

Using orientation-independent-DIC microscopy, we revealed that the density of total materials in heterochromatin was only 1.53-fold higher than that of euchromatin, whereas the DNA density was 7.5-fold higher. This surprisingly small difference may be due to the dominance of proteins and RNAs in both chromatins, which may help create a moderate barrier to heterochromatin.
Abstract
In eukaryotic cells, highly condensed inactive/silenced chromatin has long been called “heterochromatin.” However, recent research suggests that such regions are in fact not fully transcriptionally silent and that there exists only a moderate access barrier to heterochromatin. To further investigate this issue, it is critical to elucidate the physical properties of heterochromatin such as its total density in live cells. Here, using orientation-independent differential interference contrast (OI-DIC) microscopy, which is capable of mapping optical path differences, we investigated the density of the total materials in pericentric foci, a representative heterochromatin model, in live mouse NIH3T3 cells. We demonstrated that the total density of heterochromatin (208 mg/ml) was only 1.53-fold higher than that of the surrounding euchromatic regions (136 mg/ml) while the DNA density of heterochromatin was 5.5- to 7.5-fold higher. We observed similar minor differences in density in typical facultative heterochromatin, the inactive human X chromosomes. This surprisingly small difference may be due to that nonnucleosomal materials (proteins/RNAs) (∼120 mg/ml) are dominant in both chromatin regions. Monte Carlo simulation suggested that nonnucleosomal materials contribute to creating a moderate access barrier to heterochromatin, allowing minimal protein access to functional regions. Our OI-DIC imaging offers new insight into the live cellular environments.
INTRODUCTION
In eukaryotic cells, a long strand of genomic DNA is three dimensionally organized in a cell nucleus as chromatin. Growing evidence has suggested that the nucleosomes, consisting of DNA wrapped around core histones (Luger et al., 1997), are rather irregularly folded without the regular chromatin fibers (Fussner et al., 2012; Joti et al., 2012; Maeshima et al., 2014; Hsieh et al., 2015; Ricci et al., 2015; Sanborn et al., 2015; Chen et al., 2016; Ou et al., 2017). Superresolution fluorescence microscopy and chromosome conformation capture (3C) derivatives such as Hi-C have revealed the existence of various chromatin domains, such as topologically associating domain (TAD), in the cell (Markaki et al., 2010; Dixon et al., 2012; Nora et al., 2012; Sexton et al., 2012; Rao et al., 2014; Eagen et al., 2015; Boettiger et al., 2016; Cremer et al., 2017; Nozaki et al., 2017).
According to typical textbook models, chromatin can be categorized into two types (“euchromatin” and “heterochromatin”) based on its degree of compaction (Alberts et al., 2007; Pollard et al., 2016). These two types of chromatin were originally observed in 1928 by Heitz (1928) and were described as sparse and dense chromatic regions. Heterochromatin has been further grouped into constitutive and facultative types, each of which plays a distinct role in the regulation of genomic functions (Brown, 1966; Trojer and Reinberg, 2007).
Constitutive heterochromatin is typically gene poor, usually possesses AT-rich repetitive DNA sequences and has a highly condensed structure (Maison et al., 2010; Saksouk et al., 2015). It provides structural functions in areas such as chromosome centromeres or telomeres and replicates in the mid to late S phase. Masses of pericentric heterochromatin in mouse interphase cells, often called “chromocenters” (Guenatri et al., 2004), have been extensively studied as a constitutive heterochromatin model. Pericentric heterochromatin is composed of highly clustered major satellite repeat sequences (several Mb of 234 base pair units) associated with centromeres, and is highly condensed and easily detectable by DNA staining as large 4′,6-diamidino-2-phenylindole–(DAPI) dense foci (Maison et al., 2010; Saksouk et al., 2015). The signal intensity of foci by DAPI staining is sixfold higher than that in surrounding regions (Bancaud et al., 2009), and is marked with heterochromatin protein 1 (HP1) (Grewal and Jia, 2007; Maison et al., 2010), MeCP2 (Nan et al., 1996; Brero et al., 2005), and tail methylation of histone H3 (H3K9me3) (Allis and Jenuwein, 2016).
Facultative heterochromatin corresponds to genetic regions that are silenced through a mechanism involving histone modification or RNA binding (Trojer and Reinberg, 2007; Wutz, 2011). It is not repetitive, and shares some features of constitutive heterochromatin. Importantly, under specific developmental or environmental conditions, facultative heterochromatin can lose its condensed structure and become transcriptionally active. A famous process involving facultative heterochromatin is X-inactivation, through which one of the copies of the X chromosome present in female mammals is transcriptionally silenced, creating a Barr body (Wutz, 2011; Nakajima and Sado, 2014; Smeets et al., 2014; da Rocha and Heard, 2017; Jegu et al., 2017).
Although the heterochromatin is generally well condensed, it does not completely prevent protein diffusion (Cremer et al., 2015). For example, fluorescence correlation spectroscopy (FCS) imaging detected diffusion of protein molecules into heterochromatin regions (Bancaud et al., 2009; Baum et al., 2014), as reported in condensed mitotic chromosomes (Chen et al., 2005; Hihara et al., 2012). In light of other factors, including their low levels of transcription (Trojer and Reinberg, 2007; Saksouk et al., 2015) and DNA replication in mid to late S phase (Guenatri et al., 2004; Wu et al., 2006), heterochromatin regions seem to pose only a moderate barrier to protein access. To determine what is responsible for such a barrier, it is critical to not only characterize the molecular components of heterochromatin (including histone modifications, specific proteins, and RNAs) (Grewal and Jia, 2007; Maison et al., 2010; Saksouk et al., 2015; Allis and Jenuwein, 2016) but also to investigate the physical properties of heterochromatin, such as its density in live cells. We could relatively easily obtain DNA density information in the heterochromatin as described above (e.g., Bancaud et al., 2009) and also from the recent volume analyses on specific regions in heterochromatin using a combination of superresolution imaging, chromatin immunoprecipitation sequencing (ChIP-seq), and DNA-fluorescence in situ hybridization (FISH) techniques (see Results) (Boettiger et al., 2016; Wang et al., 2016). However, to understand the moderate access barrier mechanism, we also need to know the total density, including nonnucleosomal material (proteins, RNAs) in the heterochromatin of live cells.
While elucidating such physical properties in live cells is technically challenging, it can be achieved through differential interference contrast (DIC) microscopy (Inoué and Spring, 1997; Oldenbourg and Shribak, 2010), a common tool in cell biology research. DIC images are produced by the interference of two laterally displaced light beams passing through a sample (e.g., live cells), capturing information about the optical path length in the sample to reveal otherwise invisible features (Allen et al., 1969). The difference in optical path length between the two beams provides contrast to the image, which reflects local differences in the refractive index within the sample. However, contrast in DIC images depends on the direction of displacement between the two light beams and the sample, also called the “shear direction,” precluding quantitative measurement of the optical path length.
To overcome the limitations of DIC systems, an orientation-independent differential interference contrast (OI-DIC) microscopy method was developed (Shribak, 2013). OI-DIC microscopy allows the directions of displacement for the two light beams to be switched rapidly without mechanically rotating the sample or the prisms, generating a quantitative optical path difference (OPD) map. Based on the OPD value, it is possible to estimate the density of intracellular components in live cells.
Using OI-DIC microscopy, we focused on the pericentric heterochromatin foci (chromocenters) in live mouse NIH3T3 cells. We quantified the absolute density of the materials in the pericentric foci and surrounding regions, which are putatively euchromatic and free from MeCP2 and H3K9me3 heterochromatic marks. Our quantification showed that the density of the heterochromatin was 208 mg/ml, only 1.53-fold higher than that of the surrounding euchromatic regions (136 mg/ml). Surprisingly, this difference was much smaller than that obtained from fluorescence staining of genomic DNA (5.5–7.5-fold). These results, as well as further analysis, suggested that nonnucleosomal materials (proteins, RNAs), which can contribute to chromatin compaction through the macromolecular crowding effect (Asakura and Oosawa, 1954; Marenduzzo et al., 2006; Hancock, 2007), were dominant in both heterochromatin and euchromatin (∼120 mg/ml). Our computational simulation suggested that the nonnucleosomal materials help create a moderate barrier to the diffusion of proteins to heterochromatin, where they dynamically regulate heterochromatin functions. The results of this study reveal a novel aspect of heterochromatin in live cells related to its density.
