

Summary
Chronic jet lag induces spontaneous hepatocellular carcinoma (HCC) in wild-type mice following a mechanism very similar to that observed in obese humans. The process initiates with non-alcoholic fatty liver disease (NAFLD) that progresses to steatohepatitis and fibrosis before HCC detection. This pathophysiological pathway is driven by jet lag induced genome-wide gene deregulation and global liver metabolic dysfunction, with nuclear receptor-controlled cholesterol/bile acid and xenobiotic metabolism among the top deregulated pathways. Ablation of farnesoid X receptor (FXR) dramatically increases enterohepatic bile acid levels and jet lag-induced HCC, while loss of constitutive androstane receptor (CAR), a well-known liver tumor promoter that mediates toxic bile acid signaling, inhibits NAFLD-induced hepatocarcinogenesis. Circadian disruption activates CAR by promoting cholestasis, peripheral clock disruption, and sympathetic dysfunction.
Graphical abstract
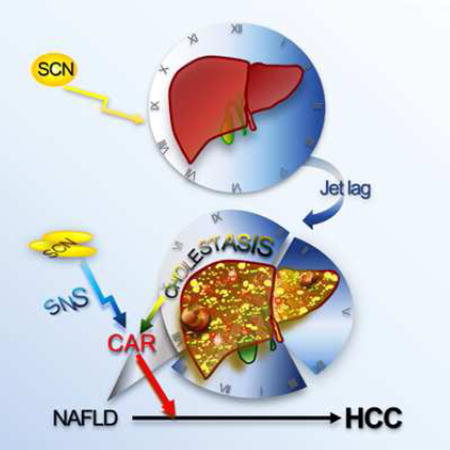
Kettner et al. show that experimental chronic jet lag induces persistent deregulation of liver gene expression and metabolism, culminating in the development of hepatocellular carcinoma. The bile acid receptor FXR and xenobiotic receptor CAR play an important role in this process.
Introduction
Hepatocellular carcinoma (HCC), the most common liver malignancy, was previously considered a rare cancer in the U.S., but a nearly 3-fold increase in incidence since the 1980s has made it the fastest rising cause of cancer-related death (El-Serag, 2011). Unlike in developing countries where hepatitis viral infection and aflatoxin contamination are major HCC risk factors, up to 30–50% of HCC diagnosed in the U.S. is resulted from liver metabolic diseases. Among these diseases, non-alcoholic fatty liver disease (NAFLD) is predicted to become the leading cause of HCC in the 21st century as a secondary consequence of the obesity pandemic (Siegel and Zhu, 2009).
NAFLD is found in 30–40% of the general population and up to 95% of those with morbid obesity (Review et al., 2014). Excessive fat accumulation induces liver injury, inflammation and regeneration, which progresses to non-alcoholic steatohepatitis (NASH) in a fraction of patients. This further stimulates the formation of scar tissue (fibrosis) and cirrhosis, both of which predispose to HCC (Michelotti et al., 2013). Although severe NAFLD can lead to hepatocarcinogenesis without NASH and fibrosis (Guzman et al., 2008), NASH and advanced fibrosis increase HCC risk by 15 and 25 folds in humans, respectively (Guzman et al., 2008; Hashimoto et al., 2009). The co-existence of NASH with other metabolic disorders such as leptin resistance and cholestasis significantly accelerates liver fibrosis (Wang et al., 2010).
Like HCC caused by other risk factors, NAFLD-induced HCC is predominantly diagnosed in males, but at older ages and earlier disease stages (Hashimoto et al., 2009). However, no significant difference in overall 5-year survival is found among HCC patients with or without NAFLD-related etiologies (Tokushige et al., 2011). The lack of proper mouse models that develop NAFLD in response to chronic metabolic stress and progress to NAFLD-initiated liver injury, inflammation, fibrosis and HCC as observed in obese human subjects has significantly impaired the study on the mechanism of metabolic syndrome-induced hepatocarcinogenesis (Michelotti et al., 2013). Despite the impending burden of NAFLD-induced HCC, no oncogene addiction loops specific to this disease have been identified (Villanueva et al., 2013).
The prevalence of obesity and NAFLD in our society is coupled with the epidemic of chronic circadian disruption, also termed “social jet lag” (Roenneberg et al., 2012). Circadian dysfunction among night-shift workers and individuals suffering from sleep dyspnea has been identified as a common risk factor of obesity, metabolic disorders, NAFLD and cancer including HCC (Bass and Takahashi, 2010; Fu and Kettner, 2013; Hu et al., 2013; Kao et al., 2012; Kim et al., 2013; Zimberg et al., 2012).
Circadian homeostasis in mammals is maintained by a central clock located in the hypothalamic suprachiasmatic nucleus that constantly synchronizes to external solar light cues and controls subordinate clocks in peripheral tissues via circadian output pathways. Among these pathways, the sympathetic nervous system (SNS) targets peripheral organs via adrenergic receptor-mediated intracellular signaling (Furness, 2006). SNS dysfunction is closely associated with metabolic syndrome, oncogenic activation, neoplastic growth and tumor initiation (Lee et al., 2010; Magnon et al., 2013; Tentolouris et al., 2006). The clock is operated by circadian genes. The current model of the molecular clock is based on the cyclic expression of the bHLH-PAS transcription factors BMAL1 and CLOCK and their downstream targets Cryptochrome (Cry1 and Cry2) and Period (Per1, Per2 and Per3), which encode the repressors of BMAL1/CLOCK heterodimer. The molecular clock also targets clock-controlled genes (CCGs) to couple diverse functions of peripheral organs with daily physical activity (Mohawk et al., 2012). Recent studies have revealed that germline or tissue-specific ablation of core circadian genes promotes genomic instability and accelerates both tumorigenesis and cancer progression in mice (Kettner et al., 2014)
The liver is the focus of circadian regulation of metabolic pathways, especially those controlling the synthesis and metabolism of glucose, lipid, cholesterol and bile acid (Bass and Takahashi, 2010). We have previously reported that mutations in both Per1 and Per2 or phase-shifts of the liver clock by restricted feeding both promote uncontrolled intrahepatic bile acid accumulation (cholestasis) and disrupt xenobiotic metabolism in mice (Ma et al., 2009), and that HCC is the second most commonly observed malignancy in circadian gene-mutant mouse models (Lee et al., 2010).
Bile acids are synthesized from cholesterol in the liver and function to promote dietary fat and lipid absorption. As detergents, bile acid intrahepatic levels are tightly controlled by a classic endocrine negative feedback loop in which bile acids act as ligands for the farnesoid X receptor (FXR, NR1H4), which then indirectly suppresses Cyp7A1, the rate limiting enzyme in bile acid biosynthesis (Goodwin et al., 2000). FXR also plays a broad role in liver function by inhibiting steatosis and inflammatory genes (Lopez-Velazquez et al., 2012). Ablation of Nr1h4 (hereafter referred to as Fxr) not only promotes cholestasis but also NAFLD, NASH and HCC in mice (Kim et al., 2007; Lu et al., 2000; Wang et al., 2008; Watanabe et al., 2004; Yang et al., 2007).
Intrahepatic cholestasis can also activate the constitutive androstane receptor (CAR, NR1I3) (Guo et al., 2003; Huang et al., 2005; Ma et al., 2009; Zhang et al., 2004), a central regulator of xenobiotic metabolism. CAR activation is the basis for phenobarbital promoted non-genotoxic or mutagen-induced hepatocarcinogenesis in mice (Huang et al., 2005; Yamamoto et al., 2004), which is strongly linked to β-catenin activation or mutation (Loeppen et al., 2002), a common molecular lesion in human HCC (Zucman-Rossi et al., 2015). We have recently shown that combined pharmacologic activation of CAR and genetic activation of β-catenin in mice is sufficient to induce liver tumors that share an extensive gene expression signature with a subset of human HCC displaying β-catenin activation (Dong et al., 2015).
Based on the discoveries described above, we hypothesize that chronic circadian disruption is sufficent to induce spontaneous hepatocarcinogenesis by driving persistant liver metabilic dysfunction and oncogenic activation.
