

Abstract
The dependence of the rate on polymer mass was examined for the reaction of four sulfhydryl-directed poly(ethylene glycol) reagents with cysteine residues located in the lumen of the staphylococcal α-hemolysin pore. The logarithms of the apparent rate constants for a particular site in the lumen were proportional to N, the number of repeat units in a polymer chain. The proportionality constant was −(a/D)5/3, where a is the persistence length of the polymer (≈3.5Å) and D is the diameter of the pore. Despite some incongruencies with the assumptions of the derivation, the result suggests that the polymers partition into the lumen of the pore according to the simple scaling law of Daoud and de Gennes, cpore/csolution = exp(−N(a/D)5/3). Therefore, the measured reaction rates yield an estimate of the diameter of the pore and might be applied to determine the approximate dimensions of cavities within other similar proteins.
The interactions of polymers with proteinaceous channels and pores has been studied extensively (1, 2). The osmotic effects of polymers on the voltage-dependent anion channel of mitochondria (VDAC) and other channels have been examined (1, 3–5). Polymer partitioning into pores from concentrated solutions has been investigated in studies of single-channel conductance (6), access resistance (7, 8), and single-channel noise (2, 8–10). The movement of nucleic acids through pores has been examined through their effects on single-channel conductance (11–14). The effects of covalently attached polymers on the properties of channels and pores have also been investigated (15–17).
Given the interest in this area, it is of fundamental importance to understand how polymers partition from dilute solution into protein pores. This problem has received attention by theoreticians. Notably, scaling theory has been used to estimate partition coefficients (18, 19). Specifically, Daoud and de Gennes (20) found the partition coefficient (Π) for polymers into a tube to be
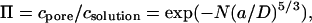 |
1 |
where c is the concentration, N is the number of units in a polymer chain, a is the persistence length of the polymer, and D is the diameter of the pore. This relation applies to a narrow tube both where the Flory radius of the polymer RF > D (20) and where RF ≈ D (21, 22). It requires that the free energy of confinement of each segment (“blob”) of the polymer within the pore is kBT, where kB is the Boltzmann constant and T the absolute temperature.
Interestingly, the scaling relation has not been subjected to extensive experimental examination. Several studies have shown that −ln Π is linearly dependent on N (18, 23), but the appropriateness of the scaling coefficient −(a/D)5/3 has received less attention. Here, we analyze how poly(ethylene glycol) (PEG) molecules partition into the pore formed by staphylococcal α-hemolysin (αHL). The dimensions of the αHL pore have been determined by crystallography (Fig. 1; ref. 24). Importantly for the present study, the transmembrane portion of the pore is a β-barrel measuring ≈20 Å in diameter. We show that the side chains of cysteine residues projecting into the lumen of the barrel react with sulfhydryl-directed PEG reagents at rates that suggest the reagents partition into the pore according to the Daoud and de Gennes relation.
Figure 1.

Sections through the αHL pore. (a) On the cis side of the bilayer, the protein has a large vestibule, which measures ≈46 Å in internal diameter. The transmembrane domain is a 14-stranded β-barrel of ≈20 Å diameter. The two domains of the lumen are separated by a constriction of ≈14 Å diameter. The cysteine mutations discussed here are marked. (b) A view from the trans side of the bilayer.
Materials and Methods
Reactivity of MePEG-OPSS Reagents in Solution.
The rates of reaction of monomethoxypoly(ethylene glycol)-o-pyridyl disulfide (MePEG-OPSS) reagents (Table 1; 1 mM) with β-mercaptoethanol (71.5 μM) were determined in 300 mM KCl/5 mM Tris⋅HCl, pH 8.5, the same buffer that was used for reaction with the cysteine mutants of αHL (25). The reactions were monitored by measuring the absorbance of the product, 2-thiopyridone, at 343 nm (26–28). The rate constants for the four reagents were not significantly different: MePEG-OPSS-1k, 4.5 × 103 M−1⋅s−1; MePEG-OPSS-1.8k, 4.5 × 103 M−1⋅s−1; MePEG-OPSS-2.5k, 4.2 × 103 M−1⋅s−1; MePEG-OPSS-5k, 4.7 × 103 M−1⋅s−1. By comparison, the rate constant for the reaction between 2-mercaptoethanol and 2,2′-dipyridyl disulfide at pH 8.1 is 4.25 × 103 M−1⋅s−1 (27).