RESULTS
Density of heterochromatin in live mouse cells was only 1.53-fold higher than that of the surrounding euchromatic regions
To estimate the density of total materials in the heterochromatin and euchromatin regions, we used an OI-DIC microscopy system; the principal schematic of this system is shown in Figure 1A. On the basis of the OPD map obtained from OI-DIC imaging (Figure 1B), as well as the measured thickness of the sample (Supplemental Figure S1, A and B) and refractive index (RI) of the surrounding medium, we were able to calculate the dry mass density of the sample (Figure 1B and Supplemental Figures S1–S3; for more details, see Materials and Methods). Next, to evaluate whether OI-DIC imaging and subsequent analysis could accurately estimate RI, we observed glass rods (diameter = 4 µm) in a mineral oil with known RI (1.54 and 1.58) and calculated the theoretical OPD (Figure 1, C and D). We found that theoretical and optically measured OPD were almost identical (Figure 1, C and D), validating the accuracy of our OI-DIC imaging for estimating the RI of samples with measured OPD.
FIGURE 1:
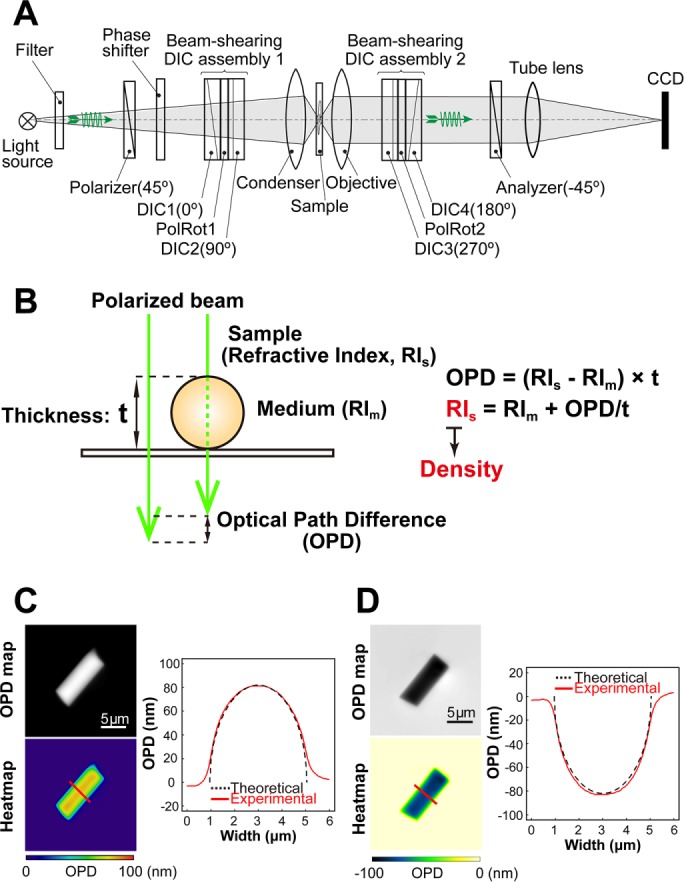
OI-DIC imaging system for biological samples. (A) Schematic of the OI-DIC setup. Details of the microcopy system are described under Materials and Methods. (B) A procedure for estimating sample (depicted as a sphere) RI. Our OI-DIC microscopy can computationally quantify optical path differences (OPDs) at each spatial point. For details, see Materials and Methods. (C, D) Validation of density imaging by OI-DIC microscopy using known glass rods and mineral oils. The RI of the glass rods was 1.56, and those of the oils were 1.54 in C and 1.58 in D. Note that the theoretical and experimental values were almost the same, ensuring the accuracy of our RI quantification. Scale bar: 5 µm.
Using the method described above (see also Figure 2A), we performed OI-DIC imaging of live mouse fibroblast NIH3T3 cells. We clearly observed cytoplasmic organelles, nuclear envelopes, and presumably nucleoli (Figure 2B, left). Subsequent analyses mainly focused on large pericentric heterochromatin foci, which are a strong constitutive heterochromatin model. To identify the precise positions of heterochromatin foci in a nucleus, we specifically labeled the foci with enhanced green fluorescent protein–(EGFP) fused MeCP2 (Figure 2B, right), which is a methylated DNA binding protein and a marker protein of constitutive heterochromatin (Nan et al., 1996; Brero et al., 2005). We confirmed that MeCP2-EGFP localized in certain regions that were strongly stained with Hoechst 33342 as foci (Figure 2B, center and right).
FIGURE 2:

Density imaging of heterochromatin in live mouse cells. (A) Simple schematic of the method to estimate the RIcy. The RIcy can be calculated based on the measured RImed and thickness t values (Supplemental Figures S1, A and B, and S3). For details, see Supplemental Figure S2 and Materials and Methods. (B) Typical images of the optical path difference (OPD) map, DNA staining, and MeCP2-EGFP signals in live NIH3T3 cells. In DNA staining and MeCP2-EGFP images, colocalized large foci (blue and cyan arrowheads) were assumed to be the pericentric heterochromatin. Note that the OPD of pericentric foci was similar to or slightly higher than that of the surrounding regions. High-intensity regions with the heart and sphere shapes in the nuclei of OPD maps were nucleoli (Supplemental Figure S4). Insets: magnified images of the foci indicated by cyan arrowheads. Scale bar: 5 µm. (C) Confocal microscope images of DNA staining (Hoechst 33342) and MeCP2-EGFP in live NIH3T3 cells. The regions surrounding the pericentric foci seem almost free of MeCP2. Scale bar: 5 µm. (D) Difference in signal intensity between the heterochromatin (Hch) visualized by MeCP2-EGFP and the surrounding euchromatin region (Ech) in live NIH3T3 cells. The fluorescence ratio between these regions is 38.6 (n = 22 cells). (E) Confocal images of DNA staining (DAPI) and immunostaining with α-H3K9me3 in a fixed NIH3T3 cell. Consistent with MeCP2-EGFP, the regions surrounding pericentric foci were almost free of α-H3K9me3 signals. Scale bar: 5 µm. (F) The signal intensity quantification of the images in E. The fluorescence ratio is 32.3 (n = 21 cells). (G) The estimated total densities of euchromatin (Ech) and pericentric heterochromatin foci (Hch) were 136 and 208 mg/ml, respectively. The median density ratio between them was 1.53. Ech, n = 13; Hch, n = 26.
After acquiring the OPD map for live NIH3T3 cells, we unexpectedly found that the OPD of the pericentric foci (arrowheads in Figure 2B) was similar to or slightly higher than that of the surrounding regions. Because the surrounding regions not only exhibited much weaker Hoechst 33342 signals (Figure 2, B, center, and C, left) but also were nearly free of MeCP2 (Figure 2, C, right, and D) and histone H3K9me3 marks (Figure 2, E, right, and F) (Allis and Jenuwein, 2016), we called them “surrounding euchromatin regions” or “euchromatin regions” (see also Discussion). Thickness data for cytoplasm, nuclei, and cells was obtained by confocal laser microscopy (Supplemental Figure S1), which we used to estimate densities at the pericentric foci labeled with MeCP2-EGFP and the surrounding euchromatin (for details, see Table 1 and Supplemental Figure S2).
TABLE 1:
Densities of cellular compartments in various cell lines/conditions (mg/ml) (median ± quartile deviation).
| Cytosol | Surrounding euchromatin | Heterochromatin | Nucleolus | |
|---|---|---|---|---|
| Live NIH3T3 | 165 ± 18.6 | 136 ± 12.9 | 208 ± 31.3 | 259 ± 24.4 |
| NIH3T3 MeOH fixed | 136 ± 17.3 | 109 ± 15.4 | 289 ± 31.3 | 292 ± 34.1 |
| NIH3T3 FA fixed | 145 ± 8.78 | 144 ± 8.05 | 174 ± 16.5 | 254 ± 20.7 |
| Live RPE-1 | 161 ± 6.91 | 137 ± 7.52 | 202 ± 25.2 (Inactive X) | 226 ± 15.9 |
Interestingly, the density of pericentric foci was 208 mg/ml, whereas that of the surrounding euchromatin was 136 mg/ml (Figure 2G and Table 1). The difference between these two densities (heterochromatin/euchromatin) was only 1.53-fold (Figure 2G). To compare the obtained total density of the pericentric foci with their DNA density, we examined the fluorescence intensity of Hoechst-stained live mouse NIH3T3 cells using confocal laser scanning microscopy (Figure 3A, left). In accordance with a previous report (Bancaud et al., 2009), the intensity of DNA staining was 7.5-fold higher than that of the surrounding regions (Figure 3B), which was much greater than the total density difference (1.53-fold). Live NIH3T3 cells expressing histone H3.1-EGFP (Figure 3A, right panel) also showed that the intensity of EGFP in the foci was 5.5-fold higher than that of the surrounding regions (Figure 3C), thus excluding possible bias resulting from the high-affinity of the Hoechst dye to the AT-rich sequences of the foci. Furthermore, this 5.5-to 7.5-fold density difference is consistent with those estimated from volumes of specific heterochromatin (inactive/repressed) and euchromatin (actively transcribed) regions based on superresolution imaging, ChIP-seq, and DNA-FISH techniques (Boettiger et al., 2016). Taken together, these results suggest that, although DNA was highly condensed at the pericentric heterochromatin foci, the total density of the regions (including nonnucleosomal materials such as proteins and RNAs) was comparable to that of the surrounding euchromatin regions (Figure 2G).
