

Summary
Noncoding RNAs (ncRNAs) regulate gene expression in all organisms. Bacterial 6S RNAs globally regulate transcription by binding RNA polymerase (RNAP) holoenzyme and competing with promoter DNA. Escherichia coli (Eco) 6S RNA interacts specifically with the housekeeping σ70-holoenzyme (Eσ70) and plays a key role in the transcriptional reprogramming upon shifts between exponential and stationary phase. Inhibition is relieved upon 6S RNA-templated RNA synthesis. We report here the 3.8 Å resolution structure of a complex between 6S RNA and Eσ70 determined by single-particle cryo-electron microscopy and validation of the structure using footprinting and crosslinking approaches. Duplex RNA segments have A-form C3′-endo sugar puckers but widened major groove widths, giving the RNA an overall architecture that mimics B-form promoter DNA. Our results help explain the specificity of Eco 6S RNA for Eσ70, and show how an ncRNA can mimic B-form DNA to directly regulate transcription by the DNA-dependent RNAP.
Keywords: 6S RNA, Cryo-electron microscopy, non-coding RNA, RNA polymerase, transcription regulation
Graphical abstract
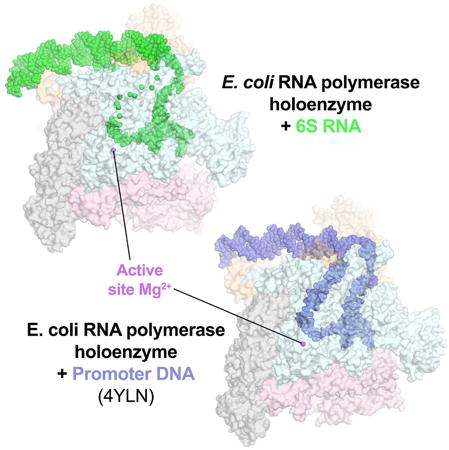
Structural analysis of an E. coli 6S RNA/RNA polymerase holoenzyme binary complex shows how a non-coding RNA can directly regulate transcription by the DNA-dependent RNA polymerase. Functional parallels between bacterial and eukaryotic non-coding RNA RNA polymerase regulators suggest that the structural principles delineated here are widely applicable.
Introduction
Noncoding RNAs (ncRNAs) play critical and diverse roles in the regulation of gene expression from bacteria to humans (Gottesman and Storz, 2011; Kaikkonen et al., 2011). Bacterial 6S RNAs globally regulate transcription by directly binding the RNA polymerase (RNAP) holoenzyme and competing with promoter DNA (Cavanagh and Wassarman, 2014; Wassarman and Storz, 2000). Escherichia coli (Eco) 6S RNA, a 184-nucleotide (nt) ncRNA, plays a key role in the transcriptional reprogramming upon shifts between exponential and stationary phase by binding with marked specificity to and inhibiting the housekeeping σ70-holoenzyme (Eσ70), allowing for the increased activity of the stationary phase-specific σ factor, σS (Steuten et al., 2014).
Although not conserved in sequence, 6S RNAs are widely conserved among bacteria through a common secondary structure that includes a transcription bubble mimic (Barrick et al., 2005; Trotochaud and Wassarman, 2005). Indeed, 6S RNA can serve as a transcription template during outgrowth from stationary phase, likely due to high NTP concentrations (Wassarman and Saecker, 2006). Synthesis of an RNA product (pRNA) induces a structural rearrangement of the RNA (Figure S1A) that provokes release of Eσ70 from 6S RNA (Beckmann et al., 2012; Panchapakesan and Unrau, 2012), providing a mechanism for rapidly recovering Eσ70 activity in response to nutrient availability (Cavanagh et al., 2012). How the ncRNA 6S RNA interacts with the DNA-dependent RNAP to outcompete promoter DNA has not been elucidated at the structural level.
Here we report the cryo-electron microscopy (cryoEM) structure of a complex between an Eco 6S RNA fragment and Eσ70 as well as validation of the structure using footprinting and crosslinking approaches. Our results help explain the specificity of Eco 6S RNA for Eσ70, and show how an ncRNA can mimic B-form DNA to directly regulate transcription by the DNA-dependent RNAP. Functional parallels between bacterial and eukaryotic ncRNA RNAP inhibitors (Wagner et al., 2013; Yakovchuk et al., 2009) suggest that the structural principles for 6S RNA binding and inhibition of Eco RNAP delineated here are widely applicable.
Results
Structure Determination and Validation
We first employed nucleic acid footprinting approaches to probe 6S RNA/Eσ70 interactions (Figures 1A, S1B). RNase footprints of wild-type Eco core RNAP containing full-length α-subunits combined with full-length σ70 and footprints of core RNAP lacking the α-subunit C-terminal domains (Twist et al., 2011) (ΔαCTD-E) combined with σ70 lacking the N-terminal region 1.1 (Δ1.1σ70; Bae et al., 2013) were identical, indicating that the αCTDs and σ701.1 did not play a role in 6S RNA interactions, at least in the final binary complex studied here. The footprinting results point to complete enclosure of the 6S RNA CB and DD (see Figure 1A for definitions of the 6S RNA structural elements) in the RNAP active site cleft. Fe2+-directed cleavage indicates loading of the transcription start site (TSS), U44, into the RNAP active site itself (Figure 1A; Wassarman and Saecker, 2006). The RNA upstream of the CB showed a pattern of alternating protection and hypersensitivity from RNases, suggesting interaction with the surface of the RNAP. RNase protection did not extend downstream beyond the DB, indicating that the CS was exposed outside of the RNAP active site cleft and not important for binding, as observed previously (Shephard et al., 2010).
Figure 1. CryoEM structure of the Eco 6S RNA/Eσ70 complex.
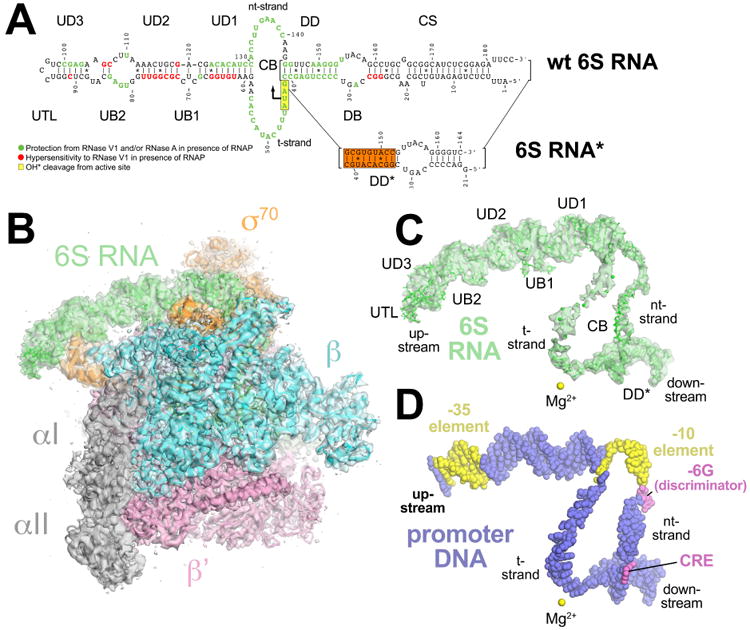
(A) (top) Secondary structure of wild-type Eco 6S RNA as observed in the cryoEM structure. Structural elements of the 6S RNA are labeled (CS, closing stem; DB, downstream bulge; DD, downstream duplex; CB, central bubble; UD1-3, upstream duplexes 1-3; UB1-2, upstream bulges 1-2; UTL, upstream terminal loop). The sequence is color-coded according to the RNase footprinting and localized hydroxyl-radical cleavage results comparing 6S RNA with and without Eσ70 (Figure S1B). RNA positions protected from RNase V1 and/or RNase A cleavage in the presence of Eσ70 are colored green. RNA positions showing hypersensitivity to RNAse V1 in the presence of Eσ70 are colored red. RNA positions efficiently cleaved by hydroxyl radicals generated from Fe2+ in place of the RNAP active site Mg2+ are boxed in yellow (Wassarman and Saecker, 2006). The position of pRNA synthesis initiation (U44) is indicated by a bent arrow.
(below) Secondary structure of 6S RNA* used in cryoEM structure determination, generated by truncating the CS and shuffling the sequence of the DD to give DD* (shuffled sequences highlighted in the orange box).
(B) The 3.8 Å resolution cryoEM density map of Eco 6S RNA/Eσ70 is rendered as a transparent surface colored as shown. Superimposed is the final refined model; the RNAP is shown as a backbone ribbon, the 6S RNA is shown in stick format.
(C) The 3.8-Å resolution cryoEM density map with the superimposed model of only the 6S RNA. Shown for reference is the RNAP active site Mg2+ ion (yellow sphere).
(D) Shown is just the promoter DNA after superimposing the Eσ70 from the crystal structure of an open promoter complex (PDB ID 4XLN) (Bae et al., 2015) onto the 6S RNA/Eσ70 structure. The promoter DNA is colored blue but the -35 and -10 elements of the DNA are colored yellow. The -6G of the discriminator and the Core Recognition Element (CRE) are colored violet. Shown for reference is the RNAP active site Mg2+ ion (yellow sphere).
See also Table S1 and Figures S1 – S4.
6S RNA is predicted to exist in two nearly isoenergetic isoforms, even in the absence of pRNA synthesis (Figure S1A). To generate a homogeneous population for structure determination we locked 6S RNA into isoform 1 by shuffling the sequence of the DD to prevent formation of isoform 2. The 6S RNA* (Figure 1A), generated by a combination of DD sequence shuffling (DD*) and truncation of much of the CS, bound Eσ70 specifically and initiated transcription from the correct TSS as efficiently as wild-type 6S RNA (Figure S1C) and was used for structure determination.
We used single-particle cryoEM to determine the structure of the 6S RNA*/ΔαCTD-E-Δ1.1σ70 (termed 6S RNA/Eσ70 here forward) binary complex (Figures 1B-C, S2, S3). The overall resolution of the cryoEM density map was 3.8 Å (Figure S2D) but local resolution calculations indicated much of the central core of the structure was determined to 3.3 – 3.8 Å resolution (Figure S4B). A model of the 6S RNA/Eσ70 complex was built and refined into the cryoEM map (Materials and Methods; Table S1; Figure S2E). The RNA density downstream of the DD*, as well as for the CB t-strand from nucleotides 48-57, was very weak (Figures 1C, S4) so these parts of the RNA were not modeled. Portions of the RNA upstream of UB2 were also difficult to model due to the low resolution of the map (Figure S4B) so were modeled with the assistance of MC-fold (Parisien and Major, 2008). The 6S RNA*/Eσ70 model was consistent with the RNA footprinting data (Figures 1A, 2A).
