

Abstract
Hepatocellular carcinoma (HCC) is one of the most lethal cancers worldwide and therapeutic agents for this malignancy are lacking. MicroRNAs play critical roles in carcinogenesis and present tremendous therapeutic potential. Here we report that microRNA-206 is a robust tumor suppressor that plays important roles in the development of HCC by regulating cell cycle progression and cMet signaling pathway. MicroRNA-206 was under-expressed in livers of two HCC mouse models, human individuals bearing HCC, and human HCC cell lines. Combining bioinformatic prediction and molecular and cellular approaches, we identified cMET (Met proto-oncogene), CCND1, and CDK6 as functional targets of microRNA-206. By inhibiting expression of cMET, CCND1 and CDK6, microRNA-206 delayed cell cycle progression, induced apoptosis and impaired proliferation of three distinct human HCC cell lines. Systemic administration of microRNA-206 completely prevented HCC development in both cMyc and AKT/Ras HCC mice, while 100% of control mice died from lethal tumor burdens. Conversely, re-introduction of cMet or Cdk6 into livers of cMyc and AKT/Ras HCC mice recovered growth of HCC inhibited by microRNA-206. These results strongly suggested that cMet and Cdk6 were two functional targets that mediated the inhibitory effect of microRNA-206 on the development of HCC. MicroRNA-206 overexpression demonstrated a profound therapeutic effect on HCC in xenograft and cMyc HCC mice. In summary, this study defines a potentially critical role of microRNA-206 in preventing the growth of HCC, and suggests its use as a potential therapeutic strategy for this malignancy.
Keywords: microRNA, hepatocellular carcinoma, therapeutic agent
HCC is a very common malignant disease with more than 700,000 new patients diagnosed per year.1 The incidence of HCC worldwide nearly matched its mortality, demonstrating the aggressive nature of this malignancy and limited therapeutic options.1, 2 Although hepatitis B (HBV) and C (HCV) infection are the major risk factors of HCC, liver damage due to NAFLD (non-alcoholic fatty liver disease) is associated with the majority of HCC patients in the Western world.3, 4 During the past 30 years, HCC patients have tripled due to the prevalence of obesity and its related morbidities, including NAFLD.1 Given the limited success with chemotherapy and the insensitivity of HCC to radiotherapy, tumor extirpation represents the only choice for long-term cure.1, 2 Unfortunately, even with successful surgical removal, the presence of NAFLD is associated with an increased recurrence of tumor.3, 4 Continuous efforts are needed to identify new targets and molecular pathways for drug development in the treatment of HCC.
MicroRNAs (miRNAs) are naturally-occurring small non-coding RNAs that function primarily by binding to the 3′-untranslated regions (3′-UTR) of specific mRNAs, which leads to either mRNA or translational pausing.5 Accumulating evidence has shown that these small molecules play important roles in carcinogenesis, lipid metabolism and development by repressing expression of their targets.6–8 MiRNA profiling of HCC tumors and its adjacent benign tissues identified a number of dysregulated miRNAs in HCC.9–11 Given their critical role in carcinogenesis,12 these miRNAs now represent novel therapeutic agents for human cancers. However, the detailed mechanism(s) by which miRNAs modulate hepatocarcinogenesis is largely unknown and miRNA therapeutic drugs for this cancer are unavailable.
Amplification and overexpression of the cMYC oncogene is a frequent event in human HCC.13, 14 In mice, overexpression of cMyc via hydrodynamic transfection leads to rapid liver tumor formation.15 In addition, coordinated activation of AKT/mTOR and RAS/MAPK cascades occurs in over 50% of all human HCCs, and is associated with biological aggressiveness and poor prognosis.16 This phenotype can be recapitulated in vivo by hydrodynamically transfecting activated forms of AKT (myr-AKT) and NRas (NRas-V12) oncogenes (AKT/Ras) into the mouse liver.16 In our previous study, we showed that all mice transfected with AKT/Ras or cMyc died by 6 to 8 weeks post-injection due to tumor burden.15 In addition, cMyc and AKT/Ras-induced HCC in rodents recapitulate, in a highly reliable way, the phases of tumor initiation and progression that occur in humans. In this study, we used cMyc and AKT/Ras mice, xenograft tumor mice and human HCC cell lines to investigate the underlying mechanisms by which miR-206 inhibits HCC, in addition to assessing its therapeutic potential for liver cancer.
Materials and Methods
Construction of Expression Vectors for miR-206, AKT, Ras, cMyc, cMet, Cdk6 and Ccnd1
A 400 bp fragment containing the miR-206 precursor was amplified from human genomic DNA, inserted into a pT3-EF1α vector and referred as to pT3-EF1α-miR-206. Construction of vectors pT3-EF1α-cMyc,15 pT3-EF1α-myr-AKT,15 nRasV12/pT2-CAGGS,15 pT3-EF1α-Ccnd1,17 pT3-EF1α-Cdk6,17 pT3-EF1α-cMet,17 and pCMV/Sleeping Beauty transposase (pCMV/SB)15 was carried out as previously described. All the plasmids used to induce HCC in wild-type FVB/N mice were purified using the Endotoxin free Maxi prep kit (Sigma, St. Louis, MO).
Construction of a Mini-circle Expression Vector for miR-206
We generated a versatile expression vector of miR-206 by cloning a human miR-206 precursor region into mini-circle vectors purchased from System Biosciences (Palo Alto, CA). A transthyretin gene (TTR) promoter was inserted upstream of the miR-206 precursor to ensure liver-specific expression of miR-206.18 This new construct was referred to as MC-TTR-miR-206. To rule out non-specific effects of the plasmid, we generated a miR-206 mis-matched-expression vector by mutating seed region of miR-206, termed MC-TTR-miR-206-MM. To prepare mini-circle vectors, parental MC-TTR-miR-206 vector was transformed into a special host E. coli bacterial strain ZYCY10P3S2T (System Biosciences, Palo Alto, CA). All mini-circles were made based on the manufacturer’s instruction.
Preparation of miR-206 Mimic
miR-206 mimic and scramble control were purchased from Creative Biogene (Shirley, NY). miR-206 mimic is chemically-modified double-strand miRNAs which can mimic mature endogenous miR-206 after transfection into cells. The antisense strand of miR-206 has 2 phosphorothioates at the 5′ end, 4 phosphorothioates, 4 cholesterol groups at the 3′ end, and full length nucleotide 2′-methoxy modification. Both miR-206 mimic and scramble control were formulated in 0.9% NaCl to a final concentration of 10 mg/ml.
Establishment of AKT/Ras and cMyc HCC Mice
Eight week old FVB/N mice (wild-type) were obtained from Charles River (Wilmington, MA). Standard hydrodynamic injections were performed as previously described.15 To determine the inhibitory effect of miR-206 on cMyc-induced HCC, mice (n=10) were hydrodynamically injected with 5 μg pT3-EF1α-cMyc, and 10 μg pT3-EF1α-miR-206 together with 0.6 μg pCMV/SB. Control mice (n=10) were injected with 4 μg pT3-EF1α-cMyc and 10 μg pT3-EF1α together with 0.6 μg pCMV/SB. To evaluate the effect of miR-206 on AKT/Ras-induced HCC, mice (n=10) were hydrodynamically injected with 4 μg pT3EF1α-myr-AKT, 4 μg NRasV12/pT2-CAGGS, and10 μg pT3-EF1α-miR-206 together with 0.72 μg pCMV/SB. Control mice (n=10) received 4 μg pT3-EF1α-myr-AKT, 4 μg NRasV12/pT2-CAGGS, and10 μg pT3-EF1α together with 0.72 μg pCMV/SB. To examine whether cMet, Cdk6 and Ccnd1 were the functional targets of miR-206, wild-type FVB/N mice were randomly separated into five groups: Group I mice (control group, n=10) were injected with a combination of 4 μg pT3-EF1α-myr-AKT, 4 μg NRasV12/pT2-CAGGS, 10 μg pT3-EF1α and 1.12 μg pCMV/SB; Group II (n=10) were injected with a combination of 4 μg pT3-EF1α-myr-AKT, 4 μg NRasV12/pT2-CAGGS, 10 μg pT3-EF1α-miR-206, and 1.12 μg pCMV/SB. This group was used to determine whether miR-206 was able to prevent HCC development; Group III-V (n=10) received 4 μg pT3-EF1α-myr-AKT, 4 μg NRasV12/pT2-CAGGS, 10 μg pT3-EF1α-miR-206 and 1.12 μg pCMV/SB together with 10 μg pT3-EF1α-cMet, pT3-EF1α-Cdk6 or pT3-EF1α-Ccnd1. This Group was used to determine whether additional treatment of Ccnd1, Cdk6 or cMet expression vector was able to recover the development of HCC inhibited by miR-206. The same strategy was used to determine whether cMet, Cdk6 and Ccnd1 mediated the inhibitory effect of miR-206 on HCC in cMyc mice. The plasmid mixtures were diluted in 2 ml saline (0.9% NaCl), filtered through 0.22 μm filter, and injected into the lateral tail vein of 6 to 8-week-old FVB/N mice in 5 to 7 seconds. Mice were housed, fed, and monitored in accordance with protocols approved by the committee for animal research at the University of California, San Francisco and the University of Minnesota.
