

Abstract
Most drug therapies distribute the agents throughout the entire body, even though the drugs are typically only needed at specific tissues. This often limits dosage and causes discomfort and harmful side‐effects. Significant research has examined nanoparticles (NPs) for use as targeted delivery vehicles for therapeutic cargo, however, major clinical success has been limited. Current work focuses mainly on liposomal and polymer‐based NPs, but emerging research is exploring the engineering of viral capsids as noninfectious protein‐based NPs—termed virus‐like particles (VLPs). This review covers the research that has been performed thus far and outlines the potential for these VLPs to become highly effective delivery vehicles that overcome the many challenges encountered for targeted delivery of therapeutic cargo.
Keywords: nanoparticle, protein engineering, virus‐like particle, targeted delivery, therapeutics
Abbreviations
- CCMV
cowpea chlorotic mottle virus
- CPMV
cowpea mosaic virus
- EDC
1‐ethyl‐3‐(3‐dimethylaminopropyl)carbodiimide hydrochloride
- EGF
epidermal growth factor
- EPR
enhanced permeability and retention
- HBVc
hepatitis B virus core
- MS2
enterobacteria phage MS2
- NHS
n‐hydroxysuccinimide
- NP
nanoparticle
- P22
Salmonella typhimurium P22
- PEG
polyethylene glycol
- Qβ
enterobacteria phage Qβ
- VLP
virus‐like particle
1. Introduction
Currently, numerous diseases lack adequate treatment, most notably cancer and various genetic disorders. In 2016, the National Cancer Institute estimates that 1,685,210 new cases of cancer will be diagnosed in the United States and 595,690 people will die from the disease—a 35% mortality rate.1 Typical cancer treatment includes chemotherapy, radiation, and surgery. However, surgery is highly invasive and often fails—especially after metastasis. Chemotherapy and radiation can be effective temporarily, but result in harsh side‐effects that drastically reduce quality of life. In particular, systemic administration of chemotherapeutic agents is usually limited by those side‐effects and the effective dose at the tumor site is only a small fraction of the administered drugs.2
In addition to cancer, the development of gene therapies for treatment of genetic disorders, such as mitochondrial disorders and Parkinson's disease has been a major focus of research. By 2012, over 1800 gene therapy clinical trials had been conducted or approved.3 However, success in clinical trials has been limited because of numerous technical barriers, including difficulty in reaching the targeted tissues. Although the U.S. FDA approved the first oncolytic viral therapy, Imlygic, recently, no virus‐derived therapies for gene delivery have been FDA‐approved according to the latest information from the FDA's website.
Targeted delivery has long been one of the most promising, but also most challenging, opportunities for improving the treatment of these diseases. Targeted delivery offers three key advantages that systemic delivery lacks: (a) the therapeutic will act primarily at the desired site‐of‐action, limiting off‐target effects such as the harmful side‐effects involved with chemotherapy; (b) the delivery vehicle can provide much higher local concentrations of the therapeutic within the diseased tissues, allowing a more effective treatment; and (c) the delivery vehicle can carry the therapeutic to sites it would not normally be able to reach, improving the efficiency of gene therapies. The first attempts at developing delivery vehicles were antibody‐drug conjugates. These carriers have been extensively developed with two FDA‐approved examples [Trastuzumab emtansine (Kadcyla) and Brentuximab vedotin (Adcetris)], and many more are in clinical trials.4 However, they suffer from several limitations including structural heterogeneity, instability, and limited solubility.4, 5 In addition, antibody‐drug conjugates typically deliver only a few drug molecules per antibody.4 In contrast, nanoparticle (NP)‐based delivery agents, including liposomal, polymer‐based, metal‐based, and protein‐based NPs, have the potential to provide safer and more effective delivery by encapsulating therapeutic cargo inside the particle with a much higher cargo/carrier ratio. This review will focus on the development of virus‐like particles (VLPs), protein‐based NPs derived from viral capsids, as targeted therapeutic delivery agents. Several previously published reviews have covered VLPs. A review from the Bundy lab excellently describes many ways to covalently attach ligands to the surface of VLPs, but lacks further information pertinent to their use as drug delivery vehicles.6 A 2014 review from the Tullman‐Ercek lab covers cargo loading, specific targeting, and application for using VLPs as delivery vehicles, but lacks specific surface modification information and loading small molecule drugs.7 Another 2014 review from the van Hest lab has an excellent discussion of surface and interior covalent attachment and genetic fusion strategies, but contains no discussion of nonspecific cargo loading or attachment techniques.8 Lastly, a recent 2016 review covers a large variety of VLPs and other protein‐based NPs, but lacks depth for each individual vehicle.9 This review, while focusing on six of the most used VLPs, attempts to combine and expand on the information within these other reviews while addressing new factors, including particle stability, expression platforms, and purification methods, that are important for the development of these vehicles as therapeutic NPs.
2. Using vlps overcomes the limitations of current np‐based therapeutics
Despite many attempts, only a few liposomal and protein‐based NPs have been approved for cancer drug delivery, including Doxil and Abraxane—and these are all passive‐targeting delivery agents relying on the enhanced permeability and retention (EPR) effect for tumor localization.5, 10, 11 Liposomal NPs are limited by particle instability, rapid clearance, and spontaneous membrane fusion with off‐target cells.12, 13 The polymer‐based NP technologies suffer from structural heterogeneity, particle instability, slow and nonuniform drug release, and potential immunogenicity.14, 15 The more stable metal‐based NPs suffer from a lack of specificity and high toxicity.16 In addition, most of these NPs suffer from clearance mediated by phagocytes and dendritic cells, including Kupffer cells in the liver. Coating NPs with polyethylene glycol (PEG) can help avoid phagocytes and extend the blood circulation time by creating “stealth” brushes.17, 18 However, PEGylation can also reduce NP uptake by the targeted cells and is potentially immunogenic.17, 18 Finally, surface functionalization of these NPs is difficult to control and nonuniform.19
An alternate type of drug delivery NP that is showing promise is the VLP.20 VLPs are self‐assembled, homogeneous NPs derived from the coat proteins of viral capsids. They typically lack their natural genome and are therefore noninfectious. VLPs are an emerging class of targeted delivery vehicles with the potential to overcome the limitations of other NPs.7, 20 In recent years, several groups have shown that VLPs can pack and deliver therapeutic cargo such as chemotherapeutic drugs, siRNA, RNA aptamers, proteins, and peptides.21, 22, 23, 24, 25, 26, 27 However, there are still challenges when using VLPs. Similar to other NPs, avoiding phagocyte‐mediated clearance is a major challenge, even with PEGylated VLPs.22, 28 In addition, VLP stability can also be an issue.29 Lastly, recent research has shown that ellipsoid NPs are able to extravasate from the blood vessel more effectively than spherical NPs.30 This ellipsoid shape is possible for conventional polymeric NPs, but is not feasible for icosahedral VLPs. However, the capability of VLP surface modification allows a variety of functional ligands to be added with the potential to address these limitations. By displaying multiple ligands with high affinity for the tight junctions between endothelial cells, VLPs may be able to efficiently extravasate from the vasculature of the blood vessels.
3. Challenges to targeted delivery using NPs
As mentioned previously, targeted drug delivery by NPs must overcome multiple challenges (Table 1).31 The ability to overcome these challenges must either be intrinsic or be imparted as the VLPs are prepared by: (a) loading the cargo inside the NP, and (b) functionalizing the NP to deliver its cargo primarily to the intended cells. While in the bloodstream and the interstitial space, the NP must remain stable, retain its cargo, and avoid nonspecific engulfment by phagocytes. Additionally, after extravasation into the extravascular tissue, the NP must specifically target the intended cells while avoiding other healthy cells to limit organ accumulation and toxicity. After adsorption to the targeted cells and internalization through receptor‐mediated endocytic pathways, NPs carrying macromolecular cargo must escape the endosome, disassemble, and release their therapeutic cargo to the cytosol (in a functional form). Even though endosomal escape may not be required when delivering small, stable molecules (since subsequent lysosomal degradation of the NP should eventually release the cargo), these other requirements still present a daunting challenge for the development of targeted delivery vehicles. However, combining surface functionalization of the VLPs with the ability to load therapeutic cargo can provide the design flexibility and complexity needed to open the door to multiple new therapies for unmet medical needs. The several attempts to overcome these challenges are outlined in the following sections.
Table 1.
Challenges to targeted drug delivery and possible solutions
| Challenge | Possible solutions | Tested VLPs |
|---|---|---|
| Stabilize nanoparticle | Stabilize with disulfide bonds | HBVc, MS2, Qβ |
| Avoid phagocytes | Display PEG or the CD47 ectodomain | MS2, Qβ, P22, CCMV, CPMV |
| Extravasate from blood vessel | — | — |
| Target specific cells | Display targeting ligands | HBVc, MS2, Qβ, CPMV |
| Escape endosome | Display cell‐penetrating peptides | MS2, P22, CPMV |
| Release cargo | Reduce stabilizing disulfide bonds in cytosol | HBVc, Qβ |
4. Commonly used vlps and their production methods
This review will focus on six of the most actively developed VLPs from: one animal virus, three bacteriophages, and two plant viruses (Figure 1).32, 33, 34, 35, 36, 37 Table 2 summarizes the properties of these VLPs.
Figure 1.
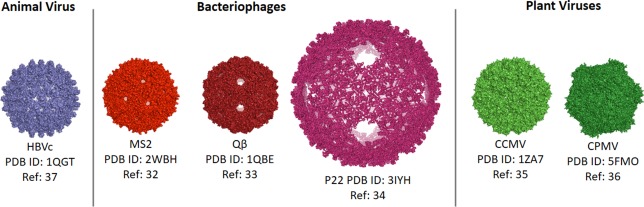
Structures of the six VLPs discussed in this review
Table 2.
