

Abstract
Hormones can transmit signals through adenosine 3′,5′-monophosphate (cAMP) to precise intracellular locations. The fidelity of these responses relies on the activation of localized protein kinase A (PKA) holoenzymes. Association of PKA regulatory type II (RII) subunits with A-kinase—anchoring proteins (AKAPs) confers location, and catalytic (C) subunits phosphorylate substrates. Single-particle electron microscopy demonstrated that AKAP79 constrains RII-C subassemblies within 150 to 250 angstroms of its targets. Native mass spectrometry established that these macromolecular assemblies incorporated stoichiometric amounts of cAMP. Chemical-biology— and live cell—imaging techniques revealed that catalytically active PKA holoenzymes remained intact within the cytoplasm. These findings indicate that the parameters of anchored PKA holoenzyme action are much more restricted than originally anticipated.
Classical in vitro biochemistry and elegant structural studies have led to a commonly held view that the active protein kinase A (PKA) catalytic (C) subunit dissociates from the regulatory subunit dimer in the presence of excess adenosine 3′,5′-monophosphate (cAMP) (1–3). Yet the utility of this rudimentary mechanism inside cells remains unclear (4). Single-particle negative-stain electron microscopy (EM) analysis of A-kinase—anchoring protein 79 with PKA (AKAP79:PKA) holoenzyme complexes of AKAP79, the regulatory II (RII) subunit, and the C subunit (AKAP79:2RII:2C) detected a range of conformationally distinct species (Fig. 1, A to D, and fig. S1). A set of ~14,000 particles was subjected to reference-free two-dimensional (2D) classification using RELION (5). Poorly aligned classes were discarded over multiple iterations to reveal an ensemble of three-lobed assemblies in which the peripheral densities are observed at different distances from each other. These range from 150 Å in the compact configuration to 250 Å in the extended conformation (Fig. 1E). The tandem peripheral lobes represent the C subunit in complex with the cAMP-binding domains of RII (6), whereas the central density represents the AKAP-RII dimer interface (Fig. 1E). Thus, AKAP79 constrains each subassembly of regulatory and C subunits to within 250 Å of substrates. We surmise that the restricted movement of the anchored C subunit in this configuration augments phosphorylation of local substrates that are either interacting with the AKAP (Fig. 1F) or in proximity to the peripheral lobes of the extended PKA holoenzyme (Fig. 1G).
Fig. 1. AKAP-PKA holoenzyme assemblies remain intact upon cAMP stimulation.
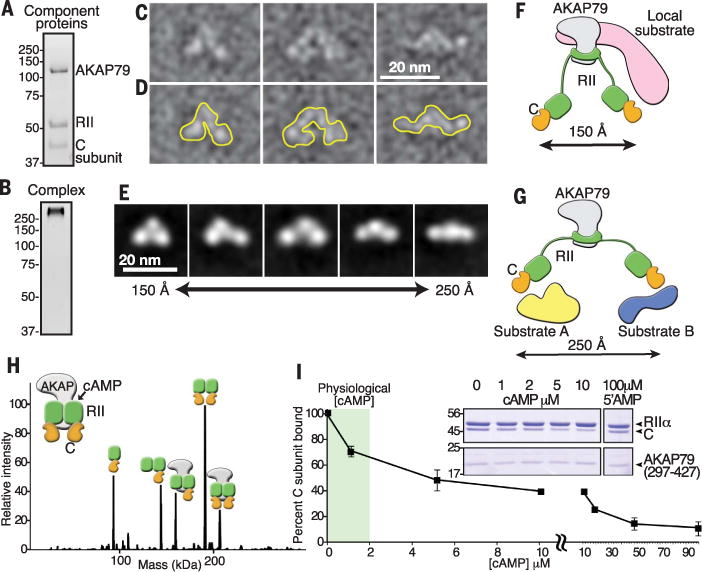
(A) SDS—polyacrylamide gel electrophoresis (SDS-PAGE) and stain-free visualization of a purified complex of AKAP79 with RII and C subunits of PKA. (B) Stain-free visualization on native gels of intact AKAP79-PKA holoenzyme complex. (C to E) Representative negative-stain EM micrographs of AKAP79:2RII:2C single particles. Particles were low-pass filtered to 20 Å. (D) Traces outlining the selected particles in (C). (E) Reference-free class averages from RELION 2D alignment and classification showing range of particle lengths. (F and G) Schematics illustrating the flexibility and range of motion in AKAP79-anchored PKA holoenzymes. A compact state may favor the phosphorylation of associated substrates (F), whereas an extended conformation may allow PKA to act on multiple distinct local substrates (G). (H) Zero-charge state native nanoelectrospray ionization mass spectrum of RII+C+AKAP (297–427) complexes. The stoichiometry of AKAP79(297–427) (grey), RII (green), and C subunit (orange) is indicated. (I) Percentage of C subunit bound to AKAP79(297–427)—PKA complexes after incubation with increasing cAMP concentrations. Data are presented as means ± SEM. (Inset) Coomassie blue staining of proteins in pull-down experiments.
