

Abstract
Background
Phosphodiesterase 10A (PDE10A) is selectively expressed in medium spiny neurons of the striatum. TAK-063 is a selective inhibitor of PDE10A in clinical development for the treatment of schizophrenia.
Objectives
Safety, tolerability, and pharmacokinetics (PK) of TAK-063 were evaluated following multiple rising oral doses, and PK/adverse event (AE) models were developed to characterize the relationship between TAK-063 exposure and incidence of specific AEs.
Methods
Healthy Japanese subjects (HJS) aged 20–55 years and subjects with stable schizophrenia (SSS) aged 18–55 years were enrolled and randomized to either TAK-063 or placebo. Study medication was administered as a tablet once daily (at night) with food over a 7-day period.
Results
TAK-063 and placebo groups consisted of 62 and 15 subjects, respectively. A majority of subjects (71 of 77) completed the study. AEs were mostly of mild or moderate severity, and no deaths were reported. The most common AE was somnolence. For equivalent doses, the rate of extrapyramidal syndromes (EPS) was higher in SSS than in HJS. PK parameters were comparable between HJS and SSS at equivalent doses. The incidence of somnolence and EPS symptoms increased with exposure, and this was described with the PK/AE model. A maximum tolerated dose was not determined.
Conclusions
Multiple doses of TAK-063 were safe and well tolerated. PK/AE models characterized the incidence of somnolence and EPS with increasing TAK-063 exposure, and simulations suggested that a once-daily dose range of up to 30 mg would be suitable for future studies.
ClinicalTrials.gov Identifier
Electronic supplementary material
The online version of this article (doi:10.1007/s40268-017-0214-8) contains supplementary material, which is available to authorized users.
Key Points
| In the fed state, pharmacokinetic parameters for TAK-063 were comparable at equivalent doses between healthy Japanese subjects and subjects with stable schizophrenia. |
| TAK-063 was safe and generally well tolerated when administered once daily over a 7-day period to healthy Japanese subjects (up to 20 mg) and subjects with stable schizophrenia (up to 100 mg). |
| Pharmacokinetic/adverse event modeling suggests that single daily doses up to 30 mg TAK-063 may be suitable for further development in schizophrenia. |
Introduction
TAK-063 is a selective inhibitor [1] of phosphodiesterase 10A (PDE10A), a dual-substrate cyclic nucleotide phosphodiesterase that regulates the levels of cyclic adenosine monophosphate and cyclic guanosine monophosphate downstream of D1 and D2 receptor signaling in medium spiny neurons of the striatum. Preclinical data suggest that targeting both pathways via PDE10A inhibition may be a viable approach to treating schizophrenia [1]. In general, current antipsychotics are antagonists or partial agonists of dopamine receptors and may cause hyperprolactinemia, weight gain, and extrapyramidal syndromes (EPS) [1–4]. In preclinical studies, TAK-063 did not increase glucose or prolactin levels, and this lack of metabolic effects may be due in part to the selective, striatal expression of PDE10A and the activity of TAK-063 downstream of the dopamine receptor in D1- and D2-receptor-expressing neurons [1, 2, 5].
Previously, a phase 1 single-rising-dose study in healthy Japanese and non-Japanese subjects evaluated the safety, tolerability, and pharmacokinetics (PK) of single doses of TAK-063 under fasting and fed conditions [6]. TAK-063 was generally safe and well tolerated, and exhibited a small increase in exposure in the presence of food [6]. The present study assessed the safety, tolerability, and PK of TAK-063 when administered as multiple daily doses in healthy Japanese subjects (HJS) and subjects with stable schizophrenia (SSS) in the fed state. The relationship between TAK-063 exposure and adverse events (AEs) was also examined via logistic regression modeling.
Methods
Subjects
This was a phase 1, randomized, double-blind, placebo-controlled, single-center, safety and tolerability study of multiple rising doses of TAK-063 in HJS and SSS. The study site was in compliance with institutional review board (IRB) regulations, Good Clinical Practice regulations and guidelines, and all applicable local regulations. The study protocol was approved by the appropriate local IRB for each site.
Subjects aged 20–55 (HJS) and 18–55 years (SSS) were included in this study. HJS and SSS were enrolled in cohorts of approximately ten subjects each and randomized (8:2) to either TAK-063 or placebo. SSS who were on stable antipsychotic monotherapy for at least 1 month before screening and had a Clinical Global Impression of Severity (CGI-S) score of ≤ 4 and a total Positive and Negative Symptom Scale (PANSS) score of ≤ 70 at screening and check-in (day − 1) were included. HJS were ineligible if they had used nicotine within 28 days or had a history of mental disorders in the past 3 years. Any subject who tested positive for illicit drugs, had a history of substance abuse, or exhibited elevated suicide risk in the previous 6 months was excluded.
Study Design
The study consisted of a screening visit followed by a washout interval, during which subjects with stable schizophrenia were required to discontinue their psychotropic medications, including antipsychotics, for approximately 5 half-lives, followed by a 7-day treatment period, study exit (day 8), and follow-up on day 14 (Supplementary Fig. 1; see the electronic supplementary material). Baseline assessments were physical examination; vital signs; weight, height, and body mass index (BMI); clinical laboratory tests; electrocardiogram (ECG); CGI-Severity of Illness; PK urine collection; cognition battery; Columbia Suicide Severity Rating Scale (C-SSRS); PANSS; Extrapyramidal Symptom Rating Scale-Abbreviated (ESRS-A); sleepiness, mood, and alertness assessments; and pretreatment events. All baseline assessments were completed on day − 1. Study medication was administered as a tablet once daily with food over a 7-day period in the clinic followed by an exit visit on day 8 and a follow-up phone call on day 14. SSS taking other non-psychotropic medications for medical conditions (e.g., hypertension) were allowed to continue on their medication.
Dose levels were based on the safety data from a single-rising-dose study of TAK-063 [6] and emerging data from the present study. The dose groups for SSS were 3, 10, 20, 30, and 100 mg, and the dose groups for HJS were 3, 10, and 20 mg. Cohorts were staggered such that at a given dose level, SSS cohorts preceded cohorts of HJS at equivalent or higher doses (Supplementary Fig. 2). Decisions to increase or decrease subsequent dose levels occurred after review of blinded safety, tolerability, and PK data.