Figure 2. Correspondence of the 6S RNA/Eσ70 Structure with Footprinting and Crosslinking Data.
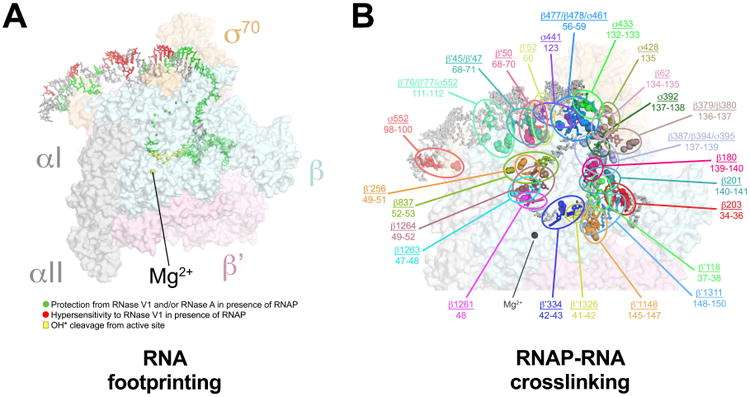
(A) Correspondence of the 6S RNA/Eσ70 structure with the RNase footprinting and localized hydroxyl-radical cleavage results (Figure 1A). The 6S RNA/Eσ70 structure is shown (RNAP as a transparent molecular surface, 6S RNA in stick format). The RNA is color-coded according to Figure 1A (green, protected from RNase cleavage by Eσ70; red, hypersensitive to RNAse V1 cleavage in the presence of Eσ70; yellow, efficiently cleaved by hydroxyl radicals generated at the RNAP active site).
(B) The results of RNAP-BPA/6S RNA crosslink mapping are superimposed on the 6S RNA/Eσ70 structure. RNAP residues substituted individually with BPA and crosslinked to the 6S RNA are shown in CPK format. The BPA-substituted RNAPs were crosslinked to the 6S RNA, followed by mapping of the crosslinks on the RNA (Figure S4). The mapped 6S RNA nucleotides are indicated and color-matched with the crosslinked BPA-RNAP residue. The matched RNAP residue and corresponding crosslinked RNA nucleotides are circled and labeled (RNAP residue/6S RNA nucleotides) in the same color. The average minimum distance between Cα-positions of crosslinked amino acids and RNA nucleotide for the 36 mapped crosslinks was 9 ± 3 Å, consistent with the ∼7.5 Å crosslink distance and accounting for molecular flexibility. The peak of the crosslink distance distribution was 7.5 Å.
See also Figures S1 and S5.
The model was further validated by an extensive dataset of mapped protein-6S RNA crosslinks from p-benzoyl-L-phenylalanine (BPA) site-specifically incorporated at 35 distinct sites in the RNAP β, β′, and σ70 subunits (Figures 2B, S5). Biochemical experiments (see below) provided further validation of the structure.
Duplex Segments of the 6S RNA show an Unusual Nucleic Acid Geometry
As expected, the CB, including the 6S RNA t-strand near the RNAP active site, mimics a promoter DNA transcription bubble (Figure 3). Strikingly, however, the overall architecture and path of the 6S RNA phosphate backbone matches that of B-form promoter open complex DNA (Bae et al., 2015) over essentially the entire length of the RNA (Figures 1C, 1D, 4A). B-form RNA duplex is highly unfavorable, and there are no duplex regions of the 6S RNA that show B-form geometry (Tables S2-S4).
Figure 3. Active-site density and comparison with promoter DNA.
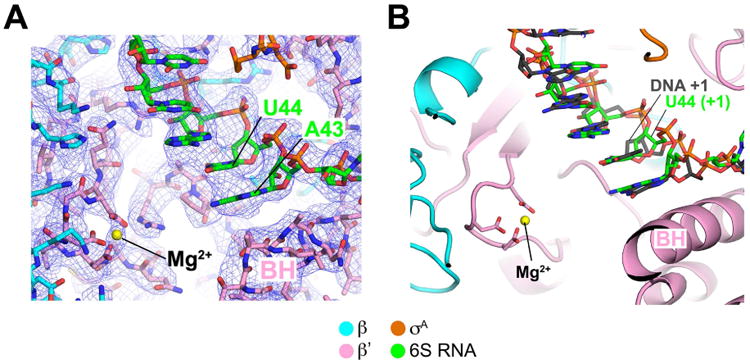
(A) View of the 6S RNA/Eσ70 cryoEM density map (blue mesh) showing the region around the RNAP active site (Mg2+ ion shown as a yellow sphere). The TSS on 6S RNA is U44.
(B) Shown is the 6S RNA/Eσ70 structure near the RNAP active site (Mg2+ ion, yellow sphere). Superimposed on the 6S RNA is the promoter DNA t-strand from the promoter open complex structure (Bae et al., 2015). The TSS on 6S RNA (U44) and promoter DNA (DNA +1) are noted.
Figure 4. 6S RNA mimics B-form promoter DNA.
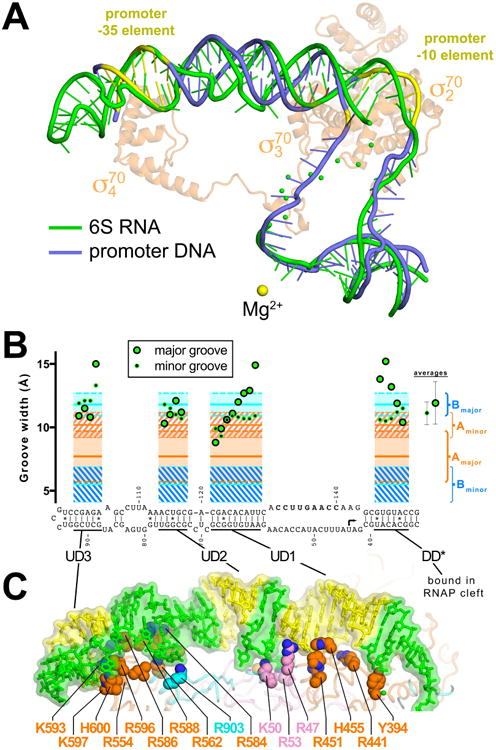
(A) Superimposition of 6S RNA (green) and promoter DNA from an open promoter complex (blue; PDB ID 4XLN) (Bae et al., 2015). The -35 and -10 elements of the promoter DNA are colored yellow. For reference the Eco σ70 from the 6S RNA/Eσ70 structure is shown (transparent light orange Cα ribbon) and the RNAP active site Mg2+ ion is shown as a yellow sphere.
(B) Major groove (large green dot with black border) and minor groove (small black dot with green border) widths (Mgw and mgw, respectively) for duplex regions of the 6S RNA, calculated using 3DNA (Lu and Olson, 2008) but with the van der Waals radii of the phosphate groups (5.8 Å) subtracted, compared with average values (± standard deviation) for B-form (blue regions) and A-form (orange regions) nucleic acids (PDB IDs: A-form - 1D4R, 1LNT, 1QC0, 1RXB; B-form – 1D23, 1D49, 1D56, 1D57, 158D). The average 6S RNA mgw (11.1 ± 0.9 Å, shown on the right under ‘averages’) is close to the average mgw for A-form nucleic acids (10.2 ± 1.0 Å), while the average Mgw of the 6S RNA (11.9 ± 1.7 Å) is closer to the average Mgw for B-form nucleic acids (11.8 ± 0.9 Å).
(C) Shown is the upstream portion of the 6S RNA (transparent molecular surface with stick model inside). Yellow indicates duplex regions with widened major grooves, other regions are colored green. Shown in CPK format are amino acid side chains of RNAP residues that interact with the RNA (< 4.5 Å; β residues, blue; β′, pink; σ70, orange). Selected residues that make polar interactions with the RNA backbone are labeled.
See also Tables S2 – S4.
Insight into the origin of the overall ‘B-form’ appearance of the 6S RNA comes from an analysis of the minor and major groove widths (mgw and Mgw, respectively). In duplex stretches of the 6S RNA long enough to measure the groove widths (DD*, UD1, UD2, and UD3)(Lu and Olson, 2008), the mgws conform to A-form nucleic acids while the Mgws conform to B-form (Figure 4B, Table S2). The sugar puckers (Table S3), intrastrand phosphate-phosphate distances (Table S4), and twist (Table S2) are characteristic of A-form nucleic acid duplex, but the rise and pitch (Table S2) are characteristic of B-form. Thus, overall the duplex regions of the 6S RNA are neither A-nor B-form, but have characteristics of both.
The segments of duplex 6S RNA with widened Mgws resembling B-form duplex correspond to regions with extensive 6S RNA/RNAP interactions, which occur almost exclusively with the 6S RNA phosphate backbone (Figure 4C). It is likely that the considerable binding free energy of the RNAP drives the distortion of the 6S RNA helix geometry, but a structure of the RNAP-interacting determinant of the 6S RNA in the absence of the RNAP will be required to assess whether the RNA structure is predisposed for RNAP binding or if the RNA structure is distorted through an induced fit mechanism.
6S RNA/Eσ70 Interactions
The duplex segments of 6S RNA are interrupted by regions comprising base-pair mismatches, non-canonical base pairs, and flipped-out bases (CB, UB1, and UB2) that are conserved in an alignment of 101 enterobacteriaceae 6S RNA sequences (Figure 5A). Base-specific 6S RNA/RNAP interactions occur nearly exclusively with ‘flipped-out’ bases from these regions (C69 of UB1; G82 of UB2; U135/G136/G143 of the CB nt-strand; Figures 5B-E).
Figure 5. Conserved Interactions and cryoEM density.
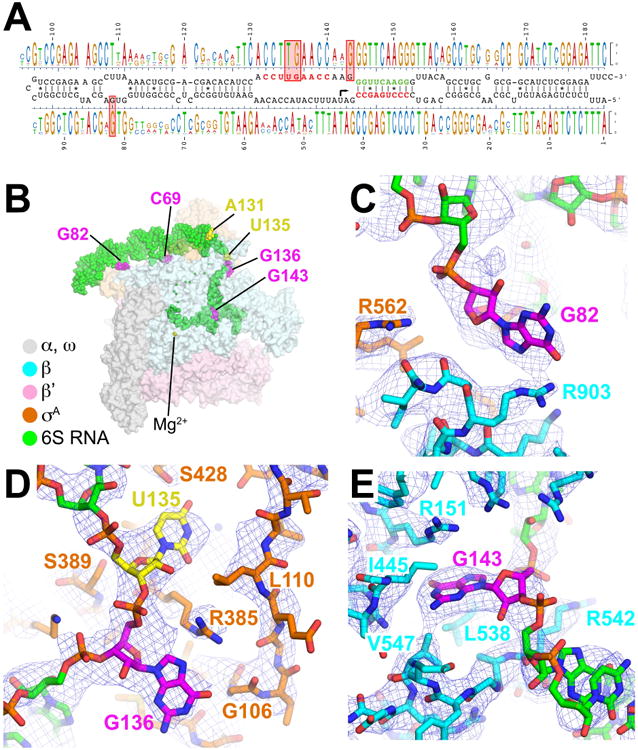
(A) Shown in the middle is the secondary structure of the wild-type Eco 6S RNA. On top and bottom is aligned a sequence logo (Schneider and Stephens, 1990) derived from a sequence alignment of 101 enterobacteriaceae 6S RNA sequences.
