

Abstract

A new approach for the synthesis of 1,4-dicarbonyl compounds is reported. Chemoselective activation of amide carbonyl functionality and subsequent umpolung viaN-oxide addition generates an electrophilic enolonium species that can be coupled with a wide range of nucleophilic enolates. The method conveys broad functional group tolerance on both components, does not suffer from formation of homocoupling byproducts and avoids the use of transition metal catalysts.
Umpolung is the German term coined for reversal of innate polarity and, as a pivotal concept in organic chemistry, can lead to streamlining of syntheses by realizing nonclassical disconnections.1 It is conventionally taught in undergraduate organic chemistry courses in conjunction with the retrosynthetic disconnection of 1,4-dicarbonyl compounds (Figure 1a). Indeed, the most convergent approach implies the combination of a “naturally polarized” enolate with an “umpoled” enolate, or enolonium, species (Figure 1a, red disconnection).
Figure 1.
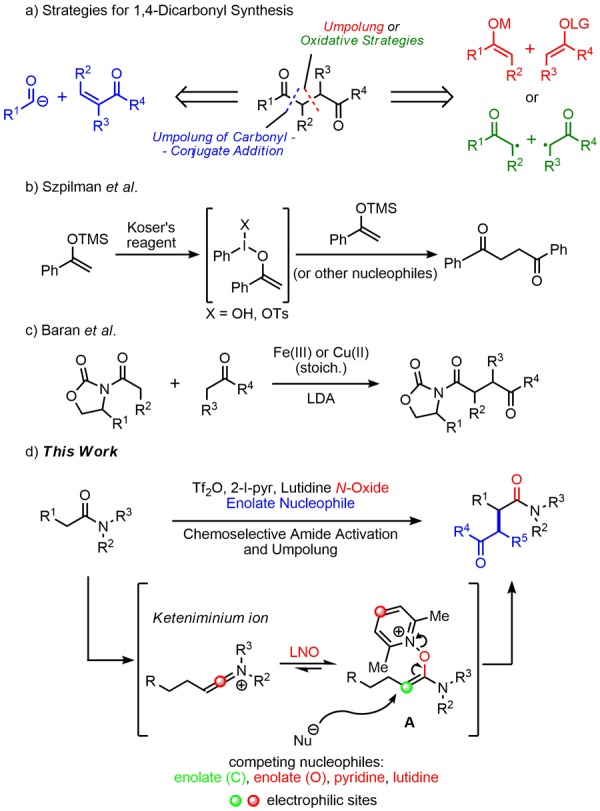
Previous approaches and our proposal.
There have been a number of reports using iodine oxidants to generate such enolonium species in situ to enable their coupling with incoming nucleophiles.2,3 Recently, Szpilman and co-workers conclusively characterized an enolonium species when using Koser’s reagent and a Lewis acid with a silyl enol ether (derived from an aryl ketone) (Figure 1b).4 Under certain conditions where no external nucleophile is present, the silyl enol ether undergoes dimerization to generate a 1,4-dicarbonyl compound. This result is of particular interest because although several different approaches have been previously described for the synthesis of this compound class,5 notably the oxidative heterocoupling of lithium enolates by Baran and co-workers (Figure 1c),6 flexible methods still remain scarce. In particular, homocoupling is a typical problem of oxidative strategies, while quaternary center formation is often not possible. We envisaged that we might be able to develop a more controlled union of enolonium and enolate species such that good yields of the cross-coupling products could be obtained to generate versatile 1,4-dicarbonyl compounds.
