

SUMMARY
Dynamic regulation of mRNA translation initiation and elongation is essential for the survival and function of neural cells. Global reductions in translation initiation resulting from mutations in the translational machinery or inappropriate activation of the integrated stress response may contribute to pathogenesis in a subset of neurodegenerative disorders. Aberrant proteins generated by non-canonical translation initiation may be a factor in the neuron death observed in the nucleotide repeat expansion diseases. Dysfunction of central components of the elongation machinery, such as the tRNAs and their associated enzymes, can cause translation infidelity and ribosome stalling, resulting in neurodegeneration. Taken together, dysregulation of mRNA translation is emerging as a unifying mechanism underlying the pathogenesis of many neurodegenerative disorders.
Keywords: integrated stress response, GTPBP2, n-Tr20, mistranslation, eIF2α phosphorylation, repeat-associated non-ATG (RAN) translation, mTOR, aminoacyl tRNA synthetase, angiogenin, n-Trtct5
The proper decoding of mRNAs into proteins is critical for cellular function. This process must be carefully regulated in all cell types, but neurons are particularly reliant on the spatial and temporal control of mRNA translation due to their striking polarized morphology and the demands of synaptic plasticity. The dynamic regulation of both translation initiation and elongation plays a critical role in learning and memory, and defects in translational control have been linked to numerous cognitive disorders (reviewed in Buffington et al., 2014; Richter and Coller, 2015). Recent evidence demonstrates that relatively subtle disruptions in translational regulation can have dramatic consequences for survival of neurons, especially in the aging brain. In this review, we examine the contribution of dysregulated cytoplasmic mRNA translation to the pathophysiology of numerous neurodegenerative diseases, as well as highlight genetic disorders resulting from mutations in components of the translational machinery. We also discuss canonical and non-canonical translation initiation events that perturb neuronal homeostasis and lead to neuronal death. Finally, we examine disruptions in translation elongation and how these disturbances can lead to neurodegeneration.
A Brief Overview of Cytoplasmic mRNA Translation
Translation is a cyclical process of initiation, elongation, and termination that is necessary for development and cellular homeostasis and function. In eukaryotes, the vast majority of protein-coding genes are encoded in the nuclear genome and translated in the cytoplasm; however, a small number of protein-coding genes (13 in humans, 8 in yeast) are transcribed from the mitochondrial genome and are translated by separate protein synthesis machinery within this organelle. Mitochondrial translation has several unique characteristics not observed in either prokaryotes or cytoplasmic translation in eukaryotes, including a distinct genetic code, uncapped mRNAs with short or non-existent 5’ untranslated regions (UTR), smaller and protein-rich ribosomes, and unique tRNAs. The topic of mitochondrial translation, and its involvement in disease has been the subject of several recent reviews, and will not be further discussed here (Boczonadi and Horvath, 2014; Kuzmenko et al., 2014; Mai et al., 2017; Pearce et al., 2013).
During initiation of cytoplasmic translation, a carefully choreographed series of steps culminates in the recognition of the mRNA start codon by elongation-competent 80S ribosomes (Figure 1). The small 40S ribosomal subunit binds to the initiator methionyl-tRNA (Met-tRNAi) in a ternary complex with GTP-bound eukaryotic initiation factor (eIF)2, with the help of multiple proteins including eIF1, eIF1A, eIF5, and eIF3, to form the 43S preinitiation complex (PIC). The 48S PIC is generated when the 43S PIC is recruited to the 5’ 7-methylguanosine (m7G) cap of the mRNA by the heterotrimeric eIF4F complex (the cap-binding protein eIF4E, the DEAD-box RNA helicase eIF4A, and the scaffold protein eIF4G) and eIF4B. In addition to binding eIF4E and eIF4A, eIF4G also binds the poly(A) binding protein (PABP), thus promoting circularization of the mRNA and possibly facilitating reinitiation of translation by terminated ribosomes. eIF4A unwinds the 5’ UTR in an ATP-dependent manner so that the PIC can scan this region base-by-base for an AUG codon in the optimal context. Recognition of the start codon triggers a series of events, including the dissociation of eIF1, the release of phosphate from the hydrolysis of eIF2-GTP to eIF2-GDP (catalyzed by the GTPase activating protein, eIF5), and conformational changes in other initiation factors. Finally, in a process catalyzed by eIF5B, the large 60S ribosomal subunit joins the PIC, leading to dissociation of several of the remaining initiation factors, and formation of a functional 80S initiation complex (reviewed in Hinnebusch, 2014; Jackson et al., 2010; Sonenberg and Hinnebusch, 2009). In addition to cap-dependent translation, a small subset of eukaryotic mRNAs undergo translational initiation through alternative means, such as by ribosome recruitment to an internal ribosome entry site (IRES) (recently reviewed in Lacerda et al., 2017).
Figure 1. Eukaryotic Cap-Dependent Translation Initiation and Key Regulatory Pathways.
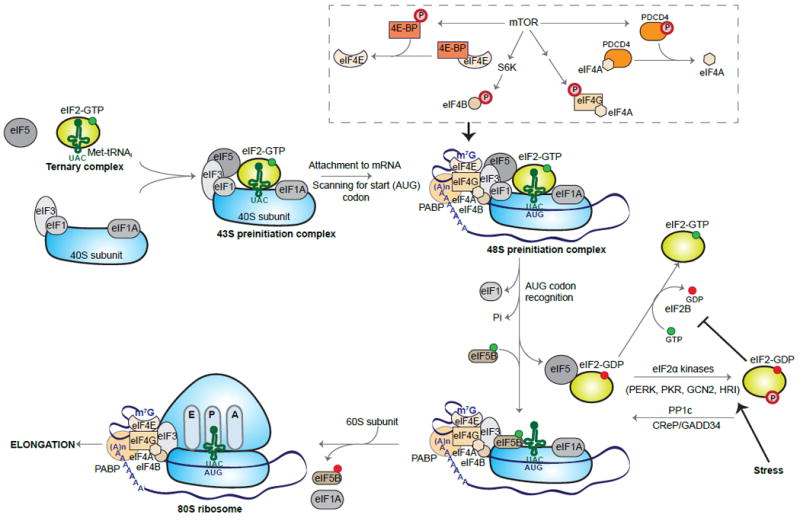
Initiation begins with the assembly of the 43S preinitiation complex (PIC), consisting of the 40S small ribosomal subunit, eIF1, eIF1A, eIF3, eIF5, and the ternary complex eIF2-GTP-Met-tRNAi. The PIC is recruited to the 5’ cap of the mRNA by the eIF4F complex (eIF4E, eIF4G, and eIF4A) and eIF4B. Circularization of the mRNA is promoted by interaction between eIF4G and PABP. The mRNA is scanned in a 5’ to 3’ direction until a start (AUG) codon is recognized, triggering the release of eIF1, inorganic phosphate (Pi), eIF5, and eIF2-GDP. The joining of the 60S ribosomal subunit to the PIC and release of several initiation factors, including eIF1A, is catalyzed by eIF5B, leading to the formation of the elongation-competent 80S ribosome. eIF2-GDP is recycled to eIF2-GTP by the exchange factor eIF2B. Under stress conditions, phosphorylation of the α-subunit of eIF2 (eIF2α) impairs recycling and ternary complex formation. Translation initiation is also regulated by mTOR, which can directly or indirectly phosphorylate eIF4G, eIF4B, 4E-BP, and PDCD4, promoting cap-dependent initiation.
In contrast to initiation, translation elongation is a more straightforward process that is highly conserved between eukaryotes and bacteria (Figure 2). After initiation, the 80S ribosome, which contains three tRNA binding sites [the aminoacyl or acceptor (A) site, the peptidyl (P) site, and the exit (E) site], is positioned on the mRNA with Met-tRNAi basepaired with the AUG start codon in the P site. The second codon of the transcript is in the A site, awaiting a cognate tRNA. This tRNA is delivered to the A site in a ternary complex with eukaryotic elongation factor (eEF)1A (the ortholog of bacterial EF-Tu) that binds to aminoacyl tRNA in a GTP-dependent manner. Cognate codon recognition by the tRNA triggers GTP hydrolysis and release of eEF1A-GDP from the tRNA. Seventy angstroms away in the peptidyl transfer center, peptide bond formation rapidly occurs. The polypeptide is transferred from the tRNA in the P site to the aminoacyl tRNA in the A site, extending the nascent chain by one amino acid. At this point, the ribosome undergoes a massive rearrangement. The small and large subunits rotate relative to each other to form the so-called hybrid state, in which the amino acid acceptor stem of the tRNAs are in the E and P sites, while their anticodon loops remain in the P and A sites. The complete and stable translocation of the ribosome along the mRNA requires the entry of the GTP-bound eEF2 (or EF-G in bacteria) into the A site. Binding of eEF2-GTP stabilizes the hybrid state, and promotes rapid GTP hydrolysis. The accompanying conformational changes reset the ribosome, with the deacylated tRNA released from the E site, the peptidyl-tRNA in the P site, and the vacant A site awaiting the next aminoacyl tRNA (reviewed in Dever and Green, 2012; Prabhakar et al., 2017).
Figure 2. Eukaryotic Translation Elongation, Termination, and Ribosome Recycling.
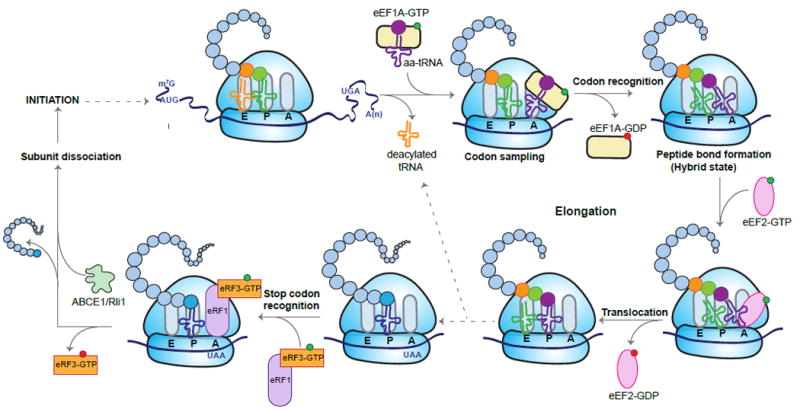
In each elongation cycle, an eEF1A-GTP-aminoacyl (aa)-tRNA ternary complex binds the ribosome, with the anticodon loop of the tRNA in contact with the mRNA codon at the A site. Recognition of the codon triggers GTP hydrolysis and the dissociation of eEF1A-GDP. Peptide bond formation leads to large conformational changes in the ribosome, and the tRNAs transition into hybrid states, with their anticodon loops in the A and P sites, and their acceptor stems shifted to the P and E sites respectively. Binding of eEF2-GTP to the A site and subsequent GTP hydrolysis promotes the translocation of the ribosome, shifting the tRNAs into the canonical P and E sites and ratcheting the mRNA by exactly one codon. The ribosome is now poised for another cycle of elongation, with binding of an appropriate eEF1A-GTP-aminoacyl-tRNA complex to the A site, and release of the deacylated tRNA from the E site. The elongation process continues until a stop codon is reached that is recognized by eRF1-eRF3-GTP. Binding of this complex to the ribosome leads to GTP hydrolysis, and the release of eRF3-GDP and the polypeptide. Finally, ABCE1 facilitates subunit dissociation and recycling, regenerating the components for another round of translation.
This cycle is repeated until the elongating ribosome encounters a stop codon (UAA, UGA, or UAG) in the A site, triggering termination of translation (Figure 2). In eukaryotes, termination requires the action of two release factors, eRF1 and eRF3. The ternary complex of eRF1 and eRF3-GTP binds to the A site of the ribosome, and recognition of the stop codon by eRF1 triggers GTP hydrolysis, resulting in a conformational change and hydrolysis of the peptidyl-tRNA to release the polypeptide. eRF1 and the ATP binding cassette protein ABCE1 (Rli in yeast) promote the physical splitting of the post-termination ribosome into the 60S and 40S subunits, the latter of which is still bound to the deacylated tRNA and mRNA. Finally, dissociation of the tRNA and mRNA from the 40S ribosome occurs via the action of numerous proteins, including some of the translation initiation factors, and the components are regenerated for another cycle of translation (reviewed in Dever and Green, 2012; Jackson et al., 2012).
Regulation of Translation Initiation
One of the major pathways that regulates translation initiation is phosphorylation of eIF2 through the integrated stress response (ISR; Figure 1). In the mammalian ISR, general translation is inhibited when eIF2 is phosphorylated by any of four stress-induced kinases: PKR (protein kinase R, EIF2AK2), PERK (PKR-like endoplasmic reticulum kinase, EIF2AK3), GCN2 (general control nonderepressible 2, EIF2AK4), and HRI (heme-regulated inhibitor, EIF2AK1). Dephosphorylation is performed by two phosphatase complexes containing the catalytic subunit PP1c (protein phosphatase 1) and one of two regulatory subunits: CReP (constitutive repressor of eIF2α phosphorylation, PPP1R15B) or GADD34 (growth arrest and DNA damage-inducible 34, PPP1R15A; reviewed in Trinh and Klann, 2013). The eIF2α kinases phosphorylate serine 51, leading to sequestration of the eIF2 guanine exchange factor eIF2B, thus inhibiting the exchange of GTP for GDP and reducing the amount of ternary complex (eIF2-GTP-Met-tRNAi) available to reinitiate translation (reviewed in Klann and Dever, 2004). This in turn reduces the translation of the vast majority of open reading frames (ORFs). However, translation increases for proteins derived from transcripts with upstream open reading frames (uORFs), such as ATF4 (activating transcription factor 4). uORFs are minor ORFs in the 5’ UTRs of mRNAs. Under basal conditions, the translation of uORFs preempts the translation of the main ORF; when eIF2α is phosphorylated and the ternary complex is limiting, translation of the uORF declines, increasing translation from the primary ORF. This allows for the translational upregulation of specific proteins needed to cope with stress (reviewed in Barbosa et al., 2013).
The other major pathway modulating translation initiation is mTOR (mechanistic target of rapamycin)-regulated phosphorylation of the initiation factors eIF4G and eIF4B, and of the initiation factor inhibitors eIF4E-binding protein (4E-BP) and programmed cell death 4 (PDCD4; Figure 1). mTOR is a serine/threonine kinase that is activated by phosphoinositide 3-kinase (PI3K) and extracellular signal-regulated kinase (ERK) pathways in response to growth factors (reviewed in Costa-Mattioli et al., 2009). mTOR-mediated phosphorylation of these proteins enhances global translation, and also specifically increases the translation of 5’ terminal oligopyrimidine (TOP) mRNAs, many of which encode proteins involved in translation (reviewed in Thoreen, 2017). The phosphorylation of these proteins increases the efficiency with which the cap-dependent translation complex (consisting of eIF4F, eIF4B, and PABP) assembles and functions. Phosphorylation of eIF4G, the scaffold of the cap-binding complex, increases both cap- and IRES-dependent translation (León et al., 2014). eIF4B, which enhances the activity of the helicase eIF4A, requires phosphorylation to associate with eIF4A (Andreou et al., 2017; Holz et al., 2005). 4E-BPs compete with eIF4G for interaction with eIF4E, the initiation factor responsible for binding the m7G cap of the mRNA (Haghighat et al., 1995). This diminishes cap-dependent translation, and may give mRNAs with particular 5’ UTR structures a competitive advantage over other mRNAs (Merrick, 2015). Phosphorylation of 4E-BPs by mTOR causes 4E-BPs to detach from eIF4E, relieving inhibition (reviewed in Klann and Dever, 2004). Analogously, binding between eIF4A and eIF4G is inhibited if eIF4A is bound by programmed cell death 4 (PDCD4; Dennis et al., 2012). Phosphorylation of PDCD4, in response to mTOR signaling, results in its degradation.
Neurodegenerative Diseases Associated with Dysregulation of eIF2
Dysregulation of eIF2 is associated with many neurodegenerative diseases. For example, biallelic mutations in any of the five subunits of eIF2B cause leukoencephalopathy with vanishing white matter (VWM), a disorder associated with the loss of oligodendrocytes, astrocytes, and axons in the central nervous system (CNS) white matter (reviewed in Bugiani et al., 2010). The progression of VWM is highly variable and rapid deterioration can be initiated by stimuli such as mild traumatic brain injury, fever, or even fright. How mutations in eIF2B cause VWM is not clear. As previously mentioned, eIF2B is responsible for facilitating the replacement of the GDP bound to eIF2 with GTP, and does this in two steps: first, it acts as a GDP dissociation inhibitor displacement factor (GDF) to strip eIF5 away from the eIF2/GDP/eIF5 complex, and second, it functions as a guanine nucleotide exchange factor (GEF), stimulating the release of GDP from eIF2 to allow binding of GTP (Jennings and Pavitt, 2014). Studies have shown that many, but not all, pathogenic eIF2B mutations decrease GEF activity. However, the recent discovery of the additional function of eIF2B as a GDF raises the possibility that some VWM mutations may disrupt GDF activity.
Given the function of eIF2B, one would predict that mutations diminishing its activity would result in decreased levels of eIF2-GTP-Met-tRNAi ternary complex, reducing global translation and increasing translation of uORF-containing transcripts. Consistent with this prediction, levels of ATF4 and its downstream target CHOP (C/EBP-homologous protein, DDIT) were increased in the brains of VWM patients (van der Voorn et al., 2005). However, activation of PERK and other branches of the unfolded protein response was also observed, suggesting that the increase in ATF4 and CHOP may not be a direct effect of reduced eIF2B activity (van Kollenburg et al., 2006; van der Voorn et al., 2005). The activation of the ISR in VWM patients raises the interesting possibility that rapid progression of the disease after febrile infection or head trauma may be due to a stress-induced increase in the level of S51-phosphorylated eIF2α (p-eIF2α) that synergizes with the otherwise relatively subtle effects of eIF2B mutations on the availability of the ternary complex. Further studies are needed to determine whether dysregulation of global translation contributes to pathogenesis in VWM.
In other neurodegenerative disorders, levels of phosphorylated eIF2α are elevated, which raises the possibility that reduced rates of translation initiation may contribute to pathology. A link between Alzheimer’s disease (AD) and reduced global translation was established in 1989, when Langstrom et al. demonstrated that polysomes from the cortices of AD patients were associated with less RNA than those from controls. Subsequently, increased levels of p-eIF2α were found in the cortex and hippocampus of AD patients and in AD mouse models (Chang et al., 2002b; Devi and Ohno, 2013; Hoozemans et al., 2009; Kim et al., 2007; Lewerenz and Maher, 2009; Ma et al., 2013; Mouton-Liger et al., 2012; O’Connor et al., 2008; Page et al., 2006; Stutzbach et al., 2013; Unterberger et al., 2006). In addition to AD, p-eIF2α levels are elevated in the brain or spinal cord of patients and mice with prion disease (Unterberger et al., 2006), amyotrophic lateral sclerosis (ALS; Ilieva et al., 2007), Parkinson’s disease (PD; Hoozemans et al., 2007, 2012), Huntington’s disease (HD; Leitman et al., 2014), and various tauopathies (Köhler et al., 2014; Nijholt et al., 2012; Radford et al., 2015; Stutzbach et al., 2013; Unterberger et al., 2006).