Table 1.
Characteristics of the MePEG-OPSS reagents used in this work
| Polymer | N | RF, Å | c, % | c*, % | ℓ, Å |
|---|---|---|---|---|---|
| MePEG-OPSS-1k | 19 | 21 | 0.34 | 3.9 | 21 |
| MePEG-OPSS-1.8k | 39 | 32 | 0.68 | 2.2 | 43 |
| MePEG-OPSS-2.5k | 56 | 39 | 0.98 | 1.7 | 61 |
| MePEG-OPSS-5k | 113 | 60 | 2.0 | 0.95 | 120 |
N, number of monomer units in polymer. The mass of the OPSS group was subtracted from Mw, and the result divided by the mass of one monomer unit (M = 44); RF, Flory radius, obtained by using RF = aN3/5, with a = 3.5 Å; c, polymer concentration in the bath (wt/vol) ignoring the contribution of the OPSS group; c*, the overlap threshold, which defines the gradual transition between dilute and semidilute regimes (37); ℓ, the predicted linear extension of a polymer in a tube of diameter 20 Å (see Eq. 5 and ref. 21).
Molecular Modeling.
A model of the αHL heptamer was generated with SPOCK 6.3 (29) with coordinates (24) from the Brookhaven Protein Data Bank (PDB ID code 7ahl). The internal diameter of the pore, at different sites in the lumen, was calculated by three methods: (i) From the coordinates of the Cα atoms in the polypeptide backbone; (ii) from the coordinates of the C-, N-, or O-terminal atoms in the amino acid side chains projecting into the lumen; and (iii) from the coordinates of the sulfur atoms in the side chains of cysteine residues, which had been used to replace wild-type side chains by using SPOCK 6.3. In all three cases, a circle was fitted to the coordinates. In ii, a few side chains that did not project toward the center of the lumen were ignored (25). The standard deviations (SD) reflect the deviations of the locations of the atoms that were used in the fit from the circles.
Results
Quantitative Analysis of Derivatization of Luminal Cysteines with Polymer Reagents.
In a previous study, we examined the reaction of four MePEG-OPSS reagents with αHL pores containing single-cysteine mutant subunits (25, 30). The masses of the MePEG chains were 1.0, 1.8, 2.5, and 5.0 kDa. A qualitative analysis of the results was used to determine the location of the constriction in the lumen of the pore. Here, a quantitative analysis of the rates is made that provides information about the diameter of the pore at various positions in the lumen. Because αHL pores are heptameric, each mutant contained seven reactive sulfhydryls. However, in most cases only one reacted (25) and the kinetics of reaction of the first sulfhydryl are discussed here.
The apparent first-order reaction rate constants (k′) for the reactions were obtained from the rate of decay of macroscopic currents in the presence of the reagents. The rate of reaction of a MePEG-OPSS with a protein pore is given by
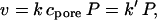 |
2 |
where k is the second-order reaction rate constant, P is the number of reactive protein pores, and cpore is the concentration of polymeric reagent in the lumen of the pore.
By substituting from Eq. 1, we obtain
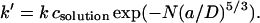 |
3 |
Therefore,
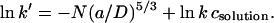 |
4 |
To test the validity of this relationship log k′ was plotted against N for positions in the transmembrane barrel (Fig. 2A). For all five positions, the plot is linear. Therefore, the results suggest that the “concentration” of the reagent in the pore is well described by a relation of the form of Eq. 1.
Figure 2.

