

Abstract
Monoclonal gammopathy of undetermined significance is a pre-malignant precursor of multiple myeloma with a 1% risk of progression per year. Although targeted analyses have shown the presence of specific genetic abnormalities such as IGH translocations, RB1 deletion, 1q gain, hyperdiploidy or RAS gene mutations, little is known about the molecular mechanism of malignant transformation. We performed whole exome sequencing together with comparative genomic hybridization plus single nucleotide polymorphism array analysis in 33 flow-cytometry-separated abnormal plasma cell samples from patients with monoclonal gammopathy of undetermined significance to describe somatic gene mutations and chromosome changes at the genome-wide level. Non-synonymous mutations and copy-number alterations were present in 97.0% and in 60.6% of cases, respectively. Importantly, the number of somatic mutations was significantly lower in monoclonal gammopathy of undetermined significance than in myeloma (P<10−4) and we identified six genes that were significantly mutated in myeloma (KRAS, NRAS, DIS3, HIST1H1E, EGR1 and LTB) within the monoclonal gammopathy of undetermined significance dataset. We also found a positive correlation with increasing chromosome changes and somatic gene mutations. IGH translocations, comprising t(4;14), t(11;14), t(14;16) and t(14;20), were present in 27.3% of cases and in a similar frequency to myeloma, consistent with the primary lesion hypothesis. MYC translocations and TP53 deletions or mutations were not detected in samples from patients with monoclonal gammopathy of undetermined significance, indicating that they may be drivers of progression to myeloma. Data from this study show that monoclonal gammopathy of undetermined significance is genetically similar to myeloma, however overall genetic abnormalities are present at significantly lower levels in monoclonal gammopathy of undetermined significant than in myeloma.
Introduction
Monoclonal gammopathy of undetermined significance (MGUS) is one of the most common pre-malignant conditions and affects 3.2% of people over 50 years old, 5.3% over 70 and 7.5% over the age of 85 years.1,2 MGUS is characterized by a serum monoclonal protein <30 g/L, <10% plasma cells in the bone marrow, and the absence of end-organ damage (CRAB: hypercalcemia, renal insufficiency, anemia, or bone lesions). The MGUS progresses to multiple myeloma (MM) in approximately 1% of patients per year.3 Risk stratification models have been proposed to assess risk of transformation using flow-cytometry and serum free light chain.4–6
Advances in molecular genetics have opened up the possibility of identifying genetic events involved in malignant transformation. Previous studies in MGUS have shown that interphase fluorescence in situ hybridization can detect known myeloma-specific chromosomal abnormalities in MGUS patients. These chromosomal abnormalities include IGH (14q32) translocations, RB1 (13q14) deletion, 1q gain and hyperdiploidy. These abnormalities are present at lower frequencies in MGUS than in myeloma.7–9 The potential prognostic significance of these abnormalities in relation to the progression of MGUS has not been specified.10 It has been shown that the incidence of these variants increases from MGUS through smoldering MM (SMM) to MM.11 Single nucleotide polymorphism (SNP) arrays have also been used to detect copy-number alterations (CNA) and these also increase in frequency from MGUS (5/patient) through SMM (7.5/patient) to MM (12/patient).12
Activation of proto-oncogenes, such as activation of KRAS, NRAS, MYC and BRAF, has been less frequently described in MGUS than in myeloma.13–15 In a previous study,16 we described the exome mutation profile of four MGUS patients which suggested that genomic complexity increased from MGUS, through SMM, MM and plasma cell leukemia. To understand the molecular pathogenesis of MGUS and the role of genetic events in relation to malignant transformation, more genome-wide studies in MGUS datasets are required and in this study we performed a comprehensive analysis of flow-sorted abnormal plasma cells from 33 MGUS patients using whole exome sequencing together with comparative genomic hybridization (CGH)+SNP arrays.
Methods
Patients’ samples
Overall, 33 MGUS patients from centers in the Czech Republic (Brno, Prague and Ostrava) were included in this study, which was approved by the University Hospital Brno Ethical Committee, after giving informed consent (Online Supplementary Table S1). Bone marrow plasma cells were isolated from the mononuclear cell fraction with a FACSAria (BD Biosciences, San Jose, CA, USA) using CD138-PE, CD19-APC and CD56-FITC antibodies (Beckman Coulter, Brea, CA, USA or Exbio, Prague, Czech Republic) to obtain a phenotypically abnormal plasma cell population (CD138+CD19−CD56+/−)17 with a median purity of 99.0% (range, 93.6–99.9%). The flow-cytometry data before and after plasma cell sorting are presented in Online Supplementary Figures S1 and S2. The median number of sorted cells was 57×103 (range, 15×103 – 480×103). Tumor DNA was isolated using a Gentra Puregene Kit, amplified using the REPLI-g Midi Kit and purified using the QIAamp DNA Mini Kit (all from Qiagen, Hilden, Germany). Previous studies had demonstrated the suitability of whole-genome-amplified DNA for array-CGH18–20 as well as next-generation sequencing21–23 analysis. Control DNA was obtained from peripheral white blood cells using a MagNA Pure System (Hoffmann-La Roche, Basel, Switzerland). The quality and quantity of DNA were measured by Qubit Fluorometer, Pico-green (both from Thermo Fisher Scientific, Waltham, MA, USA) and/or 2200 Tapestation (Agilent Technologies, Santa Clara, CA, USA).