Therapeutic Models of cMyc and AKT/Ras Mice
For MC-TTR-miR-206 experiments, FVB/N mice were injected with cMyc or AKT/Ras as described above. At 2 weeks post injection of AKT/Ras or cMyc, mice were treated with MC-TTR-miR-206 or MC-TTR-miR-206-MM at a dose of 1.5 mg/kg (i.v.) once a week for 4 weeks. For miR-206 mimic experiments, at 2 weeks post injection of AKT/Ras or cMyc, mice were treated with miR-206 mimic or control mimic at a dose of 10 mg/kg (i.v), twice a week for 4 weeks.
Global miRNA Expression Profiling of Livers of AKT/Ras and cMyc HCC Mice
A NanoString nCounter miRNA (NanoString Technologies, Seattle, WA) assay was used to perform miRNA profiling in livers of wild-type, cMyc and AKT/Ras mice. Total RNA was extracted from frozen liver tissues using miRNeasy Kit (Qiagen, Valencia, CA) according to the manufacturer’s protocol. Only RNA samples with good RNA quality as confirmed with the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA) were included for the NanoString analysis. All sample preparation and hybridization were performed according to the manufacturer’s recommendations. For platform validation using synthetic oligonucleotides, NanoString nCounter miRNA raw data were normalized for lane-to-lane variation with a dilution series of six spike-in positive controls. The sum of the six positive controls for a given lane was divided by the average sum across lanes to yield a normalization factor, which was then multiplied by the raw counts in each lane to give normalized values. Normalized miRNA array data was obtained and used for the analysis. Unsupervised clustering was performed using Genesis 1.7.6 with Euclidean distance. The comparison of miRNA profiles performed using unpaired student t-test. miRNAs with p-value < 0.001 and intensity fold change ≥ 2.0 (on either tumor vs. non-tumor or non-tumor vs. tumor) were included in the differentially-expressed miRNAs.
MiR-206 Expression Analysis in Human HCC Tumors and Normal Tissues
The dataset of GSE40744 which includes 19 normal livers and 26 HCC tumor samples was downloaded from Gene Expression Omnibus database (Pubmed). The signal intensity was normalized using robust multi-array average (RMA) analysis in R environment before conducting the expression analysis. Another 31 pairs of human HCC tumors with adjacent normal livers were analyzed for miR-206 expression using Taqman MicroRNA Assay (Invitrogen, Carlsbad, CA) (Supporting Table 1). The p values presented were calculated using student’s two sample t test.
Identification of miR-206 Targets
Identification of miR-206 target genes was conducted as previously described with minor revision.19 Breifly, we downloaded the target gene databases of miR-206 based on TargetScan,20 Pictar,21 and Starbase.22 Only hits from TargetScan or PicTar algorithm that were confirmed by Ago HITS-CLIP (high-throughput sequencing of RNAs isolated by crosslinking immunoprecipitation (HITS-CLIP) from Argonaute protein complex) were selected. These three databases were compared using Microsoft Access 2000. We then carried out Gene Ontology analysis using PathwayStudio software (Elsevier, Amsterdam Netherlands) and compared with established and potential therapeutic targets for HCC,23–25 yielding 6 potential targets of miR-206 (Supporting Table 2).
Xenograft Tumor Assay
The xenografts were established in BALB/C nude mice (Charlies River). Huh7, SNU449 or MHCC97-H was stably transfected Plenti-CMV-puromycin-miR-206 or empty vector (control). The Huh7, SNU449 and MHCC97-H cells were placed in a 6-well plate 24 hours prior to transfection. 24 hours after transfection, 5×105cells in 0.1 ml PBS were injected subcutaneously into the right flank of athymic nude mice (n=12) to establish a model of tumor-bearing mice. Tumor growth was observed every 3 days by measuring its diameter with Vernier calipers. Tumor weight was calculated by gram. Tumor volume (cm3) = d2 × D/2, where d is the shortest and D is the longest diameter, respectively. Mice were sacrificed when the tumor size reached 1.5 cm in diameter. All procedures involving mice were approved by the Institutional Animal Care Committee at the University of California San Francisco and the University of Minnesota.
Stable Cell Lines and Colony Formation Assay
Huh7, MHCC97-H, and SNU449 cells were stored in our laboratories. Plenti-CMV-puromycin-miR-206 or control plasmids together with packaging plasmids were co-transfected into human embryonic kidney 293T (HEK-293T) cells. 48h after transfection, the supernatants were harvested and filtered through a 0.45 μm filter. Huh7, MHCC97-H, and SNU449 cells were maintained as monolayer culture in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Two days later, the cells were transfected with each lentiviral stock and were selected with puromycin for 3 days. After antibiotic selection, the cells were harvested for subsequent studies. Stably transfected Huh7, MHCC97-H, and SNU449 cells were seeded in a 6-well plate at appropriate density per well in triplicate. When visible cell colonies appeared, the colonies were fixed and stained with 0.1% crystal violet dissolved in methanol for 15 min for visualization and counting.
To determine whether CCND1, CDK6 or cMET mediates the inhibitory effect of miR-206 on colony formation, miR-206-stably transfected MHCC97-H, Huh7 and SNU-449 cells (0.5 × 106 cells in 35-mm plastic dishes) were transfected with Target Protector (Exiqon, Woburn, MA) of CDK6, CCND1 or cMET or scramble control (20 nM). MHCC97-H, Huh7 and SNU449 cells that were stably transfected with empty vector served as the control. Two days after transfection, the transfected cells were suspended with 8 ml of 0.4% top agar (Sigma-Aldrich) and 2×DMEM supplemented with 20% FBS before being poured onto 6-cm tissue culture dishes coated with 3.5 ml of 0.7% bottom agar. Fourteen days later, three areas per plate were chosen randomly, and the number of visible colonies was counted.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism Software®. Data derived from cell-line experiments were presented as mean ± SEM and assessed by a two-tailed Student T-test. Statistical difference for cell cycle progression analysis was evaluated using Chi-squared test. Mann-Whitney test was used to evaluate the statistical significance for mouse experiments. All the experiments were repeated at least three times. P < 0.05 was considered to be statistically significant.
Results
MiR-206 is Under-expressed in Livers of Mouse and Human HCC tumors
To identify novel miRNAs that are critical regulators of HCC development, we carried out miRNA profiling of livers from AKT/Ras23 and cMyc24 HCC mice. We identified 12 miRNAs that were present in livers of wild-type mice but undetectable in HCC tumors of both AKT/Ras and cMyc mice (Supporting Table 3). To correlate these findings with human HCC, we measured expression of these 12 miRNAs in normal human hepatocytes and four HCC cell lines, and observed that miR-206 was the only undetectable miRNA in HCC tumors of both AKT/Ras and cMyc mice and was also reduced in human HCC cell lines compared to normal human hepatocytes (Fig. 1A–B). Database mining of miRNA profiles revealed that miR-206 was significantly reduced in human HCC tumors compared to normal liver tissues (Fig. 1C). qRT-PCR further confirmed that miR-206 was robustly reduced in another 31 human HCC tumors compared to their adjacent normal tissues (Supporting Table 1). Taken together, hepatic expression of miR-206 is decreased in both mouse and human HCC tumors, prompting us to select miR-206 for further characterization of its role in the pathogenesis of HCC.