Relevant information on the six VLPs discussed in this review
| VLP | Virus type | VLP outer diameter (nm) | VLP inner diameter (nm) | VLP geometry | VLP subunits | References |
|---|---|---|---|---|---|---|
| HBVc | Animal virus | 35 | 26 | T = 4 icosahedral | 240 coat proteins (120 dimers) | 37, 40, 48 |
| MS2 | Bacteriophage | 27 | 15 | T = 3 icosahedral | 180 coat proteins (90 dimers) | 38, 42, 49 |
| Qβ | Bacteriophage | 28 | 21 | T = 3 icosahedral | 180 coat proteins (90 dimers) | 39, 43 |
| P22 | Bacteriophage | 58‐64 | 48–50 | T = 7 icosahedral | 420 coat proteins + 100–300 removable scaffold proteins | 41, 50 |
| CCMV | Plant virus | 28 | 18 | T = 3 icosahedral | 180 coat proteins (90 dimers) | 51, 52 |
| CPMV | Plant virus | 28–31 | 22 | T = 3 icosahedral | 60 large + 60 small coat proteins | 53, 54 |
4.1. Animal virus‐based VLPs
The hepatitis B virus is comprised of an internal protein capsid and a lipid envelope containing other proteins. Two different VLPs can be produced from the virus, using either the core antigen that forms the internal capsid or the surface antigen that spontaneously combines with lipids to form NPs. We will focus on the VLP derived from the hepaitis B core (HBVc) antigen, which is formed from 240 copies of a single protein.37 These proteins first form dimers, which then assemble with pentameric or pseudo‐hexameric junctions in a T = 4 icosahedral geometry.37, 38, 40, 48 The VLP has been produced using multiple technologies including Escherichia coli cytosolic accumulation and cell‐free protein synthesis.37, 38 The assembled VLPs are typically purified using size‐exclusion chromatography or differential centrifugation.37, 38, 49 Individual coat proteins have been subsequently obtained by disassembling the VLPs with urea, which allows simultaneous cargo loading and VLP re‐assembly.37, 38, 49 Unpublished data from the Swartz group indicates that coat proteins with hexahistidine extensions can also be directly purified using immobilized metal affinity chromatography. Purifying the individual coat proteins allows control over cargo loading during VLP assembly, which in the case of HBVc is achieved by increasing the salt concentration to trigger spontaneous self‐assembly mediated primarily by hydrophobic interactions.37, 38
4.2. Bacteriophage‐based VLPs
The three bacteriophages, MS2, Qβ, and Salmonella typhimurium P22, all infect enterobacteria, most notably E. coli. Although all three are composed of only a nucleic acid‐filled viral capsid, P22 differs greatly from MS2 and Qβ. MS2 and Qβ are composed of 90 homodimers and require a specific stem‐loop hairpin secondary structure in their RNA genome to trigger VLP self‐assembly by binding to the coat proteins.38, 39, 42, 43 P22, on the other hand, is composed of up to 415 coat proteins, 100–300 scaffold proteins, and 12 portal proteins. However, the P22 VLP has been engineered to consist of 420 coat proteins and only the 100–300 scaffold proteins, which can subsequently be removed with guanidine hydrochloride, leaving only the coat proteins.41, 50 Like the HBVc VLP, these VLPs assemble with icosahedral geometry.38, 41, 43, 51, 52, 53 All three can be produced in E. coli, but Qβ can also be produced in yeast and both Qβ and MS2 can be produced using cell‐free protein synthesis.29, 38, 41, 43, 50, 54, 55, 56 MS2 VLPs have been purified using size‐exclusion chromatography, differential centrifugation, or immobilized metal affinity chromatography (for VLPs containing hexahistidine tags).38, 44, 54 Acids or urea can be used to disassemble the purified MS2 VLPs to obtain the dimers, which can then be re‐assembled after removal of the disassembly agent and the addition of the stem‐loop RNA.23, 42, 45 Qβ VLPs have been purified using size‐exclusion chromatography and the dimers can be obtained by disassembling the VLPs using acid, which can then be reassembled similar to MS2.39, 43, 57 P22 VLPs have been purified using size‐exclusion chromatography or differential centrifugation and can also be disassembled using acid to obtain the coat proteins.41, 50, 55, 56 Addition of scaffold proteins is required to reassemble the P22 VLP, but these can subsequently be removed.41 These bacteriophage‐derived VLPs differ from HBVc VLPs mainly in the assembly stimulus, using additional biomolecules (RNA or proteins) to initiate self‐assembly instead of increasing the salt concentration.
4.3. Plant virus‐based VLPs
The final two commonly‐used VLPs to be discussed are derived from plant viruses that infect the cowpea leaf: cowpea chlorotic mottle virus (CCMV) and cowpea mosaic virus (CPMV). Neither virus has a lipid envelope. Both VLPs assemble with icosahedral geometry.46, 47, 58, 59, 60 The CCMV VLPs are formed from 90 homodimers and can be produced in E. coli or yeast.46, 61 They have been purified using size‐exclusion chromatography or immobilized metal affinity chromatography, using coat proteins with hexahistidine extensions.46, 62 Dimers can be obtained by dialyzing the assembled VLPs against 0.5 M CaCl2 or by purifying hexahistidine tagged dimers directly.47, 62 Combining the dimers with RNA in a 1:6 mass ratio and lowering the pH to 4–5 induces self‐assembly.47, 62, 63, 64 CPMV, on the other hand, is formed from 60 copies of the VP60 protein which must first be proteolyzed into the L and S coat proteins (60 copies of each).59 Unfortunately, the VLP cannot be produced using E. coli or yeast; insect cells or plants must be used.58, 59 The VLPs have been purified using differential centrifugation, but the coat proteins cannot yet be obtained in usable quantities.58, 59 The inability to produce the VLP in E. coli or obtain purified coat proteins adds another challenge for targeted drug delivery; however, CPMV has been actively evaluated for therapeutic use due to the ability to easily display ligands on its surface and load cargo through association with its genome.
5. Design considerations in developing VLPs for targeted delivery
Because of their precise and repeated structures and relatively large cargo capacities, VLPs have many advantages over other types of NPs. Since they are expressed biologically and formed from multiple copies of the same protein, the VLPs are highly uniform and are easily expressed in bacteria (with some exceptions, such as CPMV). Also, they have evolved in nature to encapsidate their viral genomes, which could be advantageous for loading therapeutic cargo. Particularly in the case of MS2 and Qβ, specific stem‐loop RNA secondary structures that are required for assembly can carry other molecules into the VLP during assembly.21, 42, 65, 66 For many VLPs, peptide or protein sequences can be added directly to the primary amino acid sequence of the coat proteins either as insertions or extensions to allow presentation on either the interior or exterior surface.67, 68, 69, 70, 71 Alternatively, reactive amino acids can be used to couple cargo to the interior or ligands to the exterior of the capsids in repeated and consistent orientations.66, 72, 73, 74 VLPs are much less toxic for parenteral administration than metal NPs, more stable than liposomes, and more uniform than polymer NPs.75, 76, 77 Although a significant amount of work is still required to develop the VLPs as delivery vehicles, the current progress shows a great deal of promise.
5.1. Surface functionalization
As discussed, a delivery vehicle must provide several different functionalities. For several of these attributes, the VLP surface must be extensively modified with various biomolecules. These ligands can provide specific cellular targeting, reduce immune responses, and potentially facilitate extravasation. Most approaches require covalent attachment, however, P22 can display ligands through noncovalent interactions.78 Covalent methods take advantage of either native or nonnatural reactive amino acids (Figure 2), though genetic fusions to the primary amino acid sequence can also be used to display inserted peptides or proteins.67, 68, 69, 70, 71 Table 3 provides a summary of these surface modifications, including specific references. Although many of the published surface modifications are aimed at vaccines or other uses not related to drug delivery, the same methods can easily be applied. Furthermore, some published studies have described the attachment of ligands for targeting specific cells or avoiding the immune system.78, 84, 85, 86 These will be discussed further in later sections.
Figure 2.
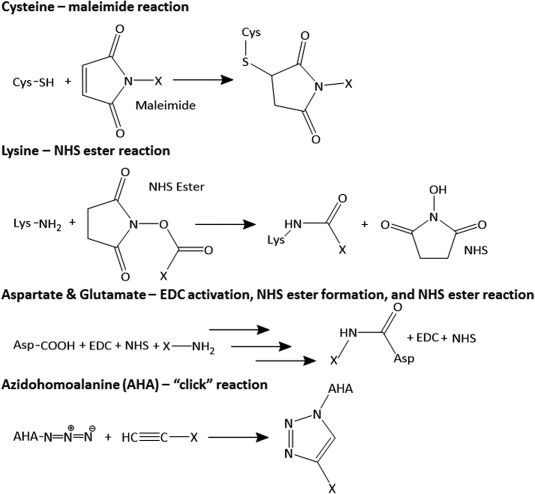
Common conjugation chemistries. Reactions used to functionalize the exterior and interior of VLPs at reactive amino acids (X is a ligand or cargo)
Table 3.
