


Abstract
GM1 ganglioside is known to promote amyloid-β (Aβ) peptide aggregation in Alzheimer’s disease. The roles of the individual saccharides and their distribution in this process are not understood. Acrylamide-based glycomonomers with either β-d-glucose or β-d-galactose pendant groups were synthesized to mimic the stereochemistry of saccharides present in GM1 and characterized via 1H NMR and electrospray ionization mass spectrometry. Glycopolymers of different molecular weights were synthesized by aqueous reversible addition–fragmentation chain transfer (aRAFT) polymerization and characterized by NMR and GPC. The polymers were used as models to investigate the effects of molecular weight and saccharide unit type on Aβ aggregation via thioflavin-T fluorescence and PAGE. High molecular weight (~350 DP) glucose-containing glycopolymers had a profound effect on Aβ aggregation, promoting formation of soluble oligomers of Aβ and limiting fibril production, while the other glycopolymers and negative control had little effect on the Aβ propagation process.
Graphical abstract
INTRODUCTION
Alzheimer’s disease (AD) is a neurodegenerative disease that accounts for approximately 60–80% of the diagnosed cases of dementia.1 Development of AD symptoms has been related to the deposition of toxic oligomers and plaques formed from amyloid-β peptides (Aβ) on the neuronal cell membranes.1,2 Among several isoforms of Aβ formed during the cleavage of the amyloid precursor protein (APP), the 40-amino-acid residue, Aβ40, is found to be the most abundant, while the 42-amino-acid variant, Aβ42, is the most aggregation prone and toxic.3–5 GM1 ganglioside, consisting of a hydrophobic tail and a hydrophilic head made of saccharide moieties, has been reported to seed Aβ aggregation.6–8 Gangliosides are positioned at the cell surface, with the hydrophobic ceramide tail embedded in the plasma membrane and the hydrophilic saccharide moieties extending well into the extracellular space, making them more accessible to the Aβ peptides.9 The aggregation mechanism of Aβ in the presence of GM1 ganglioside and the specific functions of the individual saccharide groups in the aggregation process are not yet fully understood. The observation that among the gangliosides, which differ primarily in their saccharides, GM1 strongly promotes Aβ aggregation indicates that the glycoform distribution plays an important role in the aggregation process.6,7,10–12
Glycosaminoglycans (GAGs), which are polyanionic polysaccharides, have also been reported to exhibit a strong effect on amyloid aggregation.13–18 The aggregation depends on several factors, such as the length of the polysaccharides,14,16 the nature and the degree of functionalization (e.g., sulfation),13 and the ratio of the GAGs to the amyloid.19 Short polysaccharides (DP < 5) show a very minimal effect on the rate of amyloid fibrillation, which increases with increasing chain length and ultimately reaches a maximum at high chain lengths (DP ≫ 18).16,20 Fung et al.21 reported the effect of free floating simple carbohydrates on Aβ42 aggregation and conformational changes. Glucose promoted nucleation, resulting in the formation of short and flexible protofibrils, whereas galactose promoted mature fiber formation.21 The level of β-sheet conformation increased with increasing glucose concentration, while galactose showed no such influence, indicating that the H-bonding pattern of saccharides is an important factor in determining the aggregation behavior of Aβ42.21
Matsuzaki et al. reported that gangliosides can mediate Aβ aggregation only when they remain as clusters and not when present as uniformly distributed moieties.8 It is also reported that the in vitro interactions between proteins and saccharides are substantially weaker than those observed in vivo.22,23 A physiologically relevant level of association and affinities between saccharides and proteins requires the multivalent effect of saccharides, known as the “glyco-cluster” effect.24–26 To model the glyco-cluster effect, we employed aqueous reversible addition–fragmentation chain transfer (aRAFT) polymerization to achieve high molecular weight acrylamide-based glycopolymers of desired structure and molecular weight. Pendant groups of galactose or glucose with β-stereochemistry were synthesized to model the saccharides of GM1. Acrylamide was chosen as the backbone due to its excellent water solubility, hydrolytic stability, and its stability over a wide range of pH and salt concentrations.27 These model glycopolymers were studied to investigate physiological scenarios to determine how saccharide type (galactose vs glucose) of GM1 ganglioside influences Aβ aggregation.
EXPERIMENTAL SECTION
Materials
All reagents and solvents were obtained from Sigma-Aldrich Corporation (USA) or ThermoFisher Scientific (USA) in their highest purity available. The chemicals were used without further purification unless otherwise stated. Lyophilized stocks of synthetic, wild-type Aβ42, herein referred to as Aβ (obtained from the Mayo Clinic, Rochester, MN), were stored at −20 °C. The chain transfer agent, 4-cyano-4-(ethylsulfanylthiocarbonyl) sulfanylpentanoic acid (CEP), was synthesized by adapting previously reported procedures.28,29
Characterization
Nuclear magnetic resonance (NMR) spectroscopy was performed with a Varian MercuryPLUS (300 MHz) spectrometer by taking an average of 128 scans (delay 5 s) using appropriate solvents (CDCl3 or D2O). Gel permeation chromatography (GPC) was performed on a Waters system with Waters 1525 Binary Pump and Waters 2414 differential refractive index detector utilizing two highly efficient PolySep GFC columns (elution range 3k to 400k Da). An aqueous solution containing 0.1 M NaNO3 and 0.01% (w/v) NaN3 was filtered and used as the eluent at a flow rate of 1 mL/min at 25 °C. The molecular weight calibration was performed with monodisperse linear poly(ethylene oxide) (Polymer Standard Service). For molecular weights, the entire signal of a major peak including its shoulder at a lower retention volume was integrated. Mass spectrometry was done on a ThermoFinnigan TSQ 7000 triple-quadrapole instrument that was equipped with an electrospray ionization (ESI) source. Glycomonomer samples (1 mg/mL) in a 1:1 (v/v) methanol/water solution containing sodium chloride (1 mg/mL) were injected into the ESI source at a rate of 10 µL/s. All data were analyzed using Xcalibur (FisherScientific, Inc.) software.