Study Endpoints
The primary endpoints were the percentage of subjects who experienced at least one AE after dosing and the percentage of subjects who met predefined criteria for markedly abnormal vital sign measurements, safety laboratory tests, or ECG parameters. Intensity of AEs was classified as mild, moderate, or severe, and each AE was assessed for association with the study drug. Documentation of AEs began on day 1, and AEs were assessed until the follow-up visit or call on day 14 (± 2 days). Vital signs were collected at screening, check-in, study days 1–7 (predose and 1, 4, 8, and 12 h postdose), and at study exit or early termination. Vital sign parameters were oral body temperature, respiration rate, and supine/standing blood pressure and pulse. Samples for hematology, serum chemistry, and urinalysis tests were collected following a minimum 10-h overnight fast at screening, check-in, predose on day 2, and at study exit. Samples for hormone tests (cortisol, growth hormone, prolactin, and thyroid-stimulating hormone) were collected on day 1 (predose as well as 3 and 24 h postdose), study exit, and early termination. ECGs were recorded at screening; at check-in; on days 1, 4, and 7 (predose and 1, 4, and 8 h postdose); and at study exit or early termination. Parameters measured by ECG were PR, RR, QRS, and QT intervals. Changes were considered to be clinically significant if intervention was needed or if the change exceeded the range of normal physiologic fluctuation.
Secondary endpoints included evaluation of the following plasma PK parameters: maximum observed plasma concentration (C max), time to reach maximum observed plasma concentration (t max), area under the plasma concentration–time curve from time 0 to 24 h postdose (AUC0–24), area under the plasma concentration–time curve from time 0 to last quantifiable concentration (AUCt), oral clearance (CL/F) (TAK-063 only), average plasma concentration at steady state (C av,ss), C max and AUC molar ratios between TAK-063 and the TAK-063 oxidative metabolite M-I, and accumulation ratio (AR) between day 7 and day 1 AUC0–24 for TAK-063 and M-I. Serial blood samples were collected predose as well as 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 16, and 24 h postdose on days 1 and 7 from all subjects; blood samples were collected predose on days 5 and 6 to confirm steady-state levels. Urine PK parameters included total amount excreted in urine from time 0 to 24 h (Ae24), fraction of drug excreted in urine (f e) (TAK-063 only), and renal clearance (CLR). Serial urine samples were collected at predose; postdose intervals of − 12–0 h, 0–6 h, 6–12 h, and 12–24 h on day 1; and postdose intervals of 0–6 h, 6–12 h, and 12–24 h on day 7. Plasma and urine samples were frozen at − 20 °C in duplicate sets, and plasma and urine concentrations of TAK-063 and M-I were measured by validated liquid chromatography and tandem and mass spectrometry with validated ranges of 0.5–1000 ng/mL and 5.5–2750 ng/mL, respectively, for both analytes. Elimination half-life (t ½) was not included as an endpoint, but was reported in this study.
Exploratory endpoints included assessment of change from baseline to day 7 postdose in ESRS-A scores in SSS and HJS, C-SSRS in SSS and HJS, and the change from baseline to day 7 or end of treatment in PANSS and CGI-S scores after 7 days of dosing in SSS. ESRS-A scores were evaluated for all subjects at check-in, day 1 at predose and 4 (± 1) h postdose, and day 7 at 4 (± 1) h postdose or early termination. C-SSRS was conducted at screening, at check-in, and on day 8 or early termination. PANSS and CGI-S assessments were conducted in SSS at screening, at check-in, and on day 7 postdose or early termination. In addition, assessments of cognition and mood were conducted in both HJS and SSS using a validated computerized cognition battery and computerized Bond-Lader Visual Analog Scale via the Cognitive Drug Research Computerized Assessment System. The cognition battery was conducted at check-in and on days 1, 2, 4, 6, and 7, and Bond-Lader Visual Analog Scale assessments were conducted on days 1 and 7 at predose and 2 (± 1), 6 (± 2), and 12 (± 1) h postdose or early termination. Electroencephalogram recordings, event-related potentials, and electromyographic recordings of pre-pulse inhibition were also conducted in SSS. Cognition, mood, and EEG results are reported elsewhere.
Pharmacokinetic/Adverse Event Modeling
PK/AE modeling analyses for somnolence were conducted with data from both HJS and SSS, and the EPS model was based on data from SSS. The incidence of AEs was modeled using a logistic regression model given by the expression f[P(AEi = 1)] = log[p/(1 − p)] = β + f exp, where AEi = 1 if subject i has an AE at some time during the study and AEi = 0 otherwise, using NONlinear Mixed Effects Modeling 7.2. Data processing and graphs were produced using R. The parameter β denotes the logit for subjects on placebo. The function f exp describes the exposure response relationship as a linear function (SLOPE × PK) where PK represents individual C max and AUC values. E max type [E max·PK/(PK + EC50)] functions were also used, but did not improve the fit. EC50 represents the drug concentration that yields a half-maximal response and PK represents individual Cmax and AUC values estimated with non-compartmental methods.
Statistical Analyses
All data analyses were generated using SAS Version 9.2. Unless otherwise stated, all statistical tests were two-tailed at α = 0.05; no adjustments were made for multiple comparisons. Subjects who received placebo in each cohort group were analyzed as one group for safety summaries. Dose proportionality was assessed for HJS (up to 20 mg) and SSS (up to 30 mg and over the entire range) using analysis of variance models for repeated measures. Natural log-transformed, dose-normalized plasma TAK-063 and M-I C max and AUCs were used as the responses, and treatment group, day, and day-by-treatment group interaction were used as the factors in the models. For SSS, dose proportionality was evaluated for two TAK-063 dose ranges: up to 30 mg, and the entire range, using a power model ln(PK parameter) = β 0 + β 1·ln(Dose) + ε, in which β 0 is the intercept, β 1 is the slope, and ε is the random error term. Dose proportionality was also presented graphically using box plots. PK parameters were determined using non-compartmental analysis with WinNonlin Enterprise Version 6.3 (Pharsight Corp., Mountain View, CA, USA). Plasma and urine PK parameters for TAK-063 and M-I were determined from the concentration–time profiles for all evaluable subjects.
Results
Demographics
A total of 162 subjects were screened, and 77 (30 HJS and 47 SSS) were subsequently enrolled into the study (Fig. 1). Of these enrollees, 12.5% (3 of 24) of HJS and 7.9% (3 of 38) of SSS did not complete the study, and all HJS and SSS discontinuations were reported in the TAK-063 group. The majority of subjects (HJS 90%; SSS 66%) were male, and no female HJS were randomized to the 10- or 20-mg TAK-063 group. The mean age, height, weight, and BMI were similar across HJS and SSS (Table 1). The majority of SSS were Black/African American. The first subject was screened on June 21, 2013, and the last contact of the last subject was on June 23, 2014.