In mammals, eIF2α is phosphorylated by multiple kinases and activation of these kinases is associated with autophosphorylation at particular sites. Analysis of the phosphorylated form of PKR and/or PERK (p-PKR, p-PERK) in patient brains demonstrated increased activation of these kinases in various neurodegenerative diseases (Bando et al., 2005; Nijholt et al., 2012; Stutzbach et al., 2013). For example, elevated p-PKR and p-PERK is observed in the AD brain, particularly in the hippocampus and cortex, regions associated with Aβ plaques and neurofibrillary tangles (Chang et al., 2002a; Hoozemans et al., 2009; Ma et al., 2013; Mouton-Liger et al., 2012; Onuki et al., 2004; Peel and Bredesen, 2003; Unterberger et al., 2006). p-PKR and p-PERK are also increased in the brains of patients with prion disease (Paquet et al., 2009; Unterberger et al., 2006) and PD (Bando et al., 2005; Hoozemans et al., 2007, 2012). Furthermore, increased p-PKR is observed in the brains of HD patients or spinal cords of ALS patients (Hayden et al., 2001; Hu et al., 2003), and p-PERK is increased in the brains of patients with tauopathies (Nijholt et al., 2012; Stutzbach et al., 2013; Unterberger et al., 2006).
eIF2α kinases are activated in many models of neurodegenerative disorders, allowing dissection of the contribution of these kinases to the resulting pathology. For example, experiments in AD models show that PKR, PERK, and GCN2 may all contribute to excessive eIF2α phosphorylation. Aβ increases p-PKR and p-eIF2α in cultured neurons, and a reduction in PKR diminished Aβ-associated toxicity (Chang et al., 2002a). Contextual fear learning and mGluR-dependent long-term depression, both of which are affected upon eIF2α phosphorylation dysregulation, are impaired in AD mouse models, and the reduction of PERK levels rescues these deficits (Costa-Mattioli et al., 2005, 2007; Devi and Ohno, 2014; Trinh et al., 2014; Yang et al., 2016; Zhu et al., 2011). The contribution of GCN2 to eIF2α phosphorylation in AD is less clear. While application of Aβ reduces long-term potentiation (LTP) in the wild-type hippocampus, Aβ-induced loss of LTP was not observed in the absence of Gcn2, supporting the hypothesis that GCN2 contributes to elevated eIF2α phosphorylation in AD (Ma et al., 2013). Similarly, a reduction in p-eIF2α levels and improved spatial memory were observed when Gcn2 was deleted in an AD mouse model. In contrast, GCN2 activation appears to be protective in a different AD mouse model; in this context, loss of Gcn2 increases p-PERK, increasing levels of p-eIF2α and the burden of Aβ plaques in the brain (Devi and Ohno, 2013).
Prion-infected mice also have reduced levels of global translation, which may underlie their lower levels of synaptic proteins and the accompanying synaptic dysfunction. Reduction of p-eIF2α by overexpression of the phosphatase GADD34 in prion-infected mice restored levels of synaptic proteins and reversed synaptic dysfunction, preventing neuron death without changing the levels of aggregated prion protein (Moreno et al., 2012). Further work demonstrated that small molecules that inhibit PERK or that stimulate eIF2B activity also rescued pathology in this model (Halliday et al., 2015, 2017; Moreno et al., 2013).
Additional studies in other disease models suggest that drugs that increase eIF2 activity might also be used to treat AD and tauopathy (Halliday et al., 2017; Yang et al., 2016; Zhu et al., 2013). However, while these results are potentially exciting, a careful examination of the role of eIF2α phosphorylation in ALS illustrates the importance of exercising caution in applying new treatments to patients. Superficially, ALS appears to be an excellent candidate for treatment with drugs that counteract p-eIF2α. Flies overexpressing ALS-associated TDP-43 (TAR DNA binding protein) have elevated p-eIF2α and impaired motor function. Knockdown of the fly homolog of PERK improved motor function, while knockdown of GADD34 aggravated the phenotype (Kim et al., 2014). Similarly, treatment with a PERK inhibitor diminished the toxicity of TDP-43 overexpression in rat neurons (Kim et al., 2014) and treatment of a common ALS model, the SOD1G93A (superoxide dismutase 1) mouse, with guanabenz, a GADD34 inhibitor, hastened paralysis and death in male mice (Vieira et al., 2015). However, several other studies suggest that eIF2α phosphorylation in ALS may be protective. In direct contrast to the Vieira et al. study, administration of GADD34 inhibitors, including guanabenz, delayed morbidity and death in SOD1G93A mice (Das et al., 2015; Wang et al., 2014a). Furthermore, a genetic reduction in PERK levels in a related ALS mouse model led to accelerated weight loss and lethality, and a hypomorphic GADD34 mutation slowed the disease course (Wang et al., 2011, 2014b). These divergent results may be due to the diversity of the models or time course of intervention.
In evaluating these findings, it is crucial to remember that the inhibitory effect of p-eIF2α on translation initiation may be counteracted by other perturbations in the regulation of translation; therefore the observation of increased p-eIF2α in a given disease context is not confirmation of reduced global translation. In fact, although p-eIF2α is elevated in PD patients (Hoozemans et al., 2007, 2012), evidence from Drosophila melanogaster suggests that global translation rates may be increased in PD. For example, expression of human G2019S LRRK2 (leucine rich repeat kinase 2), which causes familial PD, led to an increase in phosphorylation of the ribosomal protein S15 and widespread increased translation initiation in flies (Martin et al., 2014). Furthermore, the phenotypes of Drosophila PD models with disease-causing mutations in Pink1 and park were ameliorated by reducing translation initiation through overexpression of the fly homolog of 4E-BP (Tain et al., 2009). Whether PD patients with these mutations have increased global translation remains unknown.
Non-canonical Translation of Nucleotide Repeat Expansions
Recent evidence suggests that regulation of translation initiation is dramatically altered in the nucleotide repeat expansion diseases. Repeat expansion disorders are characterized by unusually long, tandemly reiterated blocks of three to six nucleotides and are a common cause of protein aggregation and neurodegeneration. Expression of the toxic, mutant protein or RNA and/or loss of function of the affected gene have long been hypothesized to underlie pathology in these disorders. However, several lines of evidence suggest that a broad spectrum of translation abnormalities may also contribute to disease. Mutant transcripts are often translated at higher levels than transcripts from the wild-type allele (Krauss et al., 2013). In addition, novel translation products are generated from non-canonical start sites (i.e., AUG codons in introns or other typically non-coding sequences; near-AUG codons, which differ from AUG by only one nucleotide; or non-AUG codons; Ishiguro et al., 2017; Todd et al., 2013; Zu et al., 2011). These aberrant translation events can occur on either or both the sense and antisense transcripts (Zu et al., 2013). Furthermore, frameshifting of the ribosome within the expanded repeat has been reported (Girstmair et al., 2013; Saffert et al., 2016). The scenario is further complicated by observations of multiple translational alterations from the same disease-causing locus. For example, in HD, a dominant disorder caused by a repeat expansion of at least 35 CAG repeats within the coding sequence of huntingtin (HTT), mutant HTT is translated at higher levels than wild-type HTT (Krauss et al., 2013). Translation products that are initiated from multiple frames within the HTT repeat are also observed in the brains of patients (Bañez-Coronel et al., 2015). In addition, frameshifting results in the production of proteins in which the N-terminus of HTT is fused to polyAla or polySer tracts (Girstmair et al., 2013; Saffert et al., 2016).
Frameshifting in the repeat expansion diseases was first described in 2000, in the context of CAG expansions in the coding region of ataxin 3 (ATXN3) in spinocerebellar ataxia 3 (SCA3) patients. These expansions were associated with both polyGln (0 frame) and polyAla (-1 frame) peptides (Gaspar et al., 2000). While the latter was assumed to be the product of frameshifting during elongation, in 2011, Zu et al. demonstrated that CAG and CUG repeats could be translated in all reading frames in a non-AUG-initiated manner, a process they termed repeat-associated non-ATG (RAN) translation. This discovery opened the floodgates, and soon evidence accumulated for RAN translation of CAG and other repeats in additional contexts. To date, multiple repeat-encoded proteins that are translated using non-canonical initiation sites have been identified in aggregates in patients with repeat-expansion diseases, and many other repetitive proteins can be generated from RAN translation in vitro (Table 1; (Krans et al., 2016; Todd et al., 2013; Zu et al., 2011, 2013). For example, six RAN proteins, corresponding to all frames of sense and antisense transcripts, are produced from the hexanucleotide expansion found in an intron of C9ORF72, the most common familial cause of ALS and frontotemporal dementia (FTD) (Zu et al., 2013). Furthermore, three products are made from the sense or antisense transcripts from the 55-200 CGG repeats found in the 5’UTR of FMR1 in fragile X tremor/ataxia syndrome (FXTAS; Sellier et al., 2017).
Table 1.
Repeat-Encoded Proteins Detected in Patient Tissue
| Disease | Gene | Repeat | Polypeptide | Cells/Tissues with Aggregates | Subcellular Localization | Reference |
|---|---|---|---|---|---|---|
| SCA3 | ATXN3 | CAG | Ala | Pons, lymphoblastoid cells | Nuclear | Gaspar et al., 2000 |
| SCA8 | ATXN8 | CAG | Ala | Purkinje cells | Nuclear, cytoplasmic | Zu et al., 2011 |
| DM1 | AS-DMPK | CAG | Gln | Myoblasts, skeletal muscle, leukocytes | Nuclear, cytoplasmic | Zu et al., 2011 |
| DM2 | CNBP | CCUG | Leu-Pro-Ala-Cysa | Hippocampus, cortex, striatum; mostly in gray matter | Cytoplasmic | Zu et al., 2017 |
| DM2 | AS-CNBP | CAGG | Gln-Ala-Gly-Arg | Hippocampus, cortex, striatum; mostly in white matter | Nuclear | Zu et al., 2017 |
| FXTAS | FMR1 | CGG | Gly | Hippocampus, cortex, cerebellum | Nuclear, cytoplasmic | Todd et al., 2011 |
| FXTAS | AS-FMR1 | CCG | Proa | Hippocampus, cortex, cerebellum | Nuclear, cytoplasmic | Krans et al., 2016 |
| FXTAS | AS-FMR1 | CCG | Ala | Hippocampus, cortex | Nuclear, cytoplasmic | Krans et al., 2016 |
| C9 ALS/FTD | C9ORF72 | GGGGCC | Gly-Pro | Throughout CNS | Nuclear, cytoplasmic | Mori et al., 2013a; Zu et al., 2013; Mori et al., 2013b; Ash et al., 2013; Mann et al., 2013; Schludi et al., 2015 |
| C9 ALS/FTD | C9ORF72 | GGGGCC | Gly-Ala | Throughout CNS | Nuclear, cytoplasmic | Mori et al., 2013a; Zu et al., 2013; Mori et al., 2013b; Mackenzie et al., 2013; Davidson et al., 2014; Zhang et al., 2014b; Mann et al., 2013 |
| C9 ALS/FTD | C9ORF72 | GGGGCC | Gly-Arg | Throughout CNS | Nuclear, cytoplasmic | Mori et al., 2013a; Zu et al., 2013; Mori et al., 2013b; Mann et al., 2013; Schludi et al., 2015 |
| C9 ALS/FTD | AS-C9ORF72 | GGCCCC | Pro-Ala | Throughout CNS | Nuclear, cytoplasmic | Zu et al., 2013; Mori et al., 2013b; Gendron et al., 2013; Mann et al., 2013; Gomez-Deza et al., 2015 |
| C9 ALS/FTD | AS-C9ORF72 | GGCCCC | Pro-Arga | Throughout CNS | Nuclear, cytoplasmic | Zu et al., 2013; Mori et al., 2013b; Gendron et al., 2013; Mann et al., 2013; Schludi et al., 2015 |
| C9 ALS/FTD | AS-C9ORF72 | GGCCCC | Gly-Proa | Throughout CNS | Cytoplasmic | Zu et al., 2013; Mori et al., 2013b; Gendron et al., 2013; Mann et al., 2013; Gomez-Deza et al., 2015 |
| HD | HTT | CAG | Glna | Throughout brain, absent from white matter of the striatum | Nuclear, cytoplasmic | Davies and Rubinsztein, 2006; Banez-Coronel et al., 2015; Girstmair et al., 2013; Saffert et al., 2016; Aronin et al., 1995; Zhao et al., 2016 |
| HD | HTT | CAG | Alab | Striatum (including the white matter), cortex, cerebellum | Nuclear, cytoplasmic | Davies and Rubinsztein, 2006; Banez-Coronel et al., 2015; Girstmair et al., 2013; Saffert et al., 2016 |
| HD | HTT | CAG | Serb | Striatum (including the white matter), cortex, cerebellum | Nuclear, cytoplasmic | Davies and Rubinsztein, 2006; Banez-Coronel et al., 2015; Girstmair et al., 2013; Saffert et al., 2016 |
| HD | AS-HTT | CUG | Cys | Striatum (including the white matter), cortex, cerebellum | Nuclear, cytoplasmic | Banez-Coronel et al., 2015 |
| HD | AS-HTT | CUG | Leu | Striatum (including the white matter), cortex, cerebellum | Nuclear, cytoplasmic | Banez-Coronel et al., 2015 |
| SCA31 | BEAN1 | UGGAA | Met-Glu-Trp-Asn-Glya | Purkinje cells | Nuclear, cytoplasmic | Ishiguro et al., 2017 |
ALS/FTD, amyotrophic lateral sclerosis/frontotemporal dementia; AS-DMPK, antisense dystrophia myotonica protein kinase; AS-FMR1, antisense fragile X mental retardation 1; AS-HTT, antisense huntingtin; ATXN, ataxin; BEAN1, brain expressed associated with NEDD4 1; C9ORF72, chromosome 9 open reading frame 72; CNBP, CCHC-type zinc finger nucleic acid binding protein; CNS, central nervous system; DM, myotonic dystrophy; FMR1, fragile X mental retardation 1; FXTAS, fragile X-associated tremor/ataxia syndrome; HD, Huntington’s disease; HTT, huntingtin; SCA, spinocerebellar ataxia.
AUG initiation site available;
current in vivo data does not distinguish between RAN-translated and frameshifted products.
Due to their diverse structures and expression patterns, repeat-encoded proteins may contribute to pathogenesis in distinct ways. However, disruption of proteostasis may be a common mechanism of toxicity for many of these aberrant translation products. Consistent with this hypothesis, several RAN products from C9ORF72 (polyGly-Ala, -Gly-Pro, -Gly-Arg, and -Pro-Arg) and FMR1polyGly have been linked to proteasomal dysfunction (Gupta et al., 2017; Oh et al., 2015; Yamakawa et al., 2015; Zhang et al., 2014b). C9ORF72polyPro-Arg is also associated with impaired autophagy (Gupta et al., 2017).
In addition to disrupting proteostasis, aberrant proteins from expanded repeats may sequester macromolecules. This in turn may lead to the formation of toxic oligomers and/or loss of function of sequestered proteins or RNAs. For example, Lee et al. (2016) demonstrated that C9ORF72polyGly-Arg and -Pro-Arg, but not the less toxic C9ORF72 dipeptide repeat proteins, interact with large numbers of low-complexity domain (LCD)-containing proteins such as RNA binding proteins. In doing so, these proteins change the concentration thresholds at which LCD-containing proteins undergo liquid-liquid phase separation, potentially sequestering LCD-containing proteins in inappropriate membraneless cellular compartments. Phase separation may contribute to the defects in nucleocytoplasmic transport and nucleolar function associated with C9ORF72polyGly-Arg and -Pro-Arg (Freibaum et al., 2015; Jovičić et al., 2015; Kwon et al., 2014; Tao et al., 2015). Interestingly, these proteins also interact nonspecifically with mRNAs, blocking their interactions with translation factors and thus diminishing global translation (Kanekura et al., 2016). Less is known regarding the interactomes of other repeat-encoded proteins. However, computational analyses of the human interactome revealed that proteins containing certain non-expanded homopolymeric tracts are frequently associated with other proteins containing the same tract (Pelassa and Fiumara, 2015), which suggests that proteins with repeat expansions may have similar behavior.
The mechanisms underlying the non-canonical translation events in the nucleotide expansion diseases are only just emerging. However, recent evidence demonstrates that repeat length constitutes a major determinant influencing translation. While repeat length is often positively correlated with the levels of both AUG-initiated and RAN translation (Krans et al., 2016; Krauss et al., 2013; Scoles et al., 2015; Zu et al., 2011, 2013, 2017), RAN translation is only observed from transcripts with pathogenic repeats (Gaspar et al., 2000; Mori et al., 2013a; Todd et al., 2013; Zu et al., 2011, 2013). A similar dependence on pathogenic repeat length has been observed for frameshifting (Saffert et al., 2016). However, for other RAN-translated or frameshifted proteins, the relationship between repeat length and protein levels is less clear (Kearse et al., 2016; Krans et al., 2016; Todd et al., 2013; Toulouse et al., 2005; Zu et al., 2011). The positive correlation between repeat length and non-canonical translation levels may be due to the tendency of longer repeats to form secondary structures. Repeats associated with RAN translation all form hairpins or a mixture of hairpins and G-quadruplexes (Ciesiolka et al., 2017; Sobczak et al., 2003; but see Guo and Bartel, 2016). Furthermore, RAN translation has not been observed from expanded repeats that do not form hairpins (Bañez-Coronel et al., 2015; Krauss et al., 2013; Toulouse et al., 2005; Zu et al., 2011).
One means by which hairpins in repeat expansion sequences may promote their translation is though the recruitment of the MID1 (midline 1) complex. The E3 ubiquitin ligase MID1 and the adaptor protein IGBP1 (immunoglobulin binding protein 1) induce degradation of the protein phosphatase 2A catalytic subunit (PP2Ac/PPP2CA), which dephosphorylates mTORC1 (mTOR complex 1), leading to its disassembly (Krauss et al., 2013; Liu et al., 2011). Interestingly, the MID1 complex appears to bind to expanded CAG repeats with higher affinity than to non-expanded repeats. In agreement with a role for MID1 and mTORC1 in promoting canonical translation of these transcripts, translation is increased upon inhibition of PP2Ac, and decreased upon knockdown of MID1 or inhibition of mTORC1 (Griesche et al., 2016; Krauss et al., 2013). Whether this mechanism also contributes to RAN translation is unknown.