FIGURE 3:
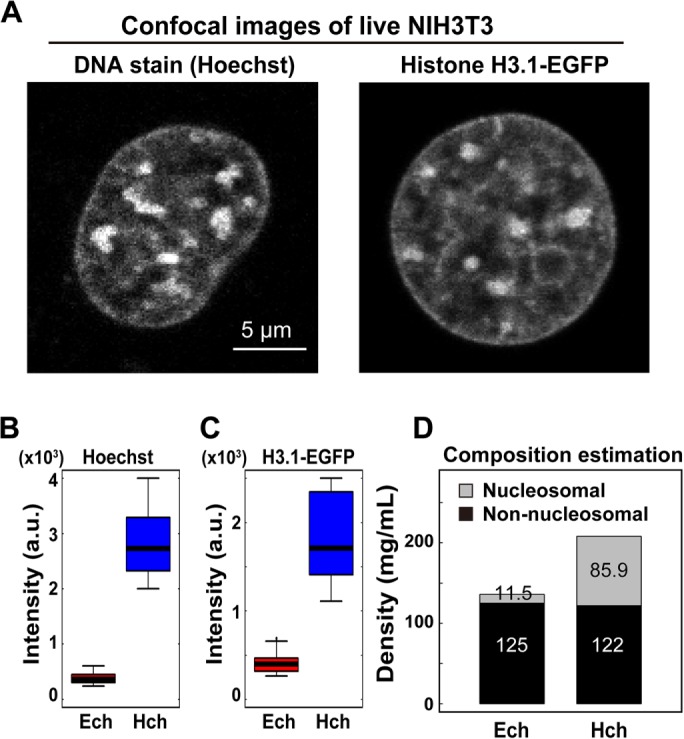
Difference in DNA density between pericentric heterochromatin foci and euchromatin. (A) Typical confocal images of Hoechst 33342 and H3.1-EGFP signals in live NIH3T3 cells. Large foci in the nuclei are pericentric heterochromatin foci. Scale bar: 5 µm. (B, C) Differences in signal intensity between the heterochromatin (Hch) and surrounding euchromatin (Ech) regions in live NIH3T3 cells. Signal intensity ratios: 7.5 (Hoechst 33342, B) and 5.5 (H3.1-EGFP, C). n = 18 for Hoechst 33342 staining and n = 16 for H3.1-EGFP. (D) Estimated composition of the pericentric foci and euchromatin in live cells. Note that nonnucleosomal materials (nonhistone proteins, RNAs) were dominant in both chromatin regions. For details, see Materials and Methods.
We also measured the total densities of cytoplasm and nucleoli in live NIH3T3 cells (Supplemental Figures S1A and S4; Table 1). These cellular features were specifically labeled in live cells using calcein (Morris, 1990) and EGFP-fibrillarin (Ochs et al., 1985), respectively. The density of cytoplasm (165 mg/ml) was slightly higher than that of the nucleoplasm, except for the pericentric foci. The density of nucleoli (259 mg/ml) was the greatest among the cellular compartments we quantified (Table 1; see also Discussion). While performing OI-DIC imaging and density estimation on live cells, we found that methanol (MeOH) fixation and formaldehyde (FA) fixation increased and decreased the density of heterochromatin foci, respectively (MeOH, 289 mg/ml; FA, 174 mg/ml) (Table 1 and Supplemental Figure S5). This result is consistent with the density of centromeric heterochromatin (165 mg/ml) in FA-fixed human cells measured by the coherent anti-Stokes Raman scattering (CARS)- based system (Pliss et al., 2010). Furthermore, the density of the cytoplasm clearly declined after each treatment (MeOH, 136 mg/ml; FA, 145 mg/ml) (Table 1 and Supplemental Figure S5). Our results show that the localization of nuclear and cytoplasmic materials was altered by the fixation processes, indicating that live-cell imaging is critical for proper density estimation.
Composition estimation of the pericentric heterochromatin foci in live cells
Given that the difference in DNA density between the foci regions and euchromatin was high (Figure 3, B and C), we were interested in determining why the difference in total density between them was so low (Figure 2G). To this end, we estimated the density compositions of the pericentric foci and euchromatin. We first calculated the average density of nucleosomes in a single nucleus on the basis of the total mouse genome size (one nucleosome/200 base pairs DNA; 2.8 G base pairs DNA in a haploid mouse cell) and nuclear volume (1000 µm3; details in Materials and Methods), and obtained a value of 11.5 mg/ml (Figure 3D). If we assume that this value is similar to the nucleosome density of the surrounding euchromatic regions, then the density of nonnucleosomal materials (proteins, RNAs) should be 125 mg/ml, because the measured total density of the region is 136 mg/ml (Figure 3D). Then, given that the DNA (or nucleosome) density of the heterochromatin foci was 7.5-fold higher than in the surrounding euchromatin regions, we calculated the densities of nucleosomes and nonnucleosomal materials in the heterochromatin foci to be 85.9 and 122 mg/ml, respectively (Figure 3D). Interestingly, these simple calculations suggested that the density of nonnucleosomal materials (proteins, RNAs) was comparable between the heterochromatin and surrounding euchromatin (Figure 3D) and that the nonnucleosomal contribution was dominant in determining these total densities.
Total density in the inactive X chromosome was only 1.48-fold higher than the surrounding regions
The pericentric foci that we investigated as described above are a model of constitutive heterochromatin. Therefore, to ascertain the generality of our findings, we examined inactivated human X chromosomes, a classic facultative heterochromatin model (Wutz, 2011; Nakajima and Sado, 2014; Smeets et al., 2014; da Rocha and Heard, 2017; Jegu et al., 2017). To this end, we used the near-diploid human cell line RPE1. RPE1 cells have brightly stained foci, often attached to the nuclear envelope (Figure 4A, right), which are very likely to be condensed inactive X chromosomes with H3K9me3 heterochromatin marks (Nozawa et al., 2013) (Figure 4B). We found that the total density of the inactive X chromosome (202 mg/ml) was just 1.48-fold higher than the surrounding regions (137 mg/ml, Figure 4C and Table 1), which were almost free of H3K9me3 heterochromatin marks (Figure 4B); cytoplasm, nucleoplasm, and nucleoli in live RPE1 cells also had similar densities to NIH3T3 (Figure 4A and Supplemental Figure S6A). These results suggest that our findings can be generalized to different types of heterochromatin.
FIGURE 4:
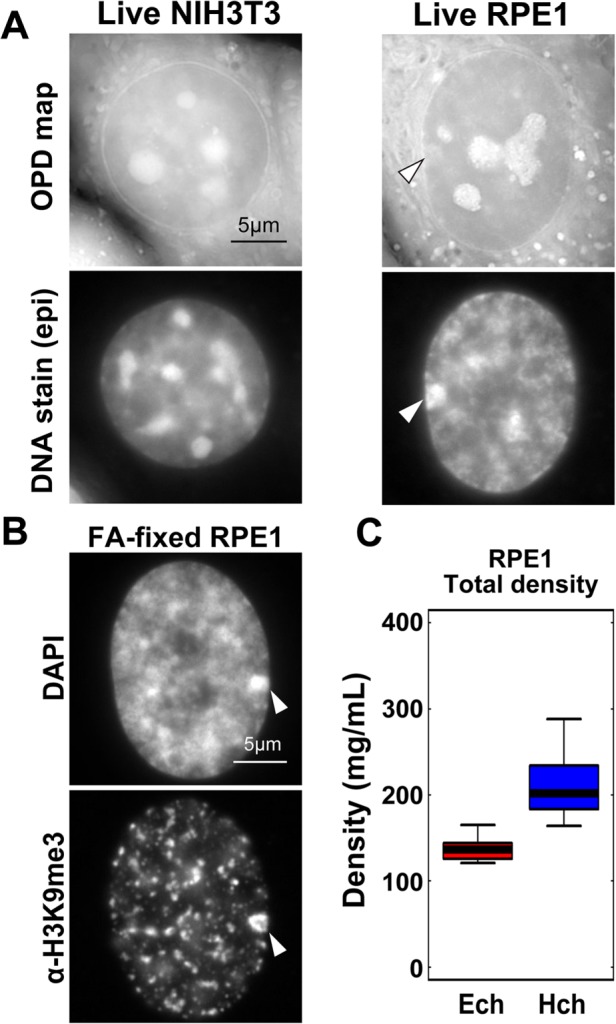
Density of condensed inactive human X chromosome. (A) OPD maps (first row) and DNA staining (second row) of live NIH3T3 cells (left) and RPE1 cells (right). Note that the RPE1 cell has a brightly stained area attached to the nuclear envelope (arrowhead), which is likely to be an inactive X chromosome. High-intensity regions in the nuclei of OPD maps are nucleoli (also see Supplemental Figure S4). (B) Fluorescence images of DNA staining (DAPI, upper) and α-H3K9me3-immunostaining of FA-fixed RPE1 cells. Regions surrounding inactive X chromosomes seem to be free of H3K9me3 marks. (C) The density of the nucleoplasm (euchromatin, Ech) and inactive X chromosome (heterochromatin, Hch) in RPE1 cells (density ratio: 1.48, n = 10 for each).
