

Abstract
During the last few decades we have become accustomed to the idea that viruses can cause tumors. It is much less considered and discussed, however, that most people infected with oncoviruses will never develop cancer. Therefore, the genetic and environmental factors that tip the scales from clearance of viral infection to development of cancer are currently an area of active investigation. Microbiota has recently emerged as a potentially critical factor that would affect this balance by increasing or decreasing the ability of viral infection to promote carcinogenesis. In this review, we provide a model of microbiome contribution to the development of oncogenic viral infections and viral associated cancers, give examples of this process in human tumors, and describe the challenges that prevent progress in the field as well as their potential solutions.
Keywords: Microbiota, Oncovirus, Microbiome, Virus-associated cancer, Transkingdom interactions
1. Introduction
As the human lifespan lengthens, the incidence of cancer worldwide is also increasing. The World Health Organization predicts the frequency of cancer occurrence to increase by 70% over the next two decades [1,2], indicating a rise in the global cancer epidemic. One of the established causes of cancer is viral infection, which is responsible for 20% of the global cancer burden [3]. Among these infections, the most common are Human Papilloma Virus (HPV) and Hepatitis C/B viruses with other less frequent contributors being Epstein - Barr virus (EBV), Human Immunodeficiency Virus (HIV), and Kaposi Sarcoma Herpesviruses (KSHV) [4]. These viruses use two different strategies to cause cancer: first, by directly affecting host cell machinery (e.g. HPV); and second, indirectly, by inhibiting the human immune system (e.g. HIV) [5,6]. It is common knowledge that the development of some cancers require viral infection, such as HPV for cervical cancer. It is less known, however, why most people infected with oncogenic viruses will never develop cancer.
A hint in solving this puzzle may come from studies demonstrating the crucial role of microbiota (collection of microorganisms living with the host) in the course of viral infections [7–10]. Moreover, microbiota have been recently implicated in different diseases associated with aberrant immune responses ranging from diabetes and autoimmunity to obesity and cancer [11,12]. For example, a recent epidemiologic study reported an association between antibiotic exposure and the development of several malignancies such as esophageal, gastric, pancreatic, lung, prostate, and breast cancers [13].
Studies thus far have placed emphasis on gastrointestinal microbiota and its role in the development and progression of gut-associated malignancies [14]. For example, Helicobacter pylori causes gastric adenocarcinoma and is a classic case of oncogenic bacteria [15]. Intestinal infections with other bacteria such as Salmonella typhi [16] and Streptococcus gallolyticus (bovis) [17] were also linked to the development of hepatobiliary and colorectal cancers, respectively. These studies represent additional support for the hypothesis that some microbiota members as an understudied environmental factor contributing to protection from or the development of virus-associated cancers. Even though the gut microbiome represents the majority of bacteria in the human microbiome [18], other body sites such as the vagina and oral cavity have been explored for their participation in cancer development and progression.
Although oncogenic properties of virus and bacteria, individually, are popular topics, the interaction between these in the context of cancer has not been well investigated. In recent years, we have witnessed an increasing number of attempts to interrogate this three-way interaction, particularly the influence of microbiota on the progression and acquisition of oncogenic viral infections. However, the question whether bacteria are beneficial or harmful in this context remains unanswered for many cancers. In this review, we provide a model of microbiome contribution to the development of oncogenic viral infections, discuss examples of this process in human tumors, and describe obstacles in the field and their potential solutions.
2. Model description
The role of bacteria (and bacterial microbiota) in viral infections leading to cancer can be assigned to two broad categories: bacteria that influence viral particles, and bacteria that affect host interaction with viral infection.
On the one hand, healthy microbiota was shown to contribute to infections by interacting with viruses directly and through bacterial byproducts [7–10]. It was reported that commensal microbiota augment the transmission of mouse mammary tumor virus [19,20], bacterial lipopolysaccharides enhance virion stability of poliovirus [21], and enteric bacteria promote norovirus infection through histo-blood group antigen-like substances [9,22]. On the other hand, healthy microbiota are critical for immune system development, especially on mucosal surfaces [23]. Antibiotic-treated mice exposed to mucosal influenza virus were observed to have impaired innate and adaptive antiviral immune responses and delayed clearance of the virus [24]. These and other studies define microbiota as a putatively important factor for the development of virus-associated cancers.
Herein we propose a model for the three-way interaction between bacteria, virus, and mammalian host, highlighting two distinct mechanisms for the contribution of microbiota to virus-associated cancers. The first involves the direct effect of interaction of bacteria and bacterial products on viruses, primarily affecting their infectivity (Fig. 1A). The second involves bacteria-host interactions leading to changes in host gene expression and subsequent activation/repression of viral expression or direct promotion of inflammation synergizing with the tumorigenic effects of a virus (Fig. 1B). There is evidence to suggest that the role of bacteria can be positive or negative in terms of disease progression with each of these cases. In this review, we discuss conventional tumor viruses and explore the role of gut, vaginal, and oral microbiota components in both of these mechanisms.
Fig. 1.
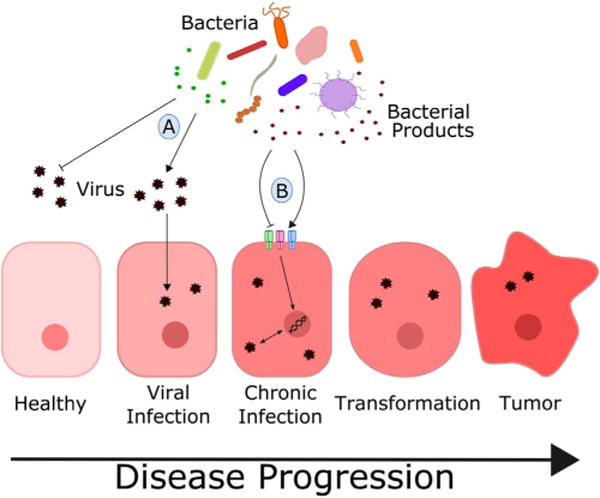
Model of bacteria-virus interactions in cancer development and progression: A) direct interaction between bacteria or bacterial by-products and virus resulting in inhibition or promotion of viral infection into host cell; B) indirect interaction between bacteria and virus mediated by host response to bacterial stimuli through activation of various pattern recognition receptors.
3. Gastrointestinal microbiome
The gut microbiome is the largest microbial community in the human body. Recent discoveries show its involvement in a variety of functions, including immune system training and metabolism regulation [25–27]. Separate members of gut microbiota as well as dysbiosis (i.e. non-specific alterations of mammalian microbial communities) have been implicated in disease development and progression. Among most prominent examples are diabetes [28], irritable bowel disease [25], and cancer [29,30]. Additionally, intestinal microbiota has been implicated in modulating the effect of different anti-cancer treatments [31–33]. Helicobacter species, in particular Helicobacter pylori, is the most well studied bacterial member of the gut community that causes cancer [15].
3.1. Hepatitis viruses
The second leading cause of cancer mortality is liver cancer [34]. The most prevalent histologic type of primary liver cancer is hepatocellular carcinoma (HCC) [35] primarily caused by chronic infection with hepatitis B (HBV) or hepatitis C (HCV) virus [36]. Although both viruses can cause cancer, HCV currently attracts stronger interest from the scientific community possibly due to the absence of a vaccine against HCV.
The pathogenicity of both HBV and HCV involves a combination of direct and indirect mechanisms. The HCV encoded core, nonstructural protein 5A (NS5A) and NS3, and HBV encoded X antigen (HBx) are able to promote host cell proliferation. Both viruses are similarly capable of blocking cell immune response, inhibiting apoptosis while promoting angiogenesis and metastasis. By contrast, chronic inflammation caused by oxidative stress also contributes to the process of carcinogenesis [6].
While HCV is oncogenic, not all patients suffering from chronic hepatitis C will develop cancer. One of the first indications that bacteria may be a critical parameter in liver cancer came with the observation that mice infected with Helicobacter were developing strong inflammatory responses leading to hepatocellular carcinoma [37]. Another group later found an association between H. pylori specific antibody levels and HCV associated hepatocellular carcinoma [38]. Helicobacter DNA was also found in liver and was associated with hepatitis C induced cirrhosis [39], which indicates the ability of H. pylori to invade the liver and putatively contribute to disease development (Fig. 2B). However, a more recent study conducted on HCV transgenic mice colonized with H. pylori found no indication of bacteria translocation into the liver and no promotion of tumorigenesis [40]. Whether this experimental system failed to promote carcinogenesis, or H. pylori does not contribute to HCV-associated liver cancer, remains unknown.
Fig. 2.
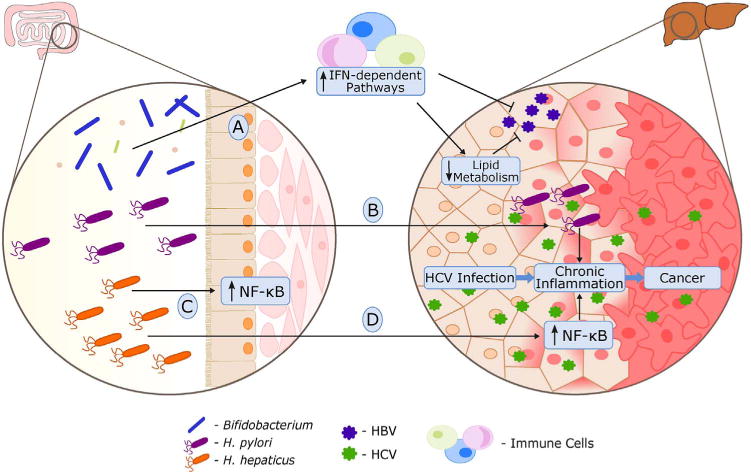
The interplay between gut microbiome, host and human hepatitis viruses (HCV and HBV): A) healthy gut microbiota stimulates host immune system resulting in HBV infection clearance; B) Helicobacter pylori invades liver and contributes to chronic inflammation induced by HCV; C, D) Helicobacter hepaticus upregulates NF-κB dependent pathways in mouse gut (C) and liver (D), synergizing with HCV-related inflammation in the development of hepatocellular carcinoma.
Another Helicobacter species, Helicobacter hepaticus, has been shown to cause chronic hepatitis and liver cancer in rodents [37]. In the following study, Fox et al. have shown that presence of H. hepaticus in the gut lumen promotes development of hepatocellular carcinoma in HCV infected mice, acting synergistically with viral infection [41]. This process did not require bacterial invasion. Fox et al. also showed that in an aflatoxin B1 HCC mouse model, H. hepaticus induced the nuclear factor-κB (NF-κB) along with downstream innate and Th1-type adaptive immunity. In a HCV transgenic mouse model, they also observed increased gene expression of an NF-kB-dependent inflammatory chemokine (CXCL9) in mice colonized with H. hepaticus [6,41,42] (Fig. 2C, D). Concordantly in both models, tumor growth was accelerated in the presence of H. hepaticus. These bacteria, detected in human intestine [43] and liver [44–46], is suspected to contribute not only to cirrhosis [47] and HCC [44], but also to a set of other conditions such as inflammatory bowel disease [43] and prostate cancer [48]. Consequently, this data suggests a synergistic relationship between H. hepaticus and HCV in human cancers.
