

Abstract
Background
We showed previously that nuclear localization of the androgen receptor (AR) and expression of the androgen-responsive gene FK506-binding protein 5 (FKBP5) in esophageal adenocarcinoma (EAC) tissues were associated with decreased patient survival, suggesting a role for androgens in this cancer.
Aim
To investigate the effect of the AR ligand 5α-dihydrotestosterone (DHT) on AR-expressing EAC cell lines in vitro.
Methods and Results
In tissue resection specimens from EAC patients, FKBP5 expression was positively associated with proliferation as measured by Ki-67 expression. We stably transduced AR into three AR-negative EAC cell lines, OE33, JH-EsoAd1, and OE19, to investigate androgen signaling in vitro. In the AR-expressing cell lines, 10 nM DHT, the concentration typically used to study AR signaling, induced changes in the expression of androgen-responsive genes and inhibited proliferation by inducing cell cycle arrest and senescence. At lower DHT concentrations near the half maximal inhibitory concentration (IC50), the AR-expressing cell lines proliferated and there were changes in the expression of androgen-responsive genes. In direct co-culture with cancer-associated fibroblast-like PShTert myofibroblasts, 10 nM DHT induced changes in the expression of androgen-responsive genes but did not inhibit proliferation.
Conclusions
This is the first study to show that EAC cell lines respond to androgen in vitro. Proliferation together with the expression of androgen-responsive genes was dependent on the concentration of DHT, or the presence of a permissive microenvironment, consistent with observations in the tissues. These findings are consistent with a role for androgen signaling in EAC.
Electronic supplementary material
The online version of this article (doi:10.1007/s10620-017-4794-5) contains supplementary material, which is available to authorized users.
Keywords: Esophageal adenocarcinoma, Androgen receptor, Fibroblast, FKBP5, Dihydrotestosterone, Direct co-culture, In vitro
Introduction
The incidence of esophageal adenocarcinoma (EAC) has increased rapidly over recent decades in Western countries [1–5]. It has a dismal prognosis, with around 65% of patients unsuitable for surgery at the time of diagnosis and an overall five-year survival rate of less than 15% [6, 7]. The major risk factors for EAC are gastro-esophageal reflux disease and obesity, leading to the only described precursor lesion for this cancer, Barrett’s esophagus. This cancer has one of the highest male-to-female ratios reported for cancers of non-reproductive organs, ranging from 7–10 to 1 [1, 2, 4, 6, 8–11], significantly higher than for the major risk factors. The gender difference appears to result from an approximate 20-year delay in onset in females of Barrett’s esophagus [12] and EAC [13]. These observations are consistent with a role for the sex steroid hormones in the biology of this cancer, with their concentrations differing between males and females, and changing over the lifespan.
The most important sex hormones in males are the androgens, and the most predominant androgen is testosterone. Testosterone passes through the cell membrane and into the cytoplasm where it, or its more physiologically effective metabolite 5α-dihydrotestosterone (DHT), binds to and activates the androgen receptor (AR). Activated AR translocates from the cytoplasm into the nucleus and binds to androgen response elements in the genome, influencing the transcription of androgen-responsive genes. The nature of the response can be modified by the relative abundance of multiple co-regulators (both co-activators and corepressors) [14].
We previously reported the immunostaining of EAC tissues for AR and the androgen-responsive gene FK506-binding protein 5 (FKBP5) [15]. We detected AR in the cancer cells of 75 of 77 cases, and in 70 it was nuclear. The expression of FKBP5 was observed in 64% of cases and only when the AR was nuclear. There was a significant association between nuclear AR and FKBP5 expression and decreased survival.
Given the association between AR localization, FKBP5 expression, and poor survival, we sought suitable cell lines to investigate the effect of androgen signaling on the behaviors of EAC cells. All available cell lines were AR negative, probably due to loss of AR expression during the establishment of cell lines from tissues [15]. We therefore stably transduced three EAC cell lines with AR. The aim of this study was to investigate factors that affect the growth of and gene expression in AR-expressing EAC cell lines in response to androgen.
Methods
Immunohistochemistry for Ki-67 and FKBP5
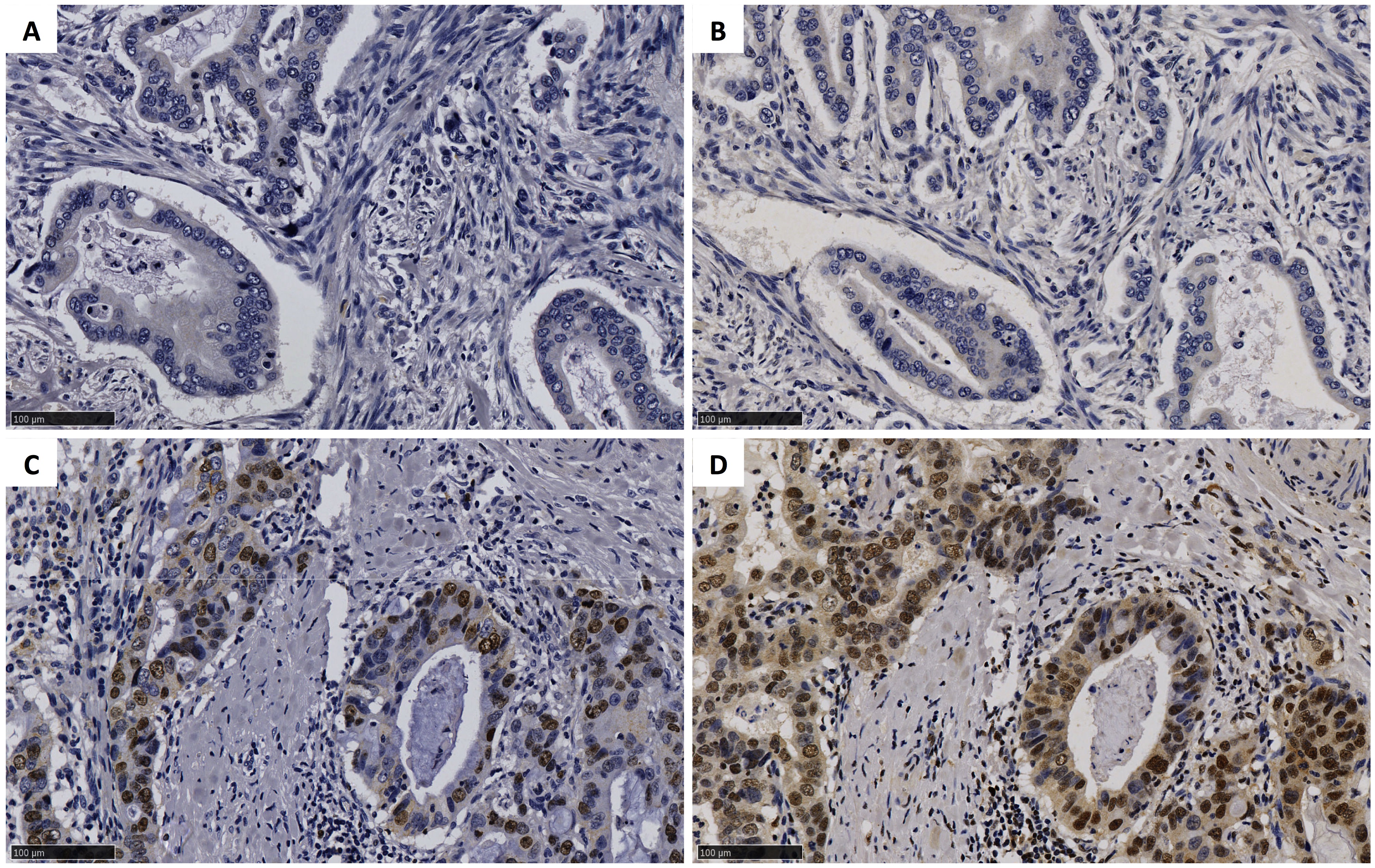
Tissue microarrays (TMAs) composed of one or more representative cores of EAC were constructed as described previously [16]. Sequential sections consisting of cores from 74 cases were immunostained for FKBP5 [15] and Ki-67 [17]. Cores were scored as the percent of epithelial cells that expressed FKBP5 or Ki-67 as follows: 0, negative; 1, < 5% (rare); 2, < 25%; 3, > 25% < 75%; 4, > 75%. The median score for FKBP5 and Ki-67 (Ki-67 index) was determined for multiple cores from each case.