Fig. 1.

Subject disposition. HJS healthy Japanese subjects, SSS subjects with stable schizophrenia. aReason for discontinuation: pretreatment event/adverse event. bReasons for discontinuations: voluntary withdrawal or other
Table 1.
Demographics and baseline characteristics
| Healthy Japanese subjects | Subjects with stable schizophrenia | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Placebo | TAK-063 | All subjects | Placebo | TAK-063 | All subjects | |||||||||
| 3 mg | 10 mg | 20 mg | Total TAK-063 | 3 mg | 10 mg | 20 mg | 30 mg | 100 mg | Total TAK-063 | |||||
| Number | n = 6 | n = 8 | n = 8 | n = 8 | n = 24 | N = 30 | n = 9 | n = 7 | n = 8 | n = 7 | n = 8 | n = 8 | n = 38 | N = 47 |
| Age, mean (SD), years | 38.5 (11.5) | 36.3 (9.8) | 25.9 (9.5) | 34.6 (10.6) | 32.3 (10.6) | 33.5 (10.9) | 43.1 (11.3) | 43.4 (4.2) | 39.6 (9.9) | 44.6 (9.0) | 45.3 (8.2) | 39.4 (8.1) | 42.4 (8.1) | 42.5 (8.7) |
| Sex, % | ||||||||||||||
| Male | 83.3 | 75.0 | 100.0 | 100.0 | 91.7 | 90.0 | 55.6 | 57.1 | 50.0 | 85.7 | 62.5 | 87.5 | 68.4 | 66.0 |
| Female | 16.7 | 25.0 | 0 | 0 | 8.3 | 10.0 | 44.4 | 42.9 | 50.0 | 14.3 | 37.5 | 12.5 | 31.6 | 34.0 |
| Racea, % | ||||||||||||||
| Asian | 100 | 100 | 100 | 100 | 100 | 100 | 0 | 0 | 0 | 14.3 | 0 | 0 | 2.6 | 2.1 |
| Blackb | 0 | 0 | 0 | 0 | 0 | 0 | 88.9 | 85.7 | 62.5 | 57.1 | 87.5 | 87.5 | 76.3 | 78.7 |
| White | 0 | 0 | 0 | 0 | 0 | 0 | 11.1 | 14.3 | 37.5 | 28.6 | 12.5 | 0 | 18.4 | 17.0 |
| BMI, mean (SD), kg/m2 | 22.2 (2.8) | 24.8 (2.0) | 22.7 (2.6) | 24.1 (2.0) | 23.9 (2.3) | 23.5 (2.4) | 28.8 (5.7) | 31.1 (3.3) | 27.0 (3.5) | 27.9 (5.5) | 32.2 (4.2) | 27.8 (4.5) | 29.2 (4.5) | 29.1 (4.7) |
BMI body mass index, SD standard deviation
aSubjects could also report as “multiracial”
bOr African American
Safety
No deaths or serious adverse events (SAEs) were reported, and of six total discontinuations, four subjects (3 SSS and 1 HJS) discontinued because of AEs. Across all subjects, the most common AE was somnolence (HJS 50.0%; SSS 55.3%) (Table 2). Other AEs occurring in more than 10% of SSS were akathisia (15.8%), orthostatic hypotension (15.8%), extrapyramidal disorder (13.2%), and headache (10.5%). Somnolence was the only AE that occurred in greater than 10% of HJS. The majority of AEs were mild [83.5% (71 of 85) in SSS and 83.9% (26 of 31) in HJS] or moderate [14.1% (12 of 85) in SSS and 16.1% (5 of 31) in HJS], and three SSS reported severe AEs, which included severe akathisia (30 mg), anxiety (100 mg), and dystonia (placebo). Among subjects treated with TAK-063, there were 25 drug-related AEs reported in HJS (placebo 3; 3 mg 7; 10 mg 8; 20 mg 10) and 66 in SSS (placebo 4; 3 mg 4; 10 mg 9; 20 mg 11; 30 mg 18; 100 mg 24). No significant changes in C-SSRS were detected.
Table 2.
Treatment-emergent AEs occurring in 2 or more subjects treated with TAK-063 in healthy Japanese subjects or subjects with stable schizophrenia
| Healthy Japanese subjects | Subjects with stable schizophrenia | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Placebo | TAK-063 | Placebo | TAK-063 | |||||||||
| 3 mg | 10 mg | 20 mg | Total TAK-063 | 3 mg | 10 mg | 20 mg | 30 mg | 100 mg | Total TAK-063 | |||
| Number | n = 6 | n = 8 | n = 8 | n = 8 | N = 24 | n = 9 | n = 7 | n = 8 | n = 7 | n = 8 | n = 8 | N = 38 |
| Subjects with any AE, n (%) | 3 (50.0) | 4 (50.0) | 5 (62.5) | 5 (62.5) | 14 (58.3) | 5 (55.6) | 4 (57.1) | 7 (87.5) | 5 (71.4) | 6 (75.0) | 8 (100.0) | 30 (78.9) |
| Somnolence | 2 (33.3) | 3 (37.5) | 4 (50.0) | 5 (62.5) | 12 (50.0) | 2 (22.2) | 2 (28.6) | 3 (37.5) | 4 (57.1) | 4 (50.0) | 8 (100.0) | 21 (55.3) |
| Akathisia | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (25.0) | 1 (14.3) | 2 (25.0) | 1 (12.5) | 6 (15.8) |
| Orthostatic hypotension | 0 | 0 | 0 | 1 (12.5) | 1 (4.2) | 0 | 0 | 2 (25.0) | 1 (14.3) | 2 (25.0) | 1 (12.5) | 6 (15.8) |
| Extrapyramidal disorder | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 (37.5) | 2 (25.0) | 5 (13.2) |
| Headache | 0 | 1 (12.5) | 0 | 1 (12.5) | 2 (8.3) | 0 | 0 | 0 | 1 (14.3) | 2 (25.0) | 1 (12.5) | 4 (10.5) |
| Anxiety | 0 | 0 | 0 | 0 | 0 | 1 (11.1) | 0 | 1 (12.5) | 0 | 0 | 2 (25.0) | 3 (7.9) |
| Oromandibular dystonia | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (14.3) | 0 | 2 (25.0) | 3 (7.9) |
| Constipation | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (12.5) | 1 (12.5) | 2 (5.3) |
| Dizziness | 0 | 0 | 1 (12.5) | 0 | 1 (4.2) | 0 | 0 | 0 | 1 (14.3) | 0 | 1 (12.5) | 2 (5.3) |
| Dizziness postural | 0 | 1 (12.5) | 0 | 0 | 1 (4.2) | 0 (0.0) | 1 (14.3) | 0 | 0 | 1 (12.5) | 0 | 2 (5.3) |
| Dyskinesia | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (25.0) | 0 | 2 (5.3) |
| Dystonia | 0 | 0 | 0 | 0 | 0 | 1 (11.1) | 0 | 0 | 2 (28.6) | 0 | 0 | 2 (5.3) |
| Nausea | 0 | 1 (12.5) | 0 | 1 (12.5) | 2 (8.3) | 0 | 0 | 1 (12.5) | 0 | 0 | 1 (12.5) | 2 (5.3) |
| Orthostatic heart rate response increased | 1 (16.7) | 0 | 1 (12.5) | 0 | 1 (4.2) | 0 | 0 | 1 (12.5) | 0 | 0 | 1 (12.5) | 2 (5.3) |
AE adverse event
None of the individual values and changes from baseline in vital signs measurements or physical examination parameters were considered clinically meaningful for either HJS or SSS, and no subject showed any clinically significant ECG parameter at any time at baseline or postdose. No clinically significant changes in serum hormone levels, hematology, or clinical laboratory tests were observed, and no trend was apparent for any hormonal or other clinical laboratory and hematology tests relative to TAK-063 dose level.