The link between HBV related oncogenesis and gut microbiota has also been reported. In 2011, Chen et al. found that enteric fungi diversity was positively correlated with a worsened disease state in chronic HBV infection [49]. More recently, Chou et al. showed that in adult C3H/HeN mice transfected with HBV, elimination of gut microbiota with antibiotics resulted in viral persistence phenotypes, including prolonged HBV surface antigens (HBsAg) presence, impaired anti-HBs production, and limited HBV core antigen (HBcAg)-specific IFN-γ–secreting splenocytes [50]. Although these results suggest that the persistence of HBV infection in antibiotic-treated mice is attributed to an ineffective adaptive humoral and cellular immune response, specific bacteria were not identified in this study. Other studies observed that a decrease in fecal Bifidobacterium populations was associated with liver disease progression of HBV infection in humans [51,52]. In addition, Bifidobacterium species have been shown to decrease the protein and transcript levels of HBsAg in a HBV-transfected human hepatoma cell line [53]. Interestingly, Bifidobacterium can also increase host gene expression of IFN-signaling components such as STAT1 [53] and lower serum cholesterol levels [54–56]. Furthermore, IFN stimulation have been shown to downregulate lipid metabolic pathways [27], known in host cells to be necessary for HBV production [57,58]. Therefore, we propose that Bifidobacterium species are able to stimulate IFN-dependent pathways which results in a downregulation of lipid metabolism and a reduction in HBV infection (Fig. 2A). Further experimentation is warranted to establish how the antiviral and antitumor effects of these bacteria contribute to overall protection against malignancy.
4. Cervicovaginal microbiome
Despite the fact that microbiota in the vagina is less diverse than in the gut it can also contribute to protection against or susceptibility to some illnesses, especially sexually transmitted diseases. Healthy stable vaginal microbiome is thought to be a first line of defense [59,60] against diseases caused by opportunistic and true pathogens: outcompeting dangerous bacteria or protecting the host with bacterial by-products, in particular lactic acid [61–64] and hydrogen peroxide [60,65,66].
Lactobacilli, commonly considered as such beneficial microbes, represent the dominant genus [67,68] in healthy vaginal microbiota. However, this does not seem to be universally applicable as Gardnerella-dominated microbiomes were observed as another frequent type in some populations [69]. Disruption of healthy vaginal microbiota (bacterial vaginosis - BV) [67] may increase risks of sexually transmitted infections (STI) [70], and even contribute to the disease progression, putatively, including cancer development.
4.1. Human papillomavirus
Human papillomavirus (HPV) is the most common STI in the United States and although the majority of HPV types are non-carcinogenic, there are at least 13 high-risk oncogenic types (hrHPV), with HPV16 and HPV18 leading the list [71,72]. One of the most common cancers caused by HPV infection remains cervical squamous cell carcinoma. However, HPV is also linked to anal, vulvar, vaginal, penile and head-and-neck carcinomas [73].
hrHPV infections remain the main predictors for cervical intraepithelial neoplasia (CIN), precursor to tumor, and cervical cancer development itself [74–76]. HPV infects basal epithelial cells. After infection HPV either exist in episomal form or can integrate into the cell genome causing genomic instability. Expression of HPV oncogenes E6 and E7 is also dependent on integration: in episomal form expression of E2 protein keeps expression levels of E6/7 low, but during integration open reading frame of E2 gene is disrupted and E2 no longer is able to control HPV oncogenes. E6, E7 and E5 HPV oncoproteins retain keratinocytes in proliferative state, avoiding apoptosis and clearance by the immune system. It was also reported that HPV is able to promote angiogenesis and deregulate cellular energetics [77].
Although hrHPV type infections are necessary for development of cervical cancer, in most cases the virus is cleared by the host. Only 0.3–1.2% of women eventually develop cervical cancer [78] which means that additional risk factors contribute to the disease progression and vaginal microbiota is possibly one of them. It appears that both, the protective role of normal vaginal microbiome and the contribution of certain pathogens, may play a role in development of cervical intraepithelial lesions and cancer.
While Lactobacillus gasseri-dominated vaginal bacterial communities are associated with faster clearance of HPV infection [79], dysbiosis, and bacterial vaginosis are associated with CIN development and progression [80,81] (Fig. 3D). Furthermore, it is unclear whether bacteria in the disrupted vaginal microbiome affect host susceptibility, virus survival, or infectivity. Some evidence (discussed below), however, points to the possible involvement of Prevotella genus that contains Prevotella bivia, a microbe known to be associated with bacterial vaginosis.
Fig. 3.

Cervicovaginal microbiota and pathogens can influence the progression of viral infection associated with cancer development: A) Lactobacillus spp., predominant bacteria in the vaginal microbiome, secretes lactic acid capable of inactivating HIV; B) butyric acid secreted by vaginal microbiota induces HIV replication in Human CD4+ T lymphocyte and macrophage/monocyte cell lines harboring latent HIV; C) Neisseria gonorrhoeae induces IL-6, IL-8 and TNFα production in genital epithelial cells and upregulates HIV gene expression in T-cells via heptose-monophosphate induced NF-kB and AP-1 pathways; D) domination of vaginal microbiota by Lactobacillus gasseri is associated with faster HPV clearance; E) Lactobacillus spp. is able to inhibit the viability of Gardnerella vaginalis and Prevotella bivia; F) Prevotella spp. produces ammonia and different amino acids that benefit G. vaginalis and Peptostreptococcus anaerobius, contributing to bacterial vaginosis; G) Chlamydia trichomatis may influence cervical cancer development, either through synergy with HPV or by contributing to local chronic inflammation.
Normally inhibited by Lactobacillus [65], Prevotella species may become abundant when the homeostasis of the vaginal microbial community is disrupted by such factors as diet or hormone status [82] (Fig. 3E). Recent findings also suggest host genetics as an important factor in Prevotella outgrowth [82]. Increasing in abundance, Prevotella species may provide nutrients (e.g. ammonia and amino acids), to other members of microbial community such as Gardnerella vaginalis and Peptostreptococcus anaerobius [83,84], and thus diversify the vaginal microbial landscape [69] (Fig. 3F). Furthermore, multiple studies of Prevotella associations with bacterial vaginosis and cervicitis [68,82,85] point to Prevotella as a conductor orchestrating the state of vaginal microbiomes. Additionally, a clear link between Prevotella genus and HPV infection [86], in particular with high risk HPV types [87], has been established. Adding to the potential importance of these microbes, Prevotella was cultured from cervical cancer samples in 1993 [88], and more recently we detected it as the most abundant genus in cervical cancer biopsies (unpublished). Finally, increased expression of NF-κB, Toll-like receptor (TLR), NOD-like receptor, and TNF-α signaling pathways in antigen presenting cells from blood, and increased levels of pro-inflammatory cytokines from vaginal lavage have been associated with microbial communities that include Prevotella [69]. Thus, it is likely that Prevotella or Prevotella-driven vaginal microbiome may act in favor of persistent HPV infection promoting cervical cancer development through upregulation of cell proliferation and chronic inflammation.
Aside from the common members of vaginal microbiota, pathogens have also been suspected in the promotion of HPV infection. For example, Chlamydia trachomatis, has been studied for some time as a potential co-factor of HPV in the process of tumorigenesis [89,90]. Investigation of the potential mechanism of how C. trachomatis infection may influence HPV infection and cancer development are underway. For example, it was demonstrated that C. trachomatis can decrease the expression of caveolin-1 (tumor suppressor) and increase C-myc mRNA levels (oncogene) [91]. A study conducted by Paba et al. found a correlation between C. trichomonas infection and upregulation of cytoplasmic and nuclear NF-kB, VEGF-c and survivin in HPV-positive CINs and cervical cancer [92], which points out to the possibility that C. trachomatis can also act through the NF-kB pathway, promoting local inflammation, cell survival and proliferation (Fig. 3G). Despite many studies and even mechanistic research, the scientific community has not yet reached a consensus about whether C. trachomatis plays a causal role in aiding HPV in carcinogenesis. Thus epidemiological studies that would investigate the temporal relationship between C. trachomatis and HPV infections are required to settle this long standing debate.
4.2. Human immunodeficiency virus
Human Immunodeficiency Virus (HIV) is a carcinogenic virus in its ability to act as a co-factor for EBV and KSHV in carcinogenesis [93]. Immunosuppression, chronic antigenic stimulation, and cytokine dysregulation are reported to contribute to HIV-associated cancer development. However, the main cause of HIV-related cancer development remains immunosuppression: the inability of the immune system to recognize and clear host cells infected with potentially oncogenic viruses enables those viruses to express oncogenic proteins, which in its turn leads to cancer development [6]. Due to the absence of vaccines and ways to eliminate HIV, understanding risk factors contributing to the pathogenesis of HIV infections becomes a priority.
Sexual transmission is one of the most common ways to acquire HIV, suggesting that vaginal microbiota may influence this process. Indeed, several studies found an association between the risk of HIV infection and bacterial vaginosis [94–96]. However, it is still unclear whether the absence or overgrowth of some bacteria in the vaginal microbiota would make women more susceptible to or protected from HIV infection.
In 2013, Aldunate et al. showed that physiological concentrations of lactic acid can inactivate different HIV subtypes [97]. Thus, Lactobacillus spp. which is known to produce lactic acid, may play an important role in protection against sexually transmitted diseases, including HIV [98] (Fig. 3A). In contrast, bacterial vaginosis predisposes females to sexually transmitted diseases [99], and can reactivate latent HIV infection [100,101]. Evidence suggests that bacteria are not acting directly on HIV, but rather via bacterial by-products (e.g. butyric acid) [105,106]. Specifically, immunosuppression caused by HIV is an important step in the establishment of lifelong latent infection and avoidance of host immune response. Latently infected cells harbor the HIV proviral DNA genome integrated into heterochromatin, allowing the persistence of transcriptionally silent proviruses [102]. Hypoacetylation of histone proteins by histone deacetylases (HDAC) is involved in the maintenance of HIV latency by repressing viral transcription [103,104]. Interestingly, butyric acid-producing bacteria (Fusobacterium nucleatum, Porphyromonas gingivalis, Clostridium cochlearium, Eubacterium multiforme, and Anaerococcus tetradius), residing in different mucosal environments (gut, vaginal, and oral cavities), are capable of reactivating latent HIV infection through host HDAC pathways [105,106] (Figs. 3B, 4B). Butyric acid is known to inhibit the enzymatic activity of HDAC by directly competing with HDAC substrate at the catalytic center of the enzyme [107] (Fig. 4A). Butyrate originating from microbiota has been shown to promote immunosuppressive/immunoregulatory effects [108]. Considering this, it is plausible that in addition to reactivating viruses, butyric acid can inhibit antiviral immunity in the host directly.
Fig. 4.