An implication of our structural model is that, although local cAMP production stimulates kinase activity, AKAP79:2RII:2C assemblies can remain intact. Native mass spectrometry (MS) allows the measurement of molecular mass and is used to determine the stoichiometry of intact protein complexes in the gas phase (7–9). In the presence of cAMP, which copurifies with RII, we observed higher-order complexes, including the intact 2RII:2C PKA holoenzyme and the pentameric AKAP79:2RII:2C assembly (Fig. 1H, fig. S1, and tables S1 and S2). From these experiments, we infer a minimal disruption of the AKAP-PKA architecture even in the presence of cAMP.
To address the relative stability of the pentameric AKAP79:2RII:2C assembly in the presence of elevated cAMP, we performed pull-down experiments with a fragment of the anchoring protein AKAP79297–427. Retention of anchored C subunits in the presence of increasing concentrations of cAMP was assessed by quantification of Coomassie blue—stained protein (Fig. 1I and inset). At physiological concentrations of cAMP (1 to 2 μM) most of the C subunit (~70 to 80%) remained associated with the AKAP79297–427-RII complex (Fig. 1I and inset). Substantial release of the C subunit was only evident at supraphysiological levels of cAMP (Fig. 1I; 10 to 90 μM), and no change in binding of RII to AKAP79297–427 was observed. In control experiments, 5′-AMP (100 μM), a degradation product of cAMP, did not alter anchored holoenzyme composition (Fig. 1I). Thus, physiological amounts of cAMP promote minimal release of the C subunit from the anchored holoenzyme in vitro.
Additional studies used high-resolution native MS to evaluate cAMP occupancy in PKA holoenzyme complexes. At basal cAMP concentrations, multiple charge states of 2RII:C subcomplexes were observed between mass/charge ratios of 5700 and 6300. These species preferentially contained two, but up to four, molecules of cAMP (fig. S1). When analysis was done at higher concentrations of cAMP (5 μM), the predominant species exhibited cyclic nucleotide occupancy of 4 mol per RII dimer (fig. S1 and table S3). Notably, the C subunit remained attached under these conditions (fig. S1). Thus, we can conclude that a substantial proportion of the PKA C subunit remained associated with the AKAP79297–427-RII dimer when cAMP-binding sites were occupied.
For a more stringent in situ test, we monitored the integrity of cellular AKAP-PKA complexes in response to ligand activation. AKAP79 or AKAP18γ complexes were immunoprecipitated from cell lysates prepared after stimulation of cells with the β-adrenergic agonist isoproterenol (Iso, 1 μM). Immunoblot analysis detected equivalent amounts of C subunit in samples from cells stimulated with isoproterenol or vehicle control (Fig. 2, A and B, top, lane 2). Local cAMP flux is controlled through a balance of second-messenger production by adenylyl cyclases and degradation by phosphodiesterases (PDEs) (10). Cells were stimulated with isoproterenol in the presence of the general PDE inhibitor 3-isobutyl-1-methylxanthine (IBMX), the selective PDE3 inhibitor milrinone, or the PDE4-specific inhibitor rolipram (11, 12). The composition of AKAP79-PKA complexes was assessed by immunoblot. Intact complexes were detected in control and Iso-treated samples (Fig. 2C, lanes 1 and 2). Inclusion of rolipram promoted full dissociation of the C subunit (Fig. 2C, lane 3). Likewise, application of IBMX induced release of the C subunit from AKAP complexes (Fig. 2C, lane 5). In contrast, inclusion of milrinone had little effect, indicating that PDE3 is nonfunctional in these AKAP79-PKA microdomains (Fig. 2C, lane 4). Kinase activation was monitored by immunoblot detection of phospho-PKA substrates (Fig. 2C, bottom panel). Similar findings were obtained upon analysis of AKAP18γ complexes and when prostaglandin E1 (PGE1) or epinephrine was used as an agonist (fig. S2). Treatment with rolipram alone did not activate PKA or release C subunits (Fig. 2C, lane 6). Thus, native production of cAMP in response to physiological effectors of GPCR signaling appears not to promote C subunit release from anchored PKA holoenzymes.
Fig. 2. Anchored PKA holoenzyme dynamics upon ligand stimulation.