In addition to the repeat itself, sequences flanking repeats can also influence non-canonical translation. Translation of most RAN products can be initiated within the expanded nucleotide repeat itself (Zu et al., 2011). However, initiation of these proteins can also occur at alternative sites upstream of the repeat. While poly(CAG) RAN translation was supported by a series of constructs with similar repeats but with unique upstream flanking sequences, the abundance and size of the RAN products varied, indicating translation initiates at multiple sites (Todd et al., 2013; Zu et al., 2011). In experiments comparing transcripts differing only in the presence or absence of an in-frame AUG codon upstream of the repeat, the presence of an AUG usually, but not always, resulted in greater translation, suggesting that some upstream sequences may influence start site selection by simply providing an in-frame AUG start codon (Bañez-Coronel et al., 2015; Kearse et al., 2016; Krans et al., 2016; Scoles et al., 2015; Todd et al., 2013; Zu et al., 2011, 2013). For RAN products that do not initiate within the repeat, upstream sequences are essential. For example, initiation of FMR1polyGly primarily relies on an ACG upstream of the expanded repeat that is translated as Met (Sellier et al., 2017). However, translation can also initiate at alternative near-AUGs if the favored ACG is deleted, or if translation of the repeat is blocked with a stop codon between the ACG and the repeat (Kearse et al., 2016; Todd et al., 2013). In the absence of a near-AUG, translation of FMR1polyGly does not occur. In addition to regulation by upstream sequences, sequence downstream of repeats may also influence levels of RAN translation (Scoles et al., 2015; Zu et al., 2011).
While the initiation site of RAN proteins is non-canonical, evidence suggests that in some cases their production depends upon canonical translational mechanisms. RAN translation of polyGly and polyAla from the 5’ UTR of FMR1 requires both the m7G mRNA cap and the helicase eIF4A (Kearse et al., 2016). This requirement suggests that RAN translation of FMR1 relies upon the 48S PIC scanning from the 5’ end of the transcript. Consistent with this hypothesis, placement of the repeat-containing FMR1 5’ UTR in the 3’ UTR of a reporter is not permissive for FMR1polyGly translation, presumably because the initiation complexes recruited at the 5’ end of the transcript dissociate at the stop codon before reaching the nucleotide repeat expansion (Todd et al., 2013). If all RAN translation were dependent upon scanning, the presence of an upstream AUG in a different frame would be predicted to diminish or prevent RAN translation. However, translation of most RAN products is unaffected by out-of-frame upstream AUGs (Bañez-Coronel et al., 2015; Krans et al., 2016; Zu et al., 2011, 2013). Furthermore, canonical cap-dependent translation is unlikely to be involved in the translation of repeats in introns or antisense transcripts.
Does repeat-associated translation cause human disease? When exogenously applied to cells in culture or when expressed in cells or in model organisms, many RAN products have been shown to be toxic. However, these proteins may be translated, trafficked, and degraded differently in patients than in cultured cells. In fact, Schludi et al., (2015) demonstrate that C9ORF72polyGly-Arg and -polyPro-Arg aggregates are paranucleolar in human patients, but nucleolar in rat hippocampal neuronal cultures, a finding which calls into question the relevance of the in vitro studies that showed that these proteins mediated nucleolar dysfunction. Furthermore, in patients with expansions in C9ORF72, the spatial distribution of most dipeptide repeat aggregates correlates poorly with cell death. C9ORF72 ALS patients experience upper and lower motor neuron death while C9ORF72 FTD patients primarily lose neurons in the frontal and temporal cortices, yet the two sets of patients have indistinguishable distributions of dipeptide repeats (Mackenzie et al., 2013). However, in contrast, aggregates of polyLeu-Pro-Ala-Cys in the brains of patients with myotonic dystrophy type 2 appear specifically in the regions with neuronal death (Zu et al., 2017).
Determining the pathogenicity of RAN translation products is complicated by the coincident presence of mutant transcripts. This problem can be addressed by experimentally altering the ratio of mutant transcript to protein. The toxicity of RAN products has been demonstrated directly by increasing the ratio of protein to RNA through exogenous application of synthetic peptides, addition of an upstream AUG to increase translation from an mRNA, or use of alternative codons that do not form RNA hairpins to encode the repetitive protein (Boeynaems et al., 2016; Freibaum et al., 2015; Kwon et al., 2014; Lee et al., 2016; May et al., 2014; Mizielinska et al., 2014; Wen et al., 2014; Yamakawa et al., 2015; Zhang et al., 2014b; Zu et al., 2017, 2013). These approaches demonstrated the toxicity of arginine-containing C9ORF72 RAN products. Furthermore, several groups have revealed the toxicity of RAN products indirectly by showing that the loss of RAN products reduces toxicity. For example, Todd et al. (2013) generated flies expressing a repeat-expanded FMR1 5’ UTR using either the complete UTR or a mutant UTR incapable of sustaining FMR1polyGly translation, and demonstrated that the presence of FMR1polyGly causes toxicity. Deployment of a similar strategy in transgenic mice revealed that FMR1polyGly production caused Purkinje cell death, various motor deficits, and premature death, while no pathology was observed in mice expressing the mutant mRNA lacking FMR1polyGly initiation sites (Sellier et al., 2017). While this approach allowed investigation of FMR1polyGly, it cannot be readily applied to all contexts, as other RAN-translated proteins can initiate within the repeat itself. A clever alternative strategy to distinguish the effects of mutant RNA from those of RAN proteins is to specifically degrade RAN proteins; this approach confirmed that arginine-containing C9ORF72 RAN products are toxic (Lee et al., 2016).
Elongation Factors and Impaired Translational Fidelity in Neurodegeneration
Accurate protein synthesis depends on the selection of the cognate aminoacyl tRNA and the precise movement of the ribosome to the next codon, maintaining the reading frame of the mRNA. Although cells grapple with a fundamental tradeoff between translation elongation accuracy and speed, the error rate under normal circumstances is quite low, and is estimated to be around one amino acid misincorporated for every 103-104 codons. Errors in ribosome translocation leading to frameshifts, which are potentially more devastating to cellular fitness, are believed to be even more infrequent (reviewed in Prabhakar et al., 2017; Zaher and Green, 2009). The elongation factors eEF1A and eEF2 play essential roles in the delivery of the cognate aminoacyl tRNA to the A site of the ribosome and in its translocation along the mRNA after peptide bond formation, respectively. Mutation of these factors in yeast, or their orthologs in bacteria, reduces translational fidelity, indicating that they play an essential role in limiting amino acid misincorporation and reading frame errors during normal translation elongation (Carr-Schmid et al., 1999; Farabaugh and Vimaladithan, 1998; Hughes et al., 1987; Ortiz et al., 2006; Sandbaken and Culbertson, 1988; Vijgenboom and Bosch, 1989).
Several studies have observed decreased expression of eEF1A and eEF2 in the brains of PD and AD patients, as well as in mouse models of these diseases; however, the role of elongation in the pathogenesis of these disorders remains unclear (Beckelman et al., 2016a, 2016b, Garcia-Esparcia et al., 2015, 2017; Hernández-Ortega et al., 2016; Li et al., 2005). A better understanding of the importance of these factors in disease is gained when we turn to genetic models and patients with mutations in these genes themselves.
Vertebrates have two eEF1A genes with distinct expression patterns. eEF1A1 is expressed ubiquitously during development, but is sharply downregulated in adult neurons and muscle, whereas eEF1A2 is restricted to neurons and muscle cells, and it undergoes significant upregulation over the course of postnatal development (Lee et al., 1993, 1995). The autosomal recessive wasted mutation in mice results in a complete loss of eEF1A2 expression, causing motor neuron degeneration and concomitant muscle wasting after weaning (Table 2; Abbott et al., 2009; Chambers et al., 1998; Newbery et al., 2007). This restricted phenotype tracks beautifully with the spatial expression of eEF1A2. Although homozygous mutations in human eEF1A have not been reported, exome sequencing has identified de novo missense mutations in eEF1A2 in multiple patients with severe epilepsy, intellectual disability, and autistic-like behavior, often with accompanying signs of neurodegeneration, such as progressive microcephaly (Inui et al., 2016; Lam et al., 2016; de Ligt et al., 2012; Lopes et al., 2016; Nakajima et al., 2015; Veeramah et al., 2013). Although these mutations disrupt highly conserved amino acids, it is unclear whether they cause a loss or gain of function. In addition, eEF1A has been reported to have several non-canonical functions, including actin regulation, further complicating our understanding of the putative mechanism in disease (Mateyak and Kinzy, 2010). Serendipitously, an eEF1A2 patient mutation (E122K) was characterized in a yeast study 30 years ago, where it was found to promote readthrough of UAG stop codons in a reporter construct, as well as allow production of a correct protein from a construct with an introduced frameshift mutation (Sandbaken and Culbertson, 1988). These studies suggest that the neuronal dysfunction observed in patients with eEF1A2 mutations could be due to deficient translational fidelity during elongation.
Table 2.
Diseases Caused by Mutations in Genes Involved in Cytoplasmic Translation Elongation
| Gene | Inheritance | Function | Disease/Clinical Presentation | References |
|---|---|---|---|---|
| Human Diseases | ||||
| EEF1A2 | De novo | Delivery of aminoacyl-tRNA (aa-tRNA) to ribosome during elongation | Progressive microcephaly with epilepsy, intellectual disability, autistic-like behavior | Inui et al., 2016; Lam et al., 2016; de Ligt et al., 2012; Lopes et al., 2016; Nakajima et al., 2015; Veeramah et al., 2013 |
| EEF2 | Dominant | Translocation of ribosome after peptide bond formation | Spinocerebellar ataxia 26 | Hekman et al., 2012 |
| AARS, GARS, HARS, MARS, YARS | Dominant | Cytoplasmic tRNA synthetases | Charcot-Marie-Tooth disease and other peripheral neuropathies | Reviewed in Meyer-Schuman and Antonellis, 2017 |
| AARS, DARS, HARS, KARS, QARS, RARS | Recessive | Cytoplasmic tRNA synthetases | Neurodegenerative disorders including peripheral neuropathy, progressive microcephaly, Usher’s syndrome, leukoencephalopathy | Kodera et al., 2015; McLaughlin et al., 2010; Meyer-Schuman and Antonellis, 2017; Nakayama et al., 2017; Puffenberger et al., 2012; Simons et al., 2015; Taft et al., 2013; Wolf et al., 2014; Zhang et al., 2014a |
| POLR1C, POLR3A, POLR3B | Recessive | RNA polymerase III subunits | Leukodystrophy with accompanying neurodegeneration | Azmanov et al., 2016; Bernard et al., 2011; Saitsu et al., 2011; Shimojima et al., 2014; Tétreault et al., 2011; Thiffault et al., 2015 |
| TRNT1 | Recessive | Nucleotidyltransferase responsible for CCA addition to 3’ end of tRNAs | Microcephaly, cortical atrophy, sensorineural deafness, photoreceptor loss | Chakraborty et al., 2014; DeLuca et al., 2016; Sasarman et al., 2015 |
| KAE1/OSGEP | Recessive | Biosynthesis of N6- threonylcarbamoyladenosine (t6A) modification on tRNAs | Microcephaly, cerebellar atrophy, renal defects | Edvardson et al., 2017 |
| CLP1, TSEN2, TSEN15, TSEN34, TSEN54 | Recessive | Splicing of intron-containing tRNAs | Pontocerebellar hypoplasia | Bierhals et al., 2013; Breuss et al., 2016; Karaca et al., 2014; Namavar et al., 2011; Schaffer et al., 2014 |
| ANG | Dominant | Stress-induced cleavage of tRNAs | Amyotrophic lateral sclerosis, Parkinson’s disease | Aparicio-Erriu and Prehn, 2012; Chen et al., 2014; Van Es et al., 2011; Fernández-Santiago et al., 2009; Greenway et al., 2006; Wu et al., 2007 |
| NSUN2 | Recessive | Cytosine-5 methylation (m5C) of tRNA | Microcephaly, cortical atrophy, intellectual disability | Abbasi-Moheb et al., 2012; Komara et al., 2015; Martinez et al., 2012) |
| GTPBP2 | Recessive | Ribosome recycling factor | Cerebellar atrophy, retinal degeneration, intellectual disability | Jaberi et al., 2015 |
| Mouse Models | ||||
| Eef1a2 | Recessive | Delivery of aa-tRNA to ribosome during elongation | wasted phenotype–motor neuron loss and muscle wasting | Chambers et al., 1998 |
| Aars | Recessive | Cytoplasmic alanyl tRNA synthetase | sticky phenotype–Purkinje cell degeneration, and unkempt appearance of fur | Lee et al., 2006 |
| Clp1 | Recessive | Splicing of intron-containing tRNAs | Clp1 kinase dead–motor neuron loss and microcephaly | Hanada et al., 2013 |
| Nsun2 | Recessive | Cytosine-5 methylation (m5C) of tRNA | Apoptosis of cortical, hippocampal, and striatal neurons; microcephaly | Blanco et al., 2014 |
| Gtpbp2, n-Tr20 | Recessive | Ribosome recycling factor, tRNAArgUCU | Ataxia; degeneration of cerebellum, cortex, hippocampus, and retina | Ishimura et al., 2014, Ishimura et al.,2016 |
| Ltn1 | Recessive | E3 ubiquitin ligase, degradation of aberrant nascent chains | lister phenotype–progressive neurodegeneration, locomotor abnormalities | Chu et al., 2009 |
The discovery of a mutation in eEF2 linked to autosomal dominant spinocerebellar ataxia (SCA26), a neurodegenerative disorder characterized by Purkinje cell loss, further bolsters a pathogenic role for impaired translation elongation in neurons. The mutated residue, a conserved proline (P596H), is located in a loop near the so-called ‘anticodon mimicry’ domain of eEF2, which interacts with the ribosomal decoding center during tRNA translocation. The histidine substitution in SCA26 patients is predicted to contribute steric hindrance that interferes with ribosome translocation (Hekman et al., 2012). In agreement with this prediction, mutation of the equivalent residue in yeast resulted in an increased rate of frameshifting on a programmed ribosomal frameshifting (PRF) reporter containing a viral sequence that promotes ribosome pausing and frameshifting. Whether PRF sequences exist in Purkinje cells is unknown. Thus, the molecular mechanism underlying degeneration in patients may involve a general increase in frameshifting that leads to mistranslation and the production of abnormal polypeptides.
Defects in Aminoacyl tRNA Synthetase Function in Neurodegeneration
In addition to the elongation factors themselves, translation elongation relies on a steady supply of charged tRNAs, produced by the aminoacyl tRNA synthetases, and mutations in these enzymes have been linked to neurological and neurodegenerative disease. Aminoacyl tRNA synthetases catalyze the reaction in which an amino acid is covalently attached to a tRNA with a specific anticodon, resulting in a charged or aminoacyl tRNA. Dominant missense mutations in a large number of cytoplasmic tRNA synthetases have been linked to Charcot-Marie-Tooth disease (CMT; Abbott et al., 2014; Meyer-Schuman and Antonellis, 2017; Park et al., 2008; Stum et al., 2011). However, the underlying molecular mechanism of these disorders has remained elusive. Many CMT-linked mutations seem to have no significant effect on the aminoacylation activity of the synthetase, and recent studies support a toxic gain of function mechanism, possibly involving the exposure of a neomorphic binding surface and resulting in aberrant protein interactions (He et al., 2015; Motley et al., 2011; Seburn et al., 2006; Sleigh et al., 2017). In contrast, other CMT-linked mutations have been reported to cause a loss of charging activity, suggesting that haploinsufficiency of the enzyme may be a possible contributing factor (Griffin et al., 2014; McLaughlin et al., 2012). Supporting this hypothesis, recessive mutations in synthetases have been linked to neurodegenerative disease, as well as many multisystem disorders involving a wider range of tissues, and many of these mutations have been found to reduce aminoacylation activity in vitro (Kodera et al., 2015; McLaughlin et al., 2010; van Meel et al., 2013; Meyer-Schuman and Antonellis, 2017; Nakayama et al., 2017; Puffenberger et al., 2012; Simons et al., 2015; Taft et al., 2013; Wolf et al., 2014; Zhang et al., 2014a). Impaired synthetase function may reduce the amount of charged tRNA available for translation elongation, with a possible increase in the levels of uncharged tRNA. Uncharged tRNAs produced as a result of amino acid deprivation have been reported to bind GCN2, leading to the activation of the ISR (Dong et al., 2000). Thus, loss of function mutations in synthetases could lead to global deficits in translation. Together, these disorders underscore the importance of these ancient enzymes in protein synthesis and neuronal function.
Accurate aminoacylation of tRNAs by their cognate synthetases is essential for faithful protein synthesis. The active sites of these enzymes distinguish cognate from non-cognate amino acids by steric hindrance; however, discrimination between structurally similar amino acids is difficult. Thus, a subset of synthetases utilize additional editing domains to proofread and remove non-cognate amino acids that are mistakenly activated by the enzyme (Guo and Schimmel, 2012; Ling et al., 2007; Schimmel, 2011). In mice, a spontaneous mutation, known as sticky because of the rough and unkempt coat of homozygotes, was found to reside in the editing domain of alanyl tRNA synthetase (AARS; Lee et al., 2006). These mice develop ataxia and have progressive cerebellar Purkinje cell degeneration. In vitro studies found that this mutation increases the mischarging of tRNAAla with serine, likely resulting in the random substitution of serine at some alanine codons during translation, introducing ambiguity in the genetic code. Treatment with serine, but not alanine, increased cell death in sticky mutant fibroblasts in a dose-dependent manner, suggesting that this mutation causes increased mistranslation in cells. In agreement, increased levels of molecular chaperones and ubiquitinated protein aggregates were observed in Purkinje cells of mutant mice. Although the defect in editing caused by this mutation is relatively mild and only increases the production of mischarged tRNAAla by about two-fold, it results in devastating neurodegeneration, highlighting the sensitivity of neurons to mischarging and consequent mistranslation.
The importance of translational fidelity in neuron survival is further supported by a study that investigated the effects of increased mischarging by a different tRNA synthetase. Mutations in the active and editing sites of the Drosophila phenylalanyl tRNA synthetase increased the mischarging of tRNAPhe with tyrosine, which resulted in misincorporation of tyrosine at phenylalanine codons. Mutant flies were sensitive to diets high in tyrosine and had increased endoplasmic reticulum stress, impaired locomotion, and neurodegeneration, a phenotype reminiscent of the sticky mice (Lu et al., 2014).
tRNA synthetases are ubiquitously expressed, as expected based on their essential role in translation. Thus, the restricted phenotype observed in the sticky mouse, and to a lesser extent in the Drosophila mutant, is unexpected. However, additional studies on a more severe AARS editing mutation revealed that other cell types, besides neurons, are vulnerable to mistranslation. Mice homozygous for a point mutation in AARS (C723A), which increases the production of tRNAAla mischarged with serine by about 15-fold over wild type, die before embryonic day 8.5 (Liu et al., 2014). Compound heterozygous mice with the sticky AARS in trans with either a loss of function allele or the severe AarsC723A allele displayed age-related cardiac fibrosis in addition to Purkinje cell loss. As previously observed in Purkinje cells in the sticky mutant mouse, ubiquitinated protein aggregates were also observed in mutant cardiomyocytes. This differential vulnerability to translation infidelity may reflect the varying capacity of the protein quality control system in different cell types or expression patterns of modifier genes.
