

ABSTRACT
Colorectal cancer (CRC) is the third most common malignancy and the fourth most common cause of cancer related death worldwide. This study was designed to find tumor suppressors involved in CRC development by performing RNA-seq. Eight CRC cell lines and 130 cases of primary CRC samples were used. RNA-seq, methylation-specific PCR (MSP), flow cytometry, transwell assays, and a xenograft mouse model were used. Reduction of TMEM176A expression was confirmed in human CRC cells by RNA-seq. TMEM176A was expressed in LS180 and SW620 cells, loss of TMEM176A expression was observed in LOVO, HCT116, RKO, and DLD1 cells, and reduced TMEM176A expression was found in HT29 and SW480 cells. Unmethylation of the TMEM176A promoter was found in LS180 and SW620 cells, whereas complete methylation was found in LOVO, HCT116, RKO, and DLD1 cells, and partial methylation was found in HT29 and SW480 cells. Promoter region methylation correlated with loss of/reduced expression of TMEM176A. Re-expression of TMEM176A was induced by 5-aza-2′-deoxycytidine. TMEM176A was methylated in 50.77% of primary colorectal cancers. Methylation of TMEM176A was associated with tumor metastasis (P<0.05) and was an independent prognostic factor for 5-year overall survival (OS) according to Cox proportional hazards model analysis (P<0.05). TMEM176A induced apoptosis and inhibited cell migration and invasion in CRC cells. TMEM176A suppressed CRC cell growth both in vitro and in vivo. Our results suggest that expression of TMEM176A is regulated by promoter region methylation. TMEM176A methylation is an independent prognostic marker for 5-year OS in CRC, and may act as a tumor suppressor in CRC.
KEYWORDS: Colorectal cancer, DNA methylation, metastasis, prognosis, TMEM176A
Introduction
Colorectal cancer (CRC) is the third most common malignancy and the fourth most common cause of cancer-related death worldwide.1 Unlike other cancers, such as lung cancer and smoking, no single risk factor accounts for most cases of colorectal cancer.2 Both genetic and epigenetic changes are regarded as important mechanisms in human cancers.3–6
TMEM176A is located in human chromosome 7q36.1, a region where there is frequent loss of heterozygosity in human esophageal and thyroid cancers.7,8 While no chromosome alterations were reported at the 7q36.1 locus in colorectal cancer, 2 tumor suppressors, XRCC2 and RARRES2, were found in this region. XRCC2 is involved in DNA repair by homologous recombination,9 and is frequently methylated in human cervical cancer.10 RARRES2 functions as a tumor suppressor by promoting β-catenin phosphorylation/degradation and inhibiting p38 phosphorylation in adrenocortical carcinoma.11 RARRES2 is frequently methylated in Ewing sarcoma.12
Human TMEM176A was first identified by screening tumor related antigens in hepatocellular carcinoma (HCC).13 However, the expression and function of TMEM176A in human cancers remains to be elucidated. In this study, we analyzed the expression of TMEM176A by RNA-seq in colorectal cancer and normal colorectal mucosa and further studied its regulation and function in human CRC.
Results
Isolation and identification of TMEM176A in human CRC
To isolate CRC-related genes, RNA-seq was performed in 4 colorectal cancer cell lines (LOVO, RKO, DLD1, HCT116) and normal human colon mucosa mixed from 15 individuals. As shown in Fig. 1A and B, the expression levels of 176 genes were reduced more than 2-fold in all 4 CRC cells compared with normal colon mucosa. TMEM176A was one of the genes found to be downregulated in all 4 CRC cell lines, as validated by RT-PCR. Searching The Cancer Genome Atlas (TCGA) database (https://cancergenome.nih.gov/) revealed that in 382 cases of CRC samples and 51 cases of unpaired adjacent colorectal tissue samples, the expression levels of TMEM176A were reduced in CRC (P=0.0006153). Only 34 cases of colorectal cancer samples had both TMEM176A expression and methylation data, which were obtained by RNA-seq and HumanMethylation450 BeadChip (450K) array, respectively. The expression of TMEM176A was inversely associated with promoter region methylation (P<0.0001; Fig. 1C, D).
Figure 1.
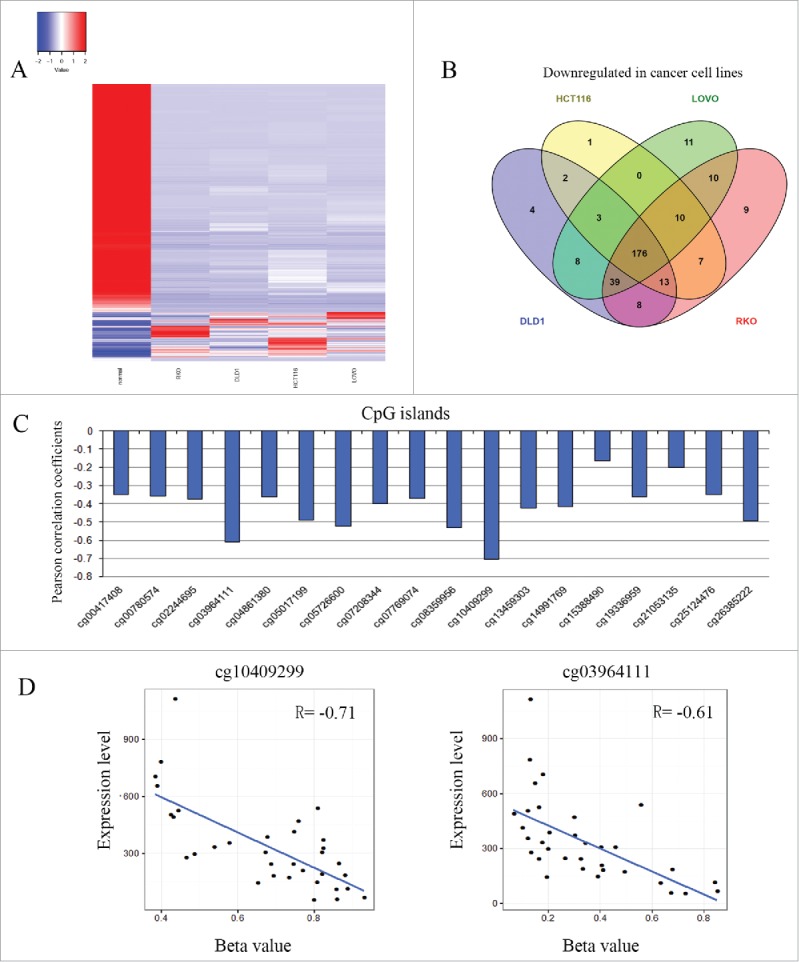
Identification of TMEM176A in human CRC and correlation between TMEM176A methylation and expression in CRC. (A) Heatmaps for the Z-scores of each differentially expressed gene's RPKM in each different cell line (Colon normal, RKO, DLD1, HCT116, LOVO). (B) Graph showing numbers of overlapping downregulated genes among RKO, DLD1, HCT116, and LOVO cancer cell lines. (C) Pearson correlation coefficient between TMEM176A methylation and expression at each CpG site. (D) Scatter-plots showing the methylation status of the 4th (4+, cg03964111) and 11th (11+, cg10409299) CpG sites, which is correlated with loss of/reduced TMEM176A expression in 34 cases of colorectal cancer (P<0.0001).
