

INTRODUCTION
“Precision dosing” focuses on the individualization of drug treatment regimens based on patient factors known to alter drug disposition and/or response. In 2015, over 8 in 10 nonfederal acute care hospitals in the United States had adopted a basic electronic health record (EHR) system,1 which will facilitate the application of precision dosing. Expanding on recent publications,2, 3 we outline the public health need, a proposed framework, and the anticipated impact for the adoption of precision dosing.
THE NEED FOR PRECISION DOSING
The concept of “precision medicine” offers the hope of optimizing disease prevention and treatment strategies by taking into account factors that contribute to interindividual variability.4 During the drug development process, several studies are performed using tools, such as pharmacokinetic/ pharmacodynamic (PK/PD) modeling, that collect useful dosing information and help to identify patient subgroups most likely to benefit from a new drug. Furthermore, genomic and nongenomic biomarker data may be used to differentiate subgroups of patients with varying drug clearance, exposure–response relationships, drug safety, and disease progression/severity.5, 6 This rich information from early development studies is used to inform phase III trial design, as well as drug dosage regimen and patient selection for these studies, and can be integral to the drug label (if the drug is eventually approved).7
In the United States the Food and Drug Administration (FDA) performs rigorous statistical and pharmacometric quantitative analyses to replicate the sponsor's analyses, and to understand better which patients will most likely receive benefit from a new drug.8, 9 Patient factors that may be considered include sex, body size, organ function, age, genotype, concomitant medications, and disease severity. The FDA may choose to change labeled doses for certain subgroups (e.g., gender, renal function, age), based on either the sponsor's or their own PK/PD analyses, when the supportive data are available. However, few options are available if the sponsor chooses not to integrate biomarker data into the design of the phase III trial. Furthermore, the dosage regimen in the approved label will either indicate quantitative or qualitative (e.g., increase/decrease) dose adjustments based on patient factors known to alter the PK and/or PD of the drug. However, it is relatively rare that dosage regimens will be recommended for patients who present with multiple characteristics known to alter drug disposition or efficacy (e.g., decreased renal function plus a drug interaction plus a polymorphic genotype). One notable example is the package insert Cerdelga (eliglustat) where dosing recommendations are provided as a function of both CYP2D6 metabolizer status and the concomitant use of CYP3A and CYP2D6 inhibitors.10
Furthermore, when the pivotal clinical trial demonstrates efficacy, the sponsor's New Drug Application (NDA) to the FDA may recommend very limited dose adjustments in the label based on patient factors (e.g., renal function, genotype, drug–drug interactions) or for special populations (e.g., pediatrics, pregnancy/lactation, geriatrics), and rarely advocate for clinical use of tools that integrate patient factors to help facilitate dose individualization. A recent “State of the Art” article extensively outlined the challenges that need to be overcome in order for “model‐informed precision dosing” to be used routinely in healthcare practices in the future.2 Precision dosing in that previous article and the present article refers to the optimization of drug dosing in individual patients with the goal of maximizing efficacy and/or minimizing toxicity. Although it can be argued that “accurate dosing” may be a more appropriate phrase, “precision dosing” has been adopted by us and others2 in order to be consistent with the widely accepted initiative focused on “precision medicine” and because it is more likely to be understood by the public.
The use of precision dosing offers the potential to overcome a common pattern observed in postregulatory approval, where the benefit‐to‐risk relationship of a drug is less favorable than what was reported in clinical trials.11 This pattern is often due to greater diversity in the patients who receive the drug postapproval, resulting in increased variability in drug exposure and response.11 A phase III clinical trial is designed with relatively narrow patient inclusion and exclusion criteria in order to maximize the likelihood of trial success and also to provide information that can inform the product label. Phase III clinical trials will usually restrict patient enrollment based on age, body size, renal and hepatic function, disease severity, and comorbidities. By definition, the phase III clinical trial population sample represents only a fraction of the market population, and understandably cannot accurately capture the diversity of patient characteristics present in a heterogeneous patient population. The lack of heterogeneity among enrolled patients is not explicitly described in the label; however, the FDA will place label restrictions for “special populations” not adequately studied (e.g., pregnancy/ lactation, pediatrics, geriatrics). While considerable effort is now made to provide dosing information for relevant drug interactions,12 children,13 and/or patients with renal or hepatic impairment,14, 15 it is uncommon for a label to include well‐defined dosing recommendations for patients with functionally impactful genetic polymorphisms, or for those who are pregnant/lactating, geriatric, or obese. It is not required that either the sponsor or the FDA qualitatively and quantitatively describe the patient diversity differences between the phase III pivotal trial(s) sample and the US anticipated market population.
The clinical and financial impact of precision dosing, while not known for most drugs, is likely to be both significant and measurable. In the United States adverse drug reactions (ADRs) are known to be a leading cause of death and cost billions of dollars in avoidable healthcare costs.16, 17, 18 Patient populations that are at risk for ADRs include patients at the extremes of age, those on multiple medications that may interact with each other, and those with end‐organ dysfunction, among others. Thus, the patients who are at greatest risk for ADRs are often those who are not well represented in phase III clinical trials. Furthermore, poor medication adherence is estimated to result in avoidable costs of $105 billion in the United States.19 The application of precision dosing principles may help to reduce the incidence of preventable ADRs related to inadequate dosage regimen selection and plausibly improve medication adherence.
Today, prescribers commonly review electronic health records (EHR) that contain information pertaining to the patient's age, body size, sex, clinical history, comorbidities, concomitant medications, and biomarkers. Ideally, the patient's clinical information would be integrated by a dosing tool into a more precise dosing regimen for consideration by the prescriber before the electronic prescription is written. This approach to drug dosing would move clinical practice towards a more patient‐centered healthcare system, which aligns with new value‐based payment models that expect providers to deliver health care that improves patient outcomes and lowers the total costs of care.20 As previously noted,2 there are numerous examples where “model‐informed precision dosing” has been used to optimize drug dosage regimens in individual patients;21, 22, 23, 24, 25, 26, 27 however, there are barriers (e.g., integration of precision dosing tools into the EHR) that need to be overcome before there is widespread dissemination of available tools.3 Also, the capability to apply precision dosing principles clinically through electronic prescribing is generally not available for new, narrow therapeutic index drugs, where serious consequences of under‐ or overdosing are present. Thus, there is a public health need to create a paradigm shift in how drug dosing is optimized in individual patients as part of the ongoing transformation of health care.
CONSEQUENCES OF IMPRECISE DOSING
The lack of optimal dose selection during development often impacts the sponsor by leading to phase III clinical trial failures, delayed market access (lost commercial opportunity), and increased development expense.3, 28 To decrease this risk, drug sponsors continue to increase their time and monetary investments in the identification and validation of predictive biomarkers and development of tools to increase the probability of phase III clinical trial successes.6, 29 Collection of additional biomarker data is at the prerogative of the sponsor and is usually not a regulatory requirement. However, biomarker and clinical end‐point PK/PD and statistical analyses enable the sponsor and regulators to gain a more precise understanding of drug disposition and response. This is particularly true when only one drug dose is studied in phase III pivotal trials, and when a new drug for a rare disease produces a weak efficacy signal. Cases exist where drugs have been approved based on the insight provided by the drug exposure–response relationship that could not otherwise be seen (e.g., extended‐release lamotrigine).9 Drug exposure–response analyses that occur during NDA review are one of the most important quantitative tools used to approve new drugs. It would be useful if these same analyses were used to help project dosing precision for the intended market population.
Individual patients may experience maximal efficacy and minimal toxicity when they receive a drug with a dosage regimen optimally tailored to achieve a drug concentration within the target therapeutic range. However, if the drug concentration achieved is too low at the prescribed dose due to poor patient adherence, large body size, or metabolizing enzyme induction, for example, drug response may be suboptimal and result in unintended outcomes (e.g., pregnancy despite appropriately prescribed oral contraception, organ rejection or graft vs. host disease despite appropriately prescribed immunosuppression), or even death, depending on the drug and disease. Alternatively, unintentional over dosage due to small body size, impaired renal function, or a loss‐of‐function polymorphism in a metabolizing enzyme may result in drug toxicities where the consequences could range from simply bothersome symptoms, to more significant morbidity, to death in extreme cases. Thus, there is an unmet need to develop novel tools and strategies to optimize drug dosage regimens with the specific intent of maximizing both drug development successes (defined as a higher rate of approvals), and also postapproval successes (defined as maximal efficacy with minimal toxicity) when the drug will be used in a more diverse patient population.