A moderate barrier of access to heterochromatin revealed by Monte Carlo simulation
Although we found that nonnucleosomal materials (proteins, RNAs) were the dominant components of heterochromatin and euchromatin, the biological significance of this finding was not immediately clear. Thus, to investigate the significance of this finding, we created a simple computational model of the heterochromatin-euchromatin boundary using Monte Carlo simulation (Metropolis et al., 1953; Morelli and ten Wolde, 2008), which can reproduce the movements of objects such as nucleosomes (Hihara et al., 2012; Nozaki et al., 2013; Maeshima et al., 2015). This type of computational modeling can be used to predict the behavior of proteins under conditions that are either difficult to directly observe by imaging or difficult to generate experimentally (Hibino et al., 2017). We examined the accessibility of the heterochromatin regions by model proteins (tracers) under various conditions (Figure 5A).
FIGURE 5:

Moderate access barrier for heterochromatin revealed by Monte Carlo simulation. (A) Simulation schemes to study the molecular access to heterochromatin in the “nucleosomes only” situation. Different numbers of crowding agents (gray spheres) were put into the left and right halves of the box at densities corresponding to the nucleosome densities of euchromatin (11.5 mg/ml) and heterochromatin (85.9 mg/ml). Then 50 tracers (red spheres) were placed in the left half of the box (left, time t = 0), and all tracer spheres were randomly moved. Later, some of the tracers (red spheres) moved into the heterochromatin region (right, time t = ∼3 ms). We analyzed the fraction of tracers in the dense half and the trajectories of the tracers. To aid in visualization, crowding agents were made transparent. (B) Typical trajectories of the tracers in the simulation corresponding to A with periodic boundaries to avoid problems caused by finite space. The trajectories were two dimensionally projected onto an x-yz plane. yz is a cylindrical coordinate; yz = (y2 + z2)1/2. The dense regions are shaded in gray, and the starting regions for the tracers are marked with yellow. Tracers with diameters of 5 or 10 nm could easily diffuse into the dense regions. (C) Table showing the percentages of various tracers (5, 10, 15, and 20 nm) that were localized in the dense half. From left to right columns: only nucleosomes condition (mean ± SD); nucleosomes/nonnucleosomes condition (mean ± SD); percentage suppression to dense regions in the nucleosomes/nonnucleosomes condition. (D) Snapshot of the simulation with additional crowding agents (nonnucleosomal materials): sparse euchromatin (left, 136 mg/ml) and dense heterochromatin (right, 208 mg/ml) regions at t = ∼3 ms. To aid in visualization, only part of the simulation space is presented (20% of the entire space, 210 × 210 × 42 nm). (E) Typical trajectories of the tracers in the simulation corresponding to D with periodic boundaries. Note that the diffusions of tracers were suppressed to a greater degree than in B. (F) Fraction of tracers localized in the dense region under various density conditions. For each tracer type (5-, 10-, 15-, and 20-nm diameter), the fraction within the dense half at equilibrium (∼50 ms) is shown. Note that the 1.53-fold higher density corresponds to the estimated density ratio between heterochromatin and euchromatin in live cells.
We defined a cubic space that had two regions, left and right halves (Figure 5, A and D). As crowding agents, spheres with 9.6 nm diameters were placed into the left (“sparse”) and right (“dense”) regions at low and high densities, respectively. For simplicity, we used the size and weight of nucleosomes as representatives of the various crowding agents including nucleosomes, proteins, RNAs, and their complexes. To maintain the density difference between the two regions, we restricted the movement of the crowding agents to within each region. To investigate accessibility into the dense half, we added spheres with various diameters to the sparse half as tracers and allowed them to move freely within the entire space. To determine whether the density difference created a barrier to the dense region, we measured the fraction of the tracers in the dense half after a period of time.
First, we placed crowding agents into the sparse and dense regions at 11.5 and 85.9 mg/ml, respectively. This represented the nucleosome densities in euchromatin and heterochromatin, respectively, and produced the 7.5-fold density difference between the two regions (Figure 5A). Under these conditions, small tracers (5–10 nm in diameter) moved quite freely within the entire space (Figure 5, B and C), although some suppression of access to the dense region (heterochromatin) by large tracers was observed, presumably due to the excluded volume effect in the dense region.
Next we added more crowding agents, representing nonnucleosomal materials, to the sparse and dense regions: 136 mg/ml in the sparse region and 208 mg/ml in the dense region (Figure 5D and Supplemental Movie 1). We observed an access suppression effect, particularly for large tracers 10–20 nm in diameter (Figure 5, C, E, and F), which is consistent with the in vivo situation (Bancaud et al., 2009; Baum et al., 2014; Cremer et al., 2015). This suggested that the dominance of nonnucleosomal materials contributed to the creation of a moderate access barrier for heterochromatin. After adding more crowding agents to the dense region to a total density of 136–340 mg/ml (1.53- to 2.5-fold density difference), the access-barrier effect became more severe (Figure 5F). With sphere tracers of 20 nm in diameter, the accessibility to heterochromatin was almost completely inhibited (Figure 5F).
DISCUSSION
To elucidate the physical properties of chromatin, we used a novel OI-DIC imaging method and subsequent computational analysis (Figures 1A and 2A; Supplemental Figures S1–S3) to calculate the absolute density of heterochromatin and adjacent euchromatin regions, including nucleosomes and nonnucleosomal materials (e.g., proteins, RNAs) in live cells (Figures 2G and 4C). On the basis of the difference in DNA density between these regions (i.e., a 5.5- to 7.5-fold difference) (Figure 3, B and C), we expected that heterochromatin would be very dense. Surprisingly, we found that the pericentric foci (208 mg/ml) and inactive X chromosomes (202 mg/ml) were only 1.53- and 1.48-fold denser, respectively, than the surrounding euchromatin (Figures 2G and 4C and Table 1). This may be because nonnucleosomal materials were dominant (∼120 mg/ml) in both regions (Figure 3D). Our study provides a novel “live” view of heterochromatin that includes not only densely packed nucleosomes but also nonnucleosomal proteins and RNAs.
The results of our simulation suggested that nucleosomes alone could not create an efficient barrier of access to heterochromatin regions (Figure 5, B and C) and that the addition of nonnucleosomal materials to both the sparse and dense regions contributed to the formation of a moderate barrier to proteins accessing dense regions (Figure 5, C, E, and F). This result is consistent with in vivo observations that a very small fraction of large fluorescent dextran (∼500 kDa; Stoke’s radius ~15 nm) can diffuse into pericentric heterochromatin in NIH3T3 cells (Bancaud et al., 2009), see also Baum et al. (2014) and Cremer et al. (2015). This type of moderate barrier, which allows minimal protein access to heterochromatin and makes the regions structurally controllable, may play an important role in heterochromatin functions such as low-level transcription (Trojer and Reinberg, 2007; Saksouk et al., 2015) and mid to late S phase DNA replication (Guenatri et al., 2004; Wu et al., 2006). On the other hand, the nucleoli, whose density was the highest (259 mg/ml) among the cellular components (Table 1 and Supplemental Figure S4), might present a much stronger barrier to protein access and require active transport systems for proteins. Consistently, it has been reported that nucleolar localization signals exist that target proteins to nucleoli (Hatanaka, 1990; Scott et al., 2010).
Besides the access barrier function of nonnucleosomal proteins and RNAs, we suggest that a high concentration (∼120 mg/ml) of the nonnucleosomal materials generates the macromolecular crowding effect/depletion attraction seen in crowded environments, where 20–30% of the volume is occupied by soluble proteins and other macromolecules (Asakura and Oosawa, 1954; Marenduzzo et al., 2006; Hancock, 2007). We further propose that this effect plays an important role in local chromatin compaction in the process of heterochromatin formation, as suggested previously (Bancaud et al., 2009; Walter et al., 2013; Golov et al., 2015). Furthermore, since recent studies using superresolution imaging or the Hi-C technique have revealed that chromatin is organized as domains (Markaki et al., 2010; Dixon et al., 2012; Nora et al., 2012; Sexton et al., 2012; Rao et al., 2014; Eagen et al., 2015; Cremer et al., 2017; Nozaki et al., 2017) with various sizes (∼100–200 nm), the macromolecular crowding effect caused by high concentrations of nonnucleosomal materials may also contribute to the formation of chromatin domains. Consistent with this notion, hypotonic treatment of live cells, which likely reduces the crowing effect, was shown to decondense the chromatin domain structure (Nozaki et al., 2017).