(B) Overall structure of Eco 6S RNA/Eσ70 complex, color-coded as shown in the legend. The RNAP is shown as a transparent molecular surface, the RNA is shown as green CPK atoms, with flipped-out bases that interact with RNAP colored magenta, and A131/U135, positions equivalent to promoter DNA A-11/T-7, colored yellow.
(C) CryoEM density (blue mesh) showing the conserved interaction between RNA nucleotide G82 (magenta) and RNAP β subunit R903.
(D) CryoEM density (blue mesh) showing RNA nucleotides U135 (yellow) and G136 (magenta) interacting with σ70 in the same manner as promoter DNA nt-strand T-7 of the -10 element and G-6 of the discriminator (Bae et al., 2015; Zhang et al., 2012).
(E) CryoEM density (blue mesh) showing RNA nucleotide G143 (magenta) interacting with a G-specific pocket of the RNAP β-subunit in the same manner as promoter DNA nt-strand G+2 (core recognition element) (Zhang et al., 2012).
See also Figure S6.
C69 is flipped out of UB1 and interacts with residues of the RNAP β′ subunit but C69 is not conserved (Figure 5A). However, the presence of the bulge UB1 seems to be important since removing it by introducing complementarity into the RNA reduces 6S RNA binding (Figure S6A).
Conserved G82 of UB2 (Figure 5A) interacts with βR903 on the flap-tip-helix (Figure 5C). The residue corresponding to Eco RNAP βR903 is 80% identical in an alignment of nearly 1,000 diverse bacterial RNAP β subunit sequences, but is 100% identical among enterobacteriaceae RNAPs (Lane and Darst, 2010). Substitution of G82 with any other nucleotide substantially disrupts 6S RNA binding to Eσ70 (Figure S6B).
U135, G136, and G143 of the CB nt-strand are all conserved (Figure 5A) and make essentially identical interactions with Eσ70 as the corresponding nucleotides of promoter DNA (Figures 5D-E). U135 interacts with the pocket of σ70 normally reserved for the nt-strand T-7 (Figure 5D), one of two nearly absolutely conserved bases of the promoter -10 element (Feklistov and Darst, 2011; Shultzaberger et al., 2007). Likewise, the interactions of G136 with σ70 mimic the interactions of promoter DNA G-6 of the discriminator element (Bae et al., 2015; Zhang et al., 2012). Finally, G143 binds in a G-specific pocket of the RNAP β subunit normally reserved for the nt-strand G+2 (the core recognition element, CRE) (Bae et al., 2015; Zhang et al., 2012). However, base substitutions at these three positions of 6S RNA have little effect on 6S RNA binding (Figure S6C), indicating that these base/RNAP interactions may be conserved for other reasons. U135/G136 are involved in the formation of 6S RNA isoform 2 (Figure S1A), suggesting their binding to RNAP may stabilize isoform 1 and help prevent the spontaneous formation of isoform 2 and premature release of 6S RNA from Eσ70, possibly reflecting the importance of the timing of RNAP release (Beckmann et al., 2012; Panchapakesan and Unrau, 2012).
Although the interactions of U135, G136, and G143 with RNAP mimic interactions of promoter DNA nt-strand T-7, G-6, and G+2, the most important and conserved promoter DNA/RNAP interaction, accommodation of the flipped out -10 element nt-strand A-11 base in a pocket of σ70 (Feklistov and Darst, 2011), is not recapitulated in 6S RNA. Although an A is conserved at the corresponding position of 6S RNA (A131, 4 nt upstream of the T-7 mimic U135; Figure 5A), A131 is not flipped out of the 6S RNA duplex base stack, does not interact with RNAP, does not crosslink with BPA-RNAP substitutions that crosslink promoter DNA near this position (σ70434) (Winkelman et al., 2015), and the σ70 W-dyad (absolutely conserved W433/W434) does not switch into the characteristic ‘chair’ conformation that stabilizes the upstream single-strand/double-strand junction of the transcription bubble (Bae et al., 2015) (Figure 6). The flipping of A-11 out of the promoter DNA duplex and capture by σ70 is thought to be the critical event that initiates transcription bubble formation (Feklistov and Darst, 2011). Since the 6S RNA CB is ‘pre-melted’, this interaction is dispensable. In support of this notion, an RNAP-holoenzyme containing a quadruple-substituted σ70 that is severely defective in initiating and maintaining the transcription bubble (σ70FYWWW; (2001) is completely inactive on T7 A1, a strong DNA promoter, but is fully active on a pre-melted T7 A1 DNA promoter template and on 6S RNA (Figure S6D, S6E). Additionally, A131C or A131G substitutions in 6S RNA do not alter RNAP affinity (Figure S6C).
Figure 6. The σ70 W-dyad and 6S RNA.
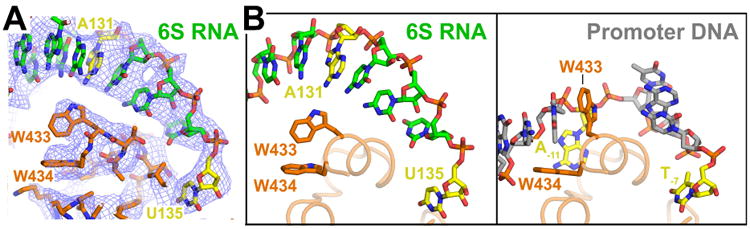
(A) View of cryoEM density (blue mesh) showing RNA nucleotide A131 (yellow) in a duplex RNA base stack passing over the σ70 W-dyad (absolutely conserved W433/W434). The Trp side chains maintain their edge-on conformation characteristic of σ70 alone (Malhotra et al., 1996) when not interacting with the double-strand/single-strand fork at the upstream edge of the transcription bubble (Bae et al., 2015).
(B) Comparison of 6S RNA (left) and promoter DNA (right) interacting with the absolutely conserved σ70 W-dyad (σ70 W433/W434). Shown is only the ‘nt-strand’ of the RNA or DNA. Corresponding nucleotides A131/U135 of 6S RNA or conserved A-11/T-7 of the promoter -10 element (Shultzaberger et al., 2007) are colored yellow. In both the RNA and DNA complexes, U135/T-7 is single-stranded and interacting with a pocket of σ70 (Feklistov and Darst, 2011). In the promoter DNA structure, the critical A-11 nucleotide is flipped out of the duplex base stack and σ70-W433 takes its place, forming a ‘chair’ conformation with W434 to stabilize the upstream edge of the newly formed transcription bubble (Bae et al., 2015). A131 remains in the base stack and the W-dyad maintains the edge-on conformation characteristic of σ70 alone (Feklistov and Darst, 2011). The 6S RNA is pre-melted (Figure 1A) and thus this interaction is dispensable.
See also Figure S6.
6S RNA Specificity for Eσ70
6S RNA shows strong specificity for Eσ70 over EσS (Trotochaud and Wassarman, 2005). The σ70 and σS are the most closely related s factors in Eco, with overlapping promoter specificity (Gaal et al., 2001). A major difference in the two s's is the non-conserved insert between conserved regions 1.2 and 2.1 (number of residues between regions 1.2 and 2.1: σ70, 248; σS, 3) (Lonetto et al., 1992), but the structure shows that 6S RNA does not interact with this region of σ70. The base-specific interactions of σ70 with flipped-out bases of the 6S RNA all involve highly conserved regions of the two σ factors (Figure 7A).
Figure 7. Structural basis for 6S RNA specificity for Eσ70 over EσS.
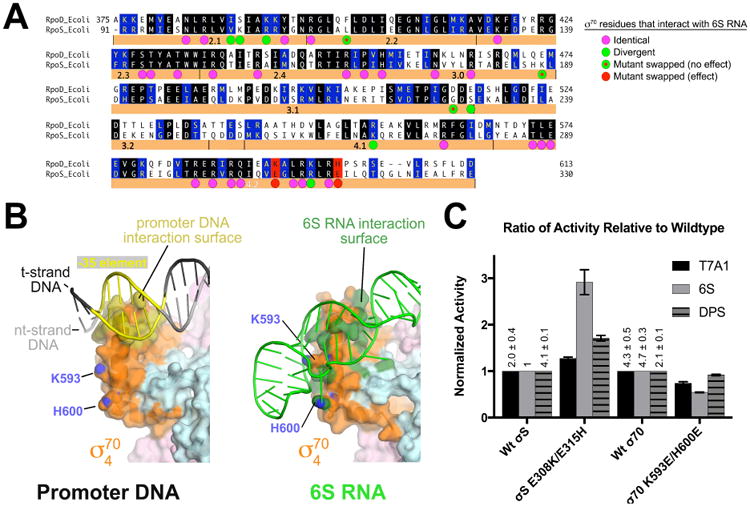
(A) Sequence alignment of Eco σ70 (RpoD) and σS (RpoS). Identical residues are shaded black, homologous residues shaded blue. Eco σ70 residues that interact with 6S RNA are denoted by a colored dot underneath. The magenta dots indicate positions that are identical between σ70/σS. Green dots indicate positions that diverge in sequence. Green dots with red dots inside are positions that were swapped between σ70 and σS but had no apparent effect on 6S RNA preference (Figure S7). Two loci, denoted in red (σ70 K593/H600; σS E308/E315), altered 6S RNA preference when swapped (see Figure 4C (Klocko and Wassarman, 2009).
(B) Interaction surfaces for promoter DNA (left; PDB ID 5WT1) (Hubin et al., 2017) and 6S RNA (right) on the σ4 domain. The RNAP holoenzyme is shown as a molecular surface (β, light cyan; β′, light pink; σ70, orange). The surface of σ4 that contacts the promoter DNA is colored dark yellow (left). Marked with their blue nitrogen atoms are K593 and H600 (using Eco σ70 numbering), which are solvent-exposed. On the right, the surface of σ704 that contacts 6S RNA is colored dark green, illustrating that K593/H600 are engaged with the 6S RNA.
