

Abstract
U1 snRNP (U1) functions in splicing introns and telescripting, which suppresses premature cleavage and polyadenylation (PCPA). Using U1 inhibition in human cells, we show that U1 telescripting is selectively required for sustaining long-distance transcription elongation in introns of large genes (median 39 kb). Evidence of widespread PCPA in the same locations in normal tissues reveals that large genes incur natural transcription attrition. Underscoring the importance of U1 telescripting as a gene-size-based mRNA-regulation mechanism, small genes were not sensitive to PCPA, and the spliced-mRNA productivity of ~1,000 small genes (median 6.8 kb) increased upon U1 inhibition. Notably, these small, upregulated genes were enriched in functions related to acute stimuli and cell-survival response, whereas genes subject to PCPA were enriched in cell-cycle progression and developmental functions. This gene size–function polarization increased in metazoan evolution by enormous intron expansion. We propose that telescripting adds an overarching layer of regulation to size–function-stratified genomes, leveraged by selective intron expansion to rapidly shift gene expression priorities.
Synthesis of full-gene-length transcripts from the majority of protein-coding genes is not the default process in metazoans; rather, it depends on the suppression of PCPA from cryptic polyadenylation signals (PASs) in nascent transcripts1,2. Although PASs at the 3′ ends of genes are most commonly used3, previous experiments have shown that blocking the 5′-end sequence of U1 using an antisense morpholino oligonucleotide (AMO) causes widespread PCPA from cryptic PASs in nascent RNA polymerase II (pol II) transcripts in metazoan cells1,2. This functional U1 inhibition revealed an additional role for U1, previously known for its role in precursor-mRNA (pre-mRNA) splicing4. We have termed this PCPA-suppression activity ‘telescripting’ to indicate that it is required for full-gene-length transcription. Similar to U1’s function in 5′-splice-site recognition, which is the first step in splicing, telescripting also depends on the base-pairing of the 5′-end sequence of U1 with pre-mRNAs. We have shown that the range of U1 telescripting is limited to about 1 kb, suggesting that U1 bound to the 5′ splice site for splicing might be insufficient to protect large (multi-kilobase) introns that are common in vertebrates. A model to address this limitation proposed that U1 would also bind extensively in introns, explaining U1’s abundance and known base pairing degeneracy. This model has recently been supported by the cross-linking of U1 to introns5. The model also proposed that PCPA and telescripting occur cotranscriptionally, but this has not yet been tested directly. U1’s role in nascent transcriptome protection has also recently been found to extend to upstream anti-sense transcripts (uaRNAs/PROMPTs), thus leading to the important observation that a high ratio of PAS to U1-binding sites in the anti-sense direction of divergent pol II promoters reinforces transcription in the protein-coding sense direction6,7.
Information about telescripting has thus far been limited by the moderate resolution of the methodologies used, including genomic tiling arrays1 and the subtractive hybridization-based HIDE-seq2. Importantly, these studies have not elucidated the potential role of U1 telescripting in gene expression regulation. Here, we applied RNA sequencing (RNA-seq) to obtain high-definition genome-wide characterization of PCPA and U1 telescripting and used pol II chromatin immunoprecipitation and sequencing (ChIP-seq) to determine U1’s role in transcription.
RESULTS
Gene-specific variations in U1 telescripting dependence
We used metabolic 30-min pulse labeling using 4-thiouridine (4-shU)8–10 to enhance detection of the effects of U1 inhibition on nascent transcripts and mRNA synthesis in HeLa cells. 4-shU labeling was carried out at 3.5 and 7.5 h post transfection (hereafter referred to 4- and 8-h transfection, respectively) with the same dose of either U1 AMO or control, nontargeting AMO. All RNAs transcribed during the labeling time window incorporate 4-shU and thus were able to be purified by a two-cycle thiol affinity selection under stringent conditions (Supplementary Fig. 1a,b). RNA-seq of these purified RNAs yielded 43–167 million mapped reads per sample (Supplementary Table 1). All samples were normalized to the same sequencing depth (RPM, reads per million). Parsing the RNA-seq reads showed that U1 AMO treatment decreased the total number of spliced junctions by 40% and changed the overall distribution of reads in genes, decreasing exon reads from 43% to 30% and increasing intron reads from 57% to 70% (Supplementary Fig. 1c–e). However, genome browser views of mapped reads in thousands of genes showed that these changes in RNA-seq reads with U1 AMO were generally not distributed uniformly throughout a gene and were inconsistent with intron retention (Fig. 1). The most common change observed with U1 AMO treatment, illustrated in representative genome browser views (Fig. 1), was the appearance of prominent peaks accumulating at the 5′ end of genes, extending from the transcription start site (TSS) and ending in one of the first introns (Fig. 1). These endpoints contained nongenomic 3′-poly(A) reads, indicating that they were produced by PCPA. Downstream of the PCPA points there was a clear reduction, and sometimes complete elimination, of reads, including in exons and the 3′ UTR. Genome browser views of representative mapped RNA-seq reads in several genes are shown in Figure 1.
Figure 1.
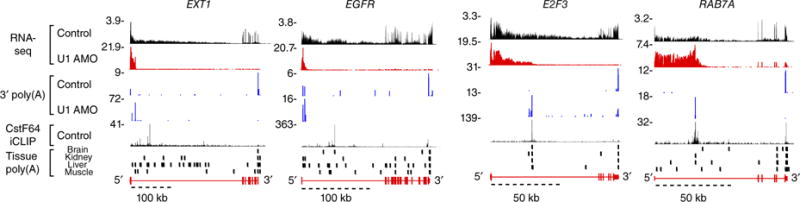
U1 inhibition causes widespread PCPA from intronic PASs. Genome browser views of representative PCPAed genes (EXT1, EGFR, E2F3, RAB7A) are shown with 4-shU labeled RNA-seq reads aligned to the human genome (hg19). HeLa cells were 4-shU labeled at 7.5 h post-transfection for 30 min with either control AMO (black) or U1 AMO (red). Values of peak heights were normalized to total mapped reads. Non-genomically-encoded 3′-poly(A) reads from poly(A)-selected RNA-seq samples are shown in blue, known binding sites for the cleavage and polyadenylation factor CstF64 (ref. 11) are shown in black, and poly(A) sites detected in various human tissues (brain, kidney, liver and muscle)12 are shown as vertical black bars. RNA-seq data are summarized in Supplementary Table 1.
The 3′-poly(A) nature of prematurely cleaved and polyadenylated (PCPAed) transcripts was further validated using 3′ rapid amplification of cDNA ends (3′ RACE) in select genes (Supplementary Fig. 2a). Frequently, multiple 3′-poly(A) peaks were detected in the same gene, including closely spaced series in the same intron, suggesting that multiple PASs can be actionable and full-length transcripts require U1 telescripting of all PASs (Fig. 1). Binding of cleavage and polyadenylation factors, such as CstF64, at or near PCPA locations in normal HeLa cells11 supports this conclusion and shows that these actionable PASs are indistinguishable from those found at the ends of full-length transcripts (Fig. 1, CstF64 iCLIP). Furthermore, as discussed below, many PCPA locations coincided with 3′-poly(A) reads that were previously detected in human tissues (Fig. 1, tissue poly(A) tracks)12.