Comparative genomic hybridization and single nucleotide polymorphism arrays
As previously described, 2–3 μg of whole-genome amplified tumor DNA and Agilent Euro Male/Female (Agilent Technologies) as control DNA were fragmented by AluI and RsaI (both from Promega, Madison, WI, USA) restriction enzymes and fluorescently labeled with the BioPrime Total for Agilent aCGH Kit (Thermo Fisher Scientific) or treated with the SureTag Complete DNA Labeling Kit (Agilent Technologies).24 After purification of labeled DNA, tumor and control DNA samples were combined with COT Human DNA (Hoffmann-La Roche) and hybridization mix (Oligo aCGH Hybridization Kit, Agilent Technologies), and co-hybridized to SurePrint G3 CGH+SNP, 4×180K (Agilent Technologies) arrays. After hybridization and washing, DNA microarrays were scanned using a Microarray Scanner (Agilent Technologies) with 3 μm resolution. Feature Extraction Software 12.0.2.2 (Agilent Technologies) was used for data extraction and quality control evaluation. Genomic Workbench 7.0.4.0 (Agilent Technologies) was used for CNA calling by the ADM-2 algorithm with the following settings: ≥100 kb size, ≥0.2 fold change of log2 ratio, ≥5 consecutive probes. CNA were manually curated and the default Database of Genomic Variants (http://www.openhelix.com) for hg19 was used to eliminate common human population copy-number variants. The array data supporting the results of this article are available at Gene Expression Omnibus (GEO), National Center for Biotechnology Information (NCBI) under the accession number GSE77979.
Exome sequencing
A previously published protocol was used for exome sequencing.25 A total of 200 ng DNA from peripheral blood and 3 μg whole-genome amplified tumor DNA were fragmented by the Covaris E-Series. Fragmented DNA was end-repaired, A-tailed and adaptors ligated by the NEBNext DNA library prep master mix set for Illumina (New England Biolabs, Ipswich, MA, USA). Modified DNA was amplified by NEBNext High-fidelity polymerase chain reaction (PCR) master mix using either eight or four PCR cycles in the case of control and tumor DNA, respectively. A total of 750 ng amplified DNA was hybridized to custom-designed RNA baits overnight (SureSelect Human All Exon V5, Agilent Technologies; enriched for IGH, IGK, IGL and MYC region capture). Captured DNA was indexed and amplified by Herculase II fusion DNA polymerase (Agilent Technologies) for eight PCR cycles. Samples were sequenced using a HiSeq 2000 (Illumina, San Diego, CA, USA) using four pooled samples per lane and 76-bp paired-ends reads. Additional information about data quality metrics and processing, somatic mutation calling and non-negative matrix factorization is given in the Online Supplementary Methods. Sequence read data for this study have been submitted to the European Genome-Phenome Archive (EGA) under accession number EGAS00001001658. The findings from the 33 MGUS patients in this study were compared to data from a cohort of 463 newly diagnosed MM (NDMM) patients from a previous study (EGAS00001001147).26
Basic statistical analysis
Data were analyzed using Statistica 12 software (StatSoft, Prague, Czech Republic) and MedCalc 14.8.1 software (MedCalc Software, Ostend, Belgium). Statistical tests were used as follows: the Fisher exact test for categorical data, the Mann-Whitney U test for continuous variables and Pearson correlation. P values ≤0.05 were considered statistically significant.
Results
Fewer copy-number changes are found in monoclonal gammopathy of undetermined significance than in multiple myeloma
Using high-density oligonucleotide CGH+SNP arrays, CNA were detected in 60.6% (20/33) of MGUS patients in comparison to 100% of MM patients, which were described in our previous study.27 A summary plot of CNA in the 33 MGUS cases is given in Online Supplementary Figure S3 and frequencies of CNA at the chromosome-arm level are listed in Online Supplementary Table S2. We found 123 CNA (42 losses and 81 gains). Although CGH+SNP arrays with higher resolution were used in this study, the median number of CNA per patient was only two (range, 0–15), fewer than the 16 (range, 1–52) found in the MM dataset.27
Numerical CNA were present in 54.5% (18/33) of cases; whole chromosome losses and whole chromosome gains were found in 39.4% (13/33) and 30.3% (10/33) of cases, respectively. Analogous to MM, we identified two distinct subgroups within MGUS: non-hyperdiploid and hyperdiploid. Non-hyperdiploidy was present in 72.7% (24/33) of patients and we distinguished subtypes within this subgroup as hypodiploid, pseudodiploid and diploid in 18.2% (6/33), 9.1% (3/33) and 45.5% (15/33) of cases, respectively. The most frequently lost chromosomes were 13 (27.3%, 9/33), X (18.2%, 6/33) and Y (12.1%, 4/33). Hyperdiploidy was detected in 27.3% (9/33) of cases and the most frequently gained chromosomes were 9 (27.3%, 9/33), 19 (27.3%, 9/33) and 3 (18.2%, 6/33). The median number of chromosomes in hyperdiploid patients was 52 (range, 48–55). Interestingly, 88.9% (8/9) of hyperdiploid patients also carried structural abnormalities in comparison to 29.2% (7/24) of non-hyperdiploid patients (P=4.39×10−3). On the other hand, 54.2% (13/24) of non-hyperdiploid patients had no CNA detected by CGH+SNP arrays.