ELECTRONIC PATIENT CARE ENVIRONMENT
Nations have invested in creating an electronic patient care environment to measure and improve healthcare quality (at both the individual and population levels), and to control costs.30 In the United States, the Health Information Technology for Economic and Clinical Health (HITECH) Act was passed in 2009, as part of the American Recovery and Reinvestment Act.31 One result of the HITECH Act, and associated funding ($27 billion over 10 years), was that by 2014, 97% of US hospitals possessed EHR software and 75% had implemented EHRs.32 Similar statistics were found among office‐based physicians; in 2013, an estimated 78% of office‐based physicians used some type of EHR, an increase from 18% in 2001.33 The EHR, which is a digital version of a patient's chart, includes information such as patient demographics, diagnoses, laboratory results, procedures, medications, results of imaging studies, and clinician notes. The EHR interfaces with applications that are important for the delivery of patient care, including a computer provider order entry (CPOE) system and clinical decision support tools. The CPOE system allows the prescriber to select the treatment regimen (drug, formulation, quantity, duration, and dosing instructions), which is then recorded in the EHR, and simultaneously sent to the pharmacy where the medicine is dispensed for the patient. Clinical decision support tools are intended to help the clinician improve the quality of care for each patient by using their personal information to improve diagnosis and treatment accuracy and effectiveness. Software components aligned with the EHR should work together seamlessly. For instance, one could imagine that when the prescriber selects a treatment regimen in the CPOE system, patient information in the EHR (e.g., diagnoses, age, weight, laboratory values) could be aligned with prior best practice knowledge embedded in the clinical decision support tools to enable the prescriber to select the optimal drug and dosage regimen, as well as provide information regarding clinically important warnings for significant risks (Figure 1). The clinician could also be reminded about the value of collecting additional information (e.g., biomarker data) to make more informed decisions. Several groups have pioneered the use of model‐based clinical decision support tools to facilitate individualization of drug dosing.21, 27, 34, 35, 36, 37, 38, 39
Figure 1.
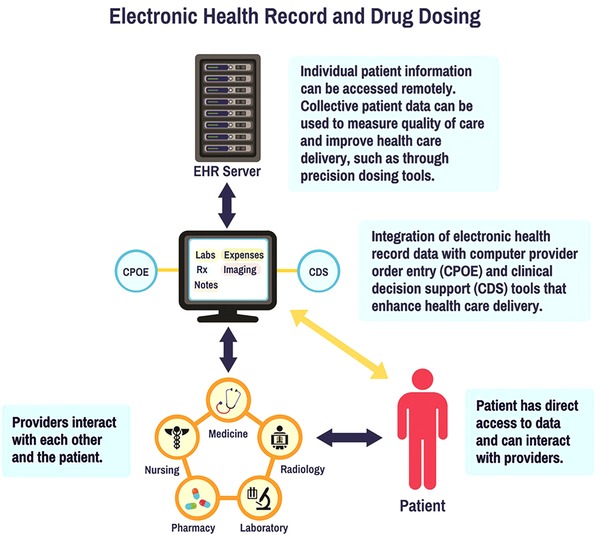
Integration of electronic health record (EHR) data with computer provider order entry (CPOE) and clinical decision support (CDS) tools.
The main goal of the HITECH Act, which was implemented in a staged manner, was to develop an electronic healthcare environment that improves individual healthcare quality and efficiency.32, 40 This Act is intended to increase patient engagement in their own health care, improve coordination between healthcare providers and the patient, and control costs. While healthcare quality is dependent on the healthcare provider's skill and expertise, the new software environment embraces best practices that should benefit patients. For example, if a prescriber is faced with an atypical patient in their practice who has a disease or personal characteristics that are not average (e.g., pregnancy, morbid obesity, combination of factors), then a warning can be provided that an “average” dose may be insufficient, which triggers dosing and monitoring options for consideration.
The functional software performance metrics have been articulated by the federal government as a condition for Medicare and Medicaid payments.31 There is competition between companies to provide software products for EHR, CPOE, and clinical decision support tools. The philosophy is that the hospitals will provide the customization needed to create the environment, dictated by local needs, policies, and values. Competition has resulted in a demand for knowledgeable professionals to build these products. However, there is still an unmet need to develop and validate precision drug dosing tools that can be integrated into these evolving technologies.3 Improving upon the new EHR environment provides a unique opportunity for clinical pharmacologists to play a pivotal role in shaping the future of precision dosing. In addition, the largescale adoption of centrally stored electronic health records, and the use of mobile applications and wearable devices, provide an opportunity for patients to be actively engaged in their health care outside the practice setting.
DRUG CHARACTERISTICS WHERE PRECISION DOSING IS IMPORTANT FOR THERAPEUTIC SUCCESS
Drugs that may benefit the most from precision dosing often have one or more of the following characteristics: a narrow therapeutic index; wide interpatient PK/PD variability; use in high‐risk patient populations; inadequate dosing that can contribute to the development of disease resistance or tachyphylaxis; and/or high drug costs that burden patients, payers, and health systems. By definition, narrow therapeutic index drugs require careful oversight, and there are examples of drugs where the doses/concentrations associated with favorable efficacy outcomes or toxicities are not vastly different.41, 42 The liability or cost associated with imprecise dosing can vary between drugs, but could include poor clinical outcomes related to sub‐ or supratherapeutic exposure, increased healthcare costs due to poor utilization of healthcare resources, and even death in extreme cases. While we have roughly estimated that there may be several hundred drug candidates for precision dosing, a more rigorous analysis will include setting criteria and a process for prioritization.43
Examples of drugs that exhibit some of these characteristics are presented in Table 1. The examples are stratified into three categories based on the level of evidence available that supports the need for precision dosing. For example, aminoglycosides are a class of antibiotics with a narrow therapeutic index, and an example of a drug class where plasma concentrations are measured routinely in order to facilitate dose individualization. Subtherapeutic aminoglycoside concentrations can result in infection progression, treatment failure, and may contribute to the emergence of resistant bacteria, while supratherapeutic concentrations can cause nephrotoxicity.44 As another example, the benefit of precision dosing has not been established clinically for most monoclonal antibodies; however, precision dosing could potentially help maximize drug efficacy and minimize toxicities, as well as reduce the treatment costs of these drugs.34, 45, 46
Table 1.
Examples of drugs where precision dosing may be beneficial
| Drug class | Drug | Precision dosing patient populations | Liability (potential consequences that may occur with supra‐ and/or subtherapeutic concentrations) |
|---|---|---|---|
| Drugs that are known to benefit from precision dosing | |||
| Aminoglycosides | Gentamicin and tobramycin44, 77, 78, 79 | Altered renal function, pediatrics, and obese |
Supratherapeutic: nephrotoxicity Subtherapeutic: emergence of resistant bacteria, lack of resolution of infection, and sepsis |
| Cardiac inotrope | Digoxin80 | Altered renal function and geriatrics |
|
| Alkylating agent (cell cycle nonspecific) | Busulfan81 |
|
|
| Calcineurin inhibitors | Cyclosporine, tacrolimus82, 83 | Pediatric and adult HSCT and solid organ transplant patients |
|
| Anticoagulant (vitamin K epoxide reductase inhibitor) | Warfarin84 | Gene variant carriers (CYP2C9, VKORC1), geriatrics, and those at risk for drug–drug interactions |
|
| Nonnucleoside analog reverse transcriptase inhibitor | Efavirenz85, 86, 87 | Gene variant carriers (CYP2B6) |
|
| Potential benefit of precision dosing has not been clinically established | |||
| mTOR inhibitors | Everolimusa, sirolimus, and temserolimus45, 88, 89 |
|
Supratherapeutic: hematologic toxicity Subtherapeutic: graft rejection |
| Polymyxins | Polymyxin B90 | Critically ill patients |
|
| Anticoagulant (direct factor Xa inhibitors) | Rivaroxaban, apixaban, and edoxaban91, 92, 93, 94 | Altered renal function and those at risk for drug–drug interactions |
|
| New drugs lacking scientific information supporting precision dosing | |||
| CDK4/6 inhibitor (oral kinase inhibitor) | Palbociclib95, 96 | Immunocompromised patients and those at risk for drug–drug interactions | Supratherapeutic: neutropenia |
| Selective BCL‐2 inhibitor | Venetoclax97, 98, 99, 100 | Those at risk for drug–drug interactions | Supratherapeutic: tumor lysis syndrome (ramp‐up phase) |
HSCT: hematopoietic stem cell transplant; VOD: veno‐occlusive disease; SOS: sinusoidal obstruction syndrome; CYP2C9: cytochrome P450 2C9; VKORC1: vitamin K epoxide reductase complex subunit 1; INR: international normalized ratio; CYP2B6: cyptochrome P450 2B6; mTOR: mammalian target of rapamycin; Advanced HR+ BC: advanced hormone receptor‐positive, HER2‐negative breast cancer; NET: advanced neuroendocrine tumors; RCC: advanced renal cell carcinoma; TSC: renal angiomyolipoma with tuberous sclerosis complex; SEGA: subependymal giant cell astrocytoma; TKI: tyrosine kinase inhibitor; CDK: Cyclin‐dependent kinase; BCL: B‐cell lymphoma.
