

ABSTRACT
Flavobacterium columnare, a member of the phylum Bacteroidetes, causes columnaris disease in wild and aquaculture-reared freshwater fish. The mechanisms responsible for columnaris disease are not known. Many members of the phylum Bacteroidetes use type IX secretion systems (T9SSs) to secrete enzymes, adhesins, and proteins involved in gliding motility. The F. columnare genome has all of the genes needed to encode a T9SS. gldN, which encodes a core component of the T9SS, was deleted in wild-type strains of F. columnare. The F. columnare ΔgldN mutants were deficient in the secretion of several extracellular proteins and lacked gliding motility. The ΔgldN mutants exhibited reduced virulence in zebrafish, channel catfish, and rainbow trout, and complementation restored virulence. PorV is required for the secretion of a subset of proteins targeted to the T9SS. An F. columnare ΔporV mutant retained gliding motility but exhibited reduced virulence. Cell-free spent media from exponentially growing cultures of wild-type and complemented strains caused rapid mortality, but spent media from ΔgldN and ΔporV mutants did not, suggesting that soluble toxins are secreted by the T9SS.
IMPORTANCE Columnaris disease, caused by F. columnare, is a major problem for freshwater aquaculture. Little is known regarding the virulence factors produced by F. columnare, and control measures are limited. Analysis of targeted gene deletion mutants revealed the importance of the type IX protein secretion system (T9SS) and of secreted toxins in F. columnare virulence. T9SSs are common in members of the phylum Bacteroidetes and likely contribute to the virulence of other animal and human pathogens.
KEYWORDS: columnaris disease, gliding motility, type IX secretion system, virulence
INTRODUCTION
Flavobacterium columnare is a common fish pathogen that causes columnaris disease (1–3). F. columnare causes epidemics in wild and cultured fish and is a major problem in freshwater aquaculture worldwide, resulting in significant mortality. Many species of freshwater fish are susceptible to columnaris disease (4, 5). The virulence mechanisms of F. columnare are incompletely understood, and current control strategies are inadequate. F. columnare isolates are genetically diverse and have been assigned to multiple genomovars (6, 7). F. columnare genomovars I and II exhibit significant differences, including host ranges (8–10). Described outbreaks in salmonid aquaculture systems have almost invariably been associated with genomovar I strains (8, 11–14), whereas epidemics in catfish and other warm-water fish have involved members of diverse genomovars (6, 7, 9, 10, 15).
Secreted enzymes, such as proteases and chondroitin sulfate lyases, have been suggested as possible F. columnare virulence factors (3). Many virulence factors of pathogenic bacteria are either secreted proteins or the secretion systems themselves (16, 17). Gram-negative bacteria use at least nine secretion systems, the type I secretion system to the type IX secretion system (T1SS to T9SS), to transport proteins across their outer membranes (16). The T9SS (previously called the Por secretion system) was originally described in the human periodontal pathogen Porphyromonas gingivalis and in the environmental bacterium Flavobacterium johnsoniae (18). T9SSs are common in, but confined to, the phylum Bacteroidetes (19, 20). In F. johnsoniae, they are involved in the secretion of cell surface components of the gliding motility apparatus, and thus, the T9SS is required for gliding motility (18, 21, 22). The F. johnsoniae T9SS is also involved in the secretion of the soluble extracellular chitinase ChiA and in the secretion of many other proteins (23, 24). The P. gingivalis T9SS secretes gingipain proteases and cell surface adhesins that are thought to function as virulence factors in periodontitis (18, 25).
Genetic analyses suggest that GldK, GldL, GldM, GldN, SprA, SprE, SprT, PorU, and PorV (Fig. 1) are T9SS components (18, 21, 24, 26, 27). Proteins secreted by T9SSs have N-terminal signal peptides that facilitate export across the cytoplasmic membrane by the Sec system. They also typically have conserved C-terminal domains (CTDs) that target them for secretion across the outer membrane by the T9SS (24, 28–34). The CTDs are often removed by the protease PorU during or after secretion (28). Some secreted proteins remain attached to the cell surface, whereas others are released in soluble form (23, 24, 34).
FIG 1.
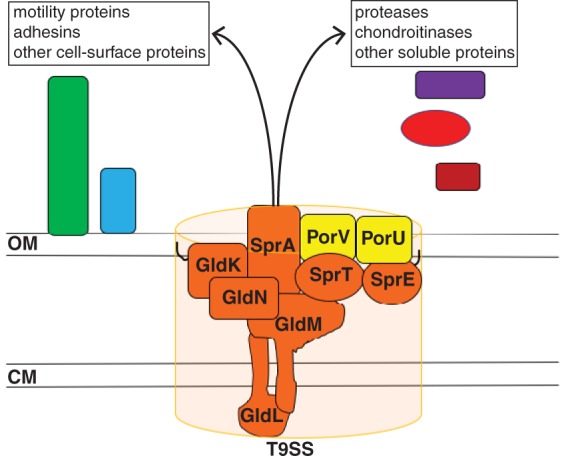
The F. columnare type IX protein secretion system. Orthologs of proteins required for secretion in Flavobacterium johnsoniae and Porphyromonas gingivalis are shown in orange. F. johnsoniae PorV is required for secretion of many but not all proteins that are targeted to the T9SS. F. johnsoniae PorU is involved in but not essential for secretion. Black lines on the lipoproteins GldK and SprE indicate lipid tails. OM, outer membrane; CM, cytoplasmic membrane.
Techniques to genetically manipulate F. columnare were recently developed (35, 36), allowing an exploration of the roles of individual genes in columnaris disease. We used these tools to generate T9SS mutants. Analysis of these mutants demonstrated that the T9SS is required for virulence and that secreted toxins have a role in pathogenesis.
RESULTS
Numerous proteins are predicted to be secreted by the F. columnare T9SS.
Bioinformatic analyses revealed that the genomes of F. columnare strain IA-S-4 (genomovar I) and F. columnare strain C#2 (genomovar II) had orthologs of the F. johnsoniae T9SS genes gldK, gldL, gldM, gldN, sprA, sprE, sprT, porU, and porV (see Table S1 in the supplemental material). The proteins encoded by these genes are thought to be components of the T9SS (19, 21, 24). Proteins secreted by T9SSs can often be identified by their conserved C-terminal domains that target them for secretion (24, 28–34, 37). Most of these CTDs belong to either the TIGRFAM protein domain family TIGR04183 (referred to as type A CTDs) or to the TIGR04131 family (referred to as type B CTDs) (37). F. columnare strains IA-S-4 and C#2 had 39 and 43 genes encoding such proteins, respectively (Table S2). Included in this list are predicted proteases, chondroitin sulfate lyases, and adhesins which might have roles in virulence.
F. columnare gldN mutants exhibit defects in gliding motility and protein secretion.
The T9SS gene gldN was deleted in F. columnare strains IA-S-4 and C#2 (Fig. 2). F. johnsoniae gldN mutants are defective in gliding motility. This is at least partially explained by the failure of the mutants to secrete motility adhesins, such as SprB, to the cell surface (18, 21). The F. columnare gldN mutants were also defective in gliding motility. They formed nonspreading colonies on agar (Fig. 3), and individual cells exhibited no movement on glass in tunnel slides (Fig. 4 and Movies S1 and S2). Complementation with pLN5 and pLN8, which carry wild-type F. columnare IA-S-4 gldN and F. columnare C#2 gldN, respectively, restored gliding motility in each case. The genome analyses described above and in Table S2 suggested that proteases and chondroitin sulfate lyases are likely secreted by the F. columnare T9SS. The F. columnare gldN mutants were defective in the digestion of proteins and chondroitin (Fig. 5), supporting the suggestion that enzymes involved in the digestion of these polymers are secreted by the T9SS.
FIG 2.

Maps of the regions containing F. columnare strain C#2 gldN (A) and porV (B) and associated deletions. Numbers below the maps refer to kbp of sequence. Binding sites for primers used in PCRs to generate deletion or complementation constructs are shown above and below the maps, with the blunt ends indicating the actual binding sites. The horizontal lines beneath the maps marked with open triangles denote regions deleted from the chromosome in the mutants. The regions of DNA carried by complementation plasmids pLN8 and pLN11 are indicated beneath the maps. The map of the gldN region of F. columnare strain IA-S-4 is identical to that shown in panel A, and F. columnare IA-S-4 gldN deletion and complementation constructs were constructed in the same way using the primers and plasmids listed in Tables 3 and 2, respectively.
FIG 3.

Photomicrographs of F. columnare colonies. Colonies grown from single cells were incubated for 48 h at 30°C on TYES agar (A), Shieh agar (B), and 10% CYE agar (C). (A) Wild-type (WT) F. columnare IA-S-4, ΔgldN mutant of strain IA-S-4 (ΔgldNIA-S-4), and ΔgldNIA-S-4 mutant complemented with wild-type gldN on pLN5. (B) Wild-type F. columnare C#2, ΔgldN mutant of strain C#2 (ΔgldNC#2), and ΔgldNC#2 mutant complemented with wild-type gldN on pLN8. (C) Wild-type F. columnare C#2, ΔporV mutant of strain C#2 (ΔporVC#2), and the ΔgldNC#2 mutant. Scale bar indicates 0.5 mm and applies to all panels.
FIG 4.