Structural abnormalities were seen in 45.5% (15/33) of MGUS samples. These related to changes in complete chromosome arms in 30.3% (10/33) of patients, mostly 1q gain (27.3%, 9/33) and 16q loss (6.1%, 2/33). Smaller interstitial changes were seen in 30.3% (10/33) of patients with a median of 0 (range, 0–4) changes per patient with a median size of 6.6 Mb (range, 0.1–88.8 Mb). We distinguished both interstitial and telomeric changes in 27.3% (9/33) and 15.2% (5/33) of patients, respectively. Recurrent deletions were detected at 1p and 6q, both in 6.1% (2/33) of samples, and at 14q in 12.1% (4/33) of patients. Only one case of homozygous deletion was found at 21q22.13, which did not include any known tumor-associated genes (Online Supplementary Figure S4). No deletions of 17p, a poor prognostic marker in MM and the location of TP53, were detected.
MYC translocations are not detected in monoclonal gammopathy of undetermined significance
The exome capture was enriched for the IGH (14q32), IGK (2p12), IGL (22q11.2) and MYC (8q24.21) loci, enabling analysis of the most frequent chromosomal translocations in MM.
We identified IGH translocations in 27.3% (9/33), consisting of t(11;14) in 12.1% (4/33), t(4;14) in 9.1% (3/33), t(14;16) in 3.0% (1/33) and t(14;20) in 3.0% (1/33) (Table 1). We defined the chromosome breakpoints on chromosomes 4, 11, 14, 16 and 20 (Online Supplementary Figure S5), and the findings did not differ from those in MM.28 All nine cases with an IGH translocation were non-hyperdiploid. Two males with a t(11;14) as well as one male with a t(14;20) did not have either numerical or structural CNA and were diploid. Similarly, two females with a t(11;14) only had loss of the X chromosome and were, therefore, pseudodiploid. All three MGUS cases with a t(4;14) had similar profiles with loss of chromosome 13 and loss of one gonosome (2 males with loss of Y and 1 female with loss of X) and were thus also hypodiploid (43, 44 and 44 chromosomes in a total). Furthermore, 66.7% (2/3) of those with t(4;14) also had 1q gain. The patient with a t(14;16) showed similarity to three cases of t(4;14) with 1q gain and 13q loss.
Table 1.
Frequency of IGH translocations in monoclonal gammopathy of undetermined significance compared to newly diagnosed multiple myeloma.
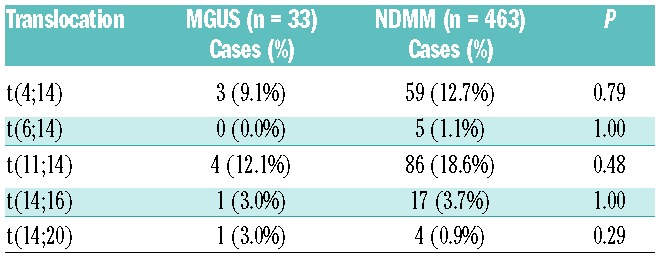
In this cohort of 33 MGUS patients we did not find any translocations involving MYC, even though they were detected in 18.4% of NDMM using the same assay.29
There are fewer mutations in monoclonal gammopathy of undetermined significance than in multiple myeloma
Acquired single nucleotide variants (SNV) were present in all (33/33) MGUS patients with a median of 89 (range, 9–315) per patient, most frequently as transition rather than transversion mutations, similarly to NDMM (Figure 1A,B). Exonic mutations and indels were found in a total of 857 genes, with 70.4% (603/857) of these being non-synonymous SNV (NS-SNV) (Figure 1C,D). These mutations were present at a significantly lower level in MGUS than in NDMM (P<10−4) (Figure 2; Online Supplementary Table S3). However, 73.5% (443/603) of genes affected by NS-SNV intersected with genes mutated in NDMM. NS-SNV were present in 97.0% (32/33) of cases with a median of 19 (range, 0–70) NS-SNV per patient.
Figure 1.

