

Abstract
The Hippo pathway is deregulated in various cancers, and the discovery of molecules that modulate this pathway may open new therapeutic avenues in oncology. TEA/ATTS domain (TEAD) transcription factors are the most distal elements of the Hippo pathway and their transcriptional activity is regulated by the Yes‐associated protein (YAP). Amongst the various possibilities for targeting this pathway, inhibition of the YAP:TEAD interaction is an attractive strategy. It has been shown recently that TEAD proteins are covalently linked via a conserved cysteine to a fatty acid molecule (palmitate) that binds to a deep hydrophobic cavity present in these proteins. This acylation of TEAD seems to be required for efficient binding to YAP, and understanding how it modulates the YAP:TEAD interaction may provide useful information on the regulation of TEAD function. In this report we have studied the effect of TEAD4 acylation on its interaction with YAP and the other co‐activator transcriptional co‐activator with PDZ‐binding motif (TAZ). We show in our biochemical and cellular assays that YAP and TAZ bind in a similar manner to acylated and non‐acylated TEAD4. This indicates that TEAD4 acylation is not a prerequisite for its interaction with YAP or TAZ. However, we observed that TEAD4 acylation significantly enhances its stability, suggesting that it may help this transcription factor to acquire and/or maintain its active conformation.
Keywords: TEAD, YAP, TAZ, Hippo pathway, Transcription factors, Protein‐protein interactions
Short abstract
PDB Code(s): 5OAQ
Introduction
TEAD (TEA/ATTS domain) transcription factors are the most distal elements of the Hippo pathway, which is essential in the control of organ size.1, 2 These proteins are unable to stimulate gene transcription unless they interact with co‐activator proteins such as YAP (Yes‐associated protein) and TAZ (transcriptional co‐activator with PDZ‐binding motif).3 The deregulation of the Hippo pathway in various cancers has recently attracted a lot of attention, and the inhibition of the YAP:TEAD interaction is foreseen as a possible new strategy for oncology.4, 5, 6, 7
The published structures of the YAP:TEAD complex8, 9 have yielded a lot of insightful information on how these two proteins interact. However, a more complex picture of TEAD structure has recently emerged. TEAD contains a deep hydrophobic cavity, called the “central pocket”, which can accommodate low‐molecular weight compounds.10 For example, flufenamic acid can bind to this pocket, inhibiting the YAP‐dependent transcription activity of TEAD.10 Furthermore, two additional publications have shown that this pocket can also be occupied by a palmitate moiety, which is covalently linked to a cysteine present at the surface of TEAD.11, 12 This cysteine is conserved in the four human TEAD paralogs, and these proteins are acylated in mammalian cells.11, 12 A reanalysis of previously published structures shows that electron densities corresponding to a fatty acid‐like molecule are also present in the electron density maps used to solve these earlier structures.12 This suggests that acylation is a general feature of TEAD and that it might be important for its function. In vitro, TEAD acylation does not require the presence of palmitoyl acyltranferases, and acylated TEAD can be readily obtained by incubation with micromolar amounts of palmitoyl‐CoA.11 Acylation was observed with the full‐length protein and the isolated YAP‐binding domain11 suggesting that in vitro the DNA‐binding domain present at the N‐terminus of the protein is dispensable for this modification to occur. The presence of a bound fatty acid enhances TEAD stability, and it does not seem to play a role in its localization and/or association to membranes.12 TEAD palmitoylation is needed for muscle differentiation in vitro, and experiments in Drosophila have shown that it is required for the physiological function of Scalloped (the homolog of human TEAD).11
Interestingly, it has also been shown that palmitoylation regulates the interaction between TEAD and its co‐activators YAP and TAZ.11 The association between these proteins and TEAD is reduced when the latter is mutated at cysteine residues to prevent its acylation. These results have been rationalized by the fact that the bound palmitate moiety rigidifies TEAD at one binding interface (Ω‐loop binding pocket) specifically used by YAP and TAZ for binding to TEAD.11 Since the inhibition of the YAP:TEAD interaction is of importance in drug discovery, we decided to study the influence of TEAD acylation on its interaction with YAP and TAZ in more detail. Under our experimental conditions, we have not observed a significant effect of TEAD4 acylation on the interaction between this protein and YAP/TAZ in different biochemical and cellular assays. However, in agreement with Noland et al.,12 we have found that the acylation of TEAD has a major impact on the stability of this protein.
