

Abstract
Human tRNALys3 serves as the primer for reverse transcription in human immunodeficiency virus type-1 (HIV-1) and anneals to the complementary primer binding site (PBS) in the genome. All tRNALys isoacceptors interact with human lysyl-tRNA synthetase (hLysRS) and are selectively packaged into virions. tRNALys3 must be released from hLysRS in order to anneal to the PBS, and this process is proposed to be facilitated by the interaction of hLysRS with a tRNA-like element (TLE) first identified in the HIV-1 5′-untranslated region (5′-UTR) of the subtype B NL4-3 virus. However, a significant subset of HIV-1 strains represented by the MAL isolate possess a different secondary structure in this region of the genome. Thus, to establish the conservation of this mechanism for primer targeting and release, we investigated the subtype A-like 5′-UTR of the MAL isolate. hLysRS bound to a 229-nt MAL RNA containing the PBS domain with high affinity (Kd = 47 nM), and to a 98-nt truncated construct with ∼10-fold reduced affinity. These results resemble previous studies using analogous NL4-3-derived RNAs. However, in contrast to studies with NL4-3, no binding was observed to smaller stem–loop elements within the MAL PBS domain. The tertiary structure of the 98-nt construct was analyzed using small-angle X-ray scattering, revealing remarkable global structural similarity to the corresponding NL4-3 PBS/TLE region. These results suggest that the tRNA-like structure within the 5′-UTR is conserved across distinct HIV-1 subtypes and that hLysRS recognition of the MAL isolate is likely not conferred by specific sequence elements but by 3D structure.
Keywords: HIV-1, primer binding site, tRNA-like element, human lysyl-tRNA synthetase, SAXS
INTRODUCTION
Human immunodeficiency virus type 1 (HIV-1) is a lentivirus that belongs to the Retroviridae family of viruses (Sonigo et al. 1985; Sharp and Hahn 2010; Thormar 2013). Like all retroviruses, HIV-1 uses a host tRNA to prime reverse transcription of its genomic RNA (gRNA) into proviral DNA (Telesnitsky and Goff 1997; Frankel and Young 1998; Freed 2001; Ganser-Pornillos et al. 2008). The specific reverse transcription primer in the case of HIV-1 is tRNALys3 (Ratner et al. 1985; Wain-Hobson et al. 1985; Litvak et al. 1994; Marquet et al. 1995). Human tRNALys3 and the other tRNALys isoacceptor, tRNALys1,2, are selectively packaged into HIV-1 virions (Jiang et al. 1993; Huang et al. 1994; Li et al. 1996). While tRNALys isoacceptors constitute 5%–6% of the total low-molecular-weight RNA population in the cytoplasm of the infected cell, this fraction increases to 50%–60% in the virion, indicating selective incorporation (Mak et al. 1994; Kleiman et al. 2010). Although both isoacceptors are packaged (∼20 molecules per virion), only tRNALys3 primes reverse transcription, as its 3′-18 nucleotides (nt) are perfectly complementary to the primer binding site (PBS) located in the 5′-untranslated region (UTR) of the HIV-1 genome (Jiang et al. 1993; Kleiman 2002).
To fulfill their canonical role in translation, tRNALys isoacceptors must be specifically recognized and aminoacylated by human lysyl-tRNA synthetase (hLysRS) (Kleiman 2002). Approximately 20–25 molecules of hLysRS are also packaged into HIV-1 virions via specific interactions with HIV-1 Gag (Cen et al. 2001, 2002; Javanbakht et al. 2003; Kovaleski et al. 2006, 2007; Dewan et al. 2014). Knockdown of endogenous hLysRS by siRNA results in reduced packaging of tRNALys, while expression of exogenous hLysRS leads to increased virus-associated tRNALys, suggesting that hLysRS is the limiting factor for tRNALys packaging (Gabor et al. 2002; Guo et al. 2003). hLysRS mutants with impaired tRNALys binding properties do not enhance primer packaging; however, hLysRS mutants with normal tRNALys binding but reduced aminoacylation activity are still able to package tRNALys at the level of wild-type (WT) hLysRS (Javanbakht et al. 2002; Cen et al. 2004). Furthermore, tRNALys3 anticodon mutations that are known to significantly weaken LysRS binding affinity, reduce virus-associated tRNALys3 levels, indicating that the anticodon plays a major role in primer packaging by allowing efficient interaction with hLysRS (Javanbakht et al. 2002).
The tRNALys:hLysRS complex is packaged into the virion via interactions with the viral Gag and GagPol polyproteins (Kleiman and Cen 2004; Kleiman et al. 2010). Gag and GagPol are large precursor proteins, which are cleaved by the viral protease to yield the structural proteins matrix (MA), capsid (CA), and nucleocapsid (NC), and the viral enzymes protease (PR), reverse transcriptase (RT), and integrase (IN) (Frankel and Young 1998; Kleiman and Cen 2004). Gag alone can package hLysRS independent of tRNALys or GagPol, but the incorporation of tRNALys into virions requires GagPol (Mak et al. 1994; Cen et al. 2001). The C-terminal domain of CA within Gag interacts with the catalytic domain of hLysRS, and the RT domain of GagPol is required for efficient packaging of tRNALys3 (Mak et al. 1994, 1997; Khorchid et al. 2000).
The HIV-1 genome is large (∼9.1 kb) and highly structured (Watts et al. 2009), and the mechanism by which tRNALys3 is released from hLysRS and specifically targeted to the PBS is not well understood. Recent studies using the HIV-1 NL4-3 virus suggested that a tRNA-like element (TLE), located proximal to the PBS, binds preferentially to hLysRS, facilitating tRNALys3 release and annealing to the PBS. These studies revealed that the TLE mimics the U-rich anticodon loop of tRNALys3 and together with other nearby elements, effectively competes with the anticodon stem–loop of tRNALys3 for binding to hLysRS (Jones et al. 2013). Nuclear magnetic resonance (NMR) spectroscopy experiments showed that the anticodon-binding domain of hLysRS interacts with the TLE in a manner that is strikingly similar to its interaction with the anticodon stem–loop of tRNALys3 (Liu et al. 2016). In addition, a small-angle X-ray scattering (SAXS) study showed that the PBS/TLE domain and tRNALys3 displayed remarkable global structural similarity (Jones et al. 2014).
The studies identifying the TLE were all carried out with a subtype B HIV-1 virus, NL4-3. In contrast to NL4-3, the PBS domain of the MAL isolate, which has a subtype A-like sequence (Robertson et al. 1995; Gao et al. 1998) (http://hiv.lanl.gov), contains a 23-nt insertion 3′ to the PBS (Fig. 1). This insertion is known to alter the secondary structure of the PBS region, which is no longer predicted to contain an exposed U-rich loop (Baudin et al. 1993; Goldschmidt et al. 2004). It has been suggested that the NL4-3 virus is structurally and functionally representative of HIV-1 strains that do not contain the 23-nt insertion, while the MAL isolate is representative of all other HIV-1 strains that contain the insertion (Goldschmidt et al. 2004).