Our group has a long-standing interest in the chemoselective activation of amides inspired by the early work from the groups of Ghosez,7 and more recently Charette,8 Movassaghi,9 and others.10 We were interested in developing a methodology that exploited our recently reported chemistry on triflic anhydride (Tf2O) mediated activation of amides with N-oxides to generate an enolonium equivalent.11,12 Herein, we report the harnessing of this discrete electrophilic entity for attack of an external carbon nucleophile as a new approach for the preparation of 1,4-dicarbonyl compounds (Figure 1d). Significantly, this amounts to a cross-coupling of different carbonyl compounds with the α-position of an amide, a transformation that has previously not been reported.13
We were well aware of a number of potential problems in executing the selective union of the electrophilic, umpoled amide species and the incoming nucleophilic enolate with the desired regioselectivity (cf. center marked with green ● in Figure 1d). First, several other competitive nucleophilic species such as 2-iodopyridine or lutidine are bound to populate the reaction mixture. Second, alternative reaction pathways such as elimination or attack at other electrophilic sites on the keteniminium ion itself or the intermediate A (see centers marked with red ● in Figure 1d) are available.
Aware of these possible pitfalls, we began our initial investigations using the sodium salt of malonate derivatives as nucleophiles, added to the in situ generated electrophilic enolonium species A. Careful optimization of the reaction conditions highlighted several key parameters (see the SI for a detailed description). 2-Iodopyridine (3 equiv used initially) was identified as the ideal base, assuring the formation and stabilization of the intermediate keteniminium ion.8a Furthermore, the use of 2,6-lutidine N-oxide (LNO) as the oxidant was found to give consistently high yields; additionally, it is the reagent of choice from an economical point of view. Though variation of the reaction time and temperature did not lead to an improved yield, we were pleased to find that a decrease of the amount of 2-iodopyridine (2.2 equiv) led to the formation of the desired product 2a in a very pleasing yield of 83% (see Scheme 1). Using our optimized conditions, we found that, in addition to unsubstituted malonates (2b and 2c), the reaction conditions also tolerated a range of functional groups on the malonate component: notably alkenes (2a), alkynes (2f), nitriles (2g), halides (2h and 2i) and acetals (2j) which allow significant opportunity for further functionalization. Furthermore, this approach allowed us to form quaternary centers adjacent to tertiary centers in good to excellent yields (2a and 2d–j). Moving beyond malonates, malonamide was a competent nucleophile (2k) and malononitrile also afforded the desired product (2l), albeit in diminished yield. Under the same conditions, sulfone product 2m and ethyl N-diphenylmethylene glycine adduct 2n could be prepared in synthetically useful yields with practical handles for further elaboration. Upon scaling up the reaction to 2 mmol scale, we were also pleased to see no decline in yield (2c).
Scheme 1. Scope of Sodium Enolates.
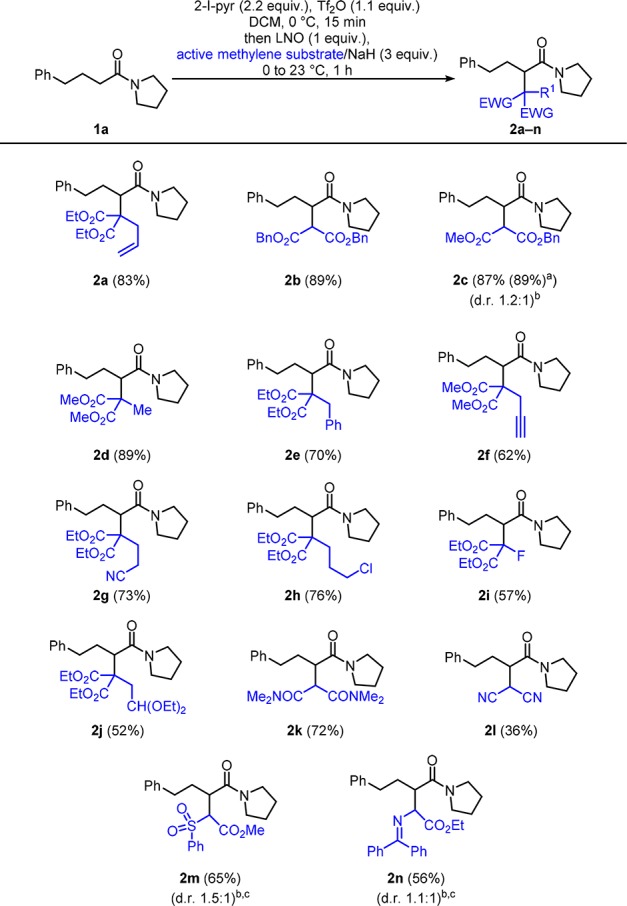
Reaction run on 2 mmol scale.