At equivalent doses, a higher rate of post-treatment EPS was observed in SSS compared with HJS (Table 3). EPS symptoms were reported in one SSS (11.1%) in the placebo group, and in 14 SSS (36.8%) treated with TAK-063, most of whom (9 of 14) were in the 30- or 100-mg dose group. Of all the HJS, one individual (4.2%) from the 20-mg TAK-063 group reported EPS symptoms. Most post-treatment EPS events in SSS were of mild or moderate severity, and one of the SSS who experienced EPS discontinued the study. There were no changes in ESRS-A subscale means for any HJS or in the placebo group in SSS. In SSS treated with TAK-063, there were no meaningful changes from baseline in ESRS-A subscales; the only absolute mean change from baseline greater than 1.0 for any ESRS-A subscale at day 7 postdose was observed in the 10-mg group for the akathisia subscale (1.43, standard deviation = 2.51). A total of nine shifts in EPS symptoms from absence at baseline to presence postdose day 7 were recorded in SSS. Of these, two occurred in the 10-mg group, five in the 30-mg group, and two in the 100-mg group. No shifts from absence to presence occurred in HJS or in the placebo group of the SSS.
Table 3.
Treatment-emergent EPS by category and preferred term for healthy Japanese subjects and subjects with stable schizophrenia
| Healthy Japanese subjects | Subjects with stable schizophrenia | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Placebo | TAK-063 | Placebo | TAK-063 | |||||||||
| 3 mg | 10 mg | 20 mg | All TAK-063 subjects | 3 mg | 10 mg | 20 mg | 30 mg | 100 mg | All TAK-063 subjects | |||
| Number | n = 6 | n = 8 | n = 8 | n = 8 | N = 24 | n = 9 | n = 7 | n = 8 | n = 7 | n = 8 | n = 8 | N = 38 |
| Subjects with any EPS, n (%) | 0 | 0 | 0 | 1 (12.5) | 1 (4.2) | 1 (11.1) | 0 | 2 (25.0) | 3 (42.9) | 5 (62.5) | 4 (50.0) | 14 (36.8) |
| Akathisia | 0 | 0 | 0 | 1 (12.5) | 1 (4.2) | 0 | 0 | 2 (25.0) | 1 (14.3) | 2 (25.0) | 1 (12.5) | 6 (15.8) |
| Restlessness | 0 | 0 | 0 | 1 (12.5) | 1 (4.2) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Dyskinesia | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (25.0) | 0 | 2 (5.3) |
| Dystonia | 0 | 0 | 0 | 0 | 0 | 1 (11.1) | 0 | 0 | 3 (42.9) | 0 | 2 (25.0) | 5 (13.2) |
| Oromandibular dystonia | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (14.3) | 0 | 2 (25.0) | 3 (7.9) |
| Dystonia | 0 | 0 | 0 | 0 | 0 | 1 (11.1) | 0 | 0 | 2 (28.6) | 0 | 0 | 2 (5.3) |
| Other EPSa | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 (50.0) | 2 (25.0) | 6 (15.8) |
| Extrapyramidal disorder | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 (37.5) | 2 (25.0) | 5 (13.2) |
| Musculoskeletal stiffness | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (12.5) | 0 | 1 (2.6) |
EPS extrapyramidal syndromes
aCode to more than one symptom domain of EPS Standardized Medical Dictionary for Regulatory Activities Queries
Pharmacokinetics
No appreciable differences in plasma PK parameters were observed between HJS and SSS at equivalent doses (Fig. 2, Table 4). No difference in C max, AR (AUC0–24 or C max), or CL/F was observed between HJS and SSS at day 7. AUC0–24 was elevated in HJS compared with SSS in the 10- and 20-mg TAK-063 groups (Table 4). TAK-063 accumulated to a minor extent on day 7, with mean ARs for C max ranging from 1.1 to 2.0 and those for AUC0–24 ranging from 1.1 to 2.1 across all subjects (Table 4).
Fig. 2.
