

Abstract
Exchange proteins directly activated by cAMP (EPACs) are critical cAMP-dependent signaling pathway mediators that play important roles in cancer, diabetes, heart failure, inflammations, infections, neurological disorders and other human diseases. EPAC specific modulators are urgently needed to explore EPAC’s physiological function, mechanism of action and therapeutic applications. On the basis of a previously identified EPAC specific inhibitor hit ESI-09, herein we have designed and synthesized a novel series of 2-substituted phenyl-N-phenyl-2-oxoacetohydrazonoyl cyanides as potent EPAC inhibitors. Compound 31 (ZL0524) has been discovered as the most potent EPAC inhibitor with IC50 values of 3.6 μM and 1.2 μM against EPAC1 and EPAC2, respectively. Molecular docking of 31 onto an active EPAC2 structure predicts that 31 occupies the hydrophobic pocket in cAMP binding domain (CBD) and also opens up new space leading to the solvent region. These findings provide inspirations for discovering next generation of EPAC inhibitors.
Keywords: Exchange proteins directly activated by cAMP, EPAC, Antagonist, Molecular docking
Graphical Abstract
Cyclic adenosine monophosphate (cAMP, 1), generated from adenosine triphosphate (ATP) by adenylyl cyclase, is a second messenger for intracellular signal transduction in many different organisms. cAMP-mediated signaling events were considered to be transduced largely by protein kinase A (PKA) until the discovery of exchange proteins directly activated by cAMP(EPACs)/cAMP regulated guanine nucleotide exchange factor (cAMP-GEF).1–4 These two intracellular receptor families mediated the major physiological effects of cAMP in mammalian cells through the cAMP binding domain (CBD) which acts as a molecular switch for controlling various cellular activities.5, 6 The identification of EPACs opens new avenues for cAMP signaling research.7 Unlike PKA, EPAC proteins do not have kinase activity and activate the Ras superfamily small GTPases Rap1 and Rap2 in response to the generation of intracellular cAMP. There are two isoforms of mammalian EPACs, EPAC1 which is more ubiquitously expressed and EPAC2 which is mainly found in CNS, pancreatic islets and adrenal gland.2 EPAC1 and EPAC2 are structurally homologous but functionally nonredundant. A number of studies have revealed that EPAC proteins are critically involved in a variety of human diseases such as cancer, inflammation, bacterial and viral infections, central nervous system disorders, energy homeostasis and obesity, and cardiac functions.8–14
Given the physiological and pathophysiological significance of EPAC proteins, the development of pharmacological EPAC modulators is needed.15 Most EPAC agonists are derivatives of cAMP,16 while non-cyclic nucleotide ligands usually display EPAC inhibitory activities, except a very recently reported small molecule partial agonist with modest potency.17 Cheng, et al developed a sensitive and robust fluorescence-based high throughput (HTS) assay,18 which led to the discovery of a series of non-cyclic nucleotide EPAC inhibitors.19 After extensive modifications by our team, dihydropyrimidine 2 (HJC0198, Fig. 1) was obtained with an IC50 value of 4.0 μM against EPAC2. It selectively blocks cAMP-induced EPAC activation without affecting cAMP-mediated PKA activation at the concentration of 25 μM.20 Diphenylamine 3 (MAY0132, Fig. 1), also developed by our team, exhibited potent and selective EPAC2 inhibitory activity with an IC50 value of 0.4 μM and inhibited cAMP-mediated EPAC2 GEF activity with IC50 of 1 μM, while displaying no significant inhibition against EPAC1 at 100 μM.21,22 Subtype selective compound will not only help understanding diverse functions of EPACs but also may contribute to therapeutic safety in the future, thus further modifications on compound 3, a good EPAC2 selective inhibitor, are undergoing in our lab. Compound 4 ((R)-CE3F4, Fig. 1), identified by Courileau and coworkers via HTS screening,23 displayed EPAC1 inhibitory activity with an IC50 value of 4.2 μM with 10-fold selectivity over EPAC2.24 In our assays, its inhibitory activities against EPAC1 and EPAC2 are 5.5 and 17 μM, respectively. Compound 5 (ESI-09, Fig. 1) is another promising hit discovered by our team, displaying micromolar inhibitory activities against both EPAC1 and EPAC2.25–27 Herein, we describe the discovery of a series of novel EPAC inhibitors such as compound 31, which displays enhanced potency against EPAC proteins in comparison with chemical lead 5.