FIGURE 1.
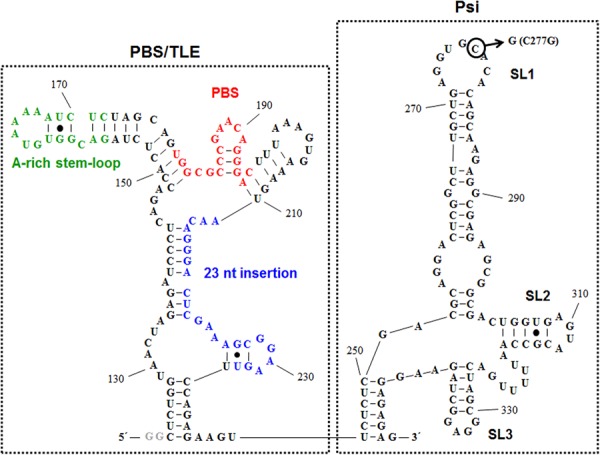
Secondary structure of MAL 123–349 predicted by mfold. The PBS/TLE (left) and Psi (ψ) (right) domains are shown in the dotted boxes. The A-rich stem–loop is shown in green, the PBS in red, and the 23-nt subtype A-specific insertion in blue. The C277G DIS mutant construct is indicated by the circled C. To facilitate in vitro transcription, two 5′ G's, not encoded by HIV-1, were added to this construct (gray).
In the present work, we aimed to determine if hLysRS binding to the HIV-1 MAL isolate PBS domain was maintained despite the lack of a U-rich loop element. We found that RNAs containing the MAL PBS domain bound to hLysRS with a similar affinity as previously measured for NL4-3. However, none of the individual stem–loops or short sequence motifs of MAL were essential for binding. SAXS analysis revealed a similar global conformation of the MAL and NL4-3 PBS domains, both of which resembled the L-shaped fold of a tRNA. Overall, these results demonstrate that these two representative HIV-1 strains share a conserved, structure-based mechanism for binding hLysRS and facilitating tRNA primer placement onto the PBS.
RESULTS
Presence of the PBS and Psi (ψ) domains in MAL RNA confer high-affinity binding to hLysRS
An N-terminally truncated hLysRS construct (hLysRSΔN65) was used in these studies to allow direct comparison to previous studies using the NL4-3 virus. The N terminus encodes a basic domain that confers nonspecific nucleic acid binding and contains a putative nuclear localization signal (Francin et al. 2002; Halwani et al. 2004). We have previously shown that both WT hLysRS and hLysRSΔN65 bind to the NL4-3 PBS/TLE domain (Jones et al. 2013). We confirmed that both of these proteins follow similar trends in their interactions with tRNALys3 and MAL-derived RNAs (Supplemental Fig. S1), suggesting that hLysRSΔN65 is suitable for our current study.
We first investigated binding of hLysRSΔN65 to a 229-nt-long MAL construct (nt 123–349) containing the PBS region and the ψ packaging signal (Fig. 1). Using fluorescence anisotropy (FA), we observed high-affinity binding of hLysRSΔN65 to WT MAL 123–349 (Kd = 47 ± 13 nM) (Table 1). A point mutation (C277G) was introduced in the SL1 dimerization initiation signal (DIS) stem–loop of MAL 123–349 to inhibit dimerization, as verified by native gel electrophoresis (Supplemental Fig. S2). MAL 123–349 C277G DIS mutant bound to hLysRSΔN65 with an approximately twofold reduced affinity relative to WT MAL 123–349 (Kd = 84 ± 21 nM). These results indicate that RNA dimerization does not significantly affect binding to hLysRSΔN65. To assess the contribution of the ψ packaging signal on hLysRS affinity, MAL 123–349 was truncated at the 3′-end to yield the 98-nt-long MAL 123–218 construct (Fig. 2). This RNA bound to hLysRSΔN65 with an ∼10-fold reduced affinity (Kd = 485 ± 133 nM) relative to MAL 123–349 (Table 1). The binding curves of these three RNAs are shown in comparison to binding to tRNALys3 in Figure 3A. These observations indicate that the presence of the ψ packaging signal enhances the MAL RNA-hLysRSΔN65 binding affinity to a similar extent as was previously observed in NL4-3 (Jones et al. 2013).
TABLE 1.
Apparent dissociation constants for MAL-derived RNAs binding to hLysRSΔN65 determined by fluorescence anisotropy
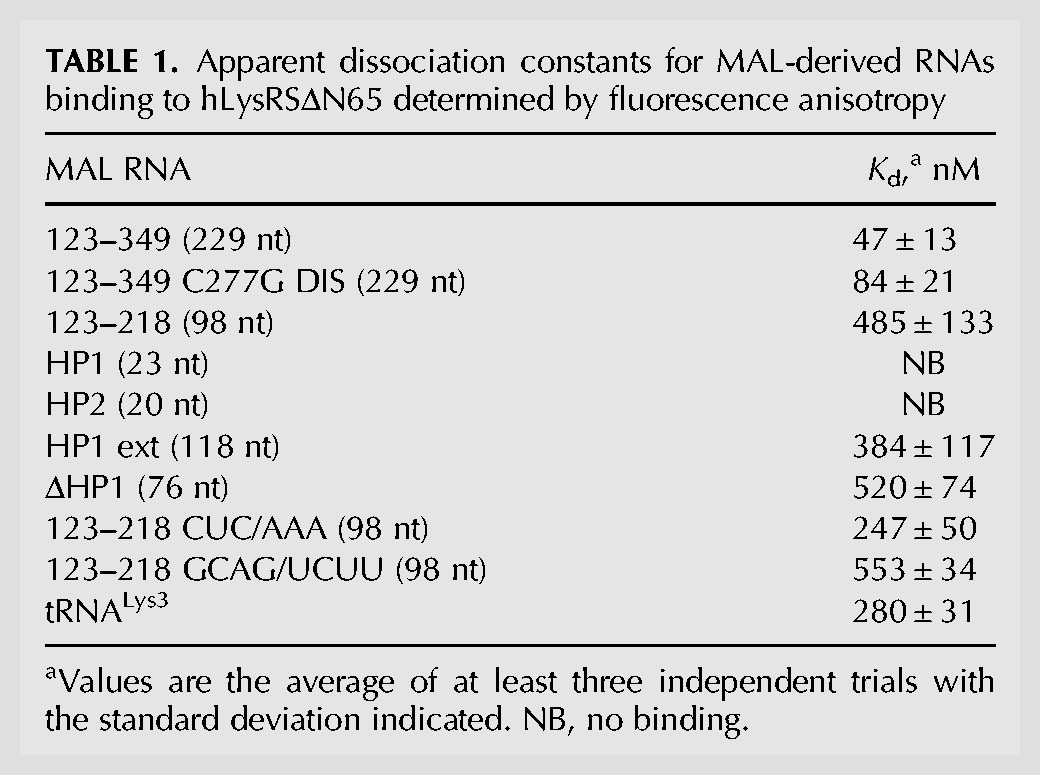
FIGURE 2.