Defects in tRNA Expression and Processing in Neurodegeneration
Growing evidence indicates that mRNA translation is exquisitely sensitive to changes in tRNA levels, and mutations in numerous genes involved in tRNA expression and processing are associated with neurodegenerative disease. tRNAs are encoded by hundreds of genes in mammals and numerous studies have found that the expression of tRNA isoacceptor (tRNAs with the same anticodon) families varies between tissues (Goodenbour and Pan, 2006; Schmitt et al., 2014), and can be altered under disease conditions such as cancer (Gingold et al., 2014; Goodarzi et al., 2016; Pavon-Eternod et al., 2009). Changes in tRNA repertoires have been hypothesized to correlate with the codon usage of genes associated with cellular differentiation state, in order to fine-tune their translation (Gingold et al., 2014; Goodarzi et al., 2016; but see Rudolph et al., 2016).
tRNAs are transcribed by RNA polymerase III (Pol III) and must undergo numerous processing events to be functional, including removal of the 5’ leader and 3’ trailer sequences, splicing of introns, addition of the non-templated 3’ CCA end necessary for aminoacylation, and the covalent modification of many nucleotides (Figure 3). Disruption of any of these steps can seriously impair the production of mature tRNAs, altering the cellular tRNA pool. Missense mutations in POLR3A and POLR3B, which encode subunits that form the catalytic core of Pol III, have been linked to a heterogeneous group of autosomal recessive disorders known as leukodystrophies (Azmanov et al., 2016; Bernard et al., 2011; Saitsu et al., 2011; Shimojima et al., 2014; Tétreault et al., 2011). These diseases are characterized by decreased white matter and neuron loss in the cerebellum (La Piana et al., 2014; Takanashi et al., 2014). Leukodystrophy has also been linked to mutations in POLR1C, which encodes a subunit of Pol III that is shared with RNA polymerase I, although Pol III assembly appears to be specifically affected by this mutation (Thiffault et al., 2015). The neural-specific effect of these Pol III mutations on the CNS is hypothesized to be due to impaired transcription of certain tRNAs that are critical in the CNS. This tRNA-centric hypothesis is bolstered by the fact that recessive mutations in aminoacyl tRNA synthetases have also been linked to white-matter disorders (Taft et al., 2013; Wolf et al., 2014). However, in addition to tRNAs, Pol III transcribes many other small non-coding RNAs, including the 5S rRNA, U6, 7SK, and RNase P RNAs. Indeed, decreased levels of multiple classes of Pol III transcripts, in addition to tRNAs, were observed in the blood of patients with one POLR3A mutation, leaving the underlying pathogenic mechanisms in these diseases murky (Azmanov et al., 2016).
Figure 3. tRNA Processing in Health and in Neurodegeneration.
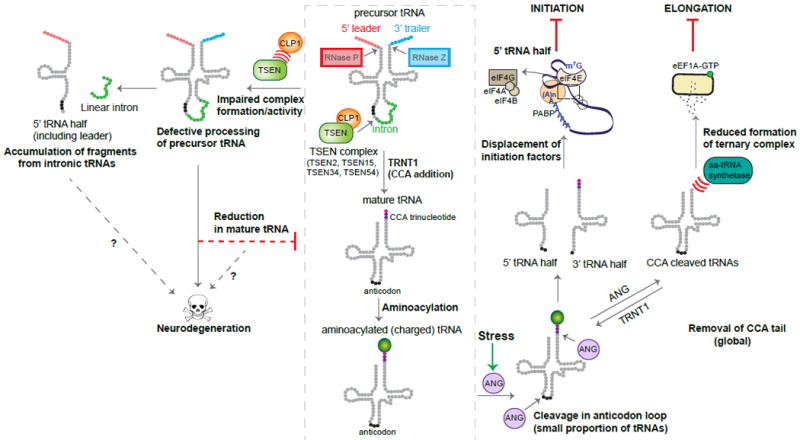
Precursor tRNAs undergo numerous processing steps in order to form mature tRNAs that can be charged by their cognate aminoacyl tRNA synthetases. A small subset of tRNAs contain introns that are removed by splicing, and the tRNA splicing endonuclease (TSEN) complex, along with the kinase CLP1, play an essential role in this process. Disease-linked mutations in TSEN subunits or in CLP1 disrupt the assembly and activity of the TSEN/CLP1 complex, impairing the processing of precursor tRNAs. This leads to the accumulation of intermediate fragments, and in some cases, a reduction in the level of mature tRNA available for charging. Mature tRNAs can be cleaved by a stress-activated ribonuclease, angiogenin (ANG). Cleavage within the anticodon loop releases tRNA halves, and specific 5’ tRNA halves can displace the eukaryotic initiation factors (eIF) 4G and eIF4A from the 7-methylguanosine (m7G) cap of the mRNA, repressing translation initiation. ANG can also remove the CCA tail (added by TRNT1) from the 3’ end of the tRNA, preventing charging by the synthetase, and thus inhibiting translation elongation.
Recent studies have linked recessive mutations in TRNT1, which encodes the nucleotidyltransferase responsible for adding the CCA trinucleotide to the 3’ end of both mitochondrial and cytoplasmic tRNAs, to diverse disorders characterized by microcephaly, cortical atrophy, sensorineural deafness and photoreceptor degeneration (Chakraborty et al., 2014; DeLuca et al., 2016; Sasarman et al., 2015). The majority of the analyzed missense mutations impaired the ability of TRNT1 to catalyze the formation of the CCA trinucleotides in vitro, and failed to complement the growth defect of CCA1 null yeast (Chakraborty et al., 2014). While TRNT1 activity should be essential for the production of all mature tRNAs, tRNAs that deviate from the canonical cloverleaf structure appear to be particularly sensitive and were the most prone to defects in CCA tail addition, suggesting that disease-associated mutations in TRNT1 may not affect the tRNA pool uniformly (Sasarman et al., 2015).
Mature tRNAs are heavily modified post-transcriptionally, and these modifications are essential for tRNA structure, stability, and function. Several human diseases, including neurological disorders, have been linked to mutations in tRNA modification enzymes, and this topic has been the subject of recent comprehensive reviews (Bednářová et al., 2017; Torres et al., 2014). Recently, homozygous mutations in KAE1 (kinase-associated endopeptidase; OSGEP), which plays a role in the biosynthesis of N6-threonylcarbamoyladenosine (t6A) modifications of tRNA, have been linked progressive cerebellar atrophy and leukodystrophy (Edvardson et al., 2017). This modification is present at nucleotide 37 in the anticodon stem loop of nearly all tRNAs decoding ANN (where N is any nucleotide) codons, and defects in t6A biosynthesis in yeast lead to leaky scanning bypass of start codons, as well as impaired reading frame maintenance (Thiaville et al., 2016). This suggests that certain hypomodified tRNAs can disrupt translation elongation and lead to neuropathology.
Approximately 6% of human tRNA genes contain introns that must be removed to produce mature tRNAs. Certain tRNA isoacceptor families are exclusively represented by intron-containing tRNAs, highlighting the importance of proper splicing. Recessive mutations in all four subunits of the tRNA splicing endonuclease complex (TSEN), as well as in CLP1 (cleavage and polyadenylation factor I subunit 1), a kinase involved in tRNA splicing, cause pontocerebellar hypoplasia (PCH), a heterogeneous group of neurodegenerative disorders with prenatal to neonatal onset characterized by cerebellar hypoplasia and microcephaly (Bierhals et al., 2013; Breuss et al., 2016; Karaca et al., 2014; Namavar et al., 2011; Schaffer et al., 2014). In addition, mice homozygous for a mutation in CLP1 that abolishes its kinase activity (Clp1K/K) display progressive loss of spinal motor neurons, and cortical microcephaly (Hanada et al., 2013; Karaca et al., 2014). Together, these disorders highlight a molecular link between tRNA splicing and neurodegeneration. tRNA splicing involves physical interaction of the TSEN complex and CLP1, and disease-associated mutations in TSEN15 and CLP1, as well as the kinase-dead mutation in CLP1, disrupt the assembly and in vitro activity of this complex. Surprisingly, although a reduction in the steady-state levels of mature tRNA was observed in CLP1 patient-derived induced neurons, no such reduction was detected in the spinal cord or fibroblasts of Clp1K/K mice, or in CLP1 patient fibroblasts. Thus, it remains unclear whether changes in the levels of mature tRNA and accompanying defects in translation elongation are involved in the disease mechanism. Alternatively, the accumulation of tRNA fragments derived from the aberrant processing of intron-containing tRNAs may impact disease pathology. Aberrant intermediates were consistently observed in both patient and Clp1K/K cells; however, the identity of the tRNA fragments differed. In the Clp1K/K mouse, 5’ tRNA halves (including the leader) were observed, while accumulation of the linear intron was detected in patient fibroblasts. Cells expressing or exposed to these aberrant intermediates were more sensitive to oxidative stress-induced cell death, though the exact molecular mechanism remains unknown.
Defects in Angiogenin-mediated tRNA Cleavage in Neurodegeneration
Accumulating evidence indicates that tRNAs may serve as a source of diverse small non-coding RNAs with a wide range of functions (Shigematsu et al., 2014; Sobala and Hutvagner, 2011). Recent studies suggest that defects in regulated tRNA cleavage are also associated with neurodegeneration. Mutations in the ribonuclease angiogenin (ANG) have been linked to familial and sporadic ALS and PD, though the penetrance of some of these mutations remains under debate (Aparicio-Erriu and Prehn, 2012; Chen et al., 2014; Corrado et al., 2007; Van Es et al., 2011; Fernández-Santiago et al., 2009; Greenway et al., 2006; Wu et al., 2007). Supporting a protective role for ANG activity, the majority of the patient mutations are predicted to disrupt its structure and function, and indeed, several patient mutations reduced its catalytic activity (Greenway et al., 2006; Wu et al., 2007). Recombinant ANG has been reported to be neuroprotective against excitotoxic and ER stress in primary motor neuron cultures, and to improve lifespan and motor performance in SOD1G93A mice (Kieran et al., 2008). ANG cleaves tRNAs in the anticodon loop, releasing 5’ and 3’ tRNA halves in response to stressors such as heat shock or arsenite treatment (Figure 3; Fu et al., 2009; Yamasaki et al., 2009). ANG activation results in a decrease in protein synthesis, yet only a small subset of tRNAs are cleaved and intact tRNA levels are not altered. However, in vitro assays indicate that specific 5’ tRNA halves can repress translational initiation by displacing eIF4G and eIF4A from the 5’ end of mRNAs (Ivanov et al., 2011). Intriguingly, a recent study found that in addition to cleaving the anticodon loop, ANG cleaves the 3’ CCA trinucleotide that is necessary for the aminoacylation of all tRNAs. This cleavage occurs far more quickly than the generation of tRNA halves in response to stress, and can rapidly and dynamically be reversed by TRNT1 (Czech et al., 2013). Thus, the molecular mechanism underlying ANG-associated neurodegeneration may involve a disruption to the regulation of both translational initiation and elongation.
The importance of the regulation of ANG-mediated cleavage of tRNAs for neuronal homeostasis is further highlighted by the neurological defects linked to mutations in a tRNA methyltransferase, NSUN2. Nsun2-deficient mice display microcephaly, elevated stress granule formation, and increased apoptosis of cortical, hippocampal, and striatal neurons (Blanco et al., 2014). In patients, loss of function mutations in NSUN2 are linked to a disorder characterized by intellectual disability, as well as microcephaly and cerebellar atrophy (Abbasi-Moheb et al., 2012; Komara et al., 2015; Martinez et al., 2012). NSUN2 catalyzes a widespread tRNA modification, cytosine-5 methylation (m5C), which decreases the affinity of ANG for tRNAs. The loss of m5C methylation in patient fibroblasts or Nsun2-deficient mice leads to an accumulation of 5’ tRNA halves. Treatment of Nsun2-deficient mice with ANG inhibitors reduced stress granule formation and rescued neuron loss, suggesting that ANG-induced tRNA fragmentation may play a key role in the pathogenesis. These studies, together with the potential role of ANG in ALS and PD, suggest that ANG activity and substrate recognition must be exquisitely balanced in neurons, and any disruption can have detrimental consequences.
Loss of a Single tRNA Gene: A Tipping Point for Neurodegeneration
As defective tRNA processing may affect levels of both tRNA fragments and mature tRNAs, the contribution of each to pathogenesis remains unclear. The recent discovery of a point mutation in a nuclear encoded tRNA in an inbred mouse strain has helped shed light on how changes in tRNA levels affect translation elongation (Ishimura et al., 2014). Ishimura et al. discovered that the commonly used strain, C57BL/6J (B6J), has a spontaneous single nucleotide mutation (C50T) in the T-stem of an arginine tRNA gene, n-Tr20 (n-Trtct5; tRNAArgUCU), that severely impairs processing of this tRNA, reducing its mature levels by about 80% (Figure 4). Mice have five tRNAArgUCU genes capable of decoding the cognate AGA codon in mRNA; however, surprisingly, n-Tr20 was specifically expressed in the brain, where it was the predominant member of this tRNA isoacceptor family. Ribosome profiling of cerebella revealed that the reduction in mature n-Tr20 in the B6J brain caused a significant increase in occupancy at arginine AGA codons, indicative of abnormal ribosome pausing. While B6J mice are healthy, the n-Tr20 mutation has devastating consequences in the presence of a second, ENU-induced, mutation in GTP-binding protein 2 (Gtpbp2). Mice that carry both mutations are profoundly ataxic, have widespread neurodegeneration in the cerebellum, cortex, hippocampus, and retina, and die by two months of age. GTPBP2 shares domain homology with a translational GTPase family that includes eukaryotic release factor eRF3 and the no-go/non-stop mRNA decay protein HBS1L. While eRF3/eRF1 acts to terminate translation at stop codons, the interaction of HBS1L with Pelota (Dom34 in yeast) has been implicated in non-canonical translation termination, and recycling ribosomes in the 3’UTR (Guydosh and Green, 2014; Pisareva et al., 2011; Shoemaker et al., 2010). GTPBP2 directly interacts with Pelota, suggesting that these proteins function as a novel ribosome-recycling complex. This hypothesis was strongly bolstered by ribosome profiling data from cerebella of the double mutant mice, in which absence of Gtpbp2 was found to dramatically increase ribosome stalling at AGA codons. Thus, while the subtle translation elongation defect caused by the reduction of n-Tr20 expression in B6J mice is normally compensated for by GTPBP2, in its absence, prolonged ribosome stalls persist and cause neurodegeneration. Deletion of the n-Tr20 gene, resulting in a complete absence of mature n-Tr20 tRNA, severely exacerbated the phenotype of the Gtpbp2-/-mice, resulting in neonatal lethality and suggesting that the residual amount of mature n-Tr20 in B6J mice is sufficient to allow postnatal survival of the double mutants (Ishimura et al., 2016). Although Gtpbp2 is ubiquitously expressed, the phenotype in double mutants is confined to the nervous system, indicating that the brain-specific expression pattern of n-Tr20 may underlie the specificity of disease. Orthologues of GTPBP2 and n-Tr20 are present in humans, and a recent study linked a mutation in GTPBP2 to a family with cerebellar atrophy, retinal degeneration, and intellectual disability (Jaberi et al., 2016). While disruption of the ribosome recycling protein GTPBP2 causes devastating neurodegeneration in both humans and mouse, as of yet, there are no known neurological disorders associated with defects in canonical cytoplasmic translational termination.
Figure 4. Ribosome stalling-mediated neurodegeneration.
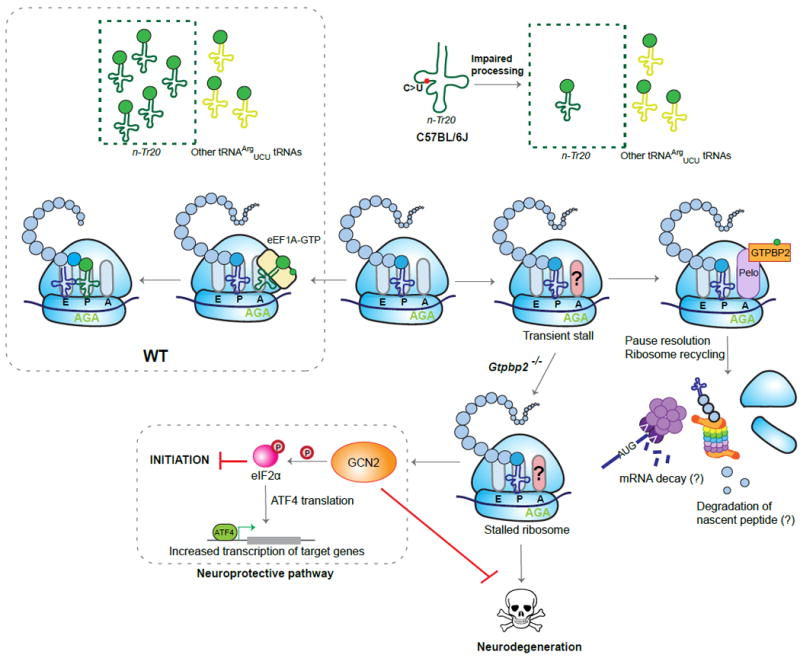
A processing mutation in n-Tr20, an arginine tRNA gene, in C57BL/6J mice significantly reduces the pool of tRNAArgUCU available for translation. This reduction leads to increased ribosome pausing on cognate AGA codons. GTPBP2 and Pelota play a role in the resolution of these paused ribosomes, and ribosome recycling may be accompanied by the degradation of the nascent protein and of the mRNA. In the absence of GTPBP2, ribosome pausing at the AGA codons is not resolved, leading to neurodegeneration. Ribosome stalling activates the integrated stress response (ISR) via the eIF2a kinase GCN2, and neurodegeneration is exacerbated in the absence of ISR activation.
Clearly, failure to recycle stalled ribosomes in mammalian neurons leads to neurodegeneration; however, the underlying molecular mechanism and signaling pathways that lead to cell death remain unknown (Ishimura et al., 2016). In investigating this question, Ishimura et al. discovered an exciting link between the dysfunction of translational elongation and regulation of initiation. Analysis of the cerebellum from mice with mutations in both Gtpbp2 and n-Tr20 prior to the onset of neurodegeneration detected increased levels of phosphorylated eIF2α and significant upregulation of genes regulated by ATF4, indicating that the ISR is activated. Genetic experiments established that the ISR was dependent on GCN2, which has been shown to be activated by uncharged tRNA (Dong et al., 2000). However, in this mouse model, activation of GCN2 occurs independently of increases in levels of uncharged tRNA, and instead, ribosome stalling itself may activate this kinase through an as of yet unknown mechanism. Intriguingly, activation of the ISR is neuroprotective, and in its absence, neurodegeneration is more widespread and accelerated.
