

The mutation that causes fibrodysplasia ossificans progressiva (FOP) creates a second skeleton of heterotopic bone for which there is no effective prevention or treatment. Clinical and basic research has unveiled hard targets for therapeutic development.
1. Brief background of fibrodysplasia ossificans progressiva
FOP (MIM#135100), a disabling disorder of progressive heterotopic endochondral ossification (HEO), is caused by heterozygous gain-of-function mutations in Activin receptor A, type I (ACVR1, also known as ALK2), a bone morphogenetic protein (BMP) type I receptor. FOP-causing mutations confer a loss of autoinhibition of Smad1/5/8 signaling and altered responsiveness to canonical (BMP) and non-canonical (Activin A) ligands. FOP progresses episodically in trauma-induced or spontaneous flare-ups or insidiously without flare-ups [1–5].
FOP affects approximately one in two million individuals worldwide across all races. Most cases are sporadic but autosomal dominant transmission with complete penetrance and variable expression are established. FOP severity differs greatly among individuals, even identical twins [reviewed in 6].
Malformations of the great toe are present at birth. Flare-ups leading to HEO begin during the first decade of life and appear spontaneously or after muscle fatigue, minor trauma, intramuscular immunizations, or influenza-like viral illnesses. Aponeuroses, fascia, tendons, ligaments and connective tissue of voluntary muscles are affected. Although some lesions regress spontaneously, most mature by an endochondral pathway to form mature HEO. Except for those following trauma, flare-ups are unpredictable. Disability is permanent. No reliable disease or stage-specific biomarkers have been identified [reviewed in 1, 2, 6]. Management is currently supportive. High dose glucocorticoids have been used in acute inflammatory flare ups, with limited efficacy [6].
FOP’s rarity, variability, and fluctuating clinical course pose substantial uncertainties when evaluating experimental therapies [1]. Knock-in mouse models recapitulate the disease phenotype and are vital in testing plausible therapeutic agents [7].
2. Targets and strategies for the prevention and treatment of FOP
A critical unmet need exists for definitive therapies for FOP. The discovery of the causative FOP gene and emerging insights into its mechanisms of action reveal four treatment strategies:
2.1 Strategy 1: Blocking activity of the mutant FOP receptor (mtACVR1)
Five approaches are being pursued: signal transduction inhibitors (STIs), blocking monoclonal antibodies against ACVR1, blocking monoclonal antibodies against Activin A, ligand traps, and mutation allele-specific inhibitory RNA.
2.1.1 Signal Transduction Inhibitors (STIs)
STIs are important molecular tools for studying BMP signaling in FOP, and have great potential for development into powerful therapeutic drugs for FOP [8]. Selective STIs for FOP will inhibit ACVR1 rather than ALK1, ALK3 or ALK6 and are being developed [9]. Broad spectrum STIs that target ALK2 are also being considered for repurposing in clinical trials.
2.1.2 Blocking Antibodies against ACVR1
Mutant ACVR1 demonstrates leaky Smad 1/5/8 signaling and ligand hyper-responsiveness, providing a rationale for using blocking antibodies to ACVR1 in the prevention and treatment of FOP. Therapeutic monoclonal antibodies specific for ACVR1 are under development [1].
2.1.3 Blocking Antibodies against Activin A
Activin A potently stimulates Smad 1/5/8 signaling from mtACVR1 but not from wild type (wt) ACVR1 [4, 5]. Activin A induces HEO in mice expressing mtACVR1 but not in mice expressing only wtACVR1. Inhibition of Activin A with a fully humanized monoclonal antibody blocks spontaneous and trauma-induced HEO in a conditional knock-in model of classic FOP [4]. Activin A is a powerful cytokine that acts as a key regulator of the immune system [reviewed in 10]. Selective activation of mtACVR1 by Activin A highlights an intriguing link between inflammation and HEO in FOP [4, 10].
The molecular, physiologic and structural basis for the sensitivity of mtACVR1 to Activin A is unknown. The unexpected discovery of Activin A in the pathogenesis of FOP identifies a therapeutic target for FOP and excavates a foundation for clinical development.
2.1.4 Ligand Traps
Ligand traps have been proposed as a therapeutic strategy in FOP. A soluble recombinant ACVR1-Fc fusion protein (the extracellular domain of human wild type ACVR1 and the Fc portion of human immunoglobulin gamma 1 abrogates dysregulated BMP signaling caused by mtACVR1 and suppresses chondro-osseous differentiation in vitro [11].