PCPA susceptibility increases with gene size
To estimate the global scope of PCPA, we implemented a computational workflow to identify genes with the characteristic RNA-seq pattern changes in U1 AMO versus control AMO as described: a significant decrease in reads in the last exon compared to the first exon (≥2-fold, P value ≤ 0.01, Poisson test) and a significant increase in reads in the first quarter compared to the last quarter of the pre-mRNA between the first 5′splice site and the last 3′splice site (P value ≤ 0.01, Poisson test). This scheme captures both single-point and complete PCPA, such as those exemplified in Figure 1, as well as partial and multipoint PCPA anywhere throughout the pre-mRNA (for example ARID1B and ABCC1, Supplementary Fig. 2b). It excludes transcription initiation downregulation, which would decrease reads throughout the transcript. The PCPA calculation was applied to the longest isoform (RefSeq) of all intron-containing expressed genes (n = 9,744, reads per million mapped reads (RPKM) ≥ 1) from the 8-h data set and identified 3,590 genes as PCPAed. Visual inspection of hundreds of genes confirmed the validity of this analysis. Nearly 90% of genes detected as PCPAed in the 4-h experiment were also called as PCPAed at 8 h post U1 AMO transfection (Supplementary Fig. 2c,d and Supplementary Table 2), suggesting rapid onset and nearly complete effectiveness for the transfected U1 AMO dose.
Comparison of PCPAed and non-PCPAed genes revealed a striking difference in gene size (Fig. 2a). The median size of PCPAed genes was 39.0 kb, much larger than the median size of all expressed genes, 22.8 kb, whereas the median size of genes that were not PCPAed was 14.2 kb. Human gene size is highly correlated with intron size in general, as shown for all expressed genes in our samples (R2 = 0.9994; Supplementary Fig. 2e). Intron number also increases with gene size; however, PCPA frequently occurred in the first or second introns (38%; see below), which in mammals are typically the largest13. Thus, increased PCPA susceptibility of large genes derives predominantly from the length rather than the number of the introns.
Figure 2.
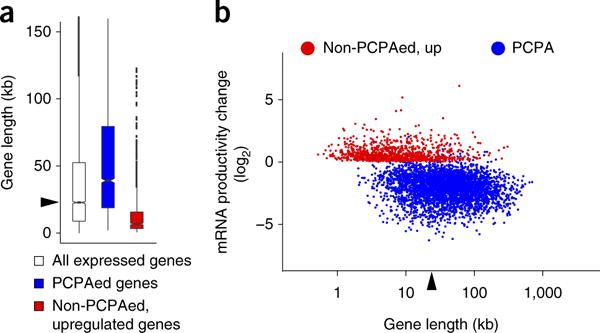
The effect of U1 inhibition and gene size on PCPA and full-length mRNA productivity. (a) Boxplots showing the gene-size distribution of PCPAed genes (n = 3,590; median 39.0 kb), non-PCPAed, upregulated genes (n = 988; median 6.8 kb) in the 8 h 4-shU-labeled RNA-seq from cells treated with U1 AMO compared to all expressed genes in control (n = 9,744; RPKM ≥ 1; median (22.8 kb) indicated by arrowhead). Boxplot midlines represent the median, box limits span the first and third quartiles, whiskers represent 1.5× the interquartile range (IQR) and points indicate outliers. (b) Scatter plot showing mRNA expression level changes in 8 h 4-shU labeled RNA-seq. mRNA productivity change was determined from the ratio of all exon reads for non-PCPAed and upregulated genes (shown in red). For the PCPAed genes (shown in blue), exon 1 reads were excluded from the calculations in both control and U1 AMO, as accumulation of PCPAed transcripts in some genes (~6%) made them appear as upregulated despite loss of full-length mRNA. The median gene length of all expressed genes (RPKM ≥ 1), 22.8 kb, is indicated by an arrowhead. The coordinates (x,y) of genes noted in the text are: EGFR (17.5, −2.3), EXT1 (18.3, −3.4), RAB7A (16.4, −0.8), E2F3 (16.5, −3.5), MYC (12.4, 1.6), CYR61 (11.6, 1.7), GAPDH (12.0, 0.05). The x,y coordinates are log2 values of gene length and fold change, respectively; for example, MYC (12.4, 1.6) corresponds to 5.4 kb and 3.0-fold increase. A list of PCPAed genes is summarized in Supplementary Table 2.
As expected, full-length mRNA production from PCPAed genes decreased (Fig. 2b and Supplementary Fig. 2d); however, numerous non-PCPAed genes were not downregulated and their transcripts continued to splice efficiently, for example GAPDH and RPL19 (Supplementary Fig. 3a,b). Importantly, many non-PCPAed genes (n = 988; median 6.8 kb) were upregulated in U1-inhibited cells (Fig. 2). Increased mRNA production and protein productivity of these upregulated genes, such as MYC, CYR61, ADAMTS1 and GADD45B, in U1 AMO–treated cells was confirmed by RT–qPCR and western blot (Fig. 3 and Supplementary Fig. 3c–e). The RT–qPCR, which included ERCC RNA spike-in controls for normalization of input RNA amount, confirmed the RNA-seq data, showing that the absolute levels of GAPDH and RPL19 mRNAs did not change after U1 AMO treatment, whereas levels of MYC, CYR61, ADAMTS1 and GADD45B mRNAs increased several fold and the transcript levels of select PCPAed genes decreased (Supplementary Fig. 3c–e). The increases in mRNA productivity were particularly striking, considering that the strong U1 inhibition was sufficient to PCPA nearly half of the expressed genes. There was no detectable PCPA in intronless genes (median 1.3 kb), which can be explained by the lower probability of a PAS arising stochastically in small genes (Supplementary Fig. 3f).
Figure 3.
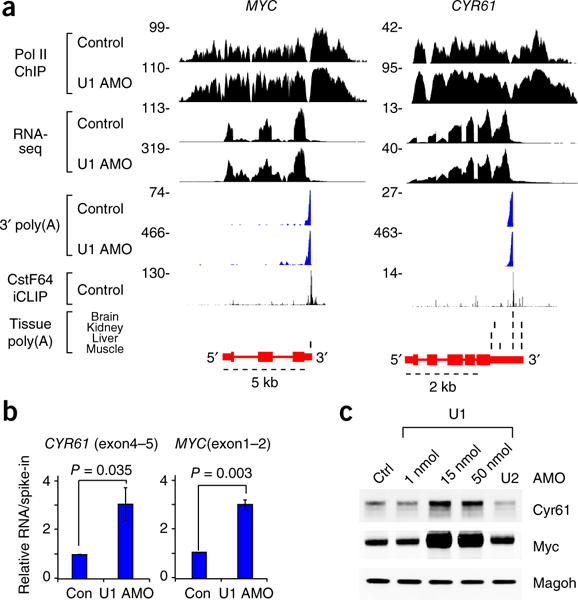
mRNA productivity upregulation in small non-PCPAed genes. (a) Representative examples of genes (MYC; CYR61) showing both PCPA resistance and increased mRNA expression. Data are presented as described for Figure 1. (b) Non-PCPAed small genes are upregulated. Purified, 4-shU-labeled RNAs from HeLa cells transfected with control or U1 AMO were used for RT-qPCR analysis. ERCC RNA spike-in controls added to each sample before rRNA depletion were used for normalization. Data are represented as mean ± s.d. (n = 3, independent cell cultures). P value was calculated with two-tailed Student’s t-test. (c) Protein expression from non-PCPAed small genes is upregulated with U1 AMO treatment. HeLa cells were transfected with control AMO, various doses of U1 AMO (1, 15 and 50 nmol) or 50 nmol of U2 AMO for 8 h and total cell extracts were used for western blot analysis. Magoh was used as a loading control. Uncropped blot images are shown in Supplementary Data Set 1.