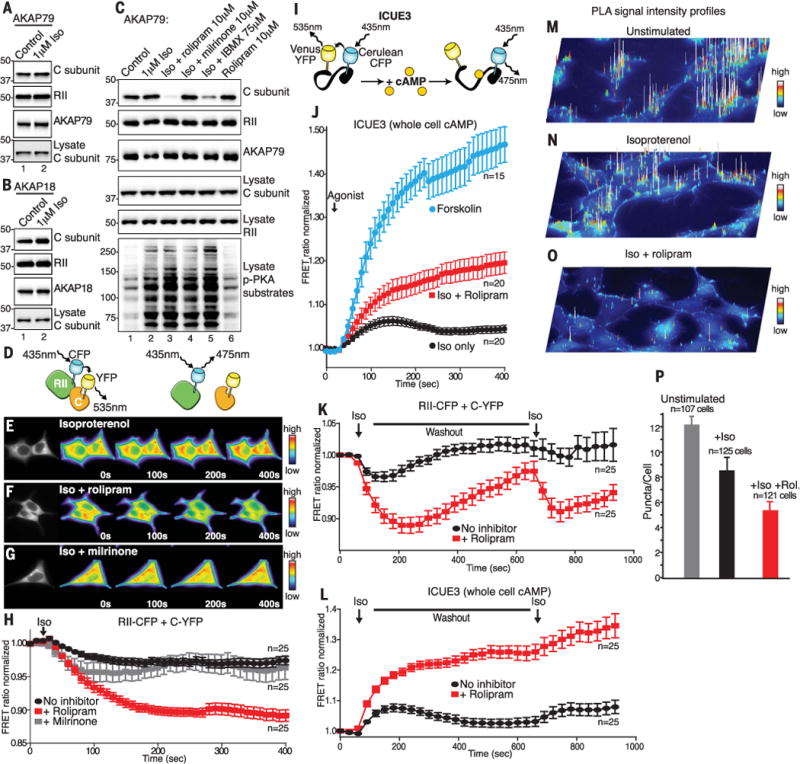
Western blot analysis of PKA C and RII subunits in (A) AKAP79 or (B) AKAP18g immunoprecipitates (IPs) after isoproterenol stimulation (Iso). (C) IP and Western blot analysis of AKAP79 complexes isolated in the presence of phosphodiesterase inhibitors. (D) Schematic depicting RII-CFP and C-YFP intermolecular FRET. (E to H) FRET analysis of holoenzyme dissociation in HEK293 cells. (E) Time course of representative cells (0 to 400 s) stimulated with isoproterenol. (F) Iso + PDE4 inhibitor rolipram. (G) Iso + PDE3 inhibitor milrinone. (H) Amalgamated data from 25 recordings under each experimental condition. (I) Schematic depicting ICUE3 cAMP FRET sensor. (J) ICUE3 FRET recordings show cAMP production in response to Iso (black), Iso + rolipram (red), and forskolin (blue). The number of experiments is indicated. (K and L) FRET analysis upon two trains of hormonal stimulation. (K) Intermolecular FRET analysis of holoenzyme dissociation in HEK293 cells upon two trains of stimulation with isoproterenol (black) and Iso + PDE4 inhibitor rolipram (red). (L) Monitoring cAMP accumulation with ICUE3 FRET sensor in response to isoproterenol (black) and Iso + PDE4 inhibitor rolipram (red). Amalgamated data from 25 recordings under each experimental condition. (M to O) Proximity ligation assay (PLA) signal-intensity projections show RII-C interactions in (M) Unstimulated, (N) Iso-stimulated, and (O) Iso- and rolipram-treated HEK293 cells. (P) Quantification of PLA puncta per cell using Fiji/ImageJ. All data are presented as means ± SEM.
Förster resonance energy transfer (FRET) was used to investigate PKA holoenzyme composition in real time (13). Initially, we used RII conjugated to cyan fluorescent protein (RII-CFP) and the C subunit conjugated to yellow fluorescent protein (C-YFP) as intermolecular FRET probes to monitor the integrity of the PKA holoenzyme after agonist stimulation of cells (Fig. 2D). Isoproterenol promoted minimal change in the CFP/YFP FRET ratio over time courses of 400 s (Fig. 2, E and H, black), consistent with negligible dissociation of the PKA holoenzyme. In contrast, preincubation of cells with rolipram before stimulation with β-adrenergic agonist triggered a pronounced and time-dependent reduction in the FRET ratio (Fig. 2, F and H, red). Thus, dissociation of the PKA holoenzyme only occurred if cAMP accumulated to supraphysiological concentrations. Pretreatment of cells with the PDE3 inhibitor milrinone had little effect on the FRET response (Fig. 2, G and H, gray). Comparable FRET recordings were obtained when PGE1 was used as the agonist (fig. S2).
The ICUE3 FRET biosensor detects agonist-responsive accumulation of cAMP (Fig. 2I) (14). Stimulation of cells with isoproterenol or PGE1 promoted transient increases in the FRET signal (Fig. 2J, black, and fig. S2). This response was enhanced by addition of rolipram (Fig. 2J, red, and fig. S2). However, the highest amounts of cAMP production (15 times physiological levels) were recorded upon application of forskolin, a direct activator of adenylyl cyclases and a common tool in cAMP research (Fig. 2J, blue). Thus, forskolin sustains supraphysiological accumulation of cAMP to concentrations far above those induced by β-agonists.