In Vitro Glycopolymer–Aβ Interactions
Freshly purified Aβ monomer (25 µM) was coincubated with 75 µM of either poly(N,N-dimethyl acrylamide) (PDMA), galactose-containing glycopolymers (PGalEAm), or glucose-containing glycopolymers (PGlcEAm). Note that the solution molarity is determined on the basis of the polymer theoretical number-average molecular weight. Thus, the concentration of pendant saccharide groups is 10-fold higher in the high molecular weight glycopolymer solutions than in the low molecular weight analogues. For all samples, 0.1 M NaNO3 was added for polymer stability and 0.01% (w/v) NaN3 was added to prevent bacterial growth. All reactions were buffered in 20 mM Tris at pH 8.0 and were carried out at 37 °C under quiescent conditions with periodic monitoring by thioflavin-T (ThT) fluorescence and immunoblotting.
Thioflavin-T (ThT) Fluorescence
Measurements were collected by mixing 70 µL of ThT (10 µM) with 5 µL of each sample. After a 1 min equilibration period, fluorescence kinetics were measured in a microcuvette with a Cary Eclipse spectrometer (Varian, Inc.) by exciting at 452 nm while monitoring emission at 482 nm over a 1 min period. Excitation and emission slits were kept constant at 10 nm.
Polyacrylamide Gel Electrophoresis
Samples were diluted into 1× Laemmli loading buffer either with (denaturing) or without (nondenaturing) 1% SDS and then loaded without heating onto either NuPAGE 4–12% Bis-Tris gels resolved in 1× MES running buffer containing 0.1% SDS (Life Technologies) for SDS-PAGE or 4–20% BioRad gels resolved in 1× Laemmli buffer for native PAGE. For SDSPAGE, prestained molecular weight (MW) markers (Novex Sharp Protein Standard, Life Technologies) were run in parallel for MW determination. Proteins were transferred to a 0.2 µm nitrocellulose membrane (BioRad) and boiled for 1 min in a microwave oven in 1× PBS followed by blocking for 1.5 h in 1× PBS containing 5% nonfat dry milk with 1% Tween 20. Blots were then probed overnight at 4 °C with a 1:6000 dilution of Ab5 monoclonal antibody, which detects amino acids 1–16 of Aβ. Blots were then incubated with a 1:6000 dilution of antimouse, horseradish peroxidase conjugated secondary antibody and developed with ECL reagent (Thermo Scientific).
Amyloid-β (Aβ) Monomer Purification and Isolation
Before the use of Aβ in any reaction, the peptide was purified by size exclusion chromatography (SEC) to remove any preformed aggregates. Briefly, 1.5–2 mg of peptide was dissolved in 0.5 mL of 10 mM aqueous NaOH and allowed to stand for 15 min at room temperature prior to SEC using a 1 × 30 cm Superdex-75 HR 10/30 column (GE Healthcare) attached to an ÄKTA FPLC system (GE Healthcare).30 The column was pre-equilibrated with 20 mM Tris–HCl (pH 8.0) at 25 °C, and the protein was eluted at a flow rate of 0.5 mL/min. Fractions of 500 µL were collected, and the concentration of Aβ was determined by UV–visible spectrometry on a Cary 50 spectrophotometer (Varian Inc.) using a molar extinction coefficient of 1450 cm−1 M−1 at 276 nm (ExPASy) corresponding to the single tyrosine residue. Peptide integrity after SEC was periodically confirmed via MALDI-ToF mass spectrometry, which showed a monoisotopic molecular mass of 4515 Da. Monomeric Aβ42 fractions were stored at 4 °C and used within 48 h of SEC purification to eliminate any possibilities of preformed aggregates in the reactions.
Glycomonomer Synthesis
The acetyl protected glycomonomers, 2′-acrylamidoethyl-2,3,4,6-tetra-O-acetyl-β-d-galactopyranoside (Ac-GalEAm) and 2′-acrylamidoethyl-2,3,4,6-tetra-O-acetyl-β-d-glucopyranoside (AcGlcEAm), were synthesized by adapting the procedures reported by Ambrosi et al.31 for the synthesis of 2′-(2,3,4,6-tetra-O-acetyl-β-d-galactosyloxy)ethyl methacrylate and 2′-(2,3,4,6-tetra-O-acetyl-β-d-glucosyloxy)ethyl methacrylate. In short, either 2,3,4,6-tetra-O-acetyl-α-d-galactopyranosyl bromide (15 g, 36.5 mmol) or 2,3,4,6-tetra-O-acetyl-α-d-glucopyranosyl bromide (15 g, 36.5 mmol) was reacted with an excess amount of N-hydroxyethyl acrylamide (HEAm) (21 g, 182.4 mmol) in anhydrous dichloromethane (400 mL) using excess silver trifluoromethanesulfonate (AgOTf) (14 g, 54.5 mmol) as a catalyst. Dry molecular sieves (20 g, 3 Å size) were added to the reaction mixture before the addition of silver trifluoromethanesulfonate to ensure that the reaction medium was completely dry. The reaction mixture was stirred for 48 h at 0 °C in a N2 atmosphere. Then, the reaction mixture was filtered, and the filtrate was washed three times with 1 M HCl and dried over sodium sulfate. A yellow colored and highly viscous product was obtained after solvent removal via rotary evaporation. Flash chromatography was performed with the crude products using silica gel as the stationary phase and a mixture of 10:1 ethyl acetate:hexane as the eluent. The eluent fractions with retardation factor (Rf) = 0.45 were collected, and the solvent was evaporated by rotary evaporation to obtain a white crystalline pure product (9.1 g, 20.43 mmol).
AcGalEAm, 1H NMR (300 MHz, CDCl3): δ [ppm] 1.99, 2.02, 2.16 (s, s, s, 12H-13,14,15,16), 3.57 (m, 2H-5), 3.72 (m, 1H-9), 3.90 (m, 2H-4), 4.13 (m, 2H-11,12), 4.46 (m, 1H-6), 5.01 (d of d, 1H-10), 5.16 (m, 1H-7), 5.37 (t, 1H-8), 5.66 (d of d, 1H-1), 6.10 (m, 1H-2), 6.31 (m, 1H-3). [1H NMR spectra, Supporting Information, Figure S1A] [ESI m/z: 445 + 23 (Na+), Supporting Information, Figure S3].
AcGlcEAm, 1H NMR (300 MHz, CDCl3): δ [ppm] 1.97, 2.03, 2.14 (s, s, s, 12H-13,14,15,16), 3.57 (m, 2H-5), 3.70 (m, 1H-9), 3.91 (m, 2H-4), 4.14 (m, 2H-11,12), 4.48 (m, 1H-6), 5.02 (d of d, 1H-10), 5.16 (m, 1H-7), 5.38 (t, 1H-8), 5.66 (d of d, 1H-1), 6.05 (m, 1H-2), 6.31 (m, 1H-3). [1H NMR spectra, Supporting Information, Figure S2A] [ESI m/z: 445 + 23 (Na+)].