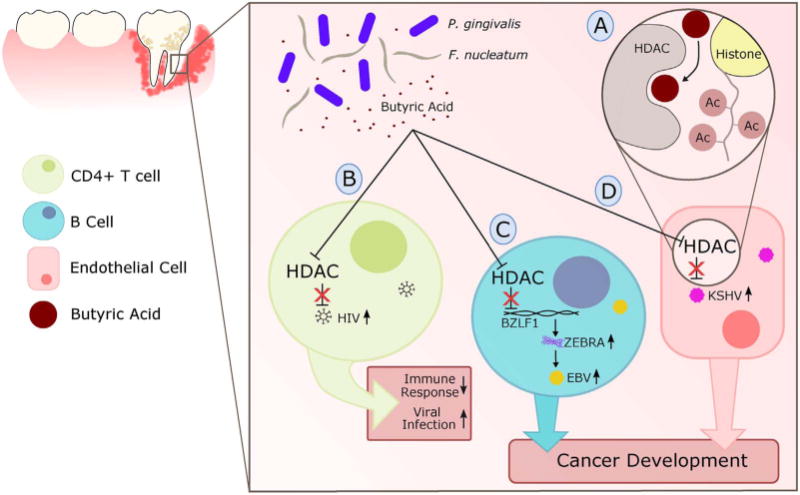
The role of periodontal disease in head and neck cancers can be mediated by the effects of HIV and herpesviruses (EBV and KSHV): A) butyric acid produced by periodontal pathogens such as Fusobacterium nucleatum and Porphyromonas gingivalis competitively inhibit Histone Deacetylases (HDACs), resulting in the reduced deacetylation of histone proteins and sustaining viral gene expression B) activation of HIV proviral gene expression through HDAC inhibition increases viral progeny production; HIV infection suppresses systemic immune responses and aides in the acquisition of other viral infections; C) inhibition of HDAC causes transcriptional activation of the BZLF1 promoter in EBV infected cells and results in production of ZEBRA, known switch for lytic cycle activation; D) inhibition of HDACs in KSHV-associated cells result in the induction of MAPK and expression of lytic genes. Both EBV (C) and KSHV (D) mechanisms promote the spread of viral infection and contributes to tumor development.
Additionally, the epidemiological synergy of sexually transmitted infections (STI) with HIV transmission was reported in multiple studies. Ferreira et al. found that stimulation of HIV infected T-cells with TLR3, 4, or 5 ligands leads to enhanced HIV-1 replication via direct activation of HIV long terminal repeats, or by inducing secreted factors that promote HIV replication [109]. Among the pathogens suspected to contribute to HIV infection and reactivation, one player stands out - Neisseria gonorrhoeae: it was found to be highly associated with HIV infection [110] and activation of HIV expression [109,111,112]. Not only does N. gonorrhoeae drives an increase of activated CD4+ T-cells [111] and pro-inflammatory cytokines in genital epithelial cells such as IL-6, IL-8 and TNFα [109], but exposure to the pathogen directly drives HIV expression in T cells in NF-kB and AP-1 dependent manner [109,111]. Malott et al. showed that N. gonorrhoeae by-product heptose-monophosphate is necessary for invoking these host responses [111] (Fig. 3C). Thus, the course of HIV infection can be altered negatively (by lactate producers) or positively (by butyrate producers or N. gonorrhoeae), preventing or favoring, respectively, development of immunodeficiency, which may increase susceptibility to EBV and KSHV infection, which are responsible for a substantial proportion of HIV-related cancers.
5. Oral microbiome
Although many studies have been devoted to HIV in the genital tract, the connection between HIV, oral microbiome, and periodontal disease has also drawn the attention of the scientific community [113–116] [105,106]. Recent works have found associations between periodontitis, a chronic inflammatory disease of the periodontium occurring in response to bacterial infection, and several types of oral cancers [117–120]. Periodontal disease is marked by a disruption in the oral microbiome and is often associated with a shift to anaerobic bacteria such as Porphyromonas gingivalis, that has also been directly implicated in cancers [121,122]. Although common risk factors for these cancers include smoking and alcohol abuse, there are an increasing number of cases where no significant smoking or drinking history has been reported. Among other risk factors are viral infections HIV, HPV, herpesviruses (EBV, HCMV, KSHV) and oral hygiene [123]. Herpes-viruses, particularly EBV and KSHV, are known to cause cancer in the context of immunodeficiency caused by HIV/AIDS [124].
5.1. Herpesviruses
Herpesviruses have recently been implicated in the progression of periodontitis in the oral cavity [125–129]. Among these viruses, KSHV, EBV, and HCMV have also been associated with head and neck cancers, primarily those found in the mouth. A hallmark of herpesviruses is their ability to establish life-long latent infection in host cells. During latent infection, certain viral genes are repressed and viral progenies are not produced. The reactivation of lytic cycle genes in virus-infected cells marks the production of viral progeny which ultimately leads to host cell lysis. Both latent and lytic cycle genes are critical for tumorigenesis and the evasion of host immune response. In addition, studies exploring co-infection and crosstalk between these viruses as well as with HIV has shown direct (virus-virus) and indirect (through host immunity) interactions, most notably affecting the transition between latent and lytic viral cycles [124,130].
Human cytomegalovirus (HCMV) is not usually regarded as an oncogenic virus. However, HCMV infections have been implicated in malignant diseases from different cancer entities and can cause fatal diseases in immunocompromised patients [131]. Some evidence also suggests its role in sustaining chronic inflammation in the progression of cancer [132]. However, in the case of oral squamous cell carcinomas, Saravani et al. reported that only 6.3% of 48 patient samples were found to have detectable HCMV [133]. Although this patient population had low incidence of HCMV detection, it does not discount the possibility of synergy between HCMV and periodontal disease (not explored) in the development of oral cancer subtypes. In fact, HCMV has been associated with active periodontal disease and P. gingivalis [126–129]. Thus, strengthening the argument to keep HCMV among potential co-factors contributing to chronic inflammation that leads to oral tumorigenesis.
Kaposi’s sarcoma-associated herpesvirus (KSHV), also known as human herpesvirus 8 (HHV-8), is a herpesvirus that has become a well-known oncovirus in immunocompromised individuals infected with HIV [134]. This virus is consistently associated with Kaposi’s sarcoma, a cancer developed from cells that line the lymph or blood vessels and usually appears as a tumor on the skin or mucosal [135]. Latent transcripts in KSHV include genes and miRNAs that favor viral persistence and replication while promoting host-cell proliferation and survival. Furthermore, lytic cycle genes favor viral replication by affecting the DNA damage response, reprogramming metabolism, promoting survival, and mediating immune evasion, all of which have a role in promoting cancer development [6]. In 2007, Morris et al. explored the effects of supernatants from cultures of different bacteria on KSHV infected BCLBL-1 cells [136]. They found that metabolic end products from pathogens (gram-negative anaerobic periodontopathogens F. nucleatum and P. gingivalis) induced lytic replication of KSHV through the activation of a stress-activated MAPK pathway in host cells. More specifically, butyric acid from bacteria supernatants were found to inhibit cellular HDACs and activate the p38 kinases pathway, resulting in hyperacetylation of histones on immediate early viral promoters (Fig. 4C). The targeting of HDACs by P. gingivalis is also seen in the reactivation of HIV as we discussed previously.
Epstein–Barr virus (EBV) was the first virus shown to cause cancer in humans and is associated with a wide range of human cancers originating from epithelial cells, lymphocytes and mesenchymal cells [137]. Infection is transmitted from host to host via salivary contact, and the virus passes through the oropharyngeal epithelium to B lymphocytes, where it establishes a lifelong latent infection. EBV has three main latency patterns, each of which have a role in avoiding host response while promoting B-cell survival/proliferation [6]. Although not deeply studied, there is increasing evidence that EBV early lytic genes, particularly those encoding homologues of Bcl-2 (BALF1), IL-10 (BCRF1) and c-fms receptor (BARF1), may be involved in oncogenesis as well as in promoting viral infection. Reactivation of the virus and production of progeny contributes to several human diseases including nasopharyngeal carcinomas and lymphomas [138]. Similar to KSHV and HIV, the latent virus was found to be reactivated upon stimulation with supernatant from P. gingivalis (containing high concentrations of butyric acid) through the inhibition of HDACs [138]. This inhibition resulted in increased acetylation of adjacent histone and transcriptional activation of the BZLF1 promoter, whose gene product ZEBRA is known to be the master regulator in EBV transition from latency to lytic state [138] (Fig. 4D). Taken together with the case of KSHV, there is compelling evidence for the ability of butyric acid producing bacteria such as P. gingivalis (main player in periodontitis) to reactivate latent herpesvirus infection and contribute to the development of cancer in the oral cavity.
As described above, butyric-acid producing bacteria (especially the periodontopathogens P. gingivalis) can regulate the viral life cycle in host cells. Periodontopathogens can induce a reactivation of HIV which in turn may lead to the opportunistic infection of herpesviruses due to immunodeficiency. Latent infection of EBV and KSHV can be reactivated by the same bacteria leading to the induction of oncogenes and transformation to malignancy. Independent of bacteria, a model of KSHV and EBV co-infection in vitro has shown complementing lytic activation [139], suggesting a more complex model of reactivation in which periodontopathogens may act in tandem with direct inter-viral interaction to regulate infection and the development of cancers. Although HCMV has not been studied extensively in the same context, its association with P. gingivalis and periodontal disease offers hints of viral-bacterial interaction potentially involving mechanisms used by other herpesviruses.
6. Human endogenous retroviruses
Endogenous retroviruses (ERVs) are endogenous viral elements located in the genome of jawed vertebrates, including humans, and are thought to be relics of ancestral infectious retroviruses [140]. Human endogenous retroviruses (HERVs) compose about 4–8% of human genome [141]. While there is not enough evidence to solidify the role of HERVs in causing human cancers, the connection between the two is constantly been researched [140]. Reactivation of HERV expression in cancers [140,142] can contribute to genome instability and is suspected to be a prominent player in disease development. Furthermore, resurrection of murine leukemia virus (MLV) in immunocompromised mice was found to be dependent on intestinal microbiota [143]. In humans, environmental and intestinal microbes were able to modulate the transcriptional activity of endogenous retroviruses [144]. It is not clear whether this process contributes to cancer development. Nonetheless, multiple studies show that upregulated HERV expression in cancerous cells can drive inflammatory responses by upregulating type I interferon pathways [145–147] and eliciting T-cell specific antitumor immunity [148]. These findings may indicate the role of the immune system in recognizing the reactivation of “sleeping” HERVs as a sign of cell transformation and clearing the potential threat before it develops into malignancies.
7. Risk factors of virus-associated cancers and microbiome
Risk factors for cancer, such as age, lifestyle, diet and genetics, have been identified but the mechanisms underlying their contribution to disease are not always well understood [149,150]. The discovery of the role of commensal microbiota and pathobionts in cancer development and progression suggests that at least some risk factors may be linked to the disease through microbiota. For example, while smoking is a risk factor for nasopharyngeal carcinoma [151], it is also associated with dysbiosis in oral microbiota [152], which in its turn can contribute to periodontal disease leading to cancer development [153]. In cervical cancer, a high number of sexual partners is a well-known risk factor. This association has been commonly attributed to increased chances to be exposed to high risk HPV [154]. However, women with multiple sexual partners also present disruption of vaginal microbiota (bacterial vaginosis) [155,156] that may facilitate chronic HPV infection and cervical cancer as discussed earlier (Section 4.1). Another example is liver cancer, for which diabetes and obesity have been identified as risk factors [149,157]. Interestingly, in obese people among many changes in the gut microbiome, a decrease in Bifidobacterium was reported [158]. Because this bacterium has been shown to play a positive role in elimination of HBV (as described in Section 3.1), this may partially explain the association between diet-induced obesity and liver cancer. Although environmental and hereditary factors make virus-bacteria-host interactions even more complex to investigate, it is crucial to remember that understanding the role of these external factors may become a powerful tool for cancer prevention and treatment.