(C) Histogram showing the normalized abortive transcription initiation activity for the indicated σ's (bottom) on the indicated templates (σ70-specific promoter T7A1; σ70-specific 6S RNA; σS-specific promoter DPS). The σS-holoenzyme activities are shown normalized with respect to wild-type σS, while the σ70-holoenzymes are shown normalized to wild-type σ70. The relative values of the wild-type σ70 and σS transcription activities on the different templates (with respect to 6S RNA with σS) are listed. The histograms represent the average of at least three independent measurements, the error bars denote S.E.M.s. Mutation of σS positions 308 and 315 to the corresponding residues in σ70 (σS E308K/E315H) increased the relative activity on 6S RNA with less effect on the relative activity on the other templates.
See also Figure S7.
The σ704 domain, responsible for recognition of the promoter DNA -35 element, is required for the 6S RNA/Eσ70 interaction and has been implicated in mediating 6S RNA specificity between σ70 and σS (Cavanagh et al., 2008; Klocko and Wassarman, 2009). We do not observe notable base-specific interactions between σ704 and 6S RNA, but the binding determinants for promoter DNA and 6S RNA on σ704 define very different surfaces on the protein (Figure 7B) (Klocko and Wassarman, 2009). The promoter DNA -35 element perches at the N-terminal end of the σ704 helix-turn-helix motif recognition helix, forming an interface with multiple DNA backbone and base-specific interactions that buries 634 Å2 of molecular surface area (Hubin et al., 2017). By contrast, the 6S RNA wraps around σ704, interacting with a large swath of the protein surface (791 Å2) (Figure 7B), consistent with BPA-RNAP crosslinks to 6S RNA (σ70552; Figure 2B) that are not observed to promoter DNA (Winkelman et al., 2015). Multiple polar interactions are established between the RNA phosphate backbone and positively charged protein side chains, mostly with residues that are conserved between σ70 and σS (Figure 7A).
Two σ704 residues that interact with the 6S RNA backbone, K593 and H600, correspond to negatively charged Glu residues in σS (E308 and E315; Figures 7A, S7A). Substitution σ70K593E is strongly defective for 6S RNA binding (Klocko and Wassarman, 2009) and substitution of these positions in σS with the corresponding residues of σ70 (σS[E308K], σS[E315H]) increases the transcription activity of the resulting σS-holoenzymes on 6S RNA ∼2-fold and 1.5-fold, respectively (Figure S7B), and a double substitution (σS[E308K/E315H]) increases the transcription activity of the resulting EσS on 6S RNA ∼3-fold. The σS single or double substitutions do not have dramatic effects on activity on a σ70-specific promoter (T7 A1) nor on a σS-specific promoter (DPS) (Figures 7C, S7B). Thus, the identity of two residues in σ704 (out of an interface that buries ∼4,500 Å2 surface area and involves at least 68 residues) account for a significant fraction of the 6S RNA preference for σ70 over σS.
Discussion
The A-form major groove of RNA duplexes is too deep and narrow for protein side chains to reach the major groove edge of base pairs for sequence-specific readout. The bovine immunodeficiency virus tat peptide forms a β-hairpin that inserts into the major groove of the TAR RNA, widening the RNA major groove to allow sequence-specific RNA recognition (Puglisi et al., 1995; Ye et al., 1995). RNAP uses a completely different mechanism to widen the 6S RNA major groove; RNAP interactions with the RNA backbone pry open the major groove, not for the purposes of sequence-specific RNA recognition but in the service of B-form mimicry (Figure 4).
Direct regulation of RNAP activity by ncRNAs is not limited to the bacterial 6S RNA. Mouse B2 and human Alu ncRNAs both repress mRNA transcription by binding to RNAP II and blocking contacts with promoter DNA (Yakovchuk et al., 2009). Remarkably, RNAP II can utilize the B2 RNA as both a transcription template and substrate, using an RNA-dependent RNAP activity to extend the 3′-end of the B2 RNA in an internally templated reaction (Wagner et al., 2013). Extension of the B2 RNA in this way destabilizes the RNA and relieves the RNAP II inhibition. The functional parallels between the bacterial and eukaryotic ncRNA RNAP inhibitors suggest that the structural principles for 6S RNA binding and inhibition of Eco RNAP delineated here are also widely applicable.
Star Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Seth Darst (darst@rockefeller.edu).
Method Details
Protein Expression and Purification
Preparation of RNase-free Eco ΔαC-terminal domain RNAP
For cryoEM, Eco core RNAP lacking the αC-Terminal Domain (αCTD) was prepared as described previously (Twist et al., 2011) but with one modification to obtain RNase-free RNAP. During Bio-rex 70 cation exchange, the protein was washed extensively with TGED buffer [10 mM Tris-HCl, pH 8.0, 5% (v/v) glycerol, 0.1 mM EDTA, 1 mM DTT] + 100 mM NaCl for 10 column volumes (CVs), then 10 CVs of TGED + 200 mM NaCl to remove contaminating RNases. The presence of RNases was detected using the RNaseAlert QC system (Ambion). The final purified RNAP was buffer-exchanged into storage buffer (10 mM Tris-HCl, pH 8.0, 150 mM KCl, 1 mM MgCl2, 2 mM DTT) and concentrated to 20 mg/mL by centrifugal filtration (Millipore) and stored at −80 °C until use.
Preparation of RNase-free Eco full-length and Δ1.1σ70
Full-length and Δ1.1 σ70 were cloned from a pET21 plasmid into a pETsumo vector using EcoRI and HindIII sites. The primers used to do this cloning were:
Sig70FL_EcoRI_for, CATCATGAATTCATGGAGCAAAACCCGCAGTCACAGCTGAAACTTCTTGTC;
Sig70delta1.1_EcoRI_for, CATCATGAATTCAACGACTGACCCGGTACGCATGTACATGCGTGAAATGG;
Sig70_HindIII_rev, ATGAAGCTTTTAATCGTCCAGGAAGCTACGCAGCACTTCAGAACGGCTC.
The pETsumo expression plasmids were transformed into Eco BL21 (DE3) cells (Invitrogen) and grown overnight on LB-agar plates containing 50 μg/mL kanamycin. Single colonies were used to inoculate 50 mL LB medium containing 50 μg/mL kanamycin and grown overnight at 37 °C. Each 2-L fl ask of LB (50 μg/mL kanamycin) was inoculated with 10 mL of cells, induced at OD600 = 0.6–0.8 by the addition of isopropyl β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 1 mM, then grown for 1 h at 30 °C. Cells were collected by cen trifugation, resuspended in lysis buffer [20 mM Tris-HCl, pH 8.0, 500 mM NaCl, 0.1 mM EDTA, 5% (v/v) glycerol, and 1 mM β-mercaptoethanol] + 5 mM imidazole and lysed in a continuous-flow French press (Avestin). The lysate was cleared by centrifugation at 27,000 × g for 30 min, then loaded onto a Hitrap IMAC HP column (GE Healthcare), and eluted with lysis buffer + 250 mM imidazole. The fractions containing full-length or Δ1.1σ70 were pooled (after analysis by SDS/PAGE), dialyzed overnight into lysis buffer + 25 mM imidazole in the presence of UlP1 protease (1 mg protease/50 mg of full-length or Δ1.1σ70). The cleaved full-length or Δ1.1σ70 were loaded onto Hitrap IMAC HP columns (GE Healthcare) to remove His6-SUMO tag. Flow-through from a subtractive IMAC column containing σ70 was dialyzed into TGED + 0.2 M NaCl. The protein was further purified on a Hitrap Heparin HP column (GE Healthcare), eluted using a gradient to TGED + 1 M NaCl to remove contaminating RNases. Fractions were assayed for RNase activity using RNaseAlert QC system (Ambion). Fractions containing no RNases were pooled and further purified by gel filtration chromatography on SuperDex 200 (GE Healthcare; equilibrated with TGED + 0.5 M NaCl). Purified full-length or Δ1.1σ70 were buffer-exchanged into storage buffer and concentrated to 10 mg/mL by centrifugal filtration (Millipore) and stored at −80 °C.
Construction of template plasmids for 6S RNA synthesis
DNA templates for in vitro T7 RNAP synthesis of 6S RNA derivatives (6S RNAΔCS and 6S RNA*) were generated by gene synthesis (GenScript) into a pUC57 vector with flanking EcoRI and BamHI restriction sites. The DNA sequences for these 6S RNA variants are as follows:
6S RNAΔCS: GGACCAGCCAGTCGGCACATGCGATATTTCATACCACAAGAATGTGGCGCTCCGCGGTTGGTGAGCATGCTCGGTCCGTCCGAGAAGCCTTAAAACTGCGACGACACATTCACCTTGAACCAAGGCGTGTACCGTTACAgcGGGTCCCGGGGATCC
6S RNA*: GGACTCCCAGTCGGCACATGCGATATTTCATACCACAAGAATGTGGCGCTCCGCGGTTGGTGAGCATGCTCGGTCCGTCCGAGAAGCCTTAAAACTGCGACGACACATTCACCTTGAACCAAGGCGTGTACCGTTACAGGGGTC
DNA templates for in vitro T3 RNAP synthesis of 6S RNA and 6S RNA-RV and derivatives were pT3-6S RNA (Trotochaud and Wassarman, 2005), and derivatives were generated by quickchange mutagenesis (Agilent). 6S RNA-RV has a 3′ extension generated from the polylinker of the parent vector (pCR-2.1, Invitrogen) from pT3-6S linearized by EcoRV to facilitate primer extension mapping. Oligos used for mutagenesis are available upon request. Plasmids were maxi-prepped (Qiagen), digested with PpuMI (NEB) and gel purified (Qiagen) for in vitro T7 RNAP transcription, or digested with SmaI (NEB) or EcoRV (NEB) and ethanol precipitated for in vitro T3 RNAP transcription.
6S RNA synthesis and purification
For cryoEM, footprinting, and transcription assays, a mutant T7 RNAP that dissociates more rapidly from the 3′-end of run-off transcription templates and therefore minimizes non-templated addition of nucleotides to the RNA transcript 3′-end (T7 RNAP*) was purified from plasmid pRC9 (a generous gift from W.T. McAllister) as previously described (He et al., 1997). In vitro transcription of 6S RNA and derivatives was performed in 30 mM Tris-HCI, pH 8.0, 10 mM DTT, 2 mM spermidine, 12.5 mM MgCI2, 20 mM each NTP, 0.01 mg/ml_T7 RNAP*, 0.2 U/μL SUPERaseln (Ambion), 1 mU/μL inorganic pyrophosphatase (Sigma), and 70 ng/μL linearized plasmid DNA. After incubating at 37 °C for three hours, the reaction was directly applied to a HiTrap Q HP column (GE Healthcare Life Sciences) pre-equilibrated with 20 mM HEPES, pH 7.5, 300 mM NaCI (Koubek et al., 2013). The following salt gradients (with 20 mM HEPES, pH 7.5) were used to purify 6S RNAs from the Q column: 300 mM NaCI (5CV), 300 mM-500 mM NaCI (5CV), 500 mM-800 mM NaCI (25CV), 800 mM-1 M NaCI (5CV), 1 M NaCI (5CV). The 6S RNAs elute within the 500 mM-800 mM NaCI gradient. RNA was precipitated with two volumes of 1:1 ethanol:isopropanol and separated through centrifugation. The RNA pellet was resuspended in stabilization buffer (10 mM Tris-HCI, pH 8.0, 1 mM MgCI2).