Consistent with the established role of U1 in splicing, there was a 40% overall decrease in spliced exon–exon junctions (Supplementary Fig. 1d) upon U1 inhibition. However, as shown in Figure 1, much of the decrease could be the result of the loss of transcription of exons downstream of PCPA points rather than splicing inhibition. Analysis of spliced exon–exon junctions showed that splicing decreased by 71% in PCPAed genes but only 26% in non-PCPAed genes. Except for genes in which PCPA occurred in the first intron, splicing inhibition (intron retention) was also evident upstream of PCPA points (Supplementary Fig. 2b). However, non-PCPAed genes showed a moderate decrease in exon–exon junctions occurring uniformly throughout the gene, and PCPAed genes had greater loss of exon–exon junctions at their 3′ ends, thus supporting the conclusion that a major fraction of the splicing decrease in U1 AMO cells is an indirect effect of loss of telescripting (Supplementary Fig. 4). Analysis of de novo–spliced reads showed a detectable increase in usage of unannotated splice sites and alternative splicing changes; however, the vast majority of splicing remained from the same previously annotated splice sites despite U1 inhibition (Supplementary Fig. 1e and Supplementary Table 3).
U1 inhibition prematurely terminates elongating pol II in gene bodies
To investigate the relationship between PCPA and pol II transcription, we performed ChIP-seq with an antibody that recognizes the N terminus of pol II. Representative patterns showed that pol II signals tracked with RNA from the TSS up to the canonical 3′ end of genes and decreased to background level within a few kb downstream of the 3′ end in control cells (Fig. 4a). This observation of the termination zone (TZ) is consistent with the torpedo termination model14–16. U1 AMO reduced pol II signal downstream of the PCPA site, whereas pol II signal from the TSS to the PCPA site remained largely unaffected. A similar pattern of pol II TZs across several kilobases past the PCPA point in gene bodies indicates that the termination mechanism at the PCPA site is the same as that of the PASs at the 3′ ends of genes. In many cases, the pol II density was markedly higher than in these same regions in the control, suggesting that transcription initiation was not inhibited and could, in some cases, be increased.
Figure 4.
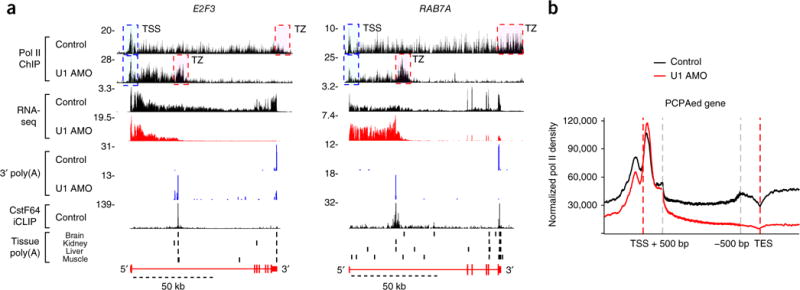
PCPA is cotranscriptional and prematurely terminates pol II elongation in gene bodies. (a) Genome browser views of pol II ChIP-seq and RNA-seq from HeLa cells 8 h after control or U1 AMO transfection. Data are presented as described for Figure 1. E2F3 and RAB7A genes show pol II signal declining after the PCPA point with U1 AMO. TSS (blue) and TZ (red) dashed boxes represent the transcription start site and termination zone, respectively. (b) Metagene plot of pol II ChIP-seq reads for PCPAed genes, identified in Figure 2 (n = 3,590), with control (black) or U1 AMO (red), relative to the TSS (−1,000 bp to +500 bp) and TES regions (transcription end site; 500 bp upstream of the annotated mRNA 3′ ends and 1,000 bp downstream). Regions between TSS + 500 bp and TES − 500 bp of each gene body were scaled to 2 kb.
By contrast, in small genes that showed increased mRNA productivity (n = 988; median 6.8 kb), such as MYC and CYR61, pol II peak heights and density were much higher with U1 AMO treatment (Fig. 3a). Pol II density increases indicate higher transcription flux, which were also observed in many PCPAed genes up to the PCPA point (Fig. 4a).
Pol II metagene plots of PCPAed genes (Fig. 4b) indicated the generality of these observations. It showed that the prominent promoter-proximal paused pol II peak (~200 bp) downstream of the sense-direction TSS was relatively unchanged, indicating that transcription initiation was not inhibited by U1 inhibition. Importantly, U1 AMO caused major polymerase losses downstream of the TSS in gene bodies, resulting in major downregulation of full-length transcription at the transcription end site (TES; Fig. 4b). The pol II signal upstream of the TSS, which represents upstream anti-sense transcription (uaRNAs/PROMPTs)6,7, was clearly decreased. RNA-seq data also showed clear PCPA in the antisense direction (Supplementary Fig. 5a), consistent with the previously described role of U1 in the bidirectional regulation of transcription from divergent promoters6,7. These data demonstrate that loss of telescripting cotranscriptionally terminated elongating polymerases prematurely in gene bodies. Pol II metagene plots for all expressed genes showed a similar pattern, indicating the impact of strong U1 inhibition on transcription throughout the genome (Supplementary Fig. 5b). TSS-proximal pol II peaks in upregulated, non-PCPAed genes increased nearly two-fold, suggesting that transcription initiation was not totally impaired.
Naturally occurring PCPA in normal human tissues causes transcription attrition in large genes
We analyzed publicly available data sets of 3′-poly(A)-seq generated by means of dedicated poly(A)-seq methodology of several human tissues, without deliberate U1 inhibition12. This analysis showed widespread usage of PASs in gene bodies under normal conditions (Figs. 1, 5a; tissue poly(A)). In many cases, U1 AMO–induced PCPA sites coincided with 3′-poly(A) locations detected in vivo (Fig. 5a). For example, in E2F3, PCPA was evident in normal tissues at the strongest CstF64-marked site in intron 1, which became more prominent with U1 AMO treatment. In other genes, such as EXT1, U1 AMO induced PCPA more proximal to the TSS, potentially from a PAS that is normally more strongly telescripted (Fig. 5a). Metagene analysis showed high coincidence of 3′-poly(A) reads in gene bodies in normal human tissues and those induced by U1 AMO in HeLa cells, suggesting that the in vivo 3′ poly(A)s are also related to U1 telescripting loss (Fig. 5b). Metagene plots confirmed the generality of CstF64-binding sites near U1 AMO–induced 3′-poly(A) reads (Figs. 1, 5c). The trend toward utilization of more TSS-proximal PASs with U1 AMO compared to normal tissues (Fig. 5d) is consistent with that of a cotranscriptional process2, in the 5′-to-3′ direction, as also demonstrated by the pol II ChIP-seq data (Fig. 4).