8. Summary
It has recently become evident that progress in the field of virus-associated cancers can be enhanced by elucidation of how bacterial microbiota contributes to virus-host interaction. Although the dissection of these transkingdom interactions is evolving [159], we are still in the early stages of this journey. Indeed, epidemiological and even some mechanistic studies are accumulating. However, the scientific community has not yet reached a unanimous agreement on whether any specific cancer requires bacteria-virus interaction for carcinogenesis.
Herein, we have described a model of bacteria-virus interactions in the development of cancer. We identify two main mechanisms; the first is accomplished by the interaction between bacteria and virus resulting in changes in the course of viral infection. The most notable example of such mechanism is the inactivation of HIV via bacteria-derived lactic acid [97]. The second mechanism is an effect of the bacteria on the host resulting in alterations of host susceptibility to viral infection. This can happen in several ways: a) bacteria inducing pro-tumor chronic inflammation (e.g. activation of NF-kB pathway [6,41,69,92,109,111]), b) commensal bacteria promoting antiviral and antitumor immunity (e.g. Bifidobacterium in HBV [53,160]), and c) bacterial metabolite reactivating oncoviruses (butyric acid inhibition of host HDAC pathways reactivating HIV [105,106], EBV [138], KSHV [136]) (Table 1).
Table 1.
Summary of findings for each virus discussed in this review. Information presented is limited to the scope of the article.
| Virus | Viral Location | Cancer Type | Bacteria-Virus Interaction | Microbiome Location | Bacteria | Bacterial By-products | Bacteria Impact on Host |
|---|---|---|---|---|---|---|---|
| HCV | liver | hepatocellular carcinoma | indirect | gut, liver |
Helicobacter pylori [37–39], Helicobacter hepaticus [37,41] |
activation of NF-κB [6,41] cirrhosis [47] HCC [44] |
|
| HBV | liver | hepatocellular carcinoma | indirect | gut | Bifidobacterium [51,52] | HBV clearance [50,53] anti-tumor immunity [160] up-regulation of IFN-dependent pathways [53] lower serum cholesterol [54–56] |
|
| HPV | vaginal | cervical | indirect | vaginal |
Lactobacillus gasseri [79] BV-associated [80,81] Chlamydia trachomatis [74–76,89,174,175] Prevotella [86,87] |
decrease caveolin-1 and increase C-myc [91] upregulation of NF-kB, VEGF-c and survivin [92] upregulation of NF-κB, Toll-like receptor (TLR), NOD-like receptor, and TNF-α signaling pathways [69] |
|
| HIV | lymphoid cells | head & neck, cervical, Kaposi’s sarcoma | direct, indirect | gut, oral, vaginal | BV-associated [100,101] Neisseria gonorrhoeae [109–112] butyric acid producing bacteria [105,106] |
lactic acid [97] heptose-monophosphate [111] butyric acid [105,106] |
TLR3,4,5 [109] activated CD4+ T-cells [111] pro-inflammatory cytokines [109] NF-kB and AP-1 [109,111] inhibit HDAC [105,106] |
| HCMV | oral | head & neck | oral | Porphyromonas gingivalis [126–129] | |||
| KSHV | oral | Kaposi’s sarcoma | indirect | oral |
Porphyromonas gingivalis [125–128,136] Fusobacterium nucleatum [136] |
butyric acid [136] | inhibit HDAC and activate p38 [136] |
| EBV | B lymphocytes | nasopharyngeal carcinoma | indirect (host) | oral | Porphyromonas gingivalis [138] | butyric acid [138] | inhibit HDAC and activate BZLF1 promoter [138] |
| HERVs | indirect | gut | upregulation of type I interferon pathways [145–147] T-cells specific antitumor immunity [148] |
In addition to proposing the model, we identified key questions that have to be answered in order to move the field forward (Box 1) and major technological/logistical solutions that are required to answer these questions (Box 2).
Finally, treatment of cancer with chemicals and radiation are currently the most popular and efficient strategies [161,162]. More recently, our armamentarium was advanced with vaccines against oncoviruses [163] and immunotherapy [164], both using the immune system to prevent or kill cancer. Unfortunately, neither of these strategies allowed us to eliminate completely any oncovirus, nor to cure some cancers.
Therefore, novel approaches are required to significantly change the status quo of this field. We believe that this change should come with methods promoting a healthy microbiome, development of next generation antibiotics targeting individual bacteria, new probiotics, and companion diagnostics that will define a course of personalized/precision medicine for each individual patient based on their respective transcriptome and microbiome.
Box 1. Current trending questions.
- Which shifts in microbial communities (presence/absence of particular bacterial species or alterations in microbial community structures) can enhance oncogenic virus infection progression or, on the contrary, help to eliminate it?
- Which host molecular pathways involved in viral infections and tumorigenesis are altered by microbiota?
- Can host genetics shape microbiota toward being anti- or pro-tumorigenic?
- How do microbiota affect the success of anticancer therapies?
- How do antitumor treatments affect microbiota? Are these effects relevant for efficient treatments and treatment-related co-morbidities?
Box 2. Emerging technologies and other solutions.
- Experimental approaches for the generation of different types of omics data that would allow simultaneous assessment of functional states of each of viruses, host and bacteria (e.g. new and improved single cell technologies for both eukaryotic and prokaryotic cells).
- Computational tools for the analysis of multi-omics datasets that would provide robust predictions of regulatory relationships between all three kingdoms (e.g. transkingdom networks [165,166], LEfSe [167]).
- Generation of new experimental models: a) humanized animal models (e.g. gnotobiotic animal models for cancer caused by oncogenic viruses harboring human microbiota and immune system); b) cell lines and tissues with virus and bacteria present (e.g. differentiated 3D cell aggregates colonized with specific bacteria [168–173]); c) next generation in vitro models such as organ-on-chip and human-on-chip microfluidic devices that allow application of robotic systems.
- Creation of multidisciplinary teams (involving oncologists with expertise in basic and clinical science, systems biologist, virologist, microbiologists, and engineers) through special funding mechanisms or even the creation of new institutions.
Acknowledgments
We would like to thank members of the Morgun and Shulzhenko laboratories for their discussions and support in the review. Special consideration to Karen DSouza, Kimberly White, and Nicholas Brown for their assistance in text editing.
Funding
This work was supported by the following grants: NIH U01 AI109695 (AM) and R01 DK103761 (NS)
References
- 1.GLOBOCAN 2012: Estimated Cancer Incidence Mortality and Prevalence Worldwide in 2012. http://globocan.iarc.fr/Pages/fact_sheets_cancer.aspx?cancer=all.
- 2.Cancer. http://www.who.int/cancer/en/
- 3.Luo GG, Ou JH. Oncogenic viruses and cancer. Virol Sin. 2015;30(2):83–84. doi: 10.1007/s12250-015-3599-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parkin DM. The global health burden of infection-associated cancers in the year 2002. Int J Cancer. 2006;118(12):3030–3044. doi: 10.1002/ijc.21731. [DOI] [PubMed] [Google Scholar]
- 5.Moore PS, Chang Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat Rev Cancer. 2010;10(12):878–889. doi: 10.1038/nrc2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mesri EA, Feitelson MA, Munger K. Human viral oncogenesis: a cancer hallmarks analysis. Cell Host Microbe. 2014;15(3):266–282. doi: 10.1016/j.chom.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Situnayake RD, Thurnham DI, Kootathep S, Chirico S, Lunec J, Davis M, McConkey B. Chain breaking antioxidant status in rheumatoid arthritis: clinical and laboratory correlates. Ann Rheum Dis. 1991;50(2):81–86. doi: 10.1136/ard.50.2.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buchli V, Pearce WB. Listening behavior in coorientational states. J Commun. 1974;24(3):62–70. doi: 10.1111/j.1460-2466.1974.tb00389.x. [DOI] [PubMed] [Google Scholar]
- 9.Konrad HR, Rattenborg CC. Combined action of laryngeal muscles. Acta Otolaryngol. 1969;67(6):646–649. doi: 10.3109/00016486909125491. [DOI] [PubMed] [Google Scholar]
- 10.Baldridge MT, Nice TJ, McCune BT, Yokoyama CC, Kambal A, Wheadon M, Diamond MS, Ivanova Y, Artyomov M, Virgin HW. Commensal microbes and interferon-lambda determine persistence of enteric murine norovirus infection. Science. 2015;347(6219):266–269. doi: 10.1126/science.1258025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Greer RL, Dong X, Moraes AC, Zielke RA, Fernandes GR, Peremyslova E, Vasquez-Perez S, Schoenborn AA, Gomes EP, Pereira AC. Akkermansia muciniphila mediates negative effects of IFNgamma on glucose metabolism. Nat Commun. 2016;7:13329. doi: 10.1038/ncomms13329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xuan C, Shamonki JM, Chung A, Dinome ML, Chung M, Sieling PA, Lee DJ. Microbial dysbiosis is associated with human breast cancer. PLoS One. 2014;9(1):e83744. doi: 10.1371/journal.pone.0083744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boursi B, Mamtani R, Haynes K, Yang YX. Recurrent antibiotic exposure may promote cancer formation–Another step in understanding the role of the human microbiota? Eur J Cancer. 2015;51(17):2655–2664. doi: 10.1016/j.ejca.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mabrok HB, Klopfleisch R, Ghanem KZ, Clavel T, Blaut M, Loh G. Lignan transformation by gut bacteria lowers tumor burden in a gnotobiotic rat model of breast cancer. Carcinogenesis. 2012;33(1):203–208. doi: 10.1093/carcin/bgr256. [DOI] [PubMed] [Google Scholar]
- 15.Polk DB, Peek RM., Jr Helicobacter pylori: gastric cancer and beyond. Nat Rev Cancer. 2010;10(6):403–414. doi: 10.1038/nrc2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Welton JC, Marr JS, Friedman SM. Association between hepatobiliary cancer and typhoid carrier state. Lancet. 1979;1(8120):791–794. doi: 10.1016/s0140-6736(79)91315-1. [DOI] [PubMed] [Google Scholar]