Cell Culture
The EAC cell lines OE33, JH-EsoAd1, and OE19 [18, 19] were obtained from the ATCC, Johns Hopkins University, and Sigma-Aldrich, respectively. They were stably transduced with AR and green fluorescent protein (GFP), or with GFP only [15]. At least six single-cell clones were established from each AR-transduced cell line, and the clone expressing the lowest amount of AR, as determined by western immunoblot, was used for all experiments unless otherwise stated. The cell lines expressing AR and GFP are referred to as OE33-AR, JH-AR, and OE19-AR, respectively. The EAC cell lines were maintained in androgen-depleted growth medium (stripped medium) consisting of phenol red-free RMPI-1640 containing l-glutamine (Life Technologies, Eugene, OR, USA), supplemented with 10% dextran charcoal stripped fetal bovine serum (Equitech-Bio, Inc., Kerrville, TX, USA), 200 U/mL penicillin, and 200 μg/mL streptomycin (Life Technologies). Stripped medium was used for experiments unless stated otherwise.
The PShTert myofibroblasts [20–22] were stably transduced with the SFG-RFP/Rluc construct to express red fluorescent protein (RFP) [23]. Neonatal foreskin fibroblasts (NFFs) and PShTert myofibroblasts were used between passages 10 and 20. Fibroblasts were maintained in DMEM containing l-glutamine (Life Technologies), supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich, St Louis, MO, USA), 200 U/mL penicillin, and 200 μg/mL streptomycin (Life Technologies).
Direct Co-culture
The NFFs and PShTert myofibroblasts were cultured in stripped medium overnight, seeded at 4 × 105 cells per well into six-well plates (BD Biosciences, Franklin Lakes, NJ, USA), and then incubated for 48 h to form confluent monolayers. Next, OE33-ARs were seeded at 1 × 105 cells per well either in monoculture or in direct co-culture with the fibroblasts. The following day (day 0), and every 48 h thereafter, the medium was replaced with stripped medium supplemented with vehicle (0 nM DHT; 0.1% ethanol) or 10 nM DHT. Cells were harvested on day 6 of treatment, unless stated otherwise.
Translocation of Androgen Receptor
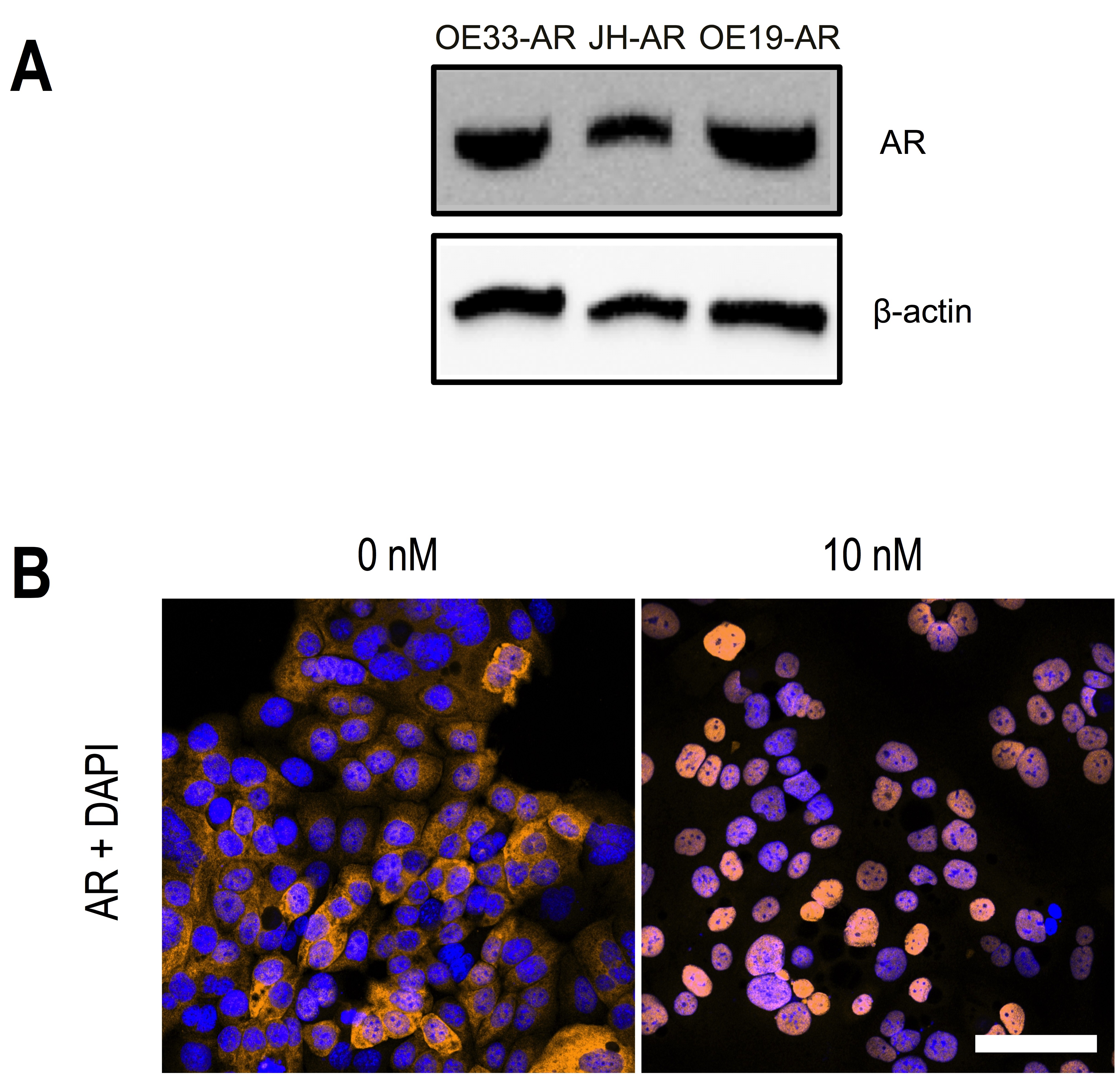
To establish direct co-cultures, fibroblasts were cultured in stripped medium overnight, then seeded at 8 × 104 fibroblasts per well in eight-well Lab-Tek Chamber Slides (Thermo Fisher Scientific, Rochester, NY, USA), and incubated for 48 h. Next, OE33-ARs (2 × 104 cells per well) were added to the wells, followed by overnight incubation. The medium, supplemented with vehicle or 10 nM DHT (day 0), was replaced then and every 48 h for 6 days.
Following treatment, the cells were washed in Dulbecco’s phosphate-buffered saline (DPBS; Life Technologies), fixed in methanol on ice for 5 min, and air-dried. The cells were blocked with 10% normal goat serum (Dako, Glostrup, Denmark) in DPBS for 20 min and labeled with rabbit antihuman AR polyclonal IgG (clone N-20; 1 µg/mL in 1.5% goat serum; Santa Cruz Biotech Inc., Santa Cruz, CA, USA) for 1 h, followed by incubation with Alexa Fluor 568 goat anti-rabbit IgG (2 µg/mL in 1.5% goat serum; Molecular Probes by Life Technologies) for 45 min. Nuclei were stained with 1 µg/mL 4′, 6-diamidino-2-phenylindole dihydrochloride (DAPI; Sigma-Aldrich) in DPBS for 15 min. Slides were mounted in fluorescent mounting medium (Dako) and stored at 4 °C in darkness. Images were captured using a Zeiss LSM 700 confocal microscope.
Cell Proliferation
To measure cell proliferation, 1 × 103 cells per well were seeded in 96-well plates and cultured for 48 h. The cells were then treated with either vehicle or various concentrations of DHT for 6–12 days, depending on the cell line. The cells were next fixed in 10% neutral buffered formalin for 30 min, stained for 10 min with 1% crystal violet (Sigma-Aldrich) in 2% ethanol, washed eight times in distilled water, and then air-dried overnight. The crystal violet was eluted using 10% acetic acid and gentle rotation of the plates. The absorbance of the eluent was measured at 595 nM using a FLUOstar Optima microplate reader (BMG Labtech, Ortenberg, Germany). To determine whether growth inhibition with DHT was mediated via AR, OE33-ARs were seeded into six-well plates (1 × 105 cells per well) and cultured for 48 h. The cells were treated for 6 days with 10 nM DHT and either vehicle (0.15% dimethyl sulfoxide; Sigma-Aldrich) or 15 µM enzalutamide (MedChem Express, Princeton, NJ, USA).