The expression of TMEM176A is regulated by promoter region methylation in colorectal cancer
We determined the expression level of TMEM176A in colorectal cancer cells (LOVO, RKO, DLD1, HT29, HCT116, SW480, SW620, LS180) by semiquantitative RT-PCR. TMEM176A expression was detected in LS180 and SW620 cells. Expression of TMEM176A was lost in LOVO, HCT116, RKO, and DLD1 cells, and reduced in HT29 and SW480 cells (Fig. 2A). Promoter region methylation was detected by methylation specific PCR (MSP). Complete methylation was found in LOVO, HCT116, RKO, and DLD1 cells, unmethylation was found in LS180 and SW620 cells, and partial methylation was found in HT29 and SW480 cells (Fig. 2B). Promoter region methylation was correlated with loss of/reduced TMEM176A expression. To further validate the efficiency of MSP primers and the methylation density in the promoter region, sodium bisulfite sequencing (BSSQ) was performed in RKO, DLD1, and SW620 cell lines. MSP results correlated with BSSQ results (Fig. 2B). Colorectal cancer cells were treated with 5-aza-2′-deoxycytidine (5-aza-dC), a DNA methylation transferase inhibitor. Upon 5-aza-dC treatment, restoration of TMEM176A expression was observed in LOVO, DLD1, RKO, and HCT116 cells, and increased expression of TMEM176A was found in HT29 and SW480 cells. No changes in TMEM176A expression were detected in LS180 and SW620 cells (Fig. 2A). These results suggest that the expression of TMEM176A is regulated by promoter region methylation in colorectal cancer.
Figure 2.
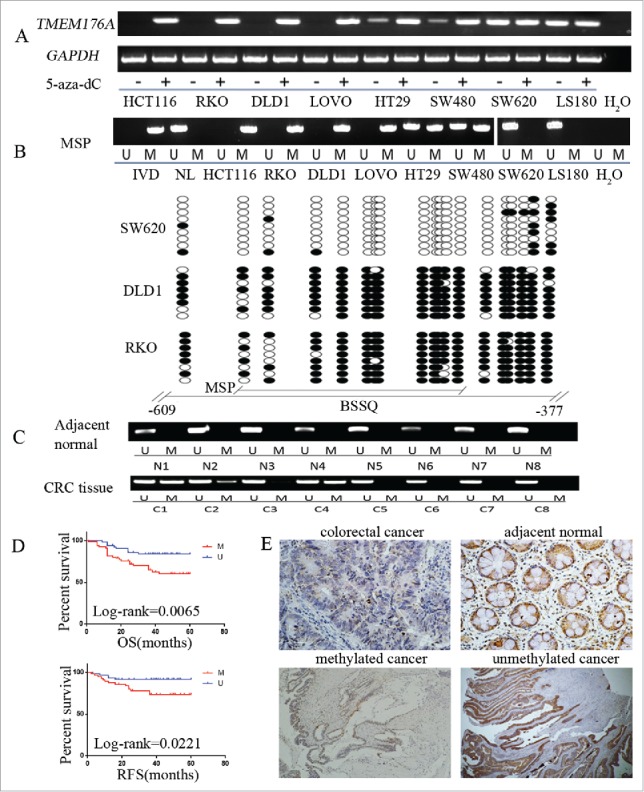
TMEM176A expression is regulated by promoter region methylation in colorectal cancer. (A) Expression of TMEM176A was detected by semiquantitative RT-PCR in 8 CRC cell lines. 5-aza-dC: 5-aza-2′-deoxycytidine. (−): untreated. (+): 5-aza-dC treated. GAPDH was used as internal control. (B) Upper: TMEM176A MSP results in CRC cell lines. IVD: in vitro methylated DNA. NL: normal lymphocyte DNA. M: methylated alleles. U: unmethylated alleles. Lower: BSSQ of TMEM176A promoter region (−609 bp to −377 bp) in RKO, DLD1, and SW620 cell lines. The region of CpG islands studied by MSP is indicated by a horizontal line marked with MSP. The region of CpG islands studied by BSSQ is indicated by a horizontal line marked with BSSQ. Filled circle: methylated CpG site. Open circle: unmethylated CpG site. (C) Representative MSP results of TMEM176A in normal colorectal mucosa (N) and primary colorectal cancer tissues (C). (D) TMEM176A methylation status is associated with RFS and OS of CRC patients. (E) IHC results of TMEM176A expression in colorectal cancer and adjacent tissue samples (200 ×, upper), and the expression of TMEM176A in unmethylated and methylated colorectal cancer (100 ×, lower).