Fig. 1.

The structures of cAMP and representative EPAC antagonists including newly identified 31 (ZL0524) reported in this paper.
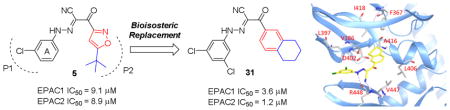
While previously reported optimizations on compound 5 focused on the phenyl ring A (Fig. 2) and substituents on isoxazole ring,28 other alternatives to replace isoxazole ring were never explored. Herein, we propose to replace the isoxazole with its bioisosteric substituted phenyl ring B (Fig. 2) to explore new chemical space and improve EPAC inhibitory potency.29 Docking studies of compound 5 into the cAMP binding domain of active EPAC2 suggested that 3-chloro phenyl ring A and tert-butylisoxazole occupy two hydrophobic pockets and play important roles for their interaction.15 Compound 6 was synthesized and gave an IC50 value of 13.3 μM, showing the phenyl bioisosteric replacement was a viable route forward.
Fig. 2.

Design strategy of 2-substituted phenyl-N-phenyl-2-oxoaceto hydrazonoyl cyanides using a bioisosteric replacement approach.
The first series of compounds were designed to keep the tert-butyl phenyl group B intact and the synthetic route is depicted in Scheme 1. Starting material methyl 4-(tert-butyl)benzoate (7) was reacted with CH3CN in the presence of CH3Li to give 3-(4-(tert-butyl)phenyl)-3-oxopropanenitrile (8).30 Anilines 9 were transformed into corresponding diazo salts and coupled with 8 in the presence of NaOAc to produce compounds 6 and 10~18 in good yields.
Scheme 1.

Synthetic route to access compounds 6 and 10~18. Reagents and conditions: (a) CH3CN, CH3Li, -78 °C, 1h, 64%; (b) i): 10% HCl, NaNO2, H2O, rt.; ii): NaOAc, EtOH, rt., 67% to quant. for two steps.
The second series of compounds were designed to replace tert-butyl group on phenyl ring B, and the synthetic protocol is depicted in Scheme 2. 3,5-Dichloroaniline 19 was transformed into its corresponding diazo salt and coupled with different 3-oxo-3-phenylpropanenitriles 21 in the presence of NaOAc to give compounds 22~33 in moderate to good yields. All the structures and purity of synthesized compounds were confirmed by 1H NMR, 13C NMR and HR-MS.31
Scheme 2.

Synthetic route of compounds 22~33. Reagents and conditions: (a) 10% HCl, NaNO2, H2O, rt; (b) NaOAc, EtOH, rt., 37% to quant.
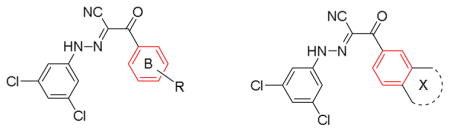
Compounds were then evaluated for their abilities to inhibit EPAC1 and EPAC2-mediated Rap1b-bGDP exchange activity using purified recombinant full-length EPAC1 and EPAC2 proteins.32 For promising compounds, the IC50 values against both EPAC1 and EPAC2 were determined (Table 1 and Table 2). Compound 5 was used as the reference compound for comparison with IC50 values of 10.8 μM and 4.4 μM against EPAC1 and EPAC2, respectively. Based on our previous SAR study, 3-Cl or 3-CF3 on phenyl ring A which resides in a hydrophobic pocket P1 is favorable for EPAC binding. Thus, for most newly designed compounds, we chose to retain these substituents. Compared to compound 6, compound 10 with an additional 5-Cl substituent has 2.5-fold improvement (IC50 = 5.4 μM) on EPAC1 inhibitory activity and 11-fold improvement (IC50 = 2.5 μM) on EPAC2 inhibitory activity. In addition, substituent at 5-position is more favorable than that with a same group at 4-positon on this phenyl ring A (compound 11 vs 12, and compound 16 vs 17). Triple substituents at 3,4,5-position displayed slightly decreased EPAC inhibition (compounds 13 and 18), but compound 18 has a 4.6-fold EPAC2/EPAC1 selectivity. 3,5-Di CF3 substituted compound 14 showed similar EPAC inhibitory activities to those of compound 10.