Cell division of OE33-AR was measured by dye dilution, using CellTrace Violet (CTV; Life Technologies). Cells were seeded in six-well plates (3 × 104 cells per well) and incubated for 24 h. Wells for time 0 were then fixed in 4% paraformaldehyde. In the other wells, the medium was replaced daily, supplemented with either vehicle or DHT at around the half maximal inhibitory concentration (IC50; 0.06 and 0.1 nM) or 10 nM. Other wells were fixed with 4% paraformaldehyde 1, 3, or 5 days following. The amount of CTV in the cells was measured using a FACSCanto II (BD Biosciences) with BD FACSDiva software. Cell doublets were excluded by doublet discrimination, based on nonlinearity of forward scatter and side scatter area versus height plots. Cells were gated based on GFP-positivity, and the median CTV intensity of this population determined using FlowJo software version 8.8.7 (Ashland, OR, USA).
Quantitative Real-Time Reverse Transcription PCR (qRT-PCR)
Total RNA was isolated from cells using TRIzol, and 1 µg was reverse-transcribed using the iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA, USA) in a final reaction volume of 20 µL. Gene expression was determined by quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR) using iQ SYBR Green Supermix (Bio-Rad Laboratories) in a final reaction volume of 10 µL, containing 0.1 µL of cDNA, and a final concentration of 0.2 µM of each forward and reverse primer (Supplementary Table S1). Reactions were performed using a CFX96 (Bio-Rad Laboratories) at 95 °C for 3 min, then 40 cycles of 95 °C for 15 s, 60 °C for 15 s, and 72 °C for 30 s, followed by a final extension of 72 °C for 1 min. Products were melted to confirm specificity. Normalized fold expression was calculated using actin beta (ACTB) as the reference gene.
Cell Cycle Analysis
OE33-ARs were seeded at 1 × 105 cells per well in stripped medium in six-well plates and incubated for 24 h. The medium was then replaced daily, supplemented with either vehicle or 0.06, 0.1, or 10 nM DHT. Wells were harvested at 0, 24, 48, or 72 h. The cells were washed, resuspended in DPBS, and fixed with a final concentration of 70% ice-cold ethanol. The cells were pelleted, resuspended with 0.25% Triton X-100 (Sigma-Aldrich) in DPBS, and incubated for 2 h with 25 μg/mL propidium iodide (Sigma-Aldrich) and 40 μg/mL bovine pancreas ribonuclease A (Sigma-Aldrich) in DPBS. The DNA content of single cells was measured using a FACSCanto II. The percentages of cells in each cell cycle phase and in sub-G1 were calculated using BD FACSDiva software.
Confocal Microscopy
To assess morphology, OE33-AR (1.72 × 103), JH-AR (5.73 × 103), or OE19-AR (5.73 × 103) cells were seeded into 96-well µ-plates (Ibidi, Martinsried, Germany) in stripped medium supplemented with either vehicle or 10 nM DHT replaced daily for 3 days. For direct co-cultures, NFFs were labeled using the CellTrace Violet (CTV) Cell Proliferation Kit according to the manufacturer’s protocol (Life Technologies). Fibroblasts were seeded at 1.14 × 104 per well, and OE33-ARs were seeded overnight at 1.43 × 103 cells per well, in either monoculture or overlying fibroblasts followed by treatment with vehicle or 10 nM DHT for 6 days, with medium replaced every 48 h. Images were captured using a Zeiss LSM 700 confocal microscope with Zen2012 SP1 (black edition) software version 8.1.
Senescence-Associated Beta-Galactosidase Assay
Cells were seeded in 24-well plates at 2 × 103 cells per well for OE33 and OE33-AR and 3 × 103 cells per well for JH-AR and OE19-AR, followed by 48-h incubation. The culture medium was then replaced daily with fresh medium supplemented with vehicle or DHT (IC50s and 10 nM). On days 0, 1, 3, and 5 for OE33 and OE33-AR and 0, 4, 6, and 8 for JH-AR and OE19-AR, wells were stained with the Senescence Cells Histochemical Staining Kit (Sigma-Aldrich). The percentage of senescence-associated beta-galactosidase (SA-β-gal) positive cells was calculated from a count of 200 cells.
Statistics
The statistical software used was Prism 6.0d for Macintosh (GraphPad Software, San Diego, CA, USA). Proliferation dose–response curves were fitted, and the IC50 determined by nonlinear regression analysis. Data are the mean ± SD of a single experiment reproduced in triplicate with p values determined by parametric unpaired Student’s t test assuming equal standard deviations (SD), unless stated otherwise. Differences were considered significant when the two-tailed p value was ≤ 0.05.
Results
EAC Tissues with a High Percentage of FKBP5-Positive Cells Had a High Proliferation Index
Previously, we reported that the expression of FKBP5, a surrogate marker for androgen signaling, was associated with reduced survival in EAC [15]. To determine the relationship between AR signaling and tumor growth in vivo, we measured the percentage of FKBP5 positive cells and the Ki-67 proliferation index in immunostained EAC resection specimens (Supplementary Fig. S1). There was a positive correlation between the FKBP5 expression and the proliferation index (nonparametric Spearman correlation r = 0.37, 95% confidence interval 0.1533–0.5628; p = 0.0010), with the proliferation index significantly higher in those tissues with higher FKBP5 expression (Fig. 1; p = 0.0002).
Fig. 1.

Ki-67 proliferation indices in EAC resection tissues with low (≤ 2) or high (> 2) FKBP5 expression. Tissue microarrays were immunostained and scored as the percentage of epithelial cells that expressed FKBP5 or Ki-67 as follows: 0, negative; 1, < 5% (rare); 2, < 25%; 3, > 25% < 75%; 4, > 75%. Data are the median score of cores from each case. p value by Mann–Whitney U test
DHT Inhibited Proliferation of AR-Expressing EAC Cell Lines In Vitro
Expression of AR protein was confirmed in the three EAC cell lines stably transduced with AR, OE33-AR, JH-AR, and OE19-AR, by western immunoblot (Supplementary Fig. S2a) and immunocytochemistry (Supplementary Fig. S2b). In the absence of DHT, AR immunoreactivity was seen by confocal microscopy to be moderate in the cytoplasm and mild to moderate in the nucleus. Exposure to 10 nM DHT induced complete nuclear localization of the AR, confirming that the transduced AR was functionally responsive to androgen.
In the AR-negative cell lines, exposure to DHT at concentrations up to 100 nM did not significantly alter proliferation (data not shown). The dose–response curves for each of the AR-expressing lines, given a single dose of DHT at the start of culture, are shown in Fig. 2a. The concentration of DHT used in most reported studies of AR signaling in vitro is 10 nM, which completely inhibited proliferation of OE33-AR and JH-AR, and almost completely of OE19-AR. The IC50s were 0.09, 0.26, and 1.3 nM for OE33-AR, JH-AR, and OE19-AR, respectively. To determine whether the differences in the DHT dose–response curves between the cell lines were due to the amount of AR expressed, we compared, within each of the transduced lines, clones with the highest and lowest expression of AR and found no significant differences in the DHT dose–response curves (data not shown). We also observed the same or similar dose–response for the uncloned OE33-AR. The addition of the AR antagonist enzalutamide (15 µM) completely blocked the growth inhibition of the AR-expressing cells induced by 10 nM DHT, confirming that the anti-proliferative effect was mediated by the AR (p < 0.0001; Fig. 2b).
Fig. 2.