Predicted secondary structure of MAL 123–218 (Goldschmidt et al. 2004). The PBS is highlighted in red, and the two 5′ G's shown in gray were added to this construct to facilitate in vitro transcription. (A–F) The various stem–loops and mutations used in the binding assays are indicated, as described in the text. In the case of HP1 and HP2, three additional closing G–C base pairs were added to stabilize the stems (gray). The apparent dissociation constants for the binding of each RNA construct to hLysRSΔN65 are indicated.
FIGURE 3.

Fluorescence anisotropy assay of (A) WT and C277G MAL 123–349, MAL 123–218, and tRNALys3 and (B) HP1, HP2, HPI ext, ΔHP1, MAL 123–218 CUC/AAA, and MAL 123–218 GCAG/UCUU binding to hLysRSΔN65. The points represent the average of three experiments, and the lines represent the best fit to a 1:1 binding equation, with standard deviations shown.
MAL PBS region is minimal domain required for binding to hLysRS
NMR experiments showed that the A-rich stem–loop of the MAL PBS region (Fig. 1) mimics the structure of the anticodon stem–loop of yeast tRNAPhe, as both loops form a U-turn motif with stacked nucleotides at the 3′-side of the loop (Puglisi and Puglisi 1998). We have also observed, using systematic evolution of ligands by exponential enrichment (SELEX) experiments, that hLysRSΔN65 prefers binding to aptamers with U-rich or A-rich single-stranded sequences (A Curtright, W Wang, K Musier-Forsyth, unpubl.). Collectively, these results suggested that the A-rich stem–loop could be the preferred site of hLysRS interaction in the MAL isolate. We prepared the same A-rich stem–loop RNA construct as was used in the NMR studies, referred to as hairpin 1 (HP1) (Fig. 2C), and tested its binding to hLysRSΔN65 (Fig. 3B). We observed that this construct did not bind to hLysRSΔN65 (Table 1), suggesting that this hairpin was not a crucial recognition element.
The MAL PBS region undergoes structural rearrangement after the annealing of tRNALys3 to the PBS, with the U-rich anticodon of tRNALys3 interacting with the A-rich loop of the MAL RNA (Goldschmidt et al. 2004). The only structural element unaltered in the PBS region is the stem–loop corresponding to hairpin 2 (HP2, Fig. 2F). This stem–loop is also U- and A-rich. However, binding assays revealed that this stem–loop also failed to bind to hLysRSΔN65 (Table 1; Fig. 3B).
An earlier alignment of HIV-1 sequences from various subtypes and circulating recombinant forms (CRFs) revealed high conservation of the UUU sequence in the loop of the NL4-3 TLE, which was essential for binding to hLysRS, and which mimicked the anticodon sequence of tRNALys3 (Jones et al. 2013). This sequence aligns with a base-paired 151CUC153 sequence in the MAL RNA (Fig. 2, green). To test the importance of this sequence in binding to hLysRSΔN65, we prepared two mutants in the context of MAL 123–218. In the CUC/AAA variant, the 151CUC153 sequence was mutated to AAA, creating an internal loop in the stem (Fig. 2D). In the GCAG/UCUU variant, compensatory mutations were made opposite to the AAA sequence to restore the base-pairing (Fig. 2E). The dissociation constants of these two variants were similar to that of WT MAL 123–218 (Kd ∼ 250–500 nM) (Table 1; Fig. 3B). This suggests that the CUC sequence is not important for binding to hLysRSΔN65, and that alteration of the secondary structure in this region does not significantly affect binding.
Next, we tested the importance of the entire A-rich stem–loop in binding to hLysRSΔN65 by preparing two mutants in the context of MAL 123-218. In one mutant, HP1 ext, the A-rich stem–loop was extended by 10 alternating A–U base pairs (Fig. 2A), while in ΔHP1 the A-rich stem–loop was replaced with a GAGA tetraloop (Fig. 2B). The binding assays indicated that extending or deleting the A-rich stem–loop did not significantly affect binding to hLysRSΔN65 (Table 1; Fig. 3B).
SAXS analysis of MAL 123–218 reveals structural similarity to the NL4-3 PBS/TLE domain and tRNALys3
We have previously shown using SAXS that the NL4-3 PBS/TLE domain mimics the overall shape of tRNALys3 (Jones et al. 2014). Therefore, we wanted to perform a similar analysis with the MAL PBS domain. MAL 123–218 possessed a similar size-exclusion chromatography (SEC) retention volume as NL4-3 PBS/TLE for its monomer and dimer species (Supplemental Fig. S3A). However, the elution profile showed that the MAL RNA has a greater tendency to oligomerize than its NL4-3 counterpart. MAL 123–218 monomer and dimer species were separable by SEC and remained monomeric and dimeric as visualized by native gel electrophoresis immediately following SEC purification (Supplemental Fig. S3B). However, within 24 h, these fractions began to reequilibrate into multiple oligomeric species. Thus, in-line SEC-SAXS was performed to isolate and analyze monomeric MAL 123–218, and compare it to the previously characterized NL4-3 PBS/TLE monomer (Jones et al. 2014).
The MAL 123–218 monomer peak generated good quality SAXS data (Fig. 4A). Inspection of the Guinier plot showed linearity at low scattering angles, indicating that the sample was free from aggregation or interparticle repulsions (Supplemental Fig. S4A). The Kratky plot showed that MAL 123–218 adopts a well-folded and elongated conformation in solution (Supplemental Fig. S4B). The radius of gyration (Rg), which represents the mass distribution about a particle's center of gravity, was calculated from both the slope of the Guinier plot (32 ± 5 Å) and the pair distance distribution [P(r)] function (32 ± 0.4 Å), a histogram of all interelectron distances in the RNA molecule (Table 2). The P(r) function (Fig. 4B) was also used to determine the maximum electron pair distance (Dmax) (104 Å) (Table 2). The Rg parameters are similar to those previously determined for NL4-3 PBS/TLE (∼34 Å), while the NL4-3 Dmax (118 Å) is modestly larger than that of the MAL RNA (Table 2). It should be noted that the NL4-3 PBS/TLE previously investigated is slightly longer (105 nt) than MAL 123-218 (98 nt).
FIGURE 4.