- 17.Abdulamir AS, Hafidh RR, Abu Bakar F. The association of Streptococcus bovis/gallolyticus with colorectal tumors: the nature and the underlying mechanisms of its etiological role. J Exp Clin Cancer Res. 2011;30:11. doi: 10.1186/1756-9966-30-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sender R, Fuchs S, Milo R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016;14(8):e1002533. doi: 10.1371/journal.pbio.1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilks J, Lien E, Jacobson AN, Fischbach MA, Qureshi N, Chervonsky AV, Golovkina TV. Mammalian lipopolysaccharide receptors incorporated into the retroviral envelope augment virus transmission. Cell Host Microbe. 2015;18(4):456–462. doi: 10.1016/j.chom.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Page Thomas DP, King B, Stephens T, Dingle JT. In vivo studies of cartilage regeneration after damage induced by catabolin/interleukin-1. Ann Rheum Dis. 1991;50(2):75–80. doi: 10.1136/ard.50.2.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robinson CM, Jesudhasan PR, Pfeiffer JK. Bacterial lipopolysaccharide binding enhances virion stability and promotes environmental fitness of an enteric virus. Cell Host Microbe. 2014;15(1):36–46. doi: 10.1016/j.chom.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilson FM, 2nd, Ostler HB. superior limbic keratoconjunctivitis. Int Ophthalmol Clin. 1986;26(4):99–112. doi: 10.1097/00004397-198602640-00010. [DOI] [PubMed] [Google Scholar]
- 23.Macpherson AJ, Harris NL. Interactions between commensal intestinal bacteria and the immune system. Nat Rev Immunol. 2004;4(6):478–485. doi: 10.1038/nri1373. [DOI] [PubMed] [Google Scholar]
- 24.Abt MC, Osborne LC, Monticelli LA, Doering TA, Alenghat T, Sonnenberg GF, Paley MA, Antenus M, Williams KL, Erikson J. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity. 2012;37(1):158–170. doi: 10.1016/j.immuni.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shreiner AB, Kao JY, Young VB. The gut microbiome in health and in disease. Curr Opin Gastroenterol. 2015;31(1):69–75. doi: 10.1097/MOG.0000000000000139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greer RL, Morgun A, Shulzhenko N. Bridging immunity and lipid metabolism by gut microbiota. J Allergy Clin Immunol. 2013;132(2):253–262. doi: 10.1016/j.jaci.2013.06.025. (quiz 263) [DOI] [PubMed] [Google Scholar]
- 27.Shulzhenko N, Morgun A, Hsiao W, Battle M, Yao M, Gavrilova O, Orandle M, Mayer L, Macpherson AJ, McCoy KD. Crosstalk between B lymphocytes, microbiota and the intestinal epithelium governs immunity versus metabolism in the gut. Nat Med. 2011;17(12):1585–1593. doi: 10.1038/nm.2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Musso G, Gambino R, Cassader M. Obesity, diabetes, and gut microbiota: the hygiene hypothesis expanded? Diabetes Care. 2010;33(10):2277–2284. doi: 10.2337/dc10-0556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Louis P, Hold GL, Flint HJ. The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol. 2014;12(10):661–672. doi: 10.1038/nrmicro3344. [DOI] [PubMed] [Google Scholar]
- 30.Tlaskalova-Hogenova H, Stepankova R, Kozakova H, Hudcovic T, Vannucci L, Tuckova L, Rossmann P, Hrncir T, Kverka M, Zakostelska Z. The role of gut microbiota (commensal bacteria) and the mucosal barrier in the pathogenesis of inflammatory and autoimmune diseases and cancer: contribution of germ-free and gnotobiotic animal models of human diseases. Cell Mol Immunol. 2011;8(2):110–120. doi: 10.1038/cmi.2010.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, Molina DA, Salcedo R, Back T, Cramer S. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science. 2013;342(6161):967–970. doi: 10.1126/science.1240527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillere R, Hannani D, Enot DP, Pfirschke C, Engblom C, Pittet MJ. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science. 2013;342(6161):971–976. doi: 10.1126/science.1240537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karin M, Jobin C, Balkwill F. Chemotherapy, immunity and microbiota–a new triumvirate? Nat Med. 2014;20(2):126–127. doi: 10.1038/nm.3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cancer Fact Sheet. http://www.who.int/mediacentre/factsheets/fs297/en/
- 35.Lafaro KJ, Demirjian AN, Pawlik TM. Epidemiology of hepatocellular carcinoma. Surg Oncol Clin N Am. 2015;24(1):1–17. doi: 10.1016/j.soc.2014.09.001. [DOI] [PubMed] [Google Scholar]
- 36.de Martel C, Maucort-Boulch D, Plummer M, Franceschi S. World-wide relative contribution of hepatitis B and C viruses in hepatocellular carcinoma. Hepatology. 2015;62(4):1190–1200. doi: 10.1002/hep.27969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ward JM, Fox JG, Anver MR, Haines DC, George CV, Collins MJ, Jr, Gorelick PL, Nagashima K, Gonda MA, Gilden RV. Chronic active hepatitis and associated liver tumors in mice caused by a persistent bacterial infection with a novel Helicobacter species. J Natl Cancer Inst. 1994;86(16):1222–1227. doi: 10.1093/jnci/86.16.1222. [DOI] [PubMed] [Google Scholar]
- 38.Leone N, Pellicano R, Brunello F, Cutufia MA, Berrutti M, Fagoonee S, Rizzetto M, Ponzetto A. Helicobacter pylori seroprevalence in patients with cirrhosis of the liver and hepatocellular carcinoma. Cancer Detect Prev. 2003;27(6):494–497. doi: 10.1016/j.cdp.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 39.Rocha M, Avenaud P, Menard A, Le Bail B, Balabaud C, Bioulac-Sage P, de Magalhaes Queiroz DM, Megraud F. Association of Helicobacter species with hepatitis C cirrhosis with or without hepatocellular carcinoma. Gut. 2005;54(3):396–401. doi: 10.1136/gut.2004.042168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garcia A, Feng Y, Parry NM, McCabe A, Mobley MW, Lertpiriyapong K, Whary MT, Fox JG. Helicobacter pylori infection does not promote hepatocellular cancer in a transgenic mouse model of hepatitis C virus pathogenesis. Gut Microbes. 2013;4(6):577–590. doi: 10.4161/gmic.26042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fox JG, Feng Y, Theve EJ, Raczynski AR, Fiala JL, Doernte AL, Williams M, McFaline JL, Essigmann JM, Schauer DB. Gut microbes define liver cancer risk in mice exposed to chemical and viral transgenic hepatocarcinogens. Gut. 2010;59(1):88–97. doi: 10.1136/gut.2009.183749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hiroi M, Ohmori Y. Constitutive nuclear factor kappaB activity is required to elicit interferon-gamma-induced expression of chemokine CXC ligand 9 (CXCL9) and CXCL10 in human tumour cell lines. Biochem J. 2003;376(Pt 2):393–402. doi: 10.1042/BJ20030842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cahill RJ, Foltz CJ, Fox JG, Dangler CA, Powrie F, Schauer DB. Inflammatory bowel disease: an immunity-mediated condition triggered by bacterial infection with Helicobacter hepaticus. Infect Immun. 1997;65(8):3126–3131. doi: 10.1128/iai.65.8.3126-3131.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang Y, Fan XG, Wang ZM, Zhou JH, Tian XF, Li N. Identification of helicobacter species in human liver samples from patients with primary hepatocellular carcinoma. J Clin Pathol. 2004;57(12):1273–1277. doi: 10.1136/jcp.2004.018556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nilsson HO, Taneera J, Castedal M, Glatz E, Olsson R, Wadstrom T. Identification of Helicobacter pylori and other Helicobacter species by PCR, hybridization, and partial DNA sequencing in human liver samples from patients with primary sclerosing cholangitis or primary biliary cirrhosis. J Clin Microbiol. 2000;38(3):1072–1076. doi: 10.1128/jcm.38.3.1072-1076.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fox JG, Dewhirst FE, Shen Z, Feng Y, Taylor NS, Paster BJ, Ericson RL, Lau CN, Correa P, Araya JC. Hepatic Helicobacter species identified in bile and gallbladder tissue from Chileans with chronic cholecystitis. Gastroenterology. 1998;114(4):755–763. doi: 10.1016/s0016-5085(98)70589-x. [DOI] [PubMed] [Google Scholar]
- 47.Rowen M, Myers M, Williamson RA. Emphysematous gastritis in a leukemic child. Med Pediatr Oncol. 1976;2(4):433–437. doi: 10.1002/mpo.2950020409. [DOI] [PubMed] [Google Scholar]
- 48.Poutahidis T, Cappelle K, Levkovich T, Lee CW, Doulberis M, Ge Z, Fox JG, Horwitz BH, Erdman SE. Pathogenic intestinal bacteria enhance prostate cancer development via systemic activation of immune cells in mice. PLoS One. 2013;8(8):e73933. doi: 10.1371/journal.pone.0073933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen Y, Chen Z, Guo R, Chen N, Lu H, Huang S, Wang J, Li L. Correlation between gastrointestinal fungi and varying degrees of chronic hepatitis B virus infection. Diagn Microbiol Infect Dis. 2011;70(4):492–498. doi: 10.1016/j.diagmicrobio.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 50.Chou HH, Chien WH, Wu LL, Cheng CH, Chung CH, Horng JH, Ni YH, Tseng HT, Wu D, Lu X. Age-related immune clearance of hepatitis B virus infection requires the establishment of gut microbiota. Proc Natl Acad Sci U S A. 2015;112(7):2175–2180. doi: 10.1073/pnas.1424775112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lu H, Wu Z, Xu W, Yang J, Chen Y, Li L. Intestinal microbiota was assessed in cirrhotic patients with hepatitis B virus infection: intestinal microbiota of HBV cirrhotic patients. Microb Ecol. 2011;61(3):693–703. doi: 10.1007/s00248-010-9801-8. [DOI] [PubMed] [Google Scholar]
- 52.Xu M, Wang B, Fu Y, Chen Y, Yang F, Lu H, Chen Y, Xu J, Li L. Changes of fecal Bifidobacterium species in adult patients with hepatitis B virus-induced chronic liver disease. Microb Ecol. 2012;63(2):304–313. doi: 10.1007/s00248-011-9925-5. [DOI] [PubMed] [Google Scholar]
- 53.Lee DK, Kang JY, Shin HS, Park IH, Ha NJ. Antiviral activity of bifidobacterium adolescentis SPM0212 against hepatitis B virus. Arch Pharm Res. 2013;36(12):1525–1532. doi: 10.1007/s12272-013-0141-3. [DOI] [PubMed] [Google Scholar]
- 54.Lee DK, Jang S, Baek EH, Kim MJ, Lee KS, Shin HS, Chung MJ, Kim JE, Lee KO, Ha NJ. Lactic acid bacteria affect serum cholesterol levels, harmful fecal enzyme activity, and fecal water content. Lipids Health Dis. 2009;8:21. doi: 10.1186/1476-511X-8-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shin HS, Park SY, Lee DK, Kim SA, An HM, Kim JR, Kim MJ, Cha MG, Lee SW, Kim KJ. Hypocholesterolemic effect of sonication-killed Bifidobacterium longum isolated from healthy adult Koreans in high cholesterol fed rats. Arch Pharm Res. 2010;33(9):1425–1431. doi: 10.1007/s12272-010-0917-7. [DOI] [PubMed] [Google Scholar]