2.1.5 Mutation Allele-Specific Inhibitory RNA
Inhibitory RNA duplexes capable of suppressing the expression of mtACVR1 in connective tissue progenitor cells from FOP patients restores dysregulated BMP signaling to levels observed in control cells and blocks chondro-osseous differentiation [reviewed in 12]. While providing proof-of-principle for allele-specific inhibition of ACVR1 in the prevention of HEO in FOP, the in vivo utility of this approach must be confirmed in mouse models of FOP. A hurdle to human application is safe, effective and durable delivery of small RNA duplexes to relevant progenitor cells [12]. While small molecule inhibitors or biologics may dominate near-term therapeutic options, opportunities on the distant horizon using targeted oligonucleotides are appreciable.
2.2 Strategy 2: Blocking Inflammatory Triggers
Clinical findings and mouse models of FOP provide strong evidence of a role for the immune system in triggering and amplifying FOP flare-ups and HEO in the setting of dysregulated BMP signaling [reviewed in 10]. Targeted ablation of mast cells, macrophages and neuro-inflammatory pathways impair HEO in mouse models [10, 13]. A child with FOP and aplastic anemia (AA) underwent bone marrow transplantation (BMT) which cured the AA but not the FOP. Subsequent graft-versus-host disease prompted a 15 year course of immunosuppression - during which time the FOP was quiescent. When immunosuppression was discontinued, flare-ups returned [14].
2.3 Strategy 3: Blocking Responding Connective Tissue Progenitor Cells
Activation of the retinoid signaling pathway inhibits chondrogenesis and HEO. Retinoic acid receptor gamma (RARγ) agonists potently down-regulate BMP signaling in pre-chondrogenic cells by promoting the degradation of BMP-pathway specific Smads [15]. The RARγ agonist palovarotene blocks trauma-induced and spontaneous HEO in a conditional FOP knock-in mouse model [15, 16] and is being used in FDA-approved clinical trials for FOP. Information can be found at: http//:clinicaltrials.gov.
2.4 Strategy 4: Blocking the Physiologic Response to Microenvironmental Factors that Promote Heterotopic Ossification
Generation of a hypoxic and inflammatory microenvironment in skeletal muscle is a critical step in the formation of HEO [17, 18]. HIF1-alpha integrates the cellular response to both hypoxia and inflammation and amplifies ligand-independent Smad 1/5/8 signaling in the presence of mtACVR1 [18]. Blocking HIF1-alpha pharmacologically with PX-478, apigenin, imatinib or rapamycin abrogates HEO in FOP mouse models [17, 18].
3. Expert opinion
Worldwide interest in FOP research skyrocketed in 2006 following the discovery of the FOP gene. Academia and the pharmaceutical and biotechnology industries have expressed keen interest in FOP and are engaged in research and development to create effective treatments and a cure for FOP.
Successful therapies for FOP will be based on blocking key genetic, molecular, cellular, and tissue targets. Comprehensive knowledge of the natural history of flare-ups and progressive disability in FOP is of paramount importance in the design of clinical trials. While robust cross-sectional natural history studies have been conducted, knowledge of the longitudinal natural history of FOP is still sparse. An annotated natural history and biomarker study has currently enrolled more than 100 patients and will follow them for over three years. Information can be found at: http//:clinicaltrials.gov.
There are several plausible scenarios for clinical trials in FOP: short-term treatment of acute flare-ups, long-term prevention of acute flare-ups, a combinatorial approach, and surgical liberation of ankylosed joints. Different medications and strategies may lend themselves to different clinical trial designs. For example (and in contrast to pre-clinical studies in FOP), the events around the onset of spontaneous flare-ups in humans are unknown. By the time a patient recognizes a flare-up, disease activity might have been smoldering for days, weeks, or even months. Thus, it is difficult to ascertain the stage of a flare-up that a patient is in or if a drug of interest would be effective at that stage. In contrast, a drug targeted to prevent acute flare-ups would require an acceptable long-term safety profile since the onset of flare-ups is unpredictable and thus preventative treatment would be chronic and life-long. This is a high hurdle for a kinase inhibitor targeted to block a highly conserved signaling pathway whose blockade may unmask unanticipated side effects. Thus, therapeutic approaches might consider partial blockade of a signaling pathway with a rescue approach targeted for breakthrough flare-ups, should they occur. Finally, due to the tremendous risk to FOP patients of stimulating more extensive HEO and resulting consequences, surgical liberation of ankylosed joints should not be undertaken until proven treatment options are established.