Figure 5.
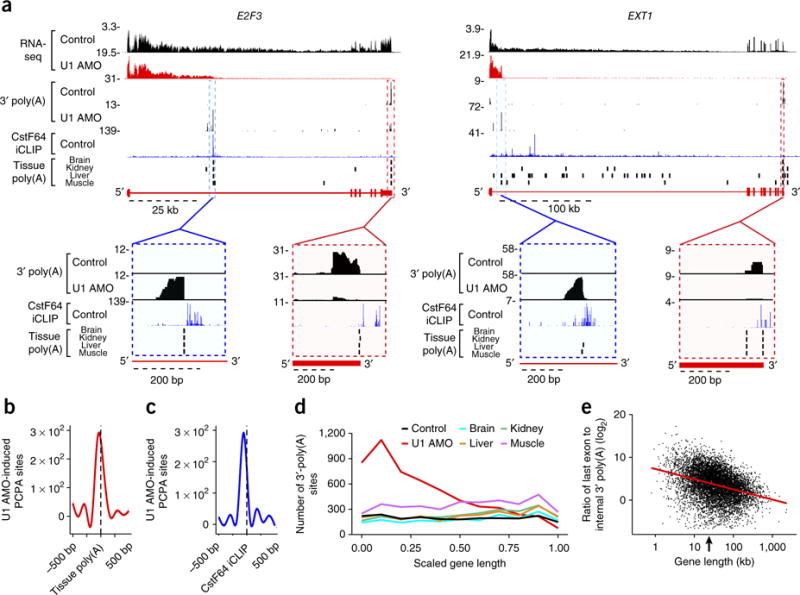
PCPA is a natural phenomenon. (a) Genome browser views of E2F3 and EXT1 genes showing colocalization of 3′-poly(A) peaks from control and U1 AMO RNA-seq and naturally occurring poly(A) sites in multiple human tissues. Blue dashed boxes show expanded views of the internal 3′-poly(A) site of a U1 AMO–induced PCPA position that has strong CstF64 and 3′-poly(A) signals from U1 AMO; red dashed boxes highlight the last exon poly(A) site typically used in normal conditions that has CstF64 and 3′-poly(A) signal from the non-U1 AMO control. (b) Smoothed metagene plot showing colocalization of internal 3′-poly(A) reads of U1 AMO RNA-seq (red line), determined by U1 AMO–induced PCPA sites, on previously defined12 3′-poly(A) sites in human tissues (black dashed line: 2,114 locations in 1,166 genes). (c) Smoothed metagene plot showing colocalization of internal 3′-poly(A) reads of U1 AMO RNA-seq (blue line) and CstF64 iCLIP-binding sites (black dashed line; top 5,000 peaks in introns). (d) Distribution of gene body 3′-poly(A) sites in control- and U1 AMO-treated cells and four human tissues. Applying the PCPA calculation to each intron and its flanking exons showed that 38% of PCPAs occurred within the first or second introns, a significant frequency compared to the expected 12% probability based on intron number (P value < 2.2 × 10−16, chi-squared test). (e) Scatter plot of the ratio of the total number of 3′-poly(A) reads in the last exon versus those in the gene body (from TSS up to, but not including, the last exon) of each expressed gene in normal human tissues; the red regression line (R2 = 0.11, P value < 1 × 10−3) indicates attrition is dependent on gene length, and the arrow on the x-axis indicates the median gene length (22.8 kb). The ratios of 3′-poly(A) reads in last exons and gene bodies are presented in Supplementary Table 4, and the raw values are plotted in Supplementary Figure 6b.
Genome browser views showed that some of the 3′ poly(A)s in introns in normal tissues corresponded to the ends of previously detected shorter, spliced isoforms (Supplementary Fig. 6a). These 3′ poly(A)s can be considered alternative polyadenylation, or PCPA (relative to the full-length) that generates alternative mRNA isoforms encoding proteins with shorter or alternative C termini (for example, homer-1, dab-1 (ref. 2) and EGFR17). However, most PCPAs did not appear to produce stable RNAs (Supplementary Fig. 6a) but nevertheless decreased the fraction of polymerases that transcribe to full length. We refer to this widespread physiological phenomenon as transcription attrition. We calculated the ratio of 3′-poly(A) reads in the last exon versus the gene body from the Derti et al.12 data sets to estimate the fraction of polymerases that transcribed and polyadenylated near the gene’s longest isoform (Fig. 5e and Supplementary Table 4). These ratios are rough estimates, because the various 3′-polyadenylated RNAs from the same gene can have different stabilities; nevertheless, a plot of each gene’s ratio versus its size clearly showed that large genes are highly susceptible to transcription attrition in human tissues (Fig. 5e and Supplementary Fig. 6b). Examples of genes with low ratios (Supplementary Fig. 6a) illustrate how transcription attrition can severely reduce full-length transcription. These observations suggest that at baseline, many large genes’ transcripts experience incomplete U1 telescripting and can potentially be shut off if the balance between U1 telescripting and PCPA shifts in favor of the latter (Supplementary Fig. 6a,c).
Gene size–function relationship
The above analyses revealed two groups of genes of particular interest in terms of full-length mRNA productivity in U1 AMO–transfected cells: genes that are PCPAed, which as a result are downregulated, and non-PCPAed and upregulated genes, which are both telescripting independent and have the ability to splice in a U1-deficient environment. Gene ontology (GO) enrichment analysis18,19 showed differential enrichment in functions encoded by these two gene groups (Fig. 6a,b and Supplementary Table 5). Upregulated, non-PCPAed genes were enriched in functions related to cell-stress response and growth-inducing stimuli, including numerous transcription, splicing, translation and signaling factors. PCPAed genes encoded diverse functions, as expected from the large number of genes in this group, but they were highly enriched in functions related to DNA replication and repair, cell cycle progression, including mitotic spindle formation, and intracellular organization and transport. PCPAed genes were also enriched for processes related to development, such as neural tube closure. These data suggest that the functions of small, PCPA-resistant, upregulated genes are particularly important for cells’ acute response to stimuli and adaptation to adverse environmental changes, during which the mRNA productivity of PCPAed genes is apparently more dispensable.
Figure 6.