- 56.Lee DK, Park SY, Jang S, Baek EH, Kim MJ, Huh SM, Choi KS, Chung MJ, Kim JE, Lee KO. The combination of mixed lactic acid bacteria and dietary fiber lowers serum cholesterol levels and fecal harmful enzyme activities in rats. Arch Pharm Res. 2011;34(1):23–29. doi: 10.1007/s12272-011-0102-7. [DOI] [PubMed] [Google Scholar]
- 57.Lin YL, Shiao MS, Mettling C, Chou CK. Cholesterol requirement of hepatitis B surface antigen (HBsAg) secretion. Virology. 2003;314(1):253–260. doi: 10.1016/s0042-6822(03)00403-3. [DOI] [PubMed] [Google Scholar]
- 58.Arenson RL, Dwyer SJ, 3rd, Huang HK, Kundel HL, Seshadri SB. Computer applications and digital imaging. Radiology. 1991;178(3):913–914. doi: 10.1148/radiology.178.3.1994451. [DOI] [PubMed] [Google Scholar]
- 59.Nardis C, Mosca L, Mastromarino P. Vaginal microbiota and viral sexually transmitted diseases. Ann Ig. 2013;25(5):443–456. doi: 10.7416/ai.2013.1946. [DOI] [PubMed] [Google Scholar]
- 60.Voravuthikunchai SP, Bilasoi S, Supamala O. Antagonistic activity against pathogenic bacteria by human vaginal lactobacilli. Anaerobe. 2006;12(5–6):221–226. doi: 10.1016/j.anaerobe.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 61.O’Hanlon DE, Moench TR, Cone RA. Vaginal pH and microbicidal lactic acid when lactobacilli dominate the microbiota. PLoS One. 2013;8(11):e80074. doi: 10.1371/journal.pone.0080074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.O’Hanlon DE, Moench TR, Cone RA. In vaginal fluid, bacteria associated with bacterial vaginosis can be suppressed with lactic acid but not hydrogen peroxide. BMC Infect Dis. 2011;11:200. doi: 10.1186/1471-2334-11-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Borgdorff H, Tsivtsivadze E, Verhelst R, Marzorati M, Jurriaans S, Ndayisaba GF, Schuren FH, van de Wijgert JH. Lactobacillus-dominated cervicovaginal microbiota associated with reduced HIV/STI prevalence and genital HIV viral load in African women. ISME J. 2014;8(9):1781–1793. doi: 10.1038/ismej.2014.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Boskey ER, Cone RA, Whaley KJ, Moench TR. Origins of vaginal acidity: high D/L lactate ratio is consistent with bacteria being the primary source. Hum Reprod. 2001;16(9):1809–1813. doi: 10.1093/humrep/16.9.1809. [DOI] [PubMed] [Google Scholar]
- 65.Atassi F, Brassart D, Grob P, Graf F, Servin AL. Lactobacillus strains isolated from the vaginal microbiota of healthy women inhibit Prevotella bivia and Gardnerella vaginalis in coculture and cell culture. FEMS Immunol Med Microbiol. 2006;48(3):424–432. doi: 10.1111/j.1574-695X.2006.00162.x. [DOI] [PubMed] [Google Scholar]
- 66.Klebanoff SJ, Hillier SL, Eschenbach DA, Waltersdorph AM. Control of the microbial flora of the vagina by H2O2-generating lactobacilli. J Infect Dis. 1991;164(1):94–100. doi: 10.1093/infdis/164.1.94. [DOI] [PubMed] [Google Scholar]
- 67.Ma B, Forney LJ, Ravel J. Vaginal microbiome: rethinking health and disease. Annu Rev Microbiol. 2012;66:371–389. doi: 10.1146/annurev-micro-092611-150157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ling Z, Kong J, Liu F, Zhu H, Chen X, Wang Y, Li L, Nelson KE, Xia Y, Xiang C. Molecular analysis of the diversity of vaginal microbiota associated with bacterial vaginosis. BMC Genomics. 2010;11:488. doi: 10.1186/1471-2164-11-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Anahtar MN, Byrne EH, Doherty KE, Bowman BA, Yamamoto HS, Soumillon M, Padavattan N, Ismail N, Moodley A, Sabatini ME. Cervicovaginal bacteria are a major modulator of host inflammatory responses in the female genital tract. Immunity. 2015;42(5):965–976. doi: 10.1016/j.immuni.2015.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brotman RM. Vaginal microbiome and sexually transmitted infections: an epidemiologic perspective. J Clin Invest. 2011;121(12):4610–4617. doi: 10.1172/JCI57172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Walboomers JM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV, Snijders PJ, Peto J, Meijer CJ, Munoz N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol. 1999;189(1):12–19. doi: 10.1002/(SICI)1096-9896(199909)189:1<12::AID-PATH431>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 72.Bosch FX, Manos MM, Munoz N, Sherman M, Jansen AM, Peto J, Schiffman MH, Moreno V, Kurman R, Shah KV. Prevalence of human papillomavirus in cervical cancer: a worldwide perspective: international biological study on cervical cancer (IBSCC) Study Group. J Natl Cancer Inst. 1995;87(11):796–802. doi: 10.1093/jnci/87.11.796. [DOI] [PubMed] [Google Scholar]
- 73.McLaughlin-Drubin ME, Meyers J, Munger K. Cancer associated human papillomaviruses. Curr Opin Virol. 2012;2(4):459–466. doi: 10.1016/j.coviro.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Remmink AJ, Walboomers JM, Helmerhorst TJ, Voorhorst FJ, Rozendaal L, Risse EK, Meijer CJ, Kenemans P. The presence of persistent high-risk HPV genotypes in dysplastic cervical lesions is associated with progressive disease: natural history up to 36 months. Int J Cancer. 1995;61(3):306–311. doi: 10.1002/ijc.2910610305. [DOI] [PubMed] [Google Scholar]
- 75.Chen HC, Schiffman M, Lin CY, Pan MH, You SL, Chuang LC, Hsieh CY, Liaw KL, Hsing AW, Chen CJ. Persistence of type-specific human papillomavirus infection and increased long-term risk of cervical cancer. J Natl Cancer Inst. 2011;103(18):1387–1396. doi: 10.1093/jnci/djr283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cuschieri KS, Cubie HA, Whitley MW, Gilkison G, Arends MJ, Graham C, McGoogan E. Persistent high risk HPV infection associated with development of cervical neoplasia in a prospective population study. J Clin Pathol. 2005;58(9):946–950. doi: 10.1136/jcp.2004.022863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer. 2010;10(8):550–560. doi: 10.1038/nrc2886. [DOI] [PubMed] [Google Scholar]
- 78.Shulzhenko N, Lyng H, Sanson GF, Morgun A. Menage a trois: an evolutionary interplay between human papillomavirus, a tumor, and a woman. Trends Microbiol. 2014;22(6):345–353. doi: 10.1016/j.tim.2014.02.009. [DOI] [PubMed] [Google Scholar]
- 79.Brotman RM, Shardell MD, Gajer P, Tracy JK, Zenilman JM, Ravel J, Gravitt PE. Interplay between the temporal dynamics of the vaginal microbiota and human papillomavirus detection. J Infect Dis. 2014;210(11):1723–1733. doi: 10.1093/infdis/jiu330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Platz-Christensen JJ, Sundstrom E, Larsson PG. Bacterial vaginosis and cervical intraepithelial neoplasia. Acta Obstet Gynecol Scand. 1994;73(7):586–588. doi: 10.3109/00016349409006278. [DOI] [PubMed] [Google Scholar]
- 81.Oh HY, Kim BS, Seo SS, Kong JS, Lee JK, Park SY, Hong KM, Kim HK, Kim MK. The association of uterine cervical microbiota with an increased risk for cervical intraepithelial neoplasia in Korea. Clin Microbiol Infect. 2015;21(7):674 e671–679. doi: 10.1016/j.cmi.2015.02.026. [DOI] [PubMed] [Google Scholar]
- 82.Si J, You HJ, Yu J, Sung J, Ko G. Prevotella as a hub for vaginal microbiota under the influence of host genetics and their association with obesity. Cell Host Microbe. 2017;21(1):97–105. doi: 10.1016/j.chom.2016.11.010. [DOI] [PubMed] [Google Scholar]
- 83.Pybus V, Onderdonk AB. Evidence for a commensal, symbiotic relationship between Gardnerella vaginalis and Prevotella bivia involving ammonia: potential significance for bacterial vaginosis. J Infect Dis. 1997;175(2):406–413. doi: 10.1093/infdis/175.2.406. [DOI] [PubMed] [Google Scholar]
- 84.Pybus V, Onderdonk AB. A commensal symbiosis between Prevotella bivia and Peptostreptococcus anaerobius involves amino acids: potential significance to the pathogenesis of bacterial vaginosis. FEMS Immunol Med Microbiol. 1998;22(4):317–327. doi: 10.1111/j.1574-695X.1998.tb01221.x. [DOI] [PubMed] [Google Scholar]
- 85.Ling Z, Liu X, Chen X, Zhu H, Nelson KE, Xia Y, Li L, Xiang C. Diversity of cervicovaginal microbiota associated with female lower genital tract infections. Microb Ecol. 2011;61(3):704–714. doi: 10.1007/s00248-011-9813-z. [DOI] [PubMed] [Google Scholar]
- 86.Lee JE, Lee S, Lee H, Song YM, Lee K, Han MJ, Sung J, Ko G. Association of the vaginal microbiota with human papillomavirus infection in a Korean twin cohort. PLoS One. 2013;8(5):e63514. doi: 10.1371/journal.pone.0063514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dareng EO, Ma B, Famooto AO, Akarolo-Anthony SN, Offiong RA, Olaniyan O, Dakum PS, Wheeler CM, Fadrosh D, Yang H. Prevalent high-risk HPV infection and vaginal microbiota in Nigerian women. Epidemiol Infect. 2016;144(1):123–137. doi: 10.1017/S0950268815000965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mikamo H, Izumi K, Ito K, Watanabe K, Ueno K, Tamaya T. Internal bacterial flora of solid uterine cervical cancer. Kansenshogaku Zasshi. 1993;67(11):1057–1061. doi: 10.11150/kansenshogakuzasshi1970.67.1057. [DOI] [PubMed] [Google Scholar]
- 89.Samoff E, Koumans EH, Markowitz LE, Sternberg M, Sawyer MK, Swan D, Papp JR, Black CM, Unger ER. Association of Chlamydia trachomatis with persistence of high-risk types of human papillomavirus in a cohort of female adolescents. Am J Epidemiol. 2005;162(7):668–675. doi: 10.1093/aje/kwi262. [DOI] [PubMed] [Google Scholar]
- 90.Smith JS, Munoz N, Herrero R, Eluf-Neto J, Ngelangel C, Franceschi S, Bosch FX, Walboomers JM, Peeling RW. Evidence for Chlamydia trachomatis as a human papillomavirus cofactor in the etiology of invasive cervical cancer in Brazil and the Philippines. J Infect Dis. 2002;185(3):324–331. doi: 10.1086/338569. [DOI] [PubMed] [Google Scholar]
- 91.Schlott T, Eiffert H, Bohne W, Landgrebe J, Brunner E, Spielbauer B, Knight B. Chlamydia trachomatis modulates expression of tumor suppressor gene caveolin-1 and oncogene C-myc in the transformation zone of non-neoplastic cervical tissue. Gynecol Oncol. 2005;98(3):409–419. doi: 10.1016/j.ygyno.2005.04.034. [DOI] [PubMed] [Google Scholar]
- 92.Paba P, Bonifacio D, Di Bonito L, Ombres D, Favalli C, Syrjanen K, Ciotti M. Co-expression of HSV2 and Chlamydia trachomatis in HPV-positive cervical cancer and cervical intraepithelial neoplasia lesions is associated with aberrations in key intracellular pathways. Intervirology. 2008;51(4):230–234. doi: 10.1159/000156481. [DOI] [PubMed] [Google Scholar]