Table 1.
IC50 values of substituted 2-(4-(tert-butyl)phenyl)-N-phenyl-2-oxoacetohydrazonoyl cyanides for inhibiting EPAC1/2 GEF activity.
| |||
|---|---|---|---|
| Entry | R | Rap1b-bGDP EPAC1 IC50 (μM)a | Rap1b-bGDP EPAC2 IC50 (μM) |
| 5 | - | 10.8 ± 1.6 | 4.4 ± 0.5 |
| 6 | 3-Cl | 13.3 ± 3.0 | 28.4 ± 5.6 |
| 10 | 3,5-di Cl | 5.4 ± 0.7 | 2.5 ± 0.6 |
| 11 | 3-Cl, 4-F | 12.4 ± 0.9 | NDb |
| 12 | 3-Cl, 5-F | 6.4 ± 1.4 | 3.4 ± 0.6 |
| 13 | 3,4,5-tri F | 24.4 ± 5.7 | 15.8 ± 3.8 |
| 14 | 3,5-di CF3 | 5.5 ± 0.7 | 6.3 ± 0.9 |
| 15 | 3-CF3, 4-Cl | 6.1 ± 1.0 | 12.4 ± 3.9 |
| 16 | 3-Cl, 5-CF3 | 5.6 ± 0.9 | 5.4 ± 1.1 |
| 17 | 3-Cl, 4-CF3 | 50.8 ± 14.6 | ND |
| 18 | 3,4,5-tri Cl | 13.9 ± 3.6 | 3.0 ± 0.4 |
The values are the mean ± SE, n = 3.
ND: not determined.
Table 2.
IC50 values of substituted 2-phenyl-N-(3,5-dichlorophenyl)-2-oxoacetohydrazonoyl cyanides for inhibiting EPAC1/2 GEF activity.
| |||
|---|---|---|---|
| Entry | R or X | Rap1b-bGDP EPAC1 IC50 (μM)a | Rap1b-bGDP EPAC2 IC50 (μM) |
| 10 | 4-tert butyl | 5.4 ± 0.7 | 2.5 ± 0.6 |
| 22 | 4-Cl | >300 | NDb |
| 23 | 3-Cl | >300 | ND |
| 24 | 4-OCF3 | 40.9 ± 6.8 | ND |
| 25 | 4-CO2Me | 29.3 ± 6.2 | ND |
| 26 | 4-Ph | >300 | ND |
| 27 | 3,4-di OMe | >300 | ND |
| 28 | 4-Piperidin-1-ylmethyl | 39.4 ± 4.7 | ND |
| 29 | 4-Morpholinomethyl | 14.6 ± 5.7 | ND |
| 30 | 4-Cyclohexane | 32.4 ± 9.5 | ND |
| 31 |
|
3.6 ± 0.3 | 1.2 ± 0.1 |
| 32 |
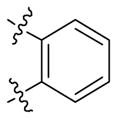
|
11.3 ± 1.4 | 2.5 ± 0.5 |
| 33 |
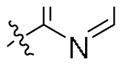
|
19.6 ± 6.7 | ND |
The values are the mean ± SE.
ND: not determined.
The first round of optimization illustrated that all compounds obtained via bioisosteric replacement of tert-butyl isoxazole ring with tert-butyl phenyl group retained both EPAC1 and EPAC2 inhibitory activities, while nearly half of them displayed more potent inhibitory activities than reference compound 5. 3,5-di Cl substituents on phenyl ring A are favored and compound 10 exhibited the most potent EPAC1 and EPAC2 inhibitory activities. Thus, we kept this fragment intact and further modified substituents on phenyl ring B which occupies the other hydrophobic pocket P2 (Table 2). We investigated 3-Cl (22) and 4-Cl (23) first, but both are detrimental to EPAC inhibition. It appears that the bulk size of substituents on phenyl ring B is critical. We then explored electron donating group 4-OCF3 (24) and electron withdrawing group 4-CO2Me (25). Interestingly, neither of them showed increased inhibition against EPAC1 compared to compound 10. Introduction of 4-Ph and 3,4-di OMe substituents on phenyl ring B led to compounds 26 and 27 which have a complete loss of EPAC inhibitory activities. The various rings including piperidine (28), morpholine (29) and cyclohexane (30) were also investigated, but none of them displayed more potent EPAC inhibition than compound 10. However, compounds 31~33 with fused rings (e.g. 1,2,3,4-tetrahydronaphthalene, naphthalene and quinoline) all displayed improved EPAC inhibitory activities when compared to aforementioned compounds that have different substituents on phenyl ring B. Among them, compound 31 exhibited the most potent EPAC inhibitory activities with IC50 values of 3.6 μM and 1.2 μM against EPAC1 and EPAC2, respectively. In Fig. 3, examples of dose-response curves are shown for compounds 5, 6, 14 and 31.