Fig. 1.
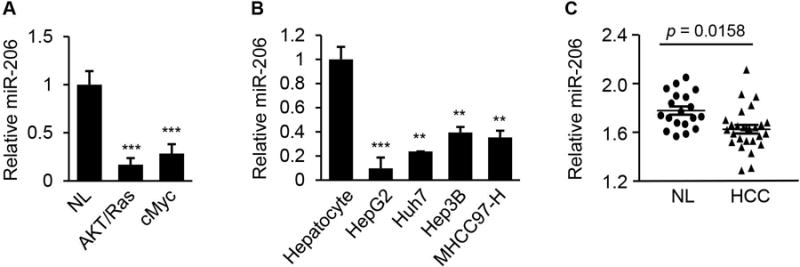
miR-206 expression was reduced in HCC tumors of human and mouse. (A) Reduced expression of miR-206 in livers of AKT/Ras (n=6) and cMyc (n=6) HCC mouse models compared to normal mice (n=6). qRT-PCR was used to determine expression of miR-206. NL: normal liver. (B) Levels of miR-206 in human HCC cell lines with divergent backgrounds compared with normal human hepatocytes as revealed by qRT-PCR. (C) Decreased expression of miR-206 in human HCC tumors (n=26) versus normal liver tissues (n=19). Mann-Whitney test was used to evaluate the statistical significance. Data represent mean ± SEM. **p < 0.01 and ***p < 0.001.
CCND1, cMET, and CDK6 are Direct Targets of miR-206
To gain insight into the function of dysregulated miR-206 in HCC, we aimed to identify those mRNAs that were targeted by miR-206 and serve as its effectors in the pathogenesis of HCC. We identified putative binding sites for miR-206 within 3′UTRs of six genes that were involved in carcinogenesis and lipid metabolism (Supporting Table 2). Among these potential targets of miR-206, cMET has been linked to tumor growth in humans and its inhibitor as a therapeutic agent for HCC has entered into clinical trials.26 CCND1 is a major cyclin controlling cell cycle progression from G1 to S phase by interacting with CDK4/CDK6.27 Our prediction results, combined with these established findings, led us to speculate that CCND1, cMET, and CDK6 were the functional targets of miR-206 and that miR-206 was able to prevent the pathogenesis of HCC by modulating the cMET pathway and cell cycle progression. MiR-206 binding sites within the 3′UTRs of these 3 genes are conserved between human and mouse (Fig. 2A). Overexpression of miR-206 in HepG2 cells robustly decreased both mRNA and protein levels of CCND1, cMET, and CDK6 (Fig. 2B), while antagonizing miR-206 led to adverse effects (Fig. 2C). To establish that miR-206 directly recognizes the predicted binding sites within the 3′ UTRs of these three genes, we cloned their 3′ UTRs into a luciferase reporter vector. As expected, inclusion of the 3′UTRs of CCND1, cMET, and CDK6 into the reporter construct reduced luciferase activity upon co-transfection with miR-206 into Hepa1,6 cells (Fig. 2D). In contrast, mutation of miR-206 binding sites within the 3′UTRs of CCND1, cMET, and CDK6 was necessary to completely offset the inhibitory effect of miR-206 on the luciferase activity (Fig. 2E). We further confirmed that miR-206 inhibited expression of mouse Ccnd1, cMet, and Cdk6 by directly interacting with their respective 3′UTRs (Supporting Fig. 1A–D). In vivo, delivery of miR-206 into livers also reduced both protein and mRNA levels of these three genes (Fig. 2F, Supporting Fig. 1E). Together, these experiments indicate that Ccnd1, cMet, and Cdk6 are direct targets of miR-206 both in vitro and in vivo.
Fig. 2.
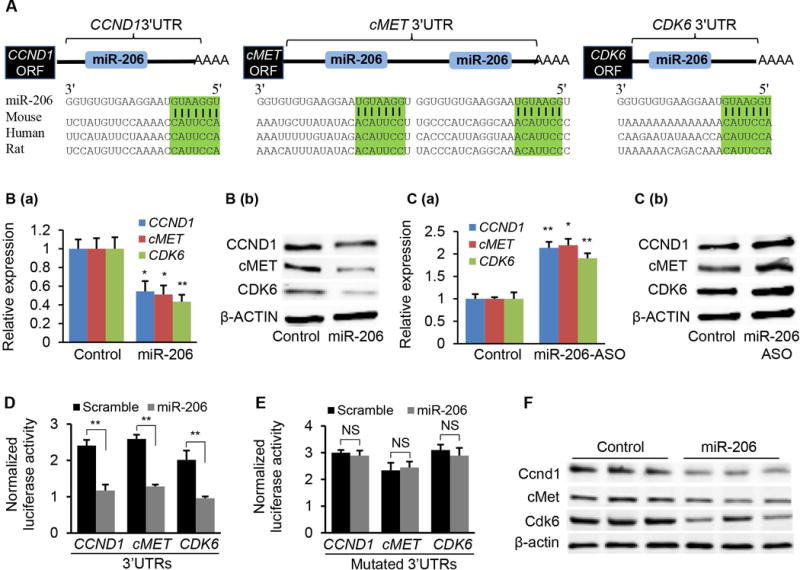
CCND1, CDK6, and cMET are direct targets of miR-206. (A) Graphic representation of the conserved miR-206 binding motifs within the 3′UTRs of CCND1, cMET and CDK6. Complementary sequences to the seed regions of miR-206 within the 3′UTRs of the three genes are conserved among 3 species (highlighted in green). (B) qRT-PCR and immunoblot analysis of CCND1, cMET and CDK6 after miR-206 mimic transfection into HepG2 cells. HepG2 cells treated with scramble served as the control. (C) qRT-PCR and Western blot analysis of CCND1, cMET and CDK6 after miR-206-ASO (Anti-sense oligonucleotide) transfection into HepG2 cells (20 nM). The control HepG2 cells received scramble (20 nM). (D–E) Luciferase activity of the luciferase reporter constructs containing either the wild-type or mutated 3′UTRs of human CCND1, cMET and CDK6 after miR-206 mimic treatment. Luciferase activity was normalized to the activity of β-galactosidase. Hepa1,6 cells transfected with scramble and the luciferase reporter constructs served as control. (F) Western blot revealing reduced protein levels of Ccnd1, cMet and Cdk6 after MC-TTR-miR-206 injection into mice (1.5 μg/g body weights). The control mice were treated with the same dose of MC-TTR-miR-206-MM. Data represent mean ± SEM. *p < 0.05; **p < 0.01; and ***p < 0.001.
MiR-206 Impairs Growth, Proliferation, Delays Cell Cycle Progression and Induces Apoptosis of Human HCC Cell Lines with Divergent Backgrounds
To determine whether miR-206 confers protection from growth of cancer cells and cell cycle progression, we increased miR-206 levels in three types of human cancer cells with divergent backgrounds including Huh7, SNU449 and MHCC97-H (Supporting Fig. 2A). Sustained expression of miR-206 robustly prevented colony formation of these three liver cancer cell lines (Fig. 3A), and significantly delayed G1/S progression of cell cycle (Fig. 3B–D). MTT assay further confirmed that miR-206 was a strong suppressor of proliferation (Fig. 3E). To determine whether CCND1, CDK6 and/or cMET mediate the inhibitory effect of miR-206 on growth and cell cycle transition of human liver cancer cells, we treated miR-206-stably transfected Huh7, SNU449 and MHCC97-H cells with TP morpholinos (Target Protector) of CCND1, CDK6 and cMET or scramble control (Supporting Fig. 2B–D). These morpholinos were complimentary to miR-206 binding sites within the 3′UTRs of CCND1, CDK6 and cMET mRNAs and prevented miR-206 from binding to the 3′UTRs.28 This design allowed us to determine whether CCND1, CDK6 and cMET mediate the effects of miR-206 on inhibiting proliferation. qRT-PCR confirmed that the TPs prevented miR-206 from binding to the 3′UTRs of CCND1, CDK6 and cMET, which subsequently impaired the ability of miR-206 to inhibit expression of CCND1, CDK6 and cMET (Supporting Fig. 2B–D). Phenotypically, treatment of cMET, CCND1 and CDK6 TPs impaired the ability of miR-206 to inhibit growth and proliferation of Huh7, SNU449 and MHCC97-H (Fig. 3F–G). In addition, miR-206 induced apoptosis of Huh7, SNU449 and MHCC97-H cells (Supporting Fig. 3A–C). The data support the notion that by interacting with CCND1, CDK6 and cMET, miR-206 delays cell cycle progression, impairs proliferation and induces apoptosis of human HCC cells lines with divergent backgrounds.