TMEM176A is frequently methylated in human primary colorectal cancer, and methylation of TMEM176A is an independent prognostic factor for 5-year overall survival
To analyze the methylation status of TMEM176A in colorectal cancer, 130 cases of primary human colorectal cancer samples and 8 cases of normal colorectal mucosa were used. TMEM176A was methylated in 50.77% (66/130) of colorectal cancer samples, while no methylation was found in normal colorectal mucosa (Fig. 2C). BRAF and KRAS genes were mutated in 3.3% (4/123) and 30.1% (37/123) of these patients, respectively. CpG island methylator phenotype (CIMP) (+) was found in 12.5% (15/120) of CRC patient samples. Methylation of TMEM176A was associated with lymph node metastasis, reduced 5-year overall survival (OS) and 5-year relapse-free survival (RFS) (all P<0.05), while no association was revealed between TMEM176A methylation and age, gender, location, differentiation, tumor stage, CIMP, BRAF, and KRAS mutations (Fig. 2D; Table 1). Further study revealed that methylation of TMEM176A was an independent prognostic factor for 5-year OS after adjusting for tumor differentiation and N stage, according to Cox proportional hazards model analysis (P<0.05; Table 2).
Table 1.
Clinicopathological features and TMEM176A methylation status in 130 patients with colorectal cancer.
|
TMEM176A methylation status |
||||
|---|---|---|---|---|
| Clinical Factor | Methylated (n = 66) | Unmethylated (n = 64) | P-value* | |
| Age | ||||
| <50 | 29 | 16 | 13 | 0.591 |
| ≥50 | 101 | 50 | 51 | |
| Gender | ||||
| M | 86 | 40 | 46 | 0.175 |
| F | 44 | 26 | 18 | |
| Tumor location | ||||
| Right-sided colon | 38 | 22 | 16 | 0.527 |
| Left-sided colon | 44 | 20 | 24 | |
| Rectum | 48 | 24 | 24 | |
| Differentiation | ||||
| Poor | 33 | 18 | 15 | 0.615 |
| Moderate and Well | 97 | 48 | 49 | |
| Tumor size (cm) | ||||
| <5 cm | 66 | 31 | 35 | 0.379 |
| ≥5 cm | 64 | 35 | 29 | |
| Lymphatic metastasis | ||||
| Negative | 75 | 32 | 43 | 0.031 |
| Positive | 55 | 34 | 21 | |
| T stage | ||||
| I-II | 70 | 30 | 40 | 0.051 |
| III-IV | 60 | 36 | 24 | |
| KRAS mutation | ||||
| Wild type | 86 | 43 | 43 | 0.891 |
| Mutation | 37 | 18 | 19 | |
| BRAF V600E | ||||
| Wild type | 119 | 59 | 60 | 1.000 |
| Mutation | 4 | 2 | 2 | |
| CIMP | ||||
| CIMP(−) | 105 | 50 | 55 | 0.535 |
| CIMP(+) | 15 | 9 | 6 | |
P values were obtained using the chi-square test. Significant difference (P<0.05) indicated in bold.
Table 2.
Univariate and multivariate analysis of TMEM176A methylation status with RFS or OS in colorectal cancer patients.
| OS |
RFS |
|||||||
|---|---|---|---|---|---|---|---|---|
| Univariate analysis |
Multivariate analysis |
Univariate analysis |
Multivariate analysis |
|||||
| HR(95% CI) | P | HR(95% CI) | P | HR(95% CI) | P | HR(95% CI) | P | |
| Age (<50 vs.≥50 y) | 1.085(0.472–2.492) | 0.848 | 1.102(0.480–2.531) | 0.819 | ||||
| Gender (male vs. female) | 1.023(0.506–2.067) | 0.95 | 1.065(0.527–2.152) | 0.861 | ||||
| Tumor location (proximal colon vs. distal colon or rectum) | 1.187(0.779–1.809) | 0.425 | 1.182(0.776–1.801) | 0.435 | ||||
| Tumor size (<5 cm vs. ≥5 cm) | 1.313(0.667–2.585) | 0.43 | 1.336(0.679–2.630) | 0.402 | ||||
| Differentiation (high or middle vs. low differentiation) | 0.381(0.193–0.751) | 0.005** | 0.549(0.269–1.122) | 0.100 | 0.384(0.194–0.758) | 0.006** | 0.554(0.273–1.124) | 0.102 |
| Pathologic T stage (T3–4 vs. T1–2) | 1.735(0.611–4.929) | 0.301 | 1.626(0.573–4.616) | 0.361 | ||||
| Pathologic N stage (N1–2 vs. N0) | 3.687(1.761–7.718) | 0.001** | 2.651(1.203–5.846) | 0.016* | 3.749(1.790–7.850) | 0.000** | 2.799(1.283–6.109) | 0.010** |
| TMEM176A (hypermethylation and unmethyaltion) | 2.646(1.265–5.536) | 0.01** | 2.160(1.021–4.568) | 0.044* | 2.503(1.197–5.236) | 0.015* | 2.014(0.953–4.256) | 0.067 |
| KRAS mutation (wild type vs. mutation) | 1.010(0.481–2.122) | 0.979 | 1.021(0.486–2.145) | 0.957 | ||||
| BRAF mutation (wild type vs. mutation) | 2.956(0.705–12.394) | 0.138 | 2.359(0.563–9.885) | 0.240 | ||||
| CIMP(−) and CIMP(+) | 0.986(0.67–1.451) | 0.943 | 0.97(0.659–1.429) | 0.878 | ||||
P<0.05.
P≤0.01.
TMEM176A expression was evaluated by immunohistochemistry (IHC) in 35 available matched colorectal cancer and adjacent tissue samples. TMEM176A staining was observed mainly in the cytoplasm and cell membranes of cells. Reduced TMEM176A expression was found in 20 cases of colorectal cancer and 2 cases of adjacent tissue samples. Expression of TMEM176A was significantly reduced in colorectal cancer compared with adjacent tissue samples (P<0.05; Fig. 2E). Among the 20 cases of cancer samples in which TMEM176A expression was reduced, 19 cases were methylated at the TMEM176A promoter. Reduced expression was associated with TMEM176A promoter region hypermethylation (P<0.05). These data suggest that TMEM176A is regulated by promoter region methylation in human primary colorectal cancer.