Plots of log k′ versus N. k′ is the apparent first-order reaction rate constant for the reaction of the first cysteine in a homoheptameric cysteine mutant with a MePEG-OPSS reagent. N is the number of monomer units in a polymer. (A) MePEG-OPSS (4 mM) was applied from the trans side of the bilayer; (B) MePEG-OPSS (4 mM) was applied from the cis side. The reagents were: MePEG-OPSS-1k (N = 19, Mw/Mn = 1.02), MePEG-OPSS-1.8k (N = 39, Mw/Mn = 1.02), MePEG-OPSS-2.5k (N = 56, Mw/Mn = 1.03), and MePEG-OPSS-5k (N = 113, Mw/Mn = 1.02). The holding potential was −40 mV, as defined (25). The electrolyte in both chambers was 300 mM KCl/5 mM Tris⋅HCl, pH 8.5.
Internal Diameter of the β-Barrel Derived from Scaling Analysis.
Given Eq. 4, the slopes of plots of ln k′ versus N are −(a/D)5/3. Because the persistence length can be taken to be a = 3.5 Å for PEG (31–33), values for D can be obtained from straight line fits. The slopes for the mutants L135C7, T117C7, and M113C7 are similar and yield, respectively, D = 21 ± 1 Å, 20 ± 1 Å, and 20 ± 0 Å (n = 5; Table 2), values that are remarkably similar to the internal diameter of the barrel measured from molecular models (Table 3, Fig. 1). By contrast with these sites, the plot for E111C7 exhibited a greater slope, corresponding to D = 16 ± 0 Å (n = 4), whereas the slope for T129C7 is more shallow, corresponding to D = 23 ± 1 Å (n = 4). When values other than 5/3 are used for the exponential, the diameters obtained are less realistic. For example, −(a/D)6/3 yields D = 31 Å and −(a/D)4/3 yields D = 15 Å at position 135.
Table 2.
Pore diameters and reaction rates extrapolated to N = 0
| Mutant | Pore diameter, D, derived from scaling analysis (Å) | Second-order rate constant, k (M−1⋅s−1), pH 8.5 |
|---|---|---|
| T129C7 | 23 ± 1 | 25 ± 2 |
| L135C7 | 21 ± 1 | 160 ± 20 |
| T117C7 | 20 ± 1 | 970 ± 30 |
| M113C7 | 20 ± 0 | 22 ± 3 |
| E111C7 | 16 ± 0 | 930 ± 10 |
| S106C7 | 27 ± 2 | 10 ± 2 |
| K8C7 | 21 ± 2 | 430 ± 30 |
The values were obtained from plots of ln k′ versus N as described in the text. The data for the plots were derived from experiments in which MePEG-OPSS was added to the trans side of the bilayer, with the exception of S106C7 and K8C7, where MePEG-OPSS was added to the cis side of the bilayer. The conditions are in Fig. 2 (legend). The values are averages ± SD from at least four experiments.
Table 3.
Internal dimensions of homoheptameric αHL pores derived from crystallographic data
| Site | Method | Diameter, Å |
|---|---|---|
| Thr-129 | WT backbone (Cα-Cα) | 28.5 ± 0.9 |
| WT side chains | 26.2 ± 0.6 | |
| Cysteine mutant | 25.7 ± 0.7 | |
| Leu-135 | WT backbone (Cα-Cα) | 24.0 ± 0.3 |
| WT side chains | 19.5 ± 0.4 | |
| Cysteine mutant | 19.5 ± 0.5 | |
| Thr-117 | WT backbone (Cα-Cα) | 24.7 ± 0.4 |
| WT side chains | 21.7 ± 0.5 | |
| Cysteine mutant | 20.5 ± 0.6 | |
| Met-113 | WT backbone (Cα-Cα) | 24.8 ± 0.3 |
| WT side chains | 16.4 ± 0.8 | |
| Cysteine mutant | 20.5 ± 0.4 | |
| Glu-111 | WT backbone (Cα-Cα) | 25.5 ± 0.3 |
| WT side chains | 18.5 ± 0.6 | |
| Cysteine mutant | 21.0 ± 0.6 | |
| Lys-147 | WT backbone (Cα-Cα) | 24.4 ± 0.2 |
| WT side chains | 14.9 ± 0.7 | |
| Cysteine mutant | 19.9 ± 0.6 | |
| Ser-106 | WT backbone (Cα-Cα) | 41.8 ± 0.4 |
| WT side chains | 37.7 ± 0.6 | |
| Cysteine mutant | 38.6 ± 0.5 | |
| Lys-8 | WT backbone (Cα-Cα) | 30.2 ± 0.1 |
| WT side chains | 24.1 ± 0.1 | |
| Cysteine mutant | 28.1 ± 0.4 |
WT, wild type. For details see Materials and Methods.