Fig. 3.
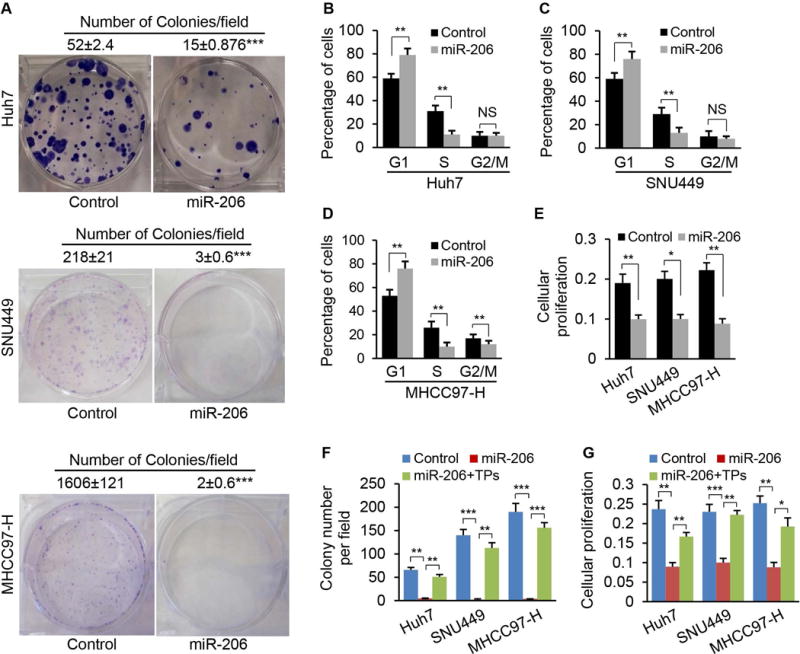
miR-206 prevented colony formation, proliferation and cell cycle progression of human HCC cell lines with divergent backgrounds. (A) Soft agar colony formation assay of Huh7, SNU449 and MHCC97-H cells stably transfected with Plenti-CMV-puromycin-miR-206 or empty plasmids (Control). The numbers represented the average colony numbers of three areas per plate. (B–D) Increased the number of cells in the G1 phase but decreased the number of cells in the S phase. Huh7, SNU449 and MHCC97-H cells were stably transfected with Plenti-CMV-puromycin-miR-206 or empty plasmids (Control). (E) Reduced proliferation of Huh7, SNU449 and MHCC97-H cells after stably transfected with Plenti-CMV-puromycin-miR-206, as revealed by MTT assay. (F–G) TPs of CCND1, cMET, and CDK6 treatment recovered colony formation and proliferation of Huh7, SNU449 and MHCC97-H cells. Huh7, SNU449 and MHCC97-H cells stably transfected with Plenti-CMV-puromycin-miR-206 were treated with TP of CCND1, cMET and CDK6 (20 nM). The Huh7, SNU449 and MHCC97-H cells treated with scramble served as the control (20 nM). NS: no significance. Data represent mean ± SEM. *p < 0.05; **p < 0.01; and ***p < 0.001.
MiR-206 Inhibits Growth of Xenograft Tumors from Different Human HCC Cell Lines
To determine whether miR-206 confers protection from human HCC tumor growth in vivo, Huh7, SNU449, and MHCC97-H cells stably transfected with miR-206 expression vector were allografted into immunocompetent male mice. Consistent with our colony formation experiments, miR-206 robustly attenuated growth of xenograft tumors from Huh7, SNU449, and MHCC97-H cells (Fig. 4A). We also determined whether CCND1, CDK6 and cMET mediated the inhibitory effect of miR-206 on growth of xenograft tumor. Specifically, we transfected TPs of CCND1, CDK6 and cMET into Huh7, SNU449, and MHCC97-H cells stably transfected with miR-206, and then performed subcutaneous xenograft of these cells into nude mice. Indeed, miR-206 robustly inhibited growth of xenograft tumor, and the TPs treatment recovered growth of the xenograft HCC tumors (Fig. 4B). Thus, the ability of miR-206 to inhibit growth of xenograft HCC tumor appears to occur by interacting with CCND1, cMET, and CDK6.
Fig. 4.
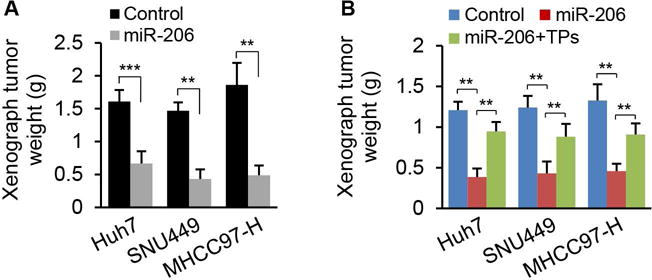
miR-206 prevented growth of xenograft tumors from three human HCC cell lines. (A) Weight of xenograft tumors from Huh7, SNU449 and MHCC97-H cells that were stably transfected with Plenti-CMV-puromycin-miR-206 (n=12) or empty vector (n=12). (B) Average weight of xenograft tumors from three groups of OD/SCID mice inoculated with (a) Huh7, SNU449 and MHCC97-H cells stably transfected with Plenti-CMV-puromycin empty plasmid (control), (b) Huh7, SNU449 and MHCC97-H cells stably transfected with Plenti-CMV-puromycin-miR-206 and scramble control (20 nM), and (c) Huh7, SNU449 and MHCC97-H cells stably transfected with Plenti-CMV-puromycin-miR-206 and TPs of CCND1, cMET and CDK6 (20 nM). Data represent mean ± SEM. **p < 0.01 and ***p < 0.001.
Delivery of miR-206 into Livers Completely Prevented HCC Development in AKT/Ras and cMyc Mice
We have previously reported that activation of AKT/Ras, AKT/cMET or cMyc triggers rapid HCC development in mice, and all mice developed lethal burden of liver tumor within 6 to 8 weeks.15, 29 Thus, we investigated the effect of manipulating miR-206 levels on HCC in livers of AKT/Ras and cMyc mice (Supporting Fig. 4A–B). Our results indicated that 100% of control mice died from lethal tumor burdens by 6 to 8 weeks post injection and all miR-206-treated AKT/Ras mice were healthy at this stage (Fig. 5A). Upon dissection, no tumor nodules were observed in livers of AKT/Ras/miR-206 mice (Fig. 5A). Mechanistically, miR-206 significantly reduced hepatic expression of Ccnd1, Cdk6 and cMet in livers of AKT/Ras HCC mice compared to control mice (Fig. 5B). The same results were obtained in the cMyc HCC mice, and 100% control mice died from lethal tumor burdens by 6 to 8 weeks post-injection; and no tumor was detected after delivery of miR-206 into livers of cMyc HCC mice (Fig. 5C). Delivery of miR-206 into cMyc HCC mice significantly reduced protein levels of Cdk6, Ccnd1 and cMet (Fig. 5D). Thus, miR-206 appears to be a robust suppressor of HCC initiation and progression via targeting Ccnd1, Cdk6 and cMet in AKR/Ras and cMyc mice.
Fig. 5.