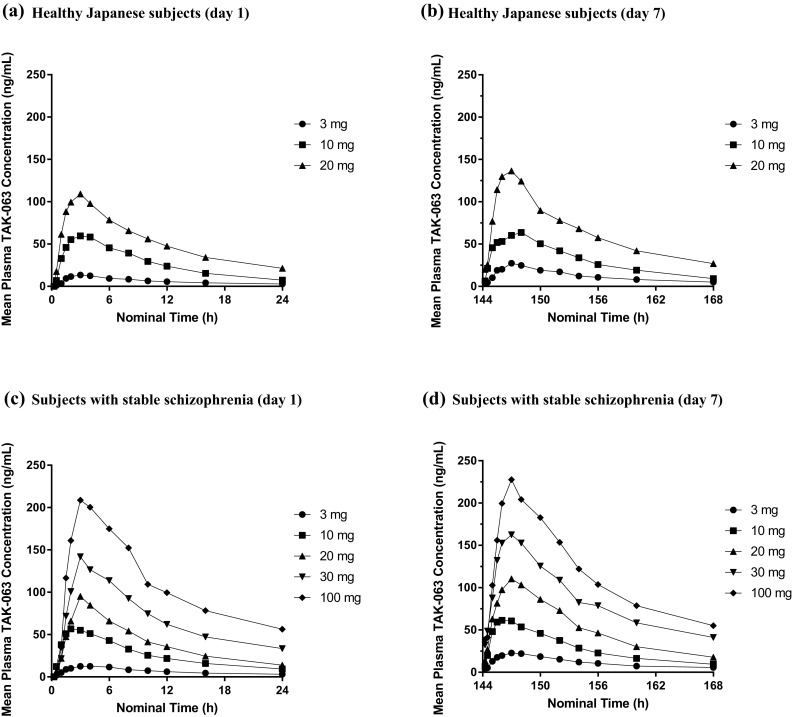
Mean concentration of TAK-063 at day 1 and day 7 following once-daily administration in healthy Japanese subjects and subjects with stable schizophrenia: a healthy Japanese subjects (day 1); b healthy Japanese subjects (day 7); c subjects with stable schizophrenia (day 1); d subjects with stable schizophrenia (day 7)
Table 4.
Plasma PK parameters for TAK-063 on days 1 and 7 following ascending multiple doses in healthy Japanese subjects and subjects with stable schizophrenia
| Parameter [mean (CV, %)] | Healthy Japanese subjects | Subjects with stable schizophrenia | ||||||
|---|---|---|---|---|---|---|---|---|
| 3 mg | 10 mg | 20 mg | 3 mg | 10 mg | 20 mg | 30 mg | 100 mg | |
| Day 1 | ||||||||
| Number | n = 8 | n = 8 | n = 8 | n = 7 | n = 8 | n = 7 | n = 8 | n = 7 |
| C max (ng/mL) | 14.9 (36) | 69.4 (19) | 116.2 (15) | 17.0 (42) | 62.8 (26) | 95.1 (32) | 151.4 (21) | 211.3 (40) |
| t max (h)a | 2.8 (1.5, 4.0) | 3.4 (2.0, 8.0) | 2.5 (1.5, 4.0) | 3.1 (1.0, 6.0) | 2.2 (1.5, 4.0) | 3.0 (3.0, 3.0) | 3.0 (2.0, 4.0) | 4.0 (2.0, 6.0) |
| t ½ (h) | 12.3 (29) | 6.9 (21) | 9.7 (19) | 12.8 (40) | 10.2 (34) | 7.9 (31) | 13.8 (38) | 13.3 (48) |
| AUC0–24 (ng·h/mL) | 148.6 (37) | 651.9 (11) | 1243.9 (22) | 159.0 (38) | 623.2 (23) | 939.3 (43) | 1619.0 (24) | 2503.2 (46) |
| CL/F (L/h) | 16.9 (31) | 13.9 (14) | 13.9 (33) | 16.1 (46) | 14.0 (31) | 21.3 (38) | 14.4 (37) | 38.1 (66) |
| Day 7 | ||||||||
| Number | n = 8 | n = 7 | n = 6 | n = 7 | n = 7 | n = 7 | n = 7 | n = 7 |
| C max (ng/mL) | 29.1 (27) | 74.6 (17) | 139.8 (18) | 25.6 (18) | 70.3 (16) | 120.0 (33) | 179.4 (19) | 228.3 (24) |
| t max (h)a | 3.0 (1.5, 4.0) | 1.5 (1.0, 6.0) | 2.5 (1.5, 4.0) | 3.0 (2.0, 6.0) | 3.0 (1.0, 6.0) | 3.0 (2.0, 4.0) | 3.0 (1.5, 3.0) | 3.0 (2.0, 3.3) |
| t ½ (h) | 11.3 (29) | 8.2 (28) | 11.0 (15) | 13.2 (16) | 9.6 (16) | 8.6 (22) | 13.7 (41) | 13.0 (43) |
| C av,ss | 12.3 (28) | 30.7 (12) | 64.0 (15) | 11.7 (22) | 28.3 (23) | 51.7 (31) | 84.6 (24) | 115.2 (26) |
| AUC0–24 (ng·h/mL) | 295.6 (28) | 736.1 (12) | 1536.4 (15) | 280.6 (22) | 680.1 (23) | 1240.8 (31) | 2030.9 (24) | 2764.0 (26) |
| CL/F (L/h) | 10.8 (26) | 13.8 (11) | 13.3 (14) | 11.2 (25) | 15.4 (25) | 17.1 (23) | 15.5 (23) | 38.5 (28) |
| AR (AUC0–24)b | 2.1 (23) | 1.1 (15) | 1.1 (16) | 1.9 (29) | 1.1 (19) | 1.4 (23) | 1.3 (10) | 1.2 (27) |
| AR Cmax (ng·h/mL)c | 2.0 (21) | 1.1 (16) | 1.1 (16) | 1.7 (33) | 1.2 (17) | 1.3 (20) | 1.3 (41) | 1.2 (24) |
AR accumulation ratio, AUC 0–24, area under the plasma concentration–time curve from time 0 to 24 h postdose, C av,ss average plasma concentration at steady state, CL/F oral clearance, C max maximum observed plasma concentration, CV coefficient of variation, PK pharmacokinetic, t ½ elimination half-life, t max time to reach maximum observed plasma concentration
a t max is presented as the median (minimum, maximum)
bDay 7 AUC0–24 (ng·h/mL)/day 1 AUC0–24 (ng·h/mL)
cDay 7 C max/day 1 C max
The rate of TAK-063 absorption was moderate and independent of dosage with median t max of 3 h in SSS and 1.5–3 h in HJS (Table 4). In SSS, the mean TAK-063 C max and AUC0–24 values on day 1 increased 9- and 10-fold, respectively, from 3 to 30 mg, suggesting a dose-proportional increase in exposure, and 12- and 16-fold, respectively, from 3 to 100 mg, indicating a less than dose-proportional increase in exposure to TAK-063 for this day 1 dose range (Table 4). On day 7, the mean TAK-063 C max and AUC0–24 values increased 7-fold from 3 to 30 mg in SSS (Table 4). For the dose range of 3–100 mg on day 7, the mean TAK-063 C max and AUC0–24 values increased 9- and 10-fold, respectively, again indicating a less than dose-proportional increase in exposure to TAK-063 on repeat dosing during this period in SSS (Fig. 3). Across the 3- to 20-mg dose range in HJS, the mean C max and AUC0–24 values increased ~ 8-fold on day 1 and ~ 5-fold on day 7, suggesting an approximately dose-proportional increase in exposure on day 1, with a less than dose-proportional increase on day 7.