Gliding of wild-type and mutant cells on glass. Cells were grown in TYES (A) or Shieh medium (B and C) at 28°C for 14 h (early stationary phase). Ten-microliter aliquots of cultures were introduced into tunnel slides and observed for motility using an Olympus BH-2 phase-contrast microscope with a heated stage set at 25°C. (A) WT F. columnare IA-S-4, ΔgldN mutant of strain IA-S-4 (ΔgldNIA-S-4), and the ΔgldNIA-S-4 mutant complemented with wild-type gldN on pLN5. (B) Wild-type F. columnare C#2, ΔgldN mutant of strain C#2 (ΔgldNC#2), and the ΔgldNC#2 mutant complemented with wild-type gldN on pLN8. (C) Wild-type F. columnare C#2 and the ΔporV mutant of strain C#2 (ΔporVC#2). In each case, a series of images were taken for 20 s using a Photometrics CoolSNAP cf2 camera. Individual frames were colored from red (time zero) to yellow, green, cyan, and finally blue (20 s) and integrated into one image, resulting in “rainbow traces” of gliding cells. The rainbow traces correspond to the first 20 s of the sequences shown in Movie S1 (top row [A]), Movie S2 (middle row [B]), and Movie S3 (bottom row [C]). White cells correspond to cells that exhibited little if any net movement. The first frame of each movie (time zero) is shown in Fig. S1. Scale bar at lower right indicates 10 μm and applies to all panels.
FIG 5.

Digestion of proteins and chondroitin sulfate by material secreted by wild-type and mutant cells. Cells examined were wild-type F. columnare IA-S-4, ΔgldN mutant in strain IA-S-4 (ΔgldNIA-S-4), ΔgldNIA-S-4 mutant complemented with plasmid pLN5, wild-type F. columnare C#2, ΔgldN mutant in strain C#2 (ΔgldNC#2), ΔgldNC#2 mutant complemented with plasmid pLN8, ΔporV mutant in strain C#2 (ΔporVC#2), and ΔporVC#2 mutant complemented with plasmid pLN11. Digestion of protein (azocasein) (A) or of chondroitin sulfate (B) by cell-free spent medium from each strain was determined as described in Materials and Methods. The values are the means and the error bars indicate standard deviations (n = 3).
Cell-free culture fluid of wild-type mutant and complemented strains of F. columnare C#2 were examined by SDS-PAGE to determine the effect on secreted proteins. Several prominent bands from the culture fluids of wild-type and complemented strains were absent in the culture fluid from the ΔgldN mutant (Fig. 6). Proteins that were present in the culture fluid of wild-type and complemented cells but absent or greatly reduced in the ΔgldN mutant were identified by liquid chromatography-mass spectrometry (LC-MS) analyses (Table 1). Soluble proteins secreted by the T9SS included potential virulence factors, such as the chondroitin sulfate lyases CslA (AX766_RS05135) and CslB (AX766_RS01510), the predicted peptidases AX766_RS05330 and AX766_RS13405, and the predicted thiol-activated cytolysins AX766_RS03975 and AX766_RS13970. F. columnare C#2 CslA and CslB exhibited 96.6% and 98.1% amino acid identity with F. columnare strain G4 CslA and CslB, respectively (35). The identification of chondroitin sulfate lyases and peptidases in the cell-free culture fluids of wild-type cells that were absent or greatly reduced in cell-free culture fluids from the gldN mutant is consistent with the enzyme activity results shown in Fig. 5. Four of the seven secreted proteins identified had recognizable T9SS CTDs that are predicted to target these proteins to the secretion system (Table 1). The other three proteins may have novel targeting sequences or may be released by a process that only indirectly involves the T9SS. Perhaps significantly, each of these three proteins that lacked obvious T9SS CTDs were predicted lipoproteins (Table 1).
FIG 6.
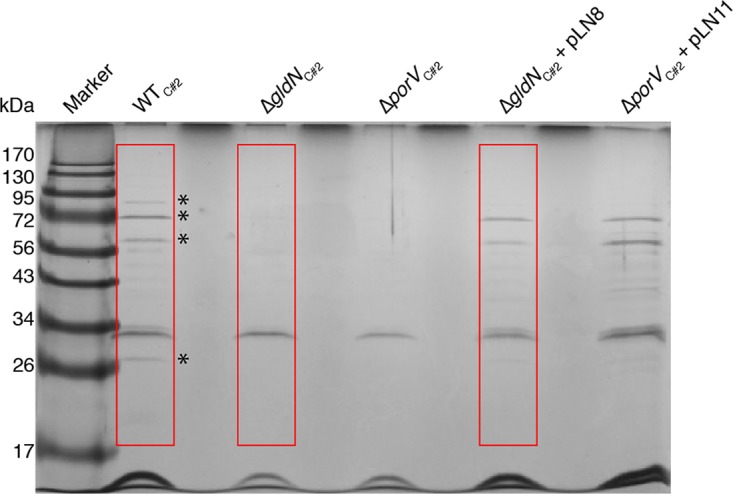
Soluble extracellular proteins of wild-type and mutant cells. Cells of wild-type F. columnare C#2, ΔgldN mutant in strain C#2 (ΔgldNC#2), ΔporV mutant in strain C#2 (ΔporVC#2), ΔgldNC#2 mutant complemented with wild-type gldN on pLN8, and ΔporVC#2 mutant complemented with wild-type porV on pLN11 were grown in Shieh medium at 25°C with shaking until cells reached an OD600 of 0.5. Equal amounts of cell-free spent media of wild-type and mutant cells were separated by SDS-PAGE, and proteins were detected by silver staining. Asterisks indicate prominent bands present in the spent media of wild-type and complemented cells that were absent or reduced in intensity in the spent media of the ΔgldNC#2 and ΔporVC#2 mutant cells. The boxed regions were subjected to LC-MS/MS analysis (Table 1). The abundant protein in all lanes at approximately 32 kDa was apparently secreted by a route other than the T9SS and verifies approximately equivalent loading of lanes.
TABLE 1.
Candidate F. columnare C#2 proteins secreted by the T9SS identified by LC-MS/MS analysis of cell-free culture fluidsa
| Locus tag/protein name | Molecular mass (kDa)b | T9SS CTDc | Lipoproteind | Predicted protein function | Spectral counts from culture fluid |
||
|---|---|---|---|---|---|---|---|
| Wild type | ΔgldN mutant | ΔgldN mutant + pLN8 | |||||
| AX766_RS05135/CslA | 86.2 | Type Ae | Chondroitin AC lyase | 145 | 3 | 131 | |
| AX766_RS03975 | 61.2 | + | Thiol-activated cytolysin | 19 | 2 | 20 | |
| AX766_RS05330 | 97.2 | Type A | Peptidase M4, thermolysin | 13 | 0 | 9 | |
| AX766_RS13970 | 61.5 | + | Thiol-activated cytolysin | 12 | 0 | 18 | |
| AX766_RS13405 | 33.9 | + | Metalloprotease | 4 | 0 | 3 | |
| AX766_RS01510/CslB | 100.2 | Type A | Chondroitin ABC lyase | 9 | 0 | 0 | |
| AX766_RS09885 | 54.0 | Type Ae | + | Unknown | 5 | 0 | 4 |
Proteins in cell-free culture fluid from wild-type F. columnare C#2, the ΔgldN mutant, and the ΔgldN mutant complemented with pLN8 were separated by SDS-PAGE and silver stained, and the regions shown in Fig. 6 spanning approximately 20 to 200 kDa were cut from the gel and analyzed by LC-MS/MS. Total/unweighted spectrum counts corresponding to the total number of spectra associated with a single protein and that are indicative of relative abundance of that protein are indicated for each of the strains analyzed. Only proteins that had at least 4 hits for the wild-type culture fluid and had at least a 5-fold reduction in the number of hits for the ΔgldN mutant are shown.
Molecular mass as calculated for full-length protein before removal of signal peptide.
T9SS CTD type identified by BLASTP analysis. Type A CTDs belong to TIGRFAM protein domain family TIGR04183.
Lipoproteins predicted by LipoP (59). + indicates a lipoprotein. Blank indicates not a lipoprotein.
The C-terminal regions of these proteins were below the trusted cutoffs for type A (TIGR04183) CTDs but exhibited more limited similarity, as detected by BLASTP analyses.
To directly assess protein secretion by wild-type and gldN mutant cells, we introduced a plasmid expressing the foreign protein mCherry carrying an N-terminal signal peptide to allow export across the cytoplasmic membrane by the Sec system, and carrying the CTD of F. johnsoniae ChiA to target the protein for secretion across the outer membrane by the T9SS. The CTD of F. johnsoniae ChiA functioned in F. columnare strain C#2, as demonstrated by the accumulation of mCherry in the spent culture fluid of wild-type cells but not of the gldN mutant (Fig. S2).
Cells of an F. columnare porV mutant are defective in protein secretion but retain gliding motility.
F. johnsoniae PorV is needed for the secretion of a subset of proteins targeted to the T9SS, but it is not needed for secretion of the gliding motility adhesin SprB (24). F. johnsoniae porV mutants thus retain the ability to glide. In order to separate F. columnare protein secretion from motility, a porV deletion mutant of wild-type strain C#2 was constructed and examined. The mutant was defective in the digestion of extracellular protein and chondroitin (Fig. 5), suggesting a protein secretion defect. Analysis of secreted proteins by SDS-PAGE demonstrated that several soluble proteins failed to be secreted in the porV mutant (Fig. 6). porV mutants retained the ability to glide (Fig. 3 and 4 and Movie S3), similar to F. johnsoniae porV mutants. The F. columnare porV mutant spread less well on agar and produced smaller colonies than did wild-type cells, but the motility of the individual ΔporV mutant and wild-type cells on glass was equivalent. The F. columnare porV mutant allowed us to partially separate motility from secretion and thus examine the roles of each on virulence.