We investigated the effect of recurrent stimulation of cAMP synthesis on PKA holoenzyme integrity with the same biosensors. After an initial pulse of isoproterenol, the drug was washed out for 500 s before application of a second stimulus (Fig. 2, K and L). Intermolecular FRET revealed that RII-CFP and C-YFP remained in proximity over the duration of these experiments (Fig. 2K, black). As expected, costimulation of cells with isoproterenol and rolipram initiated cycles of holoenzyme dissociation and reformation (Fig. 2K, red). Complementary biochemical experiments revealed that washout of rolipram and isoproterenol promoted reformation of AKAP79-PKA holoenzyme over a similar time course (fig. S2). Using the ICUE3 biosensor, we demonstrated that isoproterenol induces a transient rise in the cellular concentration of cAMP that approaches baseline during washout (Fig. 2L, black). A second application of agonist then initiates another round of cAMP production. In contrast, costimulation with rolipram produces two additive phases of intracellular cAMP accumulation (Fig. 2L, red). Collectively these experiments demonstrate that PDE4 modulates cAMP microdomains surrounding anchored and intact PKA holoenzymes
Proximity ligation assays (PLA) detect endogenous protein-protein interactions that occur within 40 to 60 nm (15). Unstimulated and agonist-treated human embryonic kidney (HEK) 293 cells (HEK293 cells) were fixed, stained with antibodies to RII and C subunits, and subjected to the PLA amplification protocol before imaging of PLA puncta (fig. S2). Image intensity profiling of PLA puncta showed the distribution of intact PKA holoenzymes (Fig. 2, M to O). The number of the puncta per cell indicated the extent of RII-C interactions under each experimental condition (Fig. 2P). Treatment with isoproterenol did not appreciably change the PLA signal as compared with unstimulated control cells (Fig. 2, M, N, and P). In contrast, treatment of cells with isoproterenol and rolipram reduced the number and intensity of PLA puncta (Fig. 2, O and P). Immunoblot detection of endogenous RII and C in HEK293 cell lysates confirmed the same trend (fig. S2). Thus, physiological agonists that mobilize cAMP signaling promote very little dissociation of native PKA holoenzymes inside cells.
To test whether covalent coupling of RII to the C subunit would alter PKA action inside cells, we generated a construct that encodes RIIα and Cα in one polypeptide, creating a nondissociable PKA fusion enzyme designated “R2C2” (Fig. 3A). To exploit this tool in a simplified genetic background, we used CRISPR-Cas9 editing to disrupt the PRKAR2A, PRKAR2B, and PRKACA genes in U2OS human osteosarcoma cells [triple-knockout (KO) U2OSR2A/R2B/CA]. Loss of protein expression due to gene-editing was confirmed by immunoblot (Fig. 3B, lane 2, top three panels). Rescue upon expression of R2C2 was confirmed by immunoblot (Fig. 3C, top, lane 3). Activity profiling using the phospho-PKA substrates antibody confirmed that isoproterenol-responsive PKA activation was reduced to baseline levels in triple-KO U2OSR2A/R2B/CA cells (fig. S3).
Fig. 3. CRISPR-Cas9 PKA triple-KO and rescue with an RII-C fusion.
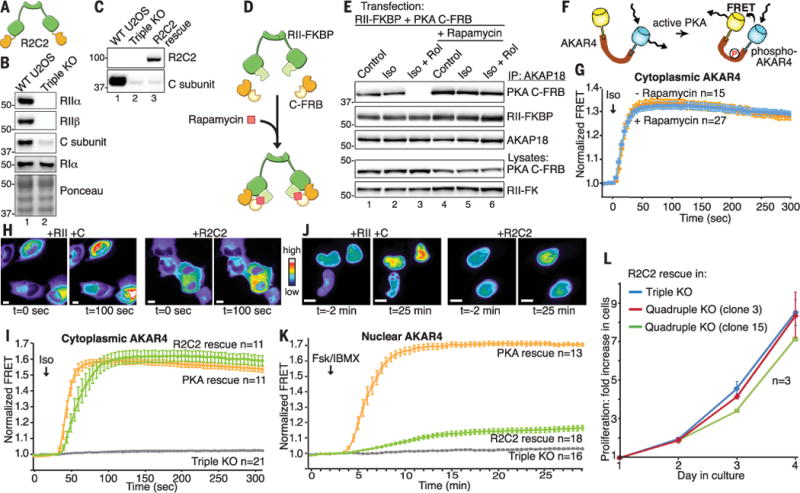
(A) Schematic depicting the R2C2 PKA fusion enzyme. (B) Immunoblots confirming knockout of PKA subunit proteins: (Top) RIIα, (top middle) RIIβ, ανδ (bottom middle) C subunit. (Bottom) Ponceau staining shows equal loading in WT and U2OSR2A/R2B/CA cells. (C) Western blots for PKA C subunit confirm expression of R2C2 fusion. (D) Schematic of rapamycin induced heterodimerization of PKA subunits. (E) Immunoblots showing that rapamycin chemically locks the PKA holoenzyme at supraphysiological concentrations of cAMP. (F) Schematic of AKAR4 PKA activity biosensor. (G) Cytoplasmic FRET recordings in response to Iso (1 μM) stimulation in U2OSR2A/R2B/CA cells expressing RII-FKBP and C-FRB in the presence (blue) or absence (orange) of rapamycin (100 nM). (H) Representative cells showing cytoplasmic AKAR4 FRET response upon rescue with (left) RIIα and Cα and (right) R2C2 fusion. (I) Cytoplasmic FRETrecordings in triple-KO U2OSR2A/R2B/CA cells (gray) and cells rescued with R2C2 fusion (green) or WT RIIα and Cα (orange). (J) Montage of cells showing nuclear AKAR4 FRET signals upon rescue with (left) RIIα and Cα or (right) R2C2 fusion. (K) Nuclear FRET recordings in U2OSR2A/R2B/CA cells (gray) and cells rescued with R2C2 fusion (green) or WT RIIα and Cα (orange). (L) Proliferation of triple KO U2OSR2A/R2B/CA cells rescued with R2C2 fusion (blue) or two different clones of quadruple KO U2OSR2A/R2B/CA/CB cells rescued with R2C2 fusion (red and green).