Glycomonomer Deprotection
Acetyl protected monomer, AcGalEAm (9.1 g) or AcGlcEAm (9.1 g), was dissolved in anhydrous methanol (46 mL) in a round-bottom flask equipped with a stir bar. The flask was sealed with a rubber septum and purged with N2 for 15 min before the dropwise addition of 25% (w/v) sodium methoxide solution in methanol (4.55 mL), and the reaction was stirred for another 45 min in a N2 atmosphere. Acetic acid was added dropwise until a neutral or a slightly acidic pH (pH ≈ 6) was achieved. The solvent was removed by rotary evaporation to obtain a highly viscous, colorless liquid which became a strongly hygroscopic, colorless solid after freeze-drying. The complete deprotection of the glycomonomers was confirmed via 1H NMR and ESI-MS.
GalEAm, 1H NMR (300 MHz, D2O): δ [ppm] 3.31–4.09 (m, 10H-4, 5, 7, 8, 9, 10, 11, 12), 4.39 (d, 1H-6), 5.72 (d of d, 1H-1), 6.07–6.36 (m, 1H-3, 1H-2). [ESI-MS m/z: 277 + 23 (Na+), Supporting Information, Figure S1B].
GlcEAm, 1H NMR (300 MHz, D2O): δ [ppm] 3.49–4.11 (m, 10H-4, 5, 7, 8, 9, 10, 11, 12), 4.39 (d, 1H-6), 5.75 (d of d, 1H-1), 6.19 (m, 1H-3, 1H-2). [1H NMR spectra, Supporting Information, Figure S2B] [ESI-MS m/z: 277 + 23 (Na+)].
General Procedure for aRAFT Polymerization of Glycomonomer
Glycopolymers with a target degree of polymerization (DP) of 35 (molecular weight = 9951 g/mol) and 350 (molecular weight = 97206 g/mol) were synthesized by aRAFT polymerization. The reaction conditions for the RAFT polymerizations were selected on the basis of previous reports for acrylate or acrylamide based polymers.32–36 The initial concentrations of the monomer to chain transfer agent and the chain transfer agent to initiator were maintained at 500:1 and 5:1, respectively, for a target DP of 35 achievable at 7% conversion and a target DP of 350 achievable at 70% conversion. The initial glycomonomer concentration in the reaction mixture was kept at 1 M. 4-Cyano-4-(ethylsulfanylthiocarbonyl) sulfanylpentanoic acid (CEP) was used as a chain transfer agent (CTA) and 4,4′-azobis(4-cyanopentanoic acid), V-501, was used as a free radical initiator for the polymerization reaction. Benzenesulfonic acid (BSA) was used as an internal standard to monitor the progress of the reactions, which were performed in a 0.1 M sodium acetate buffer solution of pH 5.0 at 70 °C. A typical procedure for glycopolymer synthesis is as follows: 7.97 mL of monomer solution (GalEAm or GlcEAm) in acetate buffer from a stock solution of 0.348 g/mL was transferred to a 10 mL graduated cylinder, and 113.5 µL of CEP solution in methanol from a 46.4 mg/mL stock solution and 22.8 µL of V-501 solution in methanol from a stock solution of 49.1 mg/mL was added to the cylinder, followed by addition of 527 µL of BSA solution in buffer from a stock solution of 150.2 mg/mL. The mixture was diluted to a total volume of 10 mL with acetate buffer. The reaction mixture was then transferred to a 25 mL round-bottom flask equipped with a stir bar. The flask was sealed with a rubber septum and parafilm and purged with high purity nitrogen gas for 40 min while stirring. An initial aliquot was taken, and the flask was placed in an oil bath heated to 70 °C. Aliquots were taken at 30 min intervals and rapidly quenched with liquid nitrogen. 1H NMR was performed with the quenched aliquots to determine the monomer conversion to polymer. Monomer conversion was determined by comparing relative integral areas of the vinyl proton peak of the monomers (5.73 ppm, 1H) to the aromatic proton peak of the BSA standard (7.77 ppm, 2H) at different reaction times. The molecular weight of the polymers formed at different time points was calculated from the NMR conversion data. The quenched solutions were transferred to dialysis tubes of molecular weight cutoff 3500 Da (Spectra/Por) and dialyzed for a period of 5 days (24 h × 5) in distilled water. The dialyzed samples were freeze-dried at −50 °C in a high vacuum (0.05 Torr) for 2 days. The samples corresponding to ~7 and ~70% conversion (~35 and ~350 DP, respectively) were tested for their molecular weight and dispersity via gel permeation chromatography (GPC). These samples with two different molecular weights (35 and 350 DP) and two different saccharide units (gal and glc) were used to further investigate their effect on Aβ aggregation.
RAFT Polymerization of Dimethyl Acrylamide
Dimethyl acrylamide (DMA) monomers were purified to remove inhibitor by passing through a column filled with basic aluminum oxide. Polymerization of DMA monomers was carried out via an aqueous RAFT polymerization technique to produce polymers with a degree of polymerization of 35 and 350, following the procedure outlined for the glycopolymer synthesis.
RESULTS AND DISCUSSION
Glycomonomer Synthesis
The acetyl protected glycomonomers were synthesized as described in the Experimental Section and characterized via 1H NMR (Figure S1, Supporting Information). The debromination reaction between the glycosyl donor, acetobromo-α-d-galactose, and the glycosyl acceptor, N-hydroxyethyl acrylamide, is evidenced by the shift in the C1 proton NMR peak from 6.69 ppm in the galactose precursor to 4.5 ppm in AcGalEAm. Complete conversion of the limiting reactant, acetobromo saccharide, was confirmed via TLC by the absence of the spot representing unreacted saccharides.
Prepolymerization deprotection of the glycomonomers was chosen over postpolymerization deprotection to minimize incomplete removal of the acetyl protecting groups, which can affect the biological properties of the sugars and their protein interactions.26,37 Complete deprotection of the protected glycomonomers was confirmed by the disappearance of the characteristic 1H NMR peak for the acetyl groups at 1.99–2.16 ppm, and by ESI-MS [m/z 445 + 23 (Na+) for AcGlcEAm and AcGalEAm and m/z 277 + 23 (Na+) for GlcEAm and GalEAm].