Results
Circadian dysfunction induces NAFLD prior to spontaneous hepatocarcinogenesis
We have reported recently that chronic circadian disruption is sufficient to induce Leptin resistance in mice independent of diet choice and the time or amount of food intake (Kettner et al., 2015). We further studied the role of prolonged circadian disruption in HCC risk by conducting a large scale survival study with C57BL/6J inbred WT and congenic mice lacking Per1 and Per2 (Per1−/−;Per2−/−), Cry1 and Cry2 (Cry1−/−;Cry2−/−), or Bmal1 in the liver (Albcre;Bmal1fl/fl), under entrained or chronic jet-lagged conditions from 4 to 90 weeks of age. Compared to control littermates maintained in steady 24 hour light/dark (24 hr LD) cycles, jet-lagged mice showed significantly reduced lifespan with disease development leading to early euthanization (Figure 1A), which include neurodegeneration, severe ulcerative dermatitis, aging, cystic renal dysplasia, and cancer (Table 1).
Figure 1. Chronic jet lag induces NAFLD and spontaneous HCC.

(A) Kaplan-Meier survival curves show life-spans of WT and circadian gene mutant mice. Ctrl: maintained in steady 24 hr LD cycles; CJ: chronically jet-lagged. p value: Ctrl vs. CJ for each mouse model, Kaplan Meier Statistics. (B) Spontaneous HCC from circadian gene mutant and jet-lagged WT mice. Top: gross image of HCC; bottom: Hematoxylin and eosin (H&E) stained tumor slides show histological diagnosis of HCC, circled by dashed green lines. Scale bars indicate 300 µm. (C) Western blots show BMAL1, CRY1 and PER2 expression in the livers of 12 week old jet-lagged WT and circadian gene mutant mice. ZT: Zeitgeber time, with light on at ZT0 and off at ZT12. (D) Summary of 3 independent Western blotting analyses on hepatic expression of BMAL1, CRY1 and PER2 (higher protein band only for PER2) with the expression level in control WT mice at ZT2 as the arbitrary unit 1 (student t test, ±SEM). *: Increase,#: decrease, comparing to WT controls at the same time, */#p<0.05, **/##p<0.01; ***/###p<0.001. (E, F) Gross liver image (E) and Oil Red O stained slides (F) showing NAFLD development in mutant and jet-lagged WT mice. (G) Representative Oil Red O stained slides show the level of fat accumulation in control (top) and jet-lagged (bottom) Albcre;Bmal1fl/fl mice at 12 weeks of age. Scale bars in all Oil Red O stained slides indicate 50 µm. Arrows indicate representative fat droplets. See also figure S1.
Table 1.
Incidence of HCC, Hepatomegaly and NAFLD in Jet-lagged WT and Mutant Mice by 90 Weeks of Age
| C57BL/6J Inbred Mouse Models |
% HCC | Age HCC found (week) |
p1* | % Hepatomegaly at 16 week |
% NAFLD at 90 week |
p2** |
|---|---|---|---|---|---|---|
| WT Ctrl (n=110) | 0 | N/A | 15.5 | 20 | ||
| WT CJ (n=80) | 8.75a | 78–90 | 0.00694 | 75 | 96.3 | 2.2E-16 |
| Cry1−/−;Cry2−/−Ctrl (n=60) | 11.7b | 51–90 | 7.87E-05 | 68.3 | 93.3 | 2.27E-16 |
| Cry1−/−;Cry2−/−CJ (n=56) | 17.9c | 43–90 | 1.26E-05 | 69.7 | 96.4 | 2.2E-16 |
| Per1−/−;Per2−/−Ctrl (n=80) | 12.5d | 50–90 | 1.34E-04 | 86.25 | 100 | 1.7E-16 |
| Per1−/−;Per2−/−CJ (n=50) | 26e | 42–90 | 1.14E-08 | 86 | 100 | 1.7E-16 |
| Albcre;Bmal1fl/fl Ctrl (n=26) | 11.53f | 74–90 | 2.3E-03 | 73.1 | 92.3 | 1.28E-09 |
| Albcre;Bmal1fl/flCJ (n=43) | 11.63g | 65–90 | 1.71E-03 | 95.35 | 100 | 7.2E-16 |
| Car−/−Ctrl (n=24) | 0 | N/A | N/A | 25 | 87.5 | 4.2E-08 |
| Car−/−CJ (n=25) | 0 | N/A | N/A | 0 | 68 | 0.0005 |
| Fxr−/−Ctrl (n=25) | 29.2 | 57–90 | 1.67E-09 | 100 | 100 | 2.87E-11 |
| Fxr−/−CJ (n=31) | 61.3 | 57–90 | 4.00E-13 | 100 | 100 | 6.46E-13 |
Non-Cancer related early euthanization: 8.8%
, 18.3%
, 17.5%
or 30.2%
early euthanization from 15–82 weeks of age due to overgrowth of teeth, severe ulcerative dermatitis, or neurological disorders.
32.1% early euthanization from 12–77 weeks of age due to overgrowth of teeth, severe ulcerative dermatitis, cystic renal dysplasia, or neurological disorders.
30% early euthanization from 35–79 weeks due to overgrowth of teeth, cystic renal dysplasia, severe ulcerative dermatitis, or neurological disorders.
15.4% early euthanization from 58–81 weeks of age due to ulcerative dermatitis.
p1: Comparing HCC incidence with WT control mice, Kaplan-Meier statistics.
p2: Comparing NAFLD incidence with WT control mice, two sample Wilcoxon test.
The major cancer types diagnosed in mutant and jet-lagged mice include pancreatic and ovarian tumors, B-lymphoma and HCC (Figures 1B and S1A), with lymphoma and HCC as the leading cause of cancer-related early euthanization. Unlike γ-irradiated mice that are especially susceptible to lymphoma (Fu et al., 2002; Lee et al., 2010), unirradiated jet-lagged WT and mutant mice displayed similar risk of lymphoma and HCC regardless of the differences in the molecular signatures of liver clock disruption in these mice, with HCC incidence specific to each mouse model by 90 weeks of age (Figures 1C, 1D and Table 1). Both sexes of mutant mice displayed increased risk of hepatocarcinogenesis, with males showed greater HCC risk than females. Under entrained condition, Per- or Cry-mutants developed fewer but larger HCCs first detected at 50 weeks of age, while Albcre;Bmal1fl/fl mice developed a large number but small HCCs first detected after 70 weeks of age. Chronic jet lag increased both numbers and sizes of tumors in Per and Cry mutants and also the size of tumor in HCC bearing Albcre;Bmal1fl/fl mice (Figures 1B, S1A–B and Table 1). Thus, chronic circadian disruption not only increases tumor incidence but also accelerates tumor progression.
Despite the fact that C57BL/6J inbred WT mice are normally resistant to spontaneous and carcinogen-induced HCC (Diwan et al., 1990; Drinkwater and Ginsler, 1986), chronically jetlagged WT mice developed HCC after 78 weeks of age. This was also male dominant and at an age correspondent to the median age of spontaneous HCC diagnosis at 67–72 years in humans (Hashimoto et al., 2009; Kunstyr and Leuenberger, 1975; Yasui et al., 2011) (Figures 1B, S1A–B and Table 1).
Liver pathology in jet-lagged WT and mutant mice, including Cry-mutants that are deficient in fat storage in adipose (Kettner et al., 2015), initiated with spontaneous NAFLD onset at a young age. Jet lag also accelerated NAFLD progression in Albcre;Bmal1fl/fl mice (Figures 1E–G and S1C). Together, these findings demonstrates that chronic circadian misalignment is sufficient to disrupt liver clock and induce NAFLD-related HCC in WT mice. Since ”social jet lag”, but not circadian gene germline or tissue-specific ablation, is the major type of circadian disruption in humans (Roenneberg, 2013), we then focused on studying the mechanism of NAFLD-induced HCC using jet-lagged WT mice, with Albcre;Bmal1fl/fl and Per1−/−;Per2−/− mice that display two distinct patterns of liver clock disruption and are easier to breed as controls to confirm the role of the clock in the pathologies observed (Figures 1C and 1D).
Circadian dysfunction induces metabolic syndrome and NAFLD to NASH and fibrosis progression
In addition to developing leptin resistance (Kettner et al., 2015), jet-lagged WT mice also displayed a decrease in serum triglycerides and free fatty acids coupled with an increase in hepatic level of triglycerides and free fatty acids. This was associated with persistent high plasma levels of glucose and insulin, characteristic of insulin resistance (Figure 2A), and a dramatic decrease in hepatic glycogen storage throughout a 24 hr period (Figures 2B and S2).