From a technical perspective, our results confirmed that OI-DIC imaging could achieve a lateral resolution of ∼300 nm and low OPD noise levels (∼0.5 nm) at a wavelength of 546 nm. The pericentric heterochromatin (Figure 2) and inactive X chromosomes (Figure 4) have micron-sized dimensions suitable for examination. On the other hand, because of the resolution limitation, we must bear in mind that the obtained total densities in the euchromatic regions that we measured in Figures 2 – 4 were the averaged values of the chromatin domains (∼100–200 nm) and surrounding domain-free regions.
In principle, any quantitative interference and phase microscopy technique, such as spatial light interference microscopy (SLIM), can be employed for density imaging of cells (Wang et al., 2011). However, OI-DIC provides better image quality and superior resolution (Supplemental Figure S7) because it employs the full numerical apertures of the condenser and the objective lenses, as well as optical subtraction of the images, followed by computation subtraction (Shribak et al., 2017). The OI-DIC technique can be easily integrated with conventional fluorescence imaging systems. Using OI-DIC, the organelles in live cells could be identified using fluorescence markers, and subsequent organelle development with density quantification could be tracked over a long time series with minimal phototoxicity.
Finally, we emphasize the importance of live cell studies to understand the biophysical nuclear properties, which govern nuclear functions and both facilitate and constrain the diffusion of protein factors and their complexes. Our study sheds new light on the need to consider not only chromatin compaction (DNA density) but also nonnucleosomal materials (proteins, RNAs) as obstacles of a free diffusion. The obtained density information can provide critical physical parameters for comprehensive computational modeling of chromatin and chromosomes (e.g., Cheng et al., 2015; Ozer et al., 2015; Shinkai et al., 2016).
MATERIALS AND METHODS
OI-DIC imaging system for biological samples
The principal schematic of the OI-DIC microscopy system is shown in Figure 1A. The microscope includes a light source with a bandpass filter, crossed linear polarizer and analyzer, phase shifter, condenser and objective lenses, tube lens, and charge-coupled device (CCD) digital camera. To overcome the limitations of conventional DIC imaging, the microscope contains two beam-shearing assemblies (Shribak, 2014). Each assembly consists of two identical DIC prisms and a 90° polarization rotator. Rotating light polarization by the rotators allows the shear directions to be rapidly switched by 90° without mechanically rotating the samples (e.g., live cells) or the prisms. We captured six raw images using OI-DIC microscopy, with two perpendicular shear directions and three biases ± 0.15λ and 0, where λ is the wavelength. The captured OI-DIC images were processed into OPD images (maps) whose intensity corresponded linearly to the OPD value. Image processing was performed using home-built software (OIDIC.exe). This and other processing algorithms were previously described (Shribak and Inoue, 2006; Shribak, 2013; Shribak et al., 2017).
To compare OI-DIC with the CellVista SLIM Pro phase-mapping microscope, manufactured by Phi Optics (Champaign, IL), we embedded 7-μm-diameter glass rods, which are used as spacers in liquid-crystal displays, in Fisher Permount mounting medium (Fisher Scientific, https://www.fishersci.com). The RIs of the glass rods and the Permount medium at wavelength 546 nm are 1.56 and 1.524, respectively. The images were obtained using 100 ×/1.4 NA oil immersion objective lenses. The CellVista SLIM captured and processed four phase-contrast images at the different biases.
Density estimation of cellular contents
To obtain calibration curves of the RIs of proteins and nucleic acids (Supplemental Figure S3), bovine serum albumin (BSA; Sigma; A-9418), and salmon sperm DNA (Fisher Scientific; BP-2514) were dissolved in cell culture medium at concentrations of 0–200 and 0–30 mg/ml, respectively. The RIs of the prepared standard solutions were measured with a refractometer, Abbé-3L (Bausch & Lomb). The measured RIs and solution densities were plotted and fitted with linear functions to obtain calibration curves.
We estimated intracellular density distribution from obtained OPD maps using the following two steps (Figure 1B). First, we calculated the RI from the OPD. Because the OPD is proportional to the thickness of a sample and the difference in RI between the sample and the surrounding solution, as shown in Figure 1B, we calculated the RI of samples on the basis of the RI of the surrounding solution and sample thickness. Second, we obtained the dry mass density (“density” for short) of the sample from its RI, because the RI of a sample is proportional to its density. For proteins and nucleic acids, which are the dominant materials in mammalian cells (>60% of dry mass) (Alberts et al., 2007), our calibration curves (Supplemental Figure S3; see above) of RI versus dry mass density using BSA and salmon sperm DNA showed that both were well fitted to linear functions and were almost identical (RI = 1.3375 + 1.4 × 10–4 × C, where C is dry mass density). Therefore, dry mass density in live cells, which consists mainly of proteins and nucleic acids, was calculated from their RI using a single calibration curve (Supplemental Figure S3). To estimate the densities of the total cell contents, we measured the average thickness of the cytoplasm and nucleus in each cell line (Supplemental Figure S1, A and B). The pericentric foci were assumed to be spherical (Supplemental Figure S1C). To obtain the RI of cytoplasm (RIcy), we used the RI of the surrounding culture medium (RImed, 1.3375) (Supplemental Figure S2). For the RIs of the nucleus and the pericentric foci, we used our calculated values of RIcy and RInuc, respectively (Supplemental Figure S2). These estimates were created using ImageJ software.
EGFP-MeCP2 construction
A plasmid containing MeCP2-EGFP was kindly provided by M.C. Cardoso (Brero et al., 2005). The moiety of MeCP2-EGFP was cut by XhoI and XbaI enzymes and blunted. The blunted fragment was inserted into the EcoRV site of the pEF5/FRT/VS-DEST Gateway Vector (Invitrogen) to generate pEF5-MeCP2-EGFP-FRT.
EGFP-mH3.1p-H3.1 construction
A mouse histone H3.1 promoter (∼840 base pairs)-H3.1 fragment was amplified from mouse embryonic stem (ES) cell genomic DNA by PCR using the following primer set:
5′-AAACATTACGAATCACCAAAGGCTCTTTTCAGAGCCACTC-3′ and
5′-AGCCCTCTCCCCGCGGATGCGGCGGGCCAGCTGGATGTCC-3′. The amplified fragment was cloned into an EGFP vector to obtain pmH3.1p-H3.1-EGFP. Then two more insert sequences were prepared. One was the H3.1promoter-H3.1-EGFP amplified from the pmH3.1p-H3.1-EGFP vector using the following primer set:
5′-TAGGCTTTTGCAAAAAGCTTAAACATTACGAATCACCAAA-3′
and 5′-TTACTTGTACAGCTCGTCCATGCCGAGAGT-3′. The other was the 3′ untranslated region of H3.1 (∼500 base pairs) amplified from the mouse ES genome using the primer pair: 5′-TGGACGAGCTGTACAAGTAAAGTTCGTCTTTCTGTGTTTTTCAAAGGCTC-3′
and 5′-GCTTGACGGGGAAAGAAGCTGCTGGTTGTAGCACTTTGGGTTGTTCTGGG-3′.
For backbone vector preparation, the region from the EF1α-promoter to the BGH polyadenylation signal sequence was removed from pEF5/V5-FRT Gateway (Invitrogen) to create the pFRT-Hygromycin vector. The pFRT-Hygromycin vector, H3.1promoter-H3.1-EGFP sequence, and 3′ untranslated region sequence were ligated by SLIC (Li and Elledge, 2007) to prepare the pmH3.1p-H3.1-EGFP-3′UTR-FRT plasmid. In addition, upstream of the H3.1 promoter, an insulator fragment (tandem cHS4 kindly provided by G. Felsenfeld) was inserted to obtain pIx2-mH3.1p-H3.1-EGFP-3′UTR-FRT.
EGFP-fibrillarin construction
To clone the fibrillarin gene, total RNA was isolated from NIH3T3 cells using the RNeasy Mini Kit (Qiagen), and first-strand cDNA was synthesized using the SuperScript III First-Strand Synthesis System (Thermo) with oligo(dT). The coding region of fibrillarin was amplified from the first-strand cDNA using the following primer pair: 5′-GGGGTACCATGAAGCCAGGTTTCAGCCC-3′ and 5′-GCGGGATCCTCAGTTCTTCACCTTGGGAG-3′. The amplified fragment was digested with KpnI and BamHI and ligated into KpnI/BamHI-digested pEGFP-C1 (Clontech, Palo Alto, CA). EGFP-fibrillarin fragments were excised, blunt-ended using T4 DNA polymerase (Takara, Japan), and inserted into EcoRV-precut pEF1-FRT to generate pEF1-EGFP-fibrillarin-FRT.