Fig. 3.
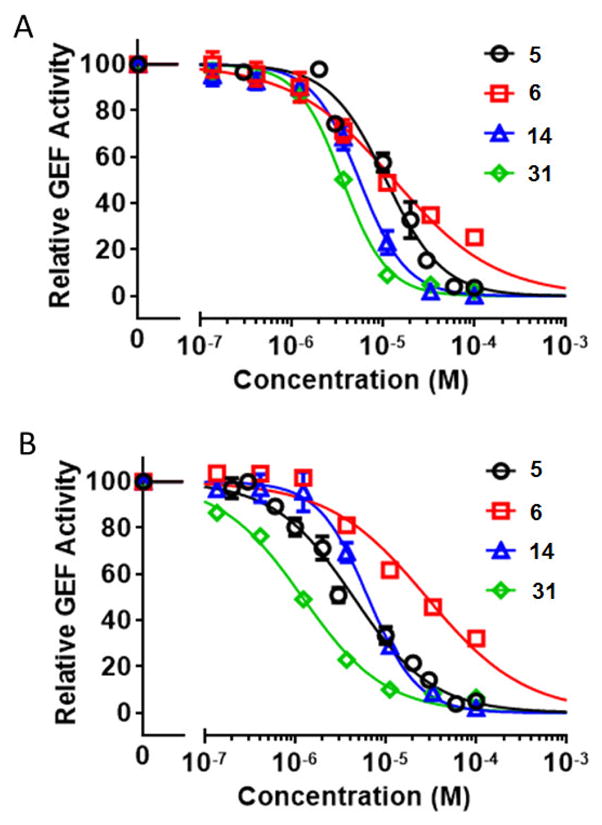
A) Relative inhibitory activity for EPAC1-mediated Rap1b-bGDP exchange. B) Relative inhibitory activity for EPAC2-mediated Rap1b-bGDP exchange. Dose-dependent inhibition of EPAC1/2 GEF activity by compounds 5, 6, 14, and 31, in the presence of 20 μM cAMP. Relative GEF activity were presented as normalized reaction rate constant (means ± SEM, n =3) described in the method.
Molecular docking study of compound 31 was performed using the Schrödinger Small-Molecule Drug Discovery Suite taking advantage of known X-ray crystal structures of activated EPAC2 protein.33 The model shows that compound 31 occupies the CBD of EPAC2 and forms hydrogen bonds with Arg448 and the cyano-hydrazine substituent (Fig. 4A). The fused tetrahydronaphthalene ring B lays in the same pocket as tert-butyl isoxazole ring of ESI-09 does and is surrounded by hydrophobic Ala416, Leu406, Val447, Val386, etc. Interestingly, ring A of compound 31 extends to the opposite direction (the solvent region) compared to ESI-09 (Fig. 4B). This docking mode suggests that there exists a large space deep pocket remaining to be explored in the CBD domain of EPAC protein for the discovery of next generation ligands.
Fig. 4.

Docking studies of compound 31. A) Predicted binding pose of antagonist 31 docking at the cAMP binding domain B (CBD) of EPAC2 (PDB Code 3CF6). Compound 31 is shown in yellow. Key residues are displayed in sticks representation. Hydrogen bond is shown in purple dotted line; B) Overlay of molecular docking pose of 31 and 5 binding at the CBD of EPAC2. Compound 31 is shown in yellow, and 5 in magenta.