Additional studies are needed to demonstrate the value of therapeutic drug monitoring in nontransplantation settings.88
As noted by Darwich et al.,2 ultimately, determination of which drugs require precision dosing should include a systematic assessment of the patient and societal implications of inadequate drug management on the patient, payers, and the health system. In addition, the data and resources required for the delivery of precision dosing should be considered. Evaluating the advantages and disadvantages of precision dosing requires input from drug sponsors who are knowledgeable about the drug, regulators who make drug approval decisions based on benefit/risk considerations, clinicians who are responsible for managing patients and prescribing the drug, and payers who must make difficult decisions about how to use limited healthcare resources most effectively. Manufacturers should have mechanisms in place to update dosing guidelines as postmarketing surveillance data become available. As previously suggested, this information can be made available through “dynamic labels” that are disseminated through available computer and mobile software platforms and can be updated as new information becomes available.2
Other obstacles that need to be considered include: the establishment of therapeutic ranges and exposure–response relationships, especially in the case of dosing multidrug combinations (e.g., combination anticancer or antiviral drug treatment) where there may be cases of overlapping therapeutic efficacy and adverse effects.2, 45 Drug plasma concentration and other biomarker assays will need to be developed for routine use. These assays will need to have acceptable accuracy and precision, a rapid turnaround of results, and a reasonable cost so that they can be used to adjust dosing. Finally, the ability to perform precision dosing may be dependent on the availability of dosage forms that could allow for dose individualization.2 For drugs where intravenous or liquid formulations do not exist, limited dosage strengths may be available, which will be a barrier to implementation. As technology advances, development of formulations that allow for individualization of drug dosing will be important in order to facilitate precision dosing.
PATIENT POPULATIONS LIKELY TO BENEFIT FROM PRECISION DOSING
The need for precision dosing requires consideration of the patient population to whom the drug is prescribed. As previously noted, drugs often are tested in clinical trials that enroll homogenous patient cohorts. If the drug is approved, there may be widespread use in patient populations where limited/no data are available to determine optimal dosing. The need for precision dosing may be greater in these patient populations because of more pronounced interpatient PK/PD variability, which could result in altered efficacy and safety profiles. Because the underlying disease process and/or exposure–response relationship of drugs used in these special populations may differ from the population originally studied, additional clinical trials are typically needed in order to assess PK/PD and develop the appropriate mathematical models that can be used to devise more precise dosage regimens. Pediatric and geriatric patients are examples of patient populations that are likely to benefit from the application of precision dosing principles, particularly for drugs with a narrow therapeutic index, due to changes in drug disposition and/or effects.47, 48 Precision dosing in special populations also requires consideration of practical and logistical factors, including low neonatal blood volumes that limit the collection of PK samples.
AVAILABLE TOOLS TO FACILITATE PRECISION DOSING
Precision dosing requires clinical tools that can translate an individual's genotypic and phenotypic characteristics into an individually tailored dosage regimen. Two fundamental elements are critical in this translation. The first element is to discover and establish genotypic and phenotypic biomarkers that can either accurately reveal a patient's disease status or reliably predict therapeutic outcomes.49 A second element is to develop and validate quantitative clinical decision support tools that translate predictive dosing models to individual patient dosing selection. These two components are both imperative for developing and implementing a precision dosing strategy for prospective evaluation, and they are interdependent on each other for aiding clinical decisions.
Genetic variants have been applied broadly as biomarkers to either stratify patients or select an appropriate therapy, dose, or regimen. Tailoring the selection and dosing of therapies for specific gene variants is a well‐recognized but an underutilized approach.50 Genetic variants in drug‐metabolizing enzymes or transporters can significantly alter PK/PD, and dosing adjustments may be advised depending on metabolizer or transporter status.51 A key challenge in validating genetic biomarkers is to identify which of the many variants are “drivers” or actionable markers. Improved evidence‐based modeling approaches are needed to make genetic information available at the time of prescribing if clinicians are to use these in routine patient care. In addition to genomic biomarkers, the use of transcriptomic, proteomic, and metabolomic methods may also allow for better tailoring of drug treatment.52, 53, 54, 55
Phenotypic biomarkers, particularly those that significantly predict drug PK/PD, are important for precision dosing. For drugs with a narrow therapeutic index, plasma or blood drug concentrations are often monitored. Therapeutic drug monitoring (TDM), also known as adaptive feedback control, is a commonly applied strategy to incorporate real‐time drug concentrations into dosing considerations to constrain concentrations within a target window.56 Also, many broader phenotypic characteristics of patients (e.g., age, renal function, body size), have been used to tailor individual therapy when relevant. While genetic biomarkers are static but variable in prevalence within a population, phenotypic biomarkers (patient characteristics) are dynamic and may change rapidly within a patient (e.g., creatinine clearance) with high inter‐ and intraindividual variabilities. This diversity and extensive variability in patient characteristics is not sufficiently represented in typical phase III trials, where there are reasonable attempts to constrain variability. Therefore, to implement precision dosing, tools should be widely developed, validated, and implemented, which enable the identification and quantitative characterization of the sources of patient variability.2, 3 For instance, if a patient has been stable while receiving digoxin to treat congestive heart failure, but serum creatinine concentrations have been increasing over recent clinic visits, then the clinical decision support system could trigger a warning to the prescriber to consider measuring the digoxin serum concentration, which could possibly lead to dose reduction and toxicity mitigation. Also, precision dosing recommendations should be simplified to focus on those phenotypic variables that are expected to have a significant impact on dosing.
There are many quantitative tools and methods available that can be used to characterize and account for sources of PK/PD variability. For example, nonlinear mixed effects modeling (i.e., population modeling),57, 58 physiologically based PK/PD (PBPK/PD) modeling,59 and Bayesian methods are used widely to characterize drug disposition and effects.2, 60 These methods are implemented in commonly used and commercially available software packages for data analysis.61, 62
Population PK/PD models create the basis for individual patient dose adjustments with the goal of achieving a target drug plasma concentration range associated with efficacy and safety. These models provide the relationship between drug concentration and effect, while identifying patient covariate characteristics from clinical studies both prior to NDA approval and thereafter.57, 58 Population PK/PD modeling can support precision dosing in several ways, including covariate‐based a priori dosing and TDM‐based a posteriori dosing. Covariate‐based a priori dosing involves dose selection based on patient factors that have been identified as important predictors of interindividual variability in PK/PD parameters.63 TDM‐based a posteriori dosing involves covariate‐based a priori dosing followed by dose individualization through assessment of drug concentrations or effects, and application of Bayesian methods to estimate individual PK/PD parameters.63, 64, 65 Various tools have been developed to facilitate dose optimization through Bayesian adaptive control methods, including InsightRx (http://insight-rx.com/), TDMx (http://www.tdmx.eu/), DoseMe (https://doseme.com.au/), and BestDose (http://www.lapk.org/bestdose.php), among others. The history and evolution of the application of Bayesian methods in clinical decision support tools has been reviewed previously.66, 67
When faced with determining dosing recommendations for new drugs in patients not studied in pivotal trials, it is possible to use prior knowledge of other drugs that have similar physicochemical and PK/PD characteristics. PBPK models represent a systems mass balance modeling approach that allows for a mechanism‐based PK analysis.59 PBPK modeling can be used to project an initial dose for these patients that will need confirmation in prospective clinical studies.2 In addition, PBPK models can be used in predicting PK when multiple characteristics are known to alter drug disposition or efficacy.10 A major advantage of PBPK modeling is the ability to integrate knowledge from human physiology, drug physicochemical properties, and enzyme and protein variability in patients to generate drug PK predictions in a given population or individual. PBPK models also have been applied broadly to guide individual dosing in special populations.68, 69 PBPK models can also support precision dosing in a variety of clinical settings, including patients with organ dysfunction, patients coadministered enzyme inhibitors and inducers, and populations with certain genetic variations.