TMEM176A inhibits cell proliferation and induces apoptosis in CRC cells
MTT cell proliferation assay and colony formation assays were used to evaluate the effects of TMEM176A on cell proliferation. For the MTT assay, optical density (OD) values before vs. after restoration of TMEM176A expression were 0.609 ± 0.018 vs. 0.531 ± 0.002 in RKO cells, and 1.4484 ± 0.081 vs. 0.9731 ± 0.061 in DLD1 cells. OD values were significantly reduced after restoration of TMEM176A expression in CRC cells (P<0.05 for both cell lines). In SW620 cells (in which TMEM176A is highly expressed), the OD values were 0.582 ± 0.031 and 0.761 ± 0.037 before and after siRNA knockdown of TMEM176A, respectively (Fig. 3A). OD values increased significantly (P<0.05) upon knockdown of TMEM176A. Colony numbers were 97 ± 4 vs. 79 ± 7 in RKO cells and 75 ± 5 vs. 52 ± 2 in DLD1 cells before and after restoration of TMEM176A expression, respectively (Fig. 3B). Colony numbers were significantly reduced after re-expression of TMEM176A (both P<0.05). These results suggest that TMEM176A inhibits cell proliferation in CRC.
Figure 3.

TMEM176A inhibits cell proliferation, colony formation, migration, and invasion, and induces apoptosis in CRC cells. (A) Effect of TMEM176A on cell proliferation measured by the MTT assay. The experiment was repeated 3 times. *: P<0.05. NC: siRNA negative control. (B) Representative results of colony formation in TMEM176A unexpressing- and TMEM176A re-expressing-RKO and TMEM176A unexpressing- and TMEM176A re-expressing-DLD1 cells. The experiment was repeated 3 times. *: P<0.05. (C) Percentage of apoptotic cells before and after restoration of TMEM176A expression in RKO and DLD1 cells before and after knockdown of TMEM176A in SW620 cells. The experiment was repeated 3 times. *: P<0.05. (D) Transwell assay showing migration and invasion results before and after restoration of TMEM176A expression in RKO and DLD1 cells and before and after knockdown of TMEM176A in SW620 cells (200 ×). The experiment was repeated 3 times. (E) Expression levels of TMEM176A, cleaved caspase-3, MMP-2, MMP-7, and MMP-9 were detected by Western blot before and after restoration of TMEM176A expression in RKO and DLD1 cells and before and after knockdown of TMEM176A in SW620 cells.
The effects of TMEM176A on CRC cell apoptosis were analyzed by flow cytometry. The ratios of apoptotic cells in TMEM176A unexpressing- and TMEM176A re-expressing-cells were 1.81 ± 0.04 vs. 3.48 ± 0.16 in RKO, and 8.28 ± 1.59 vs. 16.12 ± 1.47 in DLD1. The ratio of apoptotic cells increased significantly (with P<0.05 for both cell lines; Fig. 3C) upon re-expression of TMEM176A. In SW620 cells (in which TMEM176A is highly expressed), the ratios of apoptotic cells before and after knockdown of TMEM176A by treatment with Staurosporine, were 33.76 ± 0.74% and 24.83 ± 1.60%, respectively. The ratio of apoptotic cells decreased significantly after knockdown of TMEM176A (P<0.05). These results suggest that TMEM176A induces apoptosis in human CRC cells. As shown in Fig. 3E, the levels of cleaved caspase-3 increased after re-expression of TMEM176A in DLD1 and RKO cells, and decreased after knockdown of TMEM176A in SW620 cells, further suggesting that TMEM176A induces apoptosis in CRC cells.
TMEM176A inhibits colorectal cancer cell migration and invasion
As methylation of TMEM176A was associated with lymph node metastasis in primary colorectal cancer, we explored the effects of TMEM176A on cell migration and invasion in CRC cells. The numbers of migrated cells were 100 ± 8 vs. 40 ± 4 in RKO, and 175 ± 18 vs. 68 ± 17 in DLD1, before and after re-expression of TMEM176A. The numbers of invasive cells were 51 ± 6 vs. 25 ± 3 in RKO, and 149 ± 20 vs. 50 ± 9 in DLD1, before and after re-expression of TMEM176A. Invasive and migrated cell numbers decreased significantly (P<0.05) upon re-expression of TMEM176A. In TMEM176A-highly expressing SW620 cells, the numbers of migrated cells were 29 ± 3 vs. 56 ± 7, and the numbers of invasive cells were 20 ± 4 vs. 49 ± 4, before and after knockdown of TMEM176A. Migrated and invasive cells increased significantly after knockdown of TMEM176A in SW620 cells (P<0.05; Fig. 3D).
The mechanisms of function of TMEM176A on cell migration and invasion were further explored by detecting the expression levels of MMP2, MMP7, and MMP9 in RKO, DLD1, and SW620 cells. The expression levels of MMP2, MMP7, and MMP9 were reduced after re-expression of TMEM176A in RKO and DLD1 cells, while their expression levels increased after knockdown of TMEM176A in SW620 cells (Fig. 3E). These results further suggest that TMEM176A suppresses cell migration and invasion in CRC.
TMEM176A suppresses DLD1 cell xenograft growth in mice
To further study the function of TMEM176A in colorectal cancer, TMEM176A unexpressing-DLD1 and TMEM176A re-expressing-DLD1 cell xenograft mice models were used (Fig. 4A). Tumor volumes were 842.823 ± 223.5 vs. 305.649 ± 146.8 mm3 and tumor weights were 354.9 ± 163.8 vs. 153.9 ± 58.0 mg, respectively (Fig. 4B and C). Tumor volume and weight were significantly reduced after re-expression of TMEM176A (both P<0.05). As shown in Fig. 4D, the expression levels of TMEM176A, MMP2, MMP7, MMP9, and cleaved caspase-3 in xenografts were examined by IHC. The expression levels of MMP2, MMP7, and MMP9 decreased and the expression of cleaved caspase-3 increased in TMEM176A re-expressing-DLD1 cell xenografts compared with parental cells.