Surface ligands displayed on the VLPs
| VLP | Surface functionalization | Method | References |
|---|---|---|---|
| HBVc | Antibody fragment | Genetic fusion to coat protein | 67 |
| Green fluorescent protein | Genetic fusion to coat protein | 67 | |
| Flagellin | “Click” chemistry | 101 | |
| MS2 | Antibody fragment | “Click” chemistry | 79 |
| Transferrin | Conjugated to surface lysines | 66 | |
| DNA aptamers | paF‐based oxidative ring contraction | 84,89,100 | |
| Granulocyte macrophage colony‐stimulating factor | “Click” chemistry | 79 | |
| Nucleic acids | “Click” chemistry | 79 | |
| PEG | “Click” chemistry | 79 | |
| paF‐based oxidative ring contraction | 89,121 | ||
| Foreign epitopes (as a selection screen) | Genetic fusion to coat protein | 68 | |
| HIV‐Tat cell‐penetrating peptide | Conjugated to surface cysteines | 90,95,102 | |
| Qβ | Glycans | “Click” chemistry | 85 |
| Conjugated to surface lysines | 52,142 | ||
| Human epidermal growth factor | Genetic fusion to coat protein | 69 | |
| Antibody fragment | “Click” chemistry | 79 | |
| Transferrin | “Click” chemistry | 141 | |
| Ganglioside GM2 tumor‐associated carbohydrate antigen | “Click” chemistry | 91 | |
| Metalloporphyrin derivative | “Click” chemistry | 142 | |
| Granulocyte macrophage colony‐stimulating factor | “Click” chemistry | 79 | |
| Nucleic acids | “Click” chemistry | 79 | |
| PEG | “Click” chemistry | 79 | |
| P22 | CD47 “self‐peptide” | Genetic fusion to “decoration protein” | 78 |
| CD40L | Genetic fusion to “decoration protein” | 78 | |
| HIV‐Tat cell‐penetrating peptide | Conjugated to surface cysteines | 24 | |
| Peptide tags (for further modification) | Genetic fusion to coat protein | 70 | |
| MIANS (fluorescent probe) | Conjugated to surface cysteines | 74 | |
| CCMV | Foreign epitope (S9 peptide) | Conjugated to surface cysteines | 92 |
| Alkynes | “Click” chemistry | 105 | |
| PEG | Conjugated to surface lysines | 98 | |
| Biotin | Conjugated to surface lysines | 99 | |
| Fluorescent probes | Conjugated to cysteines, lysines, aspartates, or glutamates | 72 | |
| Peptides | Conjugated to cysteines, lysines, aspartates, or glutamates | 72 | |
| CPMV | RGD peptide (integrin‐binding) | “Click” chemistry | 96 |
| Conjugated to surface lysines | 96 | ||
| Pan‐bombesin analogue (with fluorescent probes and PEG) | “Click” chemistry | 58 | |
| Glycans | “Click” chemistry | 85 | |
| Conjugated to surface lysines | 94 | ||
| Folic acid‐PEG | “Click” chemistry | 86 | |
| Foreign epitope (peptide antigens) | Genetic fusion to coat protein | 71 | |
| Conjugated to surface cysteines | 92 | ||
| Fluorescent probes | Conjugated to surface cysteines or lysines | 73,82,135 | |
| PEG | Conjugated to surface lysines | 82,135 | |
| R5 cell‐penetrating peptides | Conjugated to surface lysines | 80 | |
| VEGFR‐1 ligand | Conjugated to surface lysines | 81 | |
| Gd‐DOTA | “Click” chemistry | 103,104 | |
| Heterologous proteins | Conjugated to surface cysteines or lysines | 93 |
5.1.1. Cysteine‐based modifications
Arguably the most commonly used reactive amino acid residue, cysteine, can be presented either naturally or by mutation on the VLP surface. Because of its free sulfhydryl group, cysteine will readily and spontaneously form disulfide bonds with other sulfhydryl‐containing ligands under oxidative conditions. However, the disulfide bond is also easily reduced and may not be ideal for surface attachments. Alternatively, a series of compounds based on maleimide readily and irreversibly form thioether linkages with cysteine residues at a pH between 6.5 and 7.5. This attachment strategy has been used to display cell‐penetrating peptides, fluorescent probes, and heterologous peptides and proteins on the surfaces of MS2, P22, CCMV, and CPMV VLPs.24, 70, 72, 73, 74, 90, 92, 93, 95, 102
5.1.2. Lysine‐based modifications
Another common amino acid residue that is easily modified is lysine because of its primary amine. Using reactions termed n‐hydroxysuccinimide (NHS) ester reactions (because NHS is released as part of the reaction), amide bonds are formed at surface‐exposed lysine residues. The reaction occurs spontaneously between pH 7.2 and 9. This attachment chemistry has been used to display transferrin on MS2, which may allow the VLP to transcytose the blood‐brain barrier, a development that could open up a new library of therapies for neurological disorders.66 Additionally, PEG, peptides, other proteins, and fluorescent probes have been displayed on Qβ, CCMV, and CPMV VLPs using the NHS reaction.72, 73, 80, 81, 82, 88, 94, 95, 98, 99
5.1.3. Aspartate‐ or glutamate‐based modifications
Although not as commonly used, the last class of reactive natural amino acid residues includes the carboxylic acids aspartate and glutamate. Unlike strategies involving cysteine and lysine, coupling to these residues requires multiple steps. First, the carboxylic acid must be activated using 1‐ethyl‐3‐(3‐dimethylaminopropyl)carbodiimide hydrochloride (EDC). Once activated, it will react with NHS to form an NHS ester. Now that the carboxylic acid side‐chain has essentially become an NHS ester, previously described with regard to lysine modifications, we can use a ligand with an exposed primary amine to form a stable amide bond. This strategy has been used primarily with CCMV to display peptides or fluorescent probes.72
5.1.4. Nonnatural amino acid‐based modifications
Beyond the 20 natural amino acids, many nonnatural amino acids have been used for site‐specific protein conjugation reactions. The two nonnatural amino acids most frequently incorporated into VLP coat proteins are azidohomoalanine (AHA) and p‐amino‐phenylalanine (pAF). These amino acids are incorporated into proteins in two ways: global methionine replacement and amber stop codon suppression. Because AHA is very similar to methionine, AHA will be incorporated at each AUG codon if the methionine supply is rate‐limiting; this is termed global methionine replacement.79 Bacteria auxotrophic for methionine or cell‐free protein synthesis can be used to limit methionine availability.38 The protein yield using global methionine replacement can be rather high from optimized procedures, but this approach will not work for pAF. Amber stop codon suppression, although more difficult and providing lower yields, will incorporate pAF. Amber stop codon suppression uses nonnative synthetases and tRNAs that do not react with the natural amino acids to incorporate the nonnatural amino acid at the amber stop codon UAG.84, 89, 100 However, the release factor protein for the amber stop signal is still present, so premature termination of the protein may also occur. Cell‐free protein synthesis offers a definite advantage here since optimized concentrations of the synthetic components can easily be added to the reaction, but premature termination still usually limits product accumulation.
Despite the drawbacks, these methods have been used to incorporate nonnatural amino acids with uniquely reactive side‐chains. AHA, displaying an azide, will participate in copper(I)‐catalyzed azide‐alkyne cycloaddition (“click” reaction) and form covalent triazole rings with alkyne‐containing ligands.79 This method has been used to display antibody fragments, folic acid, and RGD peptides on MS2 and CPMV, all of which have been shown to allow selective targeting of cancer cells.80, 86, 96 Additionally, heterologous proteins, peptides, nucleic acids, and PEG have been displayed on HBVc, MS2, Qβ, CCMV, and CPMV VLPs using this approach.79, 85, 91, 101, 103, 104, 105 pAF has been incorporated into MS2 and conjugated to ligands displaying phenylene diamines and aminophenols.84, 89, 100
5.1.5. Genetic modifications
The final covalent attachment method we will discuss is genetic modification, in which the gene for the desired surface ligand is fused to the gene for the coat protein of the VLP. While the added peptide can inhibit protein folding as well as VLP assembly, this approach has been shown to work for most VLPs, but not CCMV.67, 68, 69, 70, 71 Most of the work has fused proteins to either termini of the coat protein, but the HBVc VLP has also been shown to accept heterologous protein domains within the sequence of the coat protein itself.67 As shown in Figure 1, the HBVc VLP possesses 120 “spikes” on its surface. Protein domains have been inserted such that they are displayed at each spike, allowing optimal surface presentation. These genetic fusion methods have been used to display various heterologous proteins including antibody fragments for specific cellular targeting.67
5.1.6. Affinity‐based noncovalent modifications
P22 is unique compared to the other VLPs because of the existence of the decoration (or “dec”) protein. This protein has high affinity for the surface of P22.78 By fusing ligands to the “dec” protein, an affinity‐based noncovalent system was developed for surface display on the P22 VLP without requiring alteration of the coat proteins. This approach was used to display CD40L, derived from T cells, and the CD47 “self‐peptide,” developed by the Discher lab, which shows promise in avoiding phagocyte engulfment.106
5.2. Efficient cargo loading and retention
VLPs have been used to load a range of molecules, including small molecules (chemotherapeutics, fluorescent probes, polymers), nucleic acids, peptides, proteins, and even other NPs.21 The approaches include both covalent and noncovalent methods. Noncovalent methods are ideal as they do not require modification of the cargo, however, covalent methods typically have the advantage of more efficient encapsidation and retention of the cargo. As with surface modifications, most covalent methods for cargo loading take advantage of reactive amino acids and use the same chemistries described above (Figure 2), though some use genetic fusions to the primary amino acid sequence.25, 83, 107 Both methods will be discussed for the different types of cargo. See Table 4 for a list of cargo and loading methods, including specific references.
Table 4.
Cargo loaded by the VLPs
| VLP | Cargo | Method | References |
|---|---|---|---|
| HBVc | RNA (viral, heterologous) | Electrostatic adsorption | 111,112 |
| DNA (CpG, single‐stranded, double‐stranded) | Electrostatic adsorption | 65, 111, 113 | |
| Green fluorescent protein | Passive encapsidation | 116 | |
| Nuclease | Genetic fusion to coat protein | 107 | |
| Iron oxide NP (IONP) | Hexahistidine:NTA coordination | 49 | |
| MS2 | Taxol | Conjugated to surface cysteines | 114 |
| Alexa Fluor® 488 | Conjugated to interior cysteines | 84, 89 | |
| Porphyrin | Conjugated to interior cysteines | 100 | |
| Doxorubicin | Conjugated to stem‐loop RNA | 21 | |
| Fluorescein | Conjugated to interior tyrosines | 115 | |
| DOTA chelators | Conjugated to interior cysteines | 89, 121 | |
| RNA (messenger, micro, small‐interfering) | Genetic fusion to stem‐loop RNA | 21, 42, 66, 90, 102, 95 | |
| Ricin toxin A‐chain | Conjugated to stem‐loop RNA | 21 | |
| HIV‐1 Tat peptide | Conjugated to stem‐loop RNA | 118 | |
| Alkaline phosphatase | Electrostatic attraction to coat protein | 23, 119 | |
| Green fluorescent protein | Electrostatic attraction to coat protein | 23 | |
| Quantum dot 585 | Conjugated to stem‐loop RNA | 21 | |
| Qβ | Methacrylate (monomers, polymers) | “Click” chemistry | 22 |
| CpG DNA | Electrostatic attraction to coat protein | 65 | |
| Fluorescent proteins | Adsorption to extension on stem‐loop RNA | 52 | |
| Luciferase | Adsorption to extension on stem‐loop RNA | 39 | |
| P22 | Nickel | Conjugated to interior cysteines | 41 |
| Biotin | Conjugated to interior cysteines | 124 | |
| Fluorescein polymethacrylate | Conjugated to interior cysteines | 125 | |
| Gadopentetic acid polymethacrylate | Conjugated to interior cysteines | 125 | |
| CRISPR (Cas9 and guide RNA) | Genetic fusion to scaffold protein | 25 | |
| Green fluorescent protein or mCherry | Genetic fusion to scaffold protein | 120, 123 | |
| CellB protein | Genetic fusion to scaffold protein | 120 | |
| [NiFe] hydrogenase | Genetic fusion to scaffold protein | 117 | |
| Ziconotide peptide | Genetic fusion to scaffold protein | 24 | |
| Three enzyme cascade (genetically linked) | Genetic fusion to scaffold protein | 122 | |
| Alcohol dehydrogenase | Genetic fusion to scaffold protein | 127 | |
| CCMV | Polystyrene sulfonate | Electrostatic adsorption | 98, 126 |
| RNA | Electrostatic adsorption | 64 | |
| Green or teal fluorescent Protein | Genetic fusion | 62, 83, 128 | |
| Attraction between “leucine zipper” domains | 62, 83, 128 | ||
| Pseudozyma antarctica lipase B | Attraction between “leucine zipper” domains | 83 | |
| Horseradish peroxidase | Passive encapsidation | 46 | |
| DOTAC10 micelles with Gd(III) or Zn(II) | Electrostatic adsorption | 130 | |
| Gd(DOTA) | “Click” chemistry | 104 | |
| CPMV | Fluorescent probes | Conjugated to interior cysteines | 108 |
| Doxorubicin | Conjugated to surface aspartates or glutamates | 109 | |
| DAPI | Electrostatic adsorption | 110 | |
| Acridine orange | Electrostatic adsorption | 110 | |
| Propidium iodide | Electrostatic adsorption | 110 | |
| Proflavin | Electrostatic adsorption | 110 | |
| Iron oxide NP | Passive encapsidation | 129 | |
| Gd(III) | Coordinated by genomic RNA | 103, 104 | |
| Tb(III) | Coordinated by genomic RNA | 103, 104 |
5.2.1. Small molecules
Unfortunately, there has been no published work describing the loading of small molecules within the HBVc VLP. The bacteriophages, on the other hand, have been used extensively to load small molecules. MS2 has been shown to encapsidate chemotherapeutics (taxol and doxorubicin), fluorescent probes (Alexa Fluor® 488 and fluorescein), and other small molecules (porphyrin and DOTA chelators) through conjugations to interior cysteines (disulfide‐ and maleimide‐based linkages), interior tyrosines (diazonium coupling), and the stem‐loop RNA hairpin required for VLP assembly.21, 84, 89, 100, 114, 115, 121 Qβ VLPs with AHA incorporated using global methionine replacement have been shown to covalently conjugate methacrylate to the coat proteins using the “click” reaction and encapsidate them during VLP assembly.22, 79 Lastly, the P22 VLP has been shown to covalently load nickel ions (through iodo‐phen linkages) and derivatives of biotin, fluorescein, and gadopentetic acid (through maleimide‐based initiators) by conjugating to interior cysteines.45, 124, 125
The plant virus‐based VLPs are unique compared to the others because they allow noncovalent loading of small molecules. CCMV has been shown to load polystyrene sulfonate through noncovalent electrostatic interactions between the cargo and the coat proteins.98, 126 CPMV has been used to covalently load chemotherapeutics (doxorubicin) through conjugation to aspartates and glutamates (EDC/NHS reactions followed by esterification) and maleimide derivatives of fluorescent probes by attachment to cysteine residues.108, 109 Additionally, fluorescent probes and an antibiotic (proflavin) have been noncovalently incorporated within CPMV with the small molecules electrostatically adsorbed by CPMV's RNA genome.110
Although these molecules may not all be therapeutically relevant, the results indicate that the methods can successfully load small molecules into many of the VLPs and the approaches can easily be extended to load other small molecule drugs. However, the fact that the only noncovalent loading of small molecules uses a polymerized cargo (that is bigger than most chemotherapeutics) or adsorbs the molecule within nucleic acids shows how difficult it is to load and retain these small cargoes. This is due to the presence of pores throughout the VLP structures, as seen in Figure 1. The development of nonporous VLPs would allow more efficient noncovalent loading and retention of drugs and will be beneficial for future drug delivery strategies using VLPs.