Defects in the Degradation of Aberrant Nascent Polypeptides
Ribosomes that stall during translation elongation, and cannot proceed to canonical termination, must either be recycled in order to re-enter the cellular pool, or targeted for degradation. In addition, the mRNA transcript, associated tRNAs, and the nascent polypeptide chain must be disassembled and degraded. The exciting topic of mRNA surveillance and ribosome quality control has been the subject of several recent reviews, and will not be discussed here (Brandman and Hegde, 2016; He and Jacobson, 2015; Joazeiro, 2017). Failure to dissociate the stalled ribosomal complex can deplete the translational machinery, and this problem may be especially pronounced during local translation in neurons where this machinery is extremely limited. Emphasizing the importance of ribosome quality control in neuronal homeostasis, a recessive mutation in listerin (Ltn1), which encodes an E3 ubiquitin ligase, leads to neurodegeneration and motor dysfunction in mice (Chu et al., 2009). Listerin specifically ubiquitinates the nascent peptide chains of stalled ribosomes after ribosome splitting, and loss of Ltn1 in yeast results in the aggregation of stalled nascent polypeptides (Bengtson and Joazeiro, 2010; Choe et al., 2016; Shao et al., 2013). Thus, neurons are extremely vulnerable to disruption at every stage of mRNA translation, from initiation to ribosome recycling.
Conclusions and perspectives
Accumulating molecular, cellular, and behavioral evidence points to the delicate regulation of mRNA translation as a key pathway underlying neuronal homeostasis. Recent advances in technology and new experimental approaches have allowed us to begin to grasp the variety of molecular mechanisms that fine-tune translation, the necessity of which is highlighted by the myriad of human neurodegenerative disorders linked to mutations in translational machinery. However, despite our rapidly improving knowledge, many questions remain unanswered.
Although mRNA translation is a fundamental process in all cells, disruption of translational machinery seems to preferentially result in neurological and neurodegenerative disorders. In some cases, the analysis of mutations in what were thought to be ubiquitously expressed translation factors has led to the discovery of novel spatiotemporal regulation of these genes. However, these findings raise additional questions regarding the role for such regulation. Recent studies suggest that heterogeneity in the translational machinery, such as that observed for ribosomal proteins, may influence cell type- or developmental stage-specific translation (reviewed in Dinman, 2016; Xue and Barna, 2012). In addition, growing evidence indicates that many components of the translational machinery, including tRNA synthetases and tRNAs themselves, have a wide range of cellular functions (reviewed in Guo and Schimmel, 2013; Schimmel, 2017). An exploration of the canonical and non-canonical functions of these molecules is essential to our understanding of the phenotypes resulting from genetic mutations in translation factors.
Genetic modifiers are a particularly promising explanation for both tissue specificity and the variable penetrance of disease-linked alleles in human patients. For instance, loss of the ribosome recycling protein GTPBP2 only causes neurodegeneration in the mouse in the presence of a second mutation in a brain-specific arginine tRNA gene. This finding also demonstrates that far from being static adaptors in the translational machinery, tRNAs can play an essential role in fine-tuning translation. Indeed, synonymous or silent single nucleotide polymorphisms (sSNPs) have been linked to numerous human disorders, including neurological disease (reviewed in Sauna and Kimchi-Sarfaty, 2011), and a recent study suggests that the effect of sSNPs can be linked to the availability of the cognate tRNA (Kirchner et al., 2017). Similarly, frameshifting along an expanded poly(CAG) HTT reporter could be modulated by the expression levels of tRNAGlnCUG, as well as tRNAAlaUGC, the cognate tRNA for the frameshifted repeat (Girstmair et al., 2013). While no mutations in nuclear-encoded tRNAs have been linked to human disease as of yet, the possibility that polymorphisms in tRNA genes and their varying expression could modulate the penetrance of mutations in protein-coding genes and play a role in the genotype-phenotype relationship is intriguing.
Neurons may also be particularly susceptible to defects in proteostasis. These polarized cells require rapid and local protein synthesis for synaptic plasticity. Translation at the dendrites requires coordinated transport of the translational machinery such as tRNAs, ribosomes, and other factors as well as the mRNAs themselves. Could the supply of these factors be rate-limiting at the synapse, making translation defects in the circumscribed environment of a dendritic spine particularly devastating for synaptic function and ultimately neuronal survival? Postmitotic cells with high metabolic demands, such as neurons and cardiomyocytes, may be particularly sensitive to subtle translation defects. Indeed, protein aggregates, including those induced by altered translational fidelity, are observed in both neurodegenerative and cardiac disorders (reviewed in Willis and Patterson, 2013).
The modulation of protein synthesis plays a key role in aging, and disruption of translation regulation can alter lifespan in various species (reviewed in Gonskikh and Polacek, 2017). What translational changes occur in neurons during aging, and how distinct are these from those observed in neurodegenerative disease? Furthermore, while translational disruptions such as mistranslation and ribosomal frameshifting and pausing underlie numerous neurodegenerative conditions, growing evidence suggests that these processes also occur at low levels in healthy cells where they may serve a regulatory function (reviewed in Pan, 2013; Schwartz and Pan, 2017). Whether newly discovered translational events, such as RAN translation, are truly “non-canonical” or play a role in normal cellular function remains to be seen.
Acknowledgments
This work was supported by National Institutes of Health grants NS094637 and NS096600. S.L.A. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbasi-Moheb L, Mertel S, Gonsior M, Nouri-Vahid L, Kahrizi K, Cirak S, Wieczorek D, Motazacker MM, Esmaeeli-Nieh S, Cremer K, et al. Mutations in NSUN2 cause autosomal- Recessive intellectual disability. Am J Hum Genet. 2012;90:847–855. doi: 10.1016/j.ajhg.2012.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott CM, Newbery HJ, Squires CE, Brownstein D, Griffiths LA, Soares DC. eEF1A2 and neuronal degeneration. Biochem Soc Trans. 2009;37:1293–1297. doi: 10.1042/BST0371293. [DOI] [PubMed] [Google Scholar]
- Abbott JA, Francklyn CS, Robey-Bond SM. Transfer RNA and human disease. Front Genet. 2014;5:158. doi: 10.3389/fgene.2014.00158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreou AZ, Harms U, Klostermeier D. eIF4B stimulates eIF4A ATPase and unwinding activities by direct interaction through its 7-repeats region. RNA Biol. 2017;14:113–123. doi: 10.1080/15476286.2016.1259782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aparicio-Erriu IM, Prehn JHM. Molecular Mechanisms in Amyotrophic Lateral Sclerosis: The Role of Angiogenin, a Secreted RNase. Front Neurosci. 2012;6:167. doi: 10.3389/fnins.2012.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronin N, Chase K, Young C, Sapp E, Schwarz C, Matta N, Kornreich R, Landwehrmeyer B, Bird E, Beal MF. CAG expansion affects the expression of mutant Huntingtin in the Huntington’s disease brain. Neuron. 1995;15:1193–1201. doi: 10.1016/0896-6273(95)90106-x. [DOI] [PubMed] [Google Scholar]
- Ash PEA, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus-Hernandez M, van Blitterswijk MM, Jansen-West K, Paul JW, Rademakers R, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77:639–646. doi: 10.1016/j.neuron.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azmanov DN, Siira SJ, Chamova T, Kaprelyan A, Guergueltcheva V, Shearwood AMJ, Liu G, Morar B, Rackham O, Bynevelt M, et al. Transcriptome-wide effects of a POLR3A gene mutation in patients with an unusual phenotype of striatal involvement. Hum Mol Genet. 2016;25:4302–4314. doi: 10.1093/hmg/ddw263. [DOI] [PubMed] [Google Scholar]
- Bando Y, Onuki R, Katayama T, Manabe T, Kudo T, Taira K, Tohyama M. Double-strand RNA dependent protein kinase (PKR) is involved in the extrastriatal degeneration in Parkinson’s disease and Huntington’s disease. Neurochem Int. 2005;46:11–18. doi: 10.1016/j.neuint.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Bañez-Coronel M, Ayhan F, Tarabochia AD, Zu T, Perez BA, Tusi SK, Pletnikova O, Borchelt DR, Ross CA, Margolis RL, et al. RAN Translation in Huntington Disease. Neuron. 2015;88:667–677. doi: 10.1016/j.neuron.2015.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa C, Peixeiro I, Romão L. Gene expression regulation by upstream open reading frames and human disease. PLoS Genet. 2013;9:e1003529. doi: 10.1371/journal.pgen.1003529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckelman BC, Day S, Zhou X, Donohue M, Gouras GK, Klann E, Keene CD, Ma T. Dysregulation of Elongation Factor 1A Expression is Correlated with Synaptic Plasticity Impairments in Alzheimer’s Disease. J Alzheimers Dis. 2016a;54:669–678. doi: 10.3233/JAD-160036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckelman BC, Zhou X, Keene CD, Ma T. Impaired Eukaryotic Elongation Factor 1A Expression in Alzheimer’s Disease. Neurodegener Dis. 2016b;16:39–43. doi: 10.1159/000438925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bednářová A, Hanna M, Durham I, VanCleave T, England A, Chaudhuri A, Krishnan N. Lost in Translation: Defects in Transfer RNA Modifications and Neurological Disorders. Front Mol Neurosci. 2017;10:1–8. doi: 10.3389/fnmol.2017.00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtson MH, Joazeiro CAP. Role of a ribosome-associated E3 ubiquitin ligase in protein quality control. Nature. 2010;467:470–473. doi: 10.1038/nature09371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard G, Chouery E, Putorti ML, Tétreault M, Takanohashi A, Carosso G, Clément I, Boespflug-Tanguy O, Rodriguez D, Delague V, et al. Mutations of POLR3A encoding a catalytic subunit of RNA polymerase pol III cause a recessive hypomyelinating leukodystrophy. Am J Hum Genet. 2011;89:415–423. doi: 10.1016/j.ajhg.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierhals T, Korenke GC, Uyanik G, Kutsche K. Pontocerebellar hypoplasia type 2 and TSEN2: review of the literature and two novel mutations. Eur J Med Genet. 2013;56:325–330. doi: 10.1016/j.ejmg.2013.03.009. [DOI] [PubMed] [Google Scholar]
- Blanco S, Dietmann S, Flores JV, Hussain S, Kutter C, Humphreys P, Lukk M, Lombard P, Treps L, Popis M, et al. Aberrant methylation of tRNAs links cellular stress to neuro-developmental disorders. EMBO J. 2014;33:2020–2039. doi: 10.15252/embj.201489282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boczonadi V, Horvath R. Mitochondria: impaired mitochondrial translation in human disease. Int J Biochem Cell Biol. 2014;48:77–84. doi: 10.1016/j.biocel.2013.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeynaems S, Bogaert E, Michiels E, Gijselinck I, Sieben A, Jovičić A, De Baets G, Scheveneels W, Steyaert J, Cuijt I, et al. Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci Rep. 2016;6:20877. doi: 10.1038/srep20877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandman O, Hegde RS. Ribosome-associated protein quality control. Nat Struct Mol Biol. 2016;23:7–15. doi: 10.1038/nsmb.3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breuss MW, Sultan T, James KN, Rosti RO, Scott E, Musaev D, Furia B, Reis A, Sticht H, Al-Owain M, et al. Autosomal-Recessive Mutations in the tRNA Splicing Endonuclease Subunit TSEN15 Cause Pontocerebellar Hypoplasia and Progressive Microcephaly. Am J Hum Genet. 2016;99:228–235. doi: 10.1016/j.ajhg.2016.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffington SA, Huang W, Costa-Mattioli M. Translational control in synaptic plasticity and cognitive dysfunction. Annu Rev Neurosci. 2014;37:17–38. doi: 10.1146/annurev-neuro-071013-014100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugiani M, Boor I, Powers JM, Scheper GC, van der Knaap MS. Leukoencephalopathy with vanishing white matter: a review. J Neuropathol Exp Neurol. 2010;69:987–996. doi: 10.1097/NEN.0b013e3181f2eafa. [DOI] [PubMed] [Google Scholar]
- Carr-Schmid A, Durko N, Cavallius J, Merrick WC, Kinzy TG. Mutations in a GTP-binding motif of eukaryotic elongation factor 1A reduce both translational fidelity and the requirement for nucleotide exchange. J Biol Chem. 1999;274:30297–30302. doi: 10.1074/jbc.274.42.30297. [DOI] [PubMed] [Google Scholar]
- Chakraborty PK, Schmitz-Abe K, Kennedy EK, Mamady H, Naas T, Durie D, Campagna DR, Lau A, Sendamarai AK, Wiseman DH, et al. Mutations in TRNT1 cause congenital sideroblastic anemia with immunodeficiency, fevers, and developmental delay (SIFD) Blood. 2014;124:2867–2871. doi: 10.1182/blood-2014-08-591370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers DM, Peters J, Abbott CM. The lethal mutation of the mouse wasted (wst) is a deletion that abolishes expression of a tissue-specific isoform of translation elongation factor 1, encoded by the Eef1a2 gene. Proc Natl Acad Sci. 1998;95:4463–4468. doi: 10.1073/pnas.95.8.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang RCC, Suen KC, Ma CH, Elyaman W, Ng HK, Hugon J. Involvement of double-stranded RNA-dependent protein kinase and phosphorylation of eukaryotic initiation factor-2alpha in neuronal degeneration. J Neurochem. 2002a;83:1215–1225. doi: 10.1046/j.1471-4159.2002.01237.x. [DOI] [PubMed] [Google Scholar]
- Chang RCC, Wong AKY, Ng HK, Hugon J. Phosphorylation of eukaryotic initiation factor-2alpha (eIF2alpha) is associated with neuronal degeneration in Alzheimer’s disease. Neuroreport. 2002b;13:2429–2432. doi: 10.1097/00001756-200212200-00011. [DOI] [PubMed] [Google Scholar]
- Chen ML, Wu RM, Tai CH, Lin CH. Mutational analysis of Angiogenin gene in Parkinson’s disease. PLoS One. 2014;9:9–11. doi: 10.1371/journal.pone.0112661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe YJ, Park SH, Hassemer T, Körner R, Vincenz-Donnelly L, Hayer-Hartl M, Hartl FU. Failure of RQC machinery causes protein aggregation and proteotoxic stress. Nature. 2016;531:191–195. doi: 10.1038/nature16973. [DOI] [PubMed] [Google Scholar]
- Chu J, Hong NA, Masuda CA, Jenkins BV, Nelms KA, Goodnow CC, Glynne RJ, Wu H, Masliah E, Joazeiro CAP, et al. A mouse forward genetics screen identifies LISTERIN as an E3 ubiquitin ligase involved in neurodegeneration. Proc Natl Acad Sci U S A. 2009;106:2097–2103. doi: 10.1073/pnas.0812819106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciesiolka A, Jazurek M, Drazkowska K, Krzyzosiak WJ. Structural Characteristics of Simple RNA Repeats Associated with Disease and their Deleterious Protein Interactions. Front Cell Neurosci. 2017;11:97. doi: 10.3389/fncel.2017.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrado L, Battistini S, Penco S, Bergamaschi L, Testa L, Ricci C, Giannini F, Greco G, Patrosso MC, Pileggi S, et al. Variations in the coding and regulatory sequences of the angiogenin (ANG) gene are not associated to ALS (amyotrophic lateral sclerosis) in the Italian population. J Neurol Sci. 2007;258:123–127. doi: 10.1016/j.jns.2007.03.009. [DOI] [PubMed] [Google Scholar]
- Costa-Mattioli M, Gobert D, Harding H, Herdy B, Azzi M, Bruno M, Bidinosti M, Ben Mamou C, Marcinkiewicz E, Yoshida M, et al. Translational control of hippocampal synaptic plasticity and memory by the eIF2alpha kinase GCN2. Nature. 2005;436:1166–1173. doi: 10.1038/nature03897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, Gobert D, Stern E, Gamache K, Colina R, Cuello C, Sossin W, Kaufman R, Pelletier J, Rosenblum K, et al. eIF2alpha phosphorylation bidirectionally regulates the switch from short- to long-term synaptic plasticity and memory. Cell. 2007;129:195–206. doi: 10.1016/j.cell.2007.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czech A, Wende S, Mörl M, Pan T, Ignatova Z. Reversible and rapid transfer-RNA deactivation as a mechanism of translational repression in stress. PLoS Genet. 2013;9:e1003767. doi: 10.1371/journal.pgen.1003767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das I, Krzyzosiak A, Schneider K, Wrabetz L, D’Antonio M, Barry N, Sigurdardottir A, Bertolotti A. Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science. 2015;348:239–242. doi: 10.1126/science.aaa4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson YS, Barker H, Robinson AC, Thompson JC, Harris J, Troakes C, Smith B, Al-Saraj S, Shaw C, Rollinson S, et al. Brain distribution of dipeptide repeat proteins in frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol Commun. 2014;2:70. doi: 10.1186/2051-5960-2-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies JE, Rubinsztein DC. Polyalanine and polyserine frameshift products in Huntington’s disease. J Med Genet. 2006;43:893–896. doi: 10.1136/jmg.2006.044222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLuca AP, Whitmore SS, Barnes J, Sharma TP, Westfall TA, Anthony Scott C, Weed MC, Wiley JS, Wiley LA, Johnston RM, et al. Hypomorphic mutations in TRNT1 cause retinitis pigmentosa with erythrocytic microcytosis. Hum Mol Genet. 2016;25:44–56. doi: 10.1093/hmg/ddv446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis MD, Jefferson LS, Kimball SR. Role of p70S6K1-mediated phosphorylation of eIF4B and PDCD4 proteins in the regulation of protein synthesis. J Biol Chem. 2012;287:42890–42899. doi: 10.1074/jbc.M112.404822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dever TE, Green R. The elongation, termination, and recycling phases of translation in eukaryotes. Cold Spring Harb Perspect Biol. 2012;4:a013706. doi: 10.1101/cshperspect.a013706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi L, Ohno M. Deletion of the eIF2α Kinase GCN2 fails to rescue the memory decline associated with Alzheimer’s disease. PLoS One. 2013;8:e77335. doi: 10.1371/journal.pone.0077335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi L, Ohno M. PERK mediates eIF2α phosphorylation responsible for BACE1 elevation, CREB dysfunction and neurodegeneration in a mouse model of Alzheimer’s disease. Neurobiol Aging. 2014;35:2272–2281. doi: 10.1016/j.neurobiolaging.2014.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinman JD. Pathways to Specialized Ribosomes: The Brussels Lecture. J Mol Biol. 2016;428:2186–2194. doi: 10.1016/j.jmb.2015.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Qiu H, Garcia-Barrio M, Anderson J, Hinnebusch AG. Uncharged tRNA activates GCN2 by displacing the protein kinase moiety from a bipartite tRNA-binding domain. Mol Cell. 2000;6:269–279. doi: 10.1016/s1097-2765(00)00028-9. [DOI] [PubMed] [Google Scholar]