Ratio of isolated products.
Relative configuration not determined.
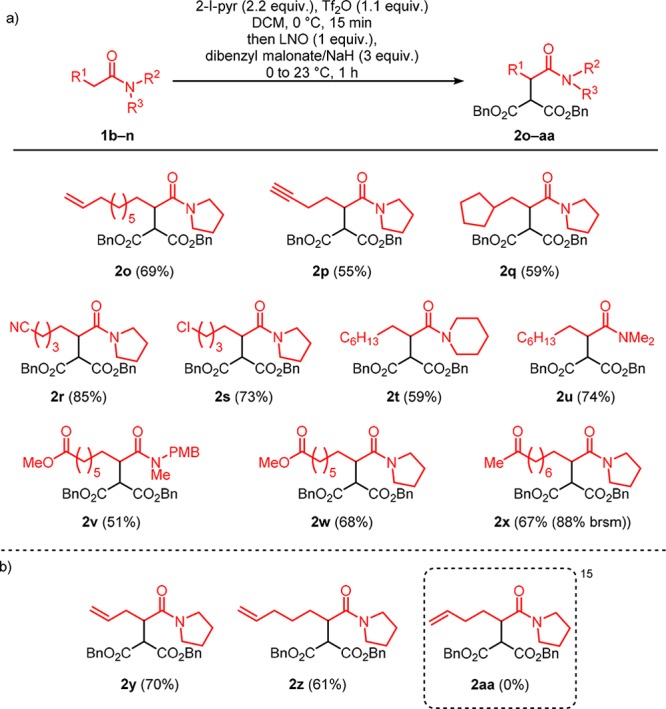
Our attention next turned to investigating the functional group tolerance and substrate scope on the amide component of the reaction (Scheme 2a). We were pleased to see that, in addition to alkenes (2o), alkynes (2p) and simple alkanes (2q),14 it was possible to include nitrile (2r) and halide (2s) moieties. Different substitution on the nitrogen atom did not overtly affect the yield (2t and 2u) and notably, a substrate with a removable PMB group also underwent the transformation (2v). Additionally, 2v serves as, to the best of our knowledge, the first example of chemoselective addition of an enolate to the α-position of an amide in the presence of another carbonyl derivative. To further showcase the unique chemoselectivity of our reaction, we expanded the scope of amides containing additional carbonyls (ester 2w and ketone 2x; here the α-proton is normally considered more reactive due to its lower pKa). In both cases, we saw no byproducts where the new C–C bond had been formed adjacent to the additional carbonyl: only next to the amide group. This unusual chemoselectivity is a hallmark of the present method. Though alkenes of varying chain-lengths were tolerated in the majority of cases (2o, 2y, 2z), the failure to form 2aa constituted a minor limitation (Scheme 2b).15
Scheme 2. Scope of Amides.