We have designed and synthesized a new series of 2-substituted phenyl-N-phenyl-2-oxoacetohydrazonoyl cyanides as EPAC inhibitors via simple chemistry with inexpensive starting material and synthetic ease suitable for scale up. Among those new molecules, compound 31 (ZL0524) was the most potent EPAC inhibitory activities with IC50 values of 3.6 μM and 1.2 μM against EPAC1 and EPAC2, respectively. Docking studies of ZL0524 with activated EPAC2 reveal that it occupies the CBD2 hydrophobic pocket, forms hydrogen bonds with Arg448 and extended to the solvent region. The findings provide us inspirations to rationally design larger molecules to reach a deeper binding pocket as next generation EPAC inhibitors. In vivo efficacy studies of 31 in infectious disease models (e.g. rickettsiosis) are under way, and the results will be reported in due course.
Acknowledgments
This work was supported by grants R01 GM106218, R01 AI111464, R01 GM066170 and R35 GM122536 from the National Institutes of Health. We want to thank Drs. Lawrence C. Sowers at the Department of Pharmacology as well as Dr. Tianzhi Wang at the NMR core facility of UTMB for the NMR spectroscopy assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Cheng X, Ji Z, Tsalkova T, Mei F. Acta Biochim Biophys Sin (Shanghai) 2008;40:651–662. doi: 10.1111/j.1745-7270.2008.00438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman DE, Graybiel AM. Science. 1998;282:2275–2279. doi: 10.1126/science.282.5397.2275. [DOI] [PubMed] [Google Scholar]
- 3.de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL. Nature. 1998;396:474–477. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- 4.Gloerich M, Bos JL. Annu Rev Pharmacol Toxicol. 2010;50:355–375. doi: 10.1146/annurev.pharmtox.010909.105714. [DOI] [PubMed] [Google Scholar]
- 5.Berman HM, Ten Eyck LF, Goodsell DS, Haste NM, Kornev A, Taylor S. Proc Natl Acad Sci U S A. 2005;102:45–50. doi: 10.1073/pnas.0408579102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rehmann H, Prakash B, Wolf E, Rueppel A, de Rooij J, Bos JL, Wittinghofer A. Nat Struct Biol. 2003;10:26–32. doi: 10.1038/nsb878. [DOI] [PubMed] [Google Scholar]
- 7.Bos JL. Nat Rev Mol Cell Biol. 2003;4:733–738. doi: 10.1038/nrm1197. [DOI] [PubMed] [Google Scholar]
- 8.Almahariq M, Mei FC, Cheng X. Acta Biochim Biophys Sin (Shanghai) 2016;48:75–81. doi: 10.1093/abbs/gmv115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singhmar P, Huo X, Eijkelkamp N, Berciano SR, Baameur F, Mei FC, Zhu Y, Cheng X, Hawke D, Mayor F, Jr, Murga C, Heijnen CJ, Kavelaars A. Proc Natl Acad Sci U S A. 2016;113:3036–3041. doi: 10.1073/pnas.1516036113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laurent AC, Breckler M, Berthouze M, Lezoualc’h F. Biochem Soc Trans. 2012;40:51–57. doi: 10.1042/BST20110642. [DOI] [PubMed] [Google Scholar]
- 11.Cazorla O, Lucas A, Poirier F, Lacampagne A, Lezoualc’h F. Proc Natl Acad Sci U S A. 2009;106:14144–14149. doi: 10.1073/pnas.0812536106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kai AK, Lam AK, Chen Y, Tai AC, Zhang X, Lai AK, Yeung PK, Tam S, Wang J, Lam KS, Vanhoutte PM, Bos JL, Chung SS, Xu A, Chung SK. FASEB J. 2013;27:4122–4135. doi: 10.1096/fj.13-230433. [DOI] [PubMed] [Google Scholar]