FACILITATING PRECISION DOSING IN NEW PATIENT POPULATIONS
Once a drug has been approved by a regulatory body, additional clinical trials may be needed in patient subgroups that are likely to benefit from a dosage regimen different from the standard dosing paradigms derived from phase III efficacy–safety outcomes data. These studies can focus on characterizing PK alone, PK/PD, efficacy, and/or safety of the drug in a new patient population.
The initial drug label at the time of FDA approval invariably possesses many gaps in drug dosing for specific patient populations, and it can oftentimes be years before they are addressed. The dosing gaps include common variables (e.g., organ failure, body size, age extremes), as well as less common variables (e.g., pregnancy, severe burns), all of which can profoundly change dosing requirements. Today, clinicians treating patients who present with a complex combination of common and uncommon variables are left to their own ingenuity to estimate dosing.
Prior knowledge of the drug can be used to inform the drug development programs in these new patient populations.70 For example, a sponsor may decide to pursue a pediatric drug development program if a drug is likely to be used in children. Full or partial extrapolation of efficacy from well‐controlled studies performed in adults may be allowed, depending on the similarity of disease processes and exposure–response relationships.13 In the case of full extrapolation, the pediatric drug development program is focused on identifying a pediatric dose that achieves comparable drug exposure to that observed in adults, and evaluating the safety of the drug at the selected dose.
Thus, in selecting the optimal strategy for developing dosing regimens for special populations, a decision should be made whether to perform a new clinical efficacy–safety trial or to match drug concentration exposure in the special patient population to historical values from the pivotal phase III trial. As shown in Figure 2, this requires consideration of similarities in the exposure–response relationship and disease processes in the special population relative to patients enrolled in the pivotal phase III trials. Since the cost is much greater to perform a new clinical trial to demonstrate efficacy and safety, bridging and application of model‐based approaches may be an acceptable strategy to aid in dose selection until evidence indicates the need to justify the costs of conducting a clinical study.2, 71
Figure 2.
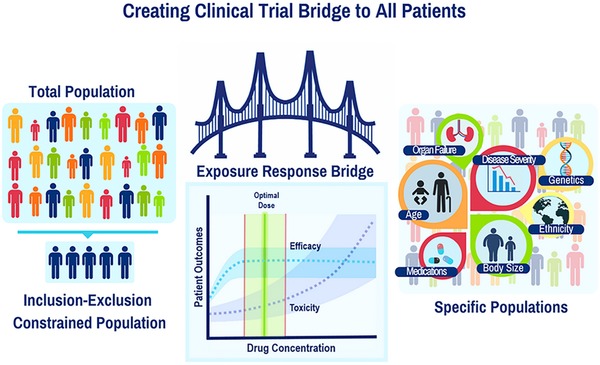
Bridging information, including exposure–response relationships, from the original patient population studied in clinical trials to specific populations that will receive the drug in real‐world clinical practice.
PROPOSED PRECISION DOSING DEVELOPMENT FRAMEWORK
A precision dosing strategy can be established for new drugs under development, or after regulatory approval. For the former, the sponsor and regulatory agencies could begin to consider whether a new drug is likely to require precision dosing at the time of approval of an Investigational New Drug (IND) application. The drug characteristics were discussed earlier in this article. We also believe that development and approval strategies (e.g., dose finding, phase III patient population, study end points) can be considered. If either the sponsor or the regulatory agency believe this new drug is a candidate for precision dosing, then the clinical development pathway can be designed to better define exposure–response relationships, and characterize responders and nonresponders using biomarkers during both early and late phase development. An early phase II regulatory decision could be made to help assure adequate biomarker and PK/PD assessments are performed in phase II/III studies.
We believe, as others have suggested, that a new postregulatory approval process is needed to support precision dosing for relevant marketed drugs.2, 3 We propose that this process could be separated into three phases: i) prior knowledge aggregation, ii) precision dosing studies and design, and iii) clinical decision support tool design, qualification, and implementation.
Prior drug‐disease knowledge needs to be aggregated to facilitate data sharing. The pharmaceutical industry has begun to make some progress on this front by outlining their commitment to the responsible sharing of clinical trial data.2, 72 Information types includes patient‐level, de‐identified phase IIb and phase III clinical trial data, previously developed population and PBPK/PD models, and information regarding relevant predictive biomarkers associated with patient outcomes. Also, the model code and biomarker assay data should be made publicly available to assure that scientists can accurately reproduce the pivotal analyses. The phase IIb and phase III data also may be used to generate a larger postmarket database, upon which subsequent precision dosing models can be created. For drugs with multiple indications, such as anticoagulants (e.g., stroke prevention in atrial fibrillation, deep vein thrombosis prophylaxis), information on all end points will be needed to generate precision dosing recommendations within various subgroups. For patient subgroups where data that would allow for precision dosing are lacking, additional data can be collected through prospective studies; for example, opportunistic protocols that capitalize on standard of care procedures for patients already receiving a drug of interest can be used to efficiently collect PK/PD data in special populations.73
Multistakeholder collaborations will be important to validate, implement, and demonstrate the value of precision dosing tools. Academic institutions and disease centric organizations, such as the American Heart Association and American Cancer Society, can be pivotal for developing precision dosing strategies, postapproval, in collaboration with the sponsor. In the United States, academic institutions affiliated with the Clinical and Translational Science Awards (CTSA) program (https://ncats.nih.gov/ctsa) can collaborate to evaluate the use of precision dosing tools across multiple sites. Funding can be pursued by these groups from the government, the pharmaceutical industry, and/or private foundations to conduct the requisite studies needed to develop, refine, and validate precision dosing strategies, and then to implement those strategies in clinical decision support tools. Unfortunately, for drugs that already have regulatory approval, there may be the perception that these necessary studies represent only incremental progress and this work may receive a low priority funding score. The justification to fund this work is to create an efficient decentralized process for rapidly selecting precision dosing candidates, and collecting the clinical information in large, diverse patient population samples upon which drug dosing models and tools can be validated and implemented. The need to create better dosing for all patients is similar to establishing the Sentinel system to provide rapid accurate postmarket safety information for medical products.74 In these studies, the number of patients enrolled can be based on an assessment of the drug's therapeutic index, expected PK/PD variability, disease severity/lethality and prevalence/penetrance, as well as the logistical factors that can affect patient enrollment and study design. The data collected from these studies can be analyzed with and without the premarket patient data to create a population PK/PD model for all patients. Based on the results of the prospective study, clinician groups with direct knowledge of the patient population under study can propose optimal dosing regimens that can then be implemented.