- Edvardson S, Prunetti L, Arraf A, Haas D, Bacusmo JM, Hu JF, Ta-Shma A, Dedon PC, de Crécy-Lagard V, Elpeleg O. tRNA N6-adenosine threonylcarbamoyltransferase defect due to KAE1/TCS3 (OSGEP) mutation manifest by neurodegeneration and renal tubulopathy. Eur J Hum Genet. 2017;25:545–551. doi: 10.1038/ejhg.2017.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Es MA, Schelhaas HJ, Van Vught PWJ, Ticozzi N, Andersen PM, Groen EJN, Schulte C, Blauw HM, Koppers M, Diekstra FP, et al. Angiogenin variants in Parkinson disease and amyotrophic lateral sclerosis. Ann Neurol. 2011;70:964–973. doi: 10.1002/ana.22611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farabaugh PJ, Vimaladithan A. Effect of frameshift-inducing mutants of elongation factor 1alpha on programmed +1 frameshifting in yeast. RNA. 1998;4:38–46. [PMC free article] [PubMed] [Google Scholar]
- Fernández-Santiago R, Hoenig S, Lichtner P, Sperfeld AD, Sharma M, Berg D, Weichenrieder O, Illig T, Eger K, Meyer T, et al. Identification of novel Angiogenin (ANG) gene missense variants in German patients with amyotrophic lateral sclerosis. J Neurol. 2009;256:1337–1342. doi: 10.1007/s00415-009-5124-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee KH, Badders N, Valentine M, Miller BL, Wong PC, et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature. 2015;525:129–133. doi: 10.1038/nature14974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H, Feng J, Liu Q, Sun F, Tie Y, Zhu J, Xing R, Sun Z, Zheng X. Stress induces tRNA cleavage by angiogenin in mammalian cells. FEBS Lett. 2009;583:437–442. doi: 10.1016/j.febslet.2008.12.043. [DOI] [PubMed] [Google Scholar]
- Garcia-Esparcia P, Hernández-Ortega K, Koneti A, Gil L, Delgado-Morales R, Castaño E, Carmona M, Ferrer I. Altered machinery of protein synthesis is region- and stage-dependent and is associated with a-synuclein oligomers in Parkinson’s disease. Acta Neuropathol Commun. 2015;3:76. doi: 10.1186/s40478-015-0257-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Esparcia P, Sideris-Lampretsas G, Hernandez-Ortega K, Grau-Rivera O, Sklaviadis T, Gelpi E, Ferrer I. Altered mechanisms of protein synthesis in frontal cortex in Alzheimer disease and a mouse model. Am J Neurodegener Dis. 2017;6:15–25. [PMC free article] [PubMed] [Google Scholar]
- Gaspar C, Jannatipour M, Dion P, Laganière J, Sequeiros J, Brais B, Rouleau GA. CAG tract of MJD-1 may be prone to frameshifts causing polyalanine accumulation. Hum Mol Genet. 2000;9:1957–1966. doi: 10.1093/hmg/9.13.1957. [DOI] [PubMed] [Google Scholar]
- Gendron TF, Bieniek KF, Zhang Y-J, Jansen-West K, Ash PEA, Caulfield T, Daughrity L, Dunmore JH, Castanedes-Casey M, Chew J, et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 2013;126:829–844. doi: 10.1007/s00401-013-1192-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingold H, Tehler D, Christoffersen NR, Nielsen MM, Asmar F, Kooistra SM, Christophersen NS, Christensen LL, Borre M, Sørensen KD, et al. A Dual Program for Translation Regulation in Cellular Proliferation and Differentiation. Cell. 2014;158:1281–1292. doi: 10.1016/j.cell.2014.08.011. [DOI] [PubMed] [Google Scholar]
- Girstmair H, Saffert P, Rode S, Czech A, Holland G, Bannert N, Ignatova Z. Depletion of cognate charged transfer RNA causes translational frameshifting within the expanded CAG stretch in huntingtin. Cell Rep. 2013;3:148–159. doi: 10.1016/j.celrep.2012.12.019. [DOI] [PubMed] [Google Scholar]
- Gomez-Deza J, Lee Y-B, Troakes C, Nolan M, Al-Sarraj S, Gallo J-M, Shaw CE. Dipeptide repeat protein inclusions are rare in the spinal cord and almost absent from motor neurons in C9ORF72 mutant amyotrophic lateral sclerosis and are unlikely to cause their degeneration. Acta Neuropathol Commun. 2015;3:38. doi: 10.1186/s40478-015-0218-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonskikh Y, Polacek N. Alterations of the translation apparatus during aging and stress response. Mech Ageing Dev. 2017:0–1. doi: 10.1016/j.mad.2017.04.003. [DOI] [PubMed] [Google Scholar]
- Goodarzi H, Nguyen HCB, Zhang S, Dill BD, Molina H, Tavazoie SF. Modulated Expression of Specific tRNAs Drives Gene Expression and Cancer Progression. Cell. 2016;165:1416–1427. doi: 10.1016/j.cell.2016.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodenbour JM, Pan T. Diversity of tRNA genes in eukaryotes. Nucleic Acids Res. 2006;34:6137–6146. doi: 10.1093/nar/gkl725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graille M, Séraphin B. Surveillance pathways rescuing eukaryotic ribosomes lost in translation. Nat Rev Mol Cell Biol. 2012;13:727–735. doi: 10.1038/nrm3457. [DOI] [PubMed] [Google Scholar]
- Greenway MJ, Andersen PM, Russ C, Ennis S, Cashman S, Donaghy C, Patterson V, Swingler R, Kieran D, Prehn J, et al. ANG mutations segregate with familial and “sporadic” amyotrophic lateral sclerosis. Nat Genet. 2006;38:411–413. doi: 10.1038/ng1742. [DOI] [PubMed] [Google Scholar]
- Griesche N, Schilling J, Weber S, Rohm M, Pesch V, Matthes F, Auburger G, Krauss S. Regulation of mRNA Translation by MID1: A Common Mechanism of Expanded CAG Repeat RNAs. Front Cell Neurosci. 2016;10:226. doi: 10.3389/fncel.2016.00226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin LB, Sakaguchi R, McGuigan D, Gonzalez MA, Searby C, Züchner S, Hou YM, Antonellis A. Impaired function is a common feature of neuropathy-associated glycyl-tRNA synthetase mutations. Hum Mutat. 2014;35:1363–1371. doi: 10.1002/humu.22681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JU, Bartel DP. RNA G-quadruplexes are globally unfolded in eukaryotic cells and depleted in bacteria. Science. 2016;353:aaf5371–aaf5371. doi: 10.1126/science.aaf5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M, Schimmel P. Structural analyses clarify the complex control of mistranslation by tRNA synthetases. Curr Opin Struct Biol. 2012;22:119–126. doi: 10.1016/j.sbi.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M, Schimmel P. Essential nontranslational functions of tRNA synthetases. Nat Chem Biol. 2013;9:145–153. doi: 10.1038/nchembio.1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R, Lan M, Mojsilovic-Petrovic J, Choi WH, Safren N, Barmada S, Lee MJ, Kalb R. The Proline/Arginine Dipeptide from Hexanucleotide Repeat Expanded C9ORF72 Inhibits the Proteasome. eNeuro. 2017;4 doi: 10.1523/ENEURO.0249-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guydosh NR, Green R. Dom34 rescues ribosomes in 3’ untranslated regions. Cell. 2014;156:950–962. doi: 10.1016/j.cell.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghighat A, Mader S, Pause A, Sonenberg N. Repression of cap-dependent translation by 4E-binding protein 1: competition with p220 for binding to eukaryotic initiation factor-4E. EMBO J. 1995;14:5701–5709. doi: 10.1002/j.1460-2075.1995.tb00257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday M, Radford H, Sekine Y, Moreno J, Verity N, le Quesne J, Ortori CA, Barrett DA, Fromont C, Fischer PM, et al. Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell Death Dis. 2015;6:e1672. doi: 10.1038/cddis.2015.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday M, Radford H, Zents KAM, Molloy C, Moreno JA, Verity NC, Smith E, Ortori CA, Barrett DA, Bushell M, et al. Repurposed drugs targeting eIF2alpha-P-mediated translational repression prevent neurodegeneration in mice. Brain. 2017;140:1768–1783. doi: 10.1093/brain/awx074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada T, Weitzer S, Mair B, Bernreuther C, Wainger BJ, Ichida J, Hanada R, Orthofer M, Cronin SJ, Komnenovic V, et al. CLP1 links tRNA metabolism to progressive motor-neuron loss. Nature. 2013;495:474–480. doi: 10.1038/nature11923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, Jacobson A. Nonsense-Mediated mRNA Decay: Degradation of Defective Transcripts Is Only Part of the Story. Annu Rev Genet. 2015;49:339–366. doi: 10.1146/annurev-genet-112414-054639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W, Bai G, Zhou H, Wei N, White NM, Lauer J, Liu H, Shi Y, Dumitru CD, Lettieri K, et al. CMT2D neuropathy is linked to the neomorphic binding activity of glycyl-tRNA synthetase. Nature. 2015;526:710–714. doi: 10.1038/nature15510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekman KE, Yu GY, Brown CD, Zhu H, Du X, Gervin K, Undlien DE, Peterson A, Stevanin G, Clark HB, et al. A conserved eEF2 coding variant in SCA26 leads to loss of translational fidelity and increased susceptibility to proteostatic insult. Hum Mol Genet. 2012;21:5472–5483. doi: 10.1093/hmg/dds392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Ortega K, Garcia-Esparcia P, Gil L, Lucas JJ, Ferrer I. Altered Machinery of Protein Synthesis in Alzheimer’s: From the Nucleolus to the Ribosome. Brain Pathol. 2016;26:593–605. doi: 10.1111/bpa.12335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch AG. The scanning mechanism of eukaryotic translation initiation. Annu Rev Biochem. 2014;83:779–812. doi: 10.1146/annurev-biochem-060713-035802. [DOI] [PubMed] [Google Scholar]
- Holz MK, Ballif BA, Gygi SP, Blenis J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell. 2005;123:569–580. doi: 10.1016/j.cell.2005.10.024. [DOI] [PubMed] [Google Scholar]
- Hoozemans JJM, van Haastert ES, Eikelenboom P, de Vos RAI, Rozemuller JM, Scheper W. Activation of the unfolded protein response in Parkinson’s disease. Biochem Biophys Res Commun. 2007;354:707–711. doi: 10.1016/j.bbrc.2007.01.043. [DOI] [PubMed] [Google Scholar]
- Hoozemans JJM, van Haastert ES, Nijholt DAT, Rozemuller AJM, Eikelenboom P, Scheper W. The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am J Pathol. 2009;174:1241–1251. doi: 10.2353/ajpath.2009.080814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoozemans JJM, van Haastert ES, Nijholt DAT, Rozemuller AJM, Scheper W. Activation of the unfolded protein response is an early event in Alzheimer’s and Parkinson’s disease. Neurodegener Dis. 2012;10:212–215. doi: 10.1159/000334536. [DOI] [PubMed] [Google Scholar]
- Hu JH, Zhang H, Wagey R, Krieger C, Pelech SL. Protein kinase and protein phosphatase expression in amyotrophic lateral sclerosis spinal cord. J Neurochem. 2003;85:432–442. doi: 10.1046/j.1471-4159.2003.01670.x. [DOI] [PubMed] [Google Scholar]
- Hughes D, Atkins JF, Thompson S. Mutants of elongation factor Tu promote ribosomal frameshifting and nonsense readthrough. EMBO J. 1987;6:4235–4239. doi: 10.1002/j.1460-2075.1987.tb02772.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilieva EV, Ayala V, Jové M, Dalfó E, Cacabelos D, Povedano M, Bellmunt MJ, Ferrer I, Pamplona R, Portero-Otín M. Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain. 2007;130:3111–3123. doi: 10.1093/brain/awm190. [DOI] [PubMed] [Google Scholar]
- Inui T, Kobayashi S, Ashikari Y, Sato R, Endo W, Uematsu M, Oba H, Saitsu H, Matsumoto N, Kure S, et al. Two cases of early-onset myoclonic seizures with continuous parietal delta activity caused by EEF1A2 mutations. Brain Dev. 2016;38:520–524. doi: 10.1016/j.braindev.2015.11.003. [DOI] [PubMed] [Google Scholar]
- Ishiguro T, Sato N, Ueyama M, Fujikake N, Sellier C, Kanegami A, Tokuda E, Zamiri B, Gall-Duncan T, Mirceta M, et al. Regulatory Role of RNA Chaperone TDP-43 for RNA Misfolding and Repeat-Associated Translation in SCA31. Neuron. 2017;94:108–124. e7. doi: 10.1016/j.neuron.2017.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimura R, Nagy G, Dotu I, Zhou H, Yang X-L, Schimmel P, Senju S, Nishimura Y, Chuang JH, Ackerman SL. RNA function. Ribosome stalling induced by mutation of a CNS-specific tRNA causes neurodegeneration. Science. 2014;345:455–459. doi: 10.1126/science.1249749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimura R, Nagy G, Dotu I, Chuang JH, Ackerman SL. Activation of GCN2 kinase by ribosome stalling links translation elongation with translation initiation. Elife. 2016;5:1–22. doi: 10.7554/eLife.14295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov P, Emara MM, Villen J, Gygi SP, Anderson P. Angiogenin-induced tRNA fragments inhibit translation initiation. Mol Cell. 2011;43:613–623. doi: 10.1016/j.molcel.2011.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaberi E, Rohani M, Shahidi GA, Nafissi S, Arefian E, Soleimani M, Rasooli P, Ahmadieh H, Daftarian N, KaramiNejadRanjbar M, et al. Identification of mutation in GTPBP2 in patients of a family with neurodegeneration accompanied by iron deposition in the brain. Neurobiol Aging. 2016;38216:e11–216. e18. doi: 10.1016/j.neurobiolaging.2015.10.034. [DOI] [PubMed] [Google Scholar]
- Jackson RJ, Hellen CUT, Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol. 2010;11:113–127. doi: 10.1038/nrm2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RJ, Hellen CUT, Pestova TV. Termination and post-termination events in eukaryotic translation. Adv Protein Chem Struct Biol. 2012;86:45–93. doi: 10.1016/B978-0-12-386497-0.00002-5. [DOI] [PubMed] [Google Scholar]
- Jennings MD, Pavitt GD. A new function and complexity for protein translation initiation factor eIF2B. Cell Cycle. 2014;13:2660–2665. doi: 10.4161/15384101.2014.948797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joazeiro CAP. Ribosomal Stalling During Translation: Providing Substrates for Ribosome-Associated Protein Quality Control. Annu Rev Cell Dev Biol. 2017:1–26. doi: 10.1146/annurev-cellbio-111315-125249. [DOI] [PubMed] [Google Scholar]
- Jovičić A, Mertens J, Boeynaems S, Bogaert E, Chai N, Yamada SB, Paul JW, Sun S, Herdy JR, Bieri G, et al. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci. 2015;18:1226–1229. doi: 10.1038/nn.4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanekura K, Yagi T, Cammack AJ, Mahadevan J, Kuroda M, Harms MB, Miller TM, Urano F. Poly-dipeptides encoded by the C9ORF72 repeats block global protein translation. Hum Mol Genet. 2016;25:1803–1813. doi: 10.1093/hmg/ddw052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaca E, Weitzer S, Pehlivan D, Shiraishi H, Gogakos T, Hanada T, Jhangiani SN, Wiszniewski W, Withers M, Campbell IM, et al. Human CLP1 Mutations Alter tRNA Biogenesis, Affecting Both Peripheral and Central Nervous System Function. Cell. 2014;157:636–650. doi: 10.1016/j.cell.2014.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse MG, Green KM, Krans A, Rodriguez CM, Linsalata AE, Goldstrohm AC, Todd PK. CGG Repeat-Associated Non-AUG Translation Utilizes a Cap-Dependent Scanning Mechanism of Initiation to Produce Toxic Proteins. Mol Cell. 2016;62:314–322. doi: 10.1016/j.molcel.2016.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieran D, Sebastia J, Greenway MJ, King MA, Connaughton D, Concannon CG, Fenner B, Hardiman O, Prehn JHM. Control of Motoneuron Survival by Angiogenin. J Neurosci. 2008;28:14056–14061. doi: 10.1523/JNEUROSCI.3399-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Raphael AR, LaDow ES, McGurk L, Weber RA, Trojanowski JQ, Lee VM, Finkbeiner S, Gitler AD, Bonini NM. Therapeutic modulation of eIF2a phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat Genet. 2014;46:152–160. doi: 10.1038/ng.2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H-S, Choi Y, Shin K-Y, Joo Y, Lee Y-K, Jung SY, Suh Y-H, Kim J-H. Swedish amyloid precursor protein mutation increases phosphorylation of eIF2alpha in vitro and in vivo. J Neurosci Res. 2007;85:1528–1537. doi: 10.1002/jnr.21267. [DOI] [PubMed] [Google Scholar]
- Kirchner S, Cai Z, Rauscher R, Kastelic N, Anding M, Czech A, Kleizen B, Ostedgaard LS, Braakman I, Sheppard DN, et al. Alteration of protein function by a silent polymorphism linked to tRNA abundance. PLoS Biol. 2017;15:e2000779. doi: 10.1371/journal.pbio.2000779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klann E, Dever TE. Biochemical mechanisms for translational regulation in synaptic plasticity. Nat Rev Neurosci. 2004;5:931–942. doi: 10.1038/nrn1557. [DOI] [PubMed] [Google Scholar]
- Kodera H, Osaka H, Iai M, Aida N, Yamashita A, Tsurusaki Y, Nakashima M, Miyake N, Saitsu H, Matsumoto N. Mutations in the glutaminyl-tRNA synthetase gene cause early-onset epileptic encephalopathy. J Hum Genet. 2015;60:97–101. doi: 10.1038/jhg.2014.103. [DOI] [PubMed] [Google Scholar]