With this initial success, we were intrigued to see how far the range of enolates could be extended beyond malonates (Scheme 3). In this regard, we began by taking the lithium enolate of acetophenone (generated by deprotonation with lithium diisopropylamide at −78 °C) and adding this as before to activated amide 1a. The product was formed in a good yield (2ab, and, as before, this could be scaled up without any ill-effect). Simple acetone could also be used, albeit giving the product in moderate yield (2ac) and β-ionone was also a successful ketone substrate (2ad). Different ester enolates (2ae and 2af) could be used with 2af being noteworthy in that it showed that a quaternary center could be formed (a crystal structure was obtained of this product to unambiguously verify the structure, CCDC 1569559; see the SI for details). Interestingly, this method also allowed access to amide–amide heterocoupled products: the lithium enolate of dimethylacetamide gave the product in good yield (2ag) and 2ah was formed in excellent yield. Furthermore, the cyclic enolate derived from N-methyl-2-piperidone yielded product 2ai with high diastereomeric ratio (a crystal structure was again obtained, CCDC 1577972; see the SI for details). Lactones also reacted smoothly (2aj) and we were delighted to isolate the product 2ak from the reaction with the enolate derived from the natural product Sclareolide. Finally, it was important to demonstrate that the use of lithium enolates was also compatible with other amides containing reactive functional groups. In light of this, we were able to show the compatibility of ester and ketone enolates with substrates containing functional groups of similar pKa (2al–2ao). This showcases how enolate–enolonium coupling is competitive with proton transfer between the incoming nucleophile and pre-existing carbonyl functionality in the enolonium partner.
Scheme 3. Scope of Lithium Enolates.
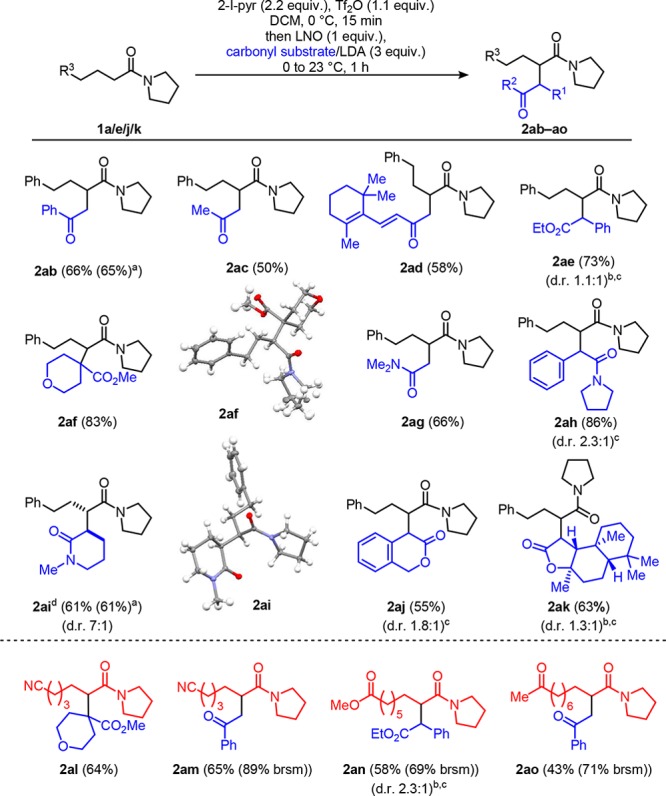
Reaction run on 1 mmol scale.
Ratio of separately isolated products.
Relative configuration not determined.
Major diastereomer depicted.
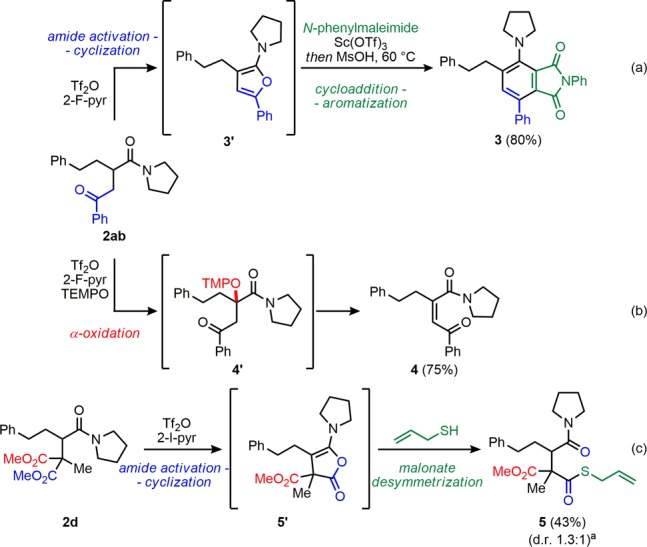
One distinct feature of our method that we were keen to take advantage of is the “traceless” nature of amide activation:16 this functional handle still remains in the products so we could again selectively activate this group over the newly installed, second carbonyl functionality. In order to demonstrate this unique reactivity, we opted to treat several 1,4-dicarbonyls with triflic anhydride (Scheme 4).