Figure 4.
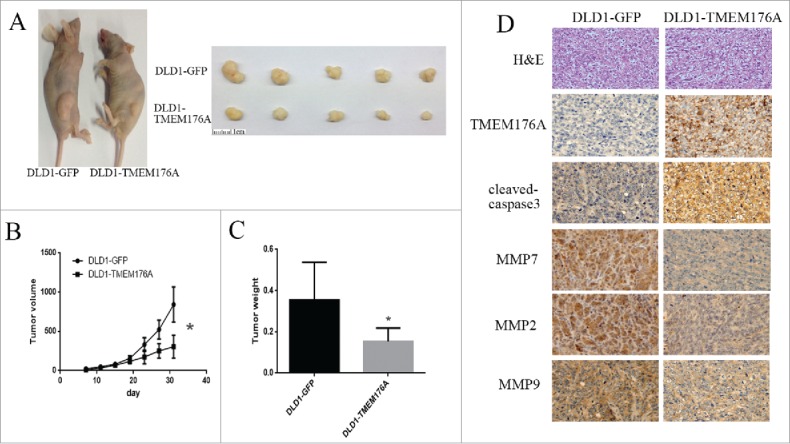
TMEM176A suppresses CRC cell xenograft tumor growth in mice. (A) Representative tumors from TMEM176A unexpressing- and TMEM176A re-expressing-DLD1 cell xenografts. (B) Tumor growth curves of TMEM176A unexpressing- and TMEM176A re-expressing-DLD1 cells. *: P<0.05. (C) Tumor weights in nude mice at the 31st day after inoculation of TMEM176A for TMEM176A unexpressing- and TMEM176A re-expressing-DLD1 cells. Bars: mean of 5 mice. *: P<0.05. (D) Images of hematoxylin and eosin staining show tumors from TMEM176A unexpressing- and TMEM176A re-expressing-DLD1 xenograft mice. IHC staining reveals the expression levels of TMEM176A, MMP2, MMP7, MMP9, and cleaved caspase-3 in TMEM176A unexpressing- and TMEM176A re-expressing-DLD1 cell xenografts (200 ×).
Discussion
Previous studies on TMEM176A mainly focused on its function in development and the immune system.14-16 Tmem176a is highly expressed in retinoid-related orphan receptor γt (RORγt)+ lymphocytes in mice.17 Tmem176a induces the expression of co-stimulatory molecules, such as cluster of differentiation (CD)80, CD86, and CD40, and is involved in the maintenance of the immature state of dendritic cells in mice.11 Significant upregulation of TMEM176A was detected in whole-blood samples of patients with multiple sclerosis.18 By screening the expression of TMEM176A in a few cases of various cancers by a fluorometric technique using tissue microarray, Cuajungcoa et al. found no TMEM176A expression differences between CRC and normal tissue samples.19 To date, no functional studies have been reported in human cancer.
Using RNA-seq, we found that the expression of TMEM176A was reduced in human colorectal cancer. It was reported that Tmem176a and its homolog, Tmem176b, are located within the same genomic locus in opposite directions and they are tightly co-regulated in various tissues in mice.15,20,21 While Drujont et al. found that the expression of Tmem176a was increased in Tmem176b−/− cells compared with wild type cells in Th17 cells in mice,17 no association was found between TMEM176A and TMEM176B expression in human CRC cells (data not shown) in our study. Further analysis suggested that TMEM176A is frequently methylated in human primary CRC and that the expression of TMEM176A is regulated by promoter region methylation. Methylation of TMEM176A may serve as a detection marker for human CRC. TMEM176A methylation is associated with CRC metastasis, and is an independent prognostic marker for 5-year OS. Our functional studies suggest that TMEM176A induces cell apoptosis, inhibits CRC metastasis, and suppresses CRC cell growth both in vitro and in vivo. In TCGA database, the expression levels of TMEM176A were reduced in CRC, and an inverse association was found between TMEM176A expression and promoter region methylation, according to 450K methylation array data. These data further support our findings.
In conclusion, TMEM176A is frequently methylated in human colorectal cancer, and methylation of TMEM176A is associated with CRC metastasis. Our study suggests that the expression of TMEM176A is regulated by promoter region methylation. TMEM176A methylation is an independent prognostic marker for 5-year OS, and may serve as a tumor suppressor in CRC.
Materials and methods
Human tissue samples and cell lines
Eight human CRC cell lines (LOVO, DLD1, RKO, HT29, HCT116, SW480, SW620, and LS180) were examined in this study. All CRC cell lines were previously established from primary colorectal cancer and maintained in 90% RPMI 1640 (Invitrogen, CA) supplemented with 10% fetal bovine serum and antibiotics. A total of 130 cases of primary colorectal cancer were surgically resected and 15 cases of normal colorectal mucosa were collected from noncancerous patients by biopsy under endoscopy. All tissues were collected from the Chinese PLA General Hospital according to the approved guidelines of the Chinese PLA General Hospital's Institutional Review Board and stored at −80°C.
Five-aza-2′-deoxycytidine treatment
CRC cell lines were split to a low density (30% confluence) 12 h before treatment. Cells were treated with 5-aza-dC (Sigma, St. Louis, MO) at a concentration of 2 μM in growth medium, which was exchanged every 24 h for a total 96 h treatment. At the end of the treatment course, RNA was isolated as described below.