Effect of DHT on the proliferation of AR-expressing EAC cell lines. a Dose–response curves for the proliferation of AR-expressing EAC cells grown for 6–12 days with vehicle or tenfold serial dilutions of DHT. Proliferation was measured by crystal violet assay. Data are the mean ± SD of six replicates, and the corresponding nonlinear regression curve, from a representative experiment for each cell line. b The effect of 15 µM enzalutamide on the proliferation of OE33-AR treated with 10 nM DHT
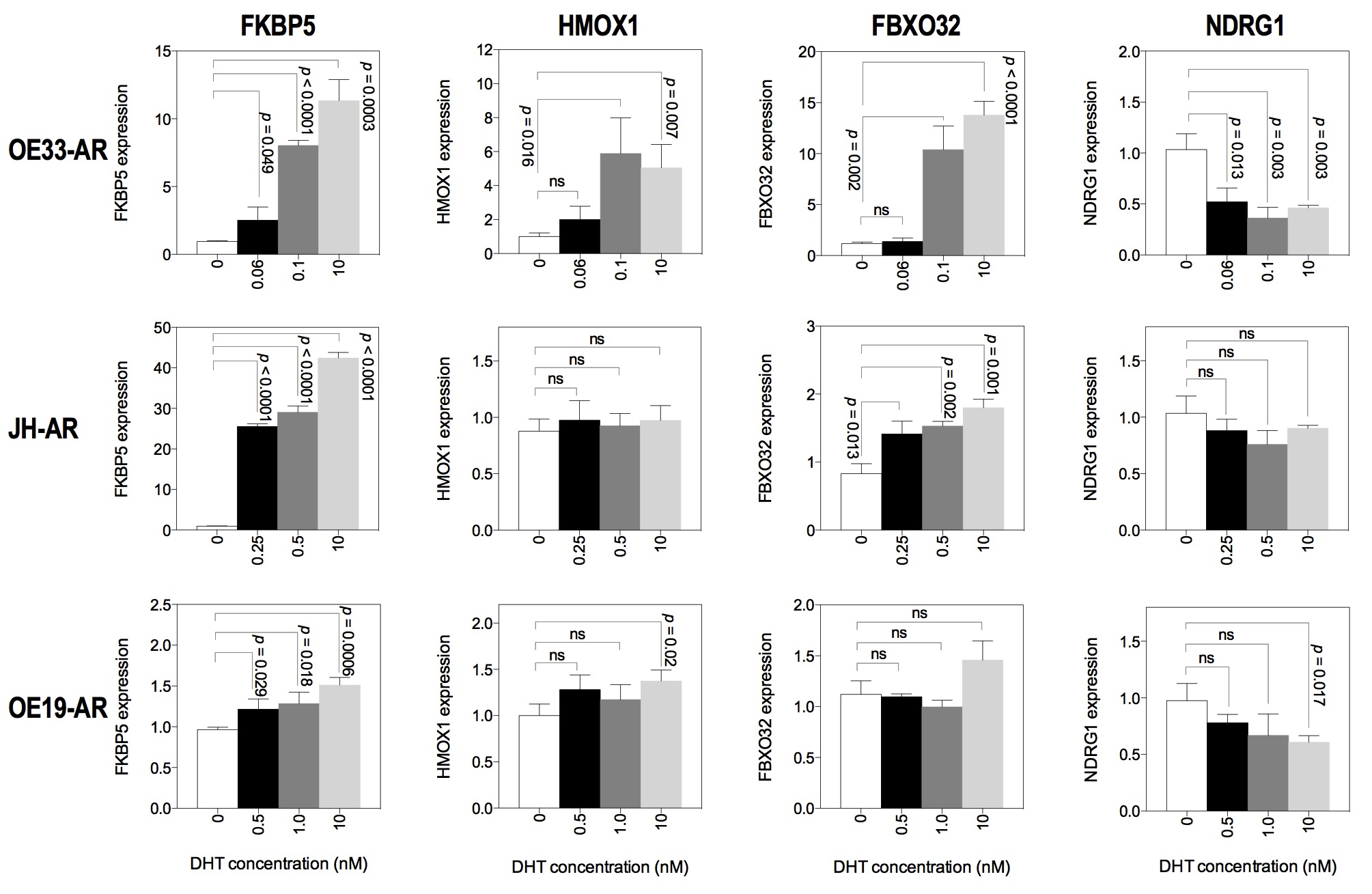
For subsequent experiments, we used two concentrations of DHT near the IC50 (0.06 and 0.1 nM for OE33-AR, 0.25 and 0.5 nM for JH-AR, and 0.5 and 1.0 nM for OE19-AR), as well as 10 nM. We next examined the possibility that proliferation may differ with daily replenishment of DHT compared to a single treatment at the start of the culture. The results in Fig. 3a show the DHT dose–response curves for OE33-AR. These were similar whether the DHT was given as a single treatment or daily in fresh medium, with IC50s of 0.08 and 0.06 nM, respectively.
Fig. 3.

Effect of single compared to daily doses of DHT on OE33-AR growth and FKBP5 expression. a Dose–response curves for the proliferation of OE33-AR grown for 5 days with vehicle or twofold serial dilutions of DHT given as a single dose or replaced daily. Data are the mean ± SD of six replicates, and the corresponding nonlinear regression curve, from a representative experiment. b Normalized fold FKBP5 expression after 3 days culture with a single dose or daily replacement of vehicle or DHT. Data are the mean ± SD of triplicate reactions for three biological replicates
DHT Induced Androgen-Responsive Gene Expression in AR-Expressing Cell Lines
In preliminary studies, we found that cell lines cultured with a single dose of 10 nM DHT expressed high levels of the androgen-responsive gene FKBP5, even though proliferation was inhibited. At lower concentrations of DHT, around the IC50, there was partial inhibition of growth but no, or very low, FKBP5 expression. This appeared to conflict with our immunostains of resection tissues where we frequently measured FKBP5 and Ki-67 expression together.
We therefore compared the effect of a single dose to daily replenishment of DHT on the induction of the androgen-responsive gene FKBP5 in OE33-AR (Fig. 3b). A single dose induced an increase in FKBP5 expression compared to vehicle of 1.2-fold for 0.06 nM DHT (p = 0.002), 2.2-fold for 0.1 nM (p = 0.0004), and 22-fold for 10 nM (p = 0.0005) after 3 days of culture. In contrast, when the DHT was replenished daily, the increase in FKBP5 expression compared to vehicle was twofold for 0.06 nM (p = 0.03), fourfold for 0.1 nM (p < 0.0001), and 16-fold for 10 nM (p = 0.0002.). The increases in FKBP5 expression for daily compared to single dosing were significantly greater for 0.06 and 0.1 nM DHT (p = 0.02 and p < 0.0001, respectively), but not for 10 nM (p = 0.639). Daily dosing, which would be expected to more closely mimic in vivo conditions, with concentrations of DHT around the IC50, permitted growth and induced significant expression of FKBP5.
Next, we measured the expression of known androgen-responsive genes in each of the three AR-expressing cell lines at the different concentrations of DHT. We found that the pattern of response was similar between the cell lines, although there were differences in the fold increases (Supplementary Fig. S3).
DHT Inhibited Cell Division and Induced Cell Cycle Arrest and Cell Senescence in AR-Expressing Cells
To understand better the inhibition of growth, we analyzed the effect of DHT on cell division, cell cycle arrest, and cell senescence. Cell division in OE33-AR, as measured by the intracellular dilution of CTV, was inhibited after 3 and 5 days of culture (Fig. 4a). The median CTV content at day 0 was 25,724 fluorescence units (FU). After 5 days of treatment with vehicle, it was reduced to 535 FU. In contrast, after 5 days of treatment with 0.06, 0.1, and 10 nM DHT the median CTV content was 621 FU, 930 FU, and 5760 FU, respectively, indicating that there was less cell division as the concentration of DHT increased.
Fig. 4.

Effect of treatment with vehicle or DHT on cell division, cell cycle stage, morphology, and senescence in AR-expressing EAC cells. OE33-ARs were treated with vehicle or DHT at IC50 doses or 10 nM. a Division of OE33-AR monitored by CellTrace Violet dye dilution on days 1, 3, and 5 post-treatment (n = 3). The peaks represent different generations of cells. b Cell cycle distribution and c normalized E2F1 expression (mean ± SD) on day 3 following treatment of OE33-AR (n = 3). d Fluorescent micrographs of OE33-AR, JH-AR and OE19-AR treated for 3 days with vehicle or 10 nM DHT. Scale bar is 75 µM. e Percentage of OE33-AR, JH-AR, and OE19-AR positive for senescence-associated beta-galactosidase (SA-β-gal) on various days post-treatment with vehicle or DHT. Data are the mean ± SD of pooled replicate experiments (OE33-AR, n = 3; JH-AR and OE19-AR, n = 2)
Next, we measured the cell cycle phase distribution by flow cytometry following 3 days of culture. The results are shown in Fig. 4b. There were no significant changes in the proportion of cells in each phase of the cell cycle following treatment with 0.06 nM DHT. Compared to vehicle, there was a 19% (p = 0.006) and 52% increase (p = 0.0004) in the proportion of cells in the G0/G1 phase following treatment with 0.1 and 10 nM DHT, respectively, a 34% (p = 0.0004) and 47% (p = 0.003) decrease in cells in the G2/M phase, and no difference and a decrease of 51% (p = 0.009) in the S phase. There was no sub-G1 population with any of the concentrations, suggesting that DHT did not induce cell death. The expression of E2F1, a transcriptional activator necessary for progression through the G1/S transition, was significantly inhibited with 0.1 nM (p = 0.0004) and 10 nM (p = 0.0002) of DHT by day 3 of culture (Fig. 4c).