The main goal for FOP treatment is prevention of progressive postnatal HEO. Thus, the battleground for FOP is childhood. Recent identification of agents such as imatinib and rapamycin that target inflammation and hypoxia-sensing pathways might be repurposed compassionately or formally evaluated by clinical trials in children, while novel therapeutics are being developed. STIs currently in non-FOP-related clinical trials that also target ALK2 might be repurposed for early entry into FOP clinical trials.
Importantly, several adults have been identified with the classic FOP mutation and congenital features of FOP but a paucity of postnatal HEO. These resilient individuals hold the key to understanding factors that trigger FOP flare-ups and amplify progression of the disease. Robust investigation is being conducted to decipher the genetic, epigenetic, environmental, and immunologic factors involved. If distinct factors can be identified in these few individuals, new robust targets for therapy are likely to emerge.
Figure 1.
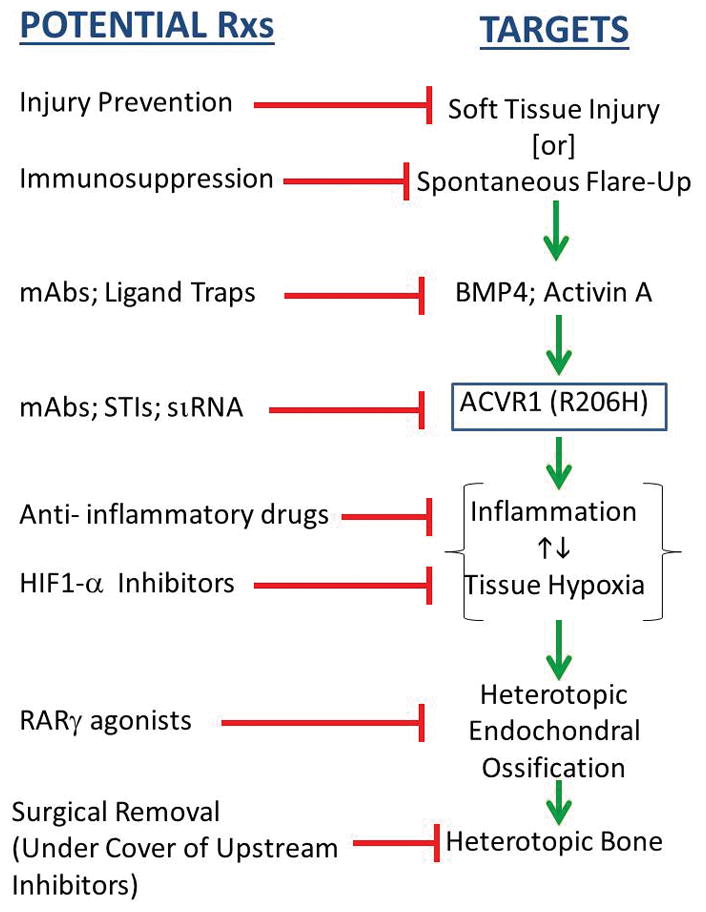
Potential Treatment Strategies for FOP Based on Identified Targets.
Key: SP= substance P, mAbs= monoclonal antibodies, STI= signal transduction inhibitors, siRNA= small inhibitory RNA
Acknowledgments
Funding
This work was supported in part by the International Fibrodysplasia Ossificans Progressiva Association (IFOPA), the Center for Research in FOP and Related Disorders, the Ian Cali Endowment for FOP Research, the Whitney Weldon Endowment for FOP Research, the Ashley Martucci FOP Research Fund, the Isaac and Rose Nassau Professorship of Orthopaedic Molecular Medicine (to FSK), the Cali-Weldon Professorship of FOP Research (to EMS), the Ian Cali FOP Clinical Scholarship (to MMA), the Kogod Professorship in Geriatrics, and the National Institutes of Health (NIH R01-AR41916).
The authors thank Mr. Robert Caron and Mrs. Kamlesh Rai for their invaluable technical and administrative assistance, and the present and past members of our research program for their work leading to our current understanding and perspectives on FOP.