- Köhler C, Dinekov M, Götz J. Granulovacuolar degeneration and unfolded protein response in mouse models of tauopathy and Aβ amyloidosis. Neurobiol Dis. 2014;71:169–179. doi: 10.1016/j.nbd.2014.07.006. [DOI] [PubMed] [Google Scholar]
- van Kollenburg B, van Dijk J, Garbern J, Thomas AAM, Scheper GC, Powers JM, van der Knaap MS. Glia-specific activation of all pathways of the unfolded protein response in vanishing white matter disease. J Neuropathol Exp Neurol. 2006;65:707–715. doi: 10.1097/01.jnen.0000228201.27539.50. [DOI] [PubMed] [Google Scholar]
- Komara M, Al-Shamsi AM, Ben-Salem S, Ali BR, Al-Gazali L. A Novel Single-Nucleotide Deletion (c.1020delA) in NSUN2 Causes Intellectual Disability in an Emirati Child. J Mol Neurosci. 2015;57:393–399. doi: 10.1007/s12031-015-0592-8. [DOI] [PubMed] [Google Scholar]
- Krans A, Kearse MG, Todd PK. Repeat-associated non-AUG translation from antisense CCG repeats in fragile X tremor/ataxia syndrome. Ann Neurol. 2016;80:871–881. doi: 10.1002/ana.24800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauss S, Griesche N, Jastrzebska E, Chen C, Rutschow D, Achmüller C, Dorn S, Boesch SM, Lalowski M, Wanker E, et al. Translation of HTT mRNA with expanded CAG repeats is regulated by the MID1-PP2A protein complex. Nat Commun. 2013;4:1511. doi: 10.1038/ncomms2514. [DOI] [PubMed] [Google Scholar]
- Kuzmenko A, Atkinson GC, Levitskii S, Zenkin N, Tenson T, Hauryliuk V, Kamenski P. Mitochondrial translation initiation machinery: Conservation and diversification. Biochimie. 2014;100:132–140. doi: 10.1016/j.biochi.2013.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon I, Xiang S, Kato M, Wu L, Theodoropoulos P, Wang T, Kim J, Yun J, Xie Y, McKnight SL. Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science. 2014;345:1139–1145. doi: 10.1126/science.1254917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacerda R, Menezes J, Romão L. More than just scanning: the importance of cap-independent mRNA translation initiation for cellular stress response and cancer. Cell Mol Life Sci. 2017;74:1659–1680. doi: 10.1007/s00018-016-2428-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam WWK, Millichap JJ, Soares DC, Chin R, McLellan A, FitzPatrick DR, Elmslie F, Lees MM, Schaefer GB, et al. DDD study. Novel de novo EEF1A2 missense mutations causing epilepsy and intellectual disability. Mol Genet Genomic Med. 2016;4:465–474. doi: 10.1002/mgg3.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langstrom NS, Anderson JP, Lindroos HG, Winblad B, Wallace WC. Alzheimer’s disease-associated reduction of polysomal mRNA translation. Brain Res Mol Brain Res. 1989;5:259–269. doi: 10.1016/0169-328x(89)90060-0. [DOI] [PubMed] [Google Scholar]
- Lee JW, Beebe K, Nangle LA, Jang J, Longo-Guess CM, Cook SA, Davisson MT, Sundberg JP, Schimmel P, Ackerman SL. Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature. 2006;443:50–55. doi: 10.1038/nature05096. [DOI] [PubMed] [Google Scholar]
- Lee K-H, Zhang P, Kim HJ, Mitrea DM, Sarkar M, Freibaum BD, Cika J, Coughlin M, Messing J, Molliex A, et al. C9orf72 Dipeptide Repeats Impair the Assembly, Dynamics, and Function of Membrane-Less Organelles. Cell. 2016;167:774–788. e17. doi: 10.1016/j.cell.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Wolfraim LA, Wang E. Differential expression of S1 and elongation factor-1a during rat development. J Biol Chem. 1993;268:24453–24459. [PubMed] [Google Scholar]
- Lee S, LeBlanc A, Duttaroy A, Wang E. Terminal differentiation-dependent alteration in the expression of translation elongation factor-1 alpha and its sister gene, S1, in neurons. Exp Cell Res. 1995;219:589–597. doi: 10.1006/excr.1995.1268. [DOI] [PubMed] [Google Scholar]
- Leitman J, Barak B, Benyair R, Shenkman M, Ashery U, Hartl FU, Lederkremer GZ. ER stress-induced eIF2-alpha phosphorylation underlies sensitivity of striatal neurons to pathogenic huntingtin. PLoS One. 2014;9:e90803. doi: 10.1371/journal.pone.0090803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- León K, Boulo T, Musnier A, Morales J, Gauthier C, Dupuy L, Heyne S, Backofen R, Poupon A, Cormier P, et al. Activation of a GPCR leads to eIF4G phosphorylation at the 5’ cap and to IRES-dependent translation. J Mol Endocrinol. 2014;52:373–382. doi: 10.1530/JME-14-0009. [DOI] [PubMed] [Google Scholar]
- Lewerenz J, Maher P. Basal levels of eIF2alpha phosphorylation determine cellular antioxidant status by regulating ATF4 and xCT expression. J Biol Chem. 2009;284:1106–1115. doi: 10.1074/jbc.M807325200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Alafuzoff I, Soininen H, Winblad B, Pei JJ. Levels of mTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer’s disease brain. FEBS J. 2005;272:4211–4220. doi: 10.1111/j.1742-4658.2005.04833.x. [DOI] [PubMed] [Google Scholar]
- de Ligt J, Willemsen MH, van Bon BWM, Kleefstra T, Yntema HG, Kroes T, Vulto-van Silfhout AT, Koolen DA, de Vries P, Gilissen C, et al. Diagnostic Exome Sequencing in Persons with Severe Intellectual Disability. N Engl J Med. 2012;367:1921–1929. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- Ling J, Roy H, Ibba M. Mechanism of tRNA-dependent editing in translational quality control. Proc Natl Acad Sci U S A. 2007;104:72–77. doi: 10.1073/pnas.0606272104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu E, Knutzen CA, Krauss S, Schweiger S, Chiang GG. Control of mTORC1 signaling by the Opitz syndrome protein MID1. Proc Natl Acad Sci U S A. 2011;108:8680–8685. doi: 10.1073/pnas.1100131108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Satz JS, Vo MN, Nangle LA, Schimmel P, Ackerman SL. Deficiencies in tRNA synthetase editing activity cause cardioproteinopathy. Proc Natl Acad Sci. 2014;111:17570–17575. doi: 10.1073/pnas.1420196111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes F, Barbosa M, Ameur A, Soares G, de Sá J, Dias AI, Oliveira G, Cabral P, Temudo T, Calado E, et al. Identification of novel genetic causes of Rett syndrome- like phenotypes. J Med Genet. 2016;53:190–199. doi: 10.1136/jmedgenet-2015-103568. [DOI] [PubMed] [Google Scholar]
- Lu J, Bergert M, Walther A, Suter B. Double-sieving-defective aminoacyl-tRNA synthetase causes protein mistranslation and affects cellular physiology and development. Nat Commun. 2014;5:5650. doi: 10.1038/ncomms6650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Trinh MA, Wexler AJ, Bourbon C, Gatti E, Pierre P, Cavener DR, Klann E. Suppression of eIF2a kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat Neurosci. 2013;16:1299–1305. doi: 10.1038/nn.3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Arzberger T, Kremmer E, Troost D, Lorenzl S, Mori K, Weng SM, Haass C, Kretzschmar HA, Edbauer D, et al. Dipeptide repeat protein pathology in C9ORF72 mutation cases: clinico-pathological correlations. Acta Neuropathol. 2013;126:859–879. doi: 10.1007/s00401-013-1181-y. [DOI] [PubMed] [Google Scholar]
- Mai N, Chrzanowska-Lightowlers ZMA, Lightowlers RN. The process of mammalian mitochondrial protein synthesis. Cell Tissue Res. 2017;367:5–20. doi: 10.1007/s00441-016-2456-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann DMA, Rollinson S, Robinson A, Bennion Callister J, Thompson JC, Snowden JS, Gendron T, Petrucelli L, Masuda-Suzukake M, Hasegawa M, et al. Dipeptide repeat proteins are present in the p62 positive inclusions in patients with frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol Commun. 2013;1:68. doi: 10.1186/2051-5960-1-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin I, Kim JW, Lee BD, Kang HC, Xu J-C, Jia H, Stankowski J, Kim M-S, Zhong J, Kumar M, et al. Ribosomal protein s15 phosphorylation mediates LRRK2 neurodegeneration in Parkinson’s disease. Cell. 2014;157:472–485. doi: 10.1016/j.cell.2014.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez FJ, Lee JH, Lee JE, Blanco S, Nickerson E, Gabriel S, Frye M, Al-Gazali L, Gleeson JG. Whole exome sequencing identifies a splicing mutation in NSUN2 as a cause of a Dubowitz-like syndrome. J Med Genet. 2012;49:380–385. doi: 10.1136/jmedgenet-2011-100686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateyak MK, Kinzy TG. eEF1A: Thinking outside the ribosome. J Biol Chem. 2010;285:21209–21213. doi: 10.1074/jbc.R110.113795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May S, Hornburg D, Schludi MH, Arzberger T, Rentzsch K, Schwenk BM, Grässer FA, Mori K, Kremmer E, Banzhaf-Strathmann J, et al. C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol. 2014;128:485–503. doi: 10.1007/s00401-014-1329-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin HM, Sakaguchi R, Liu C, Igarashi T, Pehlivan D, Chu K, Iyer R, Cruz P, Cherukuri PF, Hansen NF, et al. Compound heterozygosity for loss-of-function lysyl-tRNA synthetase mutations in a patient with peripheral neuropathy. Am J Hum Genet. 2010;87:560–566. doi: 10.1016/j.ajhg.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin HM, Sakaguchi R, Giblin W, NISC Comparative Sequencing Program. Wilson TE, Biesecker L, Lupski JR, Talbot K, Vance JM, Züchner S, et al. A recurrent loss-of-function alanyl-tRNA synthetase (AARS) mutation in patients with Charcot-Marie-Tooth disease type 2N (CMT2N) Hum Mutat. 2012;33:244–253. doi: 10.1002/humu.21635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meel E, Wegner DJ, Cliften P, Willing MC, White FV, Kornfeld S, Cole FS. Rare recessive loss-of-function methionyl-tRNA synthetase mutations presenting as a multi-organ phenotype. BMC Med Genet. 2013;14:106. doi: 10.1186/1471-2350-14-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrick WC. eIF4F: a retrospective. J Biol Chem. 2015;290:24091–24099. doi: 10.1074/jbc.R115.675280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Schuman R, Antonellis A. Emerging Mechanisms of Aminoacyl-tRNA Synthetase Mutations in Recessive and Dominant Human Disease. Hum Mol Genet. 2017 doi: 10.1093/hmg/ddx231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizielinska S, Grönke S, Niccoli T, Ridler CE, Clayton EL, Devoy A, Moens T, Norona FE, Woollacott IOC, Pietrzyk J, et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science. 2014;1192:1192–1194. doi: 10.1126/science.1256800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JA, Radford H, Peretti D, Steinert JR, Verity N, Martin MG, Halliday M, Morgan J, Dinsdale D, Ortori CA, et al. Sustained translational repression by eIF2a-P mediates prion neurodegeneration. Nature. 2012;485:507–511. doi: 10.1038/nature11058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, Ortori CA, Willis AE, Fischer PM, Barrett DA, et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci Transl Med. 2013;5:206ra138. doi: 10.1126/scitranslmed.3006767. [DOI] [PubMed] [Google Scholar]
- Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013a;339:1335–1338. doi: 10.1126/science.1232927. [DOI] [PubMed] [Google Scholar]
- Mori K, Arzberger T, Grässer FA, Gijselinck I, May S, Rentzsch K, Weng SM, Schludi MH, van der Zee J, Cruts M, et al. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 2013b;126:881–893. doi: 10.1007/s00401-013-1189-3. [DOI] [PubMed] [Google Scholar]
- Motley WW, Seburn KL, Nawaz MH, Miers KE, Cheng J, Antonellis A, Green ED, Talbot K, Yang X, Fischbeck KH, et al. Charcot-Marie-Tooth-linked mutant GARS is toxic to peripheral neurons independent of wild-type GARS levels. PLoS Genet. 2011;7:e1002399. doi: 10.1371/journal.pgen.1002399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouton-Liger F, Paquet C, Dumurgier J, Bouras C, Pradier L, Gray F, Hugon J. Oxidative stress increases BACE1 protein levels through activation of the PKR-eIF2a pathway. Biochim Biophys Acta. 2012;1822:885–896. doi: 10.1016/j.bbadis.2012.01.009. [DOI] [PubMed] [Google Scholar]
- Nakajima J, Okamoto N, Tohyama J, Kato M, Arai H, Funahashi O, Tsurusaki Y, Nakashima M, Kawashima H, Saitsu H, et al. De novo EEF1A2 mutations in patients with characteristic facial features, intellectual disability, autistic behaviors and epilepsy. Clin Genet. 2015;87:356–361. doi: 10.1111/cge.12394. [DOI] [PubMed] [Google Scholar]
- Nakayama T, Wu J, Galvin-Parton P, Weiss J, Andriola MR, Hill RS, Vaughan DJ, El-Quessny M, Barry BJ, Partlow JN, et al. Deficient activity of alanyl-tRNA synthetase underlies an autosomal recessive syndrome of progressive microcephaly, hypomyelination, and epileptic encephalopathy. Hum Mutat. 2017;38:1348–1354. doi: 10.1002/humu.23250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namavar Y, Chitayat D, Barth PG, van Ruissen F, de Wissel MB, Poll-The BT, Silver R, Baas F. TSEN54 mutations cause pontocerebellar hypoplasia type 5. Eur J Hum Genet. 2011;19:724–726. doi: 10.1038/ejhg.2011.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newbery HJ, Loh DH, O’Donoghue JE, Tomlinson VAL, Chau YY, Boyd JA, Bergmann JH, Brownstein D, Abbott CM. Translation elongation factor eEF1A2 is essential for post-weaning survival in mice. J Biol Chem. 2007;282:28951–28959. doi: 10.1074/jbc.M703962200. [DOI] [PubMed] [Google Scholar]
- Nijholt DaT, van Haastert ES, Rozemuller AJM, Scheper W, Hoozemans JJM. The unfolded protein response is associated with early tau pathology in the hippocampus of tauopathies. J Pathol. 2012;226:693–702. doi: 10.1002/path.3969. [DOI] [PubMed] [Google Scholar]
- O’Connor T, Sadleir KR, Maus E, Velliquette RA, Zhao J, Cole SL, Eimer WA, Hitt B, Bembinster LA, Lammich S, et al. Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron. 2008;60:988–1009. doi: 10.1016/j.neuron.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SY, He F, Krans A, Frazer M, Taylor JP, Paulson HL, Todd PK. RAN translation at CGG repeats induces ubiquitin proteasome system impairment in models of fragile X-associated tremor ataxia syndrome. Hum Mol Genet. 2015;24:4317–4326. doi: 10.1093/hmg/ddv165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onuki R, Bando Y, Suyama E, Katayama T, Kawasaki H, Baba T, Tohyama M, Taira K. An RNA-dependent protein kinase is involved in tunicamycin-induced apoptosis and Alzheimer’s disease. EMBO J. 2004;23:959–968. doi: 10.1038/sj.emboj.7600049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz PA, Ulloque R, Kihara GK, Zheng H, Kinzy TG. Translation elongation factor 2 anticodon mimicry domain mutants affect fidelity and diphtheria toxin resistance. J Biol Chem. 2006;281:32639–32648. doi: 10.1074/jbc.M607076200. [DOI] [PubMed] [Google Scholar]
- Page G, Rioux Bilan A, Ingrand S, Lafay-Chebassier C, Pain S, Perault Pochat MC, Bouras C, Bayer T, Hugon J. Activated double-stranded RNA-dependent protein kinase and neuronal death in models of Alzheimer’s disease. Neuroscience. 2006;139:1343–1354. doi: 10.1016/j.neuroscience.2006.01.047. [DOI] [PubMed] [Google Scholar]
- Pan T. Adaptive translation as a mechanism of stress response and adaptation. Annu Rev Genet. 2013;47:121–137. doi: 10.1146/annurev-genet-111212-133522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquet C, Bose A, Polivka M, Peoc’h K, Brouland JP, Keohane C, Hugon J, Gray F. Neuronal phosphorylated RNA-dependent protein kinase in Creutzfeldt-Jakob disease. J Neuropathol Exp Neurol. 2009;68:190–198. doi: 10.1097/NEN.0b013e318196cd7c. [DOI] [PubMed] [Google Scholar]
- Park SG, Schimmel P, Kim S. Aminoacyl tRNA synthetases and their connections to disease. Proc Natl Acad Sci. 2008;105:11043–11049. doi: 10.1073/pnas.0802862105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavon-Eternod M, Gomes S, Geslain R, Dai Q, Rosner MR, Pan T. tRNA over-expression in breast cancer and functional consequences. Nucleic Acids Res. 2009;37:7268–7280. doi: 10.1093/nar/gkp787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce S, Nezich CL, Spinazzola A. Mitochondrial diseases: translation matters. Mol Cell Neurosci. 2013;55:1–12. doi: 10.1016/j.mcn.2012.08.013. [DOI] [PubMed] [Google Scholar]
- Peel AL, Bredesen DE. Activation of the cell stress kinase PKR in Alzheimer’s disease and human amyloid precursor protein transgenic mice. Neurobiol Dis. 2003;14:52–62. doi: 10.1016/s0969-9961(03)00086-x. [DOI] [PubMed] [Google Scholar]
- Peel AL, Rao RV, Cottrell BA, Hayden MR, Ellerby LM, Bredesen DE. Double-stranded RNA-dependent protein kinase, PKR, binds preferentially to Huntington’s disease (HD) transcripts and is activated in HD tissue. Hum Mol Genet. 2001;10:1531–1538. doi: 10.1093/hmg/10.15.1531. [DOI] [PubMed] [Google Scholar]
- Pelassa I, Fiumara F. Differential Occurrence of Interactions and Interaction Domains in Proteins Containing Homopolymeric Amino Acid Repeats. Front Genet. 2015;6:345. doi: 10.3389/fgene.2015.00345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Piana R, Tonduti D, Gordish Dressman H, Schmidt JL, Murnick J, Brais B, Bernard G, Vanderver A. Brain magnetic resonance imaging (MRI) pattern recognition in Pol III-related leukodystrophies. J Child Neurol. 2014;29:214–220. doi: 10.1177/0883073813503902. [DOI] [PubMed] [Google Scholar]