- 93.Mbopi-Keou FX, Belec L, Teo CG, Scully C, Porter SR. Synergism between HIV and other viruses in the mouth. Lancet Infect Dis. 2002;2(7):416–424. doi: 10.1016/s1473-3099(02)00317-1. [DOI] [PubMed] [Google Scholar]
- 94.Myer L, Denny L, Telerant R, Souza M, Wright TC, Jr, Kuhn L. Bacterial vaginosis and susceptibility to HIV infection in South African women: a nested case-control study. J Infect Dis. 2005;192(8):1372–1380. doi: 10.1086/462427. [DOI] [PubMed] [Google Scholar]
- 95.Atashili J, Poole C, Ndumbe PM, Adimora AA, Smith JS. Bacterial vaginosis and HIV acquisition: a meta-analysis of published studies. AIDS. 2008;22(12):1493–1501. doi: 10.1097/QAD.0b013e3283021a37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sha BE, Zariffard MR, Wang QJ, Chen HY, Bremer J, Cohen MH, Spear GT. Female genital-tract HIV load correlates inversely with Lactobacillus species but positively with bacterial vaginosis and Mycoplasma hominis. J Infect Dis. 2005;191(1):25–32. doi: 10.1086/426394. [DOI] [PubMed] [Google Scholar]
- 97.Aldunate M, Tyssen D, Johnson A, Zakir T, Sonza S, Moench T, Cone R, Tachedjian G. Vaginal concentrations of lactic acid potently inactivate HIV. J Antimicrob Chemother. 2013;68(9):2015–2025. doi: 10.1093/jac/dkt156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gosmann C, Anahtar MN, Handley SA, Farcasanu M, Abu-Ali G, Bowman BA, Padavattan N, Desai C, Droit L, Moodley A. Lactobacillus-Deficient cervicovaginal bacterial communities are associated with increased HIV acquisition in young south african women. Immunity. 2017;46(1):29–37. doi: 10.1016/j.immuni.2016.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.van de Wijgert JH, Borgdorff H, Verhelst R, Crucitti T, Francis S, Verstraelen H, Jespers V. The vaginal microbiota: what have we learned after a decade of molecular characterization? PLoS One. 2014;9(8):e105998. doi: 10.1371/journal.pone.0105998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hashemi FB, Ghassemi M, Faro S, Aroutcheva A, Spear GT. Induction of human immunodeficiency virus type 1 expression by anaerobes associated with bacterial vaginosis. J Infect Dis. 2000;181(5):1574–1580. doi: 10.1086/315455. [DOI] [PubMed] [Google Scholar]
- 101.Cu-Uvin S, Hogan JW, Caliendo AM, Harwell J, Mayer KH, Carpenter CC, H.I.V.E.R. Study Association between bacterial vaginosis and expression of human immunodeficiency virus type 1 RNA in the female genital tract. Clin Infect Dis. 2001;33(6):894–896. doi: 10.1086/322613. [DOI] [PubMed] [Google Scholar]
- 102.Williams SA, Greene WC. Host factors regulating post-integration latency of HIV. Trends Microbiol. 2005;13(4):137–139. doi: 10.1016/j.tim.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 103.Williams SA, Chen LF, Kwon H, Ruiz-Jarabo CM, Verdin E, Greene WC. NF-kappaB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. EMBO J. 2006;25(1):139–149. doi: 10.1038/sj.emboj.7600900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jiang G, Espeseth A, Hazuda DJ, Margolis DM. c-Myc and Sp1 contribute to proviral latency by recruiting histone deacetylase 1 to the human immunodeficiency virus type 1 promoter. J Virol. 2007;81(20):10914–10923. doi: 10.1128/JVI.01208-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Imai K, Yamada K, Tamura M, Ochiai K, Okamoto T. Reactivation of latent HIV-1 by a wide variety of butyric acid-producing bacteria. Cell Mol Life Sci. 2012;69(15):2583–2592. doi: 10.1007/s00018-012-0936-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Imai K, Ochiai K, Okamoto T. Reactivation of latent HIV-1 infection by the periodontopathic bacterium Porphyromonas gingivalis involves histone modification. J Immunol. 2009;182(6):3688–3695. doi: 10.4049/jimmunol.0802906. [DOI] [PubMed] [Google Scholar]
- 107.Riggs MG, Whittaker RG, Neumann JR, Ingram VM. n-Butyrate causes histone modification in HeLa and Friend erythroleukaemia cells. Nature. 1977;268(5619):462–464. doi: 10.1038/268462a0. [DOI] [PubMed] [Google Scholar]
- 108.Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, Pettersson S. Host-gut microbiota metabolic interactions. Science. 2012;336(6086):1262–1267. doi: 10.1126/science.1223813. [DOI] [PubMed] [Google Scholar]
- 109.Ferreira VH, Nazli A, Khan G, Mian MF, Ashkar AA, Gray-Owen S, Kaul R, Kaushic C. Endometrial epithelial cell responses to coinfecting viral and bacterial pathogens in the genital tract can activate the HIV-1 LTR in an NF{kappa}B-and AP-1-dependent manner. J Infect Dis. 2011;204(2):299–308. doi: 10.1093/infdis/jir260. [DOI] [PubMed] [Google Scholar]
- 110.Torian LV, Makki HA, Menzies IB, Murrill CS, Benson DA, Schween FW, Weisfuse IB. High HIV seroprevalence associated with gonorrhea: new York City Department of Health, sexually transmitted disease clinics, 1990–1997. AIDS. 2000;14(2):189–195. doi: 10.1097/00002030-200001280-00015. [DOI] [PubMed] [Google Scholar]
- 111.Malott RJ, Keller BO, Gaudet RG, McCaw SE, Lai CC, Dobson-Belaire WN, Hobbs JL, St Michael F, Cox AD, Moraes TF, et al. Neisseria gonorrhoeae-derived heptose elicits an innate immune response and drives HIV-1 expression. Proc Natl Acad Sci U S A. 2013;110(25):10234–10239. doi: 10.1073/pnas.1303738110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chen A, Boulton IC, Pongoski J, Cochrane A, Gray-Owen SD. Induction of HIV-1 long terminal repeat-mediated transcription by Neisseria gonorrhoeae. AIDS. 2003;17(4):625–628. doi: 10.1097/00002030-200303070-00019. [DOI] [PubMed] [Google Scholar]
- 113.Kistler JO, Arirachakaran P, Poovorawan Y, Dahlen G, Wade WG. The oral microbiome in human immunodeficiency virus (HIV)-positive individuals. J Med Microbiol. 2015;64(9):1094–1101. doi: 10.1099/jmm.0.000128. [DOI] [PubMed] [Google Scholar]
- 114.Tenenbaum H, Elkaim R, Cuisinier F, Dahan M, Zamanian P, Lang JM. Prevalence of six periodontal pathogens detected by DNA probe method in HIV vs non-HIV periodontitis. Oral Dis. 1997;3(Suppl 1):S153–155. doi: 10.1111/j.1601-0825.1997.tb00350.x. [DOI] [PubMed] [Google Scholar]
- 115.Murray PA, Winkler JR, Peros WJ, French CK, Lippke JA. DNA probe detection of periodontal pathogens in HIV-associated periodontal lesions. Oral Microbiol Immunol. 1991;6(1):34–40. doi: 10.1111/j.1399-302x.1991.tb00449.x. [DOI] [PubMed] [Google Scholar]
- 116.Rams TE, Andriolo M, Jr, Feik D, Abel SN, McGivern TM, Slots J. Microbiological study of HIV-related periodontitis. J Periodontol. 1991;62(1):74–81. doi: 10.1902/jop.1991.62.1.74. [DOI] [PubMed] [Google Scholar]
- 117.Tezal M, Sullivan MA, Reid ME, Marshall JR, Hyland A, Loree T, Lillis C, Hauck L, Wactawski-Wende J, Scannapieco FA. Chronic periodontitis and the risk of tongue cancer. Arch Otolaryngol Head Neck Surg. 2007;133(5):450–454. doi: 10.1001/archotol.133.5.450. [DOI] [PubMed] [Google Scholar]
- 118.Tezal M, Sullivan MA, Hyland A, Marshall JR, Stoler D, Reid ME, Loree TR, Rigual NR, Merzianu M, Hauck L. Chronic periodontitis and the incidence of head and neck squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev. 2009;18(9):2406–2412. doi: 10.1158/1055-9965.EPI-09-0334. [DOI] [PubMed] [Google Scholar]
- 119.Tezal M, Grossi SG, Genco RJ. Is periodontitis associated with oral neoplasms? J Periodontol. 2005;76(3):406–410. doi: 10.1902/jop.2005.76.3.406. [DOI] [PubMed] [Google Scholar]
- 120.Kruger M, Hansen T, Kasaj A, Moergel M. The correlation between chronic periodontitis and oral cancer. Case Rep Dent. 2013;2013:262410. doi: 10.1155/2013/262410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ahn J, Segers S, Hayes RB. Periodontal disease, Porphyromonas gingivalis serum antibody levels and orodigestive cancer mortality. Carcinogenesis. 2012;33(5):1055–1058. doi: 10.1093/carcin/bgs112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ha NH, Woo BH, Kim DJ, Ha ES, Choi JI, Kim SJ, Park BS, Lee JH, Park HR. Prolonged and repetitive exposure to Porphyromonas gingivalis increases aggressiveness of oral cancer cells by promoting acquisition of cancer stem cell properties. Tumour Biol. 2015;36(12):9947–9960. doi: 10.1007/s13277-015-3764-9. [DOI] [PubMed] [Google Scholar]
- 123.Gondivkar SM, Parikh RV, Gadbail AR, Solanke V, Chole R, Mankar M, Balsaraf S. Involvement of viral factors with head and neck cancers. Oral Oncol. 2012;48(3):195–199. doi: 10.1016/j.oraloncology.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 124.da Silva SR, de Oliveira DE. HIV, EBV. and KSHV: viral cooperation in the pathogenesis of human malignancies. Cancer Lett. 2011;305(2):175–185. doi: 10.1016/j.canlet.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 125.Li H, Chen V, Chen Y, Baumgartner JC, Machida CA. Herpesviruses in endodontic pathoses: association of Epstein-Barr virus with irreversible pulpitis and apical periodontitis. J Endod. 2009;35(1):23–29. doi: 10.1016/j.joen.2008.09.017. [DOI] [PubMed] [Google Scholar]
- 126.Kubar A, Saygun I, Ozdemir A, Yapar M, Slots J. Real-time polymerase chain reaction quantification of human cytomegalovirus and Epstein-Barr virus in periodontal pockets and the adjacent gingiva of periodontitis lesions. J Periodontal Res. 2005;40(2):97–104. doi: 10.1111/j.1600-0765.2005.00770.x. [DOI] [PubMed] [Google Scholar]
- 127.Sabeti M, Valles Y, Nowzari H, Simon JH, Kermani-Arab V, Slots J. Cytomegalovirus and Epstein-Barr virus DNA transcription in endodontic symptomatic lesions. Oral Microbiol Immunol. 2003;18(2):104–108. doi: 10.1034/j.1399-302x.2003.00055.x. [DOI] [PubMed] [Google Scholar]
- 128.Saygun I, Kubar A, Ozdemir A, Yapar M, Slots J. Herpesviral-bacterial interrelationships in aggressive periodontitis. J Periodontal Res. 2004;39(4):207–212. doi: 10.1111/j.1600-0765.2004.00728.x. [DOI] [PubMed] [Google Scholar]
- 129.Slots J, Kamma JJ, Sugar C. The herpesvirus-Porphyromonas gingivalis-periodontitis axis. J Periodontal Res. 2003;38(3):318–323. doi: 10.1034/j.1600-0765.2003.00659.x. [DOI] [PubMed] [Google Scholar]