Cell culture and stable cell lines
We used RPE1, a human cell line, and NIH3T3, a mouse cell line. All of the cell lines were maintained in DMEM (Lonza) supplemented with 10% (vol/vol) fetal bovine serum (FBS) and 0.584 g/l l-glutamine (Sigma) at 37°C with 5% CO2 in air in a humidified incubator. To establish NIH3T3 cells expressing MeCP2-EGFP, EGFP-fibrillarin, or H3.1-EGFP, we used the Flp-In system (Invitrogen) as previously described (Maeshima et al., 2010; Hihara et al., 2012).
OI-DIC microscopy system
Details of the microcopy system are provided in the Supplemental Material.
Live-cell OI-DIC microscopy imaging
Cells were seeded on 24 mm × 24 mm square glass coverslips coated with poly d-lysine (Sigma) and cultured for 1–2 d. Then 30 min before imaging, 500 ng/ml Hoechst 33342 was added into the media and incubated further. The cells were mounted on a glass slide with a thin silicone spacer. We observed the mounted cells by OI-DIC and fluorescence imaging.
OI-DIC imaging of glass rods
A small number of glass rods 4 µm in diameter were suspended in two types of mineral oil with refractive indices of 1.54 and 1.58. Approximately 2 µl of the suspended solution was sandwiched between a glass slide and a coverslip, and then sealed with nail polish. The glass rods in the mineral oil were analyzed by OI-DIC microscopy using the same procedure as the live cell imaging.
Fixation
For formaldehyde fixation, cells on the square glass coverslips were washed once with phosphate-buffered saline (PBS), and then fixed in 4% formaldehyde at room temperature for 15 min. The fixed cells were washed with 10 mM HEPES-KOH, pH 7.5, 100 mM KCl, and 1 mM MgCl2 (HMK) (Maeshima et al., 2006) and permeabilized with 0.5% Triton X-100 in HMK at room temperature for 5 min. The treated cells were washed with HMK, stained with 500 ng/ml Hoechst 33342 in HMK at room temperature for 10 min, and then washed again with HMK. For MeOH fixation, cells on coverslips were fixed in ice-cold MeOH at –20°C for 30 min. Then the fixed cells were washed with HMK at room temperature for 15 min, stained with 500 ng/ml Hoechst 33342 in HMK for 10 min, and washed again with HMK. Finally, the cells were mounted on a glass slide and observed as described above.
Immunostaining of histone H3K9me3
Immunostaining was performed as previously described (Maeshima et al., 2010; Hihara et al., 2012). Cells were fixed in 2% formaldehyde (Wako) and permeabilized with Triton X-100. The primary and secondary antibodies were mouse anti-H3K9me3 (a generous gift from Hiroshi Kimura) and Alexa-Fluor-594-conjugated goat anti-mouse immunoglobulin G (Invitrogen) used at dilutions of 1:500 and 1:1000, respectively. Then DAPI (500 ng/ml) was added to the cells for 5 min, followed by washing with PBS prior to DNA staining. Images were obtained using a DeltaVision microscopy imaging system (Applied Precision) or a FLUOVIEW FV1000 confocal laser scanning microscope (OLYMPUS).
Measurements of cell thicknesses
Live RPE1 and NIH3T3 cells were seeded on 35 mm glass-bottom dishes. To fluorescently label DNA and cytoplasm, cells were incubated in culture medium containing 0.5 µg/ml Hoechst 33342 (Dojindo) and 5 µg/ml Calcein-AM (Dojindo) for 30 min. After washing out excess fluorescent dye, the stained cells were observed under an OLYMPUS FLUOVIEW FV1000 confocal laser scanning microscope equipped with a 60 ×/1.2 NA water objective. Hoechst 33342 and Calcein-AM fluorescence signals were acquired as three-dimensional image stacks (500 nm × 32 sections). The thicknesses of three regions (nucleus, cytoplasm, entire cell; see Supplemental Figure S1) were measured in each cell line from the acquired stack images by ImageJ software.
Measurement of nuclear volume of live cells
Image stacks of live NIH3T3 and RPE1 cells acquired to measure cell thickness (described above) were used. The images were converted into binary images by auto thresholding in ImageJ software. Then the volumes of binary image stacks were analyzed using the 3D Objects Counter ImageJ plugin (Bolte and Cordelieres, 2006).
Quantification of fluorescence from Hoechst-stained DNA, MeCP2-EGFP, and α-H3K9me3
Image stacks of live or immunostained NIH3T3 cells (described above) were used. The middle sections of Hoechst-stained nuclei were selected from the z-stack images. The highest intensity of a focal plane of heterochromatin and the mean intensity of the surrounding low-intensity region in a nucleus were taken as the intensities of heterochromatin and the surrounding euchromatin. These values were adjusted to account for the background intensity.
Composition estimation of euchromatin and heterochromatin in live cells
The genome size of diploid mouse cells is 5.6 × 109 base pairs. Because 1 pg of DNA is 978 × 106 base pairs (978 × 106 base pairs/pg), the mass of the whole mouse genome is 5.73 pg. If we assume that nucleosomes (core histones + DNA) form every 200 base pairs of the genome and that the mass ratio of core histones to DNA is around 1:1, the mass of the nucleosomes in a mouse nucleus is 11.5 pg (5.73 × 2). The average mouse nuclear volume was calculated to be ∼1000 µm3 (Supplemental Figure S6B), and the density of nucleosomes in mouse euchromatin was calculated to be 11.5 mg/ml. Further estimates are described under Results.
Monte Carlo simulation
Monte Carlo simulation is a computational algorithm that performs a numerical integration by making a random movement and evaluating whether the movement is acceptable based on the change in potential energy (Hibino et al., 2017). All of the molecules in the simulations were treated as hard spherical bodies. We employed a Metropolis Monte Carlo method without long-range potential or hydrodynamic interactions to determine the diffusive motion of molecules (Morelli and ten Wolde, 2008). The diameters and diffusion coefficients (Ds) of the crowding agents used in the simulations were 9.6 nm and 9 µm2 s−1, respectively, which are comparable to those of a single nucleosome molecule. The Ds of tracers (spheres with diameters of 5, 10, 15, and 20 nm) were 18, 9, 6, and 4.5 µm2 s−1, respectively. These Ds were determined by the Stokes–Einstein relationship based on parameters from the EGFP monomer, the diameter and D of which were 3.8 nm and 23.5 µm2 s−1, respectively (Hihara et al., 2012). Simulations were conducted in a 210-nm cubic box with two compartments (left and right halves) with periodic boundaries to avoid problems caused by finite space, and make the system more like an infinite one. For the “nucleosomes only” scenario, which corresponds to 11.5 mg/ml (euchromatin) and 85.9 mg/ml (heterochromatin), 134 and 968 copies of 9.6 nm spheres (crowding agents) were randomly placed in the left and right halves of the box, respectively. These crowding agents mimicked nucleosomes displaced less than 5 nm from their initial positions at t = 0 s (the “dog on a leash” model; see also Hihara et al., 2012, and Maeshima et al., 2015). Then 50 tracers that could diffuse freely were placed in the left (euchromatin) region. The motion of the molecules was iteratively simulated following previously described procedures (Hihara et al., 2012; Maeshima et al., 2015). For the second simulation (nucleosomes + nonnucleosomal materials), 1578 and 1578–3945 9.6 nm spheres (crowding agents) were randomly placed in the left and right regions of the box, respectively, to represent euchromatin and heterochromatin (1.53–2.5-fold density differences). To represent nucleosomes, 134- and 968 9.6-nm spheres were randomly placed in the left and right regions, with their behavior following the “dog on a leash” model. The rest of the spheres moved freely only in each half, to represent diffusing proteins and RNAs. Then 50 tracers were placed in the left (euchromatin) region. Although the simulation process was similar to the first simulation, we restricted the movements of crowding agents to within their regions to keep the density of each region constant. Results were obtained by averaging 150 samples from three independent trials. The simulation time step, Δt, was 10 ns.
Statistical analyses
All of the statistical analyses were performed using the two-tailed Student’s t test. p values less than 0.05 were considered statistically significant.
Supplementary Material
Acknowledgments
We are grateful to Y. Hiromi, I. Hiratani, and M. Kurusu for their critical comments on the manuscript. We thank M.C. Cardoso, H. Kimura, and G. Felsenfeld for providing research materials and A. Bancaud and J. Ellenberg for their helpful suggestions. We also thank T. Cremer and K. Saito for advice and discussion on chromatin organization and H. Araki, I. Inoue, M. Kanemaki, H. Niki, and Maeshima lab members for their helpful discussion and support. This work was supported by MEXT and Japan Society for the Promotion of Science (JSPS) grants (Nos. 23115005 and 16H04746, respectively), as well as a Japan Science and Technology Agency (JST) CREST grant (No. JPMJCR15G2). R.I. and T.N. are JSPS Fellows. R.I. was supported by the SOKENDAI Short-Stay Study Abroad Program in fiscal year 2016. Computations were partially performed on the National Institute of Genetics (NIG) supercomputer. T.T. was supported by NIG-JOINT (2016-A2). M.S. (R01-GM101701) and T.T. (R01-GM100160) were supported by the National Institute of General Medical Sciences, National Institutes of Health. The contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
Abbreviations used:
- DAPI
4′,6-diamidino-2-phenylindole
- MeCP2
methyl-CpG binding protein 2
- OI-DIC
orientation-independent differential interference contrast
- OPD
optical path difference
- RI
refractive index.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-06-0359) on August 23, 2017.