SAXS data for MAL 123–218 and comparison of the ab initio models of MAL 123–218, NL4-3 PBS/TLE, and tRNALys3. (A) Plot of intensity vs. momentum transfer for MAL 123–218. The open black circles indicate every fifth data point from the experimental SAXS curve and the black line represents the back-calculated scattering curve of the ab initio envelope. (B) The pair distance distribution [P(r)] function showing a histogram of all interelectron distances in MAL 123–218. The open black circles represent the P(r) transformation of the experimental data, while the solid black line represents the back-calculated P(r) function of the ab initio envelope. (C) The ab initio envelopes of NL4-3 PBS/TLE and tRNALys3 are shown in black. The confirmed TLE and PBS regions of the NL4-3 PBS/TLE RNA are indicated (Jones et al. 2014). The ab initio envelope of MAL 123–218 is shown in red. The envelopes were superimposed using the SUPCOMB program (Konarev et al. 2006), and in the case of tRNALys3, followed by manual adjustments to optimize the fit.
TABLE 2.
Statistics determined from analysis of the SAXS scattering curves and construction of ab initio envelopes using DAMMIN

Ab initio envelopes of MAL 123–218 were generated and the first round models possessed an average χ2 fit to the scattering data of 0.93 and a normalized spatial discrepancy (NSD) value between the 20 models of 0.83 (Table 2). NSD is a measure of the overall variance between 3D shapes (in the present case the ab initio envelopes), and values <1 are not generally considered to be significantly different (Burke and Butcher 2012). After the second round of modeling, the χ2 fit remained about the same (0.93), while the NSD value improved to 0.56 (Table 2). A second round of ab initio modeling was also performed using NL4-3 PBS/TLE SAXS data previously acquired (Jones et al. 2014), resulting in an improved χ2 fit and NSD value of 0.89 and 0.47, respectively (first round χ2 fit and NSD values were previously determined to be 0.90 and 0.76, respectively, Table 2).
The final, averaged ab initio envelopes for MAL 123–218 and NL4-3 PBS/TLE are shown separately and overlaid (Fig. 4C). Additional orientations of the overlay are shown in Supplemental Figure S5A. The NSD value comparing these two RNAs was calculated to be 0.75, indicating that they possess significantly similar shapes within the resolution of SAXS. Visual inspection of the envelopes shows a similar overall topology of the structures. Both possess an amorphous region on the right of the structure (in the orientation shown in Fig. 4C), a more well-defined tRNA-like structure on the left, and a short initiating helix shown at the bottom. Consistent with slightly smaller Rg and Dmax values, MAL 123–218 is not quite as extended as NL4-3 PBS/TLE. Finally, despite a clear similarity in overall shape, the putative MAL 123–218 TLE stem possesses a small protrusion that the NL4-3 PBS/TLE does not have (Supplemental Fig. S5A, red arrow). A comparison of the MAL 123–218 envelope and a SAXS envelope that we previously determined for tRNALys3 (Jones et al. 2014) was also made (Fig. 4C). Additional orientations of the overlay are shown in Supplemental Figure S5B. The positioning of the bend and overall size of the MAL RNA TLE stem is strikingly similar to those features in the tRNALys3 envelope. In conclusion, the SAXS models show that the MAL and NL4-3 PBS domains share a highly similar overall shape, and that the MAL PBS domain mimics the structure of tRNALys3.
DISCUSSION
In the present work, we have shown that the 5′-UTR derived from the HIV-1 MAL isolate binds to hLysRS with relatively high affinity despite the fact that it appears to lack a U-rich loop sequence, which is a major tRNALys identity element. The MAL PBS domain is required for binding to hLysRSΔN65, but the additional presence of the ψ domain enhances binding ∼10-fold (Table 1). A similar trend was previously observed in NL4-3 (Jones et al. 2013). The MAL PBS-ψ RNA (MAL 123–349) contains the DIS stem–loop, and is capable of dimerization; however, disruption of dimerization did not significantly affect binding to hLysRSΔN65 (Fig. 3A). Our previous studies showed that the TLE stem–loop present in the NL4-3 5′-UTR effectively competed with the anticodon stem–loop of tRNALys3 for binding to LysRS (Jones et al. 2013). Additionally, recent NMR chemical shift perturbation analysis showed that the anticodon-binding domain of hLysRS interacts with the NL4-3 TLE and the anticodon stem–loop of tRNALys3 in a remarkably similar manner (Liu et al. 2016). In contrast to the results obtained with the NL4-3 TLE, isolated stem–loops (HP1 and HP2) of MAL 123–218 did not bind to hLysRSΔN65 (Fig. 2). Furthermore, neither changes to the 151CUC153 sequence, which aligns with the UUU present in the TLE loop of NL4-3, nor extension or deletion of the A-rich stem–loop in MAL 123-218 led to a significant impact on binding (Fig. 2).
It is possible that the presence of an exposed U-rich stem–loop is important for hLysRS binding only in some HIV-1 strains. Multiple sequence alignment of the TLE region in various HIV-1 subtypes and CRFs showed that while the conservation of the UUU sequence is high, it is not universal, and these residues can be CUC (as is the case of MAL) or CUU (Jones et al. 2013). Furthermore, these residues are not always exposed in a hairpin loop as they would be in the tRNA anticodon loop, and in some subtypes they are base-paired in stem regions (Jones et al. 2013). Other conserved PBS domain sequence elements may play a role in hLysRS recognition, while not being sufficient to bind hLysRS alone. For example, the A-rich sequence found in a loop sequence in MAL is conserved in all HIV-1 strains (Isel et al. 1993; Goldschmidt et al. 2004) and has been proposed to interact with the annealed tRNALys3 primer anticodon sequence (Isel et al. 1993, 1995; Wilkinson et al. 2008). However, in the NL4-3 genome these residues are present in a stem bulge rather than in a hairpin loop, as is the case in MAL (Goldschmidt et al. 2004). SELEX experiments have shown that hLysRSΔN65 has a preference for binding to aptamers with U-rich or A-rich single-stranded sequences (A Curtright, W Wang, K Musier-Forsyth, unpubl.). Thus, PBS/TLE recognition may involve some combination of the U- and A-rich sequence elements present in different subtypes of HIV-1. Another possible explanation for the discrepancy between the observed high-affinity binding of hLysRSΔN65 with the MAL PBS domain, and the lack of binding observed with HP1 and HP2 is differential exposure of residues important for recognition in the different RNA constructs. For example, alterations to the secondary structure of HP1 and/or HP2 may occur in the context of the full MAL 123–218 RNA that expose A- or U-rich sequences in these hairpins that are critical for LysRS binding. Alternatively, critical RNA tertiary interactions may occur in the MAL 123–218 construct that cannot form in the individual hairpins.