Basic sequencing characteristics of the study. (A) Number of specific variants by nucleotide changes. (B) Comparison of specific variants by proportion of nucleotide changes in MGUS and NDMM. (C) Number of variants by their effect on transcription. (D) Comparison of proportion of variants by type in MGUS and NDMM.
Figure 2.

Number of single nucleotide variants in 33 patients with monoclonal gammopathy of undetermined significance compared to 463 patients with newly diagnose multiple myeloma.
We did not find any significantly mutated genes, but overall 35 genes were recurrently mutated and only three genes were mutated in more than two cases: KLHL6 (missense mutations p.L90V p.L71Q and a c.-20T>A mutation in the translation start site, a gene mutated in 13 patients in NDMM), NPIPL2 (3 cases with missense mutation p.H211R) and AKAP9 (missense mutations p.S3313N, p.N2792S and p.R1973T; mutated in 6 NDMM).
In five MGUS cases we identified SNV in six genes which were found to be significantly mutated in NDMM, including KRAS (n=2), HIST1H1E (n=2) and NRAS, DIS3, EGR1, LTB (all n=1) (Online Supplementary Table S4). When a mutation was present in one of these genes the variant allele frequency was not significantly different in MGUS compared to NDMM (Online Supplementary Figure S6), but was often lower. The only example of variant allele frequency being equivalent in MGUS and NDMM was for HIST1H1E, which was clonal, which may indicate that it is a key driver.
We found one t(11;14) MGUS patient with two mutations in CCND1 (p.K50T, p.E51D), which are associated with a negative impact on survival in patients with MM. We did not find any mutations in TP53, ATM, ATR or ZFHX4, which have been identified as unfavorable factors for patients’ survival and are involved in the DNA repair pathway.
We tested for the presence of specific mutational signatures,30 which we have previously shown to be related to the pathological activity of specific cytidine deaminases of the APOBEC family.29 The APOBEC mutational signature was not found in this cohort of 33 MGUS patients, even among those with a t(14;16) or t(14;20), possibly suggesting that APOBEC activity does not drive disease progression in the MAF subgroup at the MGUS stage and that it is, instead, acquired later in the development of MM (Online Supplementary Figure S7). With regards to the APOBEC signature, the frequency of mutations was higher in t(14;16) MM than in other subgroups.29 However, as we had only one t(14;16) case in this series we could not conclusively show a higher mutation rate in the MGUS disease stage (Online Supplementary Figure S8).
Copy-number alterations are associated with increased mutation rate
Some associations between mutations and structural changes in NDMM have been described.26 In MGUS we identified a patient with t(11;14) with two CCND1 (p.K50T, p.E51D) mutations, a case with a DIS3 (p.D488N) mutation and 13q loss and a case with an EGR1 mutation (p.M29L) with hyperdiploidy. Although, DIS3 mutations are associated with t(4;14) or t(14;16) in NDMM, the MGUS case with a DIS3 mutation did not have an IGH translocation.
An association between KRAS mutations and t(11;14) has previously been documented, but neither of the two MGUS patients with KRAS (p.Q61L and p.A146T) mutations had a t(11;14); one had no IGH translocation and the other had t(14;20). We also identified a patient with both a KRAS (p.Q61L) and an NRAS (p.G13R) mutation which, although not mutually exclusive, are negatively correlated in NDMM. This patient was also hyperdiploid, which has a positive correlation with NRAS mutations in NDMM, and did not have deletion of 13q, which is negatively correlated with NRAS mutations in NDMM. The presence of more than one Ras pathway mutation in MM is associated with intraclonal heterogeneity, where the Ras mutations are present in different subclones. Here, the presence of two Ras pathway mutations indicates that heterogeneity can occur early in the disease process. Associations of specific SNV, CNA and clinical parameters are shown in Online Supplementary Figure S9.
Interestingly, MGUS patients with CNA and/or IGH translocations (n=23) had significantly higher numbers of total SNV (P=8.17×10−5), exonic SNV (P=1.43×10–4), NS-SNV (P=1.82×10−3) and synonymous SNV (S-SNV) (P=3.75×10−4) in comparison to MGUS patients without any of these changes (n=10) (Table 2). We also found a positive correlation of increasing number of SNV and chromosomal abnormalities (Figure 3).
Table 2.
Relationship between the number of single nucleotide variants and the presence of chromosomal abnormalities.

Figure 3.

Correlation analysis of increasing number of chromosome abnormalities and single nucleotide variants per sample in 33 patients with monoclonal gammopathy of undetermined significance. Chromosome abnormalities (CHA) include CNA tested by CGH+SNP arrays and IGH translocations defined by exome sequencing. (A) CHA and total SNV. (B) CHA and exonic SNV. (C) CHA and NS-SNV. (D) CHA and S-SNV.