- Pisareva VP, Skabkin MA, Hellen CUT, Pestova TV, Pisarev AV. Dissociation by Pelota, Hbs1 and ABCE1 of mammalian vacant 80S ribosomes and stalled elongation complexes. EMBO J. 2011;30:1804–1817. doi: 10.1038/emboj.2011.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar A, Choi J, Wang J, Petrov A, Puglisi JD. Dynamic basis of fidelity and speed in translation: Coordinated multistep mechanisms of elongation and termination. Protein Sci. 2017;26:1352–1362. doi: 10.1002/pro.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puffenberger EG, Jinks RN, Sougnez C, Cibulskis K, Willert RA, Achilly NP, Cassidy RP, Fiorentini CJ, Heiken KF, Lawrence JJ, et al. Genetic mapping and exome sequencing identify variants associated with five novel diseases. PLoS One. 2012;7:e28936. doi: 10.1371/journal.pone.0028936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radford H, Moreno JA, Verity N, Halliday M, Mallucci GR. PERK inhibition prevents tau-mediated neurodegeneration in a mouse model of frontotemporal dementia. Acta Neuropathol. 2015;130:633–642. doi: 10.1007/s00401-015-1487-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter JD, Coller J. Pausing on Polyribosomes: Make Way for Elongation in Translational Control. Cell. 2015;163:292–300. doi: 10.1016/j.cell.2015.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph KLM, Schmitt BM, Villar D, White RJ, Marioni JC, Kutter C, Odom DT. Codon-Driven Translational Efficiency Is Stable across Diverse Mammalian Cell States. PLoS Genet. 2016;12:1–23. doi: 10.1371/journal.pgen.1006024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saffert P, Adamla F, Schieweck R, Atkins JF, Ignatova Z. An Expanded CAG Repeat in Huntingtin Causes +1 Frameshifting. J Biol Chem. 2016;291:18505–18513. doi: 10.1074/jbc.M116.744326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitsu H, Osaka H, Sasaki M, Takanashi J-I, Hamada K, Yamashita A, Shibayama H, Shiina M, Kondo Y, Nishiyama K, et al. Mutations in POLR3A and POLR3B encoding RNA Polymerase III subunits cause an autosomal-recessive hypomyelinating leukoencephalopathy. Am J Hum Genet. 2011;89:644–651. doi: 10.1016/j.ajhg.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandbaken MG, Culbertson MR. Mutations in elongation factor EF-1 alpha affect the frequency of frameshifting and amino acid misincorporation in Saccharomyces cerevisiae. Genetics. 1988;120:923–934. doi: 10.1093/genetics/120.4.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasarman F, Thiffault I, Weraarpachai W, Salomon S, Maftei C, Gauthier J, Ellazam B, Webb N, Antonicka H, Janer A, et al. The 3’ addition of CCA to mitochondrial tRNASer(AGY) is specifically impaired in patients with mutations in the tRNA nucleotidyl transferase TRNT1. Hum Mol Genet. 2015;24:2841–2847. doi: 10.1093/hmg/ddv044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauna ZE, Kimchi-Sarfaty C. Understanding the contribution of synonymous mutations to human disease. Nat Rev Genet. 2011;12:683–691. doi: 10.1038/nrg3051. [DOI] [PubMed] [Google Scholar]
- Schaffer AE, Eggens VRC, Caglayan AO, Reuter MS, Scott E, Coufal NG, Silhavy JL, Xue Y, Kayserili H, Yasuno K, et al. CLP1 Founder Mutation Links tRNA Splicing and Maturation to Cerebellar Development and Neurodegeneration. Cell. 2014;157:651–663. doi: 10.1016/j.cell.2014.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimmel P. Mistranslation and its control by tRNA synthetases. Philos Trans R Soc B Biol Sci. 2011;366:2965–2971. doi: 10.1098/rstb.2011.0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimmel P. The emerging complexity of the tRNA world: mammalian tRNAs beyond protein synthesis. Nat Rev Mol Cell Biol. 2017 doi: 10.1038/nrm.2017.77. [DOI] [PubMed] [Google Scholar]
- Schludi MH, May S, Grässer FA, Rentzsch K, Kremmer E, Küpper C, Klopstock T, German Consortium for Frontotemporal Lobar Degeneration Bavarian Brain Banking Alliance. Arzberger T, et al. Distribution of dipeptide repeat proteins in cellular models and C9orf72 mutation cases suggests link to transcriptional silencing. Acta Neuropathol. 2015;130:537–555. doi: 10.1007/s00401-015-1450-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt BM, Rudolph KLM, Karagianni P, Fonseca NA, White RJ, Talianidis I, Odom DT, Marioni JC, Kutter C. High-resolution mapping of transcriptional dynamics across tissue development reveals a stable mRNA-tRNA interface. Genome Res. 2014;24:1797–1807. doi: 10.1101/gr.176784.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MH, Pan T. Function and origin of mistranslation in distinct cellular contexts. Crit Rev Biochem Mol Biol. 2017;9238:1–15. doi: 10.1080/10409238.2016.1274284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scoles DR, Ho MHT, Dansithong W, Pflieger LT, Petersen LW, Thai KK, Pulst SM. Repeat Associated Non-AUG Translation (RAN Translation) Dependent on Sequence Downstream of the ATXN2 CAG Repeat. PLoS One. 2015;10:e0128769. doi: 10.1371/journal.pone.0128769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seburn KL, Nangle LA, Cox GA, Schimmel P, Burgess RW. An Active Dominant Mutation of Glycyl-tRNA Synthetase Causes Neuropathy in a Charcot-Marie-Tooth 2D Mouse Model. Neuron. 2006;51:715–726. doi: 10.1016/j.neuron.2006.08.027. [DOI] [PubMed] [Google Scholar]
- Sellier C, Buijsen RAM, He F, Natla S, Jung L, Tropel P, Gaucherot A, Jacobs H, Meziane H, Vincent A, et al. Translation of Expanded CGG Repeats into FMRpolyG Is Pathogenic and May Contribute to Fragile X Tremor Ataxia Syndrome. Neuron. 2017;93:331–347. doi: 10.1016/j.neuron.2016.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao S, Von der Malsburg K, Hegde RS. Listerin-dependent nascent protein ubiquitination relies on ribosome subunit dissociation. Mol Cell. 2013;50:637–648. doi: 10.1016/j.molcel.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigematsu M, Honda S, Kirino Y. Transfer RNA as a source of small functional RNA. J Mol Biol Mol Imaging. 2014;1:1–15. [PMC free article] [PubMed] [Google Scholar]
- Shimojima K, Shimada S, Tamasaki A, Akaboshi S, Komoike Y, Saito A, Furukawa T, Yamamoto T. Novel compound heterozygous mutations of POLR3A revealed by whole-exome sequencing in a patient with hypomyelination. Brain Dev. 2014;36:315–321. doi: 10.1016/j.braindev.2013.04.011. [DOI] [PubMed] [Google Scholar]
- Shoemaker CJ, Eyler DE, Green R. Dom34:Hbs1 promotes subunit dissociation and peptidyl-tRNA drop-off to initiate no-go decay. Science. 2010;330:369–372. doi: 10.1126/science.1192430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons C, Griffin LB, Helman G, Golas G, Pizzino A, Bloom M, Murphy JLP, Crawford J, Evans SH, Topper S, et al. Loss-of-function alanyl-tRNA synthetase mutations cause an autosomal-recessive early-onset epileptic encephalopathy with persistent myelination defect. Am J Hum Genet. 2015;96:675–681. doi: 10.1016/j.ajhg.2015.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleigh JN, Dawes JM, West SJ, Wei N, Spaulding EL, Gómez-Martín A, Zhang Q, Burgess RW, Cader MZ, Talbot K, et al. Trk receptor signaling and sensory neuron fate are perturbed in human neuropathy caused by Gars mutations. Proc Natl Acad Sci U S A. 2017;114:E3324–E3333. doi: 10.1073/pnas.1614557114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobala A, Hutvagner G. Transfer RNA-derived fragments: Origins, processing, and functions. Wiley Interdiscip Rev RNA. 2011;2:853–862. doi: 10.1002/wrna.96. [DOI] [PubMed] [Google Scholar]
- Sobczak K, de Mezer M, Michlewski G, Krol J, Krzyzosiak WJ. RNA structure of trinucleotide repeats associated with human neurological diseases. Nucleic Acids Res. 2003;31:5469–5482. doi: 10.1093/nar/gkg766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stum M, McLaughlin HM, Kleinbrink EL, Miers KE, Ackerman SL, Seburn KL, Antonellis A, Burgess RW. An assessment of mechanisms underlying peripheral axonal degeneration caused by aminoacyl-tRNA synthetase mutations. Mol Cell Neurosci. 2011;46:432–443. doi: 10.1016/j.mcn.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutzbach LD, Xie SX, Naj AC, Albin R, Gilman S, PSP Genetics Study Group. Lee VMY, Trojanowski JQ, Devlin B, Schellenberg GD. The unfolded protein response is activated in disease-affected brain regions in progressive supranuclear palsy and Alzheimer’s disease. Acta Neuropathol Commun. 2013;1:31. doi: 10.1186/2051-5960-1-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taft RJ, Vanderver A, Leventer RJ, Damiani SA, Simons C, Grimmond SM, Miller D, Schmidt J, Lockhart PJ, Pope K, et al. Mutations in DARS cause hypomyelination with brain stem and spinal cord involvement and leg spasticity. Am J Hum Genet. 2013;92:774–780. doi: 10.1016/j.ajhg.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tain LS, Mortiboys H, Tao RN, Ziviani E, Bandmann O, Whitworth AJ. Rapamycin activation of 4E-BP prevents parkinsonian dopaminergic neuron loss. Nat Neurosci. 2009;12:1129–1135. doi: 10.1038/nn.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takanashi J, Osaka H, Saitsu H, Sasaki M, Mori H, Shibayama H, Tanaka M, Nomura Y, Terao Y, Inoue K, et al. Different patterns of cerebellar abnormality and hypomyelination between POLR3A and POLR3B mutations. Brain Dev. 2014;36:259–263. doi: 10.1016/j.braindev.2013.03.006. [DOI] [PubMed] [Google Scholar]
- Tao Z, Wang H, Xia Q, Li K, Li K, Jiang X, Xu G, Wang G, Ying Z. Nucleolar stress and impaired stress granule formation contribute to C9orf72 RAN translation-induced cytotoxicity. Hum Mol Genet. 2015;24:2426–2441. doi: 10.1093/hmg/ddv005. [DOI] [PubMed] [Google Scholar]
- Tétreault M, Choquet K, Orcesi S, Tonduti D, Balottin U, Teichmann M, Fribourg S, Schiffmann R, Brais B, Vanderver A, et al. Recessive mutations in POLR3B, encoding the second largest subunit of Pol III, cause a rare hypomyelinating leukodystrophy. Am J Hum Genet. 2011;89:652–655. doi: 10.1016/j.ajhg.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiaville P, Legendre R, Rojas-Benitez D, Baudin-Baillieu A, Hatin I, Chalancon G, Glavic A, Namy O, de Crecy-Lagard V. Global translational impacts of the loss of the tRNA modification t6A in yeast. Microb Cell. 2016;3:29–45. doi: 10.15698/mic2016.01.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiffault I, Wolf NI, Forget D, Guerrero K, Tran LT, Choquet K, Lavallée-Adam M, Poitras C, Brais B, Yoon G, et al. Recessive mutations in POLR1C cause a leukodystrophy by impairing biogenesis of RNA polymerase III. Nat Commun. 2015;6:7623. doi: 10.1038/ncomms8623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoreen CC. The molecular basis of mTORC1-regulated translation. Biochem Soc Trans. 2017;45:213–221. doi: 10.1042/BST20160072. [DOI] [PubMed] [Google Scholar]
- Todd PK, Oh SY, Krans A, He F, Sellier C, Frazer M, Renoux AJ, Chen K, Scaglione KM, Basrur V, et al. CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron. 2013;78:440–455. doi: 10.1016/j.neuron.2013.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres AG, Batlle E, Ribas de Pouplana L. Role of tRNA modifications in human diseases. Trends Mol Med. 2014;20:306–314. doi: 10.1016/j.molmed.2014.01.008. [DOI] [PubMed] [Google Scholar]
- Toulouse A, Au-Yeung F, Gaspar C, Roussel J, Dion P, Rouleau Ga. Ribosomal frameshifting on MJD-1 transcripts with long CAG tracts. Hum Mol Genet. 2005;14:2649–2660. doi: 10.1093/hmg/ddi299. [DOI] [PubMed] [Google Scholar]
- Trinh MA, Klann E. Translational control by eIF2α kinases in long-lasting synaptic plasticity and long-term memory. Neurobiol Learn Mem. 2013;105:93–99. doi: 10.1016/j.nlm.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinh MA, Ma T, Kaphzan H, Bhattacharya A, Antion MD, Cavener DR, Hoeffer CA, Klann E. The eIF2α kinase PERK limits the expression of hippocampal metabotropic glutamate receptor-dependent long-term depression. Learn Mem. 2014;21:298–304. doi: 10.1101/lm.032219.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unterberger U, Höftberger R, Gelpi E, Flicker H, Budka H, Voigtländer T. Endoplasmic reticulum stress features are prominent in Alzheimer disease but not in prion diseases in vivo. J Neuropathol Exp Neurol. 2006;65:348–357. doi: 10.1097/01.jnen.0000218445.30535.6f. [DOI] [PubMed] [Google Scholar]
- Veeramah KR, Johnstone L, Karafet TM, Wolf D, Sprissler R, Salogiannis J, Barth-Maron A, Greenberg ME, Stuhlmann T, Weinert S, et al. Exome sequencing reveals new causal mutations in children with epileptic encephalopathies. Epilepsia. 2013;54:1270–1281. doi: 10.1111/epi.12201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira FG, Ping Q, Moreno AJ, Kidd JD, Thompson K, Jiang B, Lincecum JM, Wang MZ, De Zutter GS, Tassinari VR, et al. Guanabenz Treatment Accelerates Disease in a Mutant SOD1 Mouse Model of ALS. PLoS One. 2015;10:e0135570. doi: 10.1371/journal.pone.0135570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijgenboom E, Bosch L. Translational frameshifts induced by mutant species of the polypeptide chain elongation factor Tu of Escherichia coli. J Biol Chem. 1989;264:13012–13017. [PubMed] [Google Scholar]
- van der Voorn JP, van Kollenburg B, Bertrand G, Van Haren K, Scheper GC, Powers JM, van der Knaap MS. The unfolded protein response in vanishing white matter disease. J Neuropathol Exp Neurol. 2005;64:770–775. doi: 10.1097/01.jnen.0000178446.41595.3a. [DOI] [PubMed] [Google Scholar]
- Wang L, Popko B, Roos RP. The unfolded protein response in familial amyotrophic lateral sclerosis. Hum Mol Genet. 2011;20:1008–1015. doi: 10.1093/hmg/ddq546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Popko B, Tixier E, Roos RP. Guanabenz, which enhances the unfolded protein response, ameliorates mutant SOD1-induced amyotrophic lateral sclerosis. Neurobiol Dis. 2014a;71:317–324. doi: 10.1016/j.nbd.2014.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Popko B, Roos RP. An enhanced integrated stress response ameliorates mutant SOD1-induced ALS. Hum Mol Genet. 2014b;23:2629–2638. doi: 10.1093/hmg/ddt658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen X, Tan W, Westergard T, Krishnamurthy K, Markandaiah SS, Shi Y, Lin S, Shneider NA, Monaghan J, Pandey UB, et al. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate invitro and invivo neuronal death. Neuron. 2014;84:1213–1225. doi: 10.1016/j.neuron.2014.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis MS, Patterson C. Proteotoxicity and Cardiac Dysfunction — Alzheimer’s Disease of the Heart? N Engl J Med. 2013;368:455–464. doi: 10.1056/NEJMra1106180. [DOI] [PubMed] [Google Scholar]
- Wolf NI, Salomons GS, Rodenburg RJ, Pouwels PJW, Schieving JH, Derks TGJ, Fock JM, Rump P, Van Beek DM, Van Der Knaap MS, et al. Mutations in RARS cause hypomyelination. Ann Neurol. 2014;76:134–139. doi: 10.1002/ana.24167. [DOI] [PubMed] [Google Scholar]
- Wu D, Yu W, Kishikawa H, Folkerth RD, Iafrate AJ, Shen Y, Xin W, Sims K, Hu G-F. Angiogenin loss-of-function mutations in amyotrophic lateral sclerosis. Ann Neurol. 2007;62:609–617. doi: 10.1002/ana.21221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue S, Barna M. Specialized ribosomes: a new frontier in gene regulation and organismal biology. Nat Rev Mol Cell Biol. 2012;13:355–369. doi: 10.1038/nrm3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakawa M, Ito D, Honda T, Kubo K, Noda M, Nakajima K, Suzuki N. Characterization of the dipeptide repeat protein in the molecular pathogenesis of c9FTD/ALS. Hum Mol Genet. 2015;24:1630–1645. doi: 10.1093/hmg/ddu576. [DOI] [PubMed] [Google Scholar]
- Yamasaki S, Ivanov P, Hu G-F, Anderson P. Angiogenin cleaves tRNA and promotes stress-induced translational repression. J Cell Biol. 2009;185:35–42. doi: 10.1083/jcb.200811106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Zhou X, Zimmermann HR, Cavener DR, Klann E, Ma T. Repression of the eIF2a kinase PERK alleviates mGluR-LTD impairments in a mouse model of Alzheimer’s disease. Neurobiol Aging. 2016;41:19–24. doi: 10.1016/j.neurobiolaging.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaher HS, Green R. Quality control by the ribosome following peptide bond formation. Nature. 2009;457:161–166. doi: 10.1038/nature07582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Ling J, Barcia G, Jing L, Wu J, Barry BJ, Mochida GH, Hill RS, Weimer JM, Stein Q, et al. Mutations in QARS, encoding glutaminyl-trna synthetase, cause progressive microcephaly, cerebral-cerebellar atrophy, and intractable seizures. Am J Hum Genet. 2014a;94:547–558. doi: 10.1016/j.ajhg.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y-J, Jansen-West K, Xu Y-F, Gendron TF, Bieniek KF, Lin W-L, Sasaguri H, Caulfield T, Hubbard J, Daughrity L, et al. Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol. 2014b;128:505–524. doi: 10.1007/s00401-014-1336-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao T, Hong Y, Li X-J, Li S-H. Subcellular Clearance and Accumulation of Huntington Disease Protein: A Mini-Review. Front Mol Neurosci. 2016;9:27. doi: 10.3389/fnmol.2016.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu PJ, Huang W, Kalikulov D, Yoo JW, Placzek AN, Stoica L, Zhou H, Bell JC, Friedlander MJ, Krnjević K, et al. Suppression of PKR promotes network excitability and enhanced cognition by interferon- -mediated disinhibition. Cell. 2011;147:1384–1396. doi: 10.1016/j.cell.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Yan J, Jiang W, Yao X, Chen J, Chen L, Li C, Hu L, Jiang H, Shen X. Arctigenin effectively ameliorates memory impairment in Alzheimer’s disease model mice targeting both β-amyloid production and clearance. J Neurosci. 2013;33:13138–13149. doi: 10.1523/JNEUROSCI.4790-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, Margolis J, Peterson M, Markowski TW, Ingram MAC, et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci U S A. 2011;108:260–265. doi: 10.1073/pnas.1013343108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu T, Liu Y, Bañez-Coronel M, Reid T, Pletnikova O, Lewis J, Miller TM, Harms MB, Falchook AE, Subramony SH, et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci U S A. 2013;110:E4968–77. doi: 10.1073/pnas.1315438110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu T, Cleary JD, Liu Y, Bañez-Coronel M, Bubenik JL, Ayhan F, Ashizawa T, Xia G, Clark HB, Yachnis AT, et al. RAN Translation Regulated by Muscleblind Proteins in Myotonic Dystrophy Type 2. Neuron. 2017;95:1292–1305. e5. doi: 10.1016/j.neuron.2017.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]