Related studies used rapamycin-regulated heterodimerization to “pharmacologically lock” FRB domain—tagged C subunits (C-FRB) to FK506-binding protein domain—tagged RII dimers (RIIFKBP) in the context of the AKAP18 complex. In this system, the small molecule rapamycin bridges the tagged proteins to maintain a stable complex (Fig. 3D). Immunoblot analyses of AKAP18 immune complexes from cells expressing RII-FKBP and C-FRB revealed that both proteins were associated after stimulation of cells with isoproterenol in the absence of rapamycin (Fig. 3E, top, lane 2). In keeping with earlier results, administration of isoproterenol and rolipram together caused supraphysiological accumulation of cAMP and abolished this interaction (Fig. 3E, top, lane 3). Conversely, application of rapamycin locks the C-FRB/RII-FKBP subcomplex together, even when cells are treated with isoproterenol plus rolipram (Fig. 3E, top, lane 6).
We next used A-kinase activity reporter 4 (AKAR4) biosensors that measure PKA activity by changes in FRET to assess how compartmentalization affects free and pharmacologically locked PKA activity (Fig. 3F) (16). Cell-based studies in triple-KO U2OS cells confirmed that, within 30 s, cytoplasmic FRET responses to isoproterenol were robust (Fig. 3G, orange), even after pharmacological locking of this modified PKA holoenzyme with rapamycin (Fig. 3G, blue). Isoproterenol treatment of cells in the presence of rolipram has no additional effect (fig. S3). Consequently, cytoplasmic PKA signaling is sustained when the C subunit and the RII dimer are chemically constrained.
Further studies with the AKAR4 biosensor and engineered PKA holoenzymes investigated the dynamics of cytoplasmic kinase activity. Rapid increases in cytoplasmic FRET responses were recorded in U2OSR2A/R2B/CA cells rescued with murine RIIα and Cα (Fig. 3, H and I, orange). Cells expressing the R2C2 fusion protein generated isoproterenol responses of similar magnitude and duration (Fig. 3, H and I, green). There was no detectable FRET response to agonist stimulation in the U2OSR2A/R2B/CA deletion background or in cells expressing a catalytically inactive mutant of the RII-C fusion (R2C2K72A) (Fig. 3I, gray, and fig. S3). Thus, covalent coupling of the PKA subunits does not impair cAMP signaling in the cytoplasm and can reconstitute endogenous kinase activity.
As a further control, we monitored nuclear PKA activity in both wild-type (WT) U2OS and U2OSR2A/R2B/CA cells using AKAR4NLS (16). Nuclear FRET responses were slow and required supraphysiological accumulation of cAMP after stimulation of adenylyl cyclases with 20 μM forskolin in the presence of 3-isobutyl-1-methylxanthine (IBMX) for 30 min (Fig. 3, J and K, and fig. S3). Under these extreme conditions, rescue with murine RIIα and Cα resulted in a gradual increase in FRET over 15 min (Fig. 3, J and K, orange). In contrast, cells expressing R2C2 showed greatly diminished nuclear FRET responses (Fig. 3, J and K, green). Together, these results demonstrate that cytoplasmic activation of PKA is a rapid event, whereas nuclear signaling requires persistent and supraphysiological accumulation of cAMP.
Mammalian cells express four regulatory subunits and two C subunits of PKA (17). CRISPR/Cas gene editing of the PRKACB gene on a U2OSR2A/R2B/CA deletion background was consistently unsuccessful; this suggests that ablation of both catalytic genes is a lethal event. Yet quadruple-KO U2OSR2A/R2B/CA/CB cells survived and proliferated when gene editing was performed in cells stably expressing murine R2C2 (Fig. 3L and fig. S3). Thus, genetically engineered cells expressing a single copy of fused PKA are viable and respond normally to physiological stimuli that mobilize the cAMP-signaling pathway. Because intact and active PKA holoenzymes operate within a radius of 150 to 250 Å of their substrates (Fig. 1), compartmentalization by AKAPs is the critical determinant of PKA-substrate selectivity.