RNA preparation and RNA-seq
Total RNA was prepared from CRC cells and tissue samples using Trizol reagent (Life Technologies, Gaithersburg, MD). Agarose gel electrophoresis and spectrophotometric analysis were used to evaluate RNA quality and quantity. RNA was cleaved into short fragments upon mixing with fragmentation buffer. cDNA was then synthesized using the RNA fragments as templates. Short fragments were purified and resolved with elution buffer for end reparation and single nucleotide adenine addition. After that, the short fragments were connected with adapters, and the suitable fragments were selected as templates for PCR amplification. Lastly, the library was sequenced using an Illumina HiSeqTM 2000 (BGI Tech, Shenzhen, China).
Differential expression analysis
Mapped reads were counted to Gencode.v19 annotated protein coding genes with gfold V1.1.4 count function. Differential gene expression analyses between any conditions in RKO, DLD1, HCT116, LOVO, and the normal colon were performed with the gfold V1.1.4 diff function. Fold change was used to measure the expression variations between different conditions. The differential expression cutoff was set at P<0.01 and fold change >2. Heatmaps for the Z-score of each differentially expressed gene's reads per kilobase per million mapped reads (RPKM) for the 5 cell lines (normal colon mucosa, RKO, DLD1, HCT116, LOVO) were generated with heamap.2 function in gplots package.
Semiquantitative RT-PCR
Semiquantitative RT-PCR was performed as described previously.22 RT-PCR primers for TMEM176A were as follows: 5′-AGAATGTTCCCAACCAAAGGGA-3′ (F); 5′-TGGGGAAGGGGTGTAAGGAAT-3′ (R).
DNA extraction, KRAS and BRAF mutation detection, methylation-specific PCR, and bisulfite sequencing
Genomic DNA from CRC cells and tissue samples was extracted by the proteinase K method. KRAS codons 12 and 13 and BRAF codon 600 were amplified by PCR and sequenced according to previous reports.23,24 The primer sequences are listed in Table S1. MSP and BSSQ were performed as described previously.25,26 CpG island methylator phenotype (CIMP) was analyzed using the marker panel according to a report from Ogino et al.27 CIMP (+) was defined as ≥ 2 of 4 markers (CACNA1G, IGF2, MLH1, RUNX3) methylated in CRC.
Immunohistochemistry
IHC staining was performed on 4 μm thick serial sections derived from formal dehyde-fixed paraffin blocks. Rabbit polyclonal antibody against TMEM176A (Sigma, St. Louis, MO) was diluted at 1:50. IHC was performed and evaluated as described previously.26
Plasmid Construction and Transfection
The TMEM176A CDS (coding DNA sequence) region was amplified by RT-PCR. The primers were as follows: 5′-CTTAGGATCCGCCACCATGGGAACAGCCGAC-3′ (F) and 5′- ACTTAGTCGACCTAGATTCCACTCACTTCC-3′ (R). For transient transfection, TMEM176A CDS region was cloned into the pcDNA3.1(+) plasmid (Era Biotech, Shanghai, China). Transfection was performed according to the manufacturer's instructions using Lipofectamine 2000 (Invitrogen, CA).
For stable transfection, TMEM176A expressing lentiviral or GFP control vectors were packaged using the ViraPower™ lentiviral expression system (Invitrogen). Lentivirus was added to the growth medium of DLD1 and RKO cells, and cells stably expressing TMEM176A and control cells were selected by blasticidin (Invitrogen) at a concentration of 5 μg/ml.
The selected siRNAs targeting TMEM176A sequences were as follows: sense 5′-CUGUACUGCUGGAGAAUGUTT-3′ and antisense 5′-ACAUUCUUCAGCAGUACAGTT-3′. Negative control sequences were: sense 5′-ACAUUCUCCAGCAGUACAGTT-3′ and antisense 5′-ACGUGACACGUUCGGAGAATT-3′.
Colony formation assay
Cells were plated into 3.5 cm dishes and cultured for 2 weeks. Cells were then fixed with 75% ethanol for 30 min, stained with 0.2% crystal violet (Beyotime, Nanjing, China) for 20 min, and counted.
Cell viability detection
Cells were plated into 96-well plates at a density of 3 × 103 cells/well, and cell viability was measured at 24, 48, and 72 h using the MTT assay kit (Promega, Madison, Wisconsin, USA), according to the company's instructions. Absorbance was measured on a microplate reader (Thermo Multiskan MK3) at a wavelength of 490 nm.
Flow cytometry analysis of apoptosis
For apoptosis analysis, the Annexin V-PI Apoptosis Detection Kit (KeyGen Biotechnology, China) was used according to manufacturer's instructions. Each sample was analyzed by flow cytometry with a FACScan Flow Cytometer (Becton-Dickinson Biosciences, Mansfield). TMEM176A-highly expressing SW620 cells were treated with Staurosporine at 100 ng/mL for 24 h.
Transwell assay for migration
The effect of TMEM176A on cell migration was detected by the Transwell Assay System (Corning, High Wyombe, UK). Cells were harvested and suspended in serum free medium and then placed into the upper well at a concentration of 2 × 105 cells/200 µl. The complete medium containing 20% fetal bovine serum was placed into the lower well (600 µl). The chamber was incubated for 24 h. The cells remaining on the upper surface were scraped gently and washed with PBS 3 times. The cells that migrated to the lower surface of the membrane were stained with crystal violet and counted in 3 independent high-power fields (200X).
Transwell assay for invasion
Cells (2 × 105) were suspended in 200 µl of serum-free medium and loaded onto the upper compartment of an invasion chamber containing a polycarbonate membrane with an 8 µm pore size coated with a layer of extracellular matrix (ECM; Matrigel™, BD, NJ). After 48 h incubation, the invasive cells that migrated through the ECM layer to the complete medium in the lower compartment were stained with crystal violet and the numbers of invasive cells were counted in 3 independent high-power fields (200X).
Western blot
Western blot was performed as described previously.28 Antibodies were diluted according to manufacturer's instructions. Primary antibodies were as follows: anti-TMEM176A (Sigma, St. Louis, MO), anti-cleaved caspase-3 (Cell Signaling Technology), anti-MMP2 (Bioworld Tech., MN, USA), anti-MMP7 (Bioworld Tech.), anti-MMP9 (Bioworld Tech.) and anti-β-actin (Beyotime Biotech, China).