Intrinsic Reactivity of Cysteine Residues in the β-Barrel.
By extrapolating plots of ln k′ versus N, the values of the bimolecular rate constants for N = 0 (k in Eqs. 2–4) could be determined and were found to span almost two orders of magnitude (Table 2, Fig. 2). The deprotonated form of the cysteine side chains react with OPSS reagents (34) and, therefore, the range of reactivities may largely be explained by differences of the pKa values of the sulfhydryls (25, 35, 36). The values of k ranged from 22 to 970 M−1⋅s−1 (not corrected for the statistical effect arising from the presence of seven reactive cysteines per pore). By comparison, the values of k for the reaction of the four MePEG-OPSS reagents with 2-mercaptoethanol in solution, which were all similar, averaged 4.5 × 103 M−1⋅s−1 at pH 8.5, in the same buffer used for derivatizing the αHL pores.
Partition Coefficients of the Polymers into the αHL Pore.
From Eq. 1 and the data in Fig. 2, we can also derive partition coefficients for the polymer into the protein pore (Table 4). Although the results are instructive, they actually provide no additional information because the partition coefficients were used implicitly to find the internal diameters, D. In addition, the meaning of the term partition coefficient must be qualified as the polymers take up a large volume of the lumen (see below) and cannot in reality be said to be located at a particular residue.
Table 4.
Partition coefficients (Π) determined from the rates of reaction of MePEG-OPSS reagents at sites in the β-barrel
| Mutant | MePEG-OPSS-1k | MePEG-OPSS-1.8k | MePEG-OPSS-2.5k | MePEG-OPSS-5k |
|---|---|---|---|---|
| L135C7 | 0.32 ± 0.04 | 0.13 ± 0.02 | 0.058 ± 0.008 | 0.0032 ± 0.0007 |
| T117C7 | 0.28 ± 0.03 | 0.10 ± 0.01 | 0.042 ± 0.007 | 0.0012 ± 0.0004 |
| M113C7 | 0.30 ± 0.02 | 0.11 ± 0.02 | 0.048 ± 0.009 | 0.0023 ± 0.0007 |
| E111C7 | 0.17 ± 0.03 | 0.042 ± 0.008 | 0.012 ± 0.003 | NR |
NR, no observable reaction.
Internal Diameters in the Cap Domain.
Although Eq. 1 does not strictly pertain to a noncylindrical structure, we applied it to position 106 (in the large internal cavity) and position 8 (at the trans entrance) (Fig. 2B) and obtained internal diameters of 27 ± 2 Å and 21 ± 2 Å (Table 2).