To model the GM1 ganglioside, where saccharides are of β-stereo conformation, it is desirable to have the same stereochemistry in our glycomonomers and polymers. The stereochemistry was determined via 1H NMR, and the percentage of β-anomers in monomers was estimated by comparing the proton peak area at 4.5 ppm (β-anomers) to that at 5.2 ppm (α-anomers). Excellent stereospecificity was obtained (>98% β-anomers), which is attributed to neighboring group participation involving the acetate group as reported by Ambrosi et al.31 The stereospecificity of the saccharides was retained during glycomonomer deprotection and polymerization reactions, as evidenced by the unchanged 1H NMR spectra (Figure S4, Supporting Information). Yu et al. reported the synthesis of the same monomers via a different reaction pathway which produced only 84% of β-anomers.26 We adapted the procedures reported by Ambrosi et al.31 for synthesis of methacrylate glycopolymers to achieve acrylamide-based glycopolymers in high yield (~60%) and almost complete conversion to β-anomers (~98%).
Aqueous RAFT Homopolymerization of Glycomonomers
Monomer conversion was monitored via 1H NMR by comparing the relative integral areas of the vinyl proton peak of the glycomonomers (5.73 ppm, 1H) with the aromatic proton peak of benzenesulfonic acid (7.77 ppm, 2H), the internal standard used in the reactions. A linear increase of ln([M]0/[M]) as a function of time is observed for the polymerization reactions (Figure 1), where [M]0 is the initial molar concentration of the deprotected glycomonomer (GlcEAm or GalEAm) and [M] is the molar concentration of the monomer at any given time point, indicating pseudo first order kinetics.
Figure 1.

Plots of ln([M]0/[M]) vs reaction time for the aqueous RAFT polymerization of glycomonomers (GalEAm and GlcEAm) at 70 °C using CEP as a chain transfer agent indicating pseudo first order polymerization kinetics.
The aRAFT polymerization of PGalEAm exhibits an initialization period of 50 min, while that of PGlcEAm is 100 min. Similar initialization periods in aqueous RAFT polymerizations have been reported previously by our team, including Alidedeoglu et al.32 for 2-aminoethyl methacrylate monomers and McCormick et al.36 for acrylamido monomers. McLeary et al.38 investigated the initialization period observed in the RAFT polymerization of styrene with the chain transfer agent cyanoisopropyl dithiobenzoate via in situ 1H NMR. It was found that the time taken by the chain transfer agents to react with a single monomer unit is the reason for the observed delay of polymerization. Chain growth did not start until all of the CTAs were consumed. This observation was attributed to the much faster propagation rate of the CTA radicals than the radicals of the propagating chains containing a single monomer unit.38 Monomer conversions of ~7 and ~70% were achieved in 60 and 270 min, respectively, for the GalEAm reaction, whereas it took 120 and 360 min to achieve the same conversions for the GlcEAm reaction. The longer inhibition time for GlcEAm may be the result of stabilization of the macro CTA through hydrogen bonding with the pendant group, which occurs to a greater extent with the glucose derivative than with the galactose.
Aliquots were taken from the reaction mixture at 30 min intervals, and the monomer conversion was calculated via 1H NMR. The theoretical number-average molecular weight (Mnth) was calculated from 1H NMR according to eq 1, where ρ is the fractional monomer conversion, MWmon is the molecular weight of the monomer, [M]0 is the initial concentration of monomer, [CTA]0 is the initial concentration of the chain transfer agent, and MWCTA is the molecular weight of the chain transfer agent.35 The degree of polymerization was calculated from Mnth using eq 2.
| (1) |
| (2) |
The monomer conversion, molecular weight, and degree of polymerization data are summarized in Table 1.
Table 1.
Conversion and Molecular Weight Data for Glycopolymers and PDMA as Determined via NMR and GPC
| sample | target DP | % conversion | DPth | Mnth (g/mol) | Mn(GPC)a (g/mol) | Mw/Mnb |
|---|---|---|---|---|---|---|
| PGalEAm35 | 35 | 8.4 | 42 | 11800 | 2740 | 1.13 |
| PGalEAm350 | 350 | 69 | 344 | 95500 | 39100 | 3.82 |
| PGlcEAm35 | 35 | 7.8 | 39 | 11000 | 4640 | 1.16 |
| PGlcEAm350 | 350 | 69 | 344 | 95500 | 39600 | 3.82 |
| PDMA35 | 35 | 7.9 | 40 | 4220 | 3760 | 1.17 |
| PDMA350 | 350 | 69 | 346 | 34500 | 32800 | 1.24 |
Relative to PEO standards.
As determined by GPC (aqueous solution, 0.1 M NaNO3 and 0.01% (w/v) NaN3).
Figure 2 shows GPC traces for the four glycopolymers. The low molecular weight glycopolymers exhibit narrow unimodal peaks and low dispersities (PGalEAm35 ĐM = 1.13 and PGlcEAm35 ĐM = 1.16), while the high molecular weight glycopolymers show broad peaks with extended shoulders at shorter retention times. The peak broadening is attributed to aggregation, which is observed only for the high molecular weight glycopolymers. Similar association was reported by Liang et al.39 for poly[2-(β-d-glucosyloxy)ethyl acrylate in water. They reported the critical aggregation concentration (cac) to be inversely related to the glycopolymer molecular weight. They noted that, while the side chains are highly hydrophilic, the backbone is hydrophobic, and fluorescence studies indicated that the interior of the aggregates was hydrophobic. Thus, aggregation may be attributed in part to hydrophobic interactions. Other reports have attributed polysaccharide aggregation to intermolecular hydrogen bonding.40,41 It is likely that similar associations occur in our glycopolymers. Note that, in Table 1, both theoretical Mn (Mnth, determined by NMR) and relative Mn (determined by GPC) are reported. The relative Mn was calculated with respect to PEO standards, and these values are lower by a factor of approximately 2.4 in comparison to those calculated from the NMR monomer conversion data. Molecular weight trends are similar for the two sets of data, and both show a factor of 10 increase in molecular weight for the high DP glycopolymers.
Figure 2.

GPC traces for PGalEAm35, PGalEAm350, PGlcEAm35, and PGlcEAm350. Low molecular weight polymers yield narrow dispersities, while high molecular weight systems show apparent aggregation.