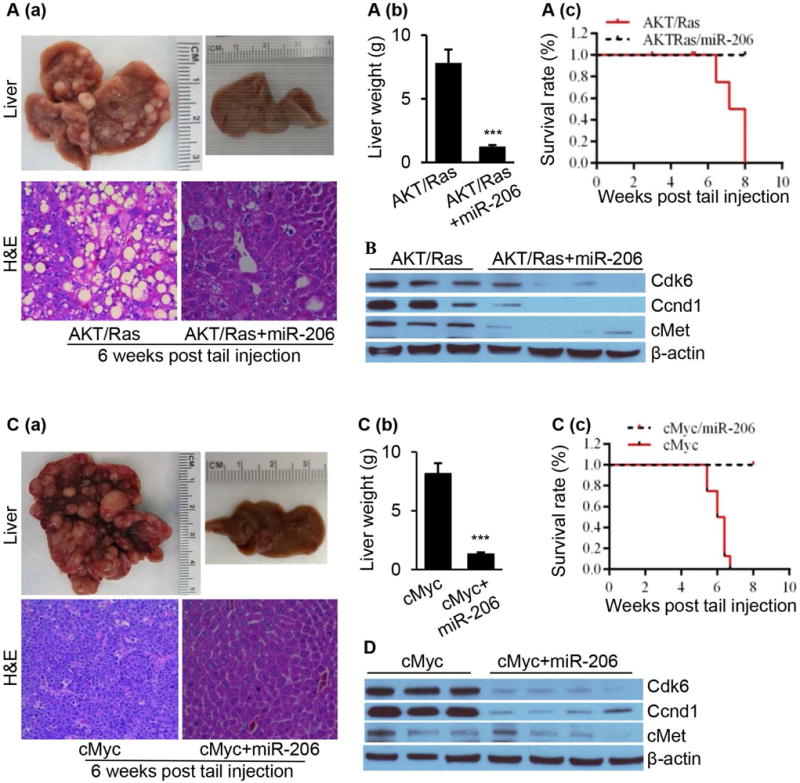
HCC was undetectable in the livers of both AKT/Ras and cMyc HCC mice after miR-206 overexpression. (A) (a) Macroscopic (upper panel) and microscopic (lower panel) appearance of livers from AKT/Ras/pT3 mice (control, n=10) and AKT/Ras/miR-206 mice (n=10) stained with H&E (100×); (b) Average liver weight of AKT/Ras/pT3 mice and AKT/Ras/miR-206 mice (n=10); and (c) Kaplan Meier survival curves of AKT/Ras/pT3 and AKT/Ras/miR-206 mouse cohort. (B) Western blot analysis of Cdk6, Ccnd1 and cMet in the livers of AKT/Ras and AKT/Ras/miR-206 mice. (C) (a) Macroscopic (upper panel) and microscopic (lower panel) appearance of livers from cMyc/pT3 mice (n=10) and cMyc/miR-206 mice (n=10) stained with H&E (100×); (b) Average liver weight of cMy/pT3 mice and cMyc/miR-206 mice; and (c) Kaplan Meier survival curve of cMyc/pT3 and cMyc/miR-206 mouse cohort. (D) Western blot analysis of Cdk6, Ccnd1 and cMet in the livers of cMyc and cMyc/miR-206 mice. Data represent mean ± SEM. ***p < 0.001.
Both Cdk6 and cMet Mediate the Inhibitory Effect of miR-206 on HCC in cMyc and AKT/Ras Mice
To determine whether cMet, Ccnd1 or Cdk6 mediates the inhibitory effect of miR-206 on HCC development, we restored hepatic expression of Ccnd1, Cdk6 or cMet in livers of AKT/Ras/miR-206 HCC mice (Supporting Fig. 5A–C). Increased expression of cMet or Cdk6 recovered growth of HCC that was completely prevented by miR-206 (Fig. 6A–C); in contrast, re-introduction of Ccnd1 into miR-206-overexpressed AKT/Ras mice failed to recover the growth of HCC tumor that was prevented by miR-206 (Fig. 6A–C). Using the same strategy, we further re-introduced Ccnd1, Cdk6 and cMet into cMyc HCC mice that were treated with miR-206 (Supporting Fig. 5D–F). As expected, all cMyc mice died eight weeks after injection and miR-206 administration completely prevented HCC development. Restoration of cMet or Cdk6 expression recovered growth of HCC, while additional treatment of Ccnd1 failed to do so (Fig. 6D–F). Thus, only cMet and Cdk6 and not Ccnd1 were able to restore growth of HCC inhibited by miR-206 in AKT/Ras and cMyc mice. Further studies are needed to investigate the contribution of the crosstalk of miR-206 with Ccnd1 in the pathogenesis of HCC.
Fig. 6.
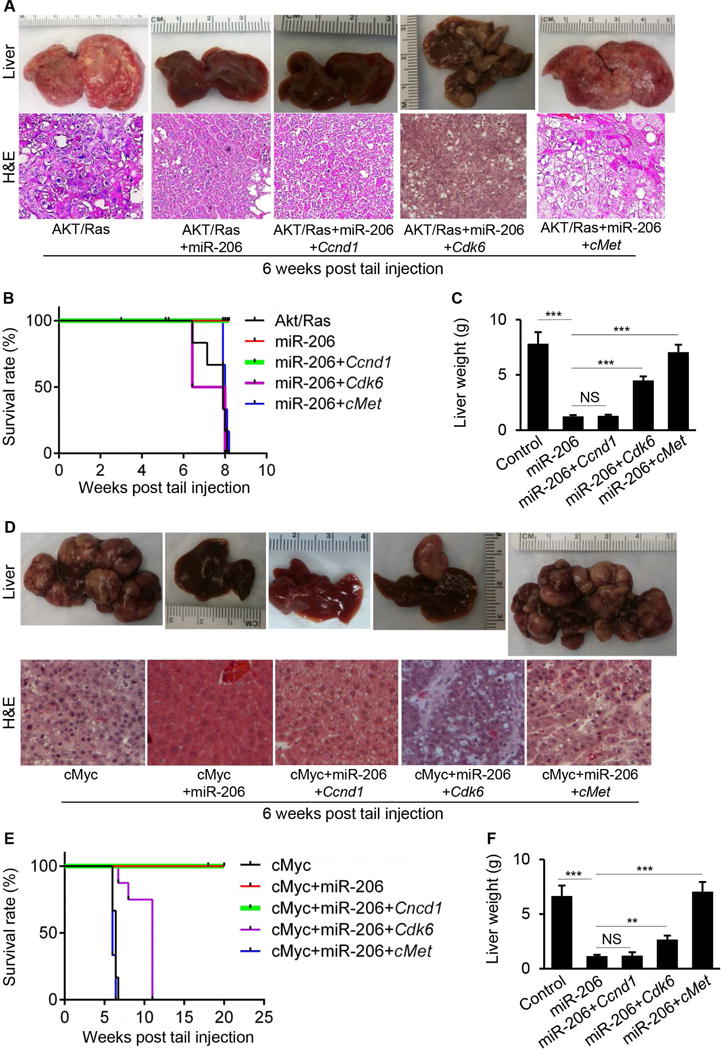
cMet and Cdk6 mediated the inhibitory effect of miR-206 on the development of HCC in both AKT/Ras and cMyc mice. (A) Macroscopic (upper panel) and microscopic (lower panel) appearance of livers from AKT/Ras (n=10), AKT/Ras/miR-206 (n=10), AKT/Ras/miR-206/Ccnd1 (n=10), AKT/Ras/miR-206/Cdk6 (n=10) and AKT/Ras/miR-206/cMet mice (n=10) stained with H&E (100×). (B) Kaplan Meier survival curves of AKT/Ras, AKT/Ras/miR-206, AKT/Ras/miR-206/Ccnd1, AKT/Ras/miR-206/Cdk6 and AKT/Ras/miR-206/cMet mice. (C) Average liver weight of AKT/Ras, AKT/Ras/miR-206, AKT/Ras/miR-206/Ccnd1, AKT/Ras/miR-206/Cdk6 and AKT/Ras/miR-206/cMet mice. (D) Macroscopic (upper panel) and microscopic (lower panel) appearance of livers from cMyc (n=10), cMyc/miR-206 (n=10), cMyc/miR-206/Ccnd1 (n=10), cMyc/miR-206/Cdk6 (n=10) and cMyc/miR-206/cMet mice (n=10) stained with H&E (100×). (E) Kaplan Meier survival curves of cMyc, cMyc/miR-206, cMyc/miR-206/Ccnd1, cMyc/miR-206/Cdk6 and cMyc/miR-206/cMet mice. (F) Average liver weight of cMyc, cMyc/miR-206, cMyc/miR-206/Ccnd1, cMyc/miR-206/Cdk6 and cMyc/miR-206/cMet mice. Data represent mean ± SEM. NS: no significance; **p < 0.01 and ***p < 0.001.