For crosslinking and 6S RNA binding experiments, 6S RNA and variants were generated by in vitro transcription with T3 RNAP from linearized pT3-6S RNA templates using T3 megascript kit (Ambion) for large scale unlabelled RNAs or with T3 RNAP (Roche) according to manufacturer protocols for 32P-labelled RNAs (Trotochaud and Wassarman, 2005). Both unlabeled and labeled RNAs were gel purified on denaturing polyacrylamide gels, visualized by UV shadowing or autoradiography, cut out and eluted in elution buffer (0.5 M ammonium acetate, 1 mM EDTA, 0.2% SDS, 10 mM Tris, pH 7.5) O/N at 37 °C. RNAs were recovered by precipitation with 2.5 volumes of ethanol and centrifugation. RNAs were resuspended in water and quantified by OD260 (unlabelled) or by cpm incorporation (labelled).
RNase footprinting
Eco Eσ70 (100 nM) was incubated with 5′- or 3′-radioactively-labeled 6S RNA (5 nM) in 10 μL of binding buffer (10 mM Tris-HCl, pH 7.0, 100 mM KCl, 10 mM MgCl2) at 37 °C for 10 min. Subsequently, 1 μL of RNase A (1 ng) or RNase V1 (0.001 U) (Ambion) was added and incubation was continued for 5 min. For the ladder (6S RNA cleaved at G-residues) 0.1 U of RNase T1 was added to the denatured RNA (according to Ambion protocol). Cleavage reactions were stopped by the addition of 20 μL of inactivation buffer (Ambion), and the transcripts were precipitated, suspended in 5 μL of a formamide-containing sample buffer and analyzed on an 8% polyacrylamide/8M urea sequencing gel run in 1× TBE.
Hydroxyl radical footprinting
Eco Eσ70 (100 nM) was mixed with 5′- or 3′-radioactively labeled 6S RNA (5 nM) in 85 μL of binding buffer and incubated for 10 min at 37 °C. The footprinting reaction was initiated by adding 5 μL of 0.6% H2O2, 5 μL of 20 mM ascorbic acid, 2.5 μL of 0.4 mM Fe(NH4)2(SO4)2×6H2O, and 2.5 μL of 0.8 mM EDTA. The reaction was stopped after 2 mins by adding 10 μL of 0.1 M thiourea. The DNA was ethanol-precipitated and dissolved in 5 μL of a formamide-containing sample buffer. DNA fragments were analyzed by PAGE as described above.
Preparation of Eco 6S RNA*/ΔαCTD-E/Δ1.1σ70 Complex for CryoEM
Purified Eco ΔαCTD-RNAP and Δ1.1σ70 were mixed at a 1:1.5 molar ratio and incubated for 15 mins at 37°C. The assembled holoenzyme was injected into a Superose6 INCREASE column (GE Healthcare) equilibrated with 10 mM Tris-HCI, pH 8.0, 150 mM KCI, 1 mM MgCI2, 5 mM DTT. The eluted protein was concentrated by centrifugal filtration (VivaScience) to 4 mg/mL protein concentration. 6S RNA* was added in a 1.2 molar excess (compared with holoenzyme) and incubated at room temperature for 15 mins. The sample was mixed with 3-([3-cholamidopropyl]dimethylammonio)-2-hydroxy-1-propanesulfonate (CHAPSO, final concentration 8 mM) prior to grid preparation.
CryoEM grid preparation
C-flat CF-1.2/1.3 400 mesh copper grids were glow-discharged for 10 seconds prior to the application of 3.5 μL of the sample (2.0 - 3.0 mg/ml protein concentration). The grids were plunge-frozen in liquid ethane using a CP3 (Gatan) with ∼80% chamber humidity at 22°C.
CryoEM of Eco 6S RNA*/ΔcαCTD-E/Δ1.1σ70
The grids were imaged using a 300 keV Titan Krios (FEI) equipped with a K2 Summit direct electron detector (Gatan). Images were recorded with Serial EM (Mastronarde, 2005) in super-resolution counting mode with a super-resolution pixel size of 0.65 Å and a defocus range of -1 μm to -3.5 μm. Data were collected with a dose of 10 electrons/physical pixel/s (1.3 Å pixel size at the specimen). Images were recorded with a 15-second exposure and 0.3-second subframes (50 total frames) to give a total dose of 90 electrons/Å2.
Dose-fractionated subframes were 2×2 binned (giving a pixel size of 1.3 Å), aligned and summed using Unblur (Grant and Grigorieff, 2015). The contrast transfer function was estimated for each summed image using CTFFIND4 (Rohou and Grigorieff, 2015). From the summed images, approximately 5,000 particles were manually picked and subjected to 2D classification in RELION (Scheres, 2012). Projection averages of the most populated classes were used as templates for automated picking in RELION. Autopicked particles were manually inspected, then subjected to 2D classification in RELION specifying 100 classes. Poorly populated classes were removed, resulting in a dataset of 808,484 particles. These particles were individually aligned across movie frames using direct-detector-alignjmbfgs software (Rubinstein and Brubaker, 2015). Aligned particles were 3D auto-refined in RELION using a model of Eco Eσ70 (PDB ID 4LK1) (Bae et al., 2013) low-pass filtered to 60 A resolution using EMAN2 (Tang et al., 2007) as an initial 3D template. 3D classification into eight classes was performed on the particles using the refined model and alignment angles. Among the 3D classes, the best-resolved class, containing 362,926 particles, was 3D auto-refined and post-processed in RELION. Local resolution calculations were performed using blocres (Cardone et al., 2013).
Model building and refinement
To build an initial model of the protein components of the complex, Eco Eσ70 (PDB ID 4LK1) (Bae et al., 2013) was manually fit into the cryoEM density map using Chimera (Pettersen et al., 2004) and real-space refined using Phenix (Adams et al., 2010). In the real-space refinement, domains of RNAP were rigid-body refined, then subsequently refined with secondary structure restraints. A model of 6S RNA* was generated using a combination of manual building using Coot (Emsley and Cowtan, 2004) and Rcrane (Keating and Pyle, 2010) as well as predicted structures (for segments of the RNA upstream of UB2) from MCfold (Parisien and Major, 2008) that matched the cryoEM density. The model was real-space refined with the assistance of ERRASER (Chou et al., 2016; 2013).
Protein/RNA crosslinking and crosslink mapping
Preparation of p-benzoyl-L-phenylalanine (BPA) substituted RNAP
For crosslinking, BPA substituted Eco core RNAP or σ70 proteins were purified as described previously (Winkelman et al., 2015). Briefly, host strains were cotransformed with overexpression plasmid variants (containing a TAG stop codon at the position for BPA substitution in β or β′ [pIA900] or σ70 [pRLG13105]) and a plasmid expressing a suppressor tRNA/tRNA synthetase pair to allow incorporation of BPA at the stop codons (Ryu and Schultz, 2006). Cotransformants were inoculated into LB containing 1 mM BPA at OD600 ∼0.3, grown for 1 hr in the dark at 37 °C (core) or 30 °C (σ70), proteins were induced by addition of 1 mM IPTG (core) or 0.2% L-arabinose (σ70) and grown for an additional 6-20 hr (core) or 1.5 hr (σ70). Cells were collected by centrifugation and proteins purified using Ni-agarose and heparin affinity chromatography (core) or Ni-agarose (σ70). σ70-holoenzyme was reconstituted from core and 4-10 fold excess σ70.
Crosslinking of 6S RNA to BPA-RNAP
6S RNA or 6S RNA-RV (2-20 nM final) was incubated with BPA-containing Eσ70 (∼100 nM final) in 20 mM HEPES, pH 7.5, 120 mM KCl, 0.5 mM MgCl2, 5% (v/v) glycerol, and 1 mM DTT for 5 min at room temperature. 25 μL reactions in 1.5 mL Eppendorf tubes were set in a rack that placed the bottom tip of the tubes directly on the surface of a 15-Watt bulb (365 nm) of a UV transilluminator. Samples were irradiated for 2 min at room temperature, tubes were removed from the light and the light turned off for 1 min to prevent overheating, and this cycle (2 mins irradiation, 1 min rest) was repeated six times (total irradiation time 12 minutes). Efficiency of crosslinking was determined by use of 32P-labelled 6S RNA and separation in 4-12% NuPage Bis-Tris polyacrylamide gels and NuPage MOPS SDS running buffer (ThermoFisher). Crosslinked RNAs ran higher in the gel that uncrosslinked RNAs and the relative crosslinking efficiency was quantified on a Typhoon phosphorimager (see Figure S4).
Enrichment of crosslinked 6S RNA for mapping
Crosslinked 6S RNAs were enriched to facilitate primer extension mapping. NaCl (1.2 M final) and total yeast RNA (20 μg) were added to crosslinking reactions to disrupt non-crosslinked 6S RNA:RNAP interations. Samples were then incubated for 20 mins at room temperature with 5 μl of Ni-NTA resin (Qiagen, prewashed 2× with high salt buffer [1.2M NaCl, 50 mM Tris-HCl, pH 7.5]) in approximately 20 μL of high salt buffer. Resin was collected by centrifugation, incubated for 5 additional mins at room temperature in 500 μL of high-salt buffer and recollected by centrifugation. This high salt wash was repeated once more, followed by a similar wash with low-salt wash buffer (0.1 M NaCl, 50 mM Tris-HCl, pH 7.5). To fragment protein crosslinked to 6S RNA, resin was then incubated in Proteinase K buffer [10 mM Tris-HCl, pH 7.5, 100 mM NaCl, 0.5% SDS, 1 mM EDTA, 0.1 mg/ml Proteinase K (Ambion), 10 μg/ml total yeast RNA] for 15 min at 65 °C followed by phenol:chloroform:isoamyl alcohol (50:50:1) extraction and ethanol precipitation. Selected RNA was resuspended in 12 μL of water. Alternatively, for many BPA σ70-6S RNA crosslinks immunoprecipitation was more efficient for enrichment. In this case, after crosslinking, holoenzyme was denatured by incubation at 85 °C in 0.5% SDS followed by immunoprecipitation with polycolonal antisera raised against Eco σ70 (WI-166) as previously described (Wassarman and Storz, 2000) to enrich UV crosslinks between 6S RNA and RNAP.