- 130.Thakker S, Verma SC. Co-infections and pathogenesis of KSHV-Associated malignancies. Front Microbiol. 2016;7:151. doi: 10.3389/fmicb.2016.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Michaelis M, Doerr HW, Cinatl J. The story of human cytomegalovirus and cancer: increasing evidence and open questions. Neoplasia. 2009;11(1):1–9. doi: 10.1593/neo.81178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Herbein G, Kumar A. The oncogenic potential of human cytomegalovirus and breast cancer. Front Oncol. 2014;4:230. doi: 10.3389/fonc.2014.00230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Saravani S, Kadeh H, Miri-Moghaddam E, Zekri A, Sanadgol N, Gholami A. Human cytomegalovirus in oral squamous cell carcinoma in southeast of Iran. Jundishapur J Microbiol. 2015;8(8):e21838. doi: 10.5812/jjm.21838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cavallin LE, Goldschmidt-Clermont P, Mesri EA. Molecular and cellular mechanisms of KSHV oncogenesis of Kaposi’s sarcoma associated with HIV/AIDS. PLoS Pathog. 2014;10(7):e1004154. doi: 10.1371/journal.ppat.1004154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137(2):289–294. doi: 10.5858/arpa.2012-0101-RS. [DOI] [PubMed] [Google Scholar]
- 136.Morris TL, Arnold RR, Webster-Cyriaque J. Signaling cascades triggered by bacterial metabolic end products during reactivation of Kaposi’s sarcoma-associated herpesvirus. J Virol. 2007;81(11):6032–6042. doi: 10.1128/JVI.02504-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ko YH. EBV and human cancer. Exp Mol Med. 2015;47:e130. doi: 10.1038/emm.2014.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Imai K, Inoue H, Tamura M, Cueno ME, Inoue H, Takeichi O, Kusama K, Saito I, Ochiai K. The periodontal pathogen Porphyromonas gingivalis induces the Epstein-Barr virus lytic switch transactivator ZEBRA by histone modification. Biochimie. 2012;94(3):839–846. doi: 10.1016/j.biochi.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 139.Spadavecchia S, Gonzalez-Lopez O, Carroll KD, Palmeri D, Lukac DM. Convergence of Kaposi’s sarcoma-associated herpesvirus reactivation with Epstein-Barr virus latency and cellular growth mediated by the notch signaling pathway in coinfected cells. J Virol. 2010;84(20):10488–10500. doi: 10.1128/JVI.00894-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kassiotis G. Endogenous retroviruses and the development of cancer. J Immunol. 2014;192(4):1343–1349. doi: 10.4049/jimmunol.1302972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 142.Lee E, Iskow R, Yang L, Gokcumen O, Haseley P, Luquette LJ, 3rd, Lohr JG, Harris CC, Ding L, Wilson RK, et al. Landscape of somatic retrotransposition in human cancers. Science. 2012;337(6097):967–971. doi: 10.1126/science.1222077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Young GR, Eksmond U, Salcedo R, Alexopoulou L, Stoye JP, Kassiotis G. Resurrection of endogenous retroviruses in antibody-deficient mice. Nature. 2012;491(7426):774–778. doi: 10.1038/nature11599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Young GR, Mavrommatis B, Kassiotis G. Microarray analysis reveals global modulation of endogenous retroelement transcription by microbes. Retrovirology. 2014;11:59. doi: 10.1186/1742-4690-11-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hung T, Pratt GA, Sundararaman B, Townsend MJ, Chaivorapol C, Bhangale T, Graham RR, Ortmann W, Criswell LA, Yeo GW. The Ro60 autoantigen binds endogenous retroelements and regulates inflammatory gene expression. Science. 2015;350(6259):455–459. doi: 10.1126/science.aac7442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, Han H, Liang G, Jones PA, Pugh TJ. DNA-Demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell. 2015;162(5):961–973. doi: 10.1016/j.cell.2015.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, Hein A, Rote NS, Cope LM, Snyder A, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. 2016;164(5):1073. doi: 10.1016/j.cell.2015.10.020. [DOI] [PubMed] [Google Scholar]
- 148.Cherkasova E, Weisman Q, Childs RW. Endogenous retroviruses as targets for antitumor immunity in renal cell cancer and other tumors. Front Oncol. 2013;3:243. doi: 10.3389/fonc.2013.00243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Danaei G, Vander Hoorn S, Lopez AD, Murray CJ, Ezzati M. Comparative Risk Assessment collaborating g: causes of cancer in the world: comparative risk assessment of nine behavioural and environmental risk factors. Lancet. 2005;366(9499):1784–1793. doi: 10.1016/S0140-6736(05)67725-2. [DOI] [PubMed] [Google Scholar]
- 150.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10(8):789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 151.Chen CJ, Liang KY, Chang YS, Wang YF, Hsieh T, Hsu MM, Chen JY, Liu MY. Multiple risk factors of nasopharyngeal carcinoma: epstein-Barr virus, malarial infection, cigarette smoking and familial tendency. Anticancer Res. 1990;10(2B):547–553. [PubMed] [Google Scholar]
- 152.Wu J, Peters BA, Dominianni C, Zhang Y, Pei Z, Yang L, Ma Y, Purdue MP, Jacobs EJ, Gapstur SM. Cigarette smoking and the oral microbiome in a large study of American adults. ISME J. 2016;10(10):2435–2446. doi: 10.1038/ismej.2016.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Grossi SG, Zambon JJ, Ho AW, Koch G, Dunford RG, Machtei EE, Norderyd OM, Genco RJ. Assessment of risk for periodontal disease: i. Risk indicators for attachment loss. J Periodontol. 1994;65(3):260–267. doi: 10.1902/jop.1994.65.3.260. [DOI] [PubMed] [Google Scholar]
- 154.Brinton LA, Hamman RF, Huggins GR, Lehman HF, Levine RS, Mallin K, Fraumeni JF., Jr Sexual and reproductive risk factors for invasive squamous cell cervical cancer. J Natl Cancer Inst. 1987;79(1):23–30. [PubMed] [Google Scholar]
- 155.Smart S, Singal A, Mindel A. Social and sexual risk factors for bacterial vaginosis. Sex Transm Infect. 2004;80(1):58–62. doi: 10.1136/sti.2003.004978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Filipp E, Raczynski P, El Midaoui A, Pawlowska A, Tarnowska-Madra U, Scholz A, Niemiec KT, Chamerski J. Chlamydia trachomatis infection in sexually active adolescents and young women. Med Wieku Rozwoj. 2005;9(1):57–64. [PubMed] [Google Scholar]
- 157.Chuang SC, La Vecchia C, Boffetta P. Liver cancer: descriptive epidemiology and risk factors other than HBV and HCV infection. Cancer Lett. 2009;286(1):9–14. doi: 10.1016/j.canlet.2008.10.040. [DOI] [PubMed] [Google Scholar]
- 158.Million M, Maraninchi M, Henry M, Armougom F, Richet H, Carrieri P, Valero R, Raccah D, Vialettes B, Raoult D. Obesity-associated gut microbiota is enriched in Lactobacillus reuteri and depleted in Bifidobacterium animalis and Methanobrevibacter smithii. Int J Obesity(2005) 2012;36(6):817–825. doi: 10.1038/ijo.2011.153. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 159.Greer R, Dong X, Morgun A, Shulzhenko N. Investigating a holobiont: microbiota perturbations and transkingdom networks. Gut Microbes. 2016;7(2):126–135. doi: 10.1080/19490976.2015.1128625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, Benyamin FW, Lei YM, Jabri B, Alegre ML. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. 2015;350(6264):1084–1089. doi: 10.1126/science.aac4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Urruticoechea A, Alemany R, Balart J, Villanueva A, Vinals F, Capella G. Recent advances in cancer therapy: an overview. Curr Pharm Des. 2010;16(1):3–10. doi: 10.2174/138161210789941847. [DOI] [PubMed] [Google Scholar]
- 162.Gaspar LE, Ding M. A review of intensity-modulated radiation therapy. Curr Oncol Rep. 2008;10(4):294–299. doi: 10.1007/s11912-008-0046-3. [DOI] [PubMed] [Google Scholar]
- 163.Schiller JT, Lowy DR. Vaccines to prevent infections by oncoviruses. Annu Rev Microbiol. 2010;64:23–41. doi: 10.1146/annurev.micro.112408.134019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480(7378):480–489. doi: 10.1038/nature10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Morgun A, Dzutsev A, Dong X, Greer RL, Sexton DJ, Ravel J, Schuster M, Hsiao W, Matzinger P, Shulzhenko N. Uncovering effects of antibiotics on the host and microbiota using transkingdom gene networks. Gut. 2015;64(11):1732–1743. doi: 10.1136/gutjnl-2014-308820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Dong X, Yambartsev A, Ramsey SA, Thomas LD, Shulzhenko N, Morgun A. Reverse enGENEering of regulatory networks from big data: a roadmap for biologists. Bioinform Biol Insights. 2015;9:61–74. doi: 10.4137/BBI.S12467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12(6):R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Radtke AL, Herbst-Kralovetz MM. Culturing and applications of rotating wall vessel bioreactor derived 3D epithelial cell models. J Vis Exp. 2012;62 doi: 10.3791/3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Doerflinger SY, Throop AL, Herbst-Kralovetz MM. Bacteria in the vaginal microbiome alter the innate immune response and barrier properties of the human vaginal epithelia in a species-specific manner. J Infect Dis. 2014;209(12):1989–1999. doi: 10.1093/infdis/jiu004. [DOI] [PubMed] [Google Scholar]
- 170.Hjelm BE, Berta AN, Nickerson CA, Arntzen CJ, Herbst-Kralovetz MM. Development and characterization of a three-dimensional organotypic human vaginal epithelial cell model. Biol Reprod. 2010;82(3):617–627. doi: 10.1095/biolreprod.109.080408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Li N, Wang D, Sui Z, Qi X, Ji L, Wang X, Yang L. Development of an improved three-dimensional in vitro intestinal mucosa model for drug absorption evaluation. Tissue Eng Part C Methods. 2013;19(9):708–719. doi: 10.1089/ten.TEC.2012.0463. [DOI] [PubMed] [Google Scholar]
- 172.Chen Y, Lin Y, Davis KM, Wang Q, Rnjak-Kovacina J, Li C, Isberg RR, Kumamoto CA, Mecsas J, Kaplan DL. Robust bioengineered 3D functional human intestinal epithelium. Sci Rep. 2015;5:13708. doi: 10.1038/srep13708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Meyers C. Organotypic (raft) epithelial tissue culture system for the differentiation-dependent replication of papillomavirus. Methods Cell Sci. 1996;18(3) [Google Scholar]
- 174.Smith JS, Bosetti C, Munoz N, Herrero R, Bosch FX, Eluf-Neto J, Meijer CJ, Van Den Brule AJ, Franceschi S, Peeling RW. Chlamydia trachomatis and invasive cervical cancer: a pooled analysis of the IARC multicentric case-control study. Int J Cancer. 2004;111(3):431–439. doi: 10.1002/ijc.20257. [DOI] [PubMed] [Google Scholar]
- 175.Madeleine MM, Anttila T, Schwartz SM, Saikku P, Leinonen M, Carter JJ, Wurscher M, Johnson LG, Galloway DA, Daling JR. Risk of cervical cancer associated with Chlamydia trachomatis antibodies by histology, HPV type and HPV cofactors. Int J Cancer. 2007;120(3):650–655. doi: 10.1002/ijc.22325. [DOI] [PMC free article] [PubMed] [Google Scholar]