Scheme 4. Derivatization of Products.
Relative configuration not determined.
We envisaged a novel heterocycle synthesis whereby activation of 2ab with triflic anhydride would lead to intramolecular trapping by the ketone to transiently afford furan 3′. To our delight, upon treatment with a suitable dienophile and acid, this underwent clean [4+2]-cycloaddition and aromatization to form phthalimide 3 in excellent yield (Scheme 4a). Next, we applied our recently developed conditions for α-oxidation of amides.17 In the event, butene-1,4-dione 4 was produced after elimination of the newly introduced OTMP moiety (Scheme 4b).
In line with the reactivity portrayed in Scheme 4a, the treatment of 2d with triflic anhydride led to the formation of putative intermediate 5′, the product of cyclization of one ester moiety onto the activated amide. Treatment of this activated ester with allyl mercaptan afforded 5. Importantly, this is the product of selective monoactivation and effective desymmetrisation of the diester moiety.18
In conclusion, we have developed a chemoselective intermolecular cross-coupling of amides. This novel approach to the construction of 1,4-dicarbonyl compounds allows the coupling of enolates derived from malonates, ketones, esters, amides, lactams and lactones to the α-position of amides via an enolonium intermediate. This unprecedented reaction allows quaternary center formation and shows broad tolerance of functionality on both the amide and the incoming nucleophile, including functional groups carrying acidic protons such as ketones, esters and nitriles. Importantly, it virtually negates the production of homocoupled products. The traceless nature of the reaction allows iterative selective amide activation, which we have taken advantage of for subsequent derivatization. We anticipate that this novel disconnection will stimulate a number of new synthetic strategies.
Acknowledgments
Generous support of this research by the FWF (Fellowship M 2274 to C.J.T.; P 30226 to N.M.), the ERC (CoG VINCAT 682002 to N.M.) and the Austrian Academy of Sciences (DOC Fellowship to D.K.) is acknowledged. We are grateful to the University of Vienna for continued support of our research programs.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b08813.
Author Contributions
‡ These authors contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- a Seebach D. Angew. Chem., Int. Ed. Engl. 1979, 18, 239. 10.1002/anie.197902393. [DOI] [Google Scholar]; b Miyata O.; Miyoshi T.; Ueda M. ARKIVOC 2013, 60. [Google Scholar]
- a Patil D. V.; Kim S. W.; Nguyen Q. H.; Kim H.; Wang S.; Hoang T.; Shin S. Angew. Chem., Int. Ed. 2017, 56, 3670. 10.1002/anie.201612471. [DOI] [PubMed] [Google Scholar]; b Targel T. A.; Kumar J. N.; Shneider O. S.; Bar S.; Fridman N.; Maximenko S.; Szpilman A. M. Org. Biomol. Chem. 2015, 13, 2546. 10.1039/C4OB02601B. [DOI] [PubMed] [Google Scholar]; c Miyoshi T.; Miyakawa T.; Ueda M.; Miyata O. Angew. Chem., Int. Ed. 2011, 50, 928. 10.1002/anie.201004374. [DOI] [PubMed] [Google Scholar]; d Quiclet-Sire B.; Tölle N.; Zafar S. N.; Zard S. Z. Org. Lett. 2011, 13, 3266. 10.1021/ol2012204. [DOI] [PubMed] [Google Scholar]