F. columnare T9SS mutants are defective in virulence toward zebrafish, rainbow trout, and channel catfish.
Wild-type F. columnare C#2, the ΔgldN mutant, and the ΔgldN mutant complemented with pLN8 (which carries wild-type gldN) were examined for ability to kill zebrafish (Fig. 7 and S3). The growth rates in cultures of wild-type, mutant, and complemented strains were similar (Fig. S4). The gldN mutant exhibited decreased virulence compared to the wild-type and complemented strains, suggesting that the T9SS has an important role in F. columnare virulence. The ΔporV mutant was also examined to begin to separate the roles of secretion and motility on virulence. The ΔporV mutant was less virulent than were the wild-type and complemented strains (Fig. 7 and S3). This suggests that a lack of secretion rather than lack of motility may account for much of the reduced virulence of T9SS mutants. For all of the zebrafish infection experiments described above, greater than 50% of the mortalities exhibited clinical signs of columnaris disease, including external lesions, tail rot, and/or damaged gills. In contrast, none of the uninfected control fish and none of the fish infected with a ΔgldN or ΔporV mutant, exhibited any of these signs or exhibited mortalities. Results similar to those described above for F. columnare strain C#2 were obtained when virulence of wild-type F. columnare strain IA-S-4 (genomovar I), the ΔgldN mutant of IA-S-4, and the complemented mutant were examined for virulence against zebrafish (data not shown).
FIG 7.
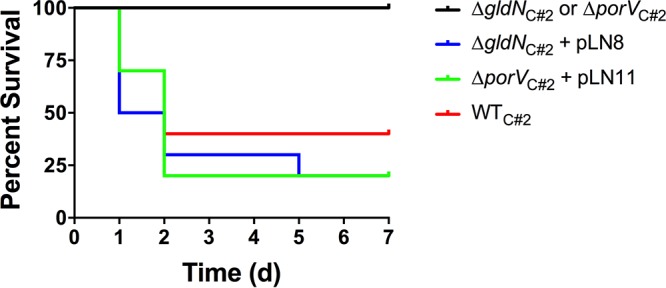
Virulence of wild-type F. columnare C#2, T9SS mutants, and complemented strains toward zebrafish. Zebrafish were exposed to F. columnare strains for 1 h at 28°C and transferred to fresh water, and percent survival was monitored for 7 days (d). Cells examined were wild-type F. columnare C#2, ΔgldN mutant in strain C#2 (ΔgldNC#2), ΔgldNC#2 mutant complemented with plasmid pLN8, ΔporV mutant in strain C#2 (ΔporVC#2), and the ΔporVC#2 mutant complemented with plasmid pLN11. The final challenge concentrations were 2.1 × 106 CFU/ml for C#2, 1.5 × 108 CFU/ml for the ΔgldNC#2 mutant, 1.5 × 107 CFU/ml for the ΔporVC#2 mutant, 2.1 × 106 CFU/ml for the ΔgldNC#2 mutant complemented with pLN8, and 2.1 × 106 CFU/ml for the ΔporVC#2 mutant complemented with pLN11. Ten fish were challenged with each strain, as indicated in Materials and Methods. Ten control fish that were exposed to an equal amount of growth medium without F. columnare cells were also included, and each of these control fish survived. Challenges at additional dilutions are shown in Fig. S3.
Wild-type F. columnare, ΔgldN mutants, and complemented mutants were also examined for ability to kill rainbow trout and channel catfish. In each case, the ΔgldN mutants exhibited decreased virulence compared to the wild-type and complemented strains (Fig. 8 and S5). For the rainbow trout challenges, clinical signs of columnaris disease, including external lesions, fin rot, and/or necrotic gills, were observed in greater than 50% of the mortalities, and such signs were observed on all channel catfish mortalities. None of the uninfected control fish and none of the fish infected with ΔgldN mutants exhibited these signs.
FIG 8.

Challenge of rainbow trout and channel catfish with F. columnare. Fish were exposed to wild-type, mutant, and complemented strains, and percent survival was monitored as described in Materials and Methods. (A) Rainbow trout were exposed to wild-type F. columnare IA-S-4 (1.2 × 105 CFU/ml), ΔgldNIA-S-4 mutant (5.5 × 106 CFU/ml), and ΔgldNIA-S-4 mutant complemented with pLN5 (2.7 × 106 CFU/ml). Sixty rainbow trout were challenged with each strain, as indicated in Materials and Methods. The results from triplicate tanks (20 fish each) were similar and were pooled to construct the figure. (B) Channel catfish were exposed to wild-type F. columnare C#2 (5.0 × 107 CFU/ml), ΔgldNC#2 mutant (4.6 × 107 CFU/ml), and ΔgldNC#2 mutant complemented with pLN8 (3.4 × 107 CFU/ml). Twenty-five channel catfish were challenged with each strain, as indicated in Materials and Methods. Rainbow trout and channel catfish were also mock challenged with growth medium lacking F. columnare cells, as described in Materials and Methods, and in each case, these fish survived. Challenges of rainbow trout and channel catfish at additional dilutions are shown in Fig. S5.
During the zebrafish, rainbow trout, and channel catfish challenges described above, reisolation of F. columnare was attempted from ∼20 to 30% of dead/moribund fish by inoculating fish tissue onto agar plates, culturing, and examining for yellow rhizoid adherent colonies. F. columnare was reisolated from all challenge mortalities, suggesting that the morbidity and mortality observed were due to F. columnare infections. For isolates from rainbow trout, 16S rRNA genes were amplified and sequenced, and each was identical to the sequence from the challenge strain F. columnare strain IA-S-4. Finally, in each study, negative-control fish that were not exposed to F. columnare experienced no mortalities.
Wild-type F. columnare but not T9SS mutants secreted materials that were toxic to fish.
The requirement of the T9SS for virulence suggested the possibility that soluble secreted proteins may be important virulence factors. Filtered cell-free spent media from cultures of wild-type, mutant, and complemented cells were examined for ability to kill zebrafish. Cell-free spent media from exponential cultures of wild-type and complemented cells caused rapid mortality, whereas spent media from the ΔgldN and ΔporV mutant cells did not (Fig. 9). The toxicity of the spent medium from wild-type cells of F. columnare strain C#2 was eliminated by heating at 60°C for 60 min and by treatment with trypsin (Fig. 10), suggesting that the toxins may be proteins. Incubation of cell-free spent culture fluid at 37°C for 12 h without trypsin resulted in apparent decreased toxicity (compare untreated trials from Fig. 10A and B). This may be the result of digestion of the toxins by secreted F. columnare proteases, although other explanations are also possible.
FIG 9.

Toxicity for zebrafish of material secreted by the T9SS. Wild-type, mutant, and complemented cells were cultured to mid-log phase (Klett units, 50 to 60) in TYES (A) or modified Shieh medium (B). Cells were removed by centrifugation, followed by filtration. Zebrafish (strain Eckwill crossed with strain Tupfel Long-fin; 6 fish per bacterial strain per trial) were exposed to cell-free spent medium (100 ml) for 5 to 7 h at 28°C, and mortalities were recorded every 30 min. Three independent trials were conducted for each strain. No mortalities occurred for control fish exposed to TYES or modified Shieh medium that had not been previously inoculated with F. columnare, or for fish exposed to cell-free culture fluids from the ΔgldN mutants or ΔporV mutant. (A) Toxicity of cell-free spent media from cultures of strains derived from F. columnare IA-S-4. Strains used were wild-type F. columnare IA-S-4, the ΔgldNIA-S-4 mutant, and the ΔgldNIA-S-4 mutant complemented with plasmid pLN5. (B) Toxicity of cell-free spent media from cultures of strains derived from F. columnare C#2. Strains used were wild-type F. columnare C#2, the ΔgldNC#2 mutant, the ΔgldNC#2 mutant complemented with plasmid pLN8, the ΔporVC#2 mutant, and the ΔporVC#2 mutant complemented with plasmid pLN11.
FIG 10.

Effect of heat and trypsin treatment on toxicity of cell-free spent culture fluid for zebrafish. Wild-type F. columnare C#2 cells were cultured to mid-log phase (Klett units, 50 to 60) in modified Shieh medium. Cells were removed by centrifugation, followed by filtration. Samples of the cell-free culture fluid were heated (60°C, 60 min) (A) or treated with trypsin (50 μg/ml, 37°C, 12 h) (B). (B) Samples without trypsin (untreated trials) were incubated at 37°C for 12 h. Zebrafish (strain Eckwill; 6 fish per bacterial strain per trial) were exposed to untreated, heat-treated, and trypsin-treated cell-free spent medium (100 ml) for 4 h at 28°C, and mortalities were recorded. Three independent trials for heat treatment and for trypsin treatment are shown. No mortalities occurred for fish exposed to modified Shieh medium that had not been previously inoculated with F. columnare (Control), or for fish exposed to cell-free spent culture fluid that had been heated or treated with trypsin (black lines).
DISCUSSION
F. columnare is an important pathogen of freshwater fish and is especially problematic under the high-density conditions often employed in aquaculture systems. Columnaris disease causes substantial economic losses and has received considerable attention. Previous studies identified possible virulence factors, including proteases and chondroitin sulfate lyases (38–40). However, definitive proof for the involvement or lack of involvement of these enzymes in virulence was lacking. Techniques for gene transfer (36) and gene deletion (35) were recently developed for F. columnare and allow experiments to test the roles of individual genes and proteins in virulence.
Genomic analysis identified the components of the F. columnare T9SS. As identified by genome analyses, T9SSs are common throughout most of the phylum Bacteroidetes and are found in all members of the genus Flavobacterium whose genomes have currently been sequenced and analyzed (20, 37). Further, recent comparative analyses identified all of the T9SS genes in the genomes of each of nine F. columnare strains examined (41, 42). For all members of the phylum Bacteroidetes that have the T9SS genes, gldK, gldL, gldM, and gldN are invariably located together in this order as an operon (20, 21). Most of the other T9SS genes are not clustered together and are not located near the gldKLMN operon. Phylogenetic analyses of T9SS genes and of 16S rRNA genes suggested that the T9SS genes are part of the core genome of most genera within the phylum Bacteroidetes (20, 21). Our genome analyses also identified dozens of proteins that have CTDs predicted to target them for secretion by the T9SS (Table S2). This is likely to be an underestimate of the actual number of T9SS-secreted proteins, because F. columnare proteins with similarity to T9SS CTDs that were slightly below the trusted TIGRFAM cutoffs were also identified, and because some proteins known to be secreted by T9SSs exhibit no obvious sequence similarity to these conserved CTD sequences (23, 24, 37). Included in the lists of predicted secreted proteins were chondroitin sulfate lyases, proteases, and adhesins, any of which could contribute to virulence.