- 13.Wang P, Liu Z, Chen H, Ye N, Cheng X, Zhou J. Bioorg Med Chem Lett. 2017;27:1633–1639. doi: 10.1016/j.bmcl.2017.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang H, Robichaux WG, Wang Z, Mei FC, Cai M, Du G, Chen J, Cheng X. Sci Rep. 2016;6:36552. doi: 10.1038/srep36552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen H, Wild C, Zhou X, Ye N, Cheng X, Zhou J. J Med Chem. 2014;57:3651–3665. doi: 10.1021/jm401425e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vliem MJ, Ponsioen B, Schwede F, Pannekoek WJ, Riedl J, Kooistra MR, Jalink K, Genieser HG, Bos JL, Rehmann H. ChemBioChem. 2008;9:2052–2054. doi: 10.1002/cbic.200800216. [DOI] [PubMed] [Google Scholar]
- 17.Parnell E, McElroy SP, Wiejak J, Baillie GL, Porter A, Adams DR, Rehmann H, Smith BO, Yarwood SJ. Sci Rep. 2017;7:294. doi: 10.1038/s41598-017-00455-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsalkova T, Mei FC, Cheng X. PloS one. 2012;7:e30441. doi: 10.1371/journal.pone.0030441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsalkova T, Mei FC, Li S, Chepurny OG, Leech CA, Liu T, Holz GG, Woods VL, Jr, Cheng X. Proc Natl Acad Sci U S A. 2012;109:18613–18618. doi: 10.1073/pnas.1210209109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen H, Tsalkova T, Mei FC, Hu Y, Cheng X, Zhou J. Bioorg Med Chem Lett. 2012;22:4038–4043. doi: 10.1016/j.bmcl.2012.04.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wild CT, Zhu Y, Na Y, Mei F, Ynalvez MA, Chen H, Cheng X, Zhou J. ACS Med Chem Lett. 2016;7:460–464. doi: 10.1021/acsmedchemlett.5b00477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen H, Tsalkova T, Chepurny OG, Mei FC, Holz GG, Cheng X, Zhou J. J Med Chem. 2013;56:952–962. doi: 10.1021/jm3014162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Courilleau D, Bisserier M, Jullian JC, Lucas A, Bouyssou P, Fischmeister R, Blondeau JP, Lezoualc’h F. J Biol Chem. 2012;287:44192–44202. doi: 10.1074/jbc.M112.422956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Courilleau D, Bouyssou P, Fischmeister R, Lezoualc’h F, Blondeau PJ. Biochem Biophys Res Commun. 2013;440:443–448. doi: 10.1016/j.bbrc.2013.09.107. [DOI] [PubMed] [Google Scholar]
- 25.Almahariq M, Tsalkova T, Mei FC, Chen H, Zhou J, Sastry SK, Schwede F, Cheng X. Mol Pharmacol. 2013;83:122–128. doi: 10.1124/mol.112.080689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu Y, Chen H, Boulton S, Mei F, Ye N, Melacini G, Zhou J, Cheng X. Sci Rep. 2015;5:9344. doi: 10.1038/srep09344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gong B, Shelite T, Mei FC, Ha T, Hu Y, Xu G, Chang Q, Wakamiya M, Ksiazek TG, Boor PJ, Bouyer DH, Popov VL, Chen J, Walker HD, Cheng X. Proc Natl Acad Sci U S A. 2013;110:19615–19620. doi: 10.1073/pnas.1314400110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ye N, Zhu Y, Chen H, Liu Z, Mei FC, Wild C, Chen H, Cheng X, Zhou J. J Med Chem. 2015;58:6033–6047. doi: 10.1021/acs.jmedchem.5b00635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Z, Yue X, Song Z, Peng X, Guo J, Ji Y, Cheng Z, Ding J, Ai J, Geng M, Zhang A. Eur J Med Chem. 2014;86:438–448. doi: 10.1016/j.ejmech.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 30.Chen H, Ding C, Wild C, Liu H, Wang T, White MA, Cheng X, Zhou J. Tetrahedron Lett. 2013;54:1546–1549. doi: 10.1016/j.tetlet.2013.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spectra data of the representative compounds: (E)-2-(4-(tert-Butyl)phenyl)-N-(3,5-dichlorophenyl)-2-oxoacetohydrazonoyl cyanide (10). Yellow solid, 69%. 