Effect of TMEM176A on DLD1 cell xenograft growth
TMEM176A re-expressed and unexpressed DLD1 cells (3 × 106 cells in 0.1 ml phosphate-buffered saline) were subcutaneously injected into the dorsal flank of 5-week-old female BABL/c nude mice (n = 5 each group). Tumor sizes were measured 7 d after inoculation for 4 weeks every 4 d. Tumor volumes were determined with the following formula: tumor volume (mm3) = [length (mm)] × [width (mm)]2 / 2. All procedures were approved by the Animal Ethics Committee of the Chinese PLA General Hospital.
Statistical analysis
Statistical analysis was performed using SPSS 17.0 software (SPSS, Chicago, IL). Chi-square or Fisher's exact tests were used to evaluate the relationship between methylation status and clinicopathological characteristics. The 2-tailed independent samples t-test was applied to determine the statistical significance of the differences between the 2 experimental groups. Survival rates were calculated by the Kaplan–Meier method, and differences in survival curves were evaluated using the log-rank test. Cox proportional hazards models were fit to determine independent associations of TMEM176A methylation with 5-year OS and 5-year relapse-free survival (RFS) outcomes. Two-sided tests were used to determine significance, and P<0.05 was considered statistically significant.
Ethics approval and consent to participate
This study was approved by the Chinese PLA General Hospital Institutional Ethical Review Board and informed consent was obtained from each patient. All animal studies were performed with the approval from the Institutional Animal Care and Use Committee of the Chinese PLA General Hospital.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Fundings
This work was supported by grants from the National Basic Research Program of China (973 Program No. 2012CB934002); National Key Scientific Instrument Special Program of China (Grant No.2011YQ03013405); National Science Foundation of China (NSFC No.81672318, U1604281, 8167100001, 81402345, 81230047, 81490753, 81121004); Beijing Science Foundation of China (BJSFC No.7171008); Translational Foundation of Chinese PLA General Hospital (2016ZHJJ-MS-GMZ).
References
- 1.Ferlay J, Shin H-R, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 2010; 127:2893-917; PMID:21351269; https://doi.org/ 10.1002/ijc.25516 [DOI] [PubMed] [Google Scholar]
- 2.Brenner H, Kloor M, Pox CP. Colorectal cancer. Lancet; 383:1490-502; https://doi.org/ 10.1016/S0140-6736(13)61649-9 [DOI] [PubMed] [Google Scholar]
- 3.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 1990; 61:759-67; PMID:2188735; https://doi.org/ 10.1016/0092-8674(90)90186-I [DOI] [PubMed] [Google Scholar]
- 4.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer Genome Landscapes. Science 2013; 339:1546-58; PMID:23539594; https://doi.org/ 10.1126/science.1235122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo M, Ren J, Brock MV, Herman JG, Carraway HE. Promoter methylation of HIN-1 in the progression to esophageal squamous cancer. Epigenetics 2008; 3:336-41; PMID:19098448; https://doi.org/ 10.4161/epi.3.6.7158 [DOI] [PubMed] [Google Scholar]
- 6.Guo M, Ren J, House MG, Qi Y, Brock MV, Herman JG. Accumulation of promoter methylation suggests epigenetic progression in squamous cell carcinoma of the esophagus. Clin Cancer Res 2006; 12:4515-22; PMID:16899597; https://doi.org/ 10.1158/1078-0432.CCR-05-2858 [DOI] [PubMed] [Google Scholar]
- 7.Riegman PH BL, Wang KK, Wink-Godschalk JC, Dinjens WN, Siersema PD, Tilanus HW, van Dekken H. Allelic imbalance of 7q32.3-q36.1 during tumorigenesis in barrett's esophagus. Cancer Res 2002; 62:1531-3; PMID:11888931 [PubMed] [Google Scholar]
- 8.Kimmel RR ZL, Nguyen D, Lee S, Aronszajn M, Cheng C, Troshin VP, Abrosimov A, Delrow J, Tuttle RM, Tsyb AF, Kopecky KJ, Davis S, Neiman PE. Microarray comparative genomic hybridizationreveals genome-wide patterns of DNA gains and losses in post-Chernobyl thyroid cancer. Radiat Res 2006; 166:519-31; PMID:16953671; https://doi.org/ 10.1667/RR0547.1 [DOI] [PubMed] [Google Scholar]
- 9.Griffin CS, Simpson PJ, Wilson CR, Thacker J. Mammalian recombination-repair genes XRCC2 and XRCC3 promote correct chromosome segregation. Nature Cell Biol 2000; 2:757-61; PMID:11025669; https://doi.org/ 10.1038/35036399 [DOI] [PubMed] [Google Scholar]
- 10.Paulikova S, Chmelarova M, Petera J, Palicka V, Paulik A. Hypermethylation of RAD51L3 and XRCC2 genes to predict late toxicity in chemoradiotherapy-treated cervical cancer patients. Folia Biol 2013; 59:240-5; PMID:24485306 [PubMed] [Google Scholar]
- 11.Liu-Chittenden Y, Jain M, Gaskins K, Wang S, Merino MJ, Kotian S, Kumar Gara S, Davis S, Zhang L, Kebebew E. RARRES2 functions as a tumor suppressor by promoting beta-catenin phosphorylation/degradation and inhibiting p38 phosphorylation in adrenocortical carcinoma. Oncogene 2017; https://doi.org/ 10.1038/onc.2016.497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alholle A, Brini AT, Gharanei S, Vaiyapuri S, Arrigoni E, Dallol A, Gentle D, Kishida T, Hiruma T, Avigad S, et al.. Functional epigenetic approach identifies frequently methylated genes in Ewing sarcoma. Epigenetics 2013; 8:1198-204; PMID:24005033; https://doi.org/ 10.4161/epi.26266 [DOI] [PubMed] [Google Scholar]