Discussion
The penetration of neutral, flexible, water-soluble polymers into pores from dilute solution is of fundamental importance for a variety of practical applications, including ultrafiltration, gel permeation chromatography, and gel electrophoresis. Here, we have examined the behavior of PEG chains by analyzing the rates of reaction of MePEG-OPSS reagents with sulfhydryl groups in the lumen of the αHL pore. The MePEG-OPSS reagents (pKa ≈ 2; refs. 25 and 28) are neutral under the conditions of the experiments analyzed here, which were conducted at pH 8.5. They are also flexible, with a persistence length a ≈3.5 Å. The pore size (D ≈ 20 Å) is comparable to or smaller than the Flory radii of the four polymers discussed here (RF = 21–60 Å; Table 1), but much larger than the persistence length. Importantly, by contrast with previous work by others on the effects of PEG on channels and pores (refs. 1 and 2 and references therein), the experiments discussed here were carried out in or close to the dilute regime, in which the interactions between individual polymer chains are minimal (Table 1; refs. 21 and 37).
We found that values of ln k′, where k′ is the apparent reaction rate constant (Eq. 2), for sites in the lumen of the β-barrel of the αHL pore were proportional to N, the number of units in a polymer chain. The proportionality constant was well fitted by −(a/D)5/3, suggesting that the polymers partition into the lumen of the pore according to the simple scaling law of Daoud and de Gennes, cpore/csolution = exp(−N(a/D)5/3, and that reaction proceeds through rapid equilibration of the reagent between the bulk solution and the barrel, followed by a slower covalent reaction with the wall. By contrast with the steep dependence of rate on polymer mass inside the pore, the rates of reaction of the four reagents with β-mercaptoethanol in bulk solution were closely similar.
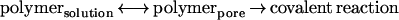 |
Because the polymers are in the dilute regime, the confinement free energy is a main determinant of partitioning into the pore. Further, Eq. 1 implies that for each segment (“blob”) of the confined polymer the free energy of confinement is equal to 1 kBT (38). It is remarkable how well the relation is obeyed given the realities of experimentation with a protein pore. In addition to the conditions of a dilute regime and water being a “good” solvent for the polymer (38, 39), the derivation assumes that the pore is a narrow cylinder with a constant cross-sectional area. The actual diameter varies between 15 Å and 24 Å, even in the β-barrel, which is the most uniform part of the structure (Table 3). The diameter of the pore should also be comparable to or smaller than the Flory radius of the polymer (i.e., D ≤ RF; refs. 20 and 22), which is the case (Table 1). The derivation also assumes no significant interactions of the polymer with the wall of the lumen, which is supported by the weak partitioning found under the conditions used here (Table 4).
Another difficulty is that the derivation of Eq. 1 assumes full penetration of the polymer into the pore. The length of a polymer in a pore, under the stated conditions, where it is envisaged as a chain of “blobs” of D = Rblob with an interblob distance of Rblob, is given by Daoud and de Gennes (20).
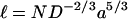 |
5 |
The MePEG-OPSS-5k reagent would then be 120 Å in length (Table 1), too long to fit in the β-barrel. The 2.5 kDa reagent would barely fit. Nonetheless, the data for MePEG-OPSS-5k do appear to scale correctly. It should also be noted that the shorter chains do not need to fully translocate into the lumen to react. Similarly, at the time of reaction, the bulk of the polymer is not located at the reaction site. Therefore, the meaning of D can be called into question. Nonetheless, the scaling relation does yield a reasonable value for the diameter at the central sites of reaction in the barrel (positions 135, 117, and 113; Tables 2 and 3). It is also notable that the scaling procedure works despite the range of reactivities at these positions, which is evident in the values of k obtained by extrapolating plots of ln k′ versus N to N = 0 (Fig. 2A, Table 2). It follows that the measurement of the rates of reaction of a single macromolecular reagent would not provide useful information about the dimensions of the pore.
The rates of reaction at positions 129 and 111 also obeyed the scaling relation, yielding diameters of 23 Å and 16 Å, respectively. The value for position 129 is consistent with the expansion of the diameter of the pore at this position to 26 Å (Table 3), although the agreement is surprising given the location of this residue at the entrance to the lumen. Because of the regular construction of the β-barrel, the internal diameter at position 111 in the mutant E111C is similar to the diameters at the central residues of the barrel. Presumably, the diameter obtained from the scaling relation is reduced because of the proximity of residue 111 to the constriction. Indeed, the value of 16 Å is close to the diameter at Lys-147 when the bulky side chains are included (15 Å).