Risk stratification of patients with monoclonal gammopathy of undetermined significance
Using a risk-stratification model,4 we divided 32 MGUS patients into low risk (n=14), intermediate-low risk (n=9) and intermediate-high risk (n=9). We found that the median number of CNA and/or IGH translocations increased from low to intermediate-low and intermediate-high risk groups: 0 (range, 0–10), 4 (range, 0–15) and 6 (range, 0–10), respectively (Table 3). Gain of 1q [present in 7.1% (1/14), 11.1% (1/9) and 66.7% (6/9) patients, in the three risk groups], as well as frequency of patients with at least one structural CNA [14.3% (2/14), 55.6% (5/9) and 77.8% (7/9), respectively], also increased with risk group. We did not find a clear increase of SNV across the risk groups (Table 3).
Table 3.
Number of chromosomal abnormalities and single nucleotide variants per case across the monoclonal gammopathy of undetermined significance risk groups.

Presence of clonal abnormalities is associated with higher risk of progression
MGUS cases were divided into six groups based on the structure of intratumor heterogeneity (Figure 4): 86.7% (13/15) of cases with at least one clonal CNA and NS-SNV showed intermediate-low/high risk, while other groups had small proportions of cases with higher risk of progression (29.4%, 5/17, P=0.002). This fact was caused by the association of clonal alterations with non-IgG variant (50.0%, 8/16; others: 11.8%, 2/17; P=0.03) and abnormal serum kappa/lambda free light chain ratio (73.3%, 11/15; others: 23.5%, 4/17; P=0.02). Chromosome abnormalities were preceded by gene mutations as a total of 63.6% (21/33) of cases showed at least one NS-SNV with a 10% or higher proportion than any CNA present. There were no examples with CNA but without NS-SNV, and no cases with a CNA at a frequency of 10% or greater than that of any NS-SNV.
Figure 4.

Intratumor heterogeneity in monoclonal gammopathy of undetermined significance. Each column shows the proportion (vertical axis) of NS-SNV (red points) and CNA (blue points) in each MGUS patient. The black line represents a frequency distribution of somatic changes. The presence of myeloma-significantly mutated genes, 1q gain and chromosome 13 loss is highlighted by dark colors. Red stars mark cases with at least one NS-SNV with 10% or higher proportion than any CNA present. Patients are divided into groups by clonal features of somatic alterations: (A) Subclonal NS-SNV and no CNA. (B) Clonal/subclonal NS-SNV and no CNA. (C) Subclonal NS-SNV and subclonal CNA. (D) Clonal/subclonal NS-SNV and subclonal CNA. (E) Clonal/sublonal NS-SNV and clonal/subclonal CNA. (F) Clonal/subclonal NS-SNV and clonal CNA. Only NS-SNV with a minimum 10% proportion are displayed. One of 33 patients is not shown as no NS-SNV and CNA were detected.
Discussion
MGUS is considered a relatively benign disease, being present in 3% of the population >50 years old but without evidence of end-organ damage. However, recent evidence indicates that nearly all cases of MM are preceded by an MGUS phase.31,32 Analysis of the genomes of MGUS samples has revealed that the genetic composition in this disorder is strikingly similar to that in MM, with the presence of IGH translocations, hyperdiploidy, gain 1q, and deletion 1p. However, these abnormalities are, in general, present at lower frequencies in the MGUS population.12 These abnormalities have been characterized in MGUS using classical cytogenetics and fluorescence in situ hybridization, as well as mapping arrays to detect changes at a higher resolution.11,12,33,34 However, exome sequencing in MGUS has only been performed in a handful of patients16 so the dataset has been too small to make meaningful conclusions. Here, we report the first comprehensive analysis of genome-wide genetic changes in 33 MGUS samples in which flow-cytometry-separated phenotypically abnormal plasma cells were analyzed, to exclude contamination by phenotypically normal plasma cells, followed by array CGH and exome sequencing.
We found that the frequency of chromosomal gains and losses, including hyperdiploidy, gain 1q, and del(13q), is lower in MGUS than in MM. Hyperdiploidy is considered as a primary myeloma lesion; however it has prognostic potential in asymptomatic stages as has been shown in SMM.35 The frequency of MGUS samples with CNA is 60.6%. the minimum size of the alterations is 100 kb and there are a median of two CNA per case. These numbers are lower compared to those for cases of MM which we previously analyzed.27 Homozygous deletion affecting genes such as FAF1/CDKN2C, BIRC2/BIRC3, RB1, TRAF3/AMN and CYLD are common in MM,27,36 but they were not present in MGUS.
The number of SNV in the samples was also significantly lower in MGUS than in NDMM; exonic, non-synonymous, synonymous and total SNV were all found at a lower frequency. Of the variants, there were no significantly mutated genes. However, there were variants present which were significant in our previous NDMM dataset, including KRAS, NRAS, HIST1H1E, DIS3, EGR1 and LTB.