RAFT Polymerization of N,N′-Dimethyl Acrylamide
To clearly establish the effects of the backbone structure and saccharide moieties of a glycopolymer on Aβ aggregation, polymers having similar backbone structures without saccharide units were synthesized. Poly(N,N-dimethyl acrylamide) (PDMA) with controlled molecular weights (~35 and ~350 DP) and low dispersities was synthesized for the Aβ aggregation studies and utilized separately from the glycopolymers. GPC and NMR data are shown in Table 1, and the dispersities are 1.17 and 1.24 for PDMA35 and PDMA350, respectively.
Investigation of Aβ42 Aggregation in the Presence of Glycopolymers
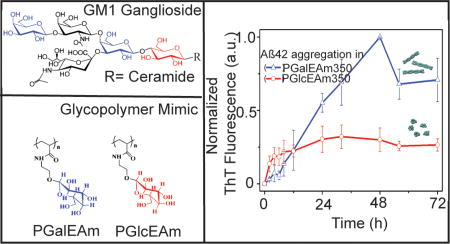
The effects of saccharide pendant group and molecular weight on Aβ aggregation were determined using ThT fluorescence by monitoring solutions of glycopolymers and the PDMA standard coincubated with Aβ42 monomer. ThT is a fluorescent dye which preferentially binds to β-sheet-rich amyloid aggregates, yielding an increase in fluorescence intensity.42,43 A 3-fold molar excess (75 µM) of polymer was incubated with Aβ (25 µM) in 20 mM Tris at pH 8.0 with 0.1 M NaNO3 and 0.01% NaN3. Samples were kept at 37 °C, and ThT fluorescence was measured periodically (Figure 3A–C). Aβ in the absence of polymer displays a short lag phase (2 h) before association (growth phase) and saturation as fibrils at 24 h of incubation (where a plateau is reached) (Figure 3A, ■). Aβ in the presence of PDMA alone (negative control) shows a small decrease in aggregation rate (increase in the lag phase); however, the saturation level is reached at 48 h (within standard experimental error) for solutions of both molecular weight polymers (Figure 3A, ○ and ▲). Samples containing PGalEAm show a marginally decreased aggregation rate in comparison to the Aβ control, particularly for the low molecular weight polymer, but the plateau region is reached at 72 h (Figure 3B, ○ and ▲). For the PGlcEAm35 solution, Aβ aggregation rate is initially reduced (Figure 3C, ○), but saturation occurs at similar intensities within 36 h. This suggests that the Aβ aggregation rate is influenced by the polymers PDMA, PGalEAm, and PGlcEAm35, but these polymers do not influence the final product of Aβ aggregation (fibrils). Fung et al.21 reported a decrease of the lag phase of Aβ42 aggregation in the presence of glucose and galactose monosaccharides, whereas Rajaram et al.44 reported a concentration dependent increase in lag phase for Aβ40 aggregation in the presence of glycoclusters made of six units of either glucose or galactose. We observed a minor decrease in lag phase for Aβ42 aggregation in the presence of PDMA, PGalEAm, and PGlcEAm35 (Figure 3A–C).
Figure 3.

Glycopolymer–Aβ aggregation studies using ThT-fluorescent (A–D). Polyacrylamide gel electrophoresis under denaturing conditions (E) and under nondenaturing conditions (F). Control sample refers to Aβ alone without any polymer. In the images in parts E and F, 1 stands for polymer with a DP of 35 and 2 stands for a DP of 350. PGlcEAm350 promotes formation of oligomers with minimal fibril production.
Aβ aggregation in the presence of PGlcEAm350, however, displays a very distinct aggregation profile (Figure 3C,▲). The ThT intensity rapidly increases and plateaus within the first 3 h of incubation (Figure 3D, ▲). The decreased fluorescence intensity as compared to other samples indicates that the size of the aggregates and/or the structure of the aggregates is different from that of the other samples. This unique aggregation profile suggests that Aβ rapidly forms smaller oligomers, but not fibrils, in the presence of PGlcEAm350. The initial rapid increase in fluorescence intensity can be attributed to the fast nucleation of Aβ in the presence of PGlcEAm350 and formation of a large number of nucleation seeds at the beginning of the interaction. Because the number of nucleation sites is higher, the number of Aβ per site is low and thus small aggregates are formed.
Fung et al. reported the formation of small aggregates of Aβ in the presence of glucose, while galactose promoted mature fibril formation.21 Glycoclusters of glucose and galactose have been reported to promote the formation of fibrils of Aβ40.44 In contrast, our ThT experiments showed that the low molecular weight glucose containing glycopolymer, PGlcEAm35, promotes Aβ fibril formation, whereas the high molecular weight PGlcEAm350 promotes formation of small aggregates (oligomers) (Figure 3C). Both low (PGalEAm35) and high (PGalEAm350) molecular weight galactose containing polymers promote formation of Aβ fibrils (Figure 3B).
To determine if this was indeed the case, polyacrylamide gel electrophoresis (PAGE) in conjunction with immunoblotting was utilized. Samples were electrophoresed under denaturing conditions (sodium dodecyl sulfate, SDS-PAGE) at 6 and 72 h of incubation (Figure 3E). At 6 h of incubation, all samples contained a band at 4.5 kDa corresponding to monomeric Aβ as well as a low molecular weight (LMW) oligomeric species (~15 kDa). High molecular weight (HMW) fibrils, which do not enter the gel, were observed in all samples with the exception of Aβ incubated with PGlcEAm350. The same analysis at 72 h revealed a small amount of Aβ monomers along with a significant concentration of HMW soluble oligomers, which is likely due to dissociation of the insoluble fibrils under denaturing conditions. However, no insoluble fibrils were observed for Aβ incubated with PGlcEAm350, and the monomer content appears to be larger than that in other samples. Together with the ThT data, this analysis confirms that PDMA, PGalEAm, and PGlcEAm35 polymers marginally affect the rate in which Aβ aggregation occurs, but the polymers do not affect the formation of the final product of fibrils. However, Aβ in the presence of PGlcEAm350 differs from the others in that oligomeric species are formed. To gain better perspective of this, nondenaturing (native) PAGE was employed at both 6 and 72 h of incubation (Figure 3F). It is important to note that the MW makers used for SDS-PAGE in panel E do not correspond to panel F, as these samples were electrophoresed under native conditions, and therefore, size estimations cannot be determined. Regardless, similar to that observed in the denaturing gel, all samples contained monomeric Aβ which had not undergone aggregation. Also, HMW fibrils were observed in all samples, except Aβ with PGlcEAm350, which exclusively formed an intermediate soluble oligomer. By 72 h of incubation, all samples (except Aβ incubated with PGlcEAm350) formed HMW fibrils with no discernible monomers or oligomers. However, the Aβ incubated with PGlcEAm350 displayed disperse soluble oligomer formation, with minimal fibril formation. From this data, it is clear that the HMW glucose containing glycopolymers show distinctly different behavior from that of other glycopolymers toward Aβ aggregation. This specific interaction produces soluble oligomers, which are reported to be the primary cause of toxicity in AD.