1H NMR (300 MHz, DMSO-d6) δ 12.43 (s, 1H), 7.81 (d, J = 8.0 Hz, 2H), 7.56 (d, J = 8.0 Hz, 2H), 7.31 (s, 2H), 1.33 (s, 9H). 13C NMR (75 MHz, DMSO-d6) δ 187.29, 166.62, 156.14, 145.04, 135.30, 133.76, 130.36, 125.25, 123.96, 115.50, 115.30, 111.49, 35.28, 31.29. HRMS (ESI): m/z (M + Na)+ calcd for C19H17Cl2N3NaO: 396.0646, found: 396.0670. (E)-2-(4-(tert-Butyl)phenyl)-N-(3-chloro-5-fluorophenyl)-2-oxoacetohydrazonoyl cyanide (12). Yellow solid, quant. 1H NMR (300 MHz, MeOD) δ 7.88 (d, J = 8.4 Hz, 2H), 7.59 (d, J = 8.4 Hz, 2H), 7.21 (s, 1H), 7.04 (d, J = 10.2 Hz, 1H), 6.94 (d, J = 8.4 Hz, 1H), 1.39 (s, 9H). 13C NMR (75 MHz, MeOD) δ 187.07, 164.99, 161.71, 156.55, 144.61, 135.93, 135.77, 133.19, 129.84, 124.80, 112.00, 111.96, 111.60, 111.26, 109.70, 101.86, 101.50, 34.61, 30.09. HRMS (ESI): m/z (M + H)+ calcd for C19H18ClFN3O: 358.1122, found: 358.1123. (E)-N-(3,5-Bis(trifluoromethyl)phenyl)-2-(4-(tert-butyl)phenyl)-2-oxoacetohydrazonoyl cyanide (14). Yellow solid, 67%. 1H NMR (300 MHz, MeOD) δ 7.88 (m, 4H), 7.69 (s, 1H), 7.62 – 7.56 (m, 2H), 1.40 (s, 9H). 13C NMR (75 MHz, MeOD) δ 187.29, 156.50, 144.04, 133.30, 133.18, 132.86, 132.41, 131.97, 129.82, 128.53, 124.92, 124.70, 121.32, 117.71, 116.96, 116.90, 116.85, 116.80, 116.75, 116.23, 115.80, 115.76, 109.47, 34.57, 30.05. HRMS (ESI): m/z (M + H)+ calcd for C21H18F6N3O: 442.1354, found: 442.1355. (E)-2-(4-(tert-Butyl)phenyl)-N-(3-chloro-5-(trifluoromethyl)phenyl )-2-oxoacetohydrazonoyl cyanide (16). Yellow solid, 76%. 1H NMR (300 MHz, DMSO-d6) δ 12.58 (s, 1H), 7.81 (d, J = 7.7 Hz, 2H), 7.56 (m, 5H), 1.32 (s, 9H). 13C NMR (75 MHz, DMSO-d6) δ 187.37, 156.13, 144.79, 135.46, 133.68, 132.39, 131.95, 130.36, 125.16, 120.80, 120.00, 115.71, 111.78, 111.33, 35.23, 31.22. HRMS (ESI): m/z (M + Na)+ calcd for C20H17F3N3ClNaO: 430.0910, found: 430.0956. (E)-N-(3,5-Dichlorophenyl)-2-oxo-2-(5,6,7,8-tetrahydronaphthalen-2-yl)acetohydrazonoyl cyanide (31). Yellow solid, 59%. 1H NMR (300 MHz, DMSO-d6) δ 12.42 (s, 1H), 7.68 (s, 1H), 7.58 (d, J = 8.0 Hz, 1H), 7.34 (s, 3H), 7.21 (d, J = 8.0 Hz, 1H), 2.81 (s, 4H), 1.78 (s, 4H). 13C NMR (75 MHz, DMSO) δ 186.93, 144.98, 142.79, 136.83, 135.34, 133.44, 131.65, 129.19, 127.36, 123.94, 115.57, 115.23, 111.44, 29.39, 29.28, 22.95, 22.84. HRMS (ESI): m/z (M + H)+ calcd for C19H16N3Cl2O: 372.0670, found: 372.0667.
- 32.Ye N, Zhu Y, Liu Z, Mei CF, Chen H, Wang P, Cheng X, Zhou J. Eur J Med Chem. 2017;134:62–71. doi: 10.1016/j.ejmech.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Molecular docking studies: The docking study was performed with Schrödinger Small-Molecule Drug Discovery Suite. The crystal structure of EPAC2 (PDB code: 3CF6) was downloaded from RCSB PDB Bank and prepared with Protein Prepared Wizard. During this step, hydrogens were added, crystal waters were removed, and partial charges were assigned using the OPLS-2005 force field. The 3D structures of ESI-09 and ZL0524 were created with Schrödinger Maestro, and the initial lowest energy conformations were calculated with LigPrep. For all dockings, the grid center was chosen on the centroid of included ligand of PDB structure CBD-B site and a 24 × 24 × 24 Å grid box size was used. All dockings were employed with Glide using the XP protocol. Docking poses were incorporated into Schrödinger Maestro for a visualization of ligand-receptor interactions and overlay analysis.