5.2.2. Nucleic acids
MS2 VLPs are particularly suited to loading RNA. They require a short stem‐loop RNA hairpin, which is typically part of their genomic RNA, to assemble into capsids.42 This sequence has been easily extended to incorporate mRNAs, micro RNAs, and small interfering RNAs.21, 42, 66, 90, 95, 102 HBVc, P22, and CCMV VLPs have all been shown to load RNA through electrostatic interactions between the nucleic acid and the coat proteins.25, 64, 111, 112 HBVc and Qβ VLPs have also used similar principles to load DNA.65, 111, 113 These nucleic acid‐loaded VLPs have been developed for various uses including vaccines and vaccine adjuvants,65 gene delivery systems,42 micro RNA delivery systems,90, 95, 102 gene knockdown systems,21, 66 and gene replacement by delivering guide RNA for the CRISPR system.25 Loading and retaining nucleic acids with VLPs is easier than for small molecules because the nucleic acids are usually much larger and the capsids have evolved to load and carry similar molecules, that is, their viral genomes.
5.2.3. Peptides and proteins
There are four main ways to load peptides or proteins into VLPs: (a) fusing the peptide or protein sequence to the amino acid sequence of the coat protein; (b) conjugating the peptide or protein to the genome; (c) engineering electrostatic interactions between the cargo and the coat protein; and (d) passive encapsidation. The first method loads hundreds of peptides or proteins per VLP. Both the first and second methods are facile, but have two major drawbacks. First, the peptide or protein must be amenable to genetic fusion or nucleic acid conjugation and still fold into an active form while also allowing the VLP subunits or nucleic acid to fold properly. Second, the peptide or protein must be able to exert its effect while fused to the coat proteins or conjugated to the nucleic acid. The third and fourth methods are less effective, though they allow loading of free peptides and proteins. Loading via electrostatic interactions is more effective than passive loading, but only works for peptides or proteins that have (or can be engineered to have) an affinity for the internal surface of the VLPs. HBVc VLPs have been shown to load proteins through genetic fusions either to the C‐terminus or within the protein sequence as well via passive encapsidation.107, 116 MS2 and Qβ VLPs have been used to encapsidate peptides and proteins after conjugating them to RNA containing the stem‐loop hairpin required for assembly.21, 39, 52, 118 MS2 loading has also been facilitated by electrostatic interactions between a poly‐anionic tag on the proteins and the capsid interior.23, 119 P22 has only been shown to load proteins and peptides by genetically fusing them to the scaffold protein, which in these cases is not removed from the VLP after assembly.24, 25, 117, 120, 122, 123, 127 CCMV loading has been accomplished using passive encapsidation, genetic fusions, and leucine zippers added to both the cargo and the coat proteins.46, 62, 83, 128 Given the difficulty in production and purification of CPMV, it is not surprising that it has not been used to load proteins yet.
5.2.4. Nanoparticles
A significant body of work has studied the use of VLPs for the development of improved contrast agents. By loading the standard NP‐based contrast agents within VLPs, the new NPs gain improved relaxivities which then give higher resolution images. Additionally, if the VLPs are further modified to target specific cells, the signal‐to‐noise ratio is increased even further giving clear images of, for example, tumors. To that end, HBVc and CPMV VLPs have been loaded with iron oxide NPs through coordination to the coat proteins or through passive encapsidation.49, 129 CPMV has also been shown to load iron oxide and gadolinium NPs through coordination to the genomic RNA.103, 104 CCMV has been used to load gadolinium derivatives through electrostatic interactions with the coat proteins or “click” chemistry.104, 130 Lastly, unrelated to MRI, MS2 was loaded with quantum dot 585 for particle tracking.21 While not immediately therapeutically relevant, using these VLPs for diagnostics could also greatly improve patient quality‐of‐life by detecting the disease at an earlier stage and more accurately assessing therapeutic efficacy. Furthermore, iron oxide NPs have the possibility of being used for radio frequency ablation to actively destroy targeted tumor cells.131
5.3. NP uniformity and stability
Unlike the metal‐based, liposomal, and polymer‐based NPs, VLPs are highly uniform. VLPs, produced with an exact number of coat proteins and arranged in a consistent geometry, will have significantly lower lot‐to‐lot variability and identical cargo release profiles. Additionally, once inside the targeted cells, the VLPs should degrade and release all of the therapeutic cargo at once—unlike polymer NPs which slowly degrade and release the cargo over time.75 While slow cargo release may occasionally be beneficial, immediate release is likely to be more effective in most cases and especially for cancer treatment.
At the same time, protein‐based design means that the VLPs are not as stable as polymer NPs. Fortunately, this drawback is known and has been studied in the hopes of making better VLPs. These studies focused on HBVc, MS2, and Qβ VLPs. The HBVc VLP forms intradimer disulfide bonds that stabilize the 120 dimers, and Qβ forms disulfide bonds that link the pentameric and hexameric subunits at the 5‐ and 3‐fold axes of symmetry.29 A mutant MS2 VLP was also designed to form disulfide bonds within the pentamers and hexamers similar to Qβ.29, 132 Upon formation of the disulfide bond networks within these VLPs, the dissociation temperatures increased: HBVc from 72–93 to 97, MS2 from 55–70 to 73, and Qβ from 40 to 85–100°C. Furthermore, a mutant HBVc designed with an additional 240 disulfide bonds that covalently link every coat protein was engineered and shown to be stable in PBS and over multiple freeze/thaw cycles, but to disassemble in reducing conditions mimicking the cytosol.40 This mutant VLP shows great promise for use as a delivery vehicle.
5.4. Pharmacokinetics and pharmacodynamics
Although there have not been in vivo biodistribution studies for HBVc and P22 VLPs to our knowledge, in‐depth studies have been performed for MS2, Qβ, CCMV, and CPMV. We focused on studies using intravenous administration into mice or rats as model systems, which are the systems likely to be studied for initial evaluation of VLP‐based targeted therapeutics.
The distribution of MS2 VLPs, labeled internally with 64Cu or 18F, was determined in mice at 24 hr and rats at 3 hr after intravenous administration. In both cases, MS2 accumulated primarily in the liver and the spleen.114, 121 PEGylation of MS2 was also studied since PEG has been shown to act as a “stealth agent” to avoid immune clearance.121 PEGylated MS2 VLPs behaved similarly, except retention in the spleen was significantly reduced.121 This ability to avoid the immune system is extremely valuable as it will likely increase the effective dose that reaches the targeted tissue. Furthermore, work has shown that the CD47 ectodomain or the CD47 “self‐peptide,” which has been displayed on VLPs, can also be used to avoid the immune system.106 Qβ, labeled externally with gadolinium, was also studied in mice at 4–5 hr after intravenous administration.97 Qβ VLPs accumulated in the liver, but unlike MS2, accumulated at lower levels in the spleen.97
The biodistribution of the plant virus‐based VLPs, CCMV, and CPMV, intravenously injected in mice at various times, are mostly similar. They primarily accumulate in the liver, spleen, kidney, and GI tract.61, 103, 133, 134, 135 CCMV, labeled with 125I, also showed significant retention by the thyroid, probably due to the iodine.61 PEGylation of CPMV VLPs greatly reduced accumulation in the liver and spleen, which suggests CCMV and CPMV could also benefit from the CD47 ectodomain displayed on the surface to avoid the immune system.106, 134
Because developing VLP‐based targeted therapies for cancer is a primary application, biodistribution studies in mice possessing tumor xenografts were also conducted. MS2 or PEGylated CPMV VLPs were injected intravenously and partially accumulated in the tumors after 24 hr. This was hypothesized to be because of the EPR effect.121, 135 We suggest that the selective accumulation in these tumors could be greatly improved using cellular targeting ligands displayed on the VLPs, which was described previously, in addition to PEG or the CD47 ectodomain to avoid phagocyte engulfment.106
5.5. Specific cellular targeting and cargo delivery
While many different cell targeting ligands have been evaluated, ranging from glycans to specific receptor‐ligands such as folate and transferrin, the most common targeting ligand is the antibody fragment, although recently RNA and DNA aptamers have been used more frequently.136, 137, 138, 139 Most research has focused on developing ligands to target cancers, such as the anti‐HER2 antibody Trastuzumab for breast cancer and the anti‐PSMA antibodies for prostate cancer.87, 88, 140 However, if an effective delivery vehicle was available, it could spur research toward identifying targets on other cells, such as those involved in mitochondrial disorders and Parkinson's disease. While P22 and CCMV have not been functionalized with targeting agents, to our knowledge, the technology used to display other ligands could be easily translated for this purpose. HBVc, MS2, Qβ, and CPMV VLPs have been functionalized with antibody fragments or other targeting ligands and the targeting of most of these has been studied using cultured cells. The proposed path for these cargo delivery vehicles is outlined in Figure 3.