Insights into the molecular timing of genetic events can be gained by analyses of known MM-specific events in MGUS cases. NRAS mutations have been detected in MGUS previously, but KRAS mutations have not previously been found in MGUS and were implicated in the transition from MGUS to MM.15 Previously, in a limited number of MGUS samples (n=20), one NRAS mutation and no KRAS mutations were detected, whereas in our 33 MGUS cases we found one patient with a clonal KRAS mutation and one with both subclonal KRAS and NRAS mutations. Neither of these two patients with RAS mutations had progressed to MM after a follow-up of 50 and 67 months. Our results confirmed that the frequency of RAS mutations in MGUS is significantly lower than in MM, but also showed that activation of this pathway does not necessarily mark the onset of disease progression. Interestingly, the presence of both a KRAS and NRAS mutation in one patient indicates early diversification and heterogeneity in this pre-malignant condition.
Another candidate for association with disease progression is del(17p) and/or mutation of the TP53 gene.37 We detected neither in this MGUS dataset, but they account for up to 11% of mutations in NDMM. Mutations in ATM and ATR were also not detected in MGUS but their prevalence in NDMM is low (<2% each) so would not have been expected in this dataset. MYC translocations were not detected in MGUS by our targeted capture of 2 Mb surrounding MYC, which has detected translocations in 18% of NDMM.26 This change at the MYC locus is consistent with data from gene expression analyses showing that MYC over-expression occurs in two-thirds of MM cases, but uncommonly in MGUS.38–41 RAS mutations, TP53 alterations and MYC translocations are all known oncogenic drivers and are likely candidates for being involved in disease progression but not initiation of the myeloma propagating cell. Moreover, del(17p)35,42 and MYC rearrangement43 have been observed as risk progression factors in SMM.
We have previously identified mutations in CCND1, associated with the t(11;14), which have a negative impact on survival. In our MGUS dataset there were four t(11;14) samples, of which one had two missense mutations in CCND1. This is consistent with NDMM, given the low numbers of t(11;14) samples, perhaps implying that there is not a link with disease progression. The frequency of IGH translocations detected by exome sequencing did not differ significantly from that found in previous fluorescence in situ hybridization-based studies, however a significantly lower t(4;14) and higher t(14;20) frequency in MGUS compared to MM was not seen due to the limited number of MGUS cases.44,45
In conclusion, MGUS is better defined by the genomic abnormalities that are absent than by the ones that are present. We show that some MM-specific structural and SNV are present in MGUS, but the overall prevalence of such lesions is significantly lower than in MM. Structural changes such as gain 1q and del(1p) are present in MGUS, at relatively high frequencies, implying they may lay the ground for progression but are not key drivers of progression. Key oncogenic drivers, namely TP53 deletion and/or mutation and MYC translocations, are noticeably absent from this MGUS dataset and are, therefore, better candidates for the onset of disease progression.
Here, we show that samples with clonal copy-number changes and mutations are associated with non-IgG heavy chain isotype and abnormal light chain ratio, which are known markers of high-risk MGUS. The clonal complexity of these samples (Figure 4E,F) is in stark contrast to the relative simplicity of those associated with a low risk of disease progression (Figure 4A–D). Follow up of these cases over time and analysis of progressive samples from MGUS, through SMM to MM will provide interesting insights into the evolutionary mechanisms of selection for aggressive malignant clones containing key driver events. These observations are consistent with a Darwinian model of myeloma evolution whereby genomic abnormalities are accumulated as disease progresses. We hypothesize that transformation from MGUS to MM is due to the acquisition of critical driver gene variants which alters the behavior of a myeloma propagating cell, which is accompanied by a wave of clonal expansion and clonal dominance resulting in crucial differences in clinical behavior.
Supplementary Material
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/9/1617
Funding: This work was supported by a Myeloma UK program grant, Cancer Research UK CTAAC sample collection grants (A12136 and A17761) and a Cancer Research UK Biomarkers and Imaging Discovery and Development grant (A14261) as well as funds from the National Institute of Health Biomedical Research Centre at the Royal Marsden Hospital. This study was partly funded by a P01 grant from the National Institutes of Health grant number CA055819. The study was also supported by the Ministry of Health of the Czech Republic (NT13492, 15-29667A) and the Ministry of Education, Youth and Sports of the Czech Republic (IRP201550).