Since secreted proteins are known virulence factors in other bacteria (16, 17), we targeted the T9SS for analysis. The deletion of gldN and porV, which encode components of the T9SS, resulted in decreased secretion of extracellular enzymes. These results are similar to those observed for F. johnsoniae and P. gingivalis and support the roles of GldN and PorV in protein secretion (18, 24, 26, 43, 44). The F. columnare T9SS mutants exhibited decreased levels of extracellular proteases, chondroitin sulfate lyases, and proteins of unknown function. The mutants also exhibited reduced virulence for zebrafish, rainbow trout, and channel catfish. In addition, whereas the cell-free culture fluid from wild-type cells was toxic for zebrafish, the culture fluids from ΔgldN and ΔporV mutants were not. The identities of the secreted toxins are not known, but they were destroyed by elevated temperature and by protease treatment, suggesting that they are proteins. The toxins may be one or more of the seven proteins that were identified in the cell-free culture fluid of wild-type cells. This possibility can now be addressed by deletion of the genes encoding these proteins, either singly or in combination.
In addition to soluble secreted proteins, such as those mentioned above, many proteins secreted by T9SSs are found on the cell surface (34). These cell surface enzymes and adhesins may also be important for virulence, and future studies are needed to address this possibility. One cell surface protein secreted by the T9SS whose function is known is F. johnsoniae SprB, which is involved in gliding motility. This adhesin is propelled rapidly along the cell surface (45, 46). The action of the gliding motor on SprB proteins that are attached to the substratum results in gliding motility. F. columnare has an SprB ortholog that is thought to be involved in gliding (42). Not surprisingly, deletion of the core F. columnare T9SS gene gldN resulted not only in secretion defects but also in a loss of motility. This motility defect may be explained by the inability to secrete SprB to the cell surface. In F. johnsoniae, the T9SS protein PorV is less essential for secretion than is GldN. F. johnsoniae PorV is required for the secretion of many proteins that are targeted to the T9SS, but it is not required for secretion of SprB and related proteins (24). For this reason, porV mutants of F. johnsoniae exhibit gliding motility. In this study, we demonstrated that an F. columnare ΔporV mutant behaved similarly. It was defective for secretion and virulence but retained gliding motility. This result partially separates the potential roles of motility and secretion in virulence. Motility may play a role in virulence, but even in the presence of a functional motility system, a secretion-defective porV mutant exhibited reduced virulence.
Secretion is important for the virulence of F. columnare, but the identities of the most important secreted virulence factors are not yet known. Recently, both secreted chondroitin sulfate lyases of F. columnare strain G4 were examined for their roles in virulence. Cells lacking both chondroitin sulfate lyases retained virulence when fish were challenged by intraperitoneal injection but exhibited decreased competitiveness during coinfection by immersion (35). Chondroitin sulfate is found in connective tissues, and the chondroitin sulfate lyases may allow the bacteria to penetrate fish tissues during infection. Genetic experiments similar to those described in this study should allow identification of the most critical secreted proteins involved in columnaris disease.
The results presented here demonstrate that the T9SS is involved in F. columnare virulence. They also suggest strategies to control this pathogen. Secreted proteins appear to be important in the disease process. Deletion of the genes encoding one or more of these proteins may result in attenuated strains that fail to cause disease but that may still interact with fish and generate a protective immune response. Such strains could potentially function as effective live attenuated vaccines. T9SSs are common in members of the phylum Bacteroidetes (20). Some of these bacteria are animal and human pathogens, and their T9SSs may play important roles in their ability to cause disease. Continued studies may result in a better understanding of the virulence mechanisms employed by these pathogens and result in strategies to control them.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
F. columnare genomovar I strain IA-S-4 and genomovar II strain C#2 (47, 48) were the wild-type strains used in this study. Strain IA-S-4 was isolated in Iowa (USA) in 2011 from the skin of a walleye with columnaris disease (B. LaFrentz, unpublished data). F. columnare strains were grown at 25 to 30°C in Shieh medium (49), modified Shieh medium (50), tryptone yeast extract salts (TYES) medium (51), or in 10% Casitone-yeast extract (CYE), which consisted of CYE medium (52) that had been diluted 10-fold with distilled water. For the solid media, 15 g of agar was added per liter. Escherichia coli strains were grown in lysogeny broth (LB) at 37°C (53). The strains and plasmids used in this study are listed in Table 2, and primers are listed in Table 3. Antibiotics were used at the following concentrations when needed: ampicillin, 100 μg/ml; tobramycin, 1 μg/ml; and tetracycline, 10 μg/ml, unless indicated otherwise.
TABLE 2.
Strains and plasmids used in this study
| Strain or plasmid | Descriptiona | Source or reference |
|---|---|---|
| Strains | ||
| E. coli | ||
| DH5αMCR | Strain used for general cloning | Life Technologies (Grand Island, NY) |
| S17-1 λpir | Strain used for conjugation | 60 |
| F. columnare | ||
| C#2 | Wild type | 47, 48 |
| FCC-2 | ΔgldN in strain C#2 | This study |
| FCC-8 | ΔporV in strain C#2 | This study |
| IA-S-4 | Wild type | B. LaFrentz |
| FCI-1 | ΔgldN in strain IA-S-4 | This study |
| Plasmids | ||
| pCP23 | E. coli-F. columnare shuttle plasmid; Apr (Tc)r | 61 |
| pMS75 | Suicide vector carrying sacB; Apr (Tcr) | 35 |
| pLN5 | Plasmid for complementation of gldN in FCI-1; IA-S-4 gldN was amplified with primers 1648 and 1649 and cloned into BamHI and SphI sites of pCP23; Apr (Tcr) | This study |
| pLN6 | 2.0-kbp region upstream of C#2 gldN amplified with primers 1618 and 1619 and cloned into BamHI and SalI sites of pMS75; Apr (Tcr) | This study |
| pLN7 | Construct used to delete C#2 gldN; 1.7-kbp region downstream of C#2 gldN amplified with primers 1620 and 1621 and cloned into SalI and SphI sites of pLN6; Apr (Tcr) | This study |
| pLN8 | Plasmid for complementation of gldN in FCC-2; C#2 gldN was amplified with primers 1682 and 1683 and cloned into BamHI and SphI sites of pCP23; Apr (Tcr) | This study |
| pLN9 | 2.0-kbp region downstream of C#2 porV amplified with primers 1722 and 1723 and cloned into BamHI and SalI sites of pMS75; Apr (Tcr) | This study |
| pLN10 | Construct used to delete C#2 porV; 2.0-kbp region upstream of C#2 porV amplified with primers 1724 and 1725 and cloned into SalI and SphI sites of pLN9; Apr (Tcr) | This study |
| pLN11 | Plasmid for complementation of porV in FCC-8; C#2 porV was amplified with primers 1747 and 1748 and cloned into BamHI and SphI sites of pCP23; Apr (Tcr) | This study |
| pLN30 | 2.0-kbp region upstream of IA-S-4 gldN amplified with primers 1893 and 1894 and cloned into BamHI and SalI sites of pMS75; Apr (Tcr) | This study |
| pLN31 | Construct used to delete IA-S-4 gldN; 1.7-kbp region downstream of IA-S-4 gldN amplified with primers 1895 and 1896 and cloned into SalI and SphI sites of pLN30; Apr (Tcr) | This study |
| pSSK52 | pCP23 carrying SPChiA-mCherry-CTDChiA; Apr (Tcr) | 23 |
| pSSK54 | pCP23 carrying SPChiA-mCherry; Apr (Tcr) | 23 |
Antibiotic resistance phenotypes: Apr, ampicillin; Tcr, tetracycline. Unless indicated otherwise, the antibiotic resistance phenotypes are those expressed in E. coli. The antibiotic resistance phenotypes given in parentheses are those expressed in F. columnare but not in E. coli.
TABLE 3.
Primers used in this study

a Underlined sequences indicate introduced restriction enzyme sites.
Growth of F. columnare in liquid media.
F. columnare IA-S-4 and C#2 strains were streaked from −80°C freezer tubes onto TYES and Shieh agar, respectively, and incubated 48 h at 30°C. All strains were restreaked on fresh agar, incubated 48 h at 30°C, and then used to inoculate broth cultures. F. columnare IA-S-4 and C#2 were grown overnight at 28°C with shaking at 200 rpm in 20 ml of TYES and Shieh liquid media, respectively (starter cultures), with 1 μg/ml tetracycline included for the complemented strains. Two milliliters of F. columnare IA-S-4 or C#2 starter cultures (normalized to optical density at 600 nm [OD600] of 0.6) was introduced into 48 ml of TYES or Shieh liquid medium, respectively, in 250-ml side-arm flasks and incubated at 28°C with shaking. Turbidity was monitored using a Klett-Summerson photoelectric colorimeter (Klett Mfg. Co., NY). Growth experiments were performed in triplicate.
Conjugative transfer of plasmids into F. columnare strains.