Figure 3.
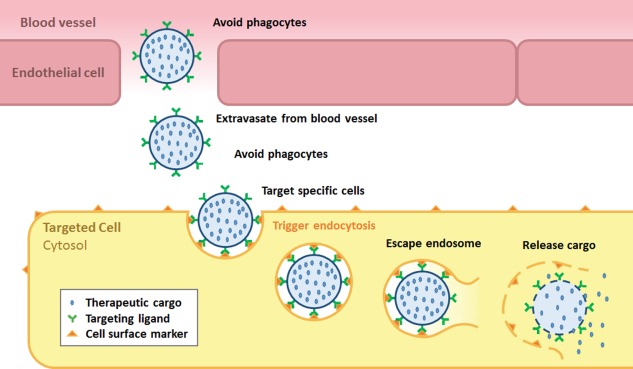
Targeted Delivery Sequence. Stabilized VLPs first extravasate from the blood vessel and then target the specific cells and trigger internalization while avoiding the immune system. Once endocytosed, the VLPs escape the endosome and then disassemble to release their cargo (italics correspond to challenges listed in Table 1)
It has been shown that single‐domain antibody fragments could be displayed on the surface of HBVc VLPs; however, no cell targeting results have been reported.67 Conversely, MS2 and Qβ have been functionalized with various targeting ligands and shown to successfully target specific cells. The Matt Francis lab has functionalized MS2 VLPs with DNA aptamers with affinity for protein tyrosine kinase 7 receptors that are expressed on Jurkat leukemia T cells. They observed efficient and selective targeting of those cells by the VLPs.84 In addition, they modified the interior surface of MS2 with porphyrins for photodynamic therapy and demonstrated that those functionalized MS2 VLPs selectively killed Jurkat cells upon illumination.100 This proved that the cargo retained its functionality after delivery by the VLPs. MS2 displaying human transferrin on its surface and carrying siRNA cargo was also shown to selectively internalize into HeLa cells through receptor‐mediated endocytosis and to deliver functional siRNA.66 Moreover, the MS2 surface has been functionalized with a peptide (SP94) that has high affinity to human hepatocellular carcinoma cells.21 These SP94‐MS2 VLPs delivered their cargo, ricin toxin A‐chain, to the targeted cells and specifically killed those cells without affecting the control cells.21 Antibody fragments also have been displayed on the MS2 surface, although they have not been tested using cell models.79
Notably, the M.G. Finn group functionalized Qβ with human transferrin and observed cellular uptake and internalization of the VLPs through clathrin‐mediated endocytosis in BSC1 cells.141 Furthermore, they displayed glycan ligands on the Qβ surface for specific targeting of cells expressing human CD22 receptors.85 Those VLPs were then loaded with either green fluorescent protein or porphyrin (for photodynamic therapy) and selectively delivered to CHO cells stably expressing human CD22.58, 142 Human epidermal growth factor (EGF) as well as a fluorescent dye were displayed on the Qβ surface, and those functionalized VLPs induced autophosphorylation of the EGF receptor and apoptosis of A431 cells.69 In addition, as with MS2, antibody fragments have been displayed on Qβ, though no cell targeting data have been reported.79
Although there has not been a specific targeting study using CCMV, CCMV VLPs containing EYFP RNA were transfected into mammalian BHK cells.143 Those VLPs were shown to protect the RNA cargo from RNases, and EYFP expression was observed in the BHK cells.143 The Finn group displayed folic acid on CPMV, and showed the specific binding and endocytosis of the functionalized CPMV VLPs by KB cells expressing folic acid receptors.86 They also produced fluorescent dye‐labeled CPMV displaying cyclic RGD ligands to target specific integrins, and those VLPs were selectively endocytosed by several different cells overexpressing the integrins (SW480, A549, and HeLa cancer cells and HEK293 cells).96 Although lacking actual targeting data, the Finn group also displayed transferrin on CPMV.144 In addition, CPMV was functionalized with intron 8, a receptor‐binding module derived from Herstatin, to target HER2 receptors.93 For tumor imaging, NIR dye‐labeled CPMV VLPs were also conjugated to a bombesin analog, and their uptake by PC‐3 prostate cancer cells was observed.58 Tumor homing was further demonstrated using human prostate tumor xenografts on the chicken chorioallantoic membrane model.58 Lastly, CPMV was functionalized with a fluorescent peptide and a VEGFR‐1 specific peptide, F56, to target endothelial cells.81 VEGFR‐1‐targeted CPMV VLPs were shown to selectively target Ea.hy926 human endothelial cells as well as HT‐29 human colon carcinoma tumor xenografts in vivo when injected intravenously.81
In some cases, VLPs have been engineered to display cell‐penetrating peptides as well—either to aid in the initial cell targeting and entry or, when used in lower concentration, to aid in escaping the endosome. The use of such agents for endosomal escape may be needed to enhance the delivery of functional therapeutic cargo. After VLPs containing macromolecular cargo are endocytosed by the targeted cells, they must escape the endosome before reaching the end of the endosomal pathway: the lysosome. The lysosome will degrade the VLPs and any nucleic acid, peptide, or protein cargo they contain. Conversely, many small molecule cargoes should remain functional after VLP degradation. Previous work has displayed three different cell‐penetrating peptides on three different VLPs: MS2 has been functionalized with the HIV‐Tat peptide and a histidine‐rich H5WYG peptide, P22 has also been functionalized with the HIV‐Tat peptide, and CPMV has been functionalized with arginine‐rich R5 peptides.21, 24, 80, 90, 95, 102 One proposed mechanism of cationic cell‐penetrating peptides (HIV‐Tat and the arginine‐rich R5 peptides) is through a direct electrostatic interaction with the negatively charged phospholipids that form the endosomal membrane. This is postulated to result in membrane destabilization and endosome lysis.31, 145 Cell‐penetrating peptides containing protonatable secondary and/or tertiary amine groups (histidine‐rich H5WYG peptide) can absorb protons across the endosomal membrane, resulting in a swelling from an influx of water and/or ions and leading to rupture of the endosomal vesicle. This is known as the “proton sponge effect.”31 Although there are some working examples of these peptides, further research is needed.
6. Conclusions and perspectives
Although VLP‐based targeted drug delivery remains a nascent technology that requires further studies to prove its clinical efficacy, significant progress has been made. Many of the initial disadvantages of using VLPs have been remedied, as shown in Table 1, and the previous studies explored in this article have laid excellent groundwork for addressing the remaining challenges. Although each VLP has advantages and disadvantages relative to each other, we believe that HBVc, Qβ, and MS2 show the most promise. The advantages of these VLPs are that they:
can load small molecules, nucleic acids, and proteins21, 22, 52, 65, 113, 126
can incorporate nonnatural amino acids for ease of surface functionalization through the “click” reaction67, 79
can be functionalized to display antibody fragments for specific cellular targeting67, 79
can be functionalized to display PEG to avoid the immune system (not shown for HBVc VLPs)79
will disassemble in the reducing conditions of the cytosol to release their cargo (not shown for MS2 VLPs)29, 40
Although it has not been experimentally proven, the disulfide bonded mutant of MS2 should behave similarly to Qβ and the disulfide bonded mutant of HBVc and disassemble in cytosolic conditions.29, 40, 132 Likewise, although HBVc has not been functionalized with PEG to our knowledge, the ease of nonnatural amino acid presentation and “click” conjugation will facilitate such experiments.79, 101 Additionally, all three can be functionalized with the CD47 ectodomain or the CD47 “self‐peptide” for potential avoidance of phagocytic clearance.106 MS2 and Qβ have also been functionalized with transferrin which may allow transcytosis across the blood‐brain barrier, allowing the VLPs to be used for neurological disorders.66, 141 While P22, CCMV, and CPMV do not currently have the same advantages as the other VLPs, we believe the same technology can be applied for them in the future.
It is also suggested that additional work focus on fully overcoming the challenges listed in Table 1. Currently, significant work has achieved loading of a variety of therapeutic cargo as well as specific cell targeting. However, efforts toward conditional VLP stabilization (including intracellular cargo release), phagocytic avoidance, and endosomal escape need to be continued. The final two relatively untouched areas where additional progress would greatly improve this technology are: (a) improving extravasation from the blood vessel to increase local concentration around the targeted cells and reduce clearance, and (b) reducing off‐target organ accumulation, mainly in the liver, kidney, and spleen. We suggest that the advances summarized here, and the suggested future directions, indicate a bright and important future for VLP‐mediated targeted drug delivery.
Literature cited
- 1. National Cancer Institute . Cancer statistics. 2016. http://www.cancer.gov/about-cancer/understanding/statistics. Accessed July 28, 2016.