An implication of this finding is that kinase inhibitor drugs operate within the confines of these AKAP nanocompartments. As proof of concept, we generated an analog-sensitive form of R2C2 that is exclusively modulated by the cell-permeable kinase inhibitor 1-napthylmethyl-PP1 (1-NM-PP1) (Fig. 4A) (18, 19). Engineering this pharmacologically tractable and fused form of PKA was achieved by replacing the gatekeeper methionine in the catalytic moiety of R2C2 with alanine (M120A). In situ characterization of this synthetic analog-sensitive kinase in U2OSR2A/R2B/CA cells was achieved by using an AKAP-derived PKA-activity reporter (AKAP18RBS-AKAR4) (Fig. 4B). In cells expressing wild-type R2C2, isoproterenol (1 μM) stimulation induced a FRET response within 10 s that was refractory to 1-NM-PP1 (Fig. 4C, green). The rate and magnitude of the FRET response was attenuated in cells expressing R2C2 M120A, a common property of analog-sensitive kinases (19). However, application of 1-NM-PP1 (2 μM) to these cells reduced the FRET signal to baseline, which indicated strong inhibition of the analog-sensitive kinase (Fig. 4C, blue). Thus, 1-NM-PP1 is an efficient inhibitor of R2C2 M120A in situ.
Fig. 4. Chemical-genetic and pharmacological control of mitochondrial PKA action.
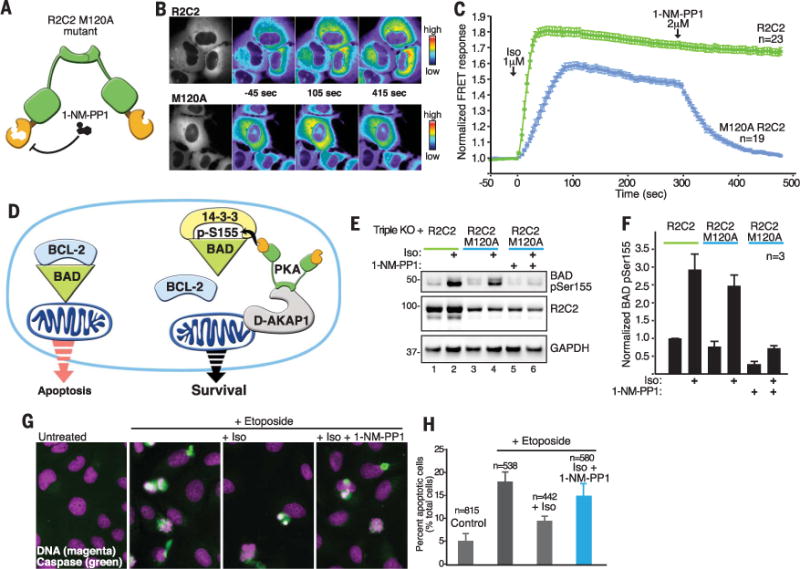
(A) Schematic depicting the introduction of an analog-sensitive kinase mutation into the R2C2 PKA fusion enzyme. The activity of this pharmacologically controlled fusion kinase was monitored in U2OSR2A/R2B/CA cells. (B) Representative cells showing cytoplasmic AKAR FRET signals at selected time points from cells expressing (top) R2C2 and (bottom) the 1-NM-PP1—sensitive R2C2 M120A mutant. The left image in both panels shows expression of the FRET reporter. The others panels show pseudocolor images of the FRET intensity at one time point before stimulation (−45 s), one time point after stimulation with isoproterenol (105 s), and a third time point after addition of 1-NM-PP1 (415 s). (C) Time courses of cytoplasmic AKAR4 FRET responses to Iso (1 μM, time zero) and 1-NM-PP1 (2 μM, 300 s) in PKA triple-KO cells expressing the (green) R2C2 fusion or (blue) the R2C2 M120A mutant. (D) Schematic depicting protective effects of anchored PKA due to phosphorylation of BAD and the resultant release of the prosurvival BCL-2 protein. (E) Inhibition of PKA directed BAD pSer155 phosphorylation by 1-NM-PP1 in cells expressing R2C2 M120A. Immunoblot detecting (top) BAD pSer155, (middle) R2C2, and (bottom) GAPDH loading controls. (F) Quantification of BAD pSer155 signal from immunoblots from three independent experiments. Data are presented as means ± SEM. (G) Fluorescent detection of apoptotic events upon treatment of cells with the apoptotic agent etoposide, plus and minus β-agonists and 1-NM-PP1. Apoptotic cells were detected by (green) activation of caspase and (magenta) condensation of DNA. (H) Quantification of the percentage of apoptotic cells from (G). The numbers of cells monitored are indicated above each column. Data are presented as means ± SEM.