- a Saito M.; Kobayashi Y.; Tsuzuki S.; Takemoto Y. Angew. Chem., Int. Ed. 2017, 56, 7653. 10.1002/anie.201703641. [DOI] [PubMed] [Google Scholar]; b Shneider O. S.; Pisarevsky E.; Fristrup P.; Szpilman A. M. Org. Lett. 2015, 17, 282. 10.1021/ol503384c. [DOI] [PubMed] [Google Scholar]; c Mizar P.; Wirth T. Angew. Chem., Int. Ed. 2014, 53, 5993. 10.1002/anie.201400405. [DOI] [PubMed] [Google Scholar]
- Arava S.; Kumar J. N.; Maksymenko S.; Iron M. A.; Parida K. N.; Fristrup P.; Szpilman A. M. Angew. Chem., Int. Ed. 2017, 56, 2599. 10.1002/anie.201610274. [DOI] [PubMed] [Google Scholar]
- a Guo F.; Clift M. D.; Thomson R. J. Eur. J. Org. Chem. 2012, 2012, 4881. 10.1002/ejoc.201200665. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Amaya T.; Maegawa Y.; Masuda T.; Osafune Y.; Hirao T. J. Am. Chem. Soc. 2015, 137, 10072. 10.1021/jacs.5b05058. [DOI] [PubMed] [Google Scholar]; c Amaya T.; Masuda T.; Maegawa Y.; Hirao T. Chem. Commun. 2014, 50, 2279. 10.1039/c3cc49624d. [DOI] [PubMed] [Google Scholar]; d Casey B. M.; Flowers R. A. J. Am. Chem. Soc. 2011, 133, 11492. 10.1021/ja205017e. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Narasaka K.; Okauchi T.; Tanaka K.; Murakami M. Chem. Lett. 1992, 21, 2099. 10.1246/cl.1992.2099. [DOI] [Google Scholar]; f Diaba F.; Montiel J. A.; Bonjoch J. Chem. Commun. 2016, 52, 14031. 10.1039/C6CC08356K. [DOI] [PubMed] [Google Scholar]
- a Baran P. S.; DeMartino M. P. Angew. Chem., Int. Ed. 2006, 45, 7083. 10.1002/anie.200603024. [DOI] [PubMed] [Google Scholar]; b DeMartino M. P.; Chen K.; Baran P. S. J. Am. Chem. Soc. 2008, 130, 11546. 10.1021/ja804159y. [DOI] [PubMed] [Google Scholar]; c Quesnelle C. A.; Gill P.; Kim S.-H.; Chen L.; Zhao Y.; Fink B. E.; Saulnier M.; Frennesson D.; DeMartino M. P.; Baran P. S.; Gavai A. V. Synlett 2016, 27, 2254. 10.1055/s-0035-1561636. [DOI] [Google Scholar]
- Falmagne J.-B.; Escudero J.; Taleb-Sahraoui S.; Ghosez L. Angew. Chem., Int. Ed. Engl. 1981, 20, 879. 10.1002/anie.198108791. [DOI] [Google Scholar]
- a Charette A. B.; Grenon M. Can. J. Chem. 2001, 79, 1694. 10.1139/v01-150. [DOI] [Google Scholar]; b Barbe G.; Charette A. B. J. Am. Chem. Soc. 2008, 130, 18. 10.1021/ja077463q. [DOI] [PubMed] [Google Scholar]; c Pelletier G.; Bechara W. S.; Charette A. B. J. Am. Chem. Soc. 2010, 132, 12817. 10.1021/ja105194s. [DOI] [PubMed] [Google Scholar]; d Bechara W. S.; Pelletier G.; Charette A. B. Nat. Chem. 2012, 4, 228. 10.1038/nchem.1268. [DOI] [PubMed] [Google Scholar]
- a Movassaghi M.; Hill M. D. J. Am. Chem. Soc. 2006, 128, 4592. 10.1021/ja060626a. [DOI] [PubMed] [Google Scholar]; b Movassaghi M.; Hill M. D. J. Am. Chem. Soc. 2006, 128, 14254. 10.1021/ja066405m. [DOI] [PubMed] [Google Scholar]; c Movassaghi M.; Hill M. D.; Ahmad O. K. J. Am. Chem. Soc. 2007, 129, 10096. 10.1021/ja073912a. [DOI] [PubMed] [Google Scholar]