MiR-206 Displayed the Therapeutic Effect on HCC in cMyc Mice
We next assessed the therapeutic potential of miR-206 for the treatment of HCC in both cMyc and AKT/Ras HCC mice. For this purpose, we generated a miR-206 in vivo expression system using a mini-circle episomal DNA vector.30 Mini-circles are episomal DNA vectors that are produced as circular expression cassettes devoid of any bacterial plasmid DNA backbone.31 Their smaller molecular size enables more efficient delivery and offers sustained expression over a period of weeks as compared to standard plasmid vectors that only work for a few days after injection into mice. The TTR promoter was used to ensure hepatocyte-specific expression of miR-206,15, 32 and the construct was referred to as MC-TTR-miR-206. FVB/N mice, which had been injected with AKT/Ras or cMyc to induce HCC for two weeks, were treated with either MC-TTR-miR-206 or MC-TTR-miR-206-MM weekly for another four weeks (Supporting Fig. 6A–B). The results showed that four weeks of MC-TTR-miR-206 treatment was markedly effective against HCC in cMyc mice (Fig. 7A–B). Surprisingly, MC-TTR-miR-206 failed to repress growth of HCC in AKT/Ras mice (Supporting Fig. 7A). To explore this observation, we determined levels of miR-206 in livers of AKT/Ras mice treated with either MC-TTR-miR-206-MM or MC-TTR-miR-206. Interestingly, MC-TTR-miR-206 treatment led to increased miR-206 levels and reduced mRNA levels of its targets including Ccnd1, Cdk6 and cMet in HCC tumors of cMyc mice compared to the control mice (Fig. 7C). In contrast, levels of miR-206 and its targets Ccnd1, Cdk6 and cMet showed no significant change in HCC tumors of AKT/Ras mice treated with MC-TTR-miR-206 (Supporting Fig. 7C). The results suggested that poor delivery of the MC-TTR-miR-206 might be responsible for the negligible therapeutic effect on HCC in ATK/Ras mice, perhaps due to rapid growth of the HCC or other unknown mechanisms.
Fig. 7.
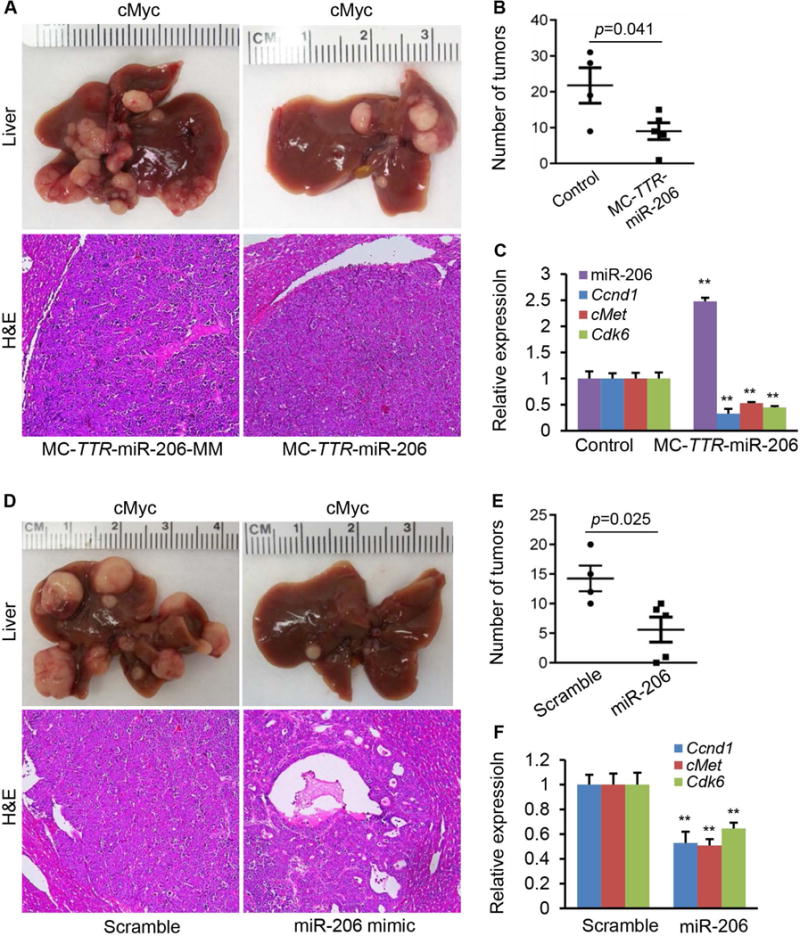
miR-206 displayed the strong therapeutic effect on HCC in cMyc mice. (A) Macroscopic (upper panel) and microscopic (lower panel) appearance of livers from cMyc mice treated with MC-TTR-miR-206 (n=6) or MC-TTR-miR-206-MM (control, n=6) stained with H&E (100×). (B) (B–C) Number of tumors and levels of miR-206, Ccnd1, Cdk6 and cMet in livers of mice treated with MC-TTR-miR-206 or MC-TTR-miR-206-MM. (D) Macroscopic (upper panel) and microscopic (lower panel) appearance of livers from cMyc mice treated with miR-206 mimic (n=6) or scramble (control, n=6) stained with H&E (100×). (E–F) Number of tumors and mRNA levels of Ccnd1, Cdk6 and cMet in livers of cMyc mice treated with miR-206 mimic or scramble. Data represent mean ± SEM. **p < 0.01.
To increase the delivery efficiency of miR-206 into HCC tumors, we generated a miR-206 double-stranded mimic whose size was much smaller than MC-TTR-miR-206. The miR-206 mimic was further modified at 2′O-methyl and labeled with four cholesterol groups to increase the stability and transfection efficiency of miR-206 mimic. The same cMyc and AKT/Ras HCC mouse models described above were used to evaluate the therapeutic effectiveness of miR-206 mimic for the treatment of HCC. Specifically, mice received a systemic treatment of miR-206 mimic twice for four weeks. When HCC tumors became invisible (about 2 weeks after AKT/Ras or cMyc injection), mice were randomized and systemically treated, via tail vein, with either miR-206 mimic or scramble control at the same dose of 10 mg/kg body weight. Following eight injections, a significant anti-tumor effect of miR-206 was observed in cMyc HCC mice (Figure 7D). Moreover, the tumor number was much smaller in cMyc mice treated with miR-206 compared to scramble treated mice (Figure 7E). We further evaluated the delivery efficiency of miR-206 by determining mRNA levels of its targets including Ccnd1, Cdk6 and cMet. As expected, we observed a significant reduction in mRNA levels of these three targets in tumors treated with miR-206 as compared to controls (Figure 7F). In AKT/Ras mice, miR-206 mimic treatment failed to stop the rapid growth of HCC tumors and did not display any therapeutic effect on HCC (Supporting Fig. 7D–E). In addition, mRNA levels of miR-206’s targets including Cncd1, Cdk6 and cMet had no change in livers of miR-206-treated mice compared to control mice (Supporting Fig. 7F).
Discussion
The incidence rate of HCC has been increasing in the United States for the past 35 years due, in large part, to the prevalence of obesity.33 Recent studies revealed that postoperative morbidity and 30-day mortality rates is significantly higher in the obesity-associated HCC patients than in those cases caused by HBV and HCV.3, 4 Although HCC has been widely investigated, the underlying mechanisms are still poorly understood due to complexity of the disease. As a result, only a single chemotherapy drug (sorafenib) is available for HCC. Unfortunately, sorafenib only improves life expectancy approximately by 3 months over placebo,34 underscoring the importance of identifying new targets and molecular pathways for the treatment of HCC. In this study, we identified a specific miRNA that can simultaneously inhibit expression of several therapeutic targets of HCC and delivery of miR-206 into livers showed the robust therapeutic effect on HCC. Our findings represent a unique basis for the role of miR-206 in the pathogenesis of HCC and its use as a potential therapeutic approach for this malignancy.