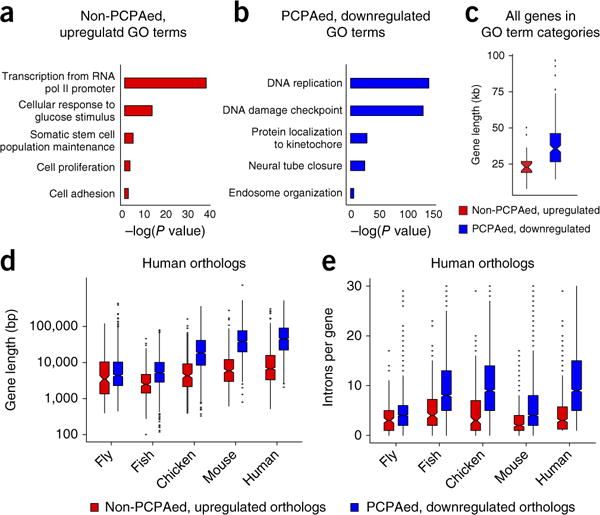
Functional and gene-size correlations of PCPAed and non-PCPAed genes that are up- or downregulated. GO analysis was performed using the XGR tool18. Enriched GO terms (P < 0.05), classified by functional category using REVIGO19, are displayed as a histogram for non-PCPAed, upregulated (a) or PCPAed, downregulated (b) genes. The top 50% of genes, ranked by fold change, are shown. (c) Boxplots showing the size distribution of human genes whose GO terms are enriched either in non-PCPAed, upregulated genes (red, median 23 kb) or PCPAed genes (blue, median 38 kb). Boxplots showing the gene size (d) or intron-per-gene distribution (e) of orthologous genes among non-PCPAed, upregulated genes (red) and PCPAed genes (blue) in five metazoans. Ortholog data across all five organisms were obtained from Ensembl33. Genes identified as PCPAed or non-PCPAed upregulated after U1 inhibition in HeLa cells were extracted from this list, and the longest isoforms were used to plot gene sizes. The non-PCPAed upregulated and PCPAed downregulated gene groups in Drosophila melanogaster had 195 (median 3.5 kb, three introns) and 1,023 (median 4.4 kb, four introns) orthologs, Takifugu rubripes had 264 (median 2.5 kb, four introns) and 1,378 (median 5.2 kb, nine introns) orthologs, Gallus gallus had 238 (median 4.3 kb, four introns) and 1,425 (median size 18.4 kb, nine introns) orthologs, and Mus musculus had 342 (median 5.9 kb, two introns) and 1,555 (median 38.0 kb, four introns) orthologs, respectively. There were 493 upregulated genes in human (hg19 GRCh38.p7) with orthologs in any of these organisms (median 6.8 kb, three introns) and 1,682 PCPAed genes with orthologs (median 45.0 kb, ten introns). Boxplot midlines represent the median, box limits span the first and third quartiles, whiskers represent 1.5× IQR and points indicate outliers. GO enrichment analysis is summarized in Supplementary Table 5.
Extending the analysis to all human genes (many of which are not expressed in HeLa cells) categorized by the same GO terms as those enriched in either group suggested a gene size–function relationship. Generally, functions related to cell survival and response to acute adverse stimuli were over-represented in small genes but under-represented in very large genes, whereas cell-division- and tissue-differentiation-related functions showed the opposite (Fig. 6c). Small and U1 AMO–upregulated genes are also more ubiquitously expressed, whereas large genes are more tissue-specific and typically expressed in differentiated cells20,21.
Comparison of orthologous genes in several metazoans showed that orthologs of the non-PCPAed genes that are upregulated with U1 AMO are consistently smaller than orthologs of PCPAed genes. Interestingly, the size gap between these gene groups progressively increased, especially in vertebrates (fish to human; Fig. 6d). However, the widening gene-size differential between the two groups could not be explained by the more moderate increase in intron (or exon) number (Fig. 6e). Calculating the ratio of intron-versus-exon length for the orthologs of the two groups in each of these organisms showed a much greater intron size expansion in PCPAed genes compared to non-PCPAed genes and upregulated genes (Supplementary Table 6). Thus, intron size expansion in the evolution of vertebrates was a primary factor in making their genomes’ size–function stratification progressively more polarized.
DISCUSSION
Our studies here show that U1 is required for selectively telescripting large introns, where it protects pol II from prematurely terminating in gene bodies. U1 telescripting averts PCPA cotranscriptionally, as originally proposed2, thus ruling out an alternative mechanism whereby the PCPA products are derived post-transcriptionally from full-length mRNA. Telescripting is thus crucial for long-range transcription elongation, and our data show that pol II termination is coupled to 3′-end cleavage wherever it occurs. PCPA points, numerous in large genes, are therefore transcription–elongation checkpoints. Traversing them depends on sufficient U1 and other factors that may play a role in telescripting. We have previously described that acute transcription upregulation can transiently create a relative U1 shortage without decreasing the U1 level in absolute terms. For example, membrane depolarization in neuronal-type cells rapidly increases overall transcription, leading to PCPA in introns of synaptogenesis genes like homer-1, thereby converting them to a shorter isoform with the opposite function to that of the full-length isoform. This switch can be antagonized by U1 overexpression and can be recapitulated by low-level U1 AMO2. Our studies show that PCPA occurs in normal tissues in humans and other organisms, indicating that it is an omnipresent barrier to full-length transcription and causes transcription attrition in thousands of genes. U1 AMO thus recapitulates and exacerbates an important physiological phenomenon and provides an experimental tool for systematically studying it.
Although not previously tested in human cells, the general splicing reduction effected by U1 inhibition was expected. However, full-length mRNA downregulation from large genes was primarily due to loss of telescripting rather than loss of splicing, because U1 inhibition terminated transcription in many large introns well before reaching the 3′ splice site (Fig. 1). Moreover, a significant portion of splicing loss was actually due to the elimination of splicing opportunities secondary to PCPA in an upstream intron. Thus, loss of splicing from lower available U1 overestimates U1’s direct role in splicing. PCPA, however, is not a secondary effect of splicing inhibition; it typically occurs at great distances upstream of the 3′splice site and is not elicited by other splicing inhibition (spliceostatin A or U2 AMO1 and Supplementary Fig. 7).
Compared to the strong effect of U1 inhibition on telescripting, U1’s well-established function in splicing was surprisingly robust in the same environment. It is possible that the residual uninhibited U1 pool is sufficient to sustain nearly normal splicing output. However, it is also possible that many small introns can splice with very little U1 or with none at all. Previous studies, albeit for only a handful of small introns in vitro, have shown that splicing can take place in U1-depleted cell extracts if the extracts are supplemented with excess SR proteins, a splicing-active group of hnRNP proteins22–24. PCPA could mimic these conditions by decreasing the number of introns that compete for the same factors.
Telescripting is an unprecedented gene regulation mechanism as it discriminates genes according to their size. The selective dependence of large nascent transcripts on telescripting stems from their long introns, which stochastically contain more PASs. This dependence in itself makes large pre-mRNAs more vulnerable to relative U1 telescripting shortage, which can be triggered by transcription upregulation2. However, the ability of many small genes to increase their spliced mRNA productivity in the same environment appears paradoxical. There are several potential and non-mutually-exclusive explanations for the upregulation of these small genes, including that U1 inhibition may generate a stress signal that activates their transcription or that PCPA knocks down factors that negatively regulate their transcription. It is also possible that, by truncating large nascent transcripts, PCPA decreases competition for transcription and other pre-mRNA-processing factors that small genes can use to boost their expression. Studies in divergent eukaryotes have shown that competition between pre-mRNAs for a variety of factors, likely minor compared to what PCPA can bring about, has a strong impact on mRNA synthesis from many genes2,25–29. Future studies will address these mechanisms.