The difference in Aβ aggregation behavior in the presence of glycopolymers of different structures can be related to H-bonding patterns.21,45 Glucose forms stronger H-bonds with Aβ, whereas galactose forms weaker bonds.21 Because the bonding between PGalEAm and Aβ is weak, more H-bonding sites are available within Aβ, which promotes self-association of Aβ and ultimately produces mature fibrils.21 Glucose has been reported to promote formation of oligomers of Aβ by forming stronger H-bonds with Aβ.21 These strong H-bonds lead to formation of more nucleating seeds and fewer H-bonding sites available within Aβ. From our ThT and PAGE experiments, it is clear that glucose-containing glycopolymers with two different molecular weights, low and high, behave differently toward Aβ aggregation. Aβ forms fibrils in the presence of PGlcEAm35, whereas it forms small aggregates or oligomers in the presence of PGlcEAm350. This suggests that H-bonding alone is not sufficient to significantly change the aggregation behavior of Aβ, and there is a concentration dependence of the glucose units (clustering effect).
It has been reported that saccharide clusters exhibit stronger H-bonding tendencies than their monosaccharide counterparts.46,47 We believe that the glucose units in the high molecular weight PGlcEAm350 form intramolecular/intermolecular clusters due to the presence of the large number of glucose units in close proximity, as reported for similar systems by others.39–41 The concentration of saccharide pendant groups is 10-fold lower in the solutions of low molecular weight PGlcEAm35, and as reported in Liang et al.,39 cac is inversely related to molecular weight. Thus, cluster formation is not observed in the low molecular weight system, presumably due to the lower concentration of saccharide units and lower H-bonding propensity. It is possible, therefore, that stronger H-bonding due to the clustering of glucose is responsible for oligomer formation by Aβ in the presence of PGlcEAm350.
CONCLUSIONS
Acrylamide based glycopolymers with β-d-glucose and β-d-galactose pendant moieties were synthesized to high (DP 350) and low (DP 35) molecular weights via aqueous RAFT polymerization for determination of the effects of saccharide structure and concentration on Aβ aggregation. Dimethylacrylamide with no pendant saccharide was polymerized to similar molecular weights as a negative control. The high molecular weight glucose containing glycopolymers exhibited a large effect on the Aβ aggregation process, inducing the formation of toxic soluble oligomers while limiting fibril formation. The other glycopolymers and PDMA caused a minor reduction in the rate of Aβ aggregation but had no effect on the ultimate extent of fibril formation. The unusual Aβ aggregation behavior in the presence of the high molecular weight glucose containing polymers may be the result of hydrogen bonding of Aβ with the glucose pendant groups and the polysaccharide cluster effect, which does not occur with the low molecular weight polymer because of its reduced concentration of saccharide. These model systems provide information about the behavior of Aβ in the presence of polysaccharides and, more importantly, demonstrate the specificity in generating low molecular weight, toxic oligomers. The report also demonstrates the potential of utilizing glycopolymer systems of controlled composition and molecular weight in determining the mechanisms of toxic oligomer and fibril formation.
Supplementary Material
Scheme 1. Reaction Scheme for the Synthesis of Glycomonomer AcGalEAm and Its Deprotection to GalEAma.

aThe same scheme applies for GlcEAm monomer synthesis.
Scheme 2. Synthetic Scheme for the Aqueous RAFT Polymerization of GalEAma.

aThe same scheme applies for the glucose containing glycomonomer GlcEAm.
Acknowledgments
Partial funding was provided by the National Science Foundation’s Experimental Program to Stimulate Competitive Research (EPSCoR) under Cooperative Agreement No. IIA1430364 and NSF OIA-1632825. The authors also wish to thank National Center for Research Resources (5P20RR016476-11) and the National Institute of General Medical Sciences (8 P20 GM103476-11) from the National Institutes of Health for funding through INBRE (to V.R.), National Institute of Aging (R15AG046915) (to V.R.), and NSF Graduate Research Fellowship Program (NSF 1445151) (to D.N.D.). Dr.V. Kozlovskaya (University of Alabama at Birmingham) is acknowledged for technical assistance.
Footnotes
ASSOCIATED CONTENT
The authors declare no competing financial interest.