Results
Characterization of recombinant TEAD4 proteins
Under our experimental conditions (see “Material and Methods” section, scheme A), the purification of wt hTEAD4217–434 expressed in Escherichia coli BL21(DE3) leads to a mixture of 3 TEAD4 species as detected by liquid chromatography—mass‐spectrometry (LC‐MS) analysis: a non‐acylated form of wt hTEAD4217–434 (MW = 29031) and 2 acylated forms corresponding to myristoylated hTEAD4217–434(MW = 29242, Myr‐TEAD4) and palmitoylated hTEAD4217–434 (MW = 29270, Palm‐TEAD4) [Fig. 1(A)]. Non‐acylated TEAD4 represented generally between 30 and 40% of the total amount of purified protein. In the remaining fraction, the relative proportion of Myr‐TEAD4 and Palm‐TEAD4 was variable, but we usually obtained more Myr‐TEAD4 than Palm‐TEAD4. Previous publications have only described recombinant Palm‐TEAD.11, 12 The difference between these studies and ours might be due to the culture media and/or growth conditions used in the different laboratories. Note that TEAD myristoylation has also been observed in mammalian cells.12
Figure 1.
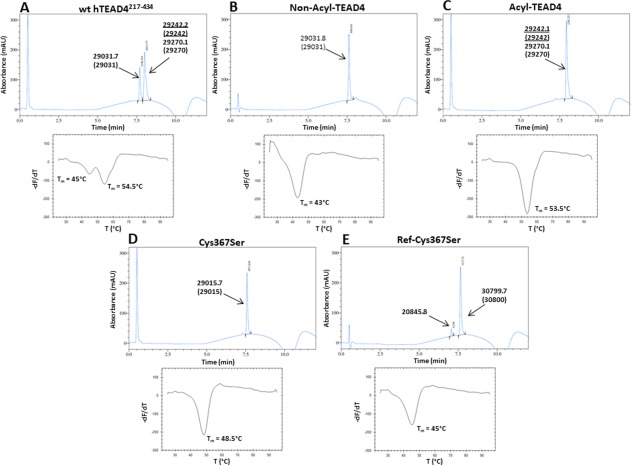
Analytics of the different TEAD4 proteins. Upper panels, representative chromatograms obtained by RP‐HPLC. The molecular weights measured in mass spectrometry of the protein species are indicated, and the theoretical molecular weights are given in brackets. Two different acylated forms were detected for wt hTEAD4217–434 (A) and Acyl‐TEAD4 (C). Underlined: Myr‐TEAD4 (major species), normal fonts: Palm‐TEAD4 (minor species). (E) The peak with a mass of 20845.8 is a minor impurity. Ref‐Cys367Ser has a larger mass than the other TEAD4 proteins, because its His6‐tag has not been cleaved off. Lower panels, representative thermograms obtained in a FTDA in the presence of 1–2 µM protein. The melting temperatures (T m) were obtained by plotting the first derivative of the fluorescence emission (F) as a function of the temperature (–dF/dT). The curve minimum corresponds to T m. The indicated T m have been measured for the experiments depicted on the figure. These T m are slightly different from the T m presented on Table II because these later are the average values obtained from several independent measurements.
The thermal stability of wt hTEAD4217–434 was measured in a fluorescence‐based thermal denaturation assay (FTDA). Two thermal transitions were observed, indicating the presence of at least 2 protein forms with distinct melting temperatures (T m) [Fig. 1(A)]. The T m values measured were 45.6 ± 0.4°C and 54.9 ± 0.2°C, revealing a substantial difference (∼9°C) in thermal stability between these TEAD4 forms. By analogy to the data published by Noland et al.,12 it can be hypothesized that the low T m form corresponds to non‐acylated TEAD and the high T m form to acylated TEAD. The high Tm form should thus be a mixture of Myr‐TEAD4 and Palm‐TEAD4 that cannot be distinguished in the FTDA.