The functions encoded by the small genes that are upregulated with U1 AMO add an important perspective. This group is ubiquitously expressed and enriched in primary-response genes that are induced during acute cell stimulation and are necessary to adapt to adverse environmental changes and stressors. Such functions are under-represented in large PCPAed genes, which are more diverse and generally expressed in differentiated tissues. Notable among the largest genes are neuronal and developmental genes20,21, which given their size would probably be highly susceptible to PCPA and could be transiently sacrificed to enhance expression of genes advantageous for cell survival, though potentially impair the organism.
In addition to a shorter transcription time, PCPA avoidance probably played a role in selection against intron expansion in genes that are crucial for enhancing cell survival prospects under adverse circumstances. The scarcity of such functions among very large genes suggests that the massive intron expansion paralleled the evolution of complex organisms30–32 and was not random but rather was selected against in such genes but permitted in others. Indeed, examination of orthologous genes in several other organisms showed that sizes of PCPAed genes increased progressively much more than those of small genes that were more refractory to U1 inhibition (Fig. 6d). The increased intron size expansion in the evolution of vertebrates, especially in mammals, suggests that a greater polarization in the sizes and functions of genes was not merely tolerated but was increasingly adopted outside of essential survival genes, suggesting that it was advantageous. The U1-inhibition experiments reveal that one such advantage of a polarized genome is that it adds a new layer of global gene-expression control. Another hitherto unexplained aspect of intron expansion is its positional bias within genes. In humans, the first or second introns are typically the largest13, extending well beyond the first quarter of the gene, where PCPA frequently occurred, followed by a cluster of smaller introns and exons. This architecture seems to be geared to maximize loss of competition for splicing resources by PCPA and potential gain for non-PCPAed genes, providing an attractive rationale for TSS-proximal intron expansion.
METHODS
Methods, including statements of data availability and any associated accession codes and references, are available in the online version of the paper.
ONLINE METHODS
Cell culture, transfection with antisense morpholino oligonucleotides (AMO), and RNA labeling
HeLa cells obtained from ATCC were maintained in DMEM supplemented with 10% FBS, 10 unit/ml penicillin and 10 μg/ml streptomycin at 37 °C and 5% CO2. Cell lines were tested for mycoplasma contamination. Control and U1 AMO sequences used were as previously described1. One million HeLa cells were transfected with the desired concentration of AMO by means of electroporation using the Neon transfection system according to the manufacturer’s recommendation (Invitrogen)1,2. Nascent RNAs were metabolically labeled in cells with 250 μM 4-thiouridine (4-shU) that was added for 30 min at 3.5–4 h and 7.5–8 h post AMO transfection.
Nascent RNA isolation, library preparation and RNA-seq
Total RNA extraction from cells was done at either 4 or 8 h after AMO transfection using Trizol (Invitrogen) followed by either poly(A) RNA purification on oligo-dT columns using Oligotex kit (Qiagen) or ribosomal RNA depletion using the Ribo-Zero kit (Illumina). For isolation of 4-shU-containing mRNAs, the free thiols on poly(A) mRNAs were reacted with 0.2 mg/ml of EZ-link biotin-HPDP (Thermo Scientific) for 1 h, then purified on M-280 streptavidin Dynabeads (Invitrogen), as previously described9. The procedure was repeated using two purification cycles to ensure very high purity. cDNA synthesis and RNA-seq library preparation were constructed using the Kapa stranded RNA-seq library preparation kit (Kapa Biosystems) according to the manufacturer’s instructions. All of the sequencing was performed on an Illumina HiSeq 2500.
Mapping RNA-seq reads
RNA-seq reads were trimmed of any adaptor sequences with the FASTX-Toolkit (version 0.0.14) and then aligned to the GRCh37/hg19 reference genome using Tophat (version 2.0.12) with default settings. For some of the further downstream analysis, Samtools (version 0.1.19) was used to remove reads with multiple mapping locations from the aligned files. Reads per exon were grouped, from which RPKM (reads per kilobase per million mapped reads)34 values were calculated using GFold35. Exon–exon-junction reads were extracted from the Tophat-assigned junction reads using Bedtools (version 2.15.0). In order to directly compare different treatment samples that may have included a different number of sequenced and mapped reads, the read coverage for each sample was normalized by dividing the signal by the total number of mapped reads in one million (RPM). This normalized value was used for visualization on a genome browser (http://genome.ucsc.edu/)36, as well as through comparison on read coverage between and throughout different genes. The longest gene isoforms were used for further global analyses.
Mapping of 3′-poly(A) reads
A workflow similar to that described in other published methods7 was used to identify and align RNA-seq reads with non-genomically-encoded 3′-poly(A) stretches. Bowtie (version0.12.7) was first used to consolidate reads that could not be aligned to the GRCh37/hg19 reference genome into a separate FASTQ file, as any nongenomic 3′-poly(A) stretch would prevent these reads from accurately aligning to the reference. These unaligned RNA-seq reads were then filtered to pool any read that contained a 3′-poly(A) stretch of ≥ 10 nucleotides. 3′-poly(A) stretches were then removed using Cutadapt (version 1.9.1)37, which were realigned to the GRCh37/hg19 reference genome. 3′-Poly(A)-site locations were subsequently aggregated with Bedtools (version 2.15.0).
Differential expression analysis
We used an exact Poisson test to compare the number of reads on different genomic locations (such as exons, exon–exon junctions, last exons) between control and U1 AMO–treated samples. A P value < 0.01 was set as the threshold of significant expression change. For a better visualization of the change in full-length mRNA expression after U1-AMO treatment, we removed the exon 1 reads from PCPAed genes in Figure 2b. Visual inspection of these genes showed that their apparent increase in RPM is due to an extremely large increase in exon 1 reads, whereas downstream exons produced little to no signal.
Alternative splicing analysis
MISO (version 0.5.2)38 analysis was performed on the aligned RNA-seq data to identify differentially spliced isoforms in experimental samples compared to controls and quantify their expression levels by computing the PSI (percent spliced isoform; Ψ) and Bayes factors. Alternative splicing events were filtered by the following criteria: (a) at least 1 inclusion read, (b) at least 1 exclusion read, such that (c) the sum of inclusion and exclusion reads is at least 10, (d) the |ΔΨ| is at least 0.3 (e) the Bayes factor is at least 10, and (a)–(e) are true in one of the samples. To calculate canonical splicing, the Tophat (version 2.0.12) assigned spliced reads were pooled, using Bedtools (version 2.15.0), into reads that overlapped exactly to known exon–exon junctions in the GRCh37/hg19 reference genome. All of the other assigned spliced reads were counted as aberrant splicing signal.