Approved precision dosing recommendations will need to be converted into a drug‐specific precision dosing tool for incorporation into clinical decision support software or the EHR itself. As noted by Darwich et al., development of a “companion tool” can begin early in the drug development process.2 We also believe that integration of precision dosing tools into the EHR itself will be critically important in order to ensure their widespread use.3 Whether the tools are open source or proprietary will depend on the funding model. During the clinical implementation processes, the functionality and usability of the tools, as well as prescriber acceptance of the dosing recommendations, must be evaluated. These implementation studies can culminate in the development of a “playbook” that would allow for widespread dissemination of the most optimal use of the precision dosing tool. Governance procedures need to be established to help assure tool quality, but also provide for local customization as needed. There should be iterative and adaptive processes for continuous refinement and quality improvement of the precision dosing tool. Because the majority of drugs are marketed globally, data pertaining to optimal drug dosing should be made publicly available to allow for widespread use of these tools, and of precision dosing strategies. For drugs developed primarily for use in only one global region (e.g., United States or Europe), it would be possible to use prior knowledge and experience to predict dosing among populations in another global region (e.g., Asia or Africa). Subsequent confirmatory studies, designed for further tool refinement, could highlight important dosing differences between distinct patient populations. For some drugs, particularly where TDM is available, precision dosing tools could evolve to allow patients to provide feedback directly to their prescriber so that enhanced patient–prescriber communications could identify a need for dose adjustments.
Depicted in Figure 3 is a systematic process for the development, validation, and implementation of precision dosing tools. The process begins with an assessment of the gap between the phase III trial and market population, which may result in conducting a large (e.g., N > 1,000) PK/PD study designed to evaluate the drug characteristics in patient subgroups not enrolled in phase III trials. The total sample size and number of patients enrolled from each relevant special population need to be quantitatively justified. These data will aid in the development of a PK/PD model that can facilitate dosage regimen individualization. As suggested by others,2, 3 this tool may need to be validated and refined prior to implementation. We recommend that this assessment culminate in the generation of a report that can help to facilitate discussions with expert clinician groups. Ultimately, the model and associated recommendations would be published for widespread evaluation and use.
Figure 3.
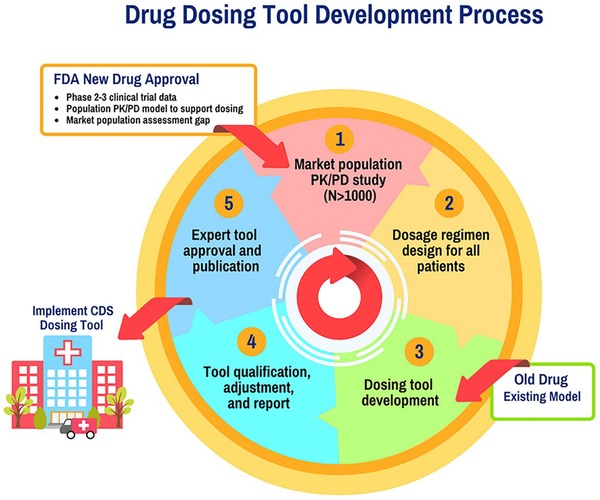
Drug dosing tool development process. Pharmacokinetic (PK) / pharmacodynamic (PD) studies in the market population can be performed, if needed. Using the information collected in these studies and/or prior information, optimal dosing regimens are designed for all patients. These dosing regimens are then implemented in clinical decision support (CDS) tools. Use of this dosing information and tool is then evaluated, and there is a process in place for continuous refinement and quality improvement of the dosing tool.
In the United States, both the FDA Center for Drug Evaluation and Research (CDER) and the Center for Devices and Radiologic Health (CDRH) may play a role in this process. Drug information and analyses performed during regulatory (CDER) approval decisions will be useful in providing a basis for recognizing which new drugs will benefit from precision dosing as well as a quantitative source for tool development. It may be that CDER scientists will wish to have access to precision dosing drug databases for subsequent labeling purposes. While CDRH can regulate medical software, there will be a need to clarify whether precision drug dosing software will require premarket approval. The precision dosing development process outlined in Figure 3, including qualification testing and a published report, will be useful for these discussions with regulators. Legal liability is uncertain for clinical decision support software and patient support software applications.75, 76 The commitments to verify software prediction accuracy and continuously improve quality may help limit liability.
ANTICIPATED IMPACT
The aforementioned proposed changes, supporting precision dosing for relevant drugs, should maximize medication efficacy and minimize medication toxicities. Precision dosing could also plausibly increase adherence as drug efficacy increases and toxicity decreases, resulting in decreased healthcare costs. Sponsors should benefit, in that their products will have an improved efficacy/safety profile. Regulatory agencies may also benefit because more patients will be better served and their internal analyses can be useful beyond the approval and labeling decisions. As recommended in this article and others,2, 3 prospective studies in real‐world patient populations, including randomized trials, should be conducted to determine the effect of the precision dosing tool on relevant patient outcomes and behaviors, such as adverse drug events and medication adherence. Further, studies also should evaluate the implementation and validation of the tool, its potential for dissemination, and its associated costs to the healthcare system. Prospective health economic analyses are needed in order to demonstrate the value of precision dosing tools. We believe that these analyses will be important as healthcare systems move toward new value‐based payment models.20
In the United States and other parts of the world, there has been an increase in the use of centrally stored electronic health records, which facilitate the implementation of precision dosing. Patient data are now available to inform precision dosing tools, and clinical decision support systems are being developed to allow prescribers to more precisely and safely use drugs. Knowledge gaps will emerge and can be quickly addressed as clinical implementation of these tools becomes routine. The system by which precision dosing is delivered should allow dosing regimens to be continuously reevaluated and improved as needed. Regulatory agencies and the sponsor can provide the necessary information for this postmarket system, while clinicians, academic scientists, and other research organizations should establish themselves as standard‐bearers for development, validation, and implementation of precision dosing tools and strategies. Ultimately, the development of these precision dosing strategies will decrease healthcare costs, improve the efficiency of drug development, and address a significant public health need.
Acknowledgment
We thank Dr. Alexander Tropsha for his insightful comments on our manuscript.
Conflict of Interest/Disclosure
D.G. receives support for research from the National Institute of Child Health and Human Development (K23HD083465), the nonprofit organization Thrasher Research Fund (www.thrasherresearch.org), and from industry (Cempra, Inc. and Jacobus Pharmaceutical Company, Inc.) for drug development in adults and children. S.C.B. has served as a consultant to Merck, Sharp and Dohme and to Luto (UK). She has received funding via her institution from Merck, Sharp and Dohme, Eli Lilly Company, AHRQ, and the National Institutes of Health for studies that use the electronic health record to improve prescribing and patient monitoring. Y.C. recieves support from the National Institute of General Medical Sciences (GM119661). K.L.R.B. receives support from the National Institute of General Medical Sciences through award numbers R01GM041935 and R35GM122576, and from industry (Otsuka Pharmaceutical Development and Commercialization, Inc., and Intercept Pharmaceuticals). K.L.R.B. is a coinventor of the sandwich‐cultured hepatocyte technology for quantification of biliary excretion (B‐CLEAR) and related technologies, which have been licensed exclusively to Qualyst Transporter Solutions, LLC. A.D.M.K. receives support for research from the National Institute of Allergy and Infectious Diseases (R01AI122319, R01AI111891, P30AI050410, R01AI116276, R01AI117739, U01AI103390, UM1AI126619), the National Center for Advancing Translational Sciences (UL1TR001111), the National Institute of Child Health and Human Development (K12HD001441), and from industry (Gilead, Merck) for clinical pharmacology and drug development. C.R.L. receives research support from the American Heart Association (16GRNT29300003). J.R.P. has served as paid consultant to Takeda, AbbVie, and Hengrui Pharmaceuticals over the past year. The remaining authors have no conflicts to disclose. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1. Henry, J. , Pylypchuk, Y. , Searcy, T. & Patel, V. Adoption of Electronic Health Record Systems among U.S. Non‐Federal Acute Care Hospitals: 2008–2015. May 2016. Accessed via: <https://dashboard.healthit.gov/evaluations/data-briefs/non-federal-acute-care-hospital-ehr-adoption-2008-2015.php>. Accessed on June 21, 2017.