Using SAXS, we demonstrated that MAL 123–218 mimics the overall 3D structure of a tRNA and has a similar global fold as the NL4-3 PBS/TLE domain (Fig. 4C). While the anticodon stem–loop sequence of tRNALys is a critical LysRS recognition element, structural models of LysRS bound to tRNALys show that the canonical L-shape of the tRNA makes extensive additional protein contacts throughout the acceptor and anticodon stems (Ofir-Birin et al. 2013). An RNA (such as the PBS domain) that adopts a similar L-shaped fold would likely be capable of making similar distributive contacts with LysRS. Despite the inability of the MAL A-rich stem–loop to bind to LysRS in isolation, it may still contribute to the overall tRNA-like nature of the MAL PBS domain as suggested by previous NMR studies (Puglisi and Puglisi 1998). As previously noted, the addition of ψ enhances LysRS affinity in both the NL4-3 and MAL strains. Although the 3D structure of the MAL ψ domain is unknown, the 3D structure of the NL4-3 ψ element has recently been reported (Jones et al. 2014; Keane et al. 2015). Given the high degree of 3D structural homology of ψ elements even from distinct retroviruses (Cantara et al. 2014), it is likely that the MAL and NL4-3 ψ domains possess similar 3D structures. RNA binding proteins often use different degrees of shape and/or sequence recognition of RNAs to achieve specificity (Stefl et al. 2005; Lunde et al. 2007), and in some instances RNA shape vs. sequence compete to modulate protein binding (Zearfoss et al. 2013).
In summary, we have shown that tRNA mimicry is conserved across two prototypical HIV-1 strains, NL4-3 and MAL. While both the U-rich loop sequence and PBS/TLE domain structure are important for hLysRS recognition in the case of NL4-3, the primary determinant of hLysRS recognition in the case of MAL is likely to be the global PBS domain structure. In both MAL and NL4-3, elements within ψ contribute to high-affinity hLysRS binding. Based on our findings, we hypothesize that tRNA mimicry in the 5′-UTR is conserved throughout HIV-1, and is used to facilitate primer tRNALys3 release from LysRS and annealing to the proximal PBS. Given the high degree of conservation of the HIV-1 5′-UTR sequence and structure (Wilkinson et al. 2008), the TLE could represent an attractive therapeutic target for future drug development.
MATERIALS AND METHODS
Preparation of proteins and nucleic acids
Plasmid phLysRSΔN65 encoding His-tagged hLysRSΔN65 was transformed into E. coli strain BL21(DE3) CP-RIL and grown overnight in 50 mL of LB media containing 100 µg/mL ampicillin and 35 µg/mL chloramphenicol at 37°C. A 10 mL aliquot of this culture was added to 1 L of LB media containing the same antibiotics and grown for 90 min at 37°C. After cooling cells to room temperature, the concentration of ampicillin was increased to 110 µg/mL and protein expression was induced as previously described (Shiba et al. 1997). Cells were harvested by centrifugation at 8000g for 8 min at 4°C, and the cell pellet was resuspended in 20 mL of lysis buffer (50 mM Na2HPO4, pH 7.5, 300 mM NaCl, 20 mM imidazole, and one protease inhibitor tablet [Roche] per liter). The cells were lysed by sonicating for 12 cycles of 15 sec of sonication followed by 45 sec of rest. Polyethylenimine (0.5% final) was added and the solution was stirred gently for 30 min at 4°C. The cellular debris was pelleted by centrifugation at 27,000g for 15 min at 4°C. Protein was precipitated by the addition of 375 mg/mL ammonium sulfate, incubated at room temperature for 15 min, and pelleted by centrifugation at 27,000g for 15 min at 4°C. The pellet was washed with 50 mL of lysis buffer supplemented with 375 mg/mL ammonium sulfate and recentrifuged at 27,000g for 15 min at 4°C. The pellet was resuspended in 20 mL of lysis buffer and further centrifugation at 27,000g for 15 min at 4°C was performed to remove insoluble debris. The supernatant was loaded onto a Ni–NTA (Sigma-Aldrich) column, which was washed with lysis buffer followed by a wash buffer containing 20 mM imidazole. Protein was eluted using increasing amounts of imidazole, and fractions containing hLysRSΔN65 were combined and concentrated using 30K MWCO centrifugal filter units (Amicon) by centrifuging at 3200g at 4°C. The concentrated protein was then dialyzed overnight in a 10K MWCO Slide-A-Lyzer dialysis cassette (Thermo Scientific) into 2× storage buffer (80 mM HEPES, pH 7.5, 300 mM NaCl, 4 mM DTT, and one protease inhibitor tablet [Roche] per liter). After dialysis, the protein solution was further concentrated using 30K MWCO centrifugal filter units (Amicon) by centrifuging at 3200g at 4°C. The concentrated protein solution was diluted with glycerol (40% final). The protein concentration was determined using the Bradford assay (Bio-Rad) and stored at −20°C. Since hLysRSΔN65 has a tendency to aggregate after extended periods of storage (>6 mo), the quality of the protein was periodically assessed by SDS–PAGE gel electrophoresis. Dynamic light scattering analysis also confirmed that the protein used in the experiments was not aggregated (data not shown).
Plasmid pJCB (a gift from Dr. Roland Marquet, Strasbourg, France) was used to obtain the MAL RNA constructs used in this study. This plasmid encodes nt 1–615 of the HIV-1 MAL isolate genome under the control of a T7 promoter to allow for T7-mediated in vitro transcription (Paillart et al. 1994). A truncated construct was generated by removing the first 122 nt of the genomic sequence from pJCB using the MAL del 1–122 forward and reverse primers (Supplemental Table S1). These primers introduce two nonnative G's at the 5′ end to ensure efficient in vitro transcription (Milligan et al. 1987). To generate homogenous 3′ termini in the DNA templates for in vitro transcription, FokI cut sites were introduced after nt 218 using MAL 218 FokI forward and reverse primers and after nt 349 using MAL 349 FokI forward and reverse primers (Supplemental Table S1) to generate plasmids pMAL 123–218 and pMAL 123–349, respectively. In vitro transcription of the digested plasmids yielded the RNAs MAL 123–218 and MAL 123–349, respectively. RNAs MAL 123–218 CUC/AAA, MAL 123–218 GCAG/UCUU, HP1 ext and ΔHP1 were obtained from pMAL 123–218, and MAL 123–349 C277G DIS mutant was obtained from pMAL 123–349 by QuikChange mutagenesis (QIAGEN). Complementary DNA oligonucleotides encoding HP1 and HP2 hairpin sequences with an upstream T7 promoter sequence followed by three nonnative G's were ordered from Integrated DNA Technologies (Supplemental Table S1). The complementary DNA strands were annealed in Milli-Q purified water by heating at 80°C for 3 min, 60°C for 3 min, and then placed on ice for at least 30 min. The annealed DNA templates were used for in vitro transcription by T7 RNA polymerase to prepare the RNAs HP1 and HP2. Unmodified human tRNALys3 was obtained from plasmid pLYSF119 as previously described (Shiba et al. 1997). The following extinction coefficients at 260 nm were used to determine the concentrations of the RNAs: HP1, 169,400 M−1cm−1; HP2, 141,400 M−1 cm−1; MAL 123–218, MAL 123–218 CUC/AAA, and MAL 123–218 GCAG/UCUU, 870,300 M−1 cm−1; MAL 123–349 and MAL 123–349 C277G DIS mutant, 2,094,500 M−1cm−1; ΔHP1, 664,700 M−1 cm−1; and HP1 ext, 1,057,200 M−1 cm−1.