When the MePEG-OPSS reagents are applied from the trans side of the bilayer, the reaction rates of the 5-kDa reagent cannot be measured at the cis residues 106 and 8, which lie beyond the constriction, and the rates of reaction of the remaining reagents do not give linear plots for log k′ versus N at these sites (data not shown). Beyond the constriction the reagents are not in equilibrium with the reagent in bulk solution on the trans side. Rather, there must be a steady state concentration in the cavity that depends on the movement of the reagent through the pore into the cis chamber, where its concentration is effectively zero. Therefore, the reaction rates are not expected to scale according to Eq. 1. Similarly, log k′ versus N plots obtained for the trans residues by reaction from the cis side were not linear and there are small but obvious anomalies in the rates—e.g., cysteines at position 106 react at k′ = 0.046 s−1 with 4 mM of the 1-kDa reagent presented from the trans chamber, and k′ = 0.021 s−1 from the cis chamber. The rate from the cis chamber would have been expected to be larger because position 106 is before the constriction in this case.
The rates of reaction at positions 106 and 8 were also measured with the reagents applied from the cis side. In this case, linear plots were obtained yielding diameters of 27 Å and 21 Å, respectively. The fits are surprising because, in the vestibule (Fig. 1), RF < D for the smaller polymers (Table 1). For example, the diameter at position 106 is 39 Å determined from the crystal structure (Table 3). The low measured diameter at position 106 may be explained in part by the fact that the larger polymers will be more highly confined by the roughly spherical cis cavity than they would be by a tube of the same diameter. The reduced value at position 8 over the value of 28 Å from the structure is less readily explained and all told it is clear that values of D obtained by scaling for the cap domain are less convincing than those from the β-barrel.
An important question is whether the approach taken here can be used to define the geometry of the lumen of channels and pores for which there is no structural data. The evidence suggests that values of D for roughly cylindrical parts of pores might be determined quite accurately by the scaling approach. Such a situation might arise, for example, where a pore is certain to be a β-barrel, but with an unknown number of strands. To pursue this possibility, additional polymers will be needed. We estimate that polymers in the mass range of 1 to 10 kDa are useful for the scaling approach when a/D ≈ 0.15. In addition to polymers with different persistence lengths, molecules of lesser or greater reactivity are required. For example, reaction rates at some positions in αHL were too low to measure (25).
In other cases (e.g., channels and pores assembled from transmembrane helices), further exploratory work will be required with proteins of known three-dimensional structure. In certain cases, the approach is clearly inapplicable. For example, for very-large-diameter pores where D ≫ L (the length of the pore), for instance that formed by streptolysin O where D ≈ 250 Å, the scaling relation of Eq. 1 is inappropriate because one blob would be of greater volume than the lumen. Furthermore, application of the approach to noncylindrical geometries must be done with caution. Here, the scaling approach appears to give a rough measure of D (e.g., the lumen is wider at position 106 than position 8), and provides an alternative to approaches such as fluorescence energy transfer, which have their own assumptions and difficulties in implementation. The residual current after derivatization with MePEG-OPSS is also a useful indicator of D (25) that complements the present approach and that might be set on a more quantitative foundation by additional experimentation and modeling.
Bezrukov and colleagues (6, 9) examined changes in unitary conductance of αHL and current noise induced by free PEG molecules of various molecular masses. Their findings were not consistent with the simple scaling theory used here. They initially concluded that PEG interacts with the walls of the lumen (9) and later (working at lower electrolyte concentrations) that the polymer molecules behave more like hard spheres with significant interparticle repulsion, rather than highly flexible chains (6). Although conciliation of these data with ours would require additional experimentation, we emphasize that our polymer solutions were in the dilute regime, which would reduce the effects of interparticle repulsion, and that our approach extends the measurements to weak partitioning (Table 4).