Fig. 3.
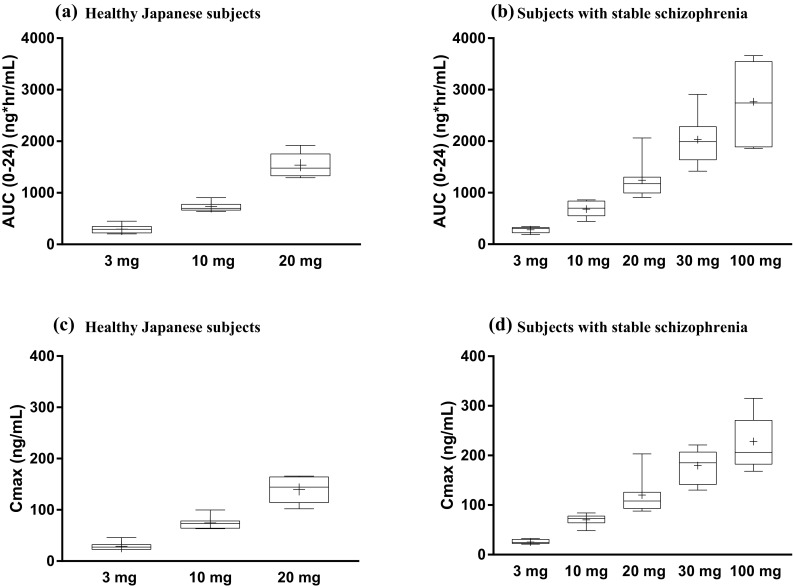
TAK-063 exposure on day 7 following once-daily oral doses: a AUC(0–24) for healthy Japanese subjects; b AUC(0–24) for subjects with stable schizophrenia; c C max for healthy Japanese subjects; d C max for subjects with stable schizophrenia. Box plots extend from the 25th to 75th percentiles, and whiskers extend from minimum to maximum values. Plus (+) sign denotes the mean. AUC (0–24) area under the plasma concentration–time curve from time 0 to 24 h postdose, C max maximum observed plasma concentration
Estimates from the power model indicated dose proportionality over the 3- to 30-mg dose range for TAK-063 and M-I in SSS on day 1 and a slightly less than dose-proportional increase in exposure at day 7. Across the entire dose range for SSS (3–100 mg), the estimate for slopes of the exposures (AUC0–24 and C max) on days 1 and 7 ranged from 0.5 to 0.8 (Fig. 2). For HJS, dose-proportionality analysis of dose-normalized C max and AUC0–24 of TAK-063 and M-I showed a slightly more than dose-proportional increase on day 1 and a less than dose-proportional increase on day 7 when comparing doses of 20 mg to doses of 3 mg (Fig. 3). Analysis of trough concentrations suggests that steady state was achieved by 5–6 days of dosing in SSS and HJS for TAK-063 and M-I. After C max was reached on days 1 and 7, plasma concentrations of TAK-063 decreased in a biphasic manner with an initial rapid phase followed by a slower terminal phase (Fig. 2).
The PK parameters for M-I were comparable to those of TAK-063 for both HJS and SSS (Supplementary Table 1). The range of median t max was 2.0–3.1 h for SSS and 2.5–3.0 h for HJS. In SSS and HJS, the concentration–time profiles of M-I were similar to that of TAK-063, and steady state was achieved by day 5 or 6, as with TAK-063. M-I accumulated to a small degree on day 7 in all dose groups in SSS and HJS, with the mean accumulation ratios for AUC0–24 ranging from 1.1 to 2.1, and the ratios for C max (day 7 to day 1) ranging from 1.0 to 1.8 across SSS and HJS (Supplementary Table 1). No difference was observed for M-I accumulation ratios (AUC0–24 and C max) between HJS and SSS for any dose groups. Upon normalization of molecular weight for M-I and TAK-063, plasma exposure in HJS was comparable at all doses, with ratios of 0.5 on day 7. C max molar ratios between TAK-063 and M-I were higher in SSS compared with HJS for the 3-, 10-, and 20-mg TAK-063 groups.
For HJS and SSS, renal clearance was a minor elimination route for TAK-063, with Ae24 between 0.02% and 0.05% of the dose on day 7 across all dose groups (range 5.21–16.2 mL/h). The fraction of TAK-063 eliminated in the urine was no larger than 0.08% for any dose group in either HJS or SSS on day 7 (Table 5), and renal clearance generally increased with TAK-063 dosage on day 1 and day 7 for TAK-063 and M-I (Table 5 and Supplementary Table 2).
Table 5.