- a Xiao K.-J.; Wang A.-E.; Huang Y.-H.; Huang P.-Q. Asian J. Org. Chem. 2012, 1, 130. 10.1002/ajoc.201200066. [DOI] [Google Scholar]; b Xiao K.-J.; Wang A.-E.; Huang P.-Q. Angew. Chem., Int. Ed. 2012, 51, 8314. 10.1002/anie.201204098. [DOI] [PubMed] [Google Scholar]; c Huang P.-Q.; Huang Y.-H.; Xiao K.-J.; Wang Y.; Xia X.-E. J. Org. Chem. 2015, 80, 2861. 10.1021/jo502929x. [DOI] [PubMed] [Google Scholar]; d Lumbroso A.; Catak S.; Sulzer-Mossé S.; De Mesmaeker A. Tetrahedron Lett. 2015, 56, 2397. 10.1016/j.tetlet.2015.02.112. [DOI] [Google Scholar]; e Lumbroso A.; Behra J.; Kolleth A.; Dakas P.-Y.; Karadeniz U.; Catak S.; Sulzer-Mossé S.; De Mesmaeker A. Tetrahedron Lett. 2015, 56, 6541. 10.1016/j.tetlet.2015.09.103. [DOI] [Google Scholar]; f Kolleth A.; Lumbroso A.; Tanriver G.; Catak S.; Sulzer-Mossé S.; De Mesmaeker A. Tetrahedron Lett. 2016, 57, 2697. 10.1016/j.tetlet.2016.04.092. [DOI] [Google Scholar]; g Huang P.-Q.; Huang Y.-H.; Xiao K.-J. J. Org. Chem. 2016, 81, 9020. 10.1021/acs.joc.6b01647. [DOI] [PubMed] [Google Scholar]; h Tona V.; de la Torre A.; Padmanaban M.; Ruider S.; González L.; Maulide N. J. Am. Chem. Soc. 2016, 138, 8348. 10.1021/jacs.6b04061. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Di Mauro G.; Maryasin B.; Kaiser D.; Shaaban S.; González L.; Maulide N. Org. Lett. 2017, 19, 3815. 10.1021/acs.orglett.7b01678. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Shaaban S.; Tona V.; Peng B.; Maulide N. Angew. Chem., Int. Ed. 2017, 56, 10938. 10.1002/anie.201703667. [DOI] [PubMed] [Google Scholar]; k Tona V.; Maryasin B.; de la Torre A.; Sprachmann J.; González L.; Maulide N. Org. Lett. 2017, 19, 2662. 10.1021/acs.orglett.7b01004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser D.; de la Torre A.; Shaaban S.; Maulide N. Angew. Chem., Int. Ed. 2017, 56, 5921. 10.1002/anie.201701538. [DOI] [PubMed] [Google Scholar]
- Da Costa R.; Gillard M.; Falmagne J. B.; Ghosez L. J. Am. Chem. Soc. 1979, 101, 4381. 10.1021/ja00509a060. [DOI] [Google Scholar]
- For examples of selective amide activation, see:; a Chen Y.; Turlik A.; Newhouse T. R. J. Am. Chem. Soc. 2016, 138, 1166. 10.1021/jacs.5b12924. [DOI] [PubMed] [Google Scholar]; b Chen M.; Dong G. J. Am. Chem. Soc. 2017, 139, 7757. 10.1021/jacs.7b04722. [DOI] [PubMed] [Google Scholar]; c Kaiser D.; Maulide N. J. Org. Chem. 2016, 81, 4421. 10.1021/acs.joc.6b00675. [DOI] [PubMed] [Google Scholar]; d Ruider S. A.; Maulide N. Angew. Chem., Int. Ed. 2015, 54, 13856. 10.1002/anie.201508536. [DOI] [PubMed] [Google Scholar]
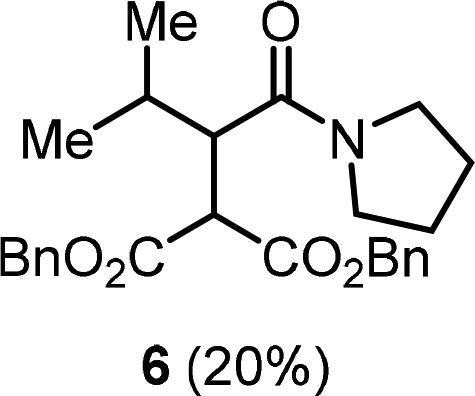
- Branching in the α- or β-position
of the amide was less well tolerated. α-Branched amides tend
to undergo desaturation (also cf. ref (12)). β-Branched amides lead to lower yields,
presumably due to a sterically congested enolonium intermediate A. In this context, isovaleramide 1n could be
transformed to product 6 in 20% yield.