Primer extension mapping of crosslink positions in 6S RNA
Different oligonucleotides (KW1466 [c157-179], KW1469 [c88-111], KW1555[c 138-161], KW1464 [c183-polylinker]) were used in primer extension reactions to cover the full length of 6S RNA. In order to map crosslinks near the 3′ end of the 6S RNA (∼130-184 nt positions in the RNA), a 6S RNA variant with an extended 3′ end was used (6S RNA-RV). Binding and crosslinking efficiency was identical between 6S RNA and 6S RNA-RV, and crosslinking positions that could be mapped on both 6S RNA and 6S RNA-RV were indistinguishable. For primer extension, oligonucleotides were 5′ end labeled with 32P by polynucleotide kinase (NEB). Labeled oligonucleotide (0.01 μg) and RNA (3 υL of crosslinked, selected RNA) were mixed in cDNA synthesis buffer (Invitrogen) in 8 μL reactions, denatured at 95 °C for 2 mins and allowed to anneal at 65 °C for 5 mins. Extension reactions were initiated by addition of 2 μL dNTP mix (25 mM DTT, 5 mM dNTPs, 0.5 μL Thermoscript Reverse Transcriptase [Invitrogen]) and incubated at 60 °C for 15 mins. Reactions were stopped by addition of 12 mL of urea load buffer {10 M urea, 1× TBE (0.089M Tris base, 0.089M boric acid, 2 mM EDTA, pH 8.3 [National Diganostics]), ∼0.01% xylene cyanol, ∼0.01% bromophenol blue}. Primer extension reactions were separated on 10% polyacrylamide, 8M urea, 1× TBE gels (National Diagnostics Urea Gel system). A salt gradient was applied to the gel to better resolve sequencing ladders over longer ranges (i.e. the “top” buffer was 0.5× TBE, the “bottom” buffer was 1× TBE with 0.3 M sodium acetate). Sequencing reactions were generated using Thermo sequenase cycle sequencing kit (Affymetrix) according to manufacturer protocols using PCR templates (amplified from pT3-6S with universal and reverse primers) and the same labeled oligonucleotide used for primer extension.
Native gel electrophoresis binding assay
32P-labelled 6S RNA, wild type (WT) or variants (as indicated) (1-10 nM final) were incubated with Eσ70 (0-40 nM final, Epicentre) in 10 μL reactions containing 20 mM HEPES, pH 7.5, 120 mM KCl, 0.5 mM MgCl2, 5% (v/v) glycerol, and 1 mM DTT for 10-15 mins at room temperature as previously described (Wassarman and Saecker, 2006). Note binding of WT 6S RNA to Eσ70 is complete by 1 min under these conditions (Klocko and Wassarman, 2009). Binding specificity was challenged by incubation for 2 mins at room temperature with heparin (100 μg/mL final), followed by addition of equal volume load buffer [50% (v′v) glycerol, 0.5× TBE, ∼0.01% xylene cyanol and bromophenol blue) and immediate loading onto a running native polyacrylamide gel (5% polyacrylamide (National Diagnostics Protogel, 37.5:1, 5% glycerol, 0.5× TBE) that had been prerun for 30 min. Electrophoresis was at 200V (15×18cm gel, 1 mm thick) for ∼2hrs at 4 °C. Following electrophoresis, gels were dried and visualized on a Typhoon phosphorimager. The percentage bound was calculated as the signal of Eσ70 bound RNA compared to the “total” RNA in the bound and the major free fraction. Some 6S RNAs formed minor alternative structures visible as additional bands in the RNA alone and were not considered in the quantification of percentage bound. Experiment shown is representative of binding observed in at least three independent experiments.
Construction and purification of σ mutations
Using the full-length σ70 pETsumo construct previously described, point mutations of σ70 were generated using site directed mutagenesis by standard PCR. The primers used for mutagenesis of σ70 were as follows:
-
RpoD_F401L, CACCAACCGTGGCTTGCAGCTGCTTGACCTGATTCAGGAAG;
RpoD_F401L_rev, CTTCCTGAATCAGGTCAAGCAGCTGCAAGCCACGGTTGGTG;
RpoD_K502E_for, GCAAAGTGCTGAAGATCGCCGAAGAGCCAATCTCCATGGAAAC;
RpoD_K502E_rev, GTTTCCATGGAGATTGGCTCTTCGGCGATCTTCAGCACTTTGC;
RpoD_D513E_for, CATGGAAACGCCGATCGGTGGCGATGAAGATTCGCATCTGG;
RpoD_D513E_rev, CCAGATGCGAATCTTCATCGCCACCGATCGGCGTTTCCATG;
RpoD_K593E_for, CGTCAGATCGAAGCGGAAGCGCTGCGCAAACTG;
RpoD_K593E_rev, CAGTTTGCGCAGCGCTTCCGCTTCGATCTGACG;
RpoD_H600E_for, GCGCTGCGCAAACTGCGTGAACCGAGCCGTTCTGAAGTG;
RpoD_H600E_rev, CACTTCAGAACGGCTCGGTTCACGCAGTTTGCGCAGCGC.
The mutant σ70 proteins were purified using the same procedure as wild-type σ70.
Point mutants of σS were made using the pET29A construct that was previously described and standard PCR mutagenesis. Primers used for this mutagenesis were as follows:
-
RpoS_E217K-rev, GTCTACCGAGGTAATGCGTTTGTTAAGACGAAGCATACG;
RpoS_E217K_for, CGTATGCTTCGTCTTAACAAACGCATTACCTCGGTAGAC;
RpoS_E308K_for, GTGTTCGCCAGATTCAGGTTAAAGGCCTGCGCCGTTTGCGC;
RpoS_E308K_rev, GCGCAAACGGCGCAGGCCTTTAACCTGAATCTGGCGAACAC;
RpoS_E315H_for, GGCCTGCGCCGTTTGCGCCACATCCTGCAAACGCAGGGG;
RpoS_E315H_rev, CCCCTGCGTTTGCAGGATGTGGCGCAAACGGCGCAGGCC;
RpoS_G228D_for, GTAGACACCCCGCTGGGTGATGATTCCGAAAAAGCGTTG;
RpoS_G228D_rev, CAACGCTTTTTCGGAATCATCACCCAGCGGGGTGTCTAC;
RpoS_L116F_for, GGCAATCGTGGTCTGGCGTTCCTGGACCTTATCGAAGAG;
RpoS_L116F_rev, CTCTTCGATAAGGTCCAGGAACGCCAGACCACGATTGCC.
Wild-type and mutant σS proteins were purified using the refolding method previously described with the exclusion of gel filtration (Nguyen and Burgess, 1996).
Abortive transcription initiation assays
The T7A1 promoter template was amplified from T7 genomic DNA using primers, t7a1 -160 (gacgccttgttgttagcc) and t7a1 +78 (gtgcgacttatcaggctgtctac). The DPS100 promoter template (Grainger et al., 2008) was amplified from a pRS vector containing the dps gene using primers DPS100for (gccacctgacgtctaagaaac) and DPS100rev (ggtctacctcaagactccag). The DNA templates were amplified using standard PCR, then sodium acetate/isopropanol-precipitated and gel-purified (Qiagen).
Eco full-length or Δ1.1σ70 or σS (2.5 pmol) was added to Eco ΔαCTD-RNAP (0.5 pmol) in transcription buffer (40 mM HEPES, pH 7.5, 100 mM KCl, 0.5 mM MgCl2, 1 mM DTT) and incubated at 37 °C for 10 mins to for m holoenzyme. 6S RNA or T7A1 (1 pmol) templates were each added and incubated at 37 °C for 5 mins to allow for binding. To initiate transcription for T7A1 and 6S RNA, 0.1 mM ApU dinucleotide primer (Trilink), 40 μM CTP, and 0.2 μCi α-P32-CTP were added together with 4.5 mM MgCI2. To initiate transcription for DPS100, the substrates 0.1 mM GpU dinucleotide primer (Trilink), 40 μM UTP, and 0.2 μCi α-P32-UTP were added together with 4.5 mM MgCl2. Final reaction volumes were 10 μL. The reactions were incubated at 37*0 for 5 mins and then quenched with 2× stop b uffer (1× TBE, pH 8.3, 7 M urea, 1% SDS, 10 mM EDTA, pH 8.0, 0.05% bromophenol blue). Samples were analyzed by denaturing PAGE and quantified by phosphoimagery using the program ImageQuant (GE Healthcare).
Quantification and Statistical Analysis
To quantify the abortive transcription assays (Figures 7C, S7B), mean values and the standard error of the mean from at least three independent measurements were calculated. The local resolution of the cryoEM map (Figure S4) was estimated using blocres (Cardone et al., 2013) with the following parameters: box size 20, verbose 5, maxresolution 2.0 Å, and cutoff 0.5. The quantification and statistical analyses for model refinement and validation were integral parts of the software algorithms used [MolProbity: (Chen et al., 2010); PHENIX: (Adams et al., 2010)].
Data and Software Availability
The accession numbers for the data reported in this paper are PDB: 5VT0.