Fluorescence anisotropy binding assay
RNAs were fluorescently labeled at their 3′ termini with fluorescein-5-semithiocarbazide (FTSC) (Invitrogen) using previously described methods (Pagano et al. 2007; Rye-McCurdy et al. 2015). Briefly, 5 nmol of the RNAs shorter than 30 nt and 2.5 nmol of RNAs longer than 30 nt were used in the labeling reactions. The RNAs were oxidized using 75 nmol of NaIO4 and then incubated with 2.5 mM FTSC overnight at 4°C. The free FTSC dye was removed from the labeled RNAs using G-25 Sephadex columns (Roche). The labeling efficiencies were calculated as previously described (Rye-McCurdy et al. 2015). A labeling efficiency of >65% was obtained for all RNAs except for MAL 123–349 (52%) and MAL 123–349 C277G DIS (37%).
The fluorescently labeled RNAs were folded in 50 mM HEPES, pH 7.5 by heating at 80°C for 2 min, 60°C for 2 min, followed by the addition of 10 mM MgCl2, and incubating on ice for at least 30 min. The FA binding assays were performed as previously described with minor modifications (Jones et al. 2013). The folded fluorescently labeled RNA (10 nM) was mixed with varying concentrations of hLysRSΔN65 in 20 mM Tris–HCl, pH 8, 15 mM NaCl, 35 mM KCl, and 1 mM MgCl2. The samples were excited at 485 nm and the emission was measured at 525 nm. The data points were fit assuming 1:1 binding (Stewart-Maynard et al. 2008). Three independent binding measurements were performed for each of the RNAs and the average value with the standard deviation was reported.
SAXS data acquisition and analysis
MAL 123–218 RNA (1 mg) was refolded in a volume of 200 µL by heating the sample in 50 mM HEPES, pH 7.4, to 80°C for 2 min, 60°C for 2 min, followed by addition of 10 mM MgCl2. The sample was then incubated at 37°C for 5 min, and on ice for at least 30 min. The folded RNA was purified by SEC on a 24-mL Superdex 200 10/300 GL Increase column (GE Healthcare) using a GE HPLC ÄKTApurifier in a buffer containing 150 mM NaCl, 50 mM HEPES, pH 7.4, 1 mM MgCl2, and 3% glycerol (wt/vol) at a flow rate of 0.5–1.1 mL/min. The absorbance of the eluate was monitored at 260 nm. Prior to SAXS analysis, SEC was performed on the folded MAL 123–218 RNA (300 µg) as described above, except that 250 µL fractions were collected and run on 8% native polyacrylamide gels containing 1 mM MgCl2 and stained with ethidium bromide to assess the homogeneity of the RNA eluted in each peak. The SAXS data were collected at station G1 at the Cornell High Energy Synchrotron Source (CHESS) using the experimental setup previously described (Acerbo et al. 2015). Briefly, the X-ray source was a 1.5 m CHESS Compact Undulator, and X-rays were detected using a Finger Lakes CCD. The beam diameter was 250 µm × 250 µm with a flux of 8.4 × 1011 photons sec−1 and an energy of 9.962 keV. X-ray exposures were taken continuously every 4 sec while the SEC eluent passed through the SAXS flow cell. Initial SAXS data processing, including radial averaging and buffer subtraction, was performed using the BioXTAS RAW software program (Nielsen et al. 2009). SAXS exposures (10 × 4 sec) corresponding to the RNA peak of interest on the SEC were averaged, and 50 × 4 sec SAXS exposures corresponding to buffer alone were averaged and then used to buffer subtract the RNA average SAXS curve (Skou et al. 2014). Subsequent data analysis steps were carried out as previously described (Jones et al. 2014) using the program PRIMUS (Konarev et al. 2006). Guinier analysis was used to calculate the radius of gyration (Rg) of the RNA, and Kratky analysis was performed to determine the extent of folding. If the Guinier plot displayed nonlinearity (a sign of poor-quality data) or Kratky analysis indicated that the RNA was not well-folded, then the data were not analyzed further. The interelectron pair distance distribution [P(r)] function was calculated using the program GNOM (Konarev et al. 2006), and the maximum interelectron distance (Dmax) and P(r)-based Rg was determined using the AutoGNOM feature. Ab initio envelopes were generated largely as previously described using the ATSAS software suite (Konarev et al. 2006; Jones et al. 2014). Briefly, 20 ab initio envelopes were generated in jagged mode with no symmetry restraints imposed, and the χ2 fits and reproducibility (NSD) values were determined. These 20 envelopes were averaged into one envelope, which was then packed with 5718 “dummy atoms” with an atomic radius of 2.0 Å. This was then used as the starting point for an additional 24 ab initio envelope calculations, generated in expert mode with no symmetry restraints imposed. These envelopes were averaged to generate the final envelope and their χ2 fits and NSD values were calculated.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Roland Marquet (Université de Strasbourg) for providing us with the plasmid pJCB. We also thank Drs. Christopher P. Jones and William A. Cantara for helpful suggestions. This work was supported by National Institutes of Health grants R01 GM113887 and P50 GM103368 (HIVE Center) to K.M.-F.; and National Institutes of Health training grant T32 GM008512 and National Institutes of Health predoctoral fellowship F31 AI120868 to E.D.O. This work is based upon research conducted at the Cornell High Energy Synchrotron Source (CHESS), which is supported by the National Science Foundation and the National Institutes of Health/National Institute of General Medical Sciences under National Science Foundation award DMR-0936384, using the Macromolecular Diffraction at CHESS (MacCHESS) facility, which is supported by award GM-103485 from the National Institutes of Health, through its National Institute of General Medical Sciences.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.062182.117.