- 2. Darwich, A.S. et al Why has model‐informed precision dosing not yet become common clinical reality? Lessons from the past and a roadmap for the future. Clin. Pharmacol. Ther. 101, 646–656 (2017). [DOI] [PubMed] [Google Scholar]
- 3. Neely, M. Scalpels not hammers: The way forward for precision drug prescription. Clin. Pharmacol. Ther. 101, 368–372 (2017). [DOI] [PubMed] [Google Scholar]
- 4. Collins, F.S. & Varmus, H. A new initiative on precision medicine. N. Engl. J. Med. 372, 793–795 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pacanowski, M. , Leptak, C. & Zineh, I. Next‐generation medicines: past regulatory experience and considerations for the future. Clin. Pharmacol. Ther. 95, 247–249 (2014). [DOI] [PubMed] [Google Scholar]
- 6. Goodsaid, F.M. et al Voluntary exploratory data submissions to the US FDA and the EMA: experience and impact. Nat. Rev. Drug Discov. 9, 435–445 (2010). [DOI] [PubMed] [Google Scholar]
- 7. Gobburu, J.V.S. & Lesko, L.J. Quantitative disease, drug, and trial models. Annu. Rev. Pharmacol. Toxicol. 49, 291–301 (2009). [DOI] [PubMed] [Google Scholar]
- 8. Zineh, I. & Woodcock, J. Clinical pharmacology and the catalysis of regulatory science: opportunities for the advancement of drug development and evaluation. Clin. Pharmacol. Ther. 93, 515–525 (2013). [DOI] [PubMed] [Google Scholar]
- 9. Lee, J.Y. et al Impact of pharmacometric analyses on new drug approval and labelling decisions: a review of 198 submissions between 2000 and 2008. Clin. Pharmacokinet. 50, 627–635 (2011). [DOI] [PubMed] [Google Scholar]
- 10. Genzyme Corporation . Cerdelga® package insert. August 2014. Accessed via: <http://www.cerdelga.com/pdf/cerdelga_prescribing_information.pdf>. Accessed on April 30, 2017.
- 11. Eichler, H.G. et al Bridging the efficacy–effectiveness gap: a regulator's perspective on addressing variability of drug response. Nat. Rev. Drug Discov. 10, 495–506 (2011). [DOI] [PubMed] [Google Scholar]
- 12. Huang, S.‐M. et al New era in drug interaction evaluation: US Food and Drug Administration update on CYP enzymes, transporters, and the guidance process. J. Clin. Pharmacol. 48, 662–670 (2008). [DOI] [PubMed] [Google Scholar]
- 13. Mulugeta, Y. et al Exposure matching for extrapolation of efficacy in pediatric drug development. J. Clin. Pharmacol. 56, 1326–1334 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huang, S.‐M. , Temple, R. , Xiao, S. , Zhang, L. & Lesko, L.J. When to conduct a renal impairment study during drug development: US Food and Drug Administration perspective. Clin. Pharmacol. Ther. 86, 475–479 (2009). [DOI] [PubMed] [Google Scholar]
- 15. Chang, Y. , Burckart, G.J. , Lesko, L.J. & Dowling, T.C. Evaluation of hepatic impairment dosing recommendations in FDA‐approved product labels. J. Clin. Pharmacol. 53, 962–966 (2013). [DOI] [PubMed] [Google Scholar]
- 16. Johnson, J.A. & Bootman, J.L. Drug‐related morbidity and mortality. A cost‐of‐illness model. Arch. Intern. Med. 155, 1949–1956 (1995). [PubMed] [Google Scholar]
- 17. Classen, D.C. , Pestotnik, S.L. , Evans, R.S. , Lloyd, J.F. & Burke, J.P. Adverse drug events in hospitalized patients. Excess length of stay, extra costs, and attributable mortality. JAMA 277, 301–306 (1997). [PubMed] [Google Scholar]
- 18. Lazarou, J. , Pomeranz, B.H. & Corey, P.N. Incidence of adverse drug reactions in hospitalized patients: a meta‐analysis of prospective studies. JAMA. 279, 1200–1205 (1998). [DOI] [PubMed] [Google Scholar]
- 19. IMS Institute for Healthcare Informatics . Avoidable costs in U.S. healthcare. June 2013. Accessed via: <http://www.imshealth.com/files/web/IMSH%20Institute/Reports/Avoidable_Costs_in%20_US_Healthcare/IHII_AvoidableCosts_2013.pdf>. Accessed on May 13, 2017.
- 20. Personalized Medicine Coalition . Paying for Personalized Medicine ‐ How Alternative Payment Models Could Help Or Hinder The Field. Accessed via: <http://www.personalizedmedicinecoalition.org/Userfiles/PMC-Corporate/file/paying_for_personalized_medicine.pdf>. Accessed on February 8, 2017.
- 21. Neely, M. et al Achieving target voriconazole concentrations more accurately in children and adolescents. Antimicrob. Agents Chemother. 59, 3090–3097 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Neely, M. et al Accurately achieving target busulfan exposure in children and adolescents with very limited sampling and the BestDose software. Ther. Drug Monit. 38, 332–342 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Krekels, E.H.J. et al Evidence‐based morphine dosing for postoperative neonates and infants. Clin. Pharmacokinet. 53, 553–563 (2014). [DOI] [PubMed] [Google Scholar]
- 24. Marsousi, N. et al Coadministration of ticagrelor and ritonavir: Toward prospective dose adjustment to maintain an optimal platelet inhibition using the PBPK approach. Clin. Pharmacol. Ther. 100, 295–304 (2016). [DOI] [PubMed] [Google Scholar]
- 25. Evans, W.E. , Relling, M.V. , Rodman, J.H. , Crom, W.R. , Boyett, J.M. & Pui, C.H. Conventional compared with individualized chemotherapy for childhood acute lymphoblastic leukemia. N. Engl. J. Med. 338, 499–505 (1998). [DOI] [PubMed] [Google Scholar]
- 26. Leroux, S. et al Clinical utility and safety of a model‐based patient‐tailored dose regimen of vancomycin in neonates. Antimicrob. Agents Chemother. 60, 2039–2042 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mccune, J.S. et al Personalized dosing of cyclophosphamide in the total body irradiation – cyclophosphamide conditioning regimen: a phase II trial in patients with hematologic malignancy. Clin. Pharmacol. Ther. 85, 615–622 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sacks, L.V. , Shamsuddin, H.H. , Yasinskaya, Y.I. , Bouri, K. , Lanthier, M.L. & Sherman, R.E. Scientific and regulatory reasons for delay and denial of FDA approval of initial applications for new drugs, 2000–2012. JAMA 311, 378–384 (2014). [DOI] [PubMed] [Google Scholar]
- 29. Peck, C.C. et al Opportunities for integration of pharmacokinetics, pharmacodynamics, and toxicokinetics in rational drug development. Clin. Pharmacol. Ther. 51, 465–473 (1992). [DOI] [PubMed] [Google Scholar]
- 30. HiMSS . Electronic Health Records: A Global Perspective. Part I. August 2010. Accessed via: <http://www.himss.org/global-ehr>. Accessed on February 4, 2017.
- 31. Blumenthal, D. Launching HITECH. N. Engl. J. Med. 362, 382–385 (2010). [DOI] [PubMed] [Google Scholar]
- 32. Gold, M. & McLaughlin, C. Assessing HITECH implementation and lessons: 5 years later. Milbank Q. 94, 654–687 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Centers for Disease Control and Prevention . Use and Characteristics of Electronic Health Record Systems Among Office‐Based Physician Practices: United States, 2001–2013. Page last updated January 17, 2014. Accessed via: <https://www.cdc.gov/nchs/data/databriefs/db143.htm#x2013;2013%3C/a>. Accessed on February 8, 2017.
- 34. Wojciechowski, J. , Upton, R.N. , Mould, D.R. , Wiese, M.D. & Foster, D.J.R. Infliximab maintenance dosing in inflammatory bowel disease: an example for in silico assessment of adaptive dosing strategies. AAPS J. 19, 1136–1147 (2017). [DOI] [PubMed] [Google Scholar]
- 35. Hope, W.W. et al Software for dosage individualization of voriconazole for immunocompromised patients. Antimicrob. Agents Chemother. 57, 1888–1894 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hennig, S. , Holthouse, F. & Staatz, C.E. Comparing dosage adjustment methods for once‐daily tobramycin in paediatric and adolescent patients with cystic fibrosis. Clin. Pharmacokinet. 54, 409–421 (2015). [DOI] [PubMed] [Google Scholar]
- 37. Abdel‐Rahman, S.M. et al Design and testing of an EHR‐integrated, busulfan pharmacokinetic decision support tool for the point‐of‐care clinician. Front. Pharmacol. 7, 1–12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Barrett, J.S. Pediatric models in motion: requirements for model‐based decision support at the bedside. Br. J. Clin. Pharmacol. 79, 85–96 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. McCune, J.S. et al Busulfan in infant to adult hematopoietic cell transplant recipients: a population pharmacokinetic model for initial and Bayesian dose personalization. Clin. Cancer Res. 20, 754–763 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. HealthIT.gov . How to Attain Meaningful Use. January 2013. Accessed via: <https://www.healthit.gov/providers-professionals/how-attain-meaningful-use>. Accessed on January 7, 2017.