To separate non‐acylated and acylated TEAD4, we modified our purification protocol. An additional ion exchange chromatography step was added to obtain non‐acylated TEAD4 (see “Material and Methods” section, scheme B). The protein preparations produced in such a way contain >95% non‐acylated wt hTEAD4217–434 [Fig. 1(B), hereafter referred to as Non‐Acyl‐TEAD4]. To produce acylated TEAD4, we incubated partially purified wt hTEAD4217–434 with myristoyl Coenzyme A during the purification process (see “Material and Methods” section, scheme C). The protein preparations obtained under such conditions contain more than 95% acylated TEAD4 [Fig. 1(C), hereafter referred to as Acyl‐TEAD4]. Note that Acyl‐TEAD4 is a mixture of Myr‐TEAD4 (major form) and Palm‐TEAD4 (minor form). In our previous reports, we used a heat treatment to produce acylated hTEAD4217–434.13, 14, 15, 16 This heat treatment allowed us to selectively unfold the low T m form of wt hTEAD4217–434 that was next eliminated by centrifugation. Heat‐treated hTEAD4217–434, which is completely acylated, behaves in a similar manner to Acyl‐TEAD4 obtained here (data not shown).
The T m values for Non‐Acyl‐TEAD4 and Acyl‐TEAD4 were 43.7 ± 0.3°C and 54.1 ± 0.2°C, respectively [Fig. 1(B,C)]. Acylated hTEAD4217–434 is therefore significantly (∼10°C) more stable than its non‐acylated counterpart. These results, which are similar to the ones obtained by Noland et al.12 for TEAD2, show that the acylation has a critical impact on TEAD stability in vitro. The T m values for Non‐Acyl‐TEAD4 and Acyl‐TEAD4 are close to those for the low and high Tm forms of wt hTEAD4217–434, respectively (Fig. 1). This validates the hypothesis suggesting that the low and high T m forms of wt hTEAD4217–434 correspond to non‐acylated and acylated TEAD4, respectively.
According to the alignment of the primary sequence of hTEAD1–4 and what we currently know about TEAD acylation,11, 12 Cys367 should be the acylation site on the TEAD4 protein. To confirm this, we co‐crystallized acylated wt hTEAD4217–434 with a peptide mimicking the region 60–100 of human YAP (YAP60–100). Our high‐resolution structure (1.95 Å, Table 1) is very similar overall to the previous ones obtained with YAP co‐crystallized with hTEAD1194–411 9 and mTEAD4210–427 17 (Supporting Information Fig. S1). However, we also unambiguously observed electron densities corresponding to the presence of a myristate moiety bound to the “central pocket” of TEAD4. The fatty acid is covalently linked to hTEAD4 Cys367, and the acylation site is close to hTEAD4 Lys344, which may enhance the reactivity of Cys367 by lowering its pKa [Fig. 2(A)]. This is in agreement with the structures recently obtained with TEAD2 and TEAD3.11, 12 A superimposition of the TEAD4 structure with those of TEAD2 and TEAD3 (pdb 5EMV and 5EMW12) reveals that the myristate (in TEAD4) and the palmitate (in TEAD2/3) moieties occupy the same pocket and bind to TEAD in a similar manner [Fig. 2(B)].
Table 1.
X‐Ray Data Collection and Refinement Statistics of the Structure of the hYAP60–100:hTEAD4217–434 Complex
| X‐ray data collection | |
| Space group | P41212 |
| Cell parameters (Å) | a = b = 59.0, c = 159.6 |
| Complexes per ASUa | 1 |
| Resolution (Å) | 1.95 (2.00–1.95) |
| Unique reflections | 21390 |
| Rmerge (%) | 0.072 (0.711) |
| I/σ(I) | 28.2 (4.3) |
| Completeness (%) | 99.8 (100.0) |
| Redundancy | 12.8 (12.8) |
| Refinement statistics | |
| Resolution (Å) | 1.95 (2.00–1.95) |
| Number of reflections used | 20319 (1446) |
| Fraction of test set for calculating R free (%) | 5.0 |
| No. of reflections in the test set | 1070 (76) |
| Rwork/Rfree | 0.229/0.252 (0.244/0.274) |
| r.m.s.d. bond lengths (Å)/bond angle (°) | 0.010/1.255 |
| Number of atoms (total/waters) | 2172/115 |
Values in brackets indicate the specific values in