Using time-lapse confocal microscopy, we observed that each of the three AR-expressing cell lines underwent extensive morphological changes over 3 days in monoculture with 10 nM DHT (Fig. 4d). The cells became discohesive, enlarged, and flattened, with the appearance of many large cytoplasmic holes. There was no microscopic evidence of extensive cell death. Because these changes were suggestive of senescence, we stained cultures for the senescence marker, senescence-associated beta-galactosidase (SA-β-gal). There was a concentration and time-dependent increase in the percentage of SA-β-gal stained cells over the duration of culture in each of the three cell lines, which was most pronounced in OE33-AR and least in OE19-AR (Fig. 4e). Together these results demonstrated that DHT inhibited the proliferation of AR-expressing EAC cell lines in vitro by inducing growth arrest and senescence in a dose-dependent manner.
The Effect of Direct Co-culture with Fibroblasts
We next explored the possibility that fibroblasts, the major cell population within the tumor microenvironment, which are known to influence the response of tumor cells to drugs [24], might modify the response of AR-expressing EAC cells to androgens. Normal neonatal foreskin fibroblasts (NFFs) and PShTert myofibroblasts representative of cancer-associated fibroblasts were used.
The morphology of OE33-AR in monoculture and direct co-culture with NFFs, vehicle, or 10 nM DHT is shown in Fig. 5a. With vehicle, there was no apparent microscopic difference between OE33-AR grown in monoculture or in direct co-culture with NFFs. They formed numerous clusters of cells with distinct cell borders, polygonal shape, and well-defined nuclei. With 10 nM DHT, growth of OE33-AR was inhibited in both the monoculture and the NFF direct co-culture. The OE33-AR were enlarged and contained numerous refractile, round bodies devoid of obvious structural content under phase microscopy and lacking GFP under fluorescence microscopy. This effect of DHT on OE33-AR growth in monoculture and direct co-culture with NFFs was also reflected in the cell counts shown in Fig. 5b.
Fig. 5.

Morphology and growth of OE33-AR in direct co-culture with fibroblasts. a Fluorescent micrographs of OE33-AR (green) in monoculture or in direct co-culture with NFFs (blue) or PShTert myofibroblasts (red) treated with vehicle or 10 nM DHT for 6 days, with the medium replaced every 48 h. Scale bar is 75 µM. b Cell counts of OE33-AR grown for 6 days with vehicle or 10 nM DHT in monoculture or direct co-culture with NFFs or PShTert myofibroblasts. c Cell counts of OE33-AR grown for 6 days with vehicle or 10 nM DHT, with or without 15 µM enzalutamide, in monoculture or direct co-culture with PShTert myofibroblasts. Cell count data are the mean ± SD of three replicates from a representative experiment
In relation to the co-culture of OE33-AR with cancer-associated fibroblast-like PShTert myofibroblasts, there were no apparent microscopic differences between the monoculture and the direct co-culture with the myofibroblasts, either with vehicle or with 10 nM DHT (Fig. 5a), and no differences between the counts of OE33-AR grown with vehicle compared to 10 nM DHT in the direct co-culture (Fig. 5b). Similar effects of the myofibroblasts on morphology (Supplementary Fig. S4) and cell counts (Supplementary Fig. S5) in the presence of 10 nM DHT were observed for the other two AR-expressing EAC cell lines, JH-AR and OE19-AR. Increasing the DHT concentration to 100 or 1000 nM resulted in complete inhibition of OE33-AR proliferation in direct co-culture with the myofibroblasts (p < 0.0001) (Supplementary Fig. S6). The addition of enzalutamide to cultures blocked the DHT mediated inhibition of OE33-AR growth in monoculture (p < 0.0001), but did not significantly alter the outcome of direct co-culture with the myofibroblasts (p = 0.544) (Fig. 5c).
Translocation of AR in Direct Co-cultures
We investigated whether nuclear translocation of AR was altered in OE33-AR in direct co-culture. The results in Fig. 6 show that the OE33-AR treated with vehicle, in either monoculture or direct co-culture, had mild AR immunoreactivity in the cytoplasm and weak immunoreactivity in the nucleus, consistent with a lack of AR activation. With 10 nM DHT, there was complete translocation of AR to the nucleus and no AR in the cytoplasm of OE33-AR in monoculture or direct co-culture with NFFs. In contrast, there was a DHT dose-dependent distribution of AR in OE33-AR directly co-cultured with the myofibroblasts. With 10 nM DHT, there was moderate immunoreactivity in the nucleus, and mild to moderate immunoreactivity in the cytoplasm. With 100 nM DHT, there was complete nuclear translocation in about 50% of OE33-AR, with mild cytoplasmic and moderate nuclear immunoreactivity in remaining cells, and with 1000 nM DHT, there was complete nuclear translocation in all cells. The finding of both nuclear and cytoplasmic AR in OE33-AR directly co-cultured with myofibroblasts was similar to our findings in EAC resection samples where AR was expressed in both the nucleus and cytoplasm in the majority of tissues.
Fig. 6.

Nuclear translocation of AR induced by DHT in OE33-AR in direct co-culture with fibroblasts. Fluorescent micrographs of OE33-AR grown for 6 days in monoculture or direct co-culture with PShTerts with vehicle or 10, 100, or 1000 nM DHT, or in direct co-culture with NFFs with vehicle or 10 nM DHT. Cells were labeled with rabbit antihuman AR polyclonal IgG (clone N-20; 1 µg/mL in 1.5% goat serum) followed by secondary antibody Alexa Fluor 568 goat anti-rabbit IgG (2 µg/mL in 1.5% goat serum) and DAPI (1 µg/mL). Merged channel images were captured using a Zeiss confocal LSM 700 microscope. Scale bar is 75 µM
DHT Induced Expression of Androgen-Responsive Genes in Co-cultures
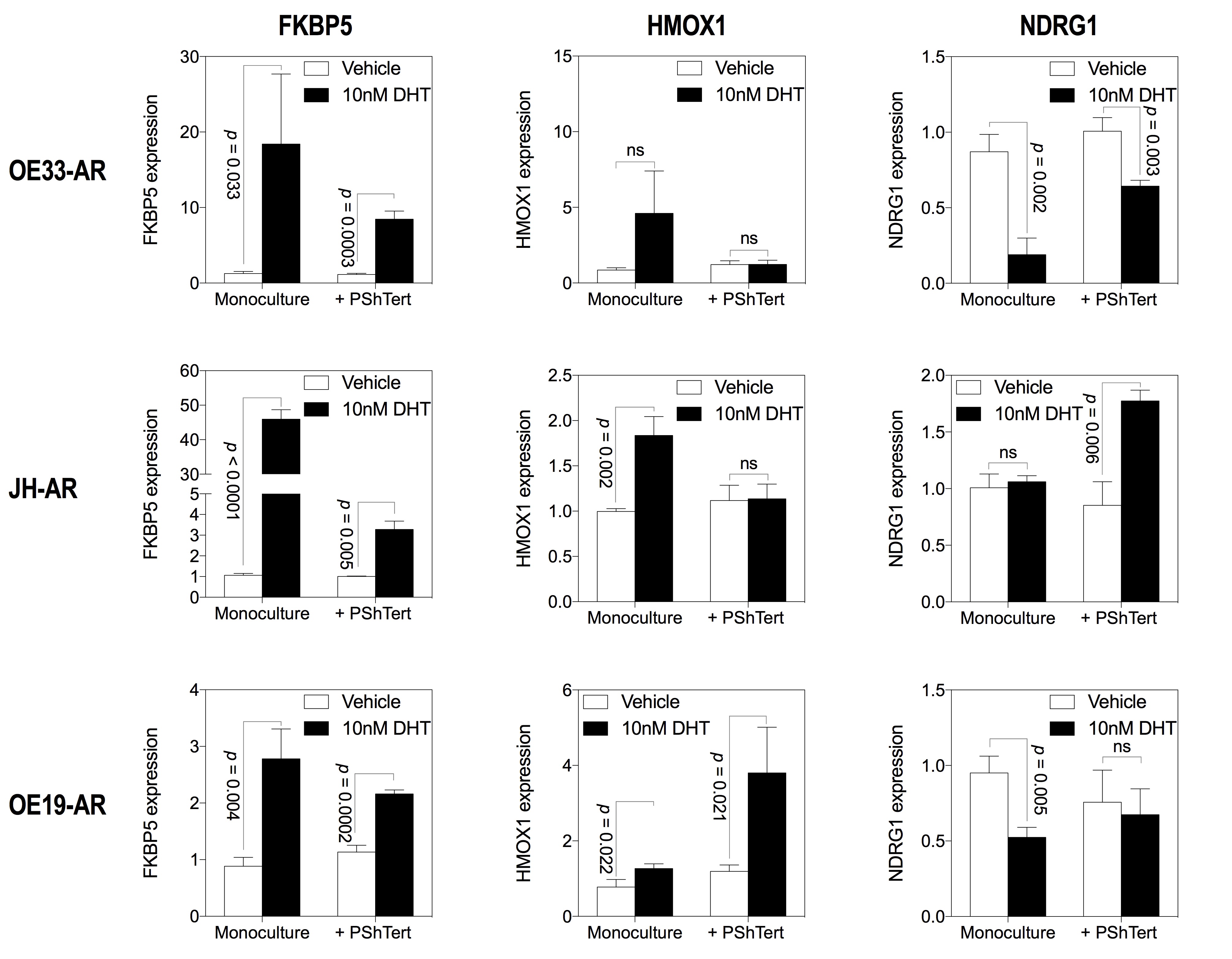
We next measured the effect of 10 nM DHT on the transcript levels of the androgen-responsive genes FKBP5, HMOX1 (heme oxygenase 1) and NDRG1 (N-myc downstream regulated 1) in OE33-AR in monoculture or direct co-culture. The OE33-ARs (GFP-positive) were sorted from NFFs (GFP-negative) or myofibroblasts (RFP-positive) in the direct co-cultures. The results in Fig. 7a show that the change in expression induced by DHT, induction, or repression was similar in the monoculture and co-cultures, although the extent of the change varied. This suggests that, although DHT did not inhibit OE33-AR growth in direct co-culture with myofibroblasts, there was still a change in the expression of androgen-responsive genes.