Plasmids were transferred from E. coli S17-1 λpir into F. columnare strains by conjugation. E. coli and F. columnare strains were incubated overnight with shaking in LB at 37°C or in Shieh broth at 30°C, respectively. Five hundred microliters of overnight culture was inoculated in fresh LB and Shieh broth and shaken at 37°C and 30°C, respectively, until the OD600 reached 0.4. The cells were collected by centrifugation at 4,200 × g for 5 min. The pellets of F. columnare were washed twice with 1 ml of Shieh medium and suspended in 50 μl of Shieh medium. The suspensions of E. coli and F. columnare were mixed and spotted on mixed cellulose ester filter membranes with a pore size of 0.45 μm (EMD Millipore, Billerica, MA) which had been placed on Shieh agar. Conjugation was allowed to proceed at 30°C for 24 h. The cells were removed from the filter membrane with a scraper and suspended in 1 ml of Shieh medium. Aliquots were spread on Shieh agar containing 1 μg/ml tobramycin and 10 μg/ml tetracycline and incubated at 30°C for 48 to 72 h.
Construction of gldN and porV deletion mutants.
Gene deletions in F. columnare were constructed essentially as previously described (35). To delete gldN from F. columnare strain C#2, a 2,024-bp product spanning gldM and including the first 144 bp of gldN was amplified by PCR using Phusion DNA polymerase (New England BioLabs, Ipswich, MA) and primers 1618 (introducing a BamHI site) and 1619 (introducing a SalI site). The product was digested with BamHI and SalI and ligated into pMS75, which had been digested with the same enzymes, to generate pLN6. A 1,723-bp product spanning AX766_RS08575 (encoding a flavin adenine dinucleotide [FAD]-dependent oxidoreductase) and the final 30 bp of gldN was amplified with primers 1620 (introducing a SalI site) and 1621 (introducing an SphI site). The product was digested with SalI and SphI and fused to the upstream region of gldN by ligation with pLN6, which had been digested with the same enzymes, to generate the deletion construct pLN7. Plasmid pLN7 was transferred by conjugation into F. columnare C#2, and colonies that had the plasmid integrated into the chromosome by recombination were obtained by selecting for tetracycline resistance. Colonies were streaked for isolation and were then inoculated into 3 ml of Shieh medium without antibiotics to allow loss of the integrated plasmid. The gldN deletion mutant (ΔgldNC#2) was obtained by selecting for sucrose resistance and was confirmed by PCR using primers 1682 and 1683, which flank the gldN coding sequence. A gldN deletion mutant of F. columnare strain IA-S-4 (ΔgldNIA-S-4) was obtained in the same way, except that primers 1893, 1894, 1895, and 1896 were used to construct deletion plasmid pLN31.
To delete F. columnare C#2 porV, a 1,967-bp product downstream of porV was amplified using primers 1722 (introducing a BamHI site) and 1723 (introducing an SalI site). The amplified product was digested with BamHI and SalI and cloned into pMS75, generating pLN9. A 2,046-bp product upstream of porV was amplified by PCR using primers 1724 (introducing an SalI site) and 1725 (introducing an SphI site). The amplified product was digested with SalI and SphI and cloned into pLN9, generating pLN10. pLN10 was introduced into F. columnare C#2 by conjugation, and the deletion mutant (ΔporVC#2) was obtained as described above.
Complementation of deletion mutants.
Plasmids carrying gldN or porV were constructed using shuttle vector pCP23. To construct a plasmid containing F. columnare C#2 gldN, primers 1682 (introducing a BamHI site) and 1683 (introducing an SphI site) were used to amplify a 1,369-bp product spanning gldN from F. columnare C#2 genomic DNA. To construct a plasmid containing F. columnare IA-S-4 gldN, primers 1648 (introducing a BamHI site) and 1649 (introducing an SphI site) were used to amplify a 1,152-bp product spanning gldN from F. columnare IA-S-4 genomic DNA. To construct a plasmid containing F. columnare C#2 porV, primers 1747 (introducing a BamHI site) and 1748 (introducing an SphI site) were used to amplify a 1,542-bp product spanning porV from F. columnare C#2 genomic DNA. The products were digested with BamHI and SphI and ligated into pCP23, which had been digested with the same enzymes, to generate pLN8, pLN5, and pLN11. The plasmids were transferred to appropriate F. columnare mutants by conjugation.
Analysis of colony spreading and cell motility.
F. columnare IA-S-4 and C#2 colonies were grown for 2 days at 30°C on TYES and Shieh agar, respectively. Colonies of wild-type, mutant, and complemented strains were examined using a Photometrics CoolSNAP cf2 camera mounted on an Olympus IMT-2 phase-contrast microscope. Gliding of individual cells was also examined microscopically. F. columnare IA-S-4 and C#2 were grown to early stationary phase (14 h) with shaking at 28°C in TYES and Shieh liquid media, respectively. Tunnel slides were constructed using double-stick tape, glass microscope slides, and glass coverslips, as previously described (22). Ten-microliter aliquots of cultures were introduced into the tunnel slides, incubated for 5 min, and observed for motility using an Olympus BH2 phase-contrast microscope at 25°C. Images were recorded with a Photometrics CoolSNAP cf2 camera and analyzed using the MetaMorph software (Molecular Devices, Downingtown, PA). Rainbow traces of cell movements were made using ImageJ version 1.45s (http://rsb.info.nih.gov/ij/) and macro Color FootPrint (45).
Measurement of protease activity.
Azocasein protease assays were performed to quantitate proteolytic activity. F. columnare strains were grown in triplicate 5-ml volumes of TYES broth for 24 h at 28°C with shaking at 175 rpm. The cultures were centrifuged at 19,980 × g for 10 min. Residual cells were removed from the supernatants by passage through 0.45 μM HT Tuffryn syringe filters (PALL Life Sciences, Ann Arbor, MI), and the cell-free culture fluids were stored at 4°C for 24 h prior to the azocasein assay. The open tubes containing the bacterial pellets were placed in an 80°C heat block for 3 h, and the dry weights of the cell pellets were determined.
Proteolytic activity for each strain was quantified as previously described (40), with some modifications. A 2% azocasein (Sigma) solution was prepared in 0.05 M Tris-HCl (pH 7.4). Cell-free supernatant (50 μl) was mixed with 50 μl of the azocasein substrate and incubated at 28°C for 2 h. Duplicate assays were performed for each supernatant sample, and negative controls were included consisting of 50 μl of sterile TYES broth. Following incubation, 130 μl of 10% trichloroacetic acid was added to each sample, mixed, and held at room temperature for 10 min. The samples were then centrifuged at 19,980 × g for 20 min to remove precipitated azocasein. One hundred microliters of the soluble fraction of each sample was removed and added to a flat-bottom 96-well plate, and 200 μl of 1 M NaOH was added to each well and mixed. The OD450 was determined using an iMark microplate reader (Bio-Rad). For each supernatant sample, the raw OD450 values obtained from the duplicate assays were averaged, and the mean negative-control OD450 was subtracted from these values. The adjusted OD450 values were converted to units of proteolytic activity by dividing by 0.001 (40). The units of proteolytic activity were multiplied by 100 to obtain the total proteolytic activity for each 5-ml culture and divided by the dry weight of the cells to obtain the proteolytic activity per milligram of dry cells. For each strain, the proteolytic activities determined for triplicate cultures were averaged.
Measurement of chondroitin sulfate lyase activity.
Chondroitin sulfate lyase activity was measured as previously described, with modifications (35, 54). Briefly, strains derived from F. columnare IA-S-4 and C#2 were grown with shaking at 200 rpm at 28°C to mid-exponential phase in TYES and Shieh broth, respectively. The cultures were normalized to the same concentration (55 Klett units, measured with a Klett colorimeter, which is equivalent to an OD600 of 0.4) and centrifuged at 16,873 × g for 10 min at 4°C, and the supernatants were harvested. Twenty microliters of supernatants and 150 μl of 0.2 mg/ml chondroitin sulfate A (Sigma-Aldrich) in 20 mM Tris buffer (pH 7.0) were added to wells in a 96-well flat-bottom microplate and incubated at 30°C for 30 min. Twenty microliters of TYES or Shieh broth was used as no-chondroitin sulfate lyase controls. Thirty microliters of 0.5% bovine serum albumin (BSA) in 0.45 M acetate buffer (pH 4.0) was added to each well, and an opaque white color developed, which was directly proportional to the amount of undigested chondroitin sulfate A present. The optical density of each well at 405 nm was measured with a microplate reader (Infinite 200 PRO; Tecan Group Ltd., Männedorf, Switzerland). The percentage of chondroitin sulfate degradation was calculated as (OD405control − OD405sample)/OD405control × 100.
Detection of recombinant mCherry secretion.
Cells of F. columnare strain C#2 or of its ΔgldN T9SS mutant were grown overnight in Shieh broth (5 μg/ml tetracycline) at 30°C with shaking. Cells carried either pSSK54, which expresses mCherry with the N-terminal signal peptide of F. johnsoniae ChiA (SP-mCherry), or pSSK52, which expresses SP-mCherry fused to the 105-amino-acid CTD of ChiA. Cells were pelleted by centrifugation at 22,000 × g for 15 min at 4°C, and the culture supernatant (spent medium) was separated. The spent medium was ultracentrifuged at 352,900 × g for 30 min at 4°C to remove residual insoluble material. For whole-cell samples, the cells were suspended in the original culture volume of phosphate-buffered saline consisting of 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 2 mM KH2PO4 (pH 7.4). Equal amounts of spent media and whole cells were boiled in SDS-PAGE loading buffer for 10 min. Proteins were separated by SDS-PAGE, and Western blot analyses were performed as previously described (26), except that polyvinylidene difluoride (PVDF) membranes were used instead of nitrocellulose. Equal amounts of each sample based on the starting material were loaded in each lane. For cell extracts, this corresponded to 10 μg of protein, whereas for spent medium, this corresponded to the equivalent volume of spent medium that contained 10 μg of cell protein before the cells were removed. For the detection of mCherry by Western blotting, commercially available antibodies against mCherry (0.5 mg per ml; BioVision Incorporated, Milpitas, CA) were used at a dilution of 1:5,000.
Analysis of secreted proteins by SDS-PAGE and LC-MS/MS.