Urine PK parameters for TAK-063 on days 1 and 7 following once-daily oral doses in healthy Japanese subjects and subjects with stable schizophrenia
| Healthy Japanese subjects | Subjects with stable schizophrenia | |||||||
|---|---|---|---|---|---|---|---|---|
| 3 mg | 10 mg | 20 mg | 3 mg | 10 mg | 20 mg | 30 mg | 100 mg | |
| Day 1 [mean (CV, %)] | ||||||||
| Number | n = 8 | n = 8 | n = 8 | n = 7 | n = 8 | n = 7 | n = 8 | n = 7 |
| f e (%) | 0.000 (NA) | 0.0359 (137) | 0.0707 (68) | 0.000 (NA) | 0.0364 (72) | 0.0222 (77) | 0.0332 (61) | 0.0349 (78) |
| Ae24 (ng) | 0.000 (NA) | 3592 (137) | 14,145 (68) | 0.000 (NA) | 3642 (72) | 4433 (77) | 9952 (61) | 34,893 (79) |
| CLR a (mL/h) | NC (NC) (n = 0) |
7.0 (100) (n = 6) |
12.9 (58) (n = 7) |
NC (NC) (n = 0) |
8.4 (46) (n = 6) |
5.2 (48) (n = 6) |
6.7 (24) (n = 7) |
16.2 (81) (n = 7) |
| Day 7 [mean (CV, %)] | ||||||||
| Number | n = 8 | n = 7 | n = 6 | n = 7 | n = 7 | n = 7 | n = 7 | n = 7 |
| f e (%) | 0.000 (NA) | 0.0476 (73) | 0.0796 (60) | 0.000 (NA) | 0.0443 (64) | 0.0399 (59) | 0.0502 (66) | 0.0355 (59) |
| Ae24 (ng) | 0.000 (NA) | 4765 (73) | 15,914 (60) | 0.000 (NA) | 4431 (64) | 7979 (59) | 15,066 (66) | 35,527 (59) |
| CLR a (mL/h) | NC (NC) (n = 0) |
7.3 (44) (n = 6) |
10.5 (61) (n = 6) |
NC (NC) (n = 0) |
6.8 (65) (n = 7) |
7.2 (15) (n = 6) |
7.3 (47) (n = 7) |
13.3 (55) (n = 7) |
Ae 0–24 total amount excreted in urine from time 0 to 24 h, CL R renal clearance, CV coefficient of variation, f e fraction of drug excreted in urine, NA not applicable, NC not calculated, PK pharmacokinetic
aFor CLR data, the number of eligible subjects is provided below the values
Pharmacokinetics/Adverse Event Modeling
PK/AE models estimating the parameters and frequencies of AEs based on these study data are presented in Fig. 4. At the 3-mg dose, the frequency of somnolence [standard error (SE)] in HJS and SSS was estimated at 28.8% (6.9%) and 29.5% (6.8%) based on C max and AUC, respectively, while for the 100-mg dose, these frequencies (SE) were 82.9% (8.9%) and 86.0% (8.5%), respectively (Fig. 4a, b; Supplementary Table 3). Based on data from the SSS, the frequency of EPS increased with increasing exposure to TAK-063. At the 3-mg dose, the frequency of EPS (SE) was estimated at 15.8% (7.1%) and 18.5% (7.4%) based on C max and AUC, respectively, while for the 100-mg dose, these frequencies (SE) were 62.0% (13.7%) and 60.1% (14.4%), respectively (Fig. 4c, d; Supplementary Table 3). Linear models exhibited suitable goodness of fit, with no extensive model improvement using E max functions.
Fig. 4.

Probability of adverse event occurrence with multiple rising doses of TAK-063 in healthy Japanese subjects and SSS: a C max vs somnolencea; b AUC vs somnolenceb; c C max vs extrapyramidal syndromesc; d AUC vs extrapyramidal syndromesd; Observed (points) and predicted (blue line) incidence of EPS (SSS only) and somnolence vs TAK-063 C max or AUC. Vertical lines indicate the mean TAK-063 C max or AUC by dose; shaded regions indicate the 95% confidence interval. AUC area under the plasma concentration–time curve, C max maximum observed plasma concentration, EPS extrapyramidal syndromes, MRD multiple rising dose, SE standard error, SSS subjects with stable schizophrenia. a β (SE) = − 1.22 (0.40); slope (SE) = 0.012 (0.0038). b β (SE) = − 1.18 (0.39); slope (SE) = 0.001 (0.0003). c β (SE) = − 1.95 (0.62); slope (SE) = 0.011 (0.0043). d β (SE) = − 1.70 (0.56); slope (SE) = 0.001 (0.0003)
Pharmacodynamics
There were no meaningful changes in PANSS or CGI-S scores from baseline to postdose day 7 across any doses in SSS.
Discussion
The results from this study suggest that TAK-063 was safe and generally well tolerated when administered once daily for 7 days at doses of up to 20 mg/day in HJS and up to 100 mg/day in SSS. No deaths or SAEs were reported with TAK-063. Only four subjects discontinued because of AEs, and the majority of AEs were of mild or moderate severity. Most drug-related AEs in SSS (42 of 66) occurred at TAK-063 doses ≥ 30 mg, although no AE observed in this study was considered to be dose-limiting. TAK-063 was shown to be safe and well tolerated in a previously conducted single-rising-dose study (single daily doses up to 1000 mg). In this study, no subjects discontinued because of AEs, and no SAEs were reported [6]. Consistent with the single-rising-dose trial, the most common AE in the present study was somnolence in HJS and SSS. In addition, there were no increases in prolactin or other hormone levels or clinically meaningful changes in clinical laboratory or hematology parameters in this study [4, 7, 8].
Antipsychotics are generally less well tolerated in healthy subjects than SSS especially with respect to EPS and somnolence [9]. Differential tolerability of antipsychotics between Western and Asian populations has also been reported, although relatively few studies have been conducted in which equivalent doses of antipsychotics are administered to both healthy subjects and SSS for direct, within-study comparisons of safety and tolerability. To our knowledge, this study is the first study of this nature conducted in Asian (Japanese) healthy subjects and Western SSS. In this study, across equivalent doses (3, 10, and 20 mg), the incidence of somnolence was relatively similar in frequency and in intensity between groups, with the majority of events being of mild severity [10]. Interestingly, at equivalent doses, the rate of EPS was higher in SSS than in HJS and only one HJS (20 mg) experienced EPS AEs. Although more frequent in SSS, EPS AEs were generally tolerable, with just a single discontinuation (SSS) because of EPS. Most of the SSS who experienced EPS (9 of 14) were in the 30-mg or 100-mg dose group. Similarly, the majority of the shifts from absence to presence of EPS in SSS occurred at 30-mg and 100-mg TAK-063 doses, indicating that doses ≥ 30 mg were associated with increased incidence of EPS. These between-group comparisons are limited by the relatively small sample size, but suggest that there may be more variation in tolerability to PDE10A inhibitors between populations than previously reported for current antipsychotics.
In the fed state, PK parameters for TAK-063 were comparable at equivalent doses between HJS and SSS, and plasma concentrations of TAK-063 decreased in a biphasic manner after C max had been reached. The rate of TAK-063 absorption did not substantially change between doses. Exposure of TAK-063 as measured by C max and AUC0–24 in SSS increased in a dose-proportional manner on day 1 at doses between 3 and 30 mg, and slightly less than dose proportional on day 7. Over the entire dose range of 3–100 mg on both days, the exposures of TAK-063 were less than dose proportional. In HJS, exposure was slightly more than dose proportional on day 1 and less than dose proportional on day 7 over a dose range of 3–20 mg. In both subject groups, the exposure to M-I increased in a similar fashion to TAK-063. Based on the present study and the phase 1, single-rising-dose study, the dose range explored included active exposures up to and beyond the likely therapeutic dose. A maximum tolerated dose was not determined in this trial.