- 41. Touw, D.J. , Neef, C. , Thomson, A.H. & Vinks, A.A. ; Cost‐Effectiveness of Therapeutic Drug Monitoring Committee of the International Association for Therapeutic Drug Monitoring and Clinical Toxicology. Cost‐effectiveness of therapeutic drug monitoring: a systematic review. Ther. Drug Monit. 27, 10–17 (2005). [DOI] [PubMed] [Google Scholar]
- 42. Tamargo, J. , Le Heuzey, J.‐Y. & Mabo, P. Narrow therapeutic index drugs: a clinical pharmacological consideration to flecainide. Eur. J. Clin. Pharmacol. 71, 549–567 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kinch, M.S. , Merkel, J. & Umlauf, S. Trends in pharmaceutical targeting of clinical indications: 1930–2013. Drug Discov. Today 19, 1682–1685 (2014). [DOI] [PubMed] [Google Scholar]
- 44. Drusano, G.L. , Ambrose, P.G. , Bhavnani, S.M. , Bertino, J.S. , Nafziger, A.N. & Louie, A. Back to the future: using aminoglycosides again and how to dose them optimally. Clin. Infect. Dis. 45, 753–760 (2007). [DOI] [PubMed] [Google Scholar]
- 45. Gao, B. , Yeap, S. , Clements, A. , Balakrishnar, B. , Wong, M. & Gurney, H. Evidence for therapeutic drug monitoring of targeted anticancer therapies. J. Clin. Oncol. 30, 4017–4025 (2012). [DOI] [PubMed] [Google Scholar]
- 46. Mould, D.R. Why therapeutic drug monitoring is needed for monoclonal antibodies and how do we implement this? Clin. Pharmacol. Ther. 99, 351–354 (2016). [DOI] [PubMed] [Google Scholar]
- 47. Kearns, G.L. , Abdel-Rahman, S.M. , Alander, S.W. , Blowey, D.L. , Leeder, J.S. & Kauffman, R.E. Developmental pharmacology — drug disposition, action, and therapy in infants and children. N. Engl. J. Med. 349, 1157–1167 (2003). [DOI] [PubMed] [Google Scholar]
- 48. Schwartz, J.B. The current state of knowledge on age, sex, and their interactions on clinical pharmacology. Clin. Pharmacol. Ther. 82, 87–96 (2007). [DOI] [PubMed] [Google Scholar]
- 49. Collins, D.C. , Sundar, R. , Lim, J.S.J. & Yap, T.A. Towards precision medicine in the clinic: from biomarker discovery to novel therapeutics. Trends Pharmacol. Sci. 38, 25–40 (2017). [DOI] [PubMed] [Google Scholar]
- 50. Blumenthal, G.M. , Mansfield, E. & Pazdur, R. Next‐generation sequencing in oncology in the era of precision medicine. JAMA Oncol. 2, 13–4 (2016). [DOI] [PubMed] [Google Scholar]
- 51. Preissner, S.C. , Hoffmann, M.F. , Preissner, R. , Dunkel, M. , Gewiess, A. & Preissner, S. Polymorphic cytochrome P450 enzymes (CYPs) and their role in personalized therapy. PLoS One 8, e82562 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Haider, S. & Pal, R. Integrated analysis of transcriptomic and proteomic data. Curr. Genomics 14, 91–110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chapal, N. , Molina, L. , Molina, F. , Laplanche, M. , Pau, B. & Petit, P. Pharmacoproteomic approach to the study of drug mode of action, toxicity, and resistance: Applications in diabetes and cancer. Fundam. Clin. Pharmacol. 18, 413–422 (2004). [DOI] [PubMed] [Google Scholar]
- 54. Witzmann, F.A. & Grant, R.A. Pharmacoproteomics in drug development. Pharmacogenomics J. 3, 69–76 (2003). [DOI] [PubMed] [Google Scholar]
- 55. Kaddurah‐Daouk, R. & Weinshilboum, R.M. ; Pharmacometabolomics Research Network. Pharmacometabolomics: implications for clinical pharmacology and systems pharmacology. Clin. Pharmacol. Ther. 95, 154–167 (2014). [DOI] [PubMed] [Google Scholar]
- 56. Beumer, J.H. Without therapeutic drug monitoring, there is no personalized cancer care. Clin. Pharmacol. Ther. 93, 228–230 (2013). [DOI] [PubMed] [Google Scholar]
- 57. Sheiner, L.B. & Ludden, T.M. Population pharmacokinetics/dynamics. Annu. Rev. Pharmacol. Toxicol. 32, 185–209 (1992). [DOI] [PubMed] [Google Scholar]
- 58. Sheiner, L.B. & Steimer, J.L. Pharmacokinetic/pharmacodynamic modeling in drug development. Annu. Rev. Pharmacol. 40, 67–95 (2000). [DOI] [PubMed] [Google Scholar]
- 59. Jones, H.M. et al Physiologically based pharmacokinetic modeling in drug discovery and development: a pharmaceutical industry perspective. Clin. Pharmacol. Ther. 97, 247–262 (2015). [DOI] [PubMed] [Google Scholar]
- 60. Sheiner, L.B. & Beal, S.L. Bayesian individualization of pharmacokinetics: simple implementation and comparison with non‐Bayesian methods. J. Pharm. Sci. 71, 1344–1348 (1982). [DOI] [PubMed] [Google Scholar]
- 61. Kiang, T.K.L. , Sherwin, C.M.T. , Spigarelli, M.G. & Ensom, M.H.H. Fundamentals of population pharmacokinetic modelling: modelling and software. Clin. Pharmacokinet. 51, 515–525 (2012). [DOI] [PubMed] [Google Scholar]
- 62. Jones, H. & Rowland‐Yeo, K. Basic concepts in physiologically based pharmacokinetic modeling in drug discovery and development. CPT Pharmacometrics Syst. Pharmacol. 2, e63 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jönsson, S. & Karlsson, M.O. A rational approach for selection of optimal covariate‐based dosing strategies. Clin. Pharmacol. Ther. 73, 7–19 (2003). [DOI] [PubMed] [Google Scholar]
- 64. Neely, M. & Jelliffe, R. Practical , individualized dosing: 21st century therapeutics and the clinical pharmacometrician. J. Clin. Pharmacol. 50, 842–847 (2010). [DOI] [PubMed] [Google Scholar]
- 65. Fukudo, M. et al Prospective evaluation of the bayesian method for individualizing tacrolimus dose early after living‐donor liver transplantation. J. Clin. Pharmacol. 49, 789–797 (2009). [DOI] [PubMed] [Google Scholar]
- 66. Mould, D.R. , D'Haens, G. & Upton, R.N. Clinical decision support tools: the evolution of a revolution. Clin. Pharmacol. Ther. 99, 405–418 (2016). [DOI] [PubMed] [Google Scholar]
- 67. Mould, D.R. & Dubinsky, M.C. Dashboard systems: Pharmacokinetic/pharmacodynamic mediated dose optimization for monoclonal antibodies. J. Clin. Pharmacol. 55 Suppl 3, S51–S59 (2015). [DOI] [PubMed] [Google Scholar]
- 68. Barrett, J.S. , Della Casa Alberighi, O. , Läer, S. & Meibohm, B. Physiologically based pharmacokinetic (PBPK) modeling in children. Clin. Pharmacol. Ther. 92, 40–49 (2012). [DOI] [PubMed] [Google Scholar]
- 69. Schlender, J.‐F. et al Development of a whole‐body physiologically based pharmacokinetic approach to assess the pharmacokinetics of drugs in elderly individuals. Clin. Pharmacokinet. 55, 1573–1589 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Jadhav, P.R. et al A proposal for scientific framework enabling specific population drug dosing recommendations. J. Clin. Pharmacol. 55, 1073–1078 (2015). [DOI] [PubMed] [Google Scholar]
- 71. Younis, I.R. et al Utility of model‐based approaches for informing dosing recommendations in specific populations: report from the public AAPS Workshop. J. Clin. Pharmacol. 57, 105–109 (2017). [DOI] [PubMed] [Google Scholar]
- 72. PhRMA Foundation . Principles for Responsible Clinical Trial Data Sharing. July 2013. Accessed via: <http://www.phrma.org/sites/default/files/pdf/PhRMAPrinciplesForResponsibleClinicalTrialDataSharing.pdf>. Accessed on April 29, 2017.