We used this chemically controllable form of fused PKA to examine phosphorylation of local extra-nuclear substrates. The mitochondrial anchoring protein D-AKAP-1 sequesters PKA at mitochondria to regulate phosphorylation of the proapoptotic protein BAD (Fig. 4D) (20). PKA phosphorylation of BAD Ser155 blocks apoptosis through a mechanism involving recruitment of 14-3-3 proteins to suppress programmed cell death (Fig. 4D) (21–23). BAD phosphorylation on Ser155 was robustly stimulated by isoproterenol in WT U2OS cells, and this effect was blunted in triple-KO cells (fig. S4). Confocal imaging confirmed that D-AKAP-1 and BAD are detected at mitochondria (fig. S4), whereas PLAs detected puncta that are indicative of an interaction between these two proteins (fig. S4). In triple-KO cells expressing the R2C2 WT and R2C2 M120A mutants, immunoblot analysis confirmed that these modified kinases effectively phosphorylated BAD Ser155 after treatment of cells with isoproterenol (Fig. 4E, top, lanes 1 to 4). Control immunoblots monitored the expression of the PKA fusion enzymes, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as loading controls (Fig. 4E, middle and bottom). Application of 1-NM-PP1 (2 μM) inhibited the activity of R2C2 M120A and blocked phosphorylation of BAD Ser155 (Fig. 4E, top, lane 6). Normalized BAD pSer155 data from three independent experiments is presented in Fig. 4F. With this tool, we could test whether local activation of PKA is inhibitory toward apoptosis.
We induced apoptosis in triple-KO cells expressing R2C2 M120A by treatment with the chemotoxic agent etoposide (Fig. 4, G and H) (24). Cell death was assessed as accumulation of a fluorescent product of caspase 3– or 7–mediated proteolysis (Fig. 4G, green) and nuclear condensation observed upon staining DNA with 4′,6-diamidino-2-phenylindole (DAPI) (magenta in Fig. 4G). Unstimulated cells exhibit low levels of apoptosis (Fig. 4G, left, and Fig. 4H). Etoposide treatment promotes cell death (Fig. 4G, left, middle, and Fig. 4H). Stimulation of R2C2 activity with β-adrenergic receptor agonists (Iso and formoterol) was protective, as it substantially reduced the apoptotic index (Fig. 4G, middle right, and Fig. 4H). Application of 1-NM-PP1 blocked this protective effect (Fig. 4G, right, and Fig. 4H, blue). Thus, fused RII and C subunits can substitute for endogenous PKA in the modulation of local cellular events.
Contrary to current dogma, our results show that even in the presence of cAMP, anchored PKA holoenzymes remain intact and proximal to anchoring sites and substrates. In addition fusion of the kinase moiety to RII supports cAMP signaling in diverse cellular contexts. These results highlight protein-protein interactions between AKAPs and PKA as the primary determent for the substrate specificity of this broad-spectrum kinase. We also shed light on another interesting facet of PKA physiology. Of the 545 genes in the human kinome, only one other protein kinase, CK2, forms a tetrameric holoenzyme (25). Our evidence that autoinhibitory and anchoring functions are provided by the constitutively associated RII dimer implies that PKA is more aligned with other members of the kinase superfamily than originally considered.
Finally, because AKAP79 constrains intact and catalytically active PKA within 150 to 250 Å, we propose that the range of anchored PKA action is restricted to substrates within the immediate vicinity (Fig. 1, F and G). This “signaling island” concept radically changes our view of how AKAP complexes operate and indicates that protein phosphorylation is much more regionally confined than previously appreciated (26–29). Another prerequisite for this new model is that the location of these active zones must coincide with microdomains where cAMP concentrations fluctuate and when substrates are available (10, 30). One exception may be PKA action in the nucleus, where cAMP responsive transcriptional events proceed through the phosphorylation of cAMP response element—binding protein (CREB) (31, 32). Nonetheless, this intricate molecular architecture helps explain how common signaling components are assembled to elicit distinct, rapid, and transient endocrine responses.
Supplementary Material
Acknowledgments
We thank K. Forbush for technical support, K.F. and N. Pollett for molecular biology and cell culture assistance, all members of the Scott Lab for critical discussions, T. Cooke for technical assistance with FRET microscopy, and M. Milnes for administrative support. This work was supported by the following grants from the NIH: 5R01DK105542 (J.D.S.), 4P01DK054441 (J.D.S.), 1R01GM120553 (D.V.); U.K. Biotechnology and Biological Sciences Research Council grant BB/L009501/1 (C.E.E.); North West Cancer Research grant CR1037 (P.A.E.); and Core funding from the Institute of Integrative Biology, Faculty of Health and Life Sciences, University of Liverpool (C.E.E. and P.A.E.). J.L.E. is the recipient of the Heart and Stroke Foundation of Canada Postdoctoral Fellowship. F.D.S. and J.D.S. conceived of and supervised the project. F.D.S., J.L.E., P.J.N., D.P.B., M.V., C.E.E., P.A.E., and J.D.S. designed the experiments. F.D.S., J.L.E., P.J.N., D.P.B., M.V., C.E.E., and P.A.E. performed experiments. F.D.S., J.L.E., P.J.N., D.P.B., M.V., C.E.E., P.A.E., and J.D.S. analyzed the data. D.V. provided guidance and software for EM experiments. I.S. provided guidance and support for MS experiments with Thermo EMR. L.K.L. designed and prepared figures. F.D.S., L.K.L., and J.D.S. wrote the manuscript.