The work described here provides a test for scaling theory, which is however flawed by the considerations outlined above. It would be interesting to elaborate on the theory to account for the departures from cylindrical geometry to see whether a better agreement can be obtained for the data corresponding to the cis entrance and the internal cavity. A better test of the simple theory might be provided by using the long uniform synthetic nanotubules under development in other laboratories (40–42).
Acknowledgments
This work was supported by grants to H.B. from the Department of Energy, National Institutes of Health, Office of Naval Research (MURI-1999), and Texas Advanced Technology Program, and to A. J. Welch from the Air Force Office of Scientific Research (MURI-1998).
Abbreviations
- αHL
staphylococcal α-hemolysin
- MePEG-OPSS
monomethoxypoly(ethylene glycol)-o-pyridyl disulfide
- PEG
poly(ethylene glycol)
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Parsegian V A, Bezrukov S M, Vodyanoy I. Biosci Rep. 1995;15:503–514. doi: 10.1007/BF01204353. [DOI] [PubMed] [Google Scholar]
- 2.Bezrukov S M. J Membr Biol. 2000;174:1–13. doi: 10.1007/s002320001026. [DOI] [PubMed] [Google Scholar]
- 3.Zimmerberg J, Parsegian V A. Nature (London) 1986;323:36–39. doi: 10.1038/323036a0. [DOI] [PubMed] [Google Scholar]
- 4.Zimmerberg J, Benzanilla F, Parsegian V A. Biophys J. 1990;57:1049–1064. doi: 10.1016/S0006-3495(90)82623-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vodyanoy I, Bezrukov S M, Parsegian V. Biophys J. 1993;65:2097–2105. doi: 10.1016/S0006-3495(93)81245-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Merzlyak P G, Yuldasheva L N, Rodrigues C G, Carneiro C M M, Krasilnikov O V, Bezrukov S M. Biophys J. 1999;77:3023–3033. doi: 10.1016/S0006-3495(99)77133-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vodyanoy I, Bezrukov S M. Biophys J. 1992;62:10–11. doi: 10.1016/S0006-3495(92)81762-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bezrukov S M, Vodyanoy I. Biophys J. 1993;64:16–25. doi: 10.1016/S0006-3495(93)81336-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bezrukov S M, Vodyanoy I, Brutyan R A, Kasianowicz J J. Macromolecules. 1996;29:8517–8522. [Google Scholar]
- 10.Bezrukov S M, Kasianowicz J J. Eur Biophys J. 1997;26:471–476. doi: 10.1007/s002490050101. [DOI] [PubMed] [Google Scholar]
- 11.Kasianowicz J J, Brandin E, Branton D, Deamer D W. Proc Natl Acad Sci USA. 1996;93:13770–13773. doi: 10.1073/pnas.93.24.13770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Akeson M, Branton D, Kasianowicz J J, Brandin E, Deamer D W. Biophys J. 1999;77:3227–3233. doi: 10.1016/S0006-3495(99)77153-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meller A, Nivon L, Brandin E, Golovchenko J, Branton D. Proc Natl Acad Sci USA. 2000;97:1079–1084. doi: 10.1073/pnas.97.3.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deamer D W, Akeson M. Trends Biotechnol. 2000;18:147–151. doi: 10.1016/s0167-7799(00)01426-8. [DOI] [PubMed] [Google Scholar]
- 15.Howorka S, Movileanu L, Lu X, Magnon M, Cheley S, Braha O, Bayley H. J Am Chem Soc. 2000;122:2411–2416. [Google Scholar]