Starter cultures of F. columnare wild-type strain C#2, the ΔgldN mutant, the ΔporV mutant, and complemented strains were grown in Shieh broth or Shieh broth containing 2.5 μg/ml tetracycline at 25°C for 20 h. For each strain, 150 μl of starter culture was inoculated into 3 ml of fresh modified Shieh broth or modified Shieh broth containing 2.5 μg/ml tetracycline and incubated at 25°C with shaking until the OD600 reached 0.5. Before collecting the spent media, the integrity of the bacterial cells for each strain was examined microscopically. Cultures were centrifuged at 16,873 × g for 10 min at 4°C. The fluid was filtered with 0.22-μm-pore-size polyvinylidene difluoride filters and stored at −80°C until needed. Proteins were separated by SDS-PAGE (12% polyacrylamide gel) and detected using the Bio-Rad (Hercules, CA) silver stain kit. Indicated regions of the gel were cut, and peptides were analyzed by enzymatic in-gel digestion and nano-liquid chromatography-tandem mass spectrometry (nano-LC-MS/MS) at the University of Wisconsin—Madison Mass Spectrometry Facility, as outlined on the website (https://www.biotech.wisc.edu/services/massspec), and as described previously (24), except that MS/MS data were searched against an F. columnare protein database instead of against an F. johnsoniae database.
Zebrafish challenges.
All procedures utilizing fish at each research facility were approved by the appropriate Institutional Animal Care and Use Committee. F. columnare wild-type, mutant, and complemented strains were grown in Shieh medium at 30°C overnight. Then, 250 μl of overnight culture was inoculated into 5 ml of fresh modified Shieh broth and shaken at 25°C until the OD600 reached 0.4. To determine the number of live cells per ml, serial dilutions were plated on Shieh agar. To test the virulence of wild-type, mutant, and complemented strains, the cultures were diluted and used to infect adult zebrafish (Danio rerio, strain Eckwill crossed with strain Tupfel Long-fin). No signs of disease were observed prior to challenge, and no indications of F. columnare or columnaris disease were observed in the uninfected control tanks or in the maintenance tanks at any time. Serial 2-fold dilutions were performed, with the highest titer being 1 ml of culture in 50 ml of water, and the lowest being 31 μl of culture in 50 ml of water. For the experiment shown in Fig. 7, the final challenge concentrations were 2.1 × 106 CFU/ml for C#2, 1.5 × 108 CFU/ml for the ΔgldNC#2 mutant, 2.1 × 106 CFU/ml for the ΔgldNC#2 mutant complemented with pLN8, 1.5 × 107 CFU/ml for the ΔporVC#2 mutant, and 2.1 × 106 CFU/ml for the ΔporVC#2 mutant complemented with pLN11. Zebrafish (10 fish per bacterial strain for each dilution) were immersed in 50 ml of water with F. columnare at 28°C for 1 h. Control fish were exposed to 1 ml of growth medium without F. columnare in 50 ml of water. After exposure, the challenged fish were transferred to 2 liters of fresh water and maintained for 7 days at 28°C. Mortalities were recorded daily. A minimum of 30% of the fish that died during this period were examined for the presence of bacteria phenotypic of F. columnare (yellow, rhizoid, adherent colonies) by swabbing gills, fins, and skin and streaking on Shieh agar.
Rainbow trout challenges.
Commercially available certified disease-free rainbow trout (Oncorhynchus mykiss) eggs were acquired from Troutlodge, Inc., Sumner, WA. Viable hatched trout were hand-fed daily to satiation using a commercially available trout feed (Ziegler, Inc., PA). Trout were maintained at the USDA-ARS National Center for Cool and Cold Water Aquaculture research facility in Kearneysville, WV, in flowthrough water at a rate of 1 liter/min at 12.5°C, until the challenge weight of ∼1.3 g was met. The fish in this facility are checked yearly for multiple diseases, including columnaris disease, and except for fish in the challenge room, they are certified disease free. No signs of disease were observed prior to challenge, and no indications of F. columnare or columnaris disease were observed in the uninfected control tanks or in the maintenance tanks at any time. Fish were moved to challenge aquaria 1 week prior to immersion challenge to acclimate to the elevated water temperature of 16°C.
The wild-type strain IA-S-4, ΔgldNIA-S-4 mutant, and ΔgldNIA-S-4 mutant complemented with plasmid pLN5 were each used for immersion challenges. Frozen bacterial stocks were stored at −80°C in 80% TYES broth and 20% glycerol. Bacterial cultures, for challenges, were grown as previously described (11). Briefly, 100 μl of frozen stocks was inoculated into 10 ml of TYES broth and incubated overnight at 30°C with shaking at 200 rpm. These starter cultures were used to inoculate Fernbach flasks containing 1 liter of TYES broth. These were incubated at 30°C with shaking at 200 rpm until an optical density at 540 nm of 0.7 to 0.75 was reached.
Challenges were performed using triplicate 4-liter tanks with restricted water flows (∼200 ml/min) at 16°C. Each tank contained 20 fish of approximately 1.35 g each. Water flows were stopped for the immersion challenge, and tanks were inoculated with bacterial cultures and incubated for 1 h, after which water flows were resumed. Control tanks were inoculated with TYES broth. Serial dilutions of water samples from each tank after inoculation were plated on TYES agar to determine the CFU per milliliter. The final challenge concentrations for the experiment shown in Fig. 8A were 1.2 × 105 CFU/ml for the wild-type IA-S-4, 5.5 × 106 CFU/ml for the ΔgldNIA-S-4 mutant, and 2.7 × 106 CFU/ml for the ΔgldNIA-S-4 mutant complemented with gldN on pLN5. Mortalities were removed and counted daily. The data for triplicate tanks of each strain were pooled, and survivor fractions for each strain were calculated. Twenty percent of the mortalities were randomly tested by homogenizing gill tissue and streaking on TYES agar plates to determine if F. columnare was present. Confirmation of F. columnare was determined by morphological observation of yellow rhizoid adherent colonies and by amplifying and sequencing 16S rRNA genes. F. columnare was detected in all mortalities. Genomovar confirmation was determined by enzymatic digestion (HaeIII) of the 16S rRNA gene, as previously described (7), and all were genomovar I, as expected for strain IA-S-4.
Channel catfish challenges.
Wild-type F. columnare strain C#2, the ΔgldNC#2 mutant, and the ΔgldNC#2 mutant complemented with wild-type gldN on pLN8 strains were grown in 100 ml of modified Shieh broth for 20 h at 28°C with shaking at 150 rpm. The optical densities at 540 nm were 1.17, 1.19, and 1.33 for the C#2, the ΔgldNC#2 mutant, and the ΔgldNC#2 mutant complemented with pLN8 strains, respectively. The number of CFU per milliliter was determined by spread plating 50 μl of serial dilutions (in duplicate) on modified Shieh agar plates. Plates were incubated for 48 h at 28°C, and colonies were counted.
Channel catfish (Ictalurus punctatus), with a mean ± standard deviation (SD) weight of 4.3 g ± 0.5 g and no history of columnaris disease, obtained from stocks held at the USDA-ARS Aquatic Animal Health Research Unit in Auburn, AL, were used for the bacterial challenges. Fish were housed in 378-liter troughs supplied with 28 ± 2°C dechlorinated municipal water prior to challenge. Fish were fed with appropriately sized fish feed (Rangen, Inc., Buhl, ID) at a rate of 3% of body weight per day prior to challenge and to satiation following challenge. Single groups of 25 fish were challenged in buckets by immersion for 15 min in 2 liters of water with three doses of each F. columnare strain (C#2, ΔgldN mutant, and ΔgldNC#2 mutant complemented with pLN8). For the experiment shown in Fig. 8B, the final challenge concentrations were 5.0 × 107 CFU/ml for C#2, 4.6 × 107 CFU/ml for the ΔgldNC#2 mutant, and 3.4 × 107 CFU/ml for the ΔgldNC#2 mutant complemented with pLN8. Following exposure to the bacteria, fish were netted from the buckets and placed into prefilled 57-liter tanks supplied with water at a flow rate of approximately 0.5 liters/min. Two additional groups of 25 fish each were mock-challenged by immersion, as described above, by adding sterile modified Shieh broth in a volume equal to the volume of bacterial culture added for the highest dose. Tanks were observed twice daily for 14 days, and dead/moribund fish were removed and recorded. Reisolation of F. columnare was attempted from 20% of the daily mortalities from each tank by inoculating head kidney tissue onto modified Shieh agar containing 1 μg/ml tobramycin (49). Plates were incubated at 28°C for 48 h and then examined for colonies phenotypic of F. columnare. For channel catfish, zebrafish, and rainbow trout challenges, survivor fractions after exposure to each bacterial strain were calculated using the product limit (Kaplan-Meier) method using GraphPad Prism (version 6.05; GraphPad Software, San Diego, CA, USA).
Toxicity of material secreted by F. columnare.
F. columnare IA-S-4 and C#2 strains were streaked from −80°C stocks onto TYES and Shieh agar, respectively, and incubated for 48 h at 30°C. Strains were restreaked on fresh plates, incubated 48 h at 30°C, and used to inoculate 20-ml starter flasks of TYES (IA-S-4 strains) or modified Shieh (C#2 strains). Starter cultures were incubated overnight, and then 6-ml volumes were used to inoculate 144 ml of medium of the same composition. Cultures were grown to mid-log phase (Klett units, 50 to 60), and cells were removed by centrifugation twice for 20 min at 3,700 × g at 4°C. The supernatants were filtered through 0.45-μm polyethersulfone (PES) filters to remove residual cells. One-tenth of a milliliter of the filtrate was plated on TYES or Shieh agar to verify that all viable cells had been removed. The filtered supernatant was kept on ice overnight before use. One hundred milliliters of supernatant placed in a 250-ml glass beaker was prewarmed to 28°C. Six zebrafish (approximately 0.35 g each) were added and incubated for 5 to 7 h. Mortalities were recorded every 30 min. Control fish were exposed to TYES (IA-S-4 strains) or modified Shieh medium (C#2 strains) instead of to cell-free spent medium. For heat inactivation experiments, cell-free spent medium was heated at 60°C for 60 min prior to zebrafish exposure. For trypsin inactivation experiments, cell-free spent medium was exposed to 50 μg/ml trypsin at 37°C for 12 h prior to zebrafish exposure. Samples without trypsin were also incubated at 37°C for 12 h for these experiments.