- 2. Srinivasarao M, Galliford CV, Low PS. Principles in the design of ligand‐targeted cancer therapeutics and imaging agents. Nat Rev Drug Discov. 2015;14(3):203–219. [DOI] [PubMed] [Google Scholar]
- 3. Ginn SL, Alexander IE, Edelstein ML, Abedi MR, Wixon J. Gene therapy clinical trials worldwide to 2012 ‐ an update. J Gene Med. 2013;15(2):65–77. [DOI] [PubMed] [Google Scholar]
- 4. Casi G, Neri D. Antibody‐drug conjugates and small molecule‐drug conjugates: opportunities and challenges for the development of selective anticancer cytotoxic agents. J Med Chem. 2015;58(22):8751–8761. [DOI] [PubMed] [Google Scholar]
- 5. Dawidczyk CM, Kim C, Park JH, et al. State‐of‐the‐art in design rules for drug delivery platforms: lessons learned from FDA‐approved nanomedicines. J Control Release. 2014;187:133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Smith MT, Hawes AK, Bundy BC. Reengineering viruses and virus‐like particles through chemical functionalization strategies. Curr Opin Biotechnol. 2013;24(4):620–626. [DOI] [PubMed] [Google Scholar]
- 7. Glasgow J, Tullman‐Ercek D. Production and applications of engineered viral capsids. Appl Microbiol Biotechnol. 2014;98(13):5847–5858. [DOI] [PubMed] [Google Scholar]
- 8. Schoonen L, van Hest JCM. Functionalization of protein‐based nanocages for drug delivery applications. Nanoscale. 2014;6(13):7124–7141. [DOI] [PubMed] [Google Scholar]
- 9. Lee EJ, Lee NK, Kim IS. Bioengineered protein‐based nanocage for drug delivery. Adv Drug Deliv Rev. 2016;106:(1):157–171. [DOI] [PubMed] [Google Scholar]
- 10. Rink JS, Plebanek MP, Tripathy S, Thaxton CS. Update on current and potential nanoparticle cancer therapies. Curr Opin Oncol. 2013;25(6):646–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pillai G. Nanomedicines for cancer therapy: an update of FDA approved and those under various stages of development. http://www.symbiosisonlinepublishing.com/pharmacy-pharmaceuticalsciences/pharmacy-pharmaceuticalsciences09.php. Accessed May 19, 2016.
- 12. Pattni BS, Chupin VV, Torchilin VP. New developments in liposomal drug delivery. Chem Rev. 2015;115(19):10938–10966. [DOI] [PubMed] [Google Scholar]
- 13. Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev. 2013;65(1):36–48. [DOI] [PubMed] [Google Scholar]
- 14. Elsabahy M, Wooley KL. Design of polymeric nanoparticles for biomedical delivery applications. Chem Soc Rev. 2012;41(7):2545–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brewer E, Coleman J, Lowman A. Emerging technologies of polymeric nanoparticles in cancer drug delivery. J Nanomater. 2011;2011:1–10. 21808638 [Google Scholar]
- 16. Wang AZ, Langer R, Farokhzad OC. Nanoparticle delivery of cancer drugs. Annu Rev Med. 2012;63:185–198. [DOI] [PubMed] [Google Scholar]
- 17. Hong RL, Huang CJ, Tseng YL, et al. Direct comparison of liposomal doxorubicin with or without polyethylene glycol coating in C‐26 tumor‐bearing mice: is surface coating with polyethylene glycol beneficial? Clin Cancer Res. 1999;5(11):3645–3652. [PubMed] [Google Scholar]
- 18. Armstrong JK, Hempel G, Koling S, et al. Antibody against poly(ethylene glycol) adversely affects PEG‐asparaginase therapy in acute lymphoblastic leukemia patients. Cancer. 2007;110(1):103–111. [DOI] [PubMed] [Google Scholar]
- 19. Vabbilisetty P, Sun X‐L. Liposome surface functionalization based on different anchoring lipids via Staudinger ligation. Org Biomol Chem. 2014;12(8):1237–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yildiz I, Shukla S, Steinmetz NF. Applications of viral nanoparticles in medicine. Curr Opin Biotechnol. 2011;22(6):901–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ashley CE, Carnes EC, Phillips GK, et al. Cell‐specific delivery of diverse cargos by bacteriophage MS2 virus‐like particles. ACS Nano. 2011;5(7):5729–5745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hovlid ML, Lau JL, Breitenkamp K, et al. Encapsidated atom‐transfer radical polymerization in Qβ virus‐like nanoparticles. ACS Nano. 2014;8(8):8003–8014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Glasgow JE, Capehart SL, Francis MB, Tullman‐Ercek D. Osmolyte‐mediated encapsulation of proteins inside MS2 viral capsids. ACS Nano. 2012;6(10):8658–8664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Anand P, O'Neil A, Lin E, Douglas T, Holford M. Tailored delivery of analgesic ziconotide across a blood brain barrier model using viral nanocontainers. Sci Rep. 2015;5:12497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Qazi S, Miettinen HM, Wilkinson RA, McCoy K, Douglas T, Wiedenheft B. Programmed self‐assembly of an active P22‐Cas9 nanocarrier system. Mol Pharm. 2016;13(3):1191–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lau JL, Baksh MM, Fiedler JD, et al. Evolution and protein packaging of small‐molecule RNA aptamers. ACS Nano. 2011;5(10):7722–7729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kelly P, Anand P, Uvaydov A, et al. Developing a dissociative nanocontainer for peptide drug delivery. Int J Environ Res Public Health. 2015;12(10):12543–12555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yildiz I, Tsvetkova I, Wen AM, et al. Engineering of brome mosaic virus for biomedical applications. RSC Adv. 2012;2(9):3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bundy BC, Swartz JR. Efficient disulfide bond formation in virus‐like particles. J Biotechnol. 2011;154(4):230–239. [DOI] [PubMed] [Google Scholar]
- 30. Champion JA, Katare YK, Mitragotri S. Particle shape: a new design parameter for micro‐ and nanoscale drug delivery carriers. J Control Release. 2007;121(1‐2):3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol. 2015;33(9):941–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Plevka P, Tars K, Liljas L. Structure and stability of icosahedral particles of a covalent coat protein dimer of bacteriophage MS2. Protein Sci. 2009;18(8):1653–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Golmohammadi R, Fridborg K, Bundule M, Valegård K, Liljas L. The crystal structure of bacteriophage Q beta at 3.5 A resolution. Structure. 1996;4(5):543–554. [DOI] [PubMed] [Google Scholar]
- 34. Parent KN, Khayat R, Tu LH, et al. P22 coat protein structures reveal a novel mechanism for capsid maturation: stability without auxiliary proteins or chemical crosslinks. Structure. 2010;18(3):390–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Speir JA, Bothner B, Qu C, Willits DA, Young MJ, Johnson JE. Enhanced local symmetry interactions globally stabilize a mutant virus capsid that maintains infectivity and capsid dynamics. J Virol. 2006;80(7):3582–3591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huynh NT, Hesketh EL, Saxena P, et al. Crystal structure and proteomics analysis of empty virus‐like particles of cowpea mosaic virus. Structure. 2016;24(4):567–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wynne SA, Crowther RA, Leslie AG. The crystal structure of the human hepatitis B virus capsid. Mol Cell. 1999;3(6):771–780. [DOI] [PubMed] [Google Scholar]
- 38. Bundy BC, Franciszkowicz MJ, Swartz JR. Escherichia coli‐based cell‐free synthesis of virus‐like particles. Biotechnol Bioeng. 2008;100(1):28–37. [DOI] [PubMed] [Google Scholar]
- 39. Fiedler JD, Brown SD, Lau JL, Finn MG. RNA‐directed packaging of enzymes within virus‐like particles. Angew Chem Int Ed Engl. 2010;49(50):9648–9651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lu Y, Chan W, Ko BY, VanLang CC, Swartz JR. Assessing sequence plasticity of a virus‐like nanoparticle by evolution toward a versatile scaffold for vaccines and drug delivery. Proc Natl Acad Sci. 2015;112(40):12360–12365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Uchida M, Morris DS, Kang S, et al. Site‐directed coordination chemistry with P22 virus‐like particles. Langmuir. 2012;28(4):1998–2006. [DOI] [PubMed] [Google Scholar]
- 42. Prel A, Caval V, Gayon R, et al. Highly efficient in vitro and in vivo delivery of functional RNAs using new versatile MS2‐chimeric retrovirus‐like particles. Mol Ther Methods Clin Dev. 2015;2:15039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fiedler JD, Higginson C, Hovlid ML, et al. Engineered mutations change the structure and stability of a virus‐like particle. Biomacromolecules. 2012;13(8):2339–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ponchon L, Catala M, Seijo B, et al. Co‐expression of RNA‐protein complexes in Escherichia coli and applications to RNA biology. Nucleic Acids Res. 2013;41(15):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rumnieks J, Ose V, Tars K, et al. Assembly of mixed rod‐like and spherical particles from group I and II RNA bacteriophage coat proteins. Virology. 2009;391(2):187–194. [DOI] [PubMed] [Google Scholar]
- 46. Comellas‐Aragonès M, Engelkamp H, Claessen VI, et al. A virus‐based single‐enzyme nanoreactor. Nat Nanotechnol. 2007;2(10):635–639. [DOI] [PubMed] [Google Scholar]
- 47. Johnson JM, Willits DA, Young MJ, Zlotnick A. Interaction with capsid protein alters RNA structure and the pathway for in vitro assembly of cowpea chlorotic mottle virus. J Mol Biol. 2004;335(2):455–464. [DOI] [PubMed] [Google Scholar]
- 48. Butler PJG, Hepatitis B. Virus (HBV): the life‐cycle and assembly of a complex virus In: Pifat‐Mrzljak G, ed. Supramolecular Structure and Function 9. Netherlands: Springer; 2007:131–151. [Google Scholar]
- 49. Shen L, Zhou J, Wang Y, et al. Efficient encapsulation of Fe3O4 nanoparticles into genetically engineered hepatitis B core virus‐like particles through a specific interaction for potential bioapplications. Small. 2015;11(9‐10):1190–1196. [DOI] [PubMed] [Google Scholar]
- 50. Yoshimura H, Edwards E, Uchida M, et al. Two‐dimensional crystallization of P22 virus‐like particles. J Phys Chem B. 2016;120(26):5938–5944. [DOI] [PubMed] [Google Scholar]
- 51. Nguyen TH, Easter N, Gutierrez L, et al. The RNA core weakly influences the interactions of the bacteriophage MS2 at key environmental interfaces. Soft Matter. 2011;7(21):10449–10456. [Google Scholar]
- 52. Rhee J‐K, Hovlid M, Fiedler JD, et al. Colorful virus‐like particles: fluorescent protein packaging by the Qβ capsid. Biomacromolecules. 2011;12(11):3977–3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bedwell GJ, Zhou Z, Uchida M, Douglas T, Gupta A, Prevelige PE. Selective biotemplated synthesis of TiO2 inside a protein cage. Biomacromolecules. 2015;16(1):214–218. [DOI] [PubMed] [Google Scholar]