PCPA calculation
The following algorithm was applied to RNA-seq data for each expressed gene with RPKM ≥ 1 in either the control or U1 AMO sample. Identification of PCPA relies on two criteria: (1) a decrease in reads of the last exon’s CDS relative to the reads in the first exon under U1 AMO conditions compared to control, and (2) an increase in reads at the first quarter (iQ1) of the region between a gene’s first 5′ splice site to the last 3′ splice site compared to the fourth quarter (iQ4) of the same region in the U1 AMO sample. We set a minimum of ten reads in iQ1 in U1-inhibited samples to avoid bias against small genes. To test for significance in the read density shift seen in the U1 AMO–treated samples compared to control, Fisher’s exact test was applied to the values for iQ1 and iQ4 in both, followed by Benjamini–Hochberg (BH) multiple testing39 with an adjusted P value ≤ 0.01. The same process was also applied to the relative decrease in last exons, and an adjusted P value ≤ 0.01 was set as the threshold of significant read-density decrease. To calculate more severe PCPAed genes, we increased the stringency by selecting those that had a last exon decrease ≥ 2-fold (P value ≤ 0.01). The significance of overlap between PCPAed genes across all samples was performed using hypergeometric testing. Additionally, we applied the same algorithm and P value testing at an individual intron level to find the PCPA location on an intron (single-intron calculation): (1) an increase in reads at the first quarter (iQ1) of a given intron (n) compared to the fourth quarter (iQ4) of the same intron in the U1 AMO sample, (2) a decrease in reads of the downstream exon (n + 1) under U1 AMO conditions compared to control. All previously mentioned statistical tests were performed using R (version 3.2.0).
Reverse transcription–qPCR quantification of 4-shU RNA and PCPA products
HeLa cells transfected with control or U1 AMO and metabolically labeled with 4-shU were used for RT–qPCR analysis. ERCC RNA spike-in controls (Ambion) were added to each sample before the rRNA-depletion process and used for normalization. cDNA was synthesized from 100 ng total RNA isolated from control- or U1 AMO–transfected cells using Transcriptor First-strand cDNA synthesis kit (Roche Applied Sciences). 2% of the cDNA was used for each qPCR reaction (Applied Biosystems 7500 Fast Real-time PCR system) using SYBR Green dye chemistry. Primer sequences are listed in Supplementary Table 7.
Metagene analysis of 3′-poly(A) reads and pol II ChIP-seq
The CGAT tool kit (version 0.2.5)40 was used to generate the metagene profiles for the 3′-poly(A) reads and ChIP-seq data. For 3′-poly(A) reads from U1 AMO RNA-seq data, downloaded nongenomic 3′-poly(A) sites from gene body regions (except last exon) in four different human tissues12 were pooled into 3′-poly(A) sites defined as internal/gene body tissue poly(A)s. To draw a metagene profile of 3′-poly(A) reads from U1 AMO RNA-seq on the internal/gene body tissue poly(A) site, genes were required to have at least one PCPA site by PCPA calculation and one tissue poly(A) site, total 1,166 genes with 2,114 unique poly(A) sites. For ChIP-seq data, the pol II reads were normalized to the IgG background, and then each gene was split into three regions where both the 1,000 bp upstream to 500 bp downstream of the TSS and 500 bp upstream to 1,000 bp downstream TES were not scaled, and the remaining interior portion of the gene body was resized to 2 kb. The unscaled TSS and TES regions were laid flanking the scaled gene body, and the read number of each nucleotide was calculated and used to draw the profile.
Pol II ChIP-seq
HeLa cells (10 million) were transfected with control AMO or 15 nmol of U1 AMO for 8 h. After formaldehyde cross-linking, nuclei were isolated using ChIP Lysis buffer (5 mM HEPES K, pH 7.9, 85 mM KCl, 0.5% NP-40, protease inhibitors), then lysed using ChIP Nuclei Lysis Buffer (50 mM Tris-HCl pH 8.0, 10 mM EDTA, pH 8.0, 1% SDS, protease inhibitors). The nuclear lysate was sheared using a Covaris sonicator and diluted. This chromatin sample was cleared by incubation with Protein A Dynabeads and then incubated with 5 μg pol II (or control IgG) antibody (Santa Cruz, sc-899 X) overnight. For IP, an antibody to pol II’s N-terminal domain was used to detect all pol II molecules on actively transcribed genes regardless of its C-terminal domain’s phosphorylation state, which could vary throughout a gene. Beads were washed extensively, then formaldehyde cross-linking was reversed overnight at 65 °C in the presence of 1% SDS and RNase A. Proteins were digested for 2 h, then DNA was extracted twice with phenol-chloroform-isoamyl alcohol and ethanol/NaCl precipitation. The DNA (5–10 ng) was used to make libraries for ChIP-seq using the Truseq ChIP kit (Illumina) according to manufacturer recommendations. Four samples were pooled and sequenced on Illumina 2000 platform (50-bp single-end sequencing). After sequencing and alignment, the background and nonspecific binding was removed for the experimental samples using the model-based analysis of ChIP-Seq (MACS) algorithm41. This program was also used to output BedGraph files for visualization on the UCSC genome browser.
Immunoblot analysis
HeLa cells transfected with control AMO, different U1 AMO doses (1, 15 and 50 nmol), 50 nmol of U2 AMO, 5 nmol of U6 AMO or 15 nmol of U12 AMO for 8 h and total cell extract were used for western blot analysis. The antibodies were specific to the CYR61 (Santa Cruz, sc-13100), c-Myc (Abcam, ab32072) and Magoh (Dreyfuss lab)42.
Gene size–function analysis
We used XGR GO term analysis (http://galahad.well.ox.ac.uk:3020)18 to classify genes into different functional categories (Supplementary Table 5) with the following criteria: (a) 10 as minimum number of genes annotated, (b) 2,000 as maximum number of genes annotated, (c) 5 as minimum overlap with your input genes, then used REVIGO (http://revigo.irb.hr)19 to summarize the long list of GO terms into a histogram (Fig. 6a,b). The number of PCPAed downregulated genes or non-PCPAed upregulated genes annotated to each GO term were summed and a Fisher’s exact test was used to check if the number of genes in the former group was significantly (P value < 0.05) higher than the latter or vice versa. To extend this analysis globally to the human genome, the median gene length was calculated for each significantly enriched GO term using all genes classified in the group from XGR, not just those expressed in HeLa, and the distribution was shown by boxplot (Fig. 6c).
Orthologs and all related information were taken from Ensembl’s Biomart (http://www.ensembl.org/)33. Nonhuman species were compared against the human genes for orthologs.
Code availability
Code for the analyses described in this study is available from the corresponding author upon request.
Data availability
All sequencing data described are available on GEO under the accession number GSE103252. A Life Sciences Reporting Summary for this article is available.
Supplementary Material
Acknowledgments
We thank members of our laboratory for helpful discussions and comments on the manuscript. This work was supported by the US National Institutes of Health (R01GM112923 to G.D.). G.D. is supported by the Howard Hughes Medical Institute.