Bioinformatic analyses.
Genome sequences of F. columnare strains C#2 (accession no. NZ_CP015107.1) (47) and IA-S-4 (B. LaFrentz, unpublished data) were analyzed. Gene clusters encoding structural components of the T9SS (Table S1) were detected using MacSyFinder (55) together with the TXSScan profile for the T9SS (56). Proteins predicted to be secreted by the T9SS were detected using HmmSearch from the HMMER suite version 3.1b1 (http://www.hmmer.org) with TIGR04183 (referred to as type A CTD) and TIGR04131 (referred to as type B CTD) HMM profiles. Hits were regarded as significant when proteins were detected above HmmSearch model-specifics trusted cutoff thresholds. N-terminal cleavage sites were predicted using the SignalP4.1 Web server (57). Proteases and glycoside hydrolases were classified using the MEROPS (https://www.ebi.ac.uk/merops/) and Carbohydrate Active enZYmes (58) databases, respectively.
Supplementary Material
ACKNOWLEDGMENTS
We thank Henry Tomasiewicz, Kris Kosteretz, Rebekah Klingler, and Michael Carvan at the University of Wisconsin—Milwaukee School of Freshwater Sciences Fish Facility for supplying zebrafish, the University of Wisconsin—Milwaukee Animal Care Program for assistance with animal care, and Greg Sabat and Greg Barret-Wilt at the University of Wisconsin—Madison Mass Spectrometry Facility for LC-MS/MS analyses.
This research was supported by funds from the USDA-ARS (CRIS project 6010-32000-026-00D), by grants to M.J.M. from the USDA-ARS (project 5090-31320-004-03S) and from the National Science Foundation (MCB-1516990), and by grants to P.N. from the Strategic Priority Research Program of the Chinese Academy of Sciences (grant XDA08010207) and from the Knowledge Innovation Program of the Chinese Academy of Sciences. It was also funded by grants to M.J.M. and D.W.H. from the University of Wisconsin Sea Grant Institute under grants from the National Sea Grant College Program, National Oceanic and Atmospheric Administration, U.S. Department of Commerce, and the State of Wisconsin, federal grant NA14OAR4170092, projects R/SFA-08 and R/SFA-11. The funders had no role in the study design, data collection and interpretation, or the decision to submit the work for publication. The USDA is an equal opportunity employer.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.01769-17.
REFERENCES
- 1.Davis HS. 1922. A new bacterial disease of freshwater fishes. Bull US Bureau Fish 38:261–280. [Google Scholar]
- 2.Ordal EJ, Rucker RR. 1944. Pathogenic myxobacteria. Exp Biol Med 56:15–18. doi: 10.3181/00379727-56-14572. [DOI] [Google Scholar]
- 3.Declercq AM, Haesebrouck F, Van den Broeck W, Bossier P, Decostere A. 2013. Columnaris disease in fish: a review with emphasis on bacterium-host interactions. Vet Res 44:27. doi: 10.1186/1297-9716-44-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Austin B, Austin DA. 2007. Bacterial fish pathogens: diseases of farmed and wild fish, 4th ed Springer, Chichester, England. [Google Scholar]
- 5.Wakabayashi H. 1991. Effect of environmental conditions on the infectivity of Flexibacter columnaris to fish. J Fish Dis 14:279–290. doi: 10.1111/j.1365-2761.1991.tb00825.x. [DOI] [Google Scholar]
- 6.Darwish AM, Ismaiel AA. 2005. Genetic diversity of Flavobacterium columnare examined by restriction fragment length polymorphism and sequencing of the 16S ribosomal RNA gene and the 16S-23S rDNA spacer. Mol Cell Probes 19:267–274. doi: 10.1016/j.mcp.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 7.LaFrentz BR, Waldbieser GC, Welch TJ, Shoemaker CA. 2014. Intragenomic heterogeneity in the 16S rRNA genes of Flavobacterium columnare and standard protocol for genomovar assignment. J Fish Dis 37:657–669. doi: 10.1111/jfd.12166. [DOI] [PubMed] [Google Scholar]
- 8.Evenhuis JP, LaFrentz BR. 2016. Virulence of Flavobacterium columnare genomovars in rainbow trout Oncorhynchus mykiss. Dis Aquat Organ 120:217–224. doi: 10.3354/dao03027. [DOI] [PubMed] [Google Scholar]
- 9.Olivares-Fuster O, Baker JL, Terhune JS, Shoemaker CA, Klesius PH, Arias CR. 2007. Host-specific association between Flavobacterium columnare genomovars and fish species. Syst Appl Microbiol 30:624–633. doi: 10.1016/j.syapm.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 10.Shoemaker CA, Olivares-Fuster O, Arias CR, Klesius PH. 2008. Flavobacterium columnare genomovar influences mortality in channel catfish (Ictalurus punctatus). Vet Microbiol 127:353–359. doi: 10.1016/j.vetmic.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 11.Evenhuis JP, LaPatra SE, Marancik D. 2014. Early life stage rainbow trout (Oncorhynchus mykiss) mortalities due to Flavobacterium columnare in Idaho, USA. Aquaculture 418:126–131. doi: 10.1016/j.aquaculture.2013.09.044. [DOI] [Google Scholar]
- 12.LaFrentz BR, LaPatra SE, Shoemaker CA, Klesius PH. 2012. Reproducible challenge model to investigate the virulence of Flavobacterium columnare genomovars in rainbow trout Oncorhynchus mykiss. Dis Aquat Organ 101:115–122. doi: 10.3354/dao02522. [DOI] [PubMed] [Google Scholar]
- 13.Welker TL, Shoemaker CA, Arias CR, Klesius PH. 2005. Transmission and detection of Flavobacterium columnare in channel catfish Ictalurus punctatus. Dis Aquat Organ 63:129–138. doi: 10.3354/dao063129. [DOI] [PubMed] [Google Scholar]
- 14.Suomalainen LR, Kunttu H, Valtonen ET, Hirvela-Koski V, Tiirola M. 2006. Molecular diversity and growth features of Flavobacterium columnare strains isolated in Finland. Dis Aquat Organ 70:55–61. doi: 10.3354/dao070055. [DOI] [PubMed] [Google Scholar]
- 15.Arias CR, Welker TL, Shoemaker CA, Abernathy JW, Klesius PH. 2004. Genetic fingerprinting of Flavobacterium columnare isolates from cultured fish. J Appl Microbiol 97:421–428. doi: 10.1111/j.1365-2672.2004.02314.x. [DOI] [PubMed] [Google Scholar]
- 16.Chagnot C, Zorgani MA, Astruc T, Desvaux M. 2013. Proteinaceous determinants of surface colonization in bacteria: bacterial adhesion and biofilm formation from a protein secretion perspective. Front Microbiol 4:303. doi: 10.3389/fmicb.2013.00303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sandkvist M. 2001. Type II secretion and pathogenesis. Infect Immun 69:3523–3535. doi: 10.1128/IAI.69.6.3523-3535.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sato K, Naito M, Yukitake H, Hirakawa H, Shoji M, McBride MJ, Rhodes RG, Nakayama K. 2010. A protein secretion system linked to bacteroidete gliding motility and pathogenesis. Proc Natl Acad Sci U S A 107:276–281. doi: 10.1073/pnas.0912010107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McBride MJ, Nakane D. 2015. Flavobacterium gliding motility and the type IX secretion system. Curr Opin Microbiol 28:72–77. doi: 10.1016/j.mib.2015.07.016. [DOI] [PubMed] [Google Scholar]
- 20.McBride MJ, Zhu Y. 2013. Gliding motility and Por secretion system genes are widespread among members of the phylum Bacteroidetes. J Bacteriol 195:270–278. doi: 10.1128/JB.01962-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shrivastava A, Johnston JJ, van Baaren JM, McBride MJ. 2013. Flavobacterium johnsoniae GldK, GldL, GldM, and SprA are required for secretion of the cell surface gliding motility adhesins SprB and RemA. J Bacteriol 195:3201–3212. doi: 10.1128/JB.00333-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shrivastava A, Rhodes RG, Pochiraju S, Nakane D, McBride MJ. 2012. Flavobacterium johnsoniae RemA is a mobile cell-surface lectin involved in gliding. J Bacteriol 194:3678–3688. doi: 10.1128/JB.00588-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kharade SS, McBride MJ. 2014. Flavobacterium johnsoniae chitinase ChiA is required for chitin utilization and is secreted by the type IX secretion system. J Bacteriol 196:961–970. doi: 10.1128/JB.01170-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kharade SS, McBride MJ. 2015. Flavobacterium johnsoniae PorV is required for secretion of a subset of proteins targeted to the type IX secretion system. J Bacteriol 197:147–158. doi: 10.1128/JB.02085-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sato K, Yukitake H, Narita Y, Shoji M, Naito M, Nakayama K. 2013. Identification of Porphyromonas gingivalis proteins secreted by the Por secretion system. FEMS Microbiol Lett 338:68–76. doi: 10.1111/1574-6968.12028. [DOI] [PubMed] [Google Scholar]