Identification of functional targets is the key step to study the function of miR-206 in the pathogenesis of HCC. With the development of deep sequencing and HITS-CLIP techniques, many targets of miR-206 have been identified.35–37 Therefore, it is important to elucidate which target mediates the function(s) of a specific miRNA. In this study, we used Target Protector Technique to prevent the interaction of miR-206 with its targets CCND1, CDK6 and cMET.38 Using this approach, we have validated CCND1, CDK6 and cMET as functional and potentially therapeutic targets of miR-206. Despite the accuracy of TP technique to prevent miRNAs from binding to their targets, the high cost of TP is a challenge for mouse experiments. CRISPR/Cas9 technique is another potential choice to study the crosstalk of miR-206 with CCND1, CDK6 and cMET.39 This approach can be used to delete or mutate the seed regions within the 3′UTRs of target genes, which will allow us to more accurately investigate whether the interaction between miR-206 and its targets CCND1, CDK6 and cMET modulates the growth of hepatic carcinoma.
In 2009, we firstly identified miR-206 as a potential tumor suppressor in liver cancer.6 During the following years, miR-206 has been identified as a under-expressed miRNA in human breast cancer,40 colorectal cancer,41 and lung squamous cell carcinoma,42 and the function of miR-206 was investigated in cancer cell lines. To date, there are no reported studies that have been performed to evaluate the therapeutic potential of miR-206 and the efficacy of miR-206 in preventing HCC in mice.43 In the present study, we validated that miR-206 was able to inhibit growth and cell cycle progression and induce apoptosis. Notably, our data provided evidence that miR-206 has the capacity to robustly prevent the development of HCC in two HCC mouse models. As described above, miR-206 is reduced and its targets are increased in other types of human cancers. Our findings, combined with those of others, suggest that miR-206 might be a broad spectrum anti-cancer agent can be applied for other types of human cancers.
To further evaluate the therapeutic potential of miR-206 on HCC, we overexpressed miR-206 in livers of cMyc and AKT/Ras mice bearing HCC tumors using the mini-circle expression vector of miR-206 or cholesterol and methyl-modified miR-206 mimic. Consistent with the strong prevention effect of miR-206 on HCC, both MC-TTR-miR-206 and miR-206 mimic treatment showed the obvious therapeutic effect on HCC in cMyc mice but not in AKT/Ras mice. We also observed that growth of HCC tumor was much faster in AKT/Ras mice than cMyc mice (Data not shown), suggesting that rapid growth of HCC might have impaired the delivery of MC-TTR-miR-206 and miR-206 mimic into the tumors of AKT/Ras mice. Indeed, qRT-PCR revealed that levels of miR-206 and its targets showed no change in livers of AKT/Ras mice treated with MC-TTR-miR-206 or miR-206 mimic compared to control mice, further indicating that the low delivery efficiency of miR-206 or other unknown mechanism(s) may contribute to negligible therapeutic effect on HCC in AKT/Ras mice. Further studies are needed to optimize the delivery efficiency of miR-206 and investigate whether miR-206 can lead to a regression of HCC tumor in AKT/Ras mice. In summary, both in vitro and in vivo data obtained in this study could contribute to the translation of miR-206 into a unique therapeutic agent for the treatment of HCC. In addition, the majority of HCC patients have elevated expression of CCND1, CDK6 and/or cMET,34, 44, 45 and some inhibitors of cMET and CCND1/CDK6 are being tested in clinical trials as therapeutic agents for HCC,26, 46 further providing evidence that miR-206 may have great therapeutic potential against a broad range of human HCCs.
The pathogenesis of HCC is very complex and the detailed mechanisms of this process remain to be investigated. We have shown that miR-206 strongly prevented the pathogenesis of HCC in two HCC mouse models. However, it cannot rule out that other targets of miR-206 might contribute to the pathogenesis of HCC. Thus, our next step is to use a modified HITS-CLIP combined by MiR-Trap,47 biotin-labeled mimics of miR-206 and RNA-Seq to identify targetome of miR-206 in livers of both AKT/Ras and cMyc HCC mice, which will allow us to provide a comprehensive and more accurate understanding for the role of miR-206 in hepatocarcinogenesis.
Supplementary Material
Acknowledgments
Financial Support
This work was supported in part by the National Institutes of Health (R01 DK102601, G.S. and R01 CA136606, XC); a Research Scholar Grant (RSG-16-210-01-RMC) from the American Cancer Society; Gilead Sciences Liver Research Program (G.S); and the Minnesota Obesity Center (G.S)
List of Abbreviations
- AKT
V-Akt murine thymoma viral oncogene homolog 1
- CDK
cyclin-dependent kinases
- CCND1
cyclin D1
- cMet
Met proto-oncogene
- CRISPR/Cas9
the clustered regularly interspaced short palindromic repeats/CRISPR associated protein 9
- HCC
hepatocellular carcinoma
- HITS-CLIP
high-throughput sequencing of RNAs isolated by crosslinking immunoprecipitation
- MAPK
mitogen-activated protein kinase
- Ras
neuroblastoma RAS viral oncogene homolog
- SB
sleeping beauty
- TP
target protector
- TTR
transthyretin
- 3′-UTR
3′-untranslated regions
Footnotes
Additional supporting information may be found in the online version of this article.
Potential conflict of interest: Nothing to report.
References
- 1.Slotta JE, Kollmar O, Ellenrieder V, et al. Hepatocellular carcinoma: s urgeon’s view on latest findings and future perspectives. World J Hepatol. 2015;7:1168–1183. doi: 10.4254/wjh.v7.i9.1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang JD, Roberts LR. Hepatocellular carcinoma: a global view. Nat Rev Gastroenterol Hepatol. 2010;7:448–458. doi: 10.1038/nrgastro.2010.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nair S, Mason A, Eason J, et al. Is obesity an independent risk factor for hepatocellular carcinoma in cirrhosis? Hepatology. 2003;36:150–155. doi: 10.1053/jhep.2002.33713. [DOI] [PubMed] [Google Scholar]
- 4.Baffy G, Brunt EM, Caldwell SH. Hepatocellular carcinoma in non-alcoholic fatty liver disease: an emerging menace. J Hepatol. 2012;56:1384–1391. doi: 10.1016/j.jhep.2011.10.027. [DOI] [PubMed] [Google Scholar]
- 5.Bartel D. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 6.Song G, Zhang Y, Wang L. MicroRNA-206 targets notch3, activates apoptosis, and inhibits tumor cell migration and focus formation. J Biol Chem. 2009;284:31921–31927. doi: 10.1074/jbc.M109.046862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yanaihara N, Caplen N, Bowman E, et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell. 2006;9:189–198. doi: 10.1016/j.ccr.2006.01.025. [DOI] [PubMed] [Google Scholar]
- 8.Kim V. MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol. 2005;6:376–385. doi: 10.1038/nrm1644. [DOI] [PubMed] [Google Scholar]
- 9.Jiang J, Gusev Y, Aderca I, et al. Association of microRNA expression in hepatocellular carcinomas with hepatitis infection, cirrhosis, and patient survival. Clin Cancer Res. 2008;14:419–429. doi: 10.1158/1078-0432.CCR-07-0523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ladeiro Y, Couchy G, Balabaud C, et al. MicroRNA profiling in hepatocellular tumors is associated with clinical features and oncogene/tumor suppressor gene mutations. Hepatology. 2008;47:1955–1963. doi: 10.1002/hep.22256. [DOI] [PubMed] [Google Scholar]