- 13.Wang Y, Han KJ, Pang XW, Vaughan HA, Qu W, Dong XY, Peng JR, Zhao HT, Rui JA, Leng XS, et al.. Large scale identification of human hepatocellular carcinoma-associated antigens by autoantibodies. Journal of immunology (Baltimore, Md: 1950) 2002; 169:1102-9; PMID:12097419; https://doi.org/ 10.4049/jimmunol.169.2.1102 [DOI] [PubMed] [Google Scholar]
- 14.Zuccolo J, Deng L, Unruh TL, Sanyal R, Bau JA, Storek J, Demetrick DJ, Luider JM, Auer-Grzesiak IA, Mansoor A, et al.. Expression of MS4A and TMEM176 Genes in Human B Lymphocytes. Front Immunol 2013; 4:195; PMID:23874341; https://doi.org/ 10.3389/fimmu.2013.00195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Condamine T, Le Texier L, Howie D, Lavault A, Hill M, Halary F, Cobbold S, Waldmann H, Cuturi MC, Chiffoleau E. Tmem176B and Tmem176A are associated with the immature state of dendritic cells. J Leukoc Biol 2010; 88:507-15; PMID:20501748; https://doi.org/ 10.1189/jlb.1109738 [DOI] [PubMed] [Google Scholar]
- 16.Grunin M, Hagbi-Levi S, Rinsky B, Smith Y, Chowers I. Transcriptome Analysis on Monocytes from Patients with Neovascular Age-Related Macular Degeneration. Sci Rep 2016; 6:29046; PMID:27374485; https://doi.org/ 10.1038/srep29046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Drujont L, Lemoine A, Moreau A, Bienvenu G, Lancien M, Cens T, Guillot F, Beriou G, Bouchet-Delbos L, Fehling HJ, et al.. RORgammat+ cells selectively express redundant cation channels linked to the Golgi apparatus. Sci Rep 2016; 6:23682; PMID:27009467; https://doi.org/ 10.1038/srep23682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nickles D, Chen HP, Li MM, Khankhanian P, Madireddy L, Caillier SJ, Santaniello A, Cree BA, Pelletier D, Hauser SL, et al.. Blood RNA profiling in a large cohort of multiple sclerosis patients and healthy controls. Hum Mol Genet 2013; 22:4194-205; PMID:23748426; https://doi.org/ 10.1093/hmg/ddt267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cuajungco MP, Podevin W, Valluri VK, Bui Q, Nguyen VH, Taylor K. Abnormal accumulation of human transmembrane (TMEM)-176A and 176B proteins is associated with cancer pathology. Acta Histochem 2012; 114:705-12; PMID:22244448; https://doi.org/ 10.1016/j.acthis.2011.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Louvet C, Chiffoleau E, Heslan M, Tesson L, Heslan JM, Brion R, Beriou G, Guillonneau C, Khalife J, Anegon I, et al.. Identification of a new member of the CD20/FcepsilonRIbeta family overexpressed in tolerated allografts. Am J Transplant 2005; 5:2143-53; PMID:16095493; https://doi.org/ 10.1111/j.1600-6143.2005.01007.x [DOI] [PubMed] [Google Scholar]
- 21.Lemoine A, Chauveau-Le Friec G, Langa F, Louvet C. Generation of a Double KO Mouse by Simultaneous Targeting of the Neighboring Genes Tmem176a and Tmem176b Using CRISPR/Cas9: Key Steps from Design to Genotyping. Journal of genetics and genomics = Yi chuan xue bao 2016; 43:329-40; PMID:27234594; https://doi.org/ 10.1016/j.jgg.2016.04.004 [DOI] [PubMed] [Google Scholar]
- 22.Yan W, Wu K, Herman JG, Brock MV, Fuks F, Yang L, Zhu H, Li Y, Yang Y, Guo M. Epigenetic regulation of DACH1, a novel Wnt signaling component in colorectal cancer. Epigenetics 2013; 8:1373-83; PMID:24149323; https://doi.org/ 10.4161/epi.26781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Samowitz WS, Sweeney C, Herrick J, Albertsen H, Levin TR, Murtaugh MA, Wolff RK, Slattery ML. Poor survival associated with the BRAF V600E mutation in microsatellite-stable colon cancers. Cancer Res 2005; 65:6063-9; PMID:16024606; https://doi.org/ 10.1158/0008-5472.CAN-05-0404 [DOI] [PubMed] [Google Scholar]
- 24.Morandi L. DETECTION OF KRAS MUTATION IN EXON 2 BY ALLELE SPECIFIC REAL TIME QUANTITATIVE PCR (AS-QPCR). WO, 2011. [Google Scholar]
- 25.Jia Y, Yang Y, Brock MV, Zhan Q, Herman JG, Guo M. Epigenetic regulation of DACT2, a key component of the Wnt signalling pathway in human lung cancer. J Pathol 2013; 230:194-204; PMID:22806826; https://doi.org/ 10.1002/path.4073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jia Y, Yang Y, Liu S, Herman JG, Lu F, Guo M. SOX17 antagonizes WNT/beta-catenin signaling pathway in hepatocellular carcinoma. Epigenetics 2010; 5:743-9; PMID:20716954; https://doi.org/ 10.4161/epi.5.8.13104 [DOI] [PubMed] [Google Scholar]
- 27.Ogino S, Kawasaki T, Kirkner GJ, Kraft P, Loda M, Fuchs CS. Evaluation of markers for CpG island methylator phenotype (CIMP) in colorectal cancer by a large population-based sample. J Mol Diagn: JMD 2007; 9:305-14; PMID:17591929; https://doi.org/ 10.2353/jmoldx.2007.060170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yan W, Wu K, Herman JG, Brock MV, Zhou Y, Lu Y, Zhang Z, Yang Y, Guo M. Epigenetic silencing of DACH1 induces the invasion and metastasis of gastric cancer by activating TGF-beta signalling. J Cell Mol Med 2014; 18:2499-511; PMID:24912879; https://doi.org/ 10.1111/jcmm.12325 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.