Supplementary Material
Key Resources Table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Polyclonal antisera against E. coli σ70 | Wassarman and Storz, 2000 | WI-166 |
| Bacterial and Virus Strains | ||
| E. coli BL21(DE3)T-H/pEcrpo(HX-)ABCZ | Twist et al., 2011 | N/A |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| ApU RNA dinucleotide | Trilink | O-31004 |
| γ-P32-ATP | Perkin Elmer/New England Nuclear | BLU502Z |
| p-Benzoylphenylalanin | Bachem | F2800 |
| Bio-Rex 70 cation exchange resin, analytical grade, 100-200 mesh | Bio-Rad | 1425842 |
| 3-([3-Cholamidopropyl]dimethylammonio)-2-hydroxy-1-propanesulfonate (CHAPSO) | Sigma-Aldrich | C4695 |
| α-P32-CTP | Perkin Elmer/New England Nucleotides | BLU508H |
| GpU RNA dinucleotide | Trilink | O-31012 |
| Heparin sodium salt from porcine intestinal mucose, grade I-A | Sigma-Aldrich | H3149-100KU |
| HiLoad 26/600 Superdex 200 pg | GE Healthcare Life Sciences | 28989336 |
| Hitrap Heparin HP | GE Healthcare Life Sciences | 17040601 |
| Hitrap IMAC HP | GE Healthcare Life Sciences | 17092003 |
| Hitrap Q HP | GE Healthcare Life Sciences | 17115301 |
| Polyethyleneimine, ∼M.N.60,000, 50% wt.% aqueous solution, branched, Acros Organics 178572500 | Fisher Scientific | AC178572500 |
| RNase A, Ambion | ThermoFisher Scientific | AM2270 |
| RNase T1, Ambion | ThermoFisher Scientific | AM2283 |
| RNase V1, Ambion | ThermoFisher Scientific | AM2275 |
| Superose 6 INCREASE 10/300 GL | GE Healthcare Life Sciences | 29091596 |
| T3 RNA polymerase | Roche | 11031163001 |
| Thermoscript Reverse Transcriptase, Invitrogen | ThermoFisher Scientific | 12236-014 |
| α-P32-UTP | Perkin Elmer/New England Nucleotides | BLU507H |
| Critical Commercial Assays | ||
| RnaseAlert QC system, Ambion | ThermoFisher Scientific | AM1966 |
| T3 Megascript kit, Ambion | ThermoFisher Scientific | Am1338 |
| Thermo sequenase cycle sequencing kit, Affymetrix | ThermoFisher Scientific | 785001KT |
| Deposited Data | ||
| Coordinates of A-form RNA | Wild et al., 1999 | PDB: 1D4R |
| Coordinates of A-form RNA | Deng et al., 2003 | PDB: 1LNT |
| Coordinates of A-form RNA | Klosterman et al., 1999 | PDB: 1QC0 |
| Coordinates of A-form RNA | Portmann et al., 1995 | PDB: 1RXB |
| Coordinates of B-form DNA | Grzeskowiak et al., 1991 | PDB: 1D23 |
| Coordinates of B-form DNA | Quintana et al., 1992 | PDB: 1D49 |
| Coordinates of B-form DNA | Yuan et al., 1992 | PDB: 1D56 |
| Coordinates of B-form DNA | Yuan et al., 1992 | PDB: 1D57 |
| Coordinates of B-form DNA | Grzeskowiak et al., 1993 | PDB: 158D |
| Coordinates of E. coli Eσ70 | Bae et al., 2013 | PDB: 4LK1 |
| Coordinates and cryoEM map of E. coli 6S RNA/Eσ70 | This paper | PDB: 5VT0 |
| Coordinates of Mycobacterium smegmatis EσA upstream fork promoter complex | Hubin et al., 2017 | PDB: 5WT1 |
| Coordinates of Thermus aquaticus EσA open promoter complex | Bae et al., 2015 | PDB: 4XLN |
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| Recombinant DNA | ||
| E. coli 6S RNA derivatives in pUC57 | GenScript | N/A |
| E. coli 6S RNA derivatives in pCR-2.1 | InVitrogen | N/A |
| pACYCDuet-1_Ec_rpoZ | Bae et al., 2015 | N/A |
| pEcrpo(HX-)ABCZ | Twist et al., 2011 | N/A |
| pETsumoEcrpoD | This paper | N/A |
| pETsumoEcΔ1.1rpoD | This paper | N/A |
| pIA900 | Winkelman et al., 2015 | N/A |
| pRC9 | He et al., 1997 | N/A |
| pRLG13105 | Winkelman et al., 2015 | N/A |
| pRS | Grainger et al., 2008 | N/A |
| RpoS_pET29A | This paper | N/A |
| Software and Algorithms | ||
| 3DNA | Lu and Olson, 2008 | http://x3dna.org/ |
| Blocres | Cardone et al., 2013 | https://lsbr.niams.nih.gov/bsoft/programs/blocres.html |
| Chimera | Pettersen et al., 2004 | https://www.cgl.ucsf.edu/chimera |
| COOT | Emsley and Cowtan, 2004 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot |
| CTFFIND4 | Rohou and Grigorieff, 2015 | http://grigoriefflab.janelia.org/ctffind4 |
| Direct-detector-align_lmbfgs | Rubinstein and Brubaker, 2015 | https://sites.google.com/site/rubinsteingroup/direct-detector-align_lmbfgs |
| EMAN2 | Tang et al., 2007 | http://blake.bcm.edu/emanwiki/EMAN2 |
| ERRASER | Chou et al, 2016 | https://www.phenix-online.org/documentation/reference/erraser.html |
| GraphPad Prism | GraphPad | https://www.graphpad.com/scientific-software/prism |
| Mcfold | Parisien and Major, 2008 | http://www.major.iric.ca/MC-Fold/ |
| MolProbity | Chen et al., 2010 | http://molprobity.biochem.duke.edu |
| PHENIX | Adams et al., 2010 | https://www.phenix-online.org |
| Pymol | PyMOL | http://www.pymol.org |
| Rcrane | Keating and Pyle, 2010 | http://pylelab.org/form/rcrane |
| RELION | Scheres, 2012 | http://www2.mrc-lmb.cam.ac.uk/relion |
| Serial EM | Mastronarde, 2005 | http://bio3d.colorado.edu/SerialEM |
| Unblur | Grant and Grigorieff, 2015 | Heep://grigoriefflab.janelia.org/unblur |
| Other | ||
| C-flat CF-1.2/1.3 400 mesh copper grids | Electron Microscopy Sciences | CF413-100 |
Highlights.
CryoEM structure of an E. coli 6S RNA/RNA polymerase complex at 3.8 Å resolution
Duplex RNA segments have A-form sugar puckers but overall architecture mimics B-form
6S RNA specificity for σ70-holo over σS-holo is largely determined by two amino acids
6S RNA mimics B-form DNA to directly regulate transcription by the DNA-dependent RNAP
Acknowledgments
We thank A.M. Pyle for help with RNA structural analysis and insightful discussions, R.M. Saecker for insightful discussions, and W.T. McAllister for advice on in vitro RNA synthesis and for the mutant T7 RNAP. We thank K. Uryu and D. Acehan at The Rockefeller University Electron Microscopy Resource Center for help with EM sample preparation, and M. Ebrahim and J. Sotiris at The Rockefeller University Cryo-EM Resource Center for help with data collection. This work was supported by National Institute of Health grants R01 GM067955 to K.M.W. and J.T.W., R01 GM37048 to R.L. Gourse, and R35 GM118130 to S.A.D.
Footnotes
Author Contributions: J.C. performed protein purification and cryoEM experiments. K.M.W. performed the RNA/protein crosslinking and crosslink mapping experiments, RNA binding and RNA mutant analyses. J.T.W. performed protein purification of BPA-RNAP variants. J.C., S.F., and K.L. performed biochemical experiments. A.F. performed the RNA footprinting analyses. Z.L. and T.W. assisted with initial cryoEM data collection and processing. J.C. and E.A.C. developed and performed in vitro RNA synthesis and purification for cryoEM. J.C., K.M.W., E.A.C. and S.A.D. analyzed data. S.A.D. prepared the manuscript with input from all co-authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae B, Davis E, Brown D, Campbell EA, Wigneshweraraj SR, Darst SA. Phage T7 Gp2 inhibition of Escherichia coli RNA polymerase involves misappropriation of σ70 domain 1.1. Proc Natl Acad Sci USA. 2013;110:19772–19777. doi: 10.1073/pnas.1314576110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae B, Feklistov A, Lass-Napiorkowska A, Landick R, Darst SA. Structure of a bacterial RNA polymerase holoenzyme open promoter complex. Elife. 2015;4:e08504. doi: 10.7554/eLife.08504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrick JE, Sudarsan N, Weinberg Z, Ruzzo WL, Breaker RR. 6S RNA is a widespread regulator of eubacterial RNA polymerase that resembles an open promoter. RNA. 2005;11:774–784. doi: 10.1261/rna.7286705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckmann BM, Hoch PG, Marz M, Willkomm DK, Salas M, Hartmann RK. A pRNA-induced structural rearrangement triggers 6S-1 RNA release from RNA polymerase in Bacillus subtilis. EMBO J. 2012;31:1727–1738. doi: 10.1038/emboj.2012.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardone G, Heymann JB, Steven AC. One number does not fit all: mapping local variations in resolution in cryo-EM reconstructions. J Struct Biol. 2013;184:226–236. doi: 10.1016/j.jsb.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh AT, Sperger JM, Wassarman KM. Regulation of 6S RNA by pRNA synthesis is required for efficient recovery from stationary phase in E. coli and B. subtilis. Nucl Acids Res. 2012;40:2234–2246. doi: 10.1093/nar/gkr1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh AT, Wassarman KM. 6S RNA, a global regulator of transcription in Escherichia coli, Bacillus subtilis, and beyond. Annu Rev Microbiol. 2014;68:45–60. doi: 10.1146/annurev-micro-092611-150135. [DOI] [PubMed] [Google Scholar]
- Cavanagh AT, Klocko AD, Liu X, Wassarman KM. Promoter specificity for 6S RNA regulation of transcription is determined by core promoter sequences and competition for region 4.2 of σ70. Mol Microbiol. 2008;67:1242–1256. doi: 10.1111/j.1365-2958.2008.06117.x. [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou FC, Echols N, Terwilliger TC, Das R. RNA Structure refinement using the ERRASER-Phenix pipeline. Methods Mol Biol. 2016;1320:269–282. doi: 10.1007/978-1-4939-2763-0_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou FC, Sripakdeevong P, Dibrov SM, Hermann T, Das R. Correcting pervasive errors in RNA crystallography through enumerative structure prediction. Nat Methods. 2013;10:74–76. doi: 10.1038/nmeth.2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egli M, Saenger W. Principles of Nucleic Acid Structure. New York, NY: Springer Science & Business Media; 2013. [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Deng J, Xiong Y, Pan B, Sundaralingam M. Structure of an RNA dodecamer containing a fragment from SRP domain IV of Escherichia coli. Acta Crystallogr D Biol Crystallogr. 2003;59:1004–1011. doi: 10.1107/s0907444903006747. [DOI] [PubMed] [Google Scholar]
- Feklistov A, Darst SA. Structural basis for promoter -10 element recognition by the bacterial RNA polymerase σ subunit. Cell. 2011;147:1257–1269. doi: 10.1016/j.cell.2011.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaal T, Ross W, Estrem ST, Nguyen LH, Burgess RR, Gourse RL. Promoter recognition and discrimination by EsigmaS RNA polymerase. Mol Microbiol. 2001;42:939–954. doi: 10.1046/j.1365-2958.2001.02703.x. [DOI] [PubMed] [Google Scholar]