Fig. 7.

Effect of co-culture with fibroblasts on DHT induced expression of androgen-responsive genes in OE33-AR. a The expression of FKBP5, HMOX1, and NDRG1 in OE33-AR grown for 6 days with vehicle or 10 nM DHT in monoculture or direct co-culture with NFFs or PShTert myofibroblasts. Expression was normalized to the reference gene ACTB and graphed relative to expression with vehicle. Data are the mean ± SD. b Expression of FKBP5 in OE33-AR grown for 6 days with vehicle or 10 nM DHT, with or without 15 µM enzalutamide, in monoculture or direct co-culture with PShTert myofibroblasts
This response of OE33-AR to DHT in direct co-culture with myofibroblasts was also confirmed in two other AR-expressing EAC cell lines, JH-AR and OE19-AR (Supplementary Fig. S7). Co-cultured cells remained unsorted since the expression of androgen-responsive genes in myofibroblasts did not increase with DHT (FKBP5, p = 0.437; HMOX1, p = 0.244; NDRG1, p = 0.339) (Supplementary Fig. S8). Unsorted OE33-AR co-cultures were used for comparison. FKBP5 was upregulated with 10 nM DHT in both monoculture and direct co-culture with myofibroblasts for all three AR-expressing EAC cell lines. HMOX1 and NDRG1 had limited androgen responsiveness for JH-AR, as shown previously in Supplementary Fig. S3. OE19-AR showed upregulation of HMOX1 and downregulation of NDRG1 with DHT in both monoculture and direct co-culture with myofibroblasts. While the use of unsorted cells may have reduced the significance of these changes, the results suggest that, for all three AR-expressing EAC cell lines, the myofibroblast blocked the growth inhibition but not the gene expression induced by DHT in monoculture.
To confirm that changes in gene expression were mediated through the AR signaling pathway, the anti-androgen enzalutamide was used. The results in Fig. 7b show the effect of enzalutamide on FKBP5 expression in OE33-AR directly co-cultured with myofibroblasts with 10 nM DHT. Again, we measured expression in unsorted cells because FKBP5 expressed by myofibroblasts did not increase with DHT. Enzalutamide reduced the expression both in monoculture (tenfold; p = 0.0002) and in direct co-culture with the myofibroblasts (17-fold; p = 0.0003), indicating that FKBP5 upregulation was mediated through AR. This confirms that, in co-culture conditions where DHT did not inhibit proliferation, DHT was still functional in regulating the expression of androgen-responsive genes through the AR signaling pathway.
Discussion
We have previously reported that nuclear localization of AR and/or expression of the androgen-responsive gene FKBP5 was associated with decreased survival in EAC, suggesting a role for androgens in this cancer [15]. Here we have extended that study to show that EAC resection tissues with a higher percentage of FKBP5 positive cells also had a higher proliferation index, showing a positive relationship between androgen signaling and cancer cell growth. We have investigated the effects of androgen on the behavior of AR-expressing EAC cell lines in vitro. The three EAC cell lines that we stably transduced with full-length human AR cDNA were responsive to androgen, as shown by DHT induced nuclear localization of the receptor, and dose-dependent changes in cell proliferation, morphology, and gene expression. The commonly used concentration of DHT, 10 nM, markedly altered the expression of androgen-responsive genes, but completely inhibited cell proliferation by inducing cell cycle arrest and senescence, which appeared to be inconsistent with our findings in patient tissues of increased proliferation and upregulation of androgen-responsive gene expression. We then found that lower concentrations of DHT around the IC50 allowed proliferation and induced significant, although smaller, changes in the expression of androgen-responsive genes. We also found that in direct co-culture with an immortalized myofibroblast cell line, PShTert, the growth inhibitory effect of 10 nM of DHT was largely nullified, but the effect on expression of androgen-responsive genes was unaltered.
Studies of the effect of androgens on cell proliferation have generated conflicting results. Inhibition of proliferation has been reported often, in normal and cancer cell lines from a range of tissues, either naturally expressing or transduced with AR [25–40]. No change or an increase in cell proliferation has also been reported [34, 40–43]. Why androgens in some cells increase and in others decrease proliferation is unclear. Many of the reported studies only used a single concentration of DHT, most commonly 10 nM. When dose–response studies have been reported, the IC50 for DHT inhibition of proliferation has been of the same order as we measured [29, 30, 33, 40]. Our finding that growth inhibition was associated with cell cycle arrest and the induction of senescence is also consistent with other reports [30, 44–46].
This is the first comprehensive study of the expression of androgen-responsive genes across a range of DHT concentrations, in parallel with measurements of proliferation, in AR-expressing cells that are growth inhibited by DHT. We showed a DHT dose-dependent alteration of the expression of these genes. This could be measured from DHT concentrations around the IC50, significantly lower than the 10 nM most commonly used for in vitro studies [27, 47, 48]. Most reported studies use a single dose of DHT given at the start of the culture period. Single doses around the IC50 resulted in small, but significant, changes in the expression of the androgen-responsive genes. Daily replenishment of the DHT increased the magnitude of the gene expression response without significantly altering proliferation. Daily replenishment, compared to a single dose, would be expected to more closely mimic the delivery of hormone in vivo. Our observations that DHT concentrations around the IC50 were sufficient to increase FKBP5 expression and allow proliferation were consistent with the association between FKBP5 and Ki-67 expression we measured in patient EAC resection specimens, which suggest our in vitro findings are clinically relevant.
The tumor microenvironment is an important determinant of the response of cancer cells to molecules such as hormones and drugs [24, 49]. We therefore examined the effect of androgens on our AR-expressing EAC cell lines in direct co-culture with fibroblasts, the primary cellular component of the microenvironment. The PShTert myofibroblast line, an immortalized activated fibroblast line which has properties typical of cancer-associated fibroblasts [20–22, 50–53], allowed the growth of AR-expressing EAC cells at concentrations of DHT that inhibited the growth of cells in monoculture, without affecting the gene expression changes induced by DHT in monoculture. These are the first reported studies of EAC cell lines in co-culture with fibroblasts or myofibroblasts. There are a number of studies of the effect on prostate cancer cell lines of co-culture with PShTert myofibroblasts, but none have investigated how fibroblasts affect the inhibition of cancer cell proliferation by androgen [20–22]. In our study, the myofibroblasts do not appear to be depleting androgen from the culture medium since there are still changes in the expression of androgen-responsive genes that can be blocked by the anti-androgen enzalutamide. This suggests that the microenvironment has the potential to modify the response of AR-expressing EAC cells to androgen in vivo. The mechanisms for this are unknown, warranting further investigation that may lead to new targets for manipulating the progression of EAC and possibly other androgen-responsive cancers.