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS
J.-M.O., I.Y., A.M.P., L.W. and G.D. conceived and designed the study. J.-M.O., C.C.V., C.A., I.Y., B.R.S. and Z.Z. performed the experiments. C.D. and C.C.V. performed the bioinformatics analysis. All authors contributed to data analysis. J.-M.O., C.C.V., C.D., J.G., I.Y. and G.D. wrote the manuscript with input from all authors. G.D. is responsible for the project’s planning and experimental design.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Kaida D, et al. U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature. 2010;468:664–668. doi: 10.1038/nature09479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berg MG, et al. U1 snRNP determines mRNA length and regulates isoform expression. Cell. 2012;150:53–64. doi: 10.1016/j.cell.2012.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shi Y, Manley JL. The end of the message: multiple protein-RNA interactions define the mRNA polyadenylation site. Genes Dev. 2015;29:889–897. doi: 10.1101/gad.261974.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lerner MR, Boyle JA, Mount SM, Wolin SL, Steitz JA. Are snRNPs involved in splicing? Nature. 1980;283:220–224. doi: 10.1038/283220a0. [DOI] [PubMed] [Google Scholar]
- 5.Engreitz JM, et al. RNA-RNA interactions enable specific targeting of noncoding RNAs to nascent Pre-mRNAs and chromatin sites. Cell. 2014;159:188–199. doi: 10.1016/j.cell.2014.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Almada AE, Wu X, Kriz AJ, Burge CB, Sharp PA. Promoter directionality is controlled by U1 snRNP and polyadenylation signals. Nature. 2013;499:360–363. doi: 10.1038/nature12349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ntini E, et al. Polyadenylation site-induced decay of upstream transcripts enforces promoter directionality. Nat Struct Mol Biol. 2013;20:923–928. doi: 10.1038/nsmb.2640. [DOI] [PubMed] [Google Scholar]
- 8.Younis I, et al. Minor introns are embedded molecular switches regulated by highly unstable U6atac snRNA. eLife. 2013;2:e00780. doi: 10.7554/eLife.00780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dölken L, et al. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA. 2008;14:1959–1972. doi: 10.1261/rna.1136108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rabani M, et al. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat Biotechnol. 2011;29:436–442. doi: 10.1038/nbt.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yao C, et al. Transcriptome-wide analyses of CstF64-RNA interactions in global regulation of mRNA alternative polyadenylation. Proc Natl Acad Sci USA. 2012;109:18773–18778. doi: 10.1073/pnas.1211101109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Derti A, et al. A quantitative atlas of polyadenylation in five mammals. Genome Res. 2012;22:1173–1183. doi: 10.1101/gr.132563.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bradnam KR, Korf I. Longer first introns are a general property of eukaryotic gene structure. PLoS One. 2008;3:e3093. doi: 10.1371/journal.pone.0003093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fong N, et al. Effects of transcription elongation rate and Xrn2 exonuclease activity on RNA polymerase II termination suggest widespread kinetic competition. Mol Cell. 2015;60:256–267. doi: 10.1016/j.molcel.2015.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Proudfoot NJ. Transcriptional termination in mammals: Stopping the RNA polymerase II juggernaut. Science. 2016;352:aad9926. doi: 10.1126/science.aad9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Connelly S, Manley JL. A functional mRNA polyadenylation signal is required for transcription termination by RNA polymerase II. Genes Dev. 1988;2:440–452. doi: 10.1101/gad.2.4.440. [DOI] [PubMed] [Google Scholar]
- 17.Vorlová S, et al. Induction of antagonistic soluble decoy receptor tyrosine kinases by intronic polyA activation. Mol Cell. 2011;43:927–939. doi: 10.1016/j.molcel.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fang H, Knezevic B, Burnham KL, Knight JC. XGR software for enhanced interpretation of genomic summary data, illustrated by application to immunological traits. Genome Med. 2016;8:129. doi: 10.1186/s13073-016-0384-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Supek F, Bošnjak M, Škunca N, Šmuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One. 2011;6:e21800. doi: 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bertagnolli NM, Drake JA, Tennessen JM, Alter O. SVD identifies transcript length distribution functions from DNA microarray data and reveals evolutionary forces globally affecting GBM metabolism. PLoS One. 2013;8:e78913. doi: 10.1371/journal.pone.0078913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gabel HW, et al. Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature. 2015;522:89–93. doi: 10.1038/nature14319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crispino JD, Blencowe BJ, Sharp PA. Complementation by SR proteins of pre-mRNA splicing reactions depleted of U1 snRNP. Science. 1994;265:1866–1869. doi: 10.1126/science.8091213. [DOI] [PubMed] [Google Scholar]
- 23.Tarn WY, Steitz JA. SR proteins can compensate for the loss of U1 snRNP functions in vitro. Genes Dev. 1994;8:2704–2717. doi: 10.1101/gad.8.22.2704. [DOI] [PubMed] [Google Scholar]
- 24.Fukumura K, Taniguchi I, Sakamoto H, Ohno M, Inoue K. U1-independent pre-mRNA splicing contributes to the regulation of alternative splicing. Nucleic Acids Res. 2009;37:1907–1914. doi: 10.1093/nar/gkp050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Munding EM, Shiue L, Katzman S, Donohue JP, Ares M., Jr Competition between pre-mRNAs for the splicing machinery drives global regulation of splicing. Mol Cell. 2013;51:338–348. doi: 10.1016/j.molcel.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cooper TA, Wan L, Dreyfuss G. RNA and disease. Cell. 2009;136:777–793. doi: 10.1016/j.cell.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller JW, et al. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 2000;19:4439–4448. doi: 10.1093/emboj/19.17.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Timchenko LT, et al. Identification of a (CUG)n triplet repeat RNA-binding protein and its expression in myotonic dystrophy. Nucleic Acids Res. 1996;24:4407–4414. doi: 10.1093/nar/24.22.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elkon R, Ugalde AP, Agami R. Alternative cleavage and polyadenylation: extent, regulation and function. Nat Rev Genet. 2013;14:496–506. doi: 10.1038/nrg3482. [DOI] [PubMed] [Google Scholar]
- 30.Catania F, Lynch M. Where do introns come from? PLoS Biol. 2008;6:e283. doi: 10.1371/journal.pbio.0060283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gelfman S, et al. Changes in exon-intron structure during vertebrate evolution affect the splicing pattern of exons. Genome Res. 2012;22:35–50. doi: 10.1101/gr.119834.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rogozin IB, Carmel L, Csuros M, Koonin EV. Origin and evolution of spliceosomal introns. Biol Direct. 2012;7:11. doi: 10.1186/1745-6150-7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yates A, et al. Ensembl 2016. Nucleic Acids Res. 2016;44:D710–D716. doi: 10.1093/nar/gkv1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 35.Feng J, et al. GFOLD: a generalized fold change for ranking differentially expressed genes from RNA-seq data. Bioinformatics. 2012;28:2782–2788. doi: 10.1093/bioinformatics/bts515. [DOI] [PubMed] [Google Scholar]
- 36.Kent WJ, et al. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet journal. 2011;17:10–12. [Google Scholar]
- 38.Katz Y, Wang ET, Airoldi EM, Burge CB. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods. 2010;7:1009–1015. doi: 10.1038/nmeth.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Benjamini Y, Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300. [Google Scholar]
- 40.Sims D, et al. CGAT: computational genomics analysis toolkit. Bioinformatics. 2014;30:1290–1291. doi: 10.1093/bioinformatics/btt756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kataoka N, Diem MD, Kim VN, Yong J, Dreyfuss G. Magoh, a human homolog of Drosophila mago nashi protein, is a component of the splicing-dependent exon-exon junction complex. EMBO J. 2001;20:6424–6433. doi: 10.1093/emboj/20.22.6424. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data described are available on GEO under the accession number GSE103252. A Life Sciences Reporting Summary for this article is available.