References
- 1.Kyle RA, Rajkumar SV. Monoclonal gammopathy of undetermined significance and smouldering multiple myeloma: emphasis on risk factors for progression. Br J Haematol. 2007;139(5):730–743. [DOI] [PubMed] [Google Scholar]
- 2.Kyle RA, Therneau TM, Rajkumar SV, et al. Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med. 2006;354(13):1362–1369. [DOI] [PubMed] [Google Scholar]
- 3.Kyle RA, Therneau TM, Rajkumar SV, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. 2002;346(8):564–569. [DOI] [PubMed] [Google Scholar]
- 4.Rajkumar SV, Kyle RA, Therneau TM, et al. Serum free light chain ratio is an independent risk factor for progression in monoclonal gammopathy of undetermined significance. Blood. 2005;106(3):812–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perez-Persona E, Mateo G, Garcia-Sanz R, et al. Risk of progression in smouldering myeloma and monoclonal gammopathies of unknown significance: comparative analysis of the evolution of monoclonal component and multiparameter flow cytometry of bone marrow plasma cells. Br J Haematol. 2010;148(1):110–114. [DOI] [PubMed] [Google Scholar]
- 6.Perez-Persona E, Vidriales MB, Mateo G, et al. New criteria to identify risk of progression in monoclonal gammopathy of uncertain significance and smoldering multiple myeloma based on multiparameter flow cytometry analysis of bone marrow plasma cells. Blood. 2007;110(7):2586–2592. [DOI] [PubMed] [Google Scholar]
- 7.Avet-Loiseau H, Facon T, Daviet A, et al. 14q32 translocations and monosomy 13 observed in monoclonal gammopathy of undetermined significance delineate a multistep process for the oncogenesis of multiple myeloma. Intergroupe Francophone du Myelome. Cancer Res. 1999;59(18):4546–4550. [PubMed] [Google Scholar]
- 8.Avet-Loiseau H, Li JY, Morineau N, et al. Monosomy 13 is associated with the transition of monoclonal gammopathy of undetermined significance to multiple myeloma. Intergroupe Francophone du Myelome. Blood. 1999;94(8):2583–2589. [PubMed] [Google Scholar]
- 9.Chng WJ, Van Wier SA, Ahmann GJ, et al. A validated FISH trisomy index demonstrates the hyperdiploid and nonhyperdiploid dichotomy in MGUS. Blood. 2005;106(6):2156–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van de Donk NW, Palumbo A, Johnsen HE, et al. The clinical relevance and management of monoclonal gammopathy of undetermined significance and related disorders: recommendations from the European Myeloma Network. Haematologica. 2014; 99(6):984–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lopez-Corral L, Gutierrez NC, Vidriales MB, et al. The progression from MGUS to smoldering myeloma and eventually to multiple myeloma involves a clonal expansion of genetically abnormal plasma cells. Clin Cancer Res. 2011;17(7):1692–1700. [DOI] [PubMed] [Google Scholar]
- 12.Lopez-Corral L, Sarasquete ME, Bea S, et al. SNP-based mapping arrays reveal high genomic complexity in monoclonal gammopathies, from MGUS to myeloma status. Leukemia. 2012;26(12):2521–2529. [DOI] [PubMed] [Google Scholar]
- 13.Chesi M, Robbiani DF, Sebag M, et al. AID-dependent activation of a MYC transgene induces multiple myeloma in a conditional mouse model of post-germinal center malignancies. Cancer Cell. 2008;13(2):167–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chng WJ, Gonzalez-Paz N, Price-Troska T, et al. Clinical and biological significance of RAS mutations in multiple myeloma. Leukemia. 2008;22(12):2280–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rasmussen T, Kuehl M, Lodahl M, Johnsen HE, Dahl IM. Possible roles for activating RAS mutations in the MGUS to MM transition and in the intramedullary to extramedullary transition in some plasma cell tumors. Blood. 2005;105(1):317–323. [DOI] [PubMed] [Google Scholar]
- 16.Walker BA, Wardell CP, Melchor L, et al. Intraclonal heterogeneity is a critical early event in the development of myeloma and precedes the development of clinical symptoms. Leukemia. 2014;28(2):384–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bataille R, Jego G, Robillard N, et al. The phenotype of normal, reactive and malignant plasma cells. Identification of “many and multiple myelomas” and of new targets for myeloma therapy. Haematologica. 2006;91(9):1234–1240. [PubMed] [Google Scholar]
- 18.Lage JM, Leamon JH, Pejovic T, et al. Whole genome analysis of genetic alterations in small DNA samples using hyper-branched strand displacement amplification and array-CGH. Genome Res. 2003;13(2):294–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Corneveaux JJ, Kruer MC, Hu-Lince D, et al. SNP-based chromosomal copy number ascertainment following multiple displacement whole-genome amplification. Biotechniques. 2007;42(1):77–83. [DOI] [PubMed] [Google Scholar]
- 20.Pugh TJ, Delaney AD, Farnoud N, et al. Impact of whole genome amplification on analysis of copy number variants. Nucleic Acids Res. 2008;36(13):e80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pinard R, de Winter A, Sarkis GJ, et al. Assessment of whole genome amplification-induced bias through high-throughput, massively parallel whole genome sequencing. BMC Genomics. 2006;7:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rykalina VN, Shadrin AA, Amstislavskiy VS, Rogaev EI, Lehrach H, Borodina TA. Exome sequencing from nanogram amounts of starting DNA: comparing three approaches. PLoS One. 2014;9(7):e101154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang L, Ma F, Chapman A, Lu S, Xie XS. Single-cell whole-genome amplification and sequencing: methodology and applications. Annu Rev Genomics Hum Genet. 2015;16:79–102. [DOI] [PubMed] [Google Scholar]