- 73. Gonzalez, D. et al Use of opportunistic clinical data and a population pharmacokinetic model to support dosing of clindamycin for premature infants to adolescents. Clin. Pharmacol. Ther. 96, 429–437 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Behrman, R.E. , Benner, J.S. , Brown, J.S. , McClellan, M. , Woodcock, J. & Platt, R. Developing the Sentinel System–a national resource for evidence development. N. Engl. J. Med. 364, 498–499 (2011). [DOI] [PubMed] [Google Scholar]
- 75. Kesselheim, A.S. , Cresswell, K. , Phansalkar, S. , Bates, D.W. & Sheikh, A. Clinical decision support systems could be modified to reduce ‘alert fatigue’ while still minimizing the risk of litigation. Health Aff (Millwood). 30, 2310–2317 (2011). [DOI] [PubMed] [Google Scholar]
- 76. Yang, Y.T. & Silverman, R.D. Mobile health applications: the patchwork of legal and liability issues suggests strategies to improve oversight. Health Aff (Millwood). 33, 222–227 (2014). [DOI] [PubMed] [Google Scholar]
- 77. Bearden, D.T. & Rodvold, K.A. Dosage adjustments for antibacterials in obese patients: applying clinical pharmacokinetics. Clin. Pharmacokinet. 38, 415–426 (2000). [DOI] [PubMed] [Google Scholar]
- 78. Gilbert, B. , Robbins, P. , Livornese, L.L. Jr . Use of antibacterial agents in renal failure. Infect. Dis. Clin. North Am. 23, 899–924 (2009). [DOI] [PubMed] [Google Scholar]
- 79. Germovsek, E. et al Development and evaluation of a gentamicin pharmacokinetic model that facilitates opportunistic gentamicin therapeutic drug monitoring in neonates and infants. Antimicrob. Agents Chemother. 60, 4869–4877 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Smith, T.W. & Haber, E. Digoxin intoxication: the relationship of clinical presentation to serum digoxin concentration. J. Clin. Invest. 49, 2377–2386 (1970). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Tesfaye, H. et al The importance of therapeutic drug monitoring (TDM) for parenteral busulfan dosing in conditioning regimen for hematopoietic stem cell transplantation (HSCT) in children. Ann. Transplant. 19, 214–224 (2014). [DOI] [PubMed] [Google Scholar]
- 82. Fahr, A. Cyclosporin clinical pharmacokinetics. Clin. Pharmacokinet. 24, 472–495 (1993). [DOI] [PubMed] [Google Scholar]
- 83. Staatz, C.E. & Tett, S.E. Clinical pharmacokinetics and pharmacodynamics of tacrolimus in solid organ transplantation. Clin. Pharmacokinet. 43, 623–653 (2004). [DOI] [PubMed] [Google Scholar]
- 84. Johnson, J.A. et al Clinical pharmacogenetics implementation consortium guidelines for CYP2C9 and VKORC1 genotypes and warfarin dosing. Clin. Pharmacol. Ther. 90, 625–629 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Masimirembwa, C. , Dandara, C. & Leutscher, P.D.C. Rolling out efavirenz for HIV precision medicine in Africa: Are we ready for pharmacovigilance and tackling neuropsychiatric adverse effects? OMICS. 20, 575–580 (2016). [DOI] [PubMed] [Google Scholar]
- 86. Schackman, B.R. , Haas, D.W. , Park, S.S. , Li, X.C. & Freedberg, K.A. Cost‐effectiveness of CYP2B6 genotyping to optimize efavirenz dosing in HIV clinical practice. Pharmacogenomics 16, 2007–2018 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Gatanaga, H. et al Successful efavirenz dose reduction in HIV type 1‐infected individuals with cytochrome P450 2B6 *6 and *26. Clin. Infect. Dis. 45, 1230–1237 (2007). [DOI] [PubMed] [Google Scholar]
- 88. Shipkova, M. et al Therapeutic drug monitoring of everolimus: a consensus report. Ther. Drug Monit. 38, 143–169 (2016). [DOI] [PubMed] [Google Scholar]
- 89. Stenton, S.B. , Partovi, N. & Ensom, M.H.H. Sirolimus: the evidence for clinical pharmacokinetic monitoring. Clin. Pharmacokinet. 44, 769–786 (2005). [DOI] [PubMed] [Google Scholar]
- 90. Onufrak, N.J. et al Critical need for clarity in polymyxin B dosing. Antimicrob. Agents Chemother. 61, 1–2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Baldin, B. et al Abstract 11598: Is therapeutic drug monitoring of direct anticoagulants really unnecessary? Circulation 132, A11598 (2015). Accessed via: <http://circ.ahajournals.org/content/132/Suppl_3/A11598>. Accessed on January 16, 2017. [Google Scholar]
- 92. Hellwig, T. & Gulseth, M. Pharmacokinetic and pharmacodynamic drug interactions with new oral anticoagulants: what do they mean for patients with atrial fibrillation? Ann. Pharmacother. 47, 1478–1487 (2013). [DOI] [PubMed] [Google Scholar]
- 93. Baglin, T. , Hillarp, A. , Tripodi, A. , Elalamy, I. , Buller, H. & Ageno, W. Measuring oral direct inhibitors of thrombin and factor Xa: A recommendation from the Subcommittee on Control of Anticoagulation of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. J. Thromb. Haemost. 11, 756–760 (2013). [DOI] [PubMed] [Google Scholar]
- 94. Parasrampuria, D.A. & Truitt, K.E. Pharmacokinetics and pharmacodynamics of edoxaban, a non‐vitamin K antagonist oral anticoagulant that inhibits clotting factor Xa. Clin. Pharmacokinet. 55, 641–655 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Tamura, K. et al Phase I study of palbociclib, a cyclin‐dependent kinase 4/6 inhibitor, in Japanese patients. Cancer Sci. 107, 755–763 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Yi, Y. , Loi, C.‐M. , Hoffman, J. & Wang, D. Physiologically based pharmacokinetic modeling of palbociclib. J. Clin. Pharmacol. 57, 173–184 (2016). [DOI] [PubMed] [Google Scholar]
- 97. Freise, K.J. et al Impact of venetoclax exposure on clinical efficacy and safety in patients with relapsed or refractory chronic lymphocytic leukemia. Clin. Pharmacokinet. 56, 515–523 (2017). [DOI] [PubMed] [Google Scholar]
- 98. Freise, K.J. , Shebley, M. & Salem, A.H. Quantitative prediction of the effect of CYP3A inhibitors and inducers on venetoclax pharmacokinetics using a physiologically based pharmacokinetic model. J. Clin. Pharmacol. 57, 796–804 (2017). [DOI] [PubMed] [Google Scholar]
- 99. Jones, A.K. , Freise, K.J. , Agarwal, S.K. , Humerickhouse, R.A. , Wong, S.L. & Salem, A.H. Clinical predictors of venetoclax pharmacokinetics in chronic leukemia and non‐Hodgkin's lymphoma patients: a pooled population pharmacokinetic analysis. AAPS J. 18, 1192–1202 (2016). [DOI] [PubMed] [Google Scholar]
- 100. Salem, A.H. , Agarwal, S.K. , Dunbar, M. , Enschede, S.L. , Humerickhouse, R.A. & Wong, S.L. Pharmacokinetics of venetoclax, a novel BCL‐2 inhibitor, in patients with relapsed or refractory chronic lymphocytic leukemia or non‐Hodgkin's lymphoma. J. Clin. Pharmacol. 57, 484–492 (2017). [DOI] [PubMed] [Google Scholar]