References
- 1.Alzheimer’s Association. 2016 Alzheimer’s disease facts and figures. Alzheimer’s Dementia. 2016;12(4):459–509. doi: 10.1016/j.jalz.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 2.Busser J, Geldmacher DS, Herrup K. Ectopic Cell Cycle Proteins Predict the Sites of Neuronal Cell Death in Alzheimer’s Disease Brain. J. Neurosci. 1998;18(8):2801–2807. doi: 10.1523/JNEUROSCI.18-08-02801.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid [beta]-peptide. Nat. Rev. Mol. Cell Biol. 2007;8(2):101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 4.Winklhofer KF, Tatzelt J, Haass C. The two faces of protein misfolding: gain- and loss-of-function in neurodegenerative diseases. EMBO J. 2008;27(2):336–349. doi: 10.1038/sj.emboj.7601930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gellermann GP, Byrnes H, Striebinger A, Ullrich K, Mueller R, Hillen H, Barghorn S. Abeta-globulomers are formed independently of the fibril pathway. Neurobiol. Dis. 2008;30(2):212–20. doi: 10.1016/j.nbd.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 6.Yanagisawa K. Role of gangliosides in Alzheimer’s disease. Biochim. Biophys. Acta, Biomembr. 2007;1768(8):1943–1951. doi: 10.1016/j.bbamem.2007.01.018. [DOI] [PubMed] [Google Scholar]
- 7.Matsuzaki K, Kato K, Yanagisawa K. Aβ polymerization through interaction with membrane gangliosides. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids. 2010;1801(8):868–877. doi: 10.1016/j.bbalip.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 8.Matsuzaki K. How Do Membranes Initiate Alzheimer’s Disease? Formation of Toxic Amyloid Fibrils by the Amyloid β-Protein on Ganglioside Clusters. Acc. Chem. Res. 2014;47(8):2397–2404. doi: 10.1021/ar500127z. [DOI] [PubMed] [Google Scholar]
- 9.Thompson JP, Schengrund C-L. Inhibition of the adherence of cholera toxin and the heat-labile enterotoxin of Escherichia coli to cell-surface GM1 by oligosaccharide-derivatized dendrimers. Biochem. Pharmacol. 1998;56(5):591–597. doi: 10.1016/s0006-2952(98)00198-1. [DOI] [PubMed] [Google Scholar]
- 10.McLaurin J, Franklin T, Fraser PE, Chakrabartty A. Structural transitions associated with the interaction of Alzheimer beta-amyloid peptides with gangliosides. J. Biol. Chem. 1998;273(8):4506–15. doi: 10.1074/jbc.273.8.4506. [DOI] [PubMed] [Google Scholar]
- 11.Ariga T, Kobayashi K, Hasegawa A, Kiso M, Ishida H, Miyatake T. Characterization of high-affinity binding between gangliosides and amyloid beta-protein. Arch. Biochem. Biophys. 2001;388(2):225–30. doi: 10.1006/abbi.2001.2304. [DOI] [PubMed] [Google Scholar]
- 12.Cebecauer M, Hof M, Amaro M. Impact of GM1 on Membrane-mediated Aggregation/Oligomerization of β-amyloid: Unifying View. Biophys. J. 2017 doi: 10.1016/j.bpj.2017.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bourgault S, Solomon JP, Reixach N, Kelly JW. Sulfated Glycosaminoglycans Accelerate Transthyretin Amyloidogenesis by Quaternary Structural Conversion. Biochemistry. 2011;50(6):1001–1015. doi: 10.1021/bi101822y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anoop A, Ranganathan S, Dhaked BD, Jha NN, Pratihar S, Ghosh S, Sahay S, Kumar S, Das S, Kombrabail M, Agarwal K, Jacob RS, Singru P, Bhaumik P, Padinhateeri R, Kumar A, Maji SK. Elucidating the Role of Disulfide Bond on Amyloid Formation and Fibril Reversibility of Somatostatin-14: RELEVANCE TO ITS STORAGE AND SECRETION. J. Biol. Chem. 2014;289(24):16884–16903. doi: 10.1074/jbc.M114.548354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stewart KL, Hughes E, Yates EA, Akien GR, Huang T-Y, Lima MA, Rudd TR, Guerrini M, Hung S-C, Radford SE, Middleton DA. Atomic Details of the Interactions of Glycosaminoglycans with Amyloid-β Fibrils. J. Am. Chem. Soc. 2016;138(27):8328–8331. doi: 10.1021/jacs.6b02816. [DOI] [PubMed] [Google Scholar]
- 16.Takase H, Tanaka M, Yamamoto A, Watanabe S, Takahashi S, Nadanaka S, Kitagawa H, Yamada T, Mukai T. Structural requirements of glycosaminoglycans for facilitating amyloid fibril formation of human serum amyloid A. Amyloid. 2016;23(2):67–75. doi: 10.3109/13506129.2016.1168292. [DOI] [PubMed] [Google Scholar]
- 17.Maji SK, Perrin MH, Sawaya MR, Jessberger S, Vadodaria K, Rissman RA, Singru PS, Nilsson KP, Simon R, Schubert D, Eisenberg D, Rivier J, Sawchenko P, Vale W, Riek R. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science. 2009;325(5938):328–32. doi: 10.1126/science.1173155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quittot N, Sebastiao M, Bourgault S. Modulation of amyloid assembly by glycosaminoglycans: from mechanism to biological significance. Biochem. Cell Biol. 2017;95(3):329–337. doi: 10.1139/bcb-2016-0236. [DOI] [PubMed] [Google Scholar]
- 19.Bazar E, Jelinek R. Divergent heparin-induced fibrillation pathways of a prion amyloidogenic determinant. ChemBioChem. 2010;11(14):1997–2002. doi: 10.1002/cbic.201000207. [DOI] [PubMed] [Google Scholar]
- 20.Jha S, Patil SM, Gibson J, Nelson CE, Alder NN, Alexandrescu AT. Mechanism of Amylin Fibrillization Enhancement by Heparin. J. Biol. Chem. 2011;286(26):22894–22904. doi: 10.1074/jbc.M110.215814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fung J, Darabie AA, McLaurin J. Contribution of simple saccharides to the stabilization of amyloid structure. Biochem. Biophys. Res. Commun. 2005;328(4):1067–1072. doi: 10.1016/j.bbrc.2005.01.068. [DOI] [PubMed] [Google Scholar]
- 22.Lindhorst T. Artificial Multivalent Sugar Ligands to Understand and Manipulate Carbohydrate-Protein Interactions. In: Penadés S, editor. Host-Guest Chemistry. Vol. 218. Springer; Berlin, Heidelberg: 2002. pp. 201–235. [Google Scholar]
- 23.Lundquist JJ, Toone EJ. The cluster glycoside effect. Chem. Rev. 2002;102(2):555–78. doi: 10.1021/cr000418f. [DOI] [PubMed] [Google Scholar]