This is the first study to show a positive association between androgen signaling and cancer cell proliferation in EAC, and that AR-expressing EAC cell lines respond to androgens in vitro. Proliferation of AR-expressing EAC cells in monoculture was inhibited by higher concentrations of DHT. Our in vitro findings suggest that, in cancer tissues in vivo, AR-expressing EAC cells at lower concentrations of DHT, or in the presence of activated fibroblasts in the microenvironment, would proliferate and DHT would alter the expression of androgen-responsive genes. Our findings are consistent with a role for androgen signaling in EAC.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
Funding was provided by Cancer Council SA (Grant No. APP1031549) and Hospital Research Foundation.
Author’s contribution
HMP performed experiments. All authors contributed to the conception and design of the experiments, data analysis, and the writing of the manuscript.
Abbreviations
- ACTB
Actin beta
- CTV
CellTrace violet
- DHT
5α-Dihydrotestosterone
- EAC
Esophageal adenocarcinoma
- E2F1
E2F transcription factor 1
- FBXO32
F-box protein 32
- FKBP5
FK506-binding protein 5
- GFP
Green fluorescent protein
- HMOX1
Heme oxygenase 1
- IC50
Half maximal inhibitory concentration
- NDRG1
N-myc downstream regulated 1
- NFF
Neonatal foreskin fibroblast
- qRT-PCR
Quantitative reverse transcription polymerase chain reaction
- RFP
Red fluorescent protein
- SA-β-gal
Senescence-associated beta-galactosidase
- SD
Standard deviation
- TERT
Telomerase reverse transcriptase
- TMA
Tissue microarray
Compliance with ethical standards
Conflict of interest
The authors declare that they have no competing interests.
Footnotes
Eric Smith and Paul Drew have contributed equally to this manuscript.
Electronic supplementary material
The online version of this article (doi:10.1007/s10620-017-4794-5) contains supplementary material, which is available to authorized users.
Contributor Information
Helen M. Palethorpe, Phone: +61 8 8133 4005, Email: helen.palethorpe@adelaide.edu.au
Paul A. Drew, Email: paul.drew@flinders.edu.au
Eric Smith, Email: eric.smith@adelaide.edu.au.
References
- 1.Chai J, Jamal MM. Esophageal malignancy: a growing concern. World J Gastroenterol. 2012;18:6521–6526. doi: 10.3748/wjg.v18.i45.6521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Edgren G, Adami HO, Weiderpass E, Nyren O. A global assessment of the oesophageal adenocarcinoma epidemic. Gut. 2013;62:1406–1414. doi: 10.1136/gutjnl-2012-302412. [DOI] [PubMed] [Google Scholar]
- 3.Hur C, Miller M, Kong CY, et al. Trends in esophageal adenocarcinoma incidence and mortality. Cancer. 2013;119:1149–1158. doi: 10.1002/cncr.27834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lagergren J, Lagergren P. Recent developments in esophageal adenocarcinoma. CA Cancer J Clin. 2013;63:232–248. doi: 10.3322/caac.21185. [DOI] [PubMed] [Google Scholar]
- 5.Lepage C, Drouillard A, Jouve JL, Faivre J. Epidemiology and risk factors for oesophageal adenocarcinoma. Dig Liver Dis. 2013;45:625–629. doi: 10.1016/j.dld.2012.12.020. [DOI] [PubMed] [Google Scholar]
- 6.Lagergren J, Mattsson F. Diverging trends in recent population-based survival rates in oesophageal and gastric cancer. PLoS ONE. 2012;7:e41352. doi: 10.1371/journal.pone.0041352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oliver SE, Robertson CS, Logan RF. Oesophageal cancer: a population-based study of survival after treatment. Br J Surg. 1992;79:1321–1325. doi: 10.1002/bjs.1800791226. [DOI] [PubMed] [Google Scholar]
- 8.Awan AK, Iftikhar SY, Morris TM, et al. Androgen receptors may act in a paracrine manner to regulate oesophageal adenocarcinoma growth. Eur J Surg Oncol. 2007;33:561–568. doi: 10.1016/j.ejso.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 9.Cooper SC, Trudgill NJ. Subjects with prostate cancer are less likely to develop esophageal cancer: analysis of SEER 9 registries database. Cancer Causes Control. 2012;23:819–825. doi: 10.1007/s10552-012-9950-9. [DOI] [PubMed] [Google Scholar]
- 10.Rutegard M, Lagergren P, Nordenstedt H, Lagergren J. Oesophageal adenocarcinoma: the new epidemic in men? Maturitas. 2011;69:244–248. doi: 10.1016/j.maturitas.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 11.Sukocheva OA, Li B, Due SL, Hussey DJ, Watson DI. Androgens and esophageal cancer: what do we know? World J Gastroenterol. 2015;21:6146–6156. doi: 10.3748/wjg.v21.i20.6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Soest EM, Siersema PD, Dieleman JP, Sturkenboom MC, Kuipers EJ. Age and sex distribution of the incidence of Barrett’s esophagus found in a Dutch primary care population. Am J Gastroenterol. 2005;100:2599–2600. doi: 10.1111/j.1572-0241.2005.00305_6.x. [DOI] [PubMed] [Google Scholar]
- 13.Derakhshan MH, Liptrot S, Paul J, Brown IL, Morrison D, McColl KE. Oesophageal and gastric intestinal-type adenocarcinomas show the same male predominance due to a 17 year delayed development in females. Gut. 2009;58:16–23. doi: 10.1136/gut.2008.161331. [DOI] [PubMed] [Google Scholar]
- 14.Leach DA, Powell SM, Bevan CL. Women in cancer thematic review: new roles for nuclear receptors in prostate cancer. Endocr Relat Cancer. 2016;23:T85–T108. doi: 10.1530/ERC-16-0319. [DOI] [PubMed] [Google Scholar]
- 15.Smith E, Palethorpe HM, Ruszkiewicz AR, et al. Androgen receptor and androgen-responsive gene FKBP5 are independent prognostic indicators for esophageal adenocarcinoma. Dig Dis Sci. 2016;61:433–443. doi: 10.1007/s10620-015-3909-0. [DOI] [PubMed] [Google Scholar]
- 16.Smith E, Ruszkiewicz AR, Jamieson GG, Drew PA. IGFBP7 is associated with poor prognosis in oesophageal adenocarcinoma and is regulated by promoter DNA methylation. Br J Cancer. 2014;110:775–782. doi: 10.1038/bjc.2013.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abu-Sneineh A, Tam W, Schoeman M, et al. The effects of high-dose esomeprazole on gastric and oesophageal acid exposure and molecular markers in Barrett’s oesophagus. Aliment Pharmacol Ther. 2010;32:1023–1030. doi: 10.1111/j.1365-2036.2010.04428.x. [DOI] [PubMed] [Google Scholar]
- 18.Alvarez H, Koorstra JB, Hong SM, et al. Establishment and characterization of a bona fide Barrett esophagus-associated adenocarcinoma cell line. Cancer Biol Ther. 2008;7:1753–1755. doi: 10.4161/cbt.7.11.6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rockett JC, Larkin K, Darnton SJ, Morris AG, Matthews HR. Five newly established oesophageal carcinoma cell lines: phenotypic and immunological characterization. Br J Cancer. 1997;75:258–263. doi: 10.1038/bjc.1997.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y, Li CX, Ye H, et al. Decrease in stromal androgen receptor associates with androgen-independent disease and promotes prostate cancer cell proliferation and invasion. J Cell Mol Med. 2008;12:2790–2798. doi: 10.1111/j.1582-4934.2008.00279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leach DA, Need EF, Toivanen R, et al. Erratum: Stromal androgen receptor regulates the composition of the microenvironment to influence prostate cancer outcome. Oncotarget. 2015;6:36923. doi: 10.18632/oncotarget.3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leach DA, Need EF, Toivanen R, et al. Stromal androgen receptor regulates the composition of the microenvironment to influence prostate cancer outcome. Oncotarget. 2015;6:16135–16150. doi: 10.18632/oncotarget.3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dobrenkov K, Olszewska M, Likar Y, et al. Monitoring the efficacy of adoptively transferred prostate cancer-targeted human T lymphocytes with PET and bioluminescence imaging. J Nucl Med. 2008;49:1162–1170. doi: 10.2967/jnumed.107.047324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klemm F, Joyce JA. Microenvironmental regulation of therapeutic response in cancer. Trends Cell Biol. 2015;25:198–213. doi: 10.1016/j.tcb.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ortmann J, Prifti S, Bohlmann MK, Rehberger-Schneider S, Strowitzki T, Rabe T. Testosterone and 5 alpha-dihydrotestosterone inhibit in vitro growth of human breast cancer cell lines. Gynecol Endocrinol. 2002;16:113–120. doi: 10.1080/gye.16.2.113.120. [DOI] [PubMed] [Google Scholar]