REFERENCES
- Acerbo AS, Cook MJ, Gillilan RE. 2015. Upgrade of MacCHESS facility for X-ray scattering of biological macromolecules in solution. J Synchrotron Radiat 22: 180–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudin F, Marquet R, Isel C, Darlix JL, Ehresmann B, Ehresmann C. 1993. Functional sites in the 5′ region of human immunodeficiency virus type 1 RNA form defined structural domains. J Mol Biol 229: 382–397. [DOI] [PubMed] [Google Scholar]
- Burke JE, Butcher SE. 2012. Nucleic acid structure characterization by small angle X-ray scattering (SAXS). Curr Protoc Nucleic Acid Chem 51: 7.18.1–7.18.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantara WA, Olson ED, Musier-Forsyth KM. 2014. Progress and outlook in structural biology of large viral RNAs. Virus Res 193: 24–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cen S, Khorchid A, Javanbakht H, Gabor J, Stello T, Shiba K, Musier-Forsyth K, Kleiman L. 2001. Incorporation of lysyl-tRNA synthetase into human immunodeficiency virus type 1. J Virol 75: 5043–5048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cen S, Javanbakht H, Kim S, Shiba K, Craven R, Rein A, Ewalt K, Schimmel P, Musier-Forsyth K, Kleiman L. 2002. Retrovirus-specific packaging of aminoacyl-tRNA synthetases with cognate primer tRNAs. J Virol 76: 13111–13115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cen S, Javanbakht H, Niu M, Kleiman L. 2004. Ability of wild-type and mutant lysyl-tRNA synthetase to facilitate tRNALys incorporation into human immunodeficiency virus type 1. J Virol 78: 1595–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewan V, Reader J, Musier-Forsyth KM. 2014. Role of aminoacyl-tRNA synthetases in infectious diseases and targets for therapeutic development. Top Curr Chem 344: 293–329. [DOI] [PubMed] [Google Scholar]
- Francin M, Kaminska M, Kerjan P, Mirande M. 2002. The N-terminal domain of mammalian lysyl-tRNA synthetase is a functional tRNA-binding domain. J Biol Chem 277: 1762–1769. [DOI] [PubMed] [Google Scholar]
- Frankel AD, Young JA. 1998. HIV-1: fifteen proteins and an RNA. Annu Rev Biochem 67: 1–25. [DOI] [PubMed] [Google Scholar]
- Freed EO. 2001. HIV-1 replication. Somat Cell Mol Genet 26: 13–33. [DOI] [PubMed] [Google Scholar]
- Gabor J, Cen S, Javanbakht H, Niu M, Kleiman L. 2002. Effect of altering the tRNALys3 concentration in human immunodeficiency virus type 1 upon its annealing to viral RNA, GagPol incorporation, and viral infectivity. J Virol 76: 9096–9102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganser-Pornillos BK, Yeager M, Sundquist WI. 2008. The structural biology of HIV assembly. Curr Opin Struct Biol 18: 203–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F, Robertson DL, Carruthers CD, Li Y, Bailes E, Kostrikis LG, Salminen MO, Bibollet-Ruche F, Peeters M, Ho DD, et al. 1998. An isolate of human immunodeficiency virus type 1 originally classified as subtype I represents a complex mosaic comprising three different group M subtypes (A, G, and I). J Virol 72: 10234–10241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldschmidt V, Paillart JC, Rigourd M, Ehresmann B, Aubertin AM, Ehresmann C, Marquet R. 2004. Structural variability of the initiation complex of HIV-1 reverse transcription. J Biol Chem 279: 35923–35931. [DOI] [PubMed] [Google Scholar]
- Guo F, Cen S, Niu M, Javanbakht H, Kleiman L. 2003. Specific inhibition of the synthesis of human lysyl-tRNA synthetase results in decreases in tRNALys incorporation, tRNA3Lys annealing to viral RNA, and viral infectivity in human immunodeficiency virus type 1. J Virol 77: 9817–9822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halwani R, Cen S, Javanbakht H, Saadatmand J, Kim S, Shiba K, Kleiman L. 2004. Cellular distribution of Lysyl-tRNA synthetase and its interaction with Gag during human immunodeficiency virus type 1 assembly. J Virol 78: 7553–7564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Mak J, Cao Q, Li Z, Wainberg MA, Kleiman L. 1994. Incorporation of excess wild-type and mutant tRNA3Lys into human immunodeficiency virus type 1. J Virol 68: 7676–7683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isel C, Marquet R, Keith G, Ehresmann C, Ehresmann B. 1993. Modified nucleotides of tRNA3Lys modulate primer/template loop-loop interaction in the initiation complex of HIV-1 reverse transcription. J Biol Chem 268: 25269–25272. [PubMed] [Google Scholar]
- Isel C, Ehresmann C, Keith G, Ehresmann B, Marquet R. 1995. Initiation of reverse transcription of HIV-1: secondary structure of the HIV-1 RNA/tRNA3Lys (template/primer). J Mol Biol 247: 236–250. [DOI] [PubMed] [Google Scholar]
- Javanbakht H, Cen S, Musier-Forsyth K, Kleiman L. 2002. Correlation between tRNALys3 aminoacylation and its incorporation into HIV-1. J Biol Chem 277: 17389–17396. [DOI] [PubMed] [Google Scholar]
- Javanbakht H, Halwani R, Cen S, Saadatmand J, Musier-Forsyth K, Gottlinger H, Kleiman L. 2003. The interaction between HIV-1 Gag and human lysyl-tRNA synthetase during viral assembly. J Biol Chem 278: 27644–27651. [DOI] [PubMed] [Google Scholar]
- Jiang M, Mak J, Ladha A, Cohen E, Klein M, Rovinski B, Kleiman L. 1993. Identification of tRNAs incorporated into wild-type and mutant human immunodeficiency virus type 1. J Virol 67: 3246–3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CP, Saadatmand J, Kleiman L, Musier-Forsyth K. 2013. Molecular mimicry of human tRNALys anti-codon domain by HIV-1 RNA genome facilitates tRNA primer annealing. RNA 19: 219–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CP, Cantara WA, Olson ED, Musier-Forsyth K. 2014. Small-angle X-ray scattering-derived structure of the HIV-1 5′ UTR reveals 3D tRNA mimicry. Proc Natl Acad Sci 111: 3395–3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane SC, Heng X, Lu K, Kharytonchyk S, Ramakrishnan V, Carter G, Barton S, Hosic A, Florwick A, Santos J, et al. 2015. RNA structure. Structure of the HIV-1 RNA packaging signal. Science 348: 917–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khorchid A, Javanbakht H, Wise S, Halwani R, Parniak MA, Wainberg MA, Kleiman L. 2000. Sequences within Pr160gag-pol affecting the selective packaging of primer tRNALys3 into HIV-1. J Mol Biol 299: 17–26. [DOI] [PubMed] [Google Scholar]
- Kleiman L. 2002. tRNALys3: the primer tRNA for reverse transcription in HIV-1. IUBMB Life 53: 107–114. [DOI] [PubMed] [Google Scholar]
- Kleiman L, Cen S. 2004. The tRNALys packaging complex in HIV-1. Int J Biochem Cell Biol 36: 1776–1786. [DOI] [PubMed] [Google Scholar]