- 16.Blaustein R O, Cole P A, Williams C, Miller C. Nat Struct Biol. 2000;7:309–311. doi: 10.1038/74076. [DOI] [PubMed] [Google Scholar]
- 17.Movileanu L, Howorka S, Braha O, Bayley H. Nat Biotechnol. 2000;18:1091–1095. doi: 10.1038/80295. [DOI] [PubMed] [Google Scholar]
- 18.Gorbunov A A, Skvortsov A M. Adv Colloid Interface Sci. 1995;62:31–108. [Google Scholar]
- 19.Grosberg A Y, Khokhlov A R. Giant Molecules. San Diego: Academic; 1997. [Google Scholar]
- 20.Daoud M, de Gennes P-G. J de Physique. 1977;38:85–93. [Google Scholar]
- 21.de Gennes P-G. Scaling Concepts in Polymer Physics. Ithaca, NY: Cornell Univ. Press; 1979. [Google Scholar]
- 22.Boyd R H, Chance R R, Ver Strate G. Macromolecules. 1996;29:1182–1190. [Google Scholar]
- 23.Haller W. Macromolecules. 1977;10:83–86. [Google Scholar]
- 24.Song L, Hobaugh M R, Shustak C, Cheley S, Bayley H, Gouaux J E. Science. 1996;274:1859–1865. doi: 10.1126/science.274.5294.1859. [DOI] [PubMed] [Google Scholar]
- 25.Movileanu L, Cheley S, Howorka S, Braha O, Bayley H. J Gen Physiol. 2001;117:239–251. doi: 10.1085/jgp.117.3.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grassetti D R, Murray J F. Arch Biochem Biophys. 1967;119:41–49. doi: 10.1016/0003-9861(67)90426-2. [DOI] [PubMed] [Google Scholar]
- 27.Brocklehurst K, Little G. FEBS Lett. 1970;9:113–116. doi: 10.1016/0014-5793(70)80327-1. [DOI] [PubMed] [Google Scholar]
- 28.Malthouse J P G, Brocklehurst K. Biochem J. 1980;185:217–222. doi: 10.1042/bj1850217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Christopher J A. SPOCK: the structural properties observation and calculation kit. Texas A&M University, College Station, TX: Center for Macromolecular Design; 1998. [Google Scholar]
- 30.Karlin A. J Gen Physiol. 2001;117:235–237. doi: 10.1085/jgp.117.3.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kenworthy A K, Hristova K, Needham D, McIntosh T J. Biophys J. 1995;68:1921–1936. doi: 10.1016/S0006-3495(95)80369-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rex S, Zuckermann M J, Lafleur M, Silvius J R. Biophys J. 1998;75:2900–2914. doi: 10.1016/S0006-3495(98)77732-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kienberger F, Pastushenko V P, Kada G, Gruber H J, Riener C, Schindler H, Hinterdorf P. Single Mol. 2000;1:123–128. [Google Scholar]
- 34.Stuchbury T, Shipton M, Norris R, Malthouse J P, Brocklehurst K, Herber J A, Suschitsky H. Biochem J. 1975;151:417–432. doi: 10.1042/bj1510417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pascual J M, Karlin A. J Gen Physiol. 1998;111:717–739. doi: 10.1085/jgp.111.6.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karlin A, Akabas M. Methods Enzymol. 1998;293:123–145. doi: 10.1016/s0076-6879(98)93011-7. [DOI] [PubMed] [Google Scholar]
- 37.Gedde U W. Polymer Physics. London: Chapman & Hall; 1995. [Google Scholar]
- 38.de Gennes P-G. Adv Polym Sci. 1999;138:91–105. [Google Scholar]
- 39.Destrée M, Lyulin A, Ryckaert J-P. Macromolecules. 1996;29:1721–1727. [Google Scholar]
- 40.Jirage K B, Hulteen J C, Martin C R. Science. 1997;278:655–658. [Google Scholar]
- 41.Sun L, Crooks R M. J Am Chem Soc. 2000;122:12340–12345. [Google Scholar]
- 42.Bayley H, Martin C R. Chem Rev. 2000;100:2575–2594. doi: 10.1021/cr980099g. [DOI] [PubMed] [Google Scholar]