- 26.Szelei J, Jimenez J, Soto AM, Luizzi MF, Sonnenschein C. Androgen-induced inhibition of proliferation in human breast cancer MCF7 cells transfected with androgen receptor. Endocrinology. 1997;138:1406–1412. doi: 10.1210/endo.138.4.5047. [DOI] [PubMed] [Google Scholar]
- 27.Kokontis JM, Lin HP, Jiang SS, et al. Androgen suppresses the proliferation of androgen receptor-positive castration-resistant prostate cancer cells via inhibition of Cdk2, CyclinA, and Skp2. PLoS ONE. 2014;9:e109170. doi: 10.1371/journal.pone.0109170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yuan S, Trachtenberg J, Mills GB, Brown TJ, Xu F, Keating A. Androgen-induced inhibition of cell proliferation in an androgen-insensitive prostate cancer cell line (PC-3) transfected with a human androgen receptor complementary DNA. Cancer Res. 1993;53:1304–1311. [PubMed] [Google Scholar]
- 29.Heisler LE, Evangelou A, Lew AM, Trachtenberg J, Elsholtz HP, Brown TJ. Androgen-dependent cell cycle arrest and apoptotic death in PC-3 prostatic cell cultures expressing a full-length human androgen receptor. Mol Cell Endocrinol. 1997;126:59–73. doi: 10.1016/S0303-7207(96)03970-6. [DOI] [PubMed] [Google Scholar]
- 30.Litvinov IV, Antony L, Dalrymple SL, Becker R, Cheng L, Isaacs JT. PC3, but not DU145, human prostate cancer cells retain the coregulators required for tumor suppressor ability of androgen receptor. Prostate. 2006;66:1329–1338. doi: 10.1002/pros.20483. [DOI] [PubMed] [Google Scholar]
- 31.Marcelli M, Haidacher SJ, Plymate SR, Birnbaum RS. Altered growth and insulin-like growth factor-binding protein-3 production in PC3 prostate carcinoma cells stably transfected with a constitutively active androgen receptor complementary deoxyribonucleic acid. Endocrinology. 1995;136:1040–1048. doi: 10.1210/endo.136.3.7532576. [DOI] [PubMed] [Google Scholar]
- 32.Shen R, Sumitomo M, Dai J, et al. Androgen-induced growth inhibition of androgen receptor expressing androgen-independent prostate cancer cells is mediated by increased levels of neutral endopeptidase. Endocrinology. 2000;141:1699–1704. doi: 10.1210/endo.141.5.7463. [DOI] [PubMed] [Google Scholar]
- 33.Chauhan S, Kunz S, Davis K, et al. Androgen control of cell proliferation and cytoskeletal reorganization in human fibrosarcoma cells: role of RhoB signaling. J Biol Chem. 2004;279:937–944. doi: 10.1074/jbc.M311325200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Birrell SN, Bentel JM, Hickey TE, et al. Androgens induce divergent proliferative responses in human breast cancer cell lines. J Steroid Biochem Mol Biol. 1995;52:459–467. doi: 10.1016/0960-0760(95)00005-K. [DOI] [PubMed] [Google Scholar]
- 35.Macedo LF, Guo Z, Tilghman SL, Sabnis GJ, Qiu Y, Brodie A. Role of androgens on MCF-7 breast cancer cell growth and on the inhibitory effect of letrozole. Cancer Res. 2006;66:7775–7782. doi: 10.1158/0008-5472.CAN-05-3984. [DOI] [PubMed] [Google Scholar]
- 36.Rossi R, Franceschetti P, Maestri I, et al. Evidence for androgen receptor gene expression in human thyroid cells and tumours. J Endocrinol. 1996;148:77–85. doi: 10.1677/joe.0.1480077. [DOI] [PubMed] [Google Scholar]
- 37.Rossi R, Zatelli MC, Franceschetti P, et al. Inhibitory effect of dihydrotestosterone on human thyroid cell growth. J Endocrinol. 1996;151:185–194. doi: 10.1677/joe.0.1510185. [DOI] [PubMed] [Google Scholar]
- 38.Vander Griend DJ, Litvinov IV, Isaacs JT. Conversion of androgen receptor signaling from a growth suppressor in normal prostate epithelial cells to an oncogene in prostate cancer cells involves a gain of function in c-Myc regulation. Int J Biol Sci. 2014;10:627–642. doi: 10.7150/ijbs.8756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Antony L, van der Schoor F, Dalrymple SL, Isaacs JT. Androgen receptor (AR) suppresses normal human prostate epithelial cell proliferation via AR/beta-catenin/TCF-4 complex inhibition of c-MYC transcription. Prostate. 2014;74:1118–1131. doi: 10.1002/pros.22828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Whitacre DC, Chauhan S, Davis T, Gordon D, Cress AE, Miesfeld RL. Androgen induction of in vitro prostate cell differentiation. Cell Growth Differ. 2002;13:1–11. [PubMed] [Google Scholar]
- 41.Yu SQ, Lai KP, Xia SJ, Chang HC, Chang C, Yeh S. The diverse and contrasting effects of using human prostate cancer cell lines to study androgen receptor roles in prostate cancer. Asian J Androl. 2009;11:39–48. doi: 10.1038/aja.2008.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhau HY, Chang SM, Chen BQ, et al. Androgen-repressed phenotype in human prostate cancer. Proc Natl Acad Sci USA. 1996;93:15152–15157. doi: 10.1073/pnas.93.26.15152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Banu KS, Govindarajulu P, Aruldhas MM. Testosterone and estradiol have specific differential modulatory effect on the proliferation of human thyroid papillary and follicular carcinoma cell lines independent of TSH action. Endocr Pathol. 2001;12:315–327. doi: 10.1385/EP:12:3:315. [DOI] [PubMed] [Google Scholar]
- 44.Mirochnik Y, Veliceasa D, Williams L, et al. Androgen receptor drives cellular senescence. PLoS ONE. 2012;7:e31052. doi: 10.1371/journal.pone.0031052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roediger J, Hessenkemper W, Bartsch S, et al. Supraphysiological androgen levels induce cellular senescence in human prostate cancer cells through the Src-Akt pathway. Mol Cancer. 2014;13:214. doi: 10.1186/1476-4598-13-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shao C, Wang Y, Yue HH, et al. Biphasic effect of androgens on prostate cancer cells and its correlation with androgen receptor coactivator dopa decarboxylase. J Androl. 2007;28:804–812. doi: 10.2164/jandrol.106.002154. [DOI] [PubMed] [Google Scholar]
- 47.Rossi R, Zatelli MC, Valentini A, et al. Evidence for androgen receptor gene expression and growth inhibitory effect of dihydrotestosterone on human adrenocortical cells. J Endocrinol. 1998;159:373–380. doi: 10.1677/joe.0.1590373. [DOI] [PubMed] [Google Scholar]
- 48.Castoria G, Giovannelli P, Di Donato M, et al. Role of non-genomic androgen signalling in suppressing proliferation of fibroblasts and fibrosarcoma cells. Cell Death Dis. 2014;5:e1548. doi: 10.1038/cddis.2014.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16:582–598. doi: 10.1038/nrc.2016.73. [DOI] [PubMed] [Google Scholar]
- 50.Leach DA, Need EF, Trotta AP, Grubisha MJ, DeFranco DB, Buchanan G. Hic-5 influences genomic and non-genomic actions of the androgen receptor in prostate myofibroblasts. Mol Cell Endocrinol. 2014;384:185–199. doi: 10.1016/j.mce.2014.01.004. [DOI] [PubMed] [Google Scholar]
- 51.Leach DA, Trotta AP, Need EF, Risbridger GP, Taylor RA, Buchanan G. The prognostic value of stromal FK506-binding protein 1 and androgen receptor in prostate cancer outcome. Prostate. 2017;77:185–195. doi: 10.1002/pros.23259. [DOI] [PubMed] [Google Scholar]
- 52.Lai KP, Huang CK, Fang LY, et al. Targeting stromal androgen receptor suppresses prolactin-driven benign prostatic hyperplasia (BPH) Mol Endocrinol. 2013;27:1617–1631. doi: 10.1210/me.2013-1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li Y, Zhang DY, Ren Q, et al. Regulation of a novel androgen receptor target gene, the cyclin B1 gene, through androgen-dependent E2F family member switching. Mol Cell Biol. 2012;32:2454–2466. doi: 10.1128/MCB.06663-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.