REFERENCES
- Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Molecular Biology of the Cell, 5th ed. New York: Garland Science: 2007. [Google Scholar]
- Allen RD, David GB, Nomarski G. The Zeiss-Nomarski differential interference equipment for transmitted-light microscopy. Z Wiss Mikrosk. 1969;69:193–221. [PubMed] [Google Scholar]
- Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet. 2016;17:487–500. doi: 10.1038/nrg.2016.59. [DOI] [PubMed] [Google Scholar]
- Asakura S, Oosawa F. On interaction betweentwo bodies immersed in a solution of macromolecules. J Chem Phys. 1954;22:1255–1256. [Google Scholar]
- Bancaud A, Huet S, Daigle N, Mozziconacci J, Beaudouin J, Ellenberg J. Molecular crowding affects diffusion and binding of nuclear proteins in heterochromatin and reveals the fractal organization of chromatin. EMBO J. 2009;28:3785–3798. doi: 10.1038/emboj.2009.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum M, Erdel F, Wachsmuth M, Rippe K. Retrieving the intracellular topology from multi-scale protein mobility mapping in living cells. Nat Commun. 2014;5:4494. doi: 10.1038/ncomms5494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettiger AN, Bintu B, Moffitt JR, Wang S, Beliveau BJ, Fudenberg G, Imakaev M, Mirny LA, Wu CT, Zhuang X. Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature. 2016;529:418–422. doi: 10.1038/nature16496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- Brero A, Easwaran HP, Nowak D, Grunewald I, Cremer T, Leonhardt H, Cardoso MC. Methyl CpG-binding proteins induce large-scale chromatin reorganization during terminal differentiation. J Cell Biol. 2005;169:733–743. doi: 10.1083/jcb.200502062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SW. Heterochromatin. Science. 1966;151:417–425. doi: 10.1126/science.151.3709.417. [DOI] [PubMed] [Google Scholar]
- Chen C, Lim HH, Shi J, Tamura S, Maeshima K, Surana U, Gan L. Budding yeast chromatin is dispersed in a crowded nucleoplasm in vivo. Mol Biol Cell. 2016;27:3357–3368. doi: 10.1091/mbc.E16-07-0506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Dundr M, Wang C, Leung A, Lamond A, Misteli T, Huang S. Condensed mitotic chromatin is accessible to transcription factors and chromatin structural proteins. J Cell Biol. 2005;168:41–54. doi: 10.1083/jcb.200407182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng TM, Heeger S, Chaleil RA, Matthews N, Stewart A, Wright J, Lim C, Bates PA, Uhlmann F. A simple biophysical model emulates budding yeast chromosome condensation. Elife. 2015;4:e05565. doi: 10.7554/eLife.05565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremer M, Schmid VJ, Kraus F, Markaki Y, Hellmann I, Maiser A, Leonhardt H, John S, Stamatoyannopoulos J, Cremer T. Initial high-resolution microscopic mapping of active and inactive regulatory sequences proves non-random 3D arrangements in chromatin domain clusters. Epigenet Chromatin. 2017;10:39. doi: 10.1186/s13072-017-0146-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremer T, Cremer M, Hubner B, Strickfaden H, Smeets D, Popken J, Sterr M, Markaki Y, Rippe K, Cremer C. The 4D nucleome: evidence for a dynamic nuclear landscape based on co-aligned active and inactive nuclear compartments. FEBS Lett. 2015;589:2931–2943. doi: 10.1016/j.febslet.2015.05.037. [DOI] [PubMed] [Google Scholar]
- da Rocha ST, Heard E. Novel players in X inactivation: insights into Xist-mediated gene silencing and chromosome conformation. Nat Struct Mol Biol. 2017;24:197–204. doi: 10.1038/nsmb.3370. [DOI] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eagen KP, Hartl TA, Kornberg RD. Stable chromosome condensation revealed by chromosome conformation capture. Cell. 2015;163:934–946. doi: 10.1016/j.cell.2015.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fussner E, Strauss M, Djuric U, Li R, Ahmed K, Hart M, Ellis J, Bazett-Jones DP. Open and closed domains in the mouse genome are configured as 10-nm chromatin fibres. EMBO Rep. 2012;13:992–996. doi: 10.1038/embor.2012.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golov AK, Gavrilov AA, Razin SV. The role of crowding forces in juxtaposing beta-globin gene domain remote regulatory elements in mouse erythroid cells. PLoS One. 2015;10:e0139855. doi: 10.1371/journal.pone.0139855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal SI, Jia S. Heterochromatin revisited. Nat Rev Genet. 2007;8:35–46. doi: 10.1038/nrg2008. [DOI] [PubMed] [Google Scholar]
- Guenatri M, Bailly D, Maison C, Almouzni G. Mouse centric and pericentric satellite repeats form distinct functional heterochromatin. J Cell Biol. 2004;166:493–505. doi: 10.1083/jcb.200403109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock R. Packing of the polynucleosome chain in interphase chromosomes: evidence for a contribution of crowding and entropic forces. Semin Cell Dev Biol. 2007;18:668–675. doi: 10.1016/j.semcdb.2007.08.006. [DOI] [PubMed] [Google Scholar]
- Hatanaka M. Discovery of the nucleolar targeting signal. Bioessays. 1990;12:143–148. doi: 10.1002/bies.950120310. [DOI] [PubMed] [Google Scholar]
- Heitz E. Das heterochromatin der moose. I Jahrb wiss Bot. 1928;69:762–818. [Google Scholar]
- Hibino K, Kaizu K, Takahashi K, Maeshima K. Combination approach of live-cell imaging and computational modeling toward further comprehensive understanding genome, chromatin and epigenetics. In: Ringrose L., editor. Epigenetics and Systems Biology. San Diego, CA: Academic Press; 2017. pp. 225–238. [Google Scholar]
- Hihara S, Pack CG, Kaizu K, Tani T, Hanafusa T, Nozaki T, Takemoto S, Yoshimi T, Yokota H, Imamoto N, et al. Local nucleosome dynamics facilitate chromatin accessibility in living mammalian cells. Cell Rep. 2012;2:1645–1656. doi: 10.1016/j.celrep.2012.11.008. [DOI] [PubMed] [Google Scholar]
- Hsieh TH, Weiner A, Lajoie B, Dekker J, Friedman N, Rando OJ. Mapping nucleosome resolution chromosome folding in yeast by Micro-C. Cell. 2015;162:108–119. doi: 10.1016/j.cell.2015.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoué S, Spring K. Video Microscopy. New York: Plenum Press; 1997. [Google Scholar]
- Jegu T, Aeby E, Lee JT. The X chromosome in space. Nat Rev Genet. 2017;18:377–389. doi: 10.1038/nrg.2017.17. [DOI] [PubMed] [Google Scholar]
- Joti Y, Hikima T, Nishino Y, Kamada F, Hihara S, Takata H, Ishikawa T, Maeshima K. Chromosomes without a 30-nm chromatin fiber. Nucleus. 2012;3:404–410. doi: 10.4161/nucl.21222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MZ, Elledge SJ. Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat Methods. 2007;4:251–256. doi: 10.1038/nmeth1010. [DOI] [PubMed] [Google Scholar]
- Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- Maeshima K, Iino H, Hihara S, Funakoshi T, Watanabe A, Nishimura M, Nakatomi R, Yahata K, Imamoto F, Hashikawa T, et al. Nuclear pore formation but not nuclear growth is governed by cyclin-dependent kinases (Cdks) during interphase. Nat Struct Mol Biol. 2010;17:1065–1071. doi: 10.1038/nsmb.1878. [DOI] [PubMed] [Google Scholar]
- Maeshima K, Imai R, Tamura S, Nozaki T. Chromatin as dynamic 10-nm fibers. Chromosoma. 2014;123:225–237. doi: 10.1007/s00412-014-0460-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeshima K, Kaizu K, Tamura S, Nozaki T, Kokubo T, Takahashi K. The physical size of transcription factors is key to transcriptional regulation in the chromatin domains. J Phys. 2015;27:064116. doi: 10.1088/0953-8984/27/6/064116. [DOI] [PubMed] [Google Scholar]
- Maeshima K, Yahata K, Sasaki Y, Nakatomi R, Tachibana T, Hashikawa T, Imamoto F, Imamoto N. Cell-cycle-dependent dynamics of nuclear pores: pore-free islands and lamins. J Cell Sci. 2006;119:4442–4451. doi: 10.1242/jcs.03207. [DOI] [PubMed] [Google Scholar]
- Maison C, Quivy JP, Probst AV, Almouzni G. Heterochromatin at mouse pericentromeres: a model for de novo heterochromatin formation and duplication during replication. Cold Spring Harb Symp Quant Biol. 2010;75:155–165. doi: 10.1101/sqb.2010.75.013. [DOI] [PubMed] [Google Scholar]