- 24.Mikulasova A, Smetana J, Wayhelova M, et al. Genome-wide profiling of copy-number alteration in monoclonal gammopathy of undetermined significance. Eur J Haematol. 2016;97(6):568–575. [DOI] [PubMed] [Google Scholar]
- 25.Kozarewa I, Rosa-Rosa JM, Wardell CP, et al. A modified method for whole exome resequencing from minimal amounts of starting DNA. PLoS One. 2012;7(3):e32617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walker BA, Boyle EM, Wardell CP, et al. Mutational spectrum, copy number changes, and outcome: results of a sequencing study of patients with newly diagnosed myeloma. J Clin Oncol. 2015;33(33):3911–3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smetana J, Frohlich J, Zaoralova R, et al. Genome-wide screening of cytogenetic abnormalities in multiple myeloma patients using array-CGH technique: a Czech multicenter experience. Biomed Res Int. 2014;2014:209670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walker BA, Wardell CP, Johnson DC, et al. Characterization of IGH locus breakpoints in multiple myeloma indicates a subset of translocations appear to occur in pregerminal center B cells. Blood. 2013;121(17): 3413–3419. [DOI] [PubMed] [Google Scholar]
- 29.Walker BA, Wardell CP, Murison A, et al. APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nat Commun. 2015;6:6997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Landgren O, Kyle RA, Pfeiffer RM, et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: a prospective study. Blood. 2009;113(22):5412–5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weiss BM, Abadie J, Verma P, Howard RS, Kuehl WM. A monoclonal gammopathy precedes multiple myeloma in most patients. Blood. 2009;113(22):5418–5422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fonseca R, Bailey RJ, Ahmann GJ, et al. Genomic abnormalities in monoclonal gammopathy of undetermined significance. Blood. 2002;100(4):1417–1424. [PubMed] [Google Scholar]
- 34.Nilsson T, Lenhoff S, Rylander L, et al. High frequencies of chromosomal aberrations in multiple myeloma and monoclonal gammopathy of undetermined significance in direct chromosome preparation. Br J Haematol. 2004;126(4):487–494. [DOI] [PubMed] [Google Scholar]
- 35.Neben K, Jauch A, Hielscher T, et al. Progression in smoldering myeloma is independently determined by the chromosomal abnormalities del(17p), t(4;14), gain 1q, hyperdiploidy, and tumor load. J Clin Oncol. 2013;31(34):4325–4332. [DOI] [PubMed] [Google Scholar]
- 36.Walker BA, Leone PE, Chiecchio L, et al. A compendium of myeloma-associated chromosomal copy number abnormalities and their prognostic value. Blood. 2010;116(15):e56–65. [DOI] [PubMed] [Google Scholar]
- 37.Ackermann J, Meidlinger P, Zojer N, et al. Absence of p53 deletions in bone marrow plasma cells of patients with monoclonal gammopathy of undetermined significance. Br J Haematol. 1998;103(4):1161–1163. [DOI] [PubMed] [Google Scholar]
- 38.Chng WJ, Huang GF, Chung TH, et al. Clinical and biological implications of MYC activation: a common difference between MGUS and newly diagnosed multiple myeloma. Leukemia. 2011;25(6):1026–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shou Y, Martelli ML, Gabrea A, et al. Diverse karyotypic abnormalities of the c-myc locus associated with c-myc dysregulation and tumor progression in multiple myeloma. Proc Natl Acad Sci USA. 2000;97(1):228–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Avet-Loiseau H, Gerson F, Magrangeas F, et al. Rearrangements of the c-myc oncogene are present in 15% of primary human multiple myeloma tumors. Blood. 2001;98(10): 3082–3086. [DOI] [PubMed] [Google Scholar]
- 41.Xiao R, Cerny J, Devitt K, et al. MYC protein expression is detected in plasma cell myeloma but not in monoclonal gammopathy of undetermined significance (MGUS). Am J Surg Pathol. 2014;38(6):776–783. [DOI] [PubMed] [Google Scholar]
- 42.Rajkumar SV, Gupta V, Fonseca R, et al. Impact of primary molecular cytogenetic abnormalities and risk of progression in smoldering multiple myeloma. Leukemia. 2013;27(8):1738–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chiecchio L, Dagrada GP, Protheroe RK, et al. Loss of 1p and rearrangement of MYC are associated with progression of smouldering myeloma to myeloma: sequential analysis of a single case. Haematologica. 2009;94(7):1024–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ross FM, Chiecchio L, Dagrada G, et al. The t(14;20) is a poor prognostic factor in myeloma but is associated with long-term stable disease in monoclonal gammopathies of undetermined significance. Haematologica. 2010;95(7):1221–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chiecchio L, Dagrada GP, Ibrahim AH, et al. Timing of acquisition of deletion 13 in plasma cell dyscrasias is dependent on genetic context. Haematologica. 2009;94(12):1708–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.