Footnotes
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Contributor Information
Frederick S. Kaplan, Isaac & Rose Nassau Professor of Orthopaedic Molecular Medicine; Professor of Orthopaedic Surgery and Medicine, Co-Director, Center for Research in FOP and Related Disorders, The Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA, USA
Robert J. Pignolo, Robert and Arlene Kogod Professor of Geriatric Medicine; Chair, Division of Geriatric Medicine & Gerontology, Mayo Clinic College of Medicine, Rochester, Minnesota, USA
Mona M. Al Mukaddam, Assistant Professor of Medicine in the Division of Endocrinology, Diabetes and Metabolism; Ian Cali Clinical Scholar, Center for Research in FOP and Related Disorders, The Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA, USA
Eileen M. Shore, Cali-Weldon Professor of FOP Research; Professor of Orthopaedic Surgery and Genetics, Co-Director, Center for Research in FOP and Related Disorders, The Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA, USA
Bibliography
Papers of special note have been highlighted as:
* of interest
** of considerable interest
- 1*.Kaplan FS, Pignolo RJ, Shore EM. From mysteries to medicines: drug development for fibrodysplasia ossificans progressive. Expert Opin Orphan Drugs. 2013;1:637–649. doi: 10.1517/21678707.2013.825208. [Previous iteration of therapeutic horizons in FOP] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2*.Pignolo RJ, Bedford-Gay C, Liljesthrom M, Durbin-Johnson BP, Shore EM, Rocke DM, Kaplan FS. The natural history of flare-ups in fibrodysplasia ossificans progressiva: a comprehensive global assessment. J Bone Miner Res. 2016;31:650–656. doi: 10.1002/jbmr.2728. [Definitive cross-sectional study on the natural history of FOP] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3**.Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho T-J, Choi IH, Connor JM, Delai P, Glaser DL, Le Merrer M, Morhart R, Rogers JG, Smith R, Triffitt JT, Urtizberea JA, Zasloff M, Brown MA, Kaplan FS. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nature Genetics. 2016;38:525–527. doi: 10.1038/ng1783. [The discovery of the FOP gene] [DOI] [PubMed] [Google Scholar]
- 4*.Hatsell SJ, Idone V, Wolken DM, Huang L, Kim HJ, Wang L, Wen X, Nannuru KC, Jimenez J, Xie L, Das N, Makhoul G, Chernomorsky R, D’Ambrosio D, Corpina RA, Schoenherr CJ, Feeley K, Yu PB, Yancopoulos GD, Murphy AJ, Economides AN. ACVR1 (R206H) receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci Transl Med. 2015;7(303):ra137. doi: 10.1126/scitranslmed.aac4358. [Monoclonal antibody against Activin A inhibits HEO in mouse model of FOP] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5*.Hino K, Ikeya M, Horigome K, Matsumoto Y, Ebise H, Nishio M, Sekiguchi K, Shibata M, Nagata S, Matsuda S, Toguchida J. Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proc Natl Acad Sci USA. 2015;112:15438–15443. doi: 10.1073/pnas.1510540112. [Mutant ACVR1 confers responsiveness to Activin A through BMP pathway signaling] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6*.Kaplan FS, Shore EM, Pignolo RJ, editors. The International Clinical Consortium on FOP. The medical management of fibrodysplasia ossificans progressiva: current treatment considerations. Clin Proc Intl Clin Consort FOP. 2011;4:1–100. Available from www.ifopa.org [A comprehensive set of guidelines on the contemporary medical management of FOP] [Google Scholar]