- 24.Mammen M, Choi S-K, Whitesides GM. Polyvalent Interactions in Biological Systems: Implications for Design and Use of Multivalent Ligands and Inhibitors. Angew. Chem., Int. Ed. 1998;37(20):2754–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 25.Matsumoto E, Yamauchi T, Fukuda T, Miura Y. Sugar microarray via click chemistry: molecular recognition with lectins and amyloid β (1–42) Sci. Technol. Adv. Mater. 2009;10(3):034605. doi: 10.1088/1468-6996/10/3/034605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu K, Kizhakkedathu JN. Synthesis of Functional Polymer Brushes Containing Carbohydrate Residues in the Pyranose Form and Their Specific and Nonspecific Interactions with Proteins. Biomacromolecules. 2010;11(11):3073–3085. doi: 10.1021/bm100882q. [DOI] [PubMed] [Google Scholar]
- 27.Ladmiral V, Melia E, Haddleton DM. Synthetic glycopolymers: an overview. Eur. Polym. J. 2004;40(3):431–449. [Google Scholar]
- 28.Convertine AJ, Benoit DSW, Duvall CL, Hoffman AS, Stayton PS. Development of a novel endosomolytic diblock copolymer for siRNA delivery. J. Controlled Release. 2009;133(3):221–229. doi: 10.1016/j.jconrel.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Le TP, Moad G, Rizzardo E, Thang SH. Polymerization with living characteristics. US7714075 B1. 2010 May 11;
- 30.Kumar A, Paslay LC, Lyons D, Morgan SE, Correia JJ, Rangachari V. Specific Soluble Oligomers of Amyloid-β Peptide Undergo Replication and Form Non-fibrillar Aggregates in Interfacial Environments. J. Biol. Chem. 2012;287(25):21253–21264. doi: 10.1074/jbc.M112.355156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ambrosi M, Batsanov AS, Cameron NR, Davis BG, Howard JAK, Hunter R. Influence of preparation procedure on polymer composition: synthesis and characterisation of polymethacrylates bearing [small beta]-D-glucopyranoside and [small beta]-D-galactopyranoside residues. J. Chem. Soc., Perkin Trans. 1. 2002;(1):45–52. [Google Scholar]
- 32.Alidedeoglu AH, York AW, McCormick CL, Morgan SE. Aqueous RAFT polymerization of 2-aminoethyl methacrylate to produce well-defined, primary amine functional homo- and copolymers. J. Polym. Sci., Part A: Polym. Chem. 2009;47(20):5405–5415. [Google Scholar]
- 33.Lowe AB, Sumerlin BS, McCormick CL. The direct polymerization of 2-methacryloxyethyl glucoside via aqueous reversible addition-fragmentation chain transfer (RAFT) polymerization. Polymer. 2003;44(22):6761–6765. [Google Scholar]
- 34.Bernard J, Hao X, Davis TP, Barner-Kowollik C, Stenzel MH. Synthesis of Various Glycopolymer Architectures via RAFT Polymerization: From Block Copolymers to Stars. Biomacromolecules. 2006;7(1):232–238. doi: 10.1021/bm0506086. [DOI] [PubMed] [Google Scholar]
- 35.Abel BA, Sims MB, McCormick CL. Tunable pH- and CO2-Responsive Sulfonamide-Containing Polymers by RAFT Polymerization. Macromolecules. 2015;48(16):5487–5495. [Google Scholar]
- 36.McCormick CL, Lowe AB. Aqueous RAFT Polymerization: Recent Developments in Synthesis of Functional Water-Soluble (Co)polymers with Controlled Structures. Acc. Chem. Res. 2004;37(5):312–325. doi: 10.1021/ar0302484. [DOI] [PubMed] [Google Scholar]
- 37.Gruber H, Knaus S. Synthetic polymers based on carbohydrates: preparation, properties and applications. Macromol. Symp. 2000;152(1):95–105. [Google Scholar]
- 38.McLeary JB, Calitz FM, McKenzie JM, Tonge MP, Sanderson RD, Klumperman B. Beyond Inhibition: A 1H NMR Investigation of the Early Kinetics of RAFT-Mediated Polymerization with the Same Initiating and Leaving Groups. Macromolecules. 2004;37(7):2383–2394. [Google Scholar]
- 39.Liang Y-Z, Li Z-C, Li F-M. Self-Association of Poly[2-(β-D-glucosyloxy) ethyl Acrylate] in Water. J. Colloid Interface Sci. 2000;224(1):84–90. doi: 10.1006/jcis.1999.6644. [DOI] [PubMed] [Google Scholar]
- 40.Balasubramanian D, Raman B, Sundari CS. Polysaccharides as amphiphiles. J. Am. Chem. Soc. 1993;115(1):74–77. [Google Scholar]
- 41.Winnik FM. Association of hydrophobic polymers in water: fluorescence studies with labeled (hydroxypropyl)celluloses. Macromolecules. 1989;22(2):734–742. [Google Scholar]
- 42.LeVine H., 3rd Thioflavine T interaction with synthetic Alzheimer’s disease beta-amyloid peptides: detection of amyloid aggregation in solution. Protein. Sci. 1993;2(3):404–10. doi: 10.1002/pro.5560020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lindberg DJ, Wranne MS, Gilbert Gatty M, Westerlund F, Esbjörner EK. Steady-state and time-resolved Thioflavin-T fluorescence can report on morphological differences in amyloid fibrils formed by Aβ(1–40) and Aβ(1–42) Biochem. Biophys. Res. Commun. 2015;458(2):418–423. doi: 10.1016/j.bbrc.2015.01.132. [DOI] [PubMed] [Google Scholar]
- 44.Rajaram H, Palanivelu MK, Arumugam TV, Rao VM, Shaw PN, McGeary RP, Ross BP. ’Click’ assembly of glycoclusters and discovery of a trehalose analogue that retards Abeta40 aggregation and inhibits Abeta40-induced neurotoxicity. Bioorg. Med. Chem. Lett. 2014;24(18):4523–8. doi: 10.1016/j.bmcl.2014.07.077. [DOI] [PubMed] [Google Scholar]
- 45.Allison SD, Chang B, Randolph TW, Carpenter JF. Hydrogen Bonding between Sugar and Protein Is Responsible for Inhibition of Dehydration-Induced Protein Unfolding. Arch. Biochem. Biophys. 1999;365(2):289–298. doi: 10.1006/abbi.1999.1175. [DOI] [PubMed] [Google Scholar]
- 46.Hayashida O, Mizuki K, Akagi K, Matsuo A, Kanamori T, Nakai T, Sando S, Aoyama Y. Macrocyclic Glycoclusters. Self-Aggregation and Phosphate-Induced Agglutination Behaviors of Calix[4]resorcarene-Based Quadruple-Chain Amphiphiles with a Huge Oligosaccharide Pool. J. Am. Chem. Soc. 2003;125(2):594–601. doi: 10.1021/ja0275663. [DOI] [PubMed] [Google Scholar]
- 47.Aoyama Y. Macrocyclic Glycoclusters: From Amphiphiles through Nanoparticles to Glycoviruses. Chem. - Eur. J. 2004;10(3):588–593. doi: 10.1002/chem.200305288. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.