- 26.Rhodes RG, Samarasam MN, Shrivastava A, van Baaren JM, Pochiraju S, Bollampalli S, McBride MJ. 2010. Flavobacterium johnsoniae gldN and gldO are partially redundant genes required for gliding motility and surface localization of SprB. J Bacteriol 192:1201–1211. doi: 10.1128/JB.01495-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rhodes RG, Samarasam MN, Van Groll EJ, McBride MJ. 2011. Mutations in Flavobacterium johnsoniae sprE result in defects in gliding motility and protein secretion. J Bacteriol 193:5322–5327. doi: 10.1128/JB.05480-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glew MD, Veith PD, Peng B, Chen YY, Gorasia DG, Yang Q, Slakeski N, Chen D, Moore C, Crawford S, Reynolds E. 2012. PG0026 is the C-terminal signal peptidase of a novel secretion system of Porphyromonas gingivalis. J Biol Chem 287:24605–24617. doi: 10.1074/jbc.M112.369223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nguyen KA, Travis J, Potempa J. 2007. Does the importance of the C-terminal residues in the maturation of RgpB from Porphyromonas gingivalis reveal a novel mechanism for protein export in a subgroup of Gram-negative bacteria? J Bacteriol 189:833–843. doi: 10.1128/JB.01530-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sato K, Sakai E, Veith PD, Shoji M, Kikuchi Y, Yukitake H, Ohara N, Naito M, Okamoto K, Reynolds EC, Nakayama K. 2005. Identification of a new membrane-associated protein that influences transport/maturation of gingipains and adhesins of Porphyromonas gingivalis J Biol Chem 280:8668–8677. [DOI] [PubMed] [Google Scholar]
- 31.Seers CA, Slakeski N, Veith PD, Nikolof T, Chen YY, Dashper SG, Reynolds EC. 2006. The RgpB C-terminal domain has a role in attachment of RgpB to the outer membrane and belongs to a novel C-terminal-domain family found in Porphyromonas gingivalis. J Bacteriol 188:6376–6386. doi: 10.1128/JB.00731-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shoji M, Sato K, Yukitake H, Kondo Y, Narita Y, Kadowaki T, Naito M, Nakayama K. 2011. Por secretion system-dependent secretion and glycosylation of Porphyromonas gingivalis hemin-binding protein 35. PLoS One 6:e21372. doi: 10.1371/journal.pone.0021372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Slakeski N, Seers CA, Ng K, Moore C, Cleal SM, Veith PD, Lo AW, Reynolds EC. 2011. C-terminal domain residues important for secretion and attachment of RgpB in Porphyromonas gingivalis. J Bacteriol 193:132–142. doi: 10.1128/JB.00773-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Veith PD, Muhammad NAN, Dashper SG, Likic VA, Gorasia DG, Chen D, Byrne SJ, Catmull DV, Reynolds EC. 2013. Protein substrates of a novel secretion system are numerous in the Bacteroidetes phylum and have in common a cleavable C-terminal secretion signal, extensive post-translational modification, and cell-surface attachment. J Proteome Res 12:4449–4461. doi: 10.1021/pr400487b. [DOI] [PubMed] [Google Scholar]
- 35.Li N, Qin T, Zhang XL, Huang B, Liu ZX, Xie HX, Zhang J, McBride MJ, Nie P. 2015. Gene deletion strategy to examine the involvement of the two chondroitin lyases in Flavobacterium columnare virulence. Appl Environ Microbiol 81:7394–7402. doi: 10.1128/AEM.01586-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Staroscik AM, Hunnicutt DW, Archibald KE, Nelson DR. 2008. Development of methods for the genetic manipulation of Flavobacterium columnare. BMC Microbiol 8:115. doi: 10.1186/1471-2180-8-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kulkarni SS, Zhu Y, Brendel CJ, McBride MJ. 2017. Diverse C-terminal sequences involved in Flavobacterium johnsoniae protein secretion. J Bacteriol 199:e00884-16. doi: 10.1128/JB.00884-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kunttu HM, Jokinen EI, Valtonen ET, Sundberg LR. 2011. Virulent and nonvirulent Flavobacterium columnare colony morphologies: characterization of chondroitin AC lyase activity and adhesion to polystyrene. J Appl Microbiol 111:1319–1326. doi: 10.1111/j.1365-2672.2011.05149.x. [DOI] [PubMed] [Google Scholar]
- 39.Suomalainen LR, Tiirola MA, Valtonen ET. 2006. Chondroitin AC lyase activity is related to virulence of fish pathogenic Flavobacterium columnare. J Fish Dis 29:757–763. doi: 10.1111/j.1365-2761.2006.00771.x. [DOI] [PubMed] [Google Scholar]
- 40.Newton JC, Wood TM, Hartley MM. 1997. Isolation and partial characterization of extracellular proteases produced by isolates of Flavobacterium columnare derived from catfish. J Aquat Anim Health 9:75–85. doi:. [DOI] [Google Scholar]
- 41.Kayansamruaj P, Dong HT, Hirono I, Kondo H, Senapin S, Rodkhum C. 2017. Comparative genome analysis of fish pathogen Flavobacterium columnare reveals extensive sequence diversity within the species. Infect Genet Evol 54:7–17. doi: 10.1016/j.meegid.2017.06.012. [DOI] [PubMed] [Google Scholar]
- 42.Tekedar HC, Karsi A, Reddy JS, Nho SW, Kalindamar S, Lawrence ML. 2017. Comparative genomics and transcriptional analysis of Flavobacterium columnare strain ATCC 49512. Front Microbiol 8:588. doi: 10.3389/fmicb.2017.00588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen YY, Peng B, Yang Q, Glew MD, Veith PD, Cross KJ, Goldie KN, Chen D, O'Brien-Simpson N, Dashper SG, Reynolds EC. 2011. The outer membrane protein LptO is essential for the O-deacylation of LPS and the co-ordinated secretion and attachment of A-LPS and CTD proteins in Porphyromonas gingivalis. Mol Microbiol 79:1380–1401. doi: 10.1111/j.1365-2958.2010.07530.x. [DOI] [PubMed] [Google Scholar]
- 44.Ishiguro I, Saiki K, Konishi K. 2009. PG27 is a novel membrane protein essential for a Porphyromonas gingivalis protease secretion system. FEMS Microbiol Lett 292:261–267. doi: 10.1111/j.1574-6968.2009.01489.x. [DOI] [PubMed] [Google Scholar]
- 45.Nakane D, Sato K, Wada H, McBride MJ, Nakayama K. 2013. Helical flow of surface protein required for bacterial gliding motility. Proc Natl Acad Sci U S A 110:11145–11150. doi: 10.1073/pnas.1219753110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nelson SS, Bollampalli S, McBride MJ. 2008. SprB is a cell surface component of the Flavobacterium johnsoniae gliding motility machinery. J Bacteriol 190:2851–2857. doi: 10.1128/JB.01904-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bartelme RP, Newton RJ, Zhu Y, Li N, LaFrentz BR, McBride MJ. 2016. Complete genome sequence of the fish pathogen Flavobacterium columnare strain C#2. Genome Announc 4(3):e00624-16. doi: 10.1128/genomeA.00624-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thomas-Jinu S, Goodwin AE. 2004. Morphological and genetic characteristics of Flavobacterium columnare isolates: correlations with virulence in fish. J Fish Dis 27:29–35. doi: 10.1046/j.1365-2761.2003.00507.x. [DOI] [PubMed] [Google Scholar]
- 49.Decostere A, Haesebrouck F, Devriese LA. 1997. Shieh medium supplemented with tobramycin for selective isolation of Flavobacterium columnare (Flexibacter columnaris) from diseased fish. J Clin Microbiol 35:322–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.LaFrentz BR, Klesius PH. 2009. Development of a culture independent method to characterize the chemotactic response of Flavobacterium columnare to fish mucus. J Microbiol Methods 77:37–40. doi: 10.1016/j.mimet.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 51.Holt JG, Krieg NR, Sneath PHA, Staley JT, Williams ST. 1994. Bergey's manual of determinative bacteriology. Williams and Wilkins, Baltimore, MD. [Google Scholar]
- 52.McBride MJ, Kempf MJ. 1996. Development of techniques for the genetic manipulation of the gliding bacterium Cytophaga johnsonae. J Bacteriol 178:583–590. doi: 10.1128/jb.178.3.583-590.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bertani G. 1951. Studies on lysogenesis I. The mode of phage liberation by lysogenic Escherichia coli. J Bacteriol 62:293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Teska JD. 1993. Assay to evaluate the reaction kinetics of chondroitin AC lyase produced by Cytophaga columnaris. J Aquat Anim Health 5:259–264. doi:. [DOI] [Google Scholar]
- 55.Abby SS, Neron B, Menager H, Touchon M, Rocha EP. 2014. MacSyFinder: a program to mine genomes for molecular systems with an application to CRISPR-Cas systems. PLoS One 9:e110726. doi: 10.1371/journal.pone.0110726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Abby SS, Cury J, Guglielmini J, Neron B, Touchon M, Rocha EP. 2016. Identification of protein secretion systems in bacterial genomes. Sci Rep 6:23080. doi: 10.1038/srep23080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Petersen TN, Brunak S, von Heijne G, Nielsen H. 2011. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 58.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. 2014. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res 42:D490–D495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Juncker AS, Willenbrock H, von Heijne G, Nielsen H, Brunak S, Krogh A. 2003. Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci 12:1652–1662. doi: 10.1110/ps.0303703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.de Lorenzo V, Timmis KN. 1994. Analysis and construction of stable phenotypes in Gram-negative bacteria with Tn5- and Tn10-derived minitransposons. Methods Enzymol 235:386–405. doi: 10.1016/0076-6879(94)35157-0. [DOI] [PubMed] [Google Scholar]
- 61.Agarwal S, Hunnicutt DW, McBride MJ. 1997. Cloning and characterization of the Flavobacterium johnsoniae (Cytophaga johnsonae) gliding motility gene, gldA. Proc Natl Acad Sci U S A 94:12139–12144. doi: 10.1073/pnas.94.22.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.