- 7*.Chakkalakal SA, Zhang D, Culbert AL, Convente MR, Caron RJ, Wright AC, Maidment AD, Kaplan FS, Shore EM. An Acvr1 R206H knock-in mouse has fibrodysplasia ossificans progressiva. J Bone Miner Res. 2012;27:1746–1756. doi: 10.1002/jbmr.1637. [The first in vivo proof that the classic FOP mutation causes FOP] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8*.Hong CC, Yu PB. Applications of small molecule BMP inhibitors in physiology and disease. Cytokine Growth Factor Rev. 2009;20:409–418. doi: 10.1016/j.cytogfr.2009.10.021. [An excellent review on STIs for BMP type I receptors] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9*.Dey D, Bagarova J, Hatsell SJ, Armstrong KA, Huang L, Ermann J, Vonner AJ, Shen Y, Mohedas AH, Lee A, Eekhoff EM, van Schie A, Demay MB, Keller C, Wagers AJ, Economides AN, Yu PB. Two tissue-resident progenitor lineages drive distinct phenotypes of heterotopic ossification. Sci Transl Med. 2016;8(366):366r. doi: 10.1126/scitranslmed.aaf1090. [An STI inhibits HEO in a mouse model of FOP] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10*.Kaplan FS, Pignolo RJ, Shore EM. Granting immunity to FOP and catching heterotopic ossification in the Act. Seminars in Cell & Developmental Biology. 2016;49:30–36. doi: 10.1016/j.semcdb.2015.12.013. [Evidence and implications of immune dysregulation in FOP] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11*.Pang J, Zuo Y, Chen Y, Song L, Zhu Q, Yu J, Shan C, Cai Z, Hao J, Kaplan FS, Shore EM, Zhang K. ACVR1-Fc suppresses BMP signaling and chondro-osseous differentiation in an in vitro model of Fibrodysplasia ossificans progressiva. Bone. 2016;92:29–36. doi: 10.1016/j.bone.2016.07.023. [Proof-of-principle of ligand traps in FOP] [DOI] [PubMed] [Google Scholar]
- 12*.Lowery JW, Rosen V. Allele-specific RNA interference in FOP: Silencing the FOP gene. Gene Ther. 2012;19:701–702. doi: 10.1038/gt.2011.190. [Excellent editorial on implications of siRNA therapeutics for FOP] [DOI] [PubMed] [Google Scholar]
- 13*.Kan L, Lounev VY, Pignolo RJ, Duan L, Liu Y, Stock SR, McGuire TL, Lu B, Gerard NP, Shore EM, Kaplan FS, Kessler JA. Substance P signaling mediates BMP-dependent heterotopic ossification. J Cell Biochem. 2011;112:2759–72. doi: 10.1002/jcb.23259. [The importance of neuro-inflammatory triggers in HEO lesion formation] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14*.Kaplan FS, Glaser DL, Shore EM, Pignolo RJ, Xu M, Zhang Y, Senitzer D, Forman SJ, Emerson SG. Hematopoietic stem-cell contribution to ectopic skeletogenesis. J Bone Joint Surg Am. 2007;89:347–357. doi: 10.2106/JBJS.F.00472. [Seminal insight on role of immune system in FOP] [DOI] [PubMed] [Google Scholar]
- 15*.Shimono K, Tung WE, Macolino C, Chi AH, Didizian JH, Mundy C, Chandraratna RA, Mishina Y, Enomoto-Iwamoto M, Pacifici M, Iwamoto M. Potent inhibition of heterotopic ossification by nuclear retinoic acid receptor-gamma agonists. Nat Med. 2011;17:454–460. doi: 10.1038/nm.2334. [A seminal study describing the efficacy and mechanism of action of RAR gamma agonists in the inhibition of HEO] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16*.Chakkalakal SA, Uchibe K, Convente MR, Zhang D, Economides AN, Kaplan FS, Pacifici M, Iwamoto M, Shore EM. Palovarotene inhibits heterotopic ossification and maintains limb mobility and growth in mice with the human ACVR1 (R206H) fibrodysplasia ossificans progressiva (FOP) mutation. J Bone Miner Res. 2016;31:1666–1675. doi: 10.1002/jbmr.2820. [Palovarotene, an RAR-gamma agonist, inhibits trauma-induced and spontaneous HEO in a mouse model of FOP] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17*.Wang H, Lindborg C, Lounev V, Kim JH, McCarrick-Walmsley R, Xu M, Mangivani L, Groppe JC, Shore EM, Schipani E, Kaplan FS, Pignolo RJ. Cellular hypoxia promotes heterotopic ossification by amplifying BMP signaling. J Bone Miner Res. 2016;31:1652–1665. doi: 10.1002/jbmr.2848. [Molecular mechanisms and therapeutic insights of hypoxia-sensing pathway in FOP] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18*.Agarwal S, Loder S, Brownley C, Cholok D, Mangiavini L, Li J, Breuler C, Sung HH, Li S, Ranganathan K, Peterson J, Tompkins R, Herndon D, Xiao W, Jumlongras D, Olsen BR, Davis TA, Mishina Y, Schipani E, Levi B. Inhibition of Hif1α prevents both trauma-induced and genetic heterotopic ossification. Proc Natl Acad Sci USA. 2016;113:E338–347. doi: 10.1073/pnas.1515397113. [Therapeutic insights of the hypoxia-sensing pathway in HEO] [DOI] [PMC free article] [PubMed] [Google Scholar]