- Kleiman L, Jones CP, Musier-Forsyth K. 2010. Formation of the tRNALys packaging complex in HIV-1. FEBS Lett 584: 359–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konarev PV, Petoukhov MV, Volkov VV, Svergun DI. 2006. ATSAS 2.1, a program package for small-angle scattering data analysis. J Appl Cryst 39: 277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovaleski BJ, Kennedy R, Hong MK, Datta SA, Kleiman L, Rein A, Musier-Forsyth K. 2006. In vitro characterization of the interaction between HIV-1 Gag and human lysyl-tRNA synthetase. J Biol Chem 281: 19449–19456. [DOI] [PubMed] [Google Scholar]
- Kovaleski BJ, Kennedy R, Khorchid A, Kleiman L, Matsuo H, Musier-Forsyth K. 2007. Critical role of helix 4 of HIV-1 capsid C-terminal domain in interactions with human lysyl-tRNA synthetase. J Biol Chem 282: 32274–32279. [DOI] [PubMed] [Google Scholar]
- Li Z, Shalom A, Huang Y, Mak J, Arts E, Wainberg MA, Kleiman L. 1996. Multiple forms of tRNALys3 in HIV-1. Biochem Biophys Res Commun 227: 530–540. [DOI] [PubMed] [Google Scholar]
- Litvak S, Sarih-Cottin L, Fournier M, Andreola M, Tarrago-Litvak L. 1994. Priming of HIV replication by tRNALys3: role of reverse transcriptase. Trends Biochem Sci 19: 114–118. [DOI] [PubMed] [Google Scholar]
- Liu S, Comandur R, Jones CP, Tsang P, Musier-Forsyth K. 2016. Anticodon-like binding of the HIV-1 tRNA-like element to human lysyl-tRNA synthetase. RNA 22: 1828–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunde BM, Moore C, Varani G. 2007. RNA-binding proteins: modular design for efficient function. Nat Rev Mol Cell Biol 8: 479–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak J, Jiang M, Wainberg MA, Hammarskjöld ML, Rekosh D, Kleiman L. 1994. Role of Pr160gag-pol in mediating the selective incorporation of tRNALys into human immunodeficiency virus type 1 particles. J Virol 68: 2065–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak J, Khorchid A, Cao Q, Huang Y, Lowy I, Parniak MA, Prasad VR, Wainberg MA, Kleiman L. 1997. Effects of mutations in Pr160gag-pol upon tRNALys3 and Pr160gag-plo incorporation into HIV-1. J Mol Biol 265: 419–431. [DOI] [PubMed] [Google Scholar]
- Marquet R, Isel C, Ehresmann C, Ehresmann B. 1995. tRNAs as primer of reverse transcriptases. Biochimie 77: 113–124. [DOI] [PubMed] [Google Scholar]
- Milligan JF, Groebe DR, Witherell GW, Uhlenbeck OC. 1987. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res 15: 8783–8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen SS, Toft KN, Snakenborg D, Jeppesen MG, Jacobsen JK, Vestergaard B, Kutter JP, Arleth L. 2009. BioXTAS RAW, a software program for high-throughput automated small-angle X-ray scattering data reduction and preliminary analysis. J Appl Crystallogr 42: 959–964. [Google Scholar]
- Ofir-Birin Y, Fang P, Bennett SP, Zhang HM, Wang J, Rachmin I, Shapiro R, Song J, Dagan A, Pozo J, et al. 2013. Structural switch of lysyl-tRNA synthetase between translation and transcription. Mol Cell 49: 30–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano JM, Farley BM, McCoig LM, Ryder SP. 2007. Molecular basis of RNA recognition by the embryonic polarity determinant MEX-5. J Biol Chem 282: 8883–8894. [DOI] [PubMed] [Google Scholar]
- Paillart JC, Marquet R, Skripkin E, Ehresmann B, Ehresmann C. 1994. Mutational analysis of the bipartite dimer linkage structure of human immunodeficiency virus type 1 genomic RNA. J Biol Chem 269: 27486–27493. [PubMed] [Google Scholar]
- Puglisi EV, Puglisi JD. 1998. HIV-1 A-rich RNA loop mimics the tRNA anticodon structure. Nat Struct Biol 5: 1033–1036. [DOI] [PubMed] [Google Scholar]
- Ratner L, Haseltine W, Patarca R, Livak KJ, Starcich B, Josephs SF, Doran ER, Rafalski JA, Whitehorn EA, Baumeister K, et al. 1985. Complete nucleotide sequence of the AIDS virus, HTLV-III. Nature 313: 277–284. [DOI] [PubMed] [Google Scholar]
- Robertson DL, Hahn BH, Sharp PM. 1995. Recombination in AIDS viruses. J Mol Evol 40: 249–259. [DOI] [PubMed] [Google Scholar]
- Rye-McCurdy T, Rouzina I, Musier-Forsyth K. 2015. Fluorescence anisotropy-based salt-titration approach to characterize protein-nucleic acid interactions. Methods Mol Biol 1259: 385–402. [DOI] [PubMed] [Google Scholar]
- Sharp PM, Hahn BH. 2010. The evolution of HIV-1 and the origin of AIDS. Philos Trans R Soc Lond B Biol Sci 365: 2487–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba K, Stello T, Motegi H, Noda T, Musier-Forsyth K, Schimmel P. 1997. Human lysyl-tRNA synthetase accepts nucleotide 73 variants and rescues Escherichia coli double-defective mutant. J Biol Chem 272: 22809–22816. [DOI] [PubMed] [Google Scholar]
- Skou S, Gillilan RE, Ando N. 2014. Synchrotron-based small-angle X-ray scattering of proteins in solution. Nat Protoc 9: 1727–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonigo P, Alizon M, Staskus K, Klatzmann D, Cole S, Danos O, Retzel E, Tiollais P, Haase A, Wain-Hobson S. 1985. Nucleotide sequence of the visna lentivirus: relationship to the AIDS virus. Cell 42: 369–382. [DOI] [PubMed] [Google Scholar]
- Stefl R, Skrisovska L, Allain FH. 2005. RNA sequence- and shape-dependent recognition by proteins in the ribonucleoprotein particle. EMBO Rep 6: 33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart-Maynard KM, Cruceanu M, Wang F, Vo MN, Gorelick RJ, Williams MC, Rouzina I, Musier-Forsyth K. 2008. Retroviral nucleocapsid proteins display nonequivalent levels of nucleic acid chaperone activity. J Virol 82: 10129–10142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telesnitsky A, Goff SP. 1997. Reverse transcriptase and the generation of retroviral DNA. In Retroviruses (ed. Coffin JM, et al. ), pp. 121–160. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [PubMed] [Google Scholar]
- Thormar H. 2013. The origin of lentivirus research: maedi-visna virus. Curr HIV Res 11: 2–9. [PubMed] [Google Scholar]
- Wain-Hobson S, Sonigo P, Danos O, Cole S, Alizon M. 1985. Nucleotide-sequence of the AIDS virus, LAV. Cell 40: 9–17. [DOI] [PubMed] [Google Scholar]
- Watts JM, Dang KK, Gorelick RJ, Leonard CW, Bess JW Jr, Swanstrom R, Burch CL, Weeks KM. 2009. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature 460: 711–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson KA, Gorelick RJ, Vasa SM, Guex N, Rein A, Mathews DH, Giddings MC, Weeks KM. 2008. High-throughput SHAPE analysis reveals structures in HIV-1 genomic RNA strongly conserved across distinct biological states. PLoS Biol 6: e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zearfoss NR, Johnson ES, Ryder SP. 2013. hnRNP A1 and secondary structure coordinate alternative splicing of Mag. RNA 19: 948–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.