- Gottesman S, Storz G. Bacterial small RNA regulators: versatile roles and rapidly evolving variations. Cold Spring Harb Perspect Biol. 2011;3:a003798. doi: 10.1101/cshperspect.a003798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grainger DC, Goldberg MD, Lee DJ, Busby SJW. Selective repression by Fis and H-NS at the Escherichia coli dps promoter. Mol Microbiol. 2008;68:1366–1377. doi: 10.1111/j.1365-2958.2008.06253.x. [DOI] [PubMed] [Google Scholar]
- Grant T, Grigorieff N. Measuring the optimal exposure for single particle cryo-EM using a 2.6 Å reconstruction of rotavirus VP6. Elife. 2015;4:e06980. doi: 10.7554/eLife.06980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzeskowiak K, Goodsell DS, Kaczor-Grzeskowiak M, Cascio D, Dickerson RE. Crystallographic analysis of C-C-A-A-G-C-T-T-G-G and its implications for bending in B-DNA. Biochemistry. 1993;32:8923–8931. doi: 10.1021/bi00085a025. [DOI] [PubMed] [Google Scholar]
- Grzeskowiak K, Yanagi K, Privé GG, Dickerson RE. The structure of B-helical C-G-A-T-C-G-A-T-C-G and comparison with C-C-A-A-C-G-T-T-G-G. The effect of base pair reversals. J Biol Chem. 1991;266:8861–8883. doi: 10.2210/pdb1d23/pdb. [DOI] [PubMed] [Google Scholar]
- He B, Rong M, Lyakhov D, Gartenstein H, Diaz G, Castagna R, McAllister WT, Durbin RK. Rapid mutagenesis and purification of phage RNA polymerases. Prot Expr Purif. 1997;9:142–151. doi: 10.1006/prep.1996.0663. [DOI] [PubMed] [Google Scholar]
- Hubin EA, Fay A, Xu C, Bean JM, Saecker RM, Glickman MS, Darst SA, Campbell EA. Structure and function of the mycobacterial transcription initiation complex with the essential regulator RbpA. Elife. 2017;6:e22520. doi: 10.7554/eLife.22520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaikkonen MU, Lam MTY, Glass CK. Non-coding RNAs as regulators of gene expression and epigenetics. Cardiovasc Res. 2011;90:430–440. doi: 10.1093/cvr/cvr097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating KS, Pyle AM. Semiautomated model building for RNA crystallography using a directed rotameric approach. Proc Natl Acad Sci USA. 2010;107:8177–8182. doi: 10.1073/pnas.0911888107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klocko AD, Wassarman KM. 6S RNA binding to Eσ70 requires a positively charged surface of σ70 region 4.2. Mol Microbiol. 2009;73:152–164. doi: 10.1111/j.1365-2958.2009.06758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klosterman PS, Shah SA, Steitz TA. Crystal structures of two plasmid copy control related RNA duplexes: An 18 base pair duplex at 1.20 Å resolution and a 19 base pair duplex at 1.55 Å resolution. Biochemistry. 1999;38:14784–14792. doi: 10.1021/bi9912793. [DOI] [PubMed] [Google Scholar]
- Koubek J, Lin KF, Chen YR, Cheng RP, Huang JJT. Strong anion-exchange fast performance liquid chromatography as a versatile tool for preparation and purification of RNA produced by in vitro transcription. RNA. 2013;19:1449–1459. doi: 10.1261/rna.038117.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane WJ, Darst SA. Molecular evolution of multisubunit RNA polymerases: sequence analysis. J Mol Biol. 2010;395:671–685. doi: 10.1016/j.jmb.2009.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonetto M, Gribskov M, Gross CA. The sigma 70 family: sequence conservation and evolutionary relationships. J Bacteriol. 1992;174:3843–3849. doi: 10.1128/jb.174.12.3843-3849.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu XJ, Olson WK. 3DNA: a versatile, integrated software system for the analysis, rebuilding and visualization of three-dimensional nucleic-acid structures. Nat Protoc. 2008;3:1213–1227. doi: 10.1038/nprot.2008.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra A, Severinova E, Darst SA. Crystal structure of a sigma 70 subunit fragment from E. coli RNA polymerase. Cell. 1996;87:127–136. doi: 10.1016/s0092-8674(00)81329-x. [DOI] [PubMed] [Google Scholar]
- Mastronarde DN. Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol. 2005;152:36–51. doi: 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Nguyen LH, Burgess RR. Overproduction and purification of sigmaS, the Escherichia coli stationary phase specific sigma transcription factor. Prot Expr Purif. 1996;8:17–22. doi: 10.1006/prep.1996.0069. [DOI] [PubMed] [Google Scholar]
- Panchapakesan SSS, Unrau PJ. E. coli 6S RNA release from RNA polymerase requires σ70 ejection by scrunching and is orchestrated by a conserved RNA hairpin. RNA. 2012;18:2251–2259. doi: 10.1261/rna.034785.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisien M, Major F. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature. 2008;452:51–55. doi: 10.1038/nature06684. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Portmann S, Usman N, Egli M. The crystal structure of r(CCCCGGGG) in two distinct lattices. Biochemistry. 1995;34:7569–7575. doi: 10.1021/bi00023a002. [DOI] [PubMed] [Google Scholar]
- Puglisi JD, Chen L, Blanchard S, Frankel AD. Solution structure of a bovine immunodeficiency virus Tat-TAR peptide-RNA complex. Science. 1995;270:1200–1203. doi: 10.1126/science.270.5239.1200. [DOI] [PubMed] [Google Scholar]
- Quintana JR, Grzeskowiak K, Yanagi K, Dickerson RE. Structure of a B-DNA decamer with a central T-A step: C-G-A-T-T-A-A-T-C-G. J Mol Biol. 1992;225:379–395. doi: 10.1016/0022-2836(92)90928-d. [DOI] [PubMed] [Google Scholar]
- Richardson JS, Schneider B, Murray LW, Kapral GJ, Immormino RM, Headd JJ, Richardson DC, Ham D, Hershkovits E, Williams LD, et al. RNA backbone: consensus all-angle conformers and modular string nomenclature (an RNA Ontology Consortium contribution) RNA. 2008;14:465–481. doi: 10.1261/rna.657708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohou A, Grigorieff N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J Struct Biol. 2015;192:216–221. doi: 10.1016/j.jsb.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal PB, Henderson R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J Mol Biol. 2003;333:721–745. doi: 10.1016/j.jmb.2003.07.013. [DOI] [PubMed] [Google Scholar]
- Rubinstein JL, Brubaker MA. Alignment of cryo-EM movies of individual particles by optimization of image translations. J Struct Biol. 2015;192:188–195. doi: 10.1016/j.jsb.2015.08.007. [DOI] [PubMed] [Google Scholar]
- Ryu Y, Schultz PG. Efficient incorporation of unnatural amino acids into proteins in Escherichia coli. Nat Methods. 2006;3:263–265. doi: 10.1038/nmeth864. [DOI] [PubMed] [Google Scholar]
- Scheres SHW. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider TD, Stephens RM. Sequence logos: a new way to display consensus sequences. Nucl Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shephard L, Dobson N, Unrau PJ. Binding and release of the 6S transcriptional control RNA. RNA. 2010;16:885–892. doi: 10.1261/rna.2036210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shultzaberger RK, Chen Z, Lewis KA, Schneider TD. Anatomy of Escherichia coli σ70 promoters. Nucl Acids Res. 2007;35:771–788. doi: 10.1093/nar/gkl956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steuten B, Hoch PG, Damm K, Schneider S, Köhler K, Wagner R, Hartmann RK. Regulation of transcription by 6S RNAs: insights from the Escherichia coli and Bacillus subtilis model systems. RNA Biol. 2014;11:508–521. doi: 10.4161/rna.28827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ. EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Tomsic M, Tsujikawa L, Panaghie G, Wang Y, Azok J, deHaseth PL. Different roles for basic and aromatic amino acids in conserved region 2 of Escherichia coli sigma(70) in the nucleation and maintenance of the single-stranded DNA bubble in open RNA polymerase-promoter complexes. J Biol Chem. 2001;276:31891–31896. doi: 10.1074/jbc.M105027200. [DOI] [PubMed] [Google Scholar]
- Trotochaud AE, Wassarman KM. A highly conserved 6S RNA structure is required for regulation of transcription. Nat Struct Mol Biol. 2005;12:313–319. doi: 10.1038/nsmb917. [DOI] [PubMed] [Google Scholar]
- Twist KA, Husnain SI, Franke JD, Jain D, Campbell EA, Nickels BE, Thomas MS, Darst SA, Westblade LF. A novel method for the production of in vivo-assembled, recombinant Escherichia coli RNA polymerase lacking the α C-terminal domain. Protein Sci. 2011;20:986–995. doi: 10.1002/pro.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner SD, Yakovchuk P, Gilman B, Ponicsan SL, Drullinger LF, Kugel JF, Goodrich JA. RNA polymerase II acts as an RNA-dependent RNA polymerase to extend and destabilize a non-coding RNA. EMBO J. 2013;32:781–790. doi: 10.1038/emboj.2013.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassarman KM, Saecker RM. Synthesis-mediated release of a small RNA inhibitor of RNA polymerase. Science. 2006;314:1601–1603. doi: 10.1126/science.1134830. [DOI] [PubMed] [Google Scholar]
- Wassarman KM, Storz G. 6S RNA regulates E. coli RNA polymerase activity. Cell. 2000;101:613–623. doi: 10.1016/s0092-8674(00)80873-9. [DOI] [PubMed] [Google Scholar]
- Wild K, Weichenrieder O, Leonard GA, Cusack S. The 2 Å structure of helix 6 of the human signal recognition particle RNA. Structure. 1999;7:1345–1352. doi: 10.1016/s0969-2126(00)80024-6. [DOI] [PubMed] [Google Scholar]
- Winkelman JT, Winkelman BT, Boyce J, Maloney MF, Chen AY, Ross W, Gourse RL. Crosslink mapping at amino acid-base resolution reveals the path of scrunched DNA in initial transcribing complexes. Mol Cell. 2015;59:768–780. doi: 10.1016/j.molcel.2015.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakovchuk P, Goodrich JA, Kugel JF. B2 RNA and Alu RNA repress transcription by disrupting contacts between RNA polymerase II and promoter DNA within assembled complexes. Proc Natl Acad Sci USA. 2009;106:5569–5574. doi: 10.1073/pnas.0810738106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, Kumar RA, Patel DJ. Molecular recognition in the bovine immunodeficiency virus Tat peptide-TAR RNA complex. Chem Biol. 1995;2:827–840. doi: 10.1016/1074-5521(95)90089-6. [DOI] [PubMed] [Google Scholar]
- Yuan H, Quintana J, Dickerson RE. Alternative structures for alternating poly(dA-dT) tracts: the structure of the B-DNA decamer C-G-A-T-A-T-A-T-C-G. Biochemistry. 1992;31:8009–8021. [PubMed] [Google Scholar]
- Zhang Y, Feng Y, Chatterjee S, Tuske S, Ho MX, Arnold E, Ebright RH. Structural basis of transcription initiation. Science. 2012;338:1076–1080. doi: 10.1126/science.1227786. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.