- 54. Lagoutte P, Mignon C, Donnat S, et al. Scalable chromatography‐based purification of virus‐like particle carrier for epitope based influenza A vaccine produced in Escherichia coli . J Virol Methods. 2016;232:8–11. [DOI] [PubMed] [Google Scholar]
- 55. Patterson DP, LaFrance B, Douglas T. Rescuing recombinant proteins by sequestration into the P22 VLP. Chem Commun. 2013;49(88):10412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Patterson DP, Schwarz B, El‐Boubbou K, van der Oost J, Prevelige PE, Douglas T. Virus‐like particle nanoreactors: programmed encapsulation of the thermostable CelB glycosidase inside the P22 capsid. Soft Matter. 2012;8(39):10158. [Google Scholar]
- 57. Bessa J, Kopf M, Bachmann MF. Cutting edge: IL‐21 and TLR signaling regulate germinal center responses in a B cell‐intrinsic manner. J Immunol. 2010;184(9):4615–4619. [DOI] [PubMed] [Google Scholar]
- 58. Steinmetz NF, Ablack AL, Hickey JL, et al. Intravital imaging of human prostate cancer using viral nanoparticles targeted to gastrin‐releasing peptide receptors. Small. 2011;7(12):1664–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Saunders K, Sainsbury F, Lomonossoff GP. Efficient generation of cowpea mosaic virus empty virus‐like particles by the proteolytic processing of precursors in insect cells and plants. Virology. 2009;393(2):329–337. [DOI] [PubMed] [Google Scholar]
- 60. Russell JT, Lin Y, Böker A, et al. Self‐assembly and cross‐linking of bionanoparticles at liquid‐liquid interfaces. Angew Chem Int Ed Engl. 2005;44(16):2420–2426. [DOI] [PubMed] [Google Scholar]
- 61. Kaiser CR, Flenniken ML, Gillitzer E, et al. Biodistribution studies of protein cage nanoparticles demonstrate broad tissue distribution and rapid clearance in vivo. Int J Nanomed. 2007;2(4):715–733. [PMC free article] [PubMed] [Google Scholar]
- 62. Rurup WF, Verbij F, Koay MST, Blum C, Subramaniam V, Cornelissen JJLM. Predicting the loading of virus‐like particles with fluorescent proteins. Biomacromolecules. 2014;15(2):558–563. [DOI] [PubMed] [Google Scholar]
- 63. Garmann RF, Comas‐Garcia M, Gopal A, Knobler CM, Gelbart WM. The assembly pathway of an icosahedral single‐stranded RNA virus depends on the strength of inter‐subunit attractions. J Mol Biol. 2014;426(5):1050–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cadena‐Nava RD, Comas‐Garcia M, Garmann RF, Rao ALN, Knobler CM, Gelbart WM. Self‐assembly of viral capsid protein and RNA molecules of different sizes: requirement for a specific high protein/RNA mass ratio. J Virol. 2012;86(6):3318–3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Storni T, Ruedl C, Schwarz K, Schwendener RA, Renner WA, Bachmann MF. Nonmethylated CG motifs packaged into virus‐like particles induce protective cytotoxic T cell responses in the absence of systemic side effects. J Immunol. 2004;172(3):1777–1785. [DOI] [PubMed] [Google Scholar]
- 66. Galaway FA, Stockley PG. MS2 viruslike particles: a robust, semisynthetic targeted drug delivery platform. Mol Pharm. 2013;10(1):59–68. [DOI] [PubMed] [Google Scholar]
- 67. Peyret H, Gehin A, Thuenemann EC, et al. Tandem fusion of hepatitis B core antigen allows assembly of virus‐like particles in bacteria and plants with enhanced capacity to accommodate foreign proteins. PLoS One. 2015;10(4):e0120751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. O'Rourke JP, Peabody DS, Chackerian B. Affinity selection of epitope‐based vaccines using a bacteriophage virus‐like particle platform. Curr Opin Virol. 2015;11:76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Pokorski JK, Hovlid ML, Finn MG. Cell targeting with hybrid Qβ virus‐like particles displaying epidermal growth factor. ChemBioChem. 2011;12(16):2441–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Servid A, Jordan P, O'Neil A, Prevelige P, Douglas T. Location of the bacteriophage P22 coat protein C‐terminus provides opportunities for the design of capsid‐based materials. Biomacromolecules. 2013;14(9):2989–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Phelps JP, Dang N, Rasochova L. Inactivation and purification of cowpea mosaic virus‐like particles displaying peptide antigens from Bacillus anthracis . J Virol Methods. 2007;141(2):146–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gillitzer E, Willits D, Young M, Douglas T. Chemical modification of a viral cage for multivalent presentation. Chem Commun. 2002;20:2390–2391. [DOI] [PubMed] [Google Scholar]
- 73. Chatterji A, Ochoa WF, Paine M, Ratna BR, Johnson JE, Lin T. New addresses on an addressable virus nanoblock: Uniquely reactive Lys residues on cowpea mosaic virus. J Chem Biol. 2004;11(6):855–863. [DOI] [PubMed] [Google Scholar]
- 74. Kang S, Lander GC, Johnson JE, Prevelige PE. Development of bacteriophage P22 a platform for molecular display: genetic and chemical modifications of the procapsid exterior surface. ChemBioChem. 2008;9(4):514–518. [DOI] [PubMed] [Google Scholar]
- 75. Steve MH, Siegel SP, Drug C, Carrier D. Poly lactic‐co‐glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers (Basel). 2012;3(3):1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Schrand AM, Rahman MF, Hussain SM, Schlager JJ, Smith DA, Syed AF. Metal‐based nanoparticles and their toxicity assessment. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2010;2(5):544–568. [DOI] [PubMed] [Google Scholar]
- 77. Narang AS, Chang RK, Hussain MA. Pharmaceutical development and regulatory considerations for nanoparticles and nanoparticulate drug delivery systems. J Pharm Sci. 2013;102(11):3867–3882. [DOI] [PubMed] [Google Scholar]
- 78. Schwarz B, Madden P, Avera J, et al. Symmetry controlled, genetic presentation of bioactive proteins on the P22 virus‐like particle using an external decoration protein. ACS Nano. 2015;9(9):9134–9147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Patel KG, Swartz JR. Surface functionalization of virus‐like particles by direct conjugation using azide ‐ alkyne click chemistry. Bioconjug Chem. 2011;22(3):376–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wu Z, Chen K, Yildiz I, et al. Development of viral nanoparticles for efficient intracellular delivery. Nanoscale. 2012;4(11):3567–3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Brunel FM, Lewis JD, Destito G, et al. Hydrazone ligation strategy to assemble multifunctional viral nanoparticles for cell imaging and tumor targeting. Nano Lett. 2010;10(3):1093–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Steinmetz NF, Manchester M. PEGylated viral nanoparticles for biomedicine: the impact of PEG chain length on VNP cell interactions in vitro and ex vivo. Biomacromolecules. 2009;10(4):784–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Minten IJ, Claessen VI, Blank K, Rowan AE, Nolte RJM, Cornelissen JJLM. Catalytic capsids: the art of confinement. Chem Sci. 2011;2(2):358. [Google Scholar]
- 84. Tong GJ, Hsiao SC, Carrico ZM, Francis MB. Viral capsid DNA aptamer conjugates as multivalent cell‐targeting vehicles. J Am Chem Soc. 2009;131(31):11174–11178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kaltgrad E, O' Reilly MK, Liao L, Han S, Paulson JC, Finn MG. On‐virus construction of polyvalent glycan ligands for cell‐surface receptors. J Am Chem Soc. 2008;130(14):4578–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Destito G, Yeh R, Rae CS, Finn MG, Manchester M. Folic acid‐mediated targeting of cowpea mosaic virus particles to tumor cells. Chem Biol. 2007;14(10):1152–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Lewis Phillips GD, Li G, Dugger DL, et al. Targeting HER2‐positive breast cancer with trastuzumab‐DM1, an antibody‐cytotoxic drug conjugate. Cancer Res. 2008;68(22):9280–9290. [DOI] [PubMed] [Google Scholar]
- 88. Goldenberg MM. Trastuzumab, a recombinant DNA‐derived humanized monoclonal antibody, a novel agent for the treatment of metastatic breast cancer. Clin Ther. 1999;21(2):309–318. [DOI] [PubMed] [Google Scholar]
- 89. Behrens CR, Hooker JM, Obermeyer AC, Romanini DW, Katz EM, Francis MB. Rapid chemoselective bioconjugation through oxidative coupling of anilines and aminophenols. J Am Chem Soc. 2011;133(41):16398–16401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Yao Y, Jia T, Pan Y, et al. Using a novel microRNA delivery system to inhibit osteoclastogenesis. Int J Mol Sci. 2015;16(4):8337–8350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Yin Z, Dulaney S, McKay CS, et al. Chemical synthesis of GM2 glycans, bioconjugation with bacteriophage Qβ, and the induction of anticancer antibodies. ChemBioChem. 2016;17(2):174–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Pomwised R, Intamaso U, Teintze M, Young M, Pincus S. Coupling peptide antigens to virus‐like particles or to protein carriers influences the Th1/Th2 polarity of the resulting immune response. Vaccines. 2016;4(2):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Chatterji A, Ochoa W, Shamieh L, et al. Chemical conjugation of heterologous proteins on the surface of cowpea mosaic virus. Bioconjug Chem. 2004;15(4):807–813. [DOI] [PubMed] [Google Scholar]
- 94. Astronomo RD, Kaltgrad E, Udit AK, et al. Defining criteria for oligomannose immunogens for HIV using icosahedral virus capsid scaffolds. Chem Biol. 2010;17(4):357–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Pan Y, Jia T, Zhang Y, et al. MS2 VLP‐based delivery of microRNA‐146a inhibits autoantibody production in lupus‐prone mice. Int J Nanomed. 2012;7:5957–5967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Hovlid ML, Steinmetz NF, Laufer B, et al. Guiding plant virus particles to integrin‐displaying cells. Nanoscale. 2012;4(12):3698–3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Prasuhn DE, Singh P, Strable E, Brown S, Manchester M, Finn MG. Plasma clearance of bacteriophage Qbeta particles as a function of surface charge. J Am Chem Soc. 2008;130(4):1328–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Comellas‐aragone M, Escosura D, Dirks ATJ, et al. Controlled integration of polymers into viral capsids. Biomacromolecules. 2009;10(11):3141–3147. [DOI] [PubMed] [Google Scholar]
