Nature. 2014;516:436–439. doi: 10.1038/nature13787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz F, Furlong EE. Transcription factors: from enhancer binding to developmental control. Nat Rev Genet. 2012;13:613–626. doi: 10.1038/nrg3207. [DOI] [PubMed] [Google Scholar]
- Teves SS, Weber CM, Henikoff S. Transcribing through the nucleosome. Trends Biochem Sci. 2014;39:577–586. doi: 10.1016/j.tibs.2014.10.004. [DOI] [PubMed] [Google Scholar]
- Thomas-Chollier M, Hufton A, Heinig M, O’Keeffe S, Masri NE, Roider HG, Manke T, Vingron M. Transcription factor binding predictions using TRAP for the analysis of ChIP-seq data and regulatory SNPs. Nature protocols. 2011;6:1860–1869. doi: 10.1038/nprot.2011.409. [DOI] [PubMed] [Google Scholar]
- Tippmann SC, Ivanek R, Gaidatzis D, Scholer A, Hoerner L, van Nimwegen E, Stadler PF, Stadler MB, Schubeler D. Chromatin measurements reveal contributions of synthesis and decay to steady-state mRNA levels. Mol Syst Biol. 2012;8:593. doi: 10.1038/msb.2012.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master transcription factors and mediator establish superenhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams LH, Fromm G, Gokey NG, Henriques T, Muse GW, Burkholder A, Fargo DC, Hu G, Adelman K. Pausing of RNA Polymerase II Regulates Mammalian Developmental Potential through Control of Signaling Networks. Mol Cell. 2015;58:311–322. doi: 10.1016/j.molcel.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wong CH, Birnbaum RY, Li G, Favaro R, Ngan CY, Lim J, Tai E, Poh HM, Wong E, et al. Chromatin connectivity maps reveal dynamic promoterenhancer long-range associations. Nature. 2013;504:306–310. doi: 10.1038/nature12716. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of IRSs and PRSs in mouse ESCs (see enclosed Excel table).
List of IRSs and PRSs in mouse bone marrow-derived macrophages (see enclosed Excel table).
List of IRSs and PRSs in mouse terminal neurons (see enclosed Excel table).
List of IRSs and PRSs in mouse trophoblast stem cells (see enclosed Excel table).
List of IRSs and PRSs in mouse murine erythroleukemia cells (see enclosed Excel table).
Primers used for PCR amplification of enhancer and control sequences, CRISPR/Cas9-mediated targeted deletion experiments, and RT-qPCR analysis.