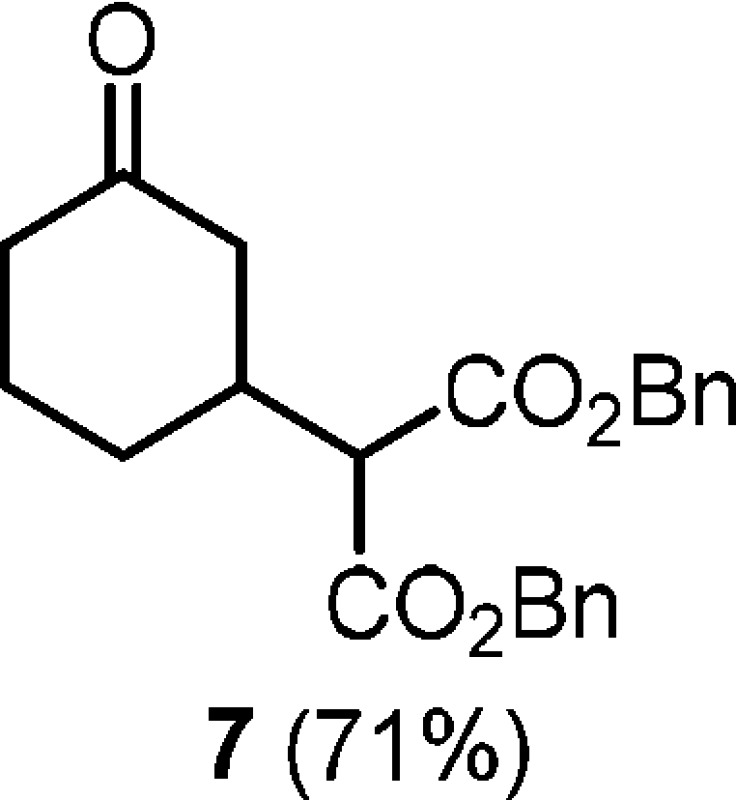
- Cyclohexanone 7 was isolated
as the sole product of the reaction of amide 1o. See
the SI for further details.
- Peng B.; Geerdink D.; Maulide N. J. Am. Chem. Soc. 2013, 135, 14968. 10.1021/ja408856p. [DOI] [PubMed] [Google Scholar]
- de la Torre A.; Kaiser D.; Maulide N. J. Am. Chem. Soc. 2017, 139, 6578. 10.1021/jacs.7b02983. [DOI] [PubMed] [Google Scholar]
- a Inoue T.; Kitagawa O.; Oda Y.; Taguchi T. J. Org. Chem. 1996, 61, 8256. 10.1021/jo961076+. [DOI] [PubMed] [Google Scholar]; b Inoue T.; Kitagawa O.; Saito A.; Taguchi T. J. Org. Chem. 1997, 62, 7384. 10.1021/jo970970d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.