Our PK/AE models estimated an increase in the frequency of somnolence and EPS with increasing TAK-063 exposure [11], and estimated frequencies of somnolence and EPS generally fell within the 95% confidence interval across doses. Model values were in agreement whether based on C max or AUC. Broadly, the slope of the curves suggested a steeper increase in somnolence and EPS frequency between the 20- and 30-mg TAK-063 doses. This change may suggest that single daily doses of ≤ 30 mg are suitable for further development in schizophrenia.
Conclusion
The data presented here are consistent with PK, safety, and tolerability findings from a phase 1, single-rising-dose study indicating that TAK-063 was well tolerated in HJS, with AEs that were non-serious and mild or moderate in intensity [6]. Overall, the data presented in this study show a favorable safety and tolerability profile of TAK-063 with multiple escalating doses up to 100 mg in SSS. The available safety, PK, and PK/AE modeling data suggest that single daily doses of < 30 mg are suitable for future clinical development of TAK-063.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
Some data included in this manuscript were presented in abstract form (Tsai M, Goldsmith P, Xie J, Macek T. Development of PK/AE Models in Subjects With Schizophrenia and in Healthy Japanese and Non-Japanese Subjects. The International Society for CNS Clinical Trials and Methodology 2015 and Goldsmith P, Affinito J, Macek T, Tsai M, Xie J, Gertsik L. Safety, Tolerability, and Pharmacokinetics of a Novel PDE10A Inhibitor–TAK-063–Following Multiple Dosing in Subjects With Stable Schizophrenia and Healthy Japanese Subjects. American Society of Clinical Psychopharmacology 2015).
Compliance with Ethical Standards
Role of funding source
The clinical study was funded by Takeda Development Center Americas, Inc. Medical writing assistance was provided by Stephanie Agbu, Ph.D, and Jake Edelstein, Ph.D, of inVentiv Medical Communications and supported by Takeda Development Center Americas, Inc.
Conflict of interest
John Affinito and Maggie McCue are employees of Takeda Development Center Americas, Inc., Deerfield, IL. At the time the study was conducted, Max Tsai, Jinhui Xie, and Thomas A. Macek were employees of Takeda Development Center Americas, Inc.; Paul Goldsmith was an employee of Takeda Development Centre Europe Ltd., London, UK; and Stefan Roepcke was an employee of Takeda Pharmaceuticals International GmbH, Zürich, Switzerland. Lev Gertsik is an employee of California Clinical Trials Medical Group/Parexel, Glendale, CA, USA. He is a consultant to Takeda.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Study registration
This study is registered as “Safety, Tolerability and Pharmacokinetic Study of Multiple Rising Doses of TAK-063 in Participants With Stable Schizophrenia and Healthy Participants” (Clinical Trials ID: NCT01879722).
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1007/s40268-017-0214-8) contains supplementary material, which is available to authorized users.
References
- 1.Suzuki K, Harada A, Shiraishi E, Kimura H. In vivo pharmacological characterization of TAK-063, a potent and selective phosphodiesterase 10A inhibitor with antipsychotic-like activity in rodents. J Pharmacol Exp Ther. 2015;352(3):471–479. doi: 10.1124/jpet.114.218552. [DOI] [PubMed] [Google Scholar]
- 2.Ginovart N, Kapur S. Role of dopamine D(2) receptors for antipsychotic activity. Handb Exp Pharmacol. 2012;212:27–52. doi: 10.1007/978-3-642-25761-2_2. [DOI] [PubMed] [Google Scholar]
- 3.Leucht S, Corves C, Arbter D, Engel RR, Li C, Davis JM. Second-generation versus first-generation antipsychotic drugs for schizophrenia: a meta-analysis. Lancet. 2009;373(9657):31–41. doi: 10.1016/S0140-6736(08)61764-X. [DOI] [PubMed] [Google Scholar]
- 4.Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):1209–1223. doi: 10.1056/NEJMoa051688. [DOI] [PubMed] [Google Scholar]
- 5.Suzuki K, Harada A, Suzuki H, Miyamoto M, Kimura H. TAK-063, a PDE10A inhibitor with balanced activation of direct and indirect pathways, provides potent antipsychotic-like effects in multiple paradigms. Neuropsychopharmacology. 2016;41(9):2252–2262. doi: 10.1038/npp.2016.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsai M, Chrones L, Xie J, Gevorkyan H, Macek TA. A phase 1 study of the safety, tolerability, pharmacokinetics, and pharmacodynamics of TAK-063, a selective PDE10A inhibitor. Psychopharmacology. 2016;233(21–22):3787–3795. doi: 10.1007/s00213-016-4412-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Henderson DC, Cagliero E, Copeland PM, Borba CP, Evins E, Hayden D, et al. Glucose metabolism in patients with schizophrenia treated with atypical antipsychotic agents: a frequently sampled intravenous glucose tolerance test and minimal model analysis. Arch Gen Psychiatry. 2005;62(1):19–28. doi: 10.1001/archpsyc.62.1.19. [DOI] [PubMed] [Google Scholar]
- 8.Remington G. Understanding antipsychotic “atypicality”: a clinical and pharmacological moving target. J Psychiatry Neurosci. 2003;28(4):275–284. [PMC free article] [PubMed] [Google Scholar]
- 9.Cutler NR. Pharmacokinetic studies of antipsychotics in healthy volunteers versus patients. J Clin Psychiatry. 2001;62(Suppl 5):10–13. [PubMed] [Google Scholar]
- 10.Okuma T. Differential sensitivity to the effects of psychotropic drugs: psychotics vs normals; Asian vs Western populations. Folia Psychiatr Neurol Jpn. 1981;35(1):79–87. doi: 10.1111/j.1440-1819.1981.tb00203.x. [DOI] [PubMed] [Google Scholar]
- 11.Tsai M, Goldsmith P, Xie J, Macek TA. Development of PK/AE models in subjects with schizophrenia and in healthy Japanese and non-Japanese Subjects. Poster presented at: International Society for CNS Clinical Trials and Methodology (ISCTM) 2015 Autumn Conference; August 27–29, 2015; Amsterdam, Netherlands.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.