- 99. Gillitzer E, Suci P, Young M, Douglas T. Controlled ligand display on a symmetrical protein‐cage architecture through mixed assembly. Small. 2006;2(8‐9):962–966. [DOI] [PubMed] [Google Scholar]
- 100. Stephanopoulos N, Tong GJ, Hsiao SC, Francis MB. Dual‐surface modified virus capsids for targeted delivery of photodynamic agents to cancer cells. ACS Nano. 2010;4(10):6014–6020. [DOI] [PubMed] [Google Scholar]
- 101. Lu Y, Welsh JP, Chan W, Swartz JR. Escherichia coli‐based cell free production of flagellin and ordered flagellin display on virus‐like particles. Biotechnol Bioeng. 2013;110(8):2073–2085. [DOI] [PubMed] [Google Scholar]
- 102. Pan Y, Zhang Y, Jia T, Zhang K, Li J, Wang L. Development of a microRNA delivery system based on bacteriophage MS2 virus‐like particles. FEBS J. 2012;279(7):1198–1208. [DOI] [PubMed] [Google Scholar]
- 103. Singh P, Prasuhn D, Yeh RM, et al. Bio‐distribution, toxicity and pathology of cowpea mosaic virus nanoparticles in vivo. J Control Release. 2007;120(1‐2):41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Prasuhn DE, Yeh RM, Obenaus A, Manchester M, Finn MG. Viral MRI contrast agents: coordination of Gd by native virions and attachment of Gd complexes by azide‐alkyne cycloaddition. Chem Commun. 2007;12:1269–1271. [DOI] [PubMed] [Google Scholar]
- 105. Hommersom CA, Matt B, van der Ham A, Cornelissen JJLM, Katsonis N. Versatile post‐functionalization of the external shell of cowpea chlorotic mottle virus by using click chemistry. Org Biomol Chem. 2014;12(24):4065–4069. [DOI] [PubMed] [Google Scholar]
- 106. Rodriguez PL, Harada T, Christian DA, Pantano DA, Tsai RK, Discher DE. Minimal “self” peptides that inhibit phagocytic clearance and enhance delivery of nanoparticles. Science. 2013;339(6122):971–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Beterams G, Böttcher B, Nassal M. Packaging of up to 240 subunits of a 17 kDa nuclease into the interior of recombinant hepatitis B virus capsids. FEBS Lett. 2000;481(2):169–176. [DOI] [PubMed] [Google Scholar]
- 108. Ochoa WF, Chatterji A, Lin T, Johnson JE. Generation and structural analysis of reactive empty particles derived from an icosahedral virus. Chem Biol. 2006;13(7):771–778. [DOI] [PubMed] [Google Scholar]
- 109. Aljabali AAA, Shukla S, Lomonossoff GP, Steinmetz NF, Evans DJ. CPMV‐DOX delivers. Mol Pharm. 2013;10(1):3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Yildiz I, Lee KL, Chen K, Shukla S, Steinmetz NF. Infusion of imaging and therapeutic molecules into the plant virus‐based carrier cowpea mosaic virus: cargo‐loading and delivery. J Control Release. 2013;172(2):568–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Strods A, Ose V, Bogans J, et al. Preparation by alkaline treatment and detailed characterisation of empty hepatitis B virus core particles for vaccine and gene therapy applications. Sci Rep. 2015;5:11639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Porterfield JZ, Dhason MS, Loeb DD, Nassal M, Stray SJ, Zlotnick A. Full‐length hepatitis B virus core protein packages viral and heterologous RNA with similarly high levels of cooperativity. J Virol. 2010;84(14):7174–7184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Dhason MS, Wang JCY, Hagan MF, Zlotnick A. Differential assembly of hepatitis B virus core protein on single‐ and double‐stranded nucleic acid suggest the dsDNA‐filled core is spring‐loaded. Virology. 2012;430(1):20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Hooker JM, O'Neil JP, Romanini DW, Taylor SE, Francis MB. Genome‐free viral capsids as carriers for positron emission tomography radiolabels. Mol Imaging Biol. 2008;10(4):182–191. [DOI] [PubMed] [Google Scholar]
- 115. Kovacs EW, Hooker JM, Romanini DW, Holder PG, Berry KE, Francis MB. Dual‐surface‐modified bacteriophage MS2 as an ideal scaffold for a viral capsid‐based drug delivery system. Bioconjug Chem. 2007;18(4):1140–1147. [DOI] [PubMed] [Google Scholar]
- 116. Lee KW, Tan WS. Recombinant hepatitis B virus core particles: association, dissociation and encapsidation of green fluorescent protein. J Virol Methods. 2008;151(2):172–180. [DOI] [PubMed] [Google Scholar]
- 117. Jordan PC, Patterson DP, Saboda KN, et al Self‐assembling biomolecular catalysts for hydrogen production. Nat Chem. 2015;8:1–7. [DOI] [PubMed] [Google Scholar]
- 118. Wei B, Wei Y, Zhang K, et al. Development of an antisense RNA delivery system using conjugates of the MS2 bacteriophage capsids and HIV‐1 TAT cell penetrating peptide. Biomed Pharmacother. 2009;63(4):313–318. [DOI] [PubMed] [Google Scholar]
- 119. Glasgow JE, Asensio MA, Jakobson CM, Francis MB, Tullman‐Ercek D. The influence of electrostatics on small molecule flux through a protein nanoreactor. ACS Synth Biol. 2015;4(9):1011–1019. [DOI] [PubMed] [Google Scholar]
- 120. Llauró A, Luque D, Edwards E, et al. Cargo‐shell and cargo‐cargo couplings govern the mechanics of artificially loaded virus‐derived cages. Nanoscale. 2016;8:9328–9336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Farkas ME, Aanei IL, Behrens CR, et al. PET imaging and biodistribution of chemically modified bacteriophage MS2. Mol Pharm. 2013;10(1):69–76. [DOI] [PubMed] [Google Scholar]
- 122. Patterson DP, Schwarz B, Waters RS, Gedeon T, Douglas T. Encapsulation of an enzyme cascade within the bacteriophage P22 virus‐like particle. ACS Chem Biol. 2014;9(2):359–365. [DOI] [PubMed] [Google Scholar]
- 123. O'Neil A, Reichhardt C, Johnson B, Prevelige PE, Douglas T. Genetically programmed in vivo packaging of protein cargo and its controlled release from bacteriophage P22. Angew Chem Int Ed. 2011;50(32):7425–7428. [DOI] [PubMed] [Google Scholar]
- 124. Kang S, Uchida M, Oneil A, Li R, Prevelige PE, Douglas T. Implementation of P22 viral capsids as nanoplatforms. Biomacromolecules. 2010;11(10):2804–2809. [DOI] [PubMed] [Google Scholar]
- 125. Lucon J, Qazi S, Uchida M, et al. Use of the interior cavity of the P22 capsid for site‐specific initiation of atom‐transfer radical polymerization with high‐density cargo loading. Nat Chem. 2012;4(10):781–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Hu Y, Zandi R, Anavitarte A, Knobler CM, Gelbart WM. Packaging of a polymer by a viral capsid: the interplay between polymer length and capsid size. Biophys J. 2008;94:1428–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Patterson DP, Prevelige PE, Douglas T. Nanoreactors by programmed enzyme encapsulation inside the capsid of the bacteriophage P22. ACS Nano. 2012;6(6):5000–5009. [DOI] [PubMed] [Google Scholar]
- 128. Minten IJ, Hendriks LJA, Nolte RJM, Cornelissen JJLM. Controlled encapsulation of multiple proteins in virus capsids. J Am Chem Soc. 2009;131(49):17771–17773. [DOI] [PubMed] [Google Scholar]
- 129. Aljabali AAA, Sainsbury F, Lomonossoff GP, Evans DJ. Cowpea mosaic virus unmodified empty viruslike particles loaded with metal and metal oxide. Small. 2010;6(7):818–821. [DOI] [PubMed] [Google Scholar]
- 130. Millán JG, Brasch M, Anaya‐Plaza E, et al. Self‐assembly triggered by self‐assembly: optically active, paramagnetic micelles encapsulated in protein cage nanoparticles. J Inorg Biochem. 2014;136:140–146. [DOI] [PubMed] [Google Scholar]
- 131. Nguyen DT, Tzou WS, Zheng L, et al. Enhanced radiofrequency ablation with magnetically directed metallic nanoparticles. Circ Arrhythm Electrophysiol. 2016;9(5):e003820. [DOI] [PubMed] [Google Scholar]
- 132. Ashcroft AE, Lago H, Macedo JMB, Horn WT, Stonehouse NJ, Stockley PG. Engineering thermal stability in RNA phage capsids via disulphide bonds. J Nanosci Nanotechnol. 2005;5(12):2034–2041. [DOI] [PubMed] [Google Scholar]
- 133. Rae CS, Wei Khor I, Wang Q, et al. Systemic trafficking of plant virus nanoparticles in mice via the oral route. Virology. 2005;343(2):224–235. [DOI] [PubMed] [Google Scholar]
- 134. Lewis JD, Destito G, Zijlstra A, et al. Viral nanoparticles as tools for intravital vascular imaging. Nat Med. 2006;12(3):354–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Shukla S, Ablack AL, Wen AM, Lee KL, Lewis JD, Steinmetz NF. Increased tumor homing and tissue penetration of the filamentous plant viral nanoparticle potato virus X. Mol Pharm. 2013;10(1):33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Ray P, White RR. Aptamers for targeted drug delivery. Pharmaceuticals. 2010;3(6):1761–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Bruno JG. A review of therapeutic aptamer conjugates with emphasis on new approaches. Pharmaceuticals. 2013;6(3):340–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Sun H, Zhu X, Lu PY, Rosato RR, Tan W, Zu Y. Oligonucleotide aptamers: new tools for targeted cancer therapy. Mol Ther Nucleic Acids. 2014;3:e182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Zhou J, Rossi JJ. Cell‐type‐specific, aptamer‐functionalized agents for targeted disease therapy. Mol Ther Nucleic Acids. 2014;3:e169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Zhang F, Shan L, Liu Y, et al. An anti‐PSMA bivalent immunotoxin exhibits specificity and efficacy for prostate cancer imaging and therapy. Adv Healthc Mater. 2013;2(5):736–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Banerjee D, Liu AP, Voss NR, Schmid SL, Finn MG. Multivalent display and receptor‐mediated endocytosis of transferrin on virus‐like particles. ChemBioChem. 2010;11(9):1273–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Rhee J‐K, Baksh M, Nycholat C, Paulson JC, Kitagishi H, Finn MG. Glycan‐targeted virus‐like nanoparticles for photodynamic therapy. Biomacromolecules. 2012;13(8):2333–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Azizgolshani O, Garmann RF, Cadena‐Nava R, Knobler CM, Gelbart WM. Reconstituted plant viral capsids can release genes to mammalian cells. Virology. 2013;441(1):12–17. [DOI] [PubMed] [Google Scholar]
- 144. Gupta SS, Kuzelka J, Singh P, Lewis WG, Manchester M, Finn MG. Accelerated bioorthogonal conjugation: a practical method for the ligation of diverse functional molecules to a polyvalent virus scaffold. Bioconjug Chem. 2005;16(6):1572–1579. [DOI] [PubMed] [Google Scholar]
- 145. Erazo‐Oliveras A, Muthukrishnan N, Baker R, Wang TY, Pellois JP. Improving the endosomal escape of cell‐penetrating peptides and their cargos: strategies and challenges. Pharmaceuticals. 2012;5(11):1177–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]