Footnotes
SUPPLEMENTARY MATERIALS
www.sciencemag.org/content/356/6344/1288/suppl/DC1)
Materials and Methods
References (33–37)
REFERENCES AND NOTES
- 1.Walsh DA, Perkins JP, Krebs EG. J Biol Chem. 1968;243:3763–3765. [PubMed] [Google Scholar]
- 2.Potter RL, Taylor SS. J Biol Chem. 1979;254:2413–2418. [PubMed] [Google Scholar]
- 3.Knighton DR, et al. Science. 1991;253:407–414. doi: 10.1126/science.1862342. [DOI] [PubMed] [Google Scholar]
- 4.Langeberg LK, Scott JD. Nat Rev Mol Cell Biol. 2015;16:232–244. doi: 10.1038/nrm3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scheres SH. J Struct Biol. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith FD, et al. eLife. 2013;2:e01319. doi: 10.7554/eLife.01319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heck AJ. Nat Methods. 2008;5:927–933. doi: 10.1038/nmeth.1265. [DOI] [PubMed] [Google Scholar]
- 8.Gold MG, et al. Proc Natl Acad Sci USA. 2011;108:6426–6431. doi: 10.1073/pnas.1014400108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Byrne DP, et al. Biochem J. 2016;473:3159–3175. doi: 10.1042/BCJ20160648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zaccolo M, Pozzan T. Science. 2002;295:1711–1715. doi: 10.1126/science.1069982. [DOI] [PubMed] [Google Scholar]
- 11.Silver PJ. Am J Cardiol. 1989;63:2A–8A. doi: 10.1016/0002-9149(89)90384-6. [DOI] [PubMed] [Google Scholar]
- 12.Jin SL, Bushnik T, Lan L, Conti M. J Biol Chem. 1998;273:19672–19678. doi: 10.1074/jbc.273.31.19672. [DOI] [PubMed] [Google Scholar]
- 13.Zhang J, Campbell RE, Ting AY, Tsien RY. Nat Rev Mol Cell Biol. 2002;3:906–918. doi: 10.1038/nrm976. [DOI] [PubMed] [Google Scholar]
- 14.DiPilato LM, Zhang J. Mol Biosyst. 2009;5:832–837. doi: 10.1039/b823243a. [DOI] [PubMed] [Google Scholar]
- 15.Söderberg O, et al. Genet Eng (NY) 2007;28:85–93. doi: 10.1007/978-0-387-34504-8_5. [DOI] [PubMed] [Google Scholar]
- 16.Herbst KJ, Allen MD, Zhang J. Mol Cell Biol. 2011;31:4063–4075. doi: 10.1128/MCB.05459-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taylor SS, Ilouz R, Zhang P, Kornev AP. Nat Rev Mol Cell Biol. 2012;13:646–658. doi: 10.1038/nrm3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morgan DJ, et al. Proc Natl Acad Sci USA. 2008;105:20740–20745. doi: 10.1073/pnas.0810971105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lopez MS, Kliegman JI, Shokat KM. Methods Enzymol. 2014;548:189–213. doi: 10.1016/B978-0-12-397918-6.00008-2. [DOI] [PubMed] [Google Scholar]
- 20.Harada H, et al. Mol Cell. 1999;3:413–422. doi: 10.1016/s1097-2765(00)80469-4. [DOI] [PubMed] [Google Scholar]
- 21.Lizcano JM, Morrice N, Cohen P. Biochem J. 2000;349:547–557. doi: 10.1042/0264-6021:3490547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tan Y, Demeter MR, Ruan H, Comb MJ. J Biol Chem. 2000;275:25865–25869. doi: 10.1074/jbc.M004199200. [DOI] [PubMed] [Google Scholar]
- 23.Virdee K, Parone PA, Tolkovsky AM. Curr Biol. 2000;10:R883. doi: 10.1016/s0960-9822(00)00843-5. [DOI] [PubMed] [Google Scholar]
- 24.Cheng EH, Sheiko TV, Fisher JK, Craigen WJ, Korsmeyer SJ. Science. 2003;301:513–517. doi: 10.1126/science.1083995. [DOI] [PubMed] [Google Scholar]
- 25.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 26.Scott JD, Dessauer CW, Taskén K. Annu Rev Pharmacol Toxicol. 2013;53:187–210. doi: 10.1146/annurev-pharmtox-011112-140204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoshi N, Langeberg LK, Gould CM, Newton AC, Scott JD. Mol Cell. 2010;37:541–550. doi: 10.1016/j.molcel.2010.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoshi N, Langeberg LK, Scott JD. Nat Cell Biol. 2005;7:1066–1073. doi: 10.1038/ncb1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hehnly H, et al. eLife. 2015;4:e09384. doi: 10.7554/eLife.09384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lygren B, et al. EMBO Rep. 2007;8:1061–1067. doi: 10.1038/sj.embor.7401081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chrivia JC, et al. Nature. 1993;365:855–859. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- 32.Hagiwara M, et al. Mol Cell Biol. 1993;13:4852–4859. doi: 10.1128/mcb.13.8.4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.