Figure 2. Chronic jet lag induces metabolic syndrome and the progression from NAFLD to NASH and fibrosis.

(A) The levels of serum and hepatic biomarkers in control and jet-lagged WT mice. ALT: alanine aminotransferase; TG: triglycerides; FFA: free fatty acids (Student t test). (B) Daily hepatic glycogen storage detected by Periodic Acid Schiff (PAS) staining in control and jetlagged WT mice. Values indicate average levels of glycogen storage detected at ZT2, 10 and 18 for each mouse model, with that in control WT mice as the arbitrary unit 1 (Image J/Color Deconvolution plugin quantification). (C) Serum liver biomarkers in 12 week old jet-lagged WT mice. AST: aspartate transaminase, ALP: alkaline phosphatase, LDH: lactate dehydrogenase (Student t test). (D) The ratios of liver vs body weight of circadian gene mutant and WT mice (Student t test). (E) Cytokeratin 19 (CK19) and H&E staining show incidence of intrahepatic bile duct proliferation and inflammation in control and jet-lagged WT mice. Arrows indicate representative proliferating bile ducts (CK19) and areas of hematopoietic cell infiltration (H&E). (F) Ki67, TUNEL and Sirus Red staining show incidence of hepatocyte proliferation and death, and liver fibrosis in control and jet-lagged WT mice. Arrows indicate representative Ki67+ or TUNEL+ hepatocytes (Ki67 or TUNEL), and liver fibrosis (Sirus Red). Scale bars are 60 µm (CK19), 50 µm (HE), 60 µm (Ki67), 50 µm (TUNEL) and 100 µm (Sirus Red) in correspondent slides. *: Increase,#: decrease, comparing to WT controls at the same time or age */#p<0.05, **/##p<0.01; ***/###p<0.001, ±SEM. See also figures S2 and S3.
The development of metabolic syndrome in jet-lagged WT mice is coupled with persistent liver damage as indicated by elevated and deregulated plasma liver parameters, such as alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), lactate dehydrogenase (LDH) and total bilirubin (Figures 2A and 2C). This is accompanied by hepatomegaly (Figure 2D), increased bile duct proliferation and chronic liver inflammation (Figure 2E), accelerated hepatocyte proliferation and death, and the appearance of fibrosis long before HCC detection (Figure 2F). Thus, chronic circadian disruption not only induces NAFLD but also persistent liver damage and fibrosis long before spontaneous hepatocarcinogenesis. Per1−/−;Per2−/− and Albcre;Bmal1fl/fl mice also displayed similar deregulation of serum and hepatic metabolic parameters and liver pathological changes in addition to NAFLD prior to HCC detection (Figure S3).
Circadian disruption induces global metabolic disruption in the liver
To study the mechanism of circadian disruption-induced NAFLD, we conducted a large scale circadian metabolomics study on serum and hepatic carnitines, lipids and prostaglandins, CoA’s, and tricarboxylic acid cycle (TCA) metabolites in WT mice at 12 and 30 weeks of age. The majority of metabolites studied displayed robust circadian rhythms in control WT mice, but were deregulated in jet-lagged WT mice. Especially, jet-lagged WT mice displayed increased serum and hepatic levels of lactate and pyruvate, coupled with elevated levels of essential bioprecursors for amino acids, nucleotides, triglyceride and cholesterol synthesis, such as α-ketoglutarate, glycerol 3 phosphate, glutamine, ribose 5 phosphate, acetyl CoA, malate and fumarate, etc. These were associated with deregulation of fatty acid transporting carnitines and accumulation of TCA cycle intermediates, such as isocitrate, succinate, fumarate and malate, an indication of mitochondrial electron transport chain disruption and redox deregulation (Figures 3 and S4–5).
Figure 3. Global disruption of liver metabolism in jet-lagged WT mice.

Hierarchical clustering heat maps show serum (left panel) and hepatic (right panel) carnitines, lipids and prostaglandins, CoA’s and TCA metabolites in control WT mice and the persistent deregulation of these metabolites in jet-lagged WT mice. See also figures S4 and S5.
Thus, chronic jet lag induces a global shift in liver metabolism to promote lipid synthesis and storage via accelerating cytoplasmic glycolysis. This is also coupled with elevated intracellular oxidative stress that induces liver damage and increased biosynthetic intermediates that support rapid cell division (Figures 1E–F, 2A–B, S1C, S2 and S4–5), a pattern of metabolic adaptation closely reminiscent of that well-characterized for most types of cancer cells (Warburg, 1956).
Jet lag induces genome-wide gene deregulation with hepatic cholesterol, bile acid and xenobiotic metabolism as top deregulated pathways
To define the mechanism of jet lag induced global disruption of liver metabolism and pathology, we carried out a large scale microarray analysis using total liver RNA prepared from 12 and 30 week old control and jet-lagged WT mice at ZT2, 10 and 18. As expected, chronic jet lag induced persistent and genome-wide gene deregulation in mouse livers (Figure 4A and Table S1–2). This includes overexpression of genes promoting lipid, amino acids and nucleotides biosynthesis and storage, cytoplasmic glycolysis, glycogenolysis, oxidative stress, hepatocyte proliferation and death, cholestasis, and fibrosis, suppression of genes stimulating fatty acid mitochondria transportation and β-oxidation, glycogen synthesis, and tumor suppression, and deregulation of genes controlling cell cycle checkpoints, DNA damage repair, and both innate and adaptive inflammatory responses (Table S3).
Figure 4. Chronic jet lag induces genome-wide gene deregulation in mouse livers.

(A) Hierarchical clustering heat map shows expression of hepatic genes in the livers of control and jet-lagged WT mice. (B) Venn diagrams showing persistent overlap of deregulated liver transcriptomic signatures in jet-lagged WT mice with that of human HCC from 12 to 30 weeks of age (hypergeometric test). (C) The summary of 3 independent RT-PCR studies on the expression of core circadian genes, Myc and Fxr in the livers of 12 week-old circadian gene mutant and WT mice, with levels detected in control WT mice at ZT2 as the arbitrary unit 1. (D–F) Western blots show the expression of Ser552 phospho-β-catenin (pβ-catenin), total β-catenin (T-β-catenin), c-Myc and p53 in (D), FXR, CAR, Cyp2B10 and Cyp7A1 in (E), and SREBP1 and PPARγ in (F) in the livers of 12 week old WT mice. (G) Serum and hepatic bile acid levels over a 24 hr period in circadian gene-mutant and WT mice at 7, 12 and 30 weeks of age. *: Increase,#: decrease, comparing to WT controls at the same time, */#p<0.05, **/##p<0.01; ***/###p<0.001, student t test, ±SEM. See also figure S6 and Table S1–6.
Importantly, the jet lag induced gene deregulation signature significantly overlaps with that found in human HCC (Figure 4B), and also includes pathways frequently deregulated in other human cancers, such as pancreatic adenocarcinoma and bladder, small cell lung, prostate, and estrogen-dependent and hereditary breast cancers (Table S4–5) (Hoshida et al., 2008; Huang et al., 2011; Huang et al., 2012; Lee et al., 2006; Woo et al., 2008; Ye et al., 2003; Zucman-Rossi et al., 2015).
The deregulation of key genes, including circadian genes Bmal1, Clock, Per1, Per2, Cry1 and Nr1d1 (encoding REV-ERBα), was validated at the mRNA level by RT-PCR (Figure 4C). Together with the results of protein expression studies shown in figure1C and 1D, these results demonstrated that chronic jet lag is sufficient to disrupt the liver clock independent of circadian gene mutations. Surprisingly, in the livers of Albcre;Bmal1fl/fl mice, Per2 mRNA still displayed a shifted and dampened circadian expression profile, while Cry1 mRNA was constitutively overexpressed. These findings were consistent with a previous report on Cry1 mRNA hepatic overexpression in Albcre;Bmal1fl/fl mice (Lamia et al., 2008), and with our finding that the negative loop proteins PER2 and CRY1 still display a robust circadian expression in the livers of these mice under entrained condition (Figures 1C–D and 4C).
Dysregulation of homologs of molecular markers of human HCCs, such as Ctnnb1 (encoding β-catenin), Myc and Trp53 (Zucman-Rossi et al., 2015), was also found at the mRNA and/or protein levels in the livers of jet-lagged WT and circadian gene-mutant mice (Figures 4C–D and S6A–B). However, unlike in mouse thymus where circadian disruption is sufficient to suppress p53 (Lee et al., 2010), chronic jet lag induced a coupled activation of p53 and c-Myc in the livers of WT mice at a young age. Thus, the p53 response to Myc oncogenic activation was still intact in the liver at the initial stage of circadian disruption (Figure 4D). This finding agrees with previous reports that the complete loss of p53 function is associated with the progression but not initiation of hepatitis B virus or aflatoxin B1-induced HCC in humans (Hosono et al., 1993; Nose et al., 1993).
Strikingly, nuclear receptor-controlled cholesterol, bile acid and xenobiotic metabolism were among the top deregulated pathways in the livers of jet-lagged WT mice at all ages studied (Table S6). Among these nuclear receptors, the suppression of FXR and induction of CAR, as reported for human HCC (Hoshida et al., 2008; Lee et al., 2006), were found in jet-lagged WT and circadian gene mutant mice, coupled with upregulation of transcription factors stimulating cell proliferation and steatosis such as β-catenin, c-Myc, SREBP1 and PPARγ (encoded by the Nr1c3), as well as Cyp2B10, a direct target of CAR activation, and Cyp7A1, the rate limiting enzyme for bile acid synthesis negatively regulated by FXR (Figures 4D–F and S6A–D).
Further analysis revealed that all HCC-prone mouse models in our study displayed significantly elevated serum and hepatic bile acid levels, characteristic of intrahepatic cholestasis, regardless of their molecular mechanisms of liver clock disruption and overall metabolic phenotypes (Figures 1C–D, 4G and S6E). Thus, the coexistence of intrahepatic cholestasis with NAFLD may play a key role in spontaneous hepatocarcinogenesis.
CAR is a CCG that promotes spontaneous HCC by stimulating NAFLD to NASH transition
We and others have previously demonstrated that elevated bile acids, as observed in mice lacking circadian homeostasis, activates CAR (Guo et al., 2003; Huang et al., 2005; Ma et al., 2009; Zhang et al., 2004), which promotes hepatocarcinogenesis independent of exogenous mutagens (Huang et al., 2005; Yamamoto et al., 2004). To define the role of cholestasis and CAR in NAFLD-induced HCC, we studied HCC risk in Fxr−/− and Nr1i3−/− (here after Car−/−) C57BL/6J mice. Under entrained conditions, Fxr−/− mice displayed the highest levels of intrahepatic bile acids and triglycerides throughout a 24 hr period among all mouse models studied, and also the highest risk of NAFLD and HCC, as expected. Chronic jet lag further accelerated NAFLD development and led to a greater than 2-fold increase in HCC incidence in Fxr−/− mice by 90 weeks of age (Figure 5A and Table 1). In contrast, although Car−/− mice were also prone to NAFLD and circadian disruption-induced cholestasis and glycogen storage disease, they showed a dramatically decreased risk of jet lag induced hepatomegaly, inflammation, hepatocyte proliferation and necrosis, fibrosis, and were completely resistant to spontaneous HCC (Figures 5, S7 and Table 1).
Figure 5. CAR stimulates NAFLD to NASH and fibrosis progression.

(A) Circadian profiles of serum and hepatic bile acids and triglyceride (TG) in 12 week old control and jet-lagged WT, Car−/− and Fxr−/− mice (Student t test). (B) Daily hepatic glycogen storage in control and jet-lagged WT and Car−/− mice detected by PAS staining. Values indicate average levels of hepatic glycogen storage detected at ZT2, 10 and 18 for each mouse model, with that in control WT mice as the arbitrary unit 1 (Image J/Color Deconvolution plugin quantification). (C) The ratios of liver vs. body weight of WT, Car−/− and Fxr−/− mice at 30 weeks of age (Student t test). (D) H&E and Ki67 staining detect liver inflammation and hepatocyte proliferation in control and jet-lagged WT, Car−/−, Fxr−/− mice. Arrows indicate representative areas of hematopoietic cell infiltration in H&E and Ki67+ hepatocytes in Ki67 slides. Scale bars indicate 50 and 60 µm in the H&E and Ki67 slides, respectively. (E) TUNEL and Sirus Red staining show incidence of hepatocyte death (TUNEL) and fibrosis (Sirus Red) in the livers of WT, Car−/−, Fxr−/− mice. Arrows indicate representative TUNEL+ hepatocytes (TUNEL) and areas of liver fibrosis (Sirus Red). Scale bars indicate 50 and 100 µm in the TUNEL and Sirus Red slides, respectively. (F) The level of bile duct and hepatocyte proliferation (Student t test), hepatocyte necrosis (Student t test), and liver fibrosis (Image J) in 30 week old control and jetlagged WT and mutant mice. All analyzed at 10× magnification with CK19, Ki67 and TUNEL signals detected in the livers of control WT mice as the arbitrary unit 1. (G) Gross image (top) and histological diagnosis of HCC by H&E staining (bottom) show NAFLD in jet-lagged WT, Car−/− and Fxr−/− mice and HCCs found in jet-lagged WT and Fxr−/− mice. Kidneys that do not show significant changes in size among different mouse models are included in gross images. Tumors in H&E slides are circled by dashed green lines. Arrows indicate representative fat droplets in H&E slides. Scale bars indicate 200 µm. *: Increase,#: decrease, comparing to WT control samples at the same age or time, */#p<0.05, **/##p<0.01; ***/##p<0.001, ±SEM. See also figure S7.
Under entrained conditions, Car−/− mice had an intact liver clock and maintained a circadian profile of FXR and Cyp7A1 expression (Figures 6A–B). However, they lacked β-catenin nuclear activation and showed dramatically dampened c-Myc and Cyp2B10 protein expression in the liver (Figures 6B–D). CAR mRNA and protein levels also displayed a robust circadian rhythm in 24 hr LD cycles, but were arrhythmic and elevated at most times in the livers of jet-lagged WT mice, coupled with constitutive overexpression of Cyp2b10 mRNA, a direct consequence of CAR-directed transcriptional activation (Figure 6E). Chronic jet lag disrupted the liver clock in both Car−/− and WT mice but failed to fully activate oncogenic, steatotic and inflammatory genes in Car−/− mice as compared to jet-lagged WT controls. This, again, was coupled with the lack of direct CAR target Cyp2b10 mRNA expression in jet-lagged Car mutants, which confirmed the ability of jet lag to activate CAR (Figures 6A and 6E). Thus, Car is a CCG. Its circadian dysregulation stimulates the pathophysiological progression from NAFLD to HCC.
Figure 6. Car is a clock-controlled gene.

(A) The summary of 3–6 independent RT-PCR studies on core circadian gene expression in WT and Car−/− mouse livers at 12 weeks of age, with the expression level in control WT mice at ZT2 as the arbitrary unit 1. (B, C) Western blots show the expression of BMAL1, CRY1, PER2, FXR, Cyp7A1 and Cyp2B10 (B), and CAR, p53, c-Myc, phospho-β-catenin (pβ-catenin) and total β-catenin (T-β-catenin) (C) in WT and Car−/− mouse livers at 12 weeks of age. (D) The summary of 3 independent Western blotting on hepatic expression of FXR, CAR, c-Myc, pβ-catenin, p53, Cyp7A1 and Cyp2B10 in 12 week old WT and Car−/− mice, with the expression level in control WT mice at ZT2 as the arbitrary unit 1. (E) The summary of 3–6 independent RT-PCR studies on the expression of Car, Cyp2b10, Fxr, Cyp7a1, Myc, Fos, Pparγ, Srebp1c, Tnf and Il6 in the livers of 12 week-old WT, Car−/− and Albcre;Bmal1fl/fl mice, with the expression level in control WT mice at ZT2 as the arbitrary unit 1. *: Increase,#: decrease, comparing to WT control samples at the same time, */#p<0.05, **/##p<0.01; ***/###p<0.001, student t test, ±SEM.
Sympathetic dysfunction promotes CAR constitutive overexpression
Previous studies have suggested that BMAL1/CLOCK and the PAR-domain basic leucine zipper transcription factor DBP may co-activate Car over a 24 hr period in the liver (Gachon et al., 2006; Koike et al., 2012; Ripperger and Schibler, 2006). However, Bmal1 and Dbp were both suppressed by circadian disruption (Figures 1C–D and 4C). Thus, jet lag induced CAR overexpression in WT mice appeared to be controlled by a previously unknown mechanism. We found that apart from multiple D- and E-boxes potentially recognized by DBP and BMAL1/CLOCK, human and mouse CAR promoters contain conserved AP1 and CRE motifs potentially activated by SNS signaling (Figures 7A). In vitro transfection assays showed that the Car promoter was moderately stimulated by low levels but suppressed by higher levels of AP1 or CREB expression. However, co-expression of AP1 and CREB, even at a moderate level, dramatically stimulated Car promoter activity (Figures 7B). We also found that chronic jet lag significantly increased SNS tone in the sleep phase. This was coupled with a constitutive AP1 and Ser133 phospho-CREB (pCREB) nuclear accumulation in mouse livers, which is a well-known consequence of cellular response to SNS-ADRβ-c-AMP-PKA signaling (Figures 7C–E). Ablation of all three β-adrenergic receptors in mice (β-less) completely inhibited AP1 and pCREB nuclear accumulation and also Car, Fos, Myc and Cyp2b10 expression in mouse livers under both entrained and jet lag conditions (Figures 7F–G), which was associated with strong HCC resistance (Figure S8A). Thus, jet lag induced SNS dysfunction may directly deregulate Car expression via activation of AP1 and CREB.
Figure 7. SNS dysfunction induces Car activation.

(A) The schematic illustration of conserved E-boxes, AP1 (A1 and A2) and CRE (C1 and C2) binding motifs and ChIP qPCR primers in the Car promoter. (B) The summary of 4–5 independent co-transfection assays studying the role of AP1 and CREB in Car promoter activation. (C) Urine catecholamine assays show sympathetic tone over a 24 hr period in control and jet-lagged WT mice at 12 weeks of age. (D) Western blots show nuclear expression of c-FOS and S133 phopho-CREB (pCREB) (top panel), and total CREB (T-CREB) (bottom panel) levels in liver nuclear extracts of WT and β-less mice at 12 weeks of age. (E) The summary of 3 independent Western blotting studying the hepatic expression of c-FOS, pCREB and T-CREB in WT and β-less mice at 12 weeks of age. F. Western blots show CAR and c-Myc expression in the livers of 12 week old β-less mice. G. The summary of 3 independent RT-PCR studies on hepatic expression of Fos, Myc, Car and Cyp2b10 mRNAs in control and jet-lagged WT and β-less mice at 12 weeks of age. (H) BMAL1, c-FOS and pCREB ChIP signals on Car and Per1 promoters in the livers of WT, β-less and AlbCre;Bmal1fl/fl mice at 12 week of age, with ChIP signals for each transcription factor detected in control WT mice at ZT2 as the arbitrary unit 1. Negative control IgGs and qPCR primers are explained in Supplemental Experimental Procedures. *: Increase,#: decrease, comparing to WT control mice at the same time, */#p<0.05, **/##p<0.01; ***/###p<0.001, student t test, ±SEM. (I) A model for the role of chronic circadian disruption in NAFLD-induced hepatocarcinogenesis. See also figure S8.
To test this hypothesis, we performed in vivo ChIP using anti-BMAL1, c-FOS and pCREB antibodies (Figures S8B–D), primers flanking the AP1, CRE, or E-box 8 sequences in the Car promoter, E-box 3, AP1 or CRE motifs in the Per1 promoter, and liver nuclear extracts from 12 week old WT, Albcre;Bmal1fl/fl and β-less mice at ZT2, 6 and 18. In the livers of WT mice, BMAL1/CLOCK transcriptional activity is low at ZT2 and 18 but peaks at ZT6 (Koike et al., 2012), while nuclear accumulation of AP1 and pCREB is lowest at ZT6, slightly elevated at ZT2 and peaks ZT18 (Figures 7D–E). Thus, ZT2, 6 and 18 are the best times in a circadian cycle to study the interplay between SNS-controlled cell signaling and the liver clock in controlling Car expression.
We found that under entrained conditions, pCREB and BMAL1 displayed a coupled and robust rhythmic interaction with both Car and Per1 promoters, which peaked at ZT2 for pCREB and ZT6 for BMAL1. Although c-FOS binding to the Per1 promoter displayed a strong circadian rhythm and peaked at ZT2, its binding to Car promoter was constitutively low. Chronic jet lag abolished BMAL1 binding to both Car and Per1 promoters, significantly dampened c-FOS and pCREB interaction with the Per1 promoter, but induced a coupled and dramatically increased binding of c-FOS and pCREB to the Car promoter at all times studied (Figure 7H), which, as shown by promoter functional analysis, strongly activated Car transcription (Figure 7B). As expected, the binding of BMAL1 or c-FOS and pCREB to Per1 and Car promoters was not detected in livers of Albcre;Bmal1fl/fl or β-less mice, respectively (Figure 7H). Thus, jet lag induced SNS dysfunction and liver clock disruption are sufficient to promote Car dysregulation.
Discussion
Lifestyle changes, but not germline mutations, drive increased cancer risk in modern societies (Anand et al., 2008). One of the major lifestyle changes is chronic social jet lag (Roenneberg, 2013). Human epidemiological studies have revealed a strong link between chronic circadian disruption and cancer risk. However, the diversity of lifestyles, heterogeneity of cancer etiologies and long latency of spontaneous cancer development impose significant difficulties for defining the role of circadian dysfunction in human cancer initiation. Although mouse models with germline or tissue-specific ablation of circadian genes display increased risk of genomic instability, neoplastic growth and cancer (Kettner et al., 2014), the role of circadian dysfunction in spontaneous cancer initiation in humans cannot be closely modeled by these mutant mice as they do not display the same circadian dynamics as normal human individuals in response to changes in environmental cues (Kettner et al., 2015).
We have previously reported that chronic jet lag significantly increases the risk of γ-radiation induced cancer in mice (Lee et al., 2010). In this study, we show that chronic circadian misalignment is sufficient to induce spontaneous hepatocarcinogenesis in mice in the absence of dietary manipulation, exogenous genotoxic stress or germline gene mutations. We found that in addition to leptin resistance, circadian dysfunction also induces hyperinsulinemia, hyperglycemia and dyslipidemia at the organismal level and liver metabolic disorders including NAFLD. Such combined systemic and liver metabolic disorders are also frequently observed among human night-shift workers (Zimberg et al., 2012). We show that jet lag induces a global shift in liver metabolism in mice, which not only promotes fat synthesis and storage via accelerated cytoplasmic glycolysis but also increases oxidative stress and the synthesis of bioprecursors supporting rapid cell division. The majority of jet-lagged WT mice display NAFLD progression to NASH and then fibrosis at a relatively young age. This is likely due to jet lag induced persistent liver injury and inflammation that triggers a chronic regenerative wound-healing process, a common mechanism of tumor initiation (Michelotti et al., 2013). Overall, this pathophysiological progression closely mimics that described for NAFLD-induced spontaneous hepatocarcinogenesis in humans.
The complex liver pathology in jet-lagged WT mice is driven by global hepatic gene deregulation, which can be detected soon after the initiation of jet lag and displays a pattern overlapping significantly with a human HCC transcriptomic signature, including the induction of key human HCC molecular markers such as Trp53, Myc and β-catenin. Thus, jet-lagged WT mice likely also develop spontaneous HCC following a molecular mechanism very similar to that in obese humans.
We found that nuclear receptor-controlled cholesterol, bile acid and xenobiotic metabolism are among top deregulated hepatic pathways in jet-lagged WT mice, and all mouse models studied here displayed cholestasis in addition to NAFLD prior to HCC detection. Especially, HCC prone Fxr−/− mice that display extremely high intrahepatic bile acid levels showed an additional more than 2-fold increase in HCC incidence under jet lag condition. Thus, we suggest that loss of bile acid homeostasis is a direct cause of increased HCC risk in response to persistent circadian disruption.
Two broad mechanisms could link cholestasis to hepatocarcinogenesis. First, as strong hydrophobic detergents, bile acids can induce hepatocyte injury, mitochondrial damage, oxidative stress, DNA damage, and hepatocyte necrosis and apoptosis, all of which drive the progression of NAFLD to NASH, fibrosis and HCC (Audard et al., 2007; Perez and Briz, 2009). Second, and more directly, elevated hepatic bile acid levels, together with an increase in hepatic toxic bile acid species due to microbiota activity associated with circadian disruption (Voigt et al., 2014), activate CAR, which is well known as a potent tumor promoter that drives non-genotoxic hepatocarcinogenesis (Guo et al., 2003; Huang et al., 2005; Yamamoto et al., 2004; Zhang et al., 2004).
The tumor promoting function of CAR is tightly associated with β-catenin activation or mutation (Dong et al., 2015; Loeppen et al., 2002). Jet lag activated CAR, as measured by constitutive overexpression of the CAR target gene Cyp2b10 over a 24 hr period, was coupled with β-catenin and c-Myc overexpression. Ablation of Car completely inhibited nuclear β-catenin expression and suppressed jet lag induced activation of oncogenic, steatotic and inflammatory genes, leading to strong HCC resistance.
In the hierarchical circadian system, peripheral organs rely on signaling from circadian output pathways to maintain synchrony with the central clock. We found that chronic jet lag promotes sympathetic dysfunction and peripheral clock disruption, both of which have been linked to oncogenic activation and increased risk of cancer (Kettner NM, 2016; Lee et al., 2010; Magnon et al., 2013). We demonstrated that jet lag induced sympathetic dysfunction and peripheral clock suppression are sufficient to promote AP1 and CREB oncogenic activation independent of somatic mutations, which drives Car overexpression throughout a 24 hr period to mediate persistent toxic bile acid signaling in jet-lagged WT mice.
We propose that chronic circadian disruption induces neuroendocrine dysfunction, leading to a coupled peripheral clock disruption, genome-wide gene deregulation, oncogenic activation and global metabolic dysfunction in the liver. Together, these induce NAFLD and also the progression from NAFLD to HCC. Among top deregulated gene pathways, the nuclear receptor CAR is constitutively activated by a combination of overexpression, in response to SNS dysfunction and peripheral clock disruption, and transactivation, in response to elevated intrahepatic bile acids, to promote spontaneous hepatocarcinogenesis by stimulating the progression from NALFD to NASH and fibrosis (Figure 7I). Inverse agonists to suppress human CAR and agonists for activation of human FXR are both pharmaceutically available (Hirschfield et al., 2015; Huang et al., 2004). Since ablation of CAR prevents spontaneous HCC and loss of FXR increases intrahepatic bile acid levels and stimulates hepatocarcinogenesis, our studies suggest that restoration of bile acid homeostasis and inhibition of CAR activation provide promising complementary strategies for prevention of metabolic syndrome-induced HCC.
Experimental Procedures
Animal Maintenance
All animal experiments were approved by the Institutional Animal Care and Use Committee at Baylor College of Medicine (BCM). Detailed experimental procedures for mouse survival, pathological studies and tissue isolation are described in Supplemental Experimental Procedures.
ELISA
Mouse serum, urine and livers were collected and analyzed by ELISA as described in Supplemental Experimental Procedures.
RNA Analysis
Total RNA was isolated from mouse livers and used for RT-PCR. Detailed procedures and RT-PCR primers are described in Supplemental Experimental Procedures.
Plasmid Construction and Reporter Assays
A 3-Kb mouse Car promoter was cloned by PCR amplification of C57BL/6J WT mouse genomic DNA, inserted upstream of the luciferase cDNA in the pGL-3 vector, and used for cotransfection assays. Detailed procedures are described in Supplemental Experimental Procedures.
Chromatin immunoprecipitation (ChIP)
Nuclear extracts were prepared from mouse livers at ZT2, 6 and 18. Antibodies against BMAL1, c-FOS or pCREB were used for immunoprecipitation. qPCRs were performed to detect BMAL1, c-FOS and pCREB ChIP signals. Detailed procedures, antibodies and qPCR primers are described in Supplemental Experimental Procedures.
Protein Expression Studies
Western blotting using total liver or nuclear extracts were performed as described in Supplemental Experimental Procedures.
Metabolomics
Targeted measurement of metabolites was carried out by the BCM Dan L Duncan Cancer Center CPRIT Cancer Proteomic and Metabolomic Core using serum and liver samples collected from 12 or 30 week-old WT mice at ZT2, 10 and 18 as described in Supplemental Experimental Procedures.
Microarray
Total liver RNA isolated from 12 and 30 week old WT mice was used for Agilent gene expression microarray at the BCM Genomic and RNA Profiling Core. Detailed procedures are described in Supplemental Experimental Procedures.
Histology and Immunohistochemistry (IHC)
Mouse liver and tumor samples were processed for histological or IHC staining at the Texas Medical Center (TMC) Digestive Disease Center following standard procedures as described in Supplemental Experimental Procedures.
Statistical Analysis
The methods for statistically analyzing data obtained from all experiments performed are described in Supplemental Experimental Procedures. In all analyses, p < 0.05 was considered statistically significant.
Supplementary Material
Significance.
Non-alcoholic fatty liver disease (NAFLD) is predicted to become the leading cause of hepatocellular carcinoma (HCC) due to the prevalence of obesity and chronic circadian disruption, but the underlying mechanisms are poorly understood. We found that circadian dysfunction promotes NAFLD-induced hepatocarcinogenesis by maintaining persistent liver gene deregulation and metabolic disruption, closely mimicking that observed in obese humans. We demonstrate that the profound circadian dysregulation of nuclear receptor-controlled hepatoprotective pathways, especially those controlled by the bile acid receptor FXR and xenobiotic receptor CAR, plays an essential role in NAFLD-induced HCC. Thus, circadian dysfunction is an independent risk factor of HCC, and restoration of bile acid homeostasis and inhibition of CAR are promising complementary strategies for prevention of metabolic syndrome-induced HCC.
Highlights.
-
►
Chronic circadian disruption induces NAFLD and spontaneous hepatocarcinogenesis
-
►
Circadian dysfunction promotes global gene deregulation and metabolic disruption
-
►
The nuclear receptor CAR drives NAFLD to NASH, fibrosis and HCC progression
-
►
Circadian disruption activates CAR via sympathetic dysfunction and Cholestasis
Acknowledgments
We thank D Bolinger, S Khan and P Patel for technical assistance, Drs. LD White for assisting microarray analysis and B Lowell for providing β-less mice. This work is funded by grants from NIH/NCI (R01 CA137019-01A) and USDA/ARS (6250-51000-055) to L.F., and CRPIT (MIRA RP150587) to D.D.M and L.F., and partially by NIH/NIDDK (R01 DK46546) to D.D.M., NIH/NIDDK (5T32 DK007696-20) to N.M.K., R01CA137019-05S to C.A.K., the NIH/NCI (P30CA125123) to BCM Genomic and RNA Profiling Core, the Public Health Service Grant DK56338 to TMC Digestive Diseases Center, the CPRIT Core Facility Support Award (RP120092), the NCI Shared Resources funds (2P30CA125123-09) and funds from the BCM Dan L. Duncan Cancer Center to the BCM Metabolomics Core, NSF DMS-1161759 and funds from Alkek Center for Molecular Discovery and the BCM Agilent Technologies Center of Excellence to A.S. and N.P.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
ACCESSION NUMBERS
The Gene Expression Omnibus (GEO) accession number for microarray analysis in this study is GSE75475.
Author Contributions
Conceptualization, L.F. and D.D.M.; Methodology, L.F., D.D.M., N.M.K. M.J.F., A.S. and N.P; Investigation, N.M.K., L.F., N.P. and C.A.K.; Statistics, H.V., C.C., N.M.K. and L.F.; Pathology, M.J.F.; Writing-Original Draft, L.F, D.D.M. N.M.K., H.V. and N.P.; Writing–Review & Editing, L.F., D.D.M. and N.M.K.; Resources, C.L. and A.S.; Supervision, L.F.; Funding Acquisition, L.F. and D.D.M.
The authors have no conflicts of interest to declare.
References
- Anand P, Kunnumakkara AB, Sundaram C, Harikumar KB, Tharakan ST, Lai OS, Sung B, Aggarwal BB. Cancer is a preventable disease that requires major lifestyle changes. Pharmaceutical research. 2008;25:2097–2116. doi: 10.1007/s11095-008-9661-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audard V, Grimber G, Elie C, Radenen B, Audebourg A, Letourneur F, Soubrane O, Vacher-Lavenu MC, Perret C, Cavard C, Terris B. Cholestasis is a marker for hepatocellular carcinomas displaying beta-catenin mutations. The Journal of pathology. 2007;212:345–352. doi: 10.1002/path.2169. [DOI] [PubMed] [Google Scholar]
- Bass J, Takahashi JS. Circadian integration of metabolism and energetics. Science. 2010;330:1349–1354. doi: 10.1126/science.1195027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diwan BA, Rice JM, Ward JM. Strain-dependent effects of phenobarbital on liver tumor promotion in inbred mice. Progress in clinical and biological research. 1990;331:69–83. [PubMed] [Google Scholar]
- Dong B, Lee JS, Park YY, Yang F, Xu G, Huang W, Finegold MJ, Moore DD. Activating CAR and beta-catenin induces uncontrolled liver growth and tumorigenesis. Nature communications. 2015;6:5944. doi: 10.1038/ncomms6944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drinkwater NR, Ginsler JJ. Genetic control of hepatocarcinogenesis in C57BL/6J and C3H/HeJ inbred mice. Carcinogenesis. 1986;7:1701–1707. doi: 10.1093/carcin/7.10.1701. [DOI] [PubMed] [Google Scholar]
- El-Serag HB. Hepatocellular carcinoma. The New England journal of medicine. 2011;365:1118–1127. doi: 10.1056/NEJMra1001683. [DOI] [PubMed] [Google Scholar]
- Fu L, Kettner NM. The circadian clock in cancer development and therapy. Progress in molecular biology and translational science. 2013;119:221–282. doi: 10.1016/B978-0-12-396971-2.00009-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L, Pelicano H, Liu J, Huang P, Lee C. The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell. 2002;111:41–50. doi: 10.1016/s0092-8674(02)00961-3. [DOI] [PubMed] [Google Scholar]
- Furness JB. The organisation of the autonomic nervous system: peripheral connections. Auton Neurosci. 2006;130:1–5. doi: 10.1016/j.autneu.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Gachon F, Olela FF, Schaad O, Descombes P, Schibler U. The circadian PAR-domain basic leucine zipper transcription factors DBP, TEF, and HLF modulate basal and inducible xenobiotic detoxification. Cell metabolism. 2006;4:25–36. doi: 10.1016/j.cmet.2006.04.015. [DOI] [PubMed] [Google Scholar]
- Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, Galardi C, Wilson JG, Lewis MC, Roth ME, et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Molecular cell. 2000;6:517–526. doi: 10.1016/s1097-2765(00)00051-4. [DOI] [PubMed] [Google Scholar]
- Guo GL, Lambert G, Negishi M, Ward JM, Brewer HB, Jr, Kliewer SA, Gonzalez FJ, Sinal CJ. Complementary roles of farnesoid X receptor, pregnane X receptor, and constitutive androstane receptor in protection against bile acid toxicity. The Journal of biological chemistry. 2003;278:45062–45071. doi: 10.1074/jbc.M307145200. [DOI] [PubMed] [Google Scholar]
- Guzman G, Brunt EM, Petrovic LM, Chejfec G, Layden TJ, Cotler SJ. Does nonalcoholic fatty liver disease predispose patients to hepatocellular carcinoma in the absence of cirrhosis? Archives of pathology & laboratory medicine. 2008;132:1761–1766. doi: 10.5858/132.11.1761. [DOI] [PubMed] [Google Scholar]
- Hashimoto E, Yatsuji S, Tobari M, Taniai M, Torii N, Tokushige K, Shiratori K. Hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Journal of gastroenterology. 2009;44(Suppl 19):89–95. doi: 10.1007/s00535-008-2262-x. [DOI] [PubMed] [Google Scholar]
- Hirschfield GM, Mason A, Luketic V, Lindor K, Gordon SC, Mayo M, Kowdley KV, Vincent C, Bodhenheimer HC, Jr, Pares A, et al. Efficacy of obeticholic acid in patients with primary biliary cirrhosis and inadequate response to ursodeoxycholic acid. Gastroenterology. 2015;148:751–761. e758. doi: 10.1053/j.gastro.2014.12.005. [DOI] [PubMed] [Google Scholar]
- Hoshida Y, Villanueva A, Kobayashi M, Peix J, Chiang DY, Camargo A, Gupta S, Moore J, Wrobel MJ, Lerner J, et al. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. The New England journal of medicine. 2008;359:1995–2004. doi: 10.1056/NEJMoa0804525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosono S, Chou MJ, Lee CS, Shih C. Infrequent mutation of p53 gene in hepatitis B virus positive primary hepatocellular carcinomas. Oncogene. 1993;8:491–496. [PubMed] [Google Scholar]
- Hu LY, Chen PM, Hu YW, Shen CC, Perng CL, Su TP, Yen SH, Tzeng CH, Chiou TJ, Yeh CM, et al. The risk of cancer among patients with sleep disturbance: a nationwide retrospective study in Taiwan. Annals of epidemiology. 2013;23:757–761. doi: 10.1016/j.annepidem.2013.09.002. [DOI] [PubMed] [Google Scholar]
- Huang Q, Lin B, Liu H, Ma X, Mo F, Yu W, Li L, Li H, Tian T, Wu D, et al. RNA-Seq analyses generate comprehensive transcriptomic landscape and reveal complex transcript patterns in hepatocellular carcinoma. PloS one. 2011;6:e26168. doi: 10.1371/journal.pone.0026168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Zhang J, Washington M, Liu J, Parant JM, Lozano G, Moore DD. Xenobiotic stress induces hepatomegaly and liver tumors via the nuclear receptor constitutive androstane receptor. Molecular endocrinology. 2005;19:1646–1653. doi: 10.1210/me.2004-0520. [DOI] [PubMed] [Google Scholar]
- Huang W, Zhang J, Wei P, Schrader WT, Moore DD. Meclizine is an agonist ligand for mouse constitutive androstane receptor (CAR) and an inverse agonist for human CAR. Molecular endocrinology. 2004;18:2402–2408. doi: 10.1210/me.2004-0046. [DOI] [PubMed] [Google Scholar]
- Huang Y, Chen HC, Chiang CW, Yeh CT, Chen SJ, Chou CK. Identification of a two-layer regulatory network of proliferation-related microRNAs in hepatoma cells. Nucleic acids research. 2012;40:10478–10493. doi: 10.1093/nar/gks789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao CH, Sun LM, Liang JA, Chang SN, Sung FC, Muo CH. Relationship of zolpidem and cancer risk: a Taiwanese population-based cohort study. Mayo Clinic proceedings. 2012;87:430–436. doi: 10.1016/j.mayocp.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettner NM, Katchy CA, Fu L. Circadian gene variants in cancer. Annals of medicine. 2014;46:208–220. doi: 10.3109/07853890.2014.914808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettner NM, Mayo SA, Hua J, Lee C, Moore DD, Fu L. Circadian Dysfunction Induces Leptin Resistance in Mice. Cell metabolism. 2015;22:448–459. doi: 10.1016/j.cmet.2015.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettner NM, V H, Coarfa C, Sreekumar A, Putluri N, Finegold MJ, Katchy CA, Lee C, Moore DD, Fu L. Circadian Homeostasis of Liver Metabolism Suppresses Hepatocarcinogenesis. Cancer cell. 2016 doi: 10.1016/j.ccell.2016.10.007. Accepted for Publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CW, Yun KE, Jung HS, Chang Y, Choi ES, Kwon MJ, Lee EH, Woo EJ, Kim NH, Shin H, Ryu S. Sleep duration and quality in relation to non-alcoholic fatty liver disease in middle-aged workers and their spouses. Journal of hepatology. 2013;59:351–357. doi: 10.1016/j.jhep.2013.03.035. [DOI] [PubMed] [Google Scholar]
- Kim I, Morimura K, Shah Y, Yang Q, Ward JM, Gonzalez FJ. Spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice. Carcinogenesis. 2007;28:940–946. doi: 10.1093/carcin/bgl249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike N, Yoo SH, Huang HC, Kumar V, Lee C, Kim TK, Takahashi JS. Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science. 2012;338:349–354. doi: 10.1126/science.1226339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunstyr I, Leuenberger HG. Gerontological data of C57BL/6J mice. I. Sex differences in survival curves. Journal of gerontology. 1975;30:157–162. doi: 10.1093/geronj/30.2.157. [DOI] [PubMed] [Google Scholar]
- Lamia KA, Storch KF, Weitz CJ. Physiological significance of a peripheral tissue circadian clock. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:15172–15177. doi: 10.1073/pnas.0806717105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Heo J, Libbrecht L, Chu IS, Kaposi-Novak P, Calvisi DF, Mikaelyan A, Roberts LR, Demetris AJ, Sun Z, et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nature medicine. 2006;12:410–416. doi: 10.1038/nm1377. [DOI] [PubMed] [Google Scholar]
- Lee S, Donehower LA, Herron AJ, Moore DD, Fu L. Disrupting circadian homeostasis of sympathetic signaling promotes tumor development in mice. PloS one. 2010;5:e10995. doi: 10.1371/journal.pone.0010995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeppen S, Schneider D, Gaunitz F, Gebhardt R, Kurek R, Buchmann A, Schwarz M. Overexpression of glutamine synthetase is associated with beta-catenin-mutations in mouse liver tumors during promotion of hepatocarcinogenesis by phenobarbital. Cancer research. 2002;62:5685–5688. [PubMed] [Google Scholar]
- Lopez-Velazquez JA, Carrillo-Cordova LD, Chavez-Tapia NC, Uribe M, Mendez-Sanchez N. Nuclear receptors in nonalcoholic Fatty liver disease. Journal of lipids. 2012;2012:139875. doi: 10.1155/2012/139875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu TT, Makishima M, Repa JJ, Schoonjans K, Kerr TA, Auwerx J, Mangelsdorf DJ. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Molecular cell. 2000;6:507–515. doi: 10.1016/s1097-2765(00)00050-2. [DOI] [PubMed] [Google Scholar]
- Ma K, Xiao R, Tseng HT, Shan L, Fu L, Moore DD. Circadian dysregulation disrupts bile acid homeostasis. PloS one. 2009;4:e6843. doi: 10.1371/journal.pone.0006843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnon C, Hall SJ, Lin J, Xue X, Gerber L, Freedland SJ, Frenette PS. Autonomic nerve development contributes to prostate cancer progression. Science. 2013;341:1236361. doi: 10.1126/science.1236361. [DOI] [PubMed] [Google Scholar]
- Michelotti GA, Machado MV, Diehl AM. NAFLD, NASH and liver cancer. Nature reviews Gastroenterology & hepatology. 2013;10:656–665. doi: 10.1038/nrgastro.2013.183. [DOI] [PubMed] [Google Scholar]
- Mohawk JA, Green CB, Takahashi JS. Central and peripheral circadian clocks in mammals. Annu Rev Neurosci. 2012;35:445–462. doi: 10.1146/annurev-neuro-060909-153128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nose H, Imazeki F, Ohto M, Omata M. p53 gene mutations and 17p allelic deletions in hepatocellular carcinoma from Japan. Cancer. 1993;72:355–360. doi: 10.1002/1097-0142(19930715)72:2<355::aid-cncr2820720208>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Perez MJ, Briz O. Bile-acid-induced cell injury and protection. World journal of gastroenterology. 2009;15:1677–1689. doi: 10.3748/wjg.15.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Review T, LaBrecque DR, Abbas Z, Anania F, Ferenci P, Khan AG, Goh KL, Hamid SS, Isakov V, Lizarzabal M, et al. World Gastroenterology Organisation global guidelines: Nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Journal of clinical gastroenterology. 2014;48:467–473. doi: 10.1097/MCG.0000000000000116. [DOI] [PubMed] [Google Scholar]
- Ripperger JA, Schibler U. Rhythmic CLOCK-BMAL1 binding to multiple E-box motifs drives circadian Dbp transcription and chromatin transitions. Nature genetics. 2006;38:369–374. doi: 10.1038/ng1738. [DOI] [PubMed] [Google Scholar]
- Roenneberg T. Chronobiology: the human sleep project. Nature. 2013;498:427–428. doi: 10.1038/498427a. [DOI] [PubMed] [Google Scholar]
- Roenneberg T, Allebrandt KV, Merrow M, Vetter C. Social jetlag and obesity. Current biology : CB. 2012;22:939–943. doi: 10.1016/j.cub.2012.03.038. [DOI] [PubMed] [Google Scholar]
- Siegel AB, Zhu AX. Metabolic syndrome and hepatocellular carcinoma: two growing epidemics with a potential link. Cancer. 2009;115:5651–5661. doi: 10.1002/cncr.24687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tentolouris N, Liatis S, Katsilambros N. Sympathetic system activity in obesity and metabolic syndrome. Annals of the New York Academy of Sciences. 2006;1083:129–152. doi: 10.1196/annals.1367.010. [DOI] [PubMed] [Google Scholar]
- Tokushige K, Hashimoto E, Horie Y, Taniai M, Higuchi S. Hepatocellular carcinoma in Japanese patients with nonalcoholic fatty liver disease, alcoholic liver disease, and chronic liver disease of unknown etiology: report of the nationwide survey. Journal of gastroenterology. 2011;46:1230–1237. doi: 10.1007/s00535-011-0431-9. [DOI] [PubMed] [Google Scholar]
- Villanueva A, Hernandez-Gea V, Llovet JM. Medical therapies for hepatocellular carcinoma: a critical view of the evidence. Nature reviews Gastroenterology & hepatology. 2013;10:34–42. doi: 10.1038/nrgastro.2012.199. [DOI] [PubMed] [Google Scholar]
- Voigt RM, Forsyth CB, Green SJ, Mutlu E, Engen P, Vitaterna MH, Turek FW, Keshavarzian A. Circadian disorganization alters intestinal microbiota. PloS one. 2014;9:e97500. doi: 10.1371/journal.pone.0097500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SN, Lee KT, Ker CG. Leptin in hepatocellular carcinoma. World journal of gastroenterology : WJG. 2010;16:5801–5809. doi: 10.3748/wjg.v16.i46.5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YD, Chen WD, Wang M, Yu D, Forman BM, Huang W. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology. 2008;48:1632–1643. doi: 10.1002/hep.22519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA, Moore DD, Auwerx J. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. The Journal of clinical investigation. 2004;113:1408–1418. doi: 10.1172/JCI21025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo HG, Park ES, Cheon JH, Kim JH, Lee JS, Park BJ, Kim W, Park SC, Chung YJ, Kim BG, et al. Gene expression-based recurrence prediction of hepatitis B virus-related human hepatocellular carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008;14:2056–2064. doi: 10.1158/1078-0432.CCR-07-1473. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Moore R, Goldsworthy TL, Negishi M, Maronpot RR. The orphan nuclear receptor constitutive active/androstane receptor is essential for liver tumor promotion by phenobarbital in mice. Cancer research. 2004;64:7197–7200. doi: 10.1158/0008-5472.CAN-04-1459. [DOI] [PubMed] [Google Scholar]
- Yang F, Huang X, Yi T, Yen Y, Moore DD, Huang W. Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer research. 2007;67:863–867. doi: 10.1158/0008-5472.CAN-06-1078. [DOI] [PubMed] [Google Scholar]
- Yasui K, Hashimoto E, Komorizono Y, Koike K, Arii S, Imai Y, Shima T, Kanbara Y, Saibara T, Mori T, et al. Characteristics of patients with nonalcoholic steatohepatitis who develop hepatocellular carcinoma. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2011;9:428–433. doi: 10.1016/j.cgh.2011.01.023. quiz e450. [DOI] [PubMed] [Google Scholar]
- Ye QH, Qin LX, Forgues M, He P, Kim JW, Peng AC, Simon R, Li Y, Robles AI, Chen Y, et al. Predicting hepatitis B virus-positive metastatic hepatocellular carcinomas using gene expression profiling and supervised machine learning. Nature medicine. 2003;9:416–423. doi: 10.1038/nm843. [DOI] [PubMed] [Google Scholar]
- Zhang J, Huang W, Qatanani M, Evans RM, Moore DD. The constitutive androstane receptor and pregnane X receptor function coordinately to prevent bile acid-induced hepatotoxicity. The Journal of biological chemistry. 2004;279:49517–49522. doi: 10.1074/jbc.M409041200. [DOI] [PubMed] [Google Scholar]
- Zimberg IZ, Fernandes Junior SA, Crispim CA, Tufik S, de Mello MT. Metabolic impact of shift work. Work. 2012;41(Suppl 1):4376–4383. doi: 10.3233/WOR-2012-0733-4376. [DOI] [PubMed] [Google Scholar]
- Zucman-Rossi J, Villanueva A, Nault JC, Llovet JM. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology. 2015;149:1226–1239. e1224. doi: 10.1053/j.gastro.2015.05.061. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.