- Marenduzzo D, Finan K, Cook PR. The depletion attraction: an underappreciated force driving cellular organization. J Cell Biol. 2006;175:681–686. doi: 10.1083/jcb.200609066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markaki Y, Gunkel M, Schermelleh L, Beichmanis S, Neumann J, Heidemann M, Leonhardt H, Eick D, Cremer C, Cremer T. Functional nuclear organization of transcription and DNA replication: a topographical marriage between chromatin domains and the interchromatin compartment. Cold Spring Harb Symp Quant Biol. 2010;75:475–492. doi: 10.1101/sqb.2010.75.042. [DOI] [PubMed] [Google Scholar]
- Metropolis N, Rosenbluth AW, MN R, Teller AH, Teller E. Equation of state calculations by fast computing machines. J Chem Phys. 1953;21:1087. (1086 pages) [Google Scholar]
- Morelli MJ, ten Wolde PR. Reaction Brownian dynamics and the effect of spatial fluctuations on the gain of a push-pull network. J Chem Phys. 2008;129:054112. doi: 10.1063/1.2958287. [DOI] [PubMed] [Google Scholar]
- Morris SJ. Real-time multi-wavelength fluorescence imaging of living cells. Biotechniques. 1990;8:296–308. [PubMed] [Google Scholar]
- Nakajima T, Sado T. Current view of the potential roles of proteins enriched on the inactive X chromosome. Genes Genet Syst. 2014;89:151–157. doi: 10.1266/ggs.89.151. [DOI] [PubMed] [Google Scholar]
- Nan X, Tate P, Li E, Bird A. DNA methylation specifies chromosomal localization of MeCP2. Mol Cell Biol. 1996;16:414–421. doi: 10.1128/mcb.16.1.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berlum NL, Meisig J, Sedat JW, et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012;485:381–385. doi: 10.1038/nature11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozaki T, Imai R, Tanbo M, Nagashima R, Tamura S, Tani T, Joti Y, Tomita M, Hibino K, Kanemaki MT, et al. Dynamic organization of chromatin domains revealed by super-resolution live-cell imaging. Mol Cell. 2017;67:282–293.e7. doi: 10.1016/j.molcel.2017.06.018. [DOI] [PubMed] [Google Scholar]
- Nozaki T, Kaizu K, Pack CG, Tamura S, Tani T, Hihara S, Nagai T, Takahashi K, Maeshima K. Flexible and dynamic nucleosome fiber in living mammalian cells. Nucleus. 2013;4:349–356. doi: 10.4161/nucl.26053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozawa RS, Nagao K, Igami KT, Shibata S, Shirai N, Nozaki N, Sado T, Kimura H, Obuse C. Human inactive X chromosome is compacted through a PRC2-independent SMCHD1-HBiX1 pathway. Nat Struct Mol Biol. 2013;20:566–573. doi: 10.1038/nsmb.2532. [DOI] [PubMed] [Google Scholar]
- Ochs RL, Lischwe MA, Spohn WH, Busch H. Fibrillarin: a new protein of the nucleolus identified by autoimmune sera. Biol Cell. 1985;54:123–133. doi: 10.1111/j.1768-322x.1985.tb00387.x. [DOI] [PubMed] [Google Scholar]
- Oldenbourg R, Shribak M. Microscopes. In: Bass M, editor. Handbook of Optics. New York: McGraw-Hill; 2010. pp. 28.1–28.62. [Google Scholar]
- Ou HD, Phan S, Deerinck TJ, Thor A, Ellisman MH, O’Shea CC. ChromEMT: Visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Science. 2017;357, eaag0025 doi: 10.1126/science.aag0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozer G, Luque A, Schlick T. The chromatin fiber: multiscale problems and approaches. Curr Opin Struct Biol. 2015;31:124–139. doi: 10.1016/j.sbi.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pliss A, Kuzmin AN, Kachynski AV, Prasad PN. Nonlinear optical imaging and Raman microspectrometry of the cell nucleus throughout the cell cycle. Biophys J. 2010;99:3483–3491. doi: 10.1016/j.bpj.2010.06.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD, Earnshaw WC, Lippincott-Schwartz J, Johnson G. Cell Biology, 3rd ed. 2016.
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, Aiden EL. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci MA, Manzo C, Garcia-Parajo MF, Lakadamyali M, Cosma MP. Chromatin fibers are formed by heterogeneous groups of nucleosomes in vivo. Cell. 2015;160:1145–1158. doi: 10.1016/j.cell.2015.01.054. [DOI] [PubMed] [Google Scholar]
- Saksouk N, Simboeck E, Dejardin J. Constitutive heterochromatin formation and transcription in mammals. Epigenet Chromatin. 2015;8:3. doi: 10.1186/1756-8935-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanborn AL, Rao SS, Huang SC, Durand NC, Huntley MH, Jewett AI, Bochkov ID, Chinnappan D, Cutkosky A, Li J, et al. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc Natl Acad Sci USA. 2015;112:E6456–E6465. doi: 10.1073/pnas.1518552112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott MS, Boisvert FM, McDowall MD, Lamond AI, Barton GJ. Characterization and prediction of protein nucleolar localization sequences. Nucleic Acids Res. 2010;38:7388–7399. doi: 10.1093/nar/gkq653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sexton T, Yaffe E, Kenigsberg E, Bantignies F, Leblanc B, Hoichman M, Parrinello H, Tanay A, Cavalli G. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell. 2012;148:458–472. doi: 10.1016/j.cell.2012.01.010. [DOI] [PubMed] [Google Scholar]
- Shinkai S, Nozaki T, Maeshima K, Togashi Y. Dynamic nucleosome movement tells Structural information of topological chromatin domains in human cells. PLoS Comput Biol. 2016;12:e1005136. doi: 10.1371/journal.pcbi.1005136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shribak M. Quantitative orientation-independent differential interference contrast microscope with fast switching shear direction and bias modulation. J Opt Soc Am A Opt Image Sci Vis. 2013;30:769–782. doi: 10.1364/JOSAA.30.000769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shribak M. Compact orientation-independent differential interference contrast (OI-DIC) microscope designed for high resolution and high sensitivity mapping of optical path and optical path gradient. Microsc Microanal. 2014;20:1350–1351. doi: 10.1017/S1431927614008484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shribak M, Inoue S. Orientation-independent differential interference contrast microscopy. Appl Opt. 2006;45:460–469. doi: 10.1364/ao.45.000460. [DOI] [PubMed] [Google Scholar]
- Shribak M, Larkin KG, Biggs D. Mapping optical path length and image enhancement using quantitative orientation-independent differential interference contrast microscopy. J Biomed Opt. 2017;22:16006. doi: 10.1117/1.JBO.22.1.016006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeets D, Markaki Y, Schmid VJ, Kraus F, Tattermusch A, Cerase A, Sterr M, Fiedler S, Demmerle J, Popken J, et al. Three-dimensional super-resolution microscopy of the inactive X chromosome territory reveals a collapse of its active nuclear compartment harboring distinct Xist RNA foci. Epigenet Chromatin. 2014;7:8. doi: 10.1186/1756-8935-7-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojer P, Reinberg D. Facultative heterochromatin: is there a distinctive molecular signature. Mol Cell. 2007;28:1–13. doi: 10.1016/j.molcel.2007.09.011. [DOI] [PubMed] [Google Scholar]
- Walter A, Chapuis C, Huet S, Ellenberg J. Crowded chromatin is not sufficient for heterochromatin formation and not required for its maintenance. J Struct Biol. 2013;184:445–453. doi: 10.1016/j.jsb.2013.10.004. [DOI] [PubMed] [Google Scholar]
- Wang S, Su JH, Beliveau BJ, Bintu B, Moffitt JR, Wu CT, Zhuang X. Spatial organization of chromatin domains and compartments in single chromosomes. Science. 2016;353:598–602. doi: 10.1126/science.aaf8084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Millet L, Mir M, Ding H, Unarunotai S, Rogers J, Gillette MU, Popescu G. Spatial light interference microscopy (SLIM) Opt Express. 2011;19:1016–1026. doi: 10.1364/OE.19.001016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R, Singh PB, Gilbert DM. Uncoupling global and fine-tuning replication timing determinants for mouse pericentric heterochromatin. J Cell Biol. 2006;174:185–194. doi: 10.1083/jcb.200601113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wutz A. Gene silencing in X-chromosome inactivation: advances in understanding facultative heterochromatin formation. Nat Rev Genet. 2011;12:542–553. doi: 10.1038/nrg3035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.