- 11.Meng F, Henson R, Wehbe-Janek H, et al. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;133:647–658. doi: 10.1053/j.gastro.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hayes J, Peruzzi PP, Lawler S. MicroRNAs in cancer: biomarkers, functions and therapy. Trends Mol Med. 2014;20:460–469. doi: 10.1016/j.molmed.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y, Wu MC, Sham JS, et al. Prognostic significance of c-myc and AIB1 amplification in hepatocellular carcinoma. Cancer. 2002;95:2346–2352. doi: 10.1002/cncr.10963. [DOI] [PubMed] [Google Scholar]
- 14.Abou-Elella A, Gramlich T, Fritsch C, et al. c-myc amplification in hepatocellular carcinoma predicts unfavorable prognosis. Mod Pathol. 1996;9:95–98. [PubMed] [Google Scholar]
- 15.Tao J, Ji J, Li X, et al. Distinct anti-oncogenic effect of various microRNAs in different mouse models of liver cancer. Oncotarget. 2015;6:6977–6988. doi: 10.18632/oncotarget.3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ho C, Wang C, Mattu S, et al. AKT (v-akt murine thymoma viral oncogene homolog 1) and N-Ras (neuroblastoma ras viral oncogene homolog) coactivation in the mouse liver promotes rapid carcinogenesis by way of mTOR (mammalian target of rapamycin complex 1), FOXM1 (forkhead box M1)/SKP2, and c-Myc pathways. Hepatology. 2012;55:833–845. doi: 10.1002/hep.24736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patil MA, Lee SA, Macias E, et al. Role of cyclin D1 as a mediator of c-Met- and beta-catenin-induced hepatocarcinogenesis. Cancer Res. 2009;69:253–261. doi: 10.1158/0008-5472.CAN-08-2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jayandharan GR, Zhong L, Sack BK, et al. Optimized Adeno-Associated Virus (AAV)–protein phosphatase-5 helper viruses for efficient liver transduction by single-stranded AAV vectors: therapeutic expression of factor IX at reduced vector doses. Hum Gene Ther. 2010;21:271–283. doi: 10.1089/hum.2009.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ng R, Wu H, Xiao H, et al. Inhibition of microRNA-24 expression in liver prevents hepatic lipid accumulation and hyperlipidemia. Hepatology. 2014;60:554–564. doi: 10.1002/hep.27153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friedman R, Farh K, Burge C, et al. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krek A, Grün D, Poy M, et al. Combinatorial microRNA target predictions. Nat Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 22.Yang J-H, Li J-H, Shao P, et al. starBase: a database for exploring microRNA–mRNA interaction maps from Argonaute CLIP-Seq and Degradome-Seq data. Nucleic Acids Res. 2011;39:D202–D209. doi: 10.1093/nar/gkq1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Villanueva A, Llovet JM. Targeted therapies for hepatocellular carcinoma. Gastroenterology. 2011;140:1410–1426. doi: 10.1053/j.gastro.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frau M, Biasi F, Feo F, et al. Prognostic markers and putative therapeutic targets for hepatocellular carcinoma. Mol Aspects Med. 2010;31:179–193. doi: 10.1016/j.mam.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 25.Thomas MB, Abbruzzese JL. Opportunities for targeted therapies in hepatocellular carcinoma. J Clin Oncol. 2005;23:8093–8108. doi: 10.1200/JCO.2004.00.1537. [DOI] [PubMed] [Google Scholar]
- 26.Santoro A, Rimassa L, Borbath I, et al. Tivantinib for second-line treatment of advanced hepatocellular carcinoma: a randomised, placebo-controlled phase 2 study. Lancet Oncol. 2013;14:55–63. doi: 10.1016/S1470-2045(12)70490-4. [DOI] [PubMed] [Google Scholar]
- 27.Sherr CJ. Cancer cell cycles. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- 28.Staton AA, Giraldez AJ. Use of target protector morpholinos to analyze the physiological roles of specific miRNA-mRNA pairs in vivo. Nat Protoc. 2011;6:2035–2049. doi: 10.1038/nprot.2011.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu J, Che L, Li L, et al. Co-activation of AKT and c-Met triggers rapid hepatocellular carcinoma development via the mTORC1/FASN pathway in mice. Sci Rep. 2016;6:20484. doi: 10.1038/srep20484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mayrhofer P, Schleef M, Jechlinger W. Gene Therapy of Cancer. Springer; 2009. Use of minicircle plasmids for gene therapy; pp. 87–104. [DOI] [PubMed] [Google Scholar]
- 31.Chen Z-Y, He C-Y, Kay MA. Improved production and purification of minicircle DNA vector free of plasmid bacterial sequences and capable of persistent transgene expression in vivo. Hum Gene Ther. 2005;16:126–131. doi: 10.1089/hum.2005.16.126. [DOI] [PubMed] [Google Scholar]
- 32.Wu H, Zhang T, Pan F, et al. MicroRNA-206 prevents hepatosteatosis and hyperglycemia by facilitating insulin signaling and impairing lipogenesis. J Hepatol. 2017;66(4):816–824. doi: 10.1016/j.jhep.2016.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Petrick JL, Kelly SP, Altekruse SF, et al. Future of hepatocellular carcinoma incidence in the United States forecast through 2030. J Clin Oncol. 2016;34:1787–1794. doi: 10.1200/JCO.2015.64.7412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Llovet JM, Bruix J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology. 2008;48:1312–1327. doi: 10.1002/hep.22506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kondo N, Toyama T, Sugiura H, et al. miR-206 expression is down-regulated in estrogen receptor α–positive human breast cancer. Cancer Res. 2008;68:5004–5008. doi: 10.1158/0008-5472.CAN-08-0180. [DOI] [PubMed] [Google Scholar]
- 36.Yan D, Dong XDE, Chen X, et al. MicroRNA-1/206 targets c-Met and inhibits rhabdomyosarcoma development. J Biol Chem. 2009;284:29596–29604. doi: 10.1074/jbc.M109.020511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang T, Liu M, Wang C, et al. Down-regulation of miR-206 promotes proliferation and invasion of laryngeal cancer by regulating VEGF expression. Anticancer Res. 2011;31:3859–3863. [PubMed] [Google Scholar]
- 38.Staton AA, Giraldez AJ. Use of target protector morpholinos to analyze the physiological roles of specific miRNA-mRNA pairs in vivo. Nat Prot. 2011;6:2035–2049. doi: 10.1038/nprot.2011.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ran FA, Hsu PD, Wright J, et al. Genome engineering using the CRISPR-Cas9 system. Nat Prot. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Amir S, Simion C, Umeh-Garcia M, et al. Regulation of the T-box transcription factor Tbx3 by the tumour suppressor microRNA-206 in breast cancer. Br J Cancer. 2016;114:1125–1134. doi: 10.1038/bjc.2016.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang XW, Xi XQ, Wu J, et al. MicroRNA-206 attenuates tumor proliferation and migration involving the downregulation of NOTCH3 in colorectal cancer. Oncol Rep. 2015;33:1402–1410. doi: 10.3892/or.2015.3731. [DOI] [PubMed] [Google Scholar]
- 42.Mataki H, Seki N, Chiyomaru T, et al. Tumor-suppressive microRNA-206 as a dual inhibitor of MET and EGFR oncogenic signaling in lung squamous cell carcinoma. Int J Oncol. 2015;46:1039–1050. doi: 10.3892/ijo.2014.2802. [DOI] [PubMed] [Google Scholar]
- 43.Yunqiao L, Vanke H, Jun X, et al. MicroRNA-206, down-regulated in hepatocellular carcinoma, suppresses cell proliferation and promotes apoptosis. Hepatogastroenterology. 2014;61:1302–1307. [PubMed] [Google Scholar]
- 44.Zhang YJ, Chen SY, Chen CJ, et al. Polymorphisms in cyclin D1 gene and hepatocellular carcinoma. Mol Carcinog. 2002;33:125–129. [PubMed] [Google Scholar]
- 45.Boix L, Rosa JL, Ventura F, et al. c-met mRNA overexpression in human hepatocellular carcinoma. Hepatology. 1994;19:88–91. [PubMed] [Google Scholar]
- 46.Goyal L, Muzumdar MD, Zhu AX. Targeting the HGF/c-MET pathway in hepatocellular carcinoma. Clin Cancer Res. 2013;19:2310–2318. doi: 10.1158/1078-0432.CCR-12-2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baigude H, Li Z, Zhou Y, et al. miR-TRAP: A benchtop chemical biology strategy to identify microRNA targets. Angew Chem. 2012;124:5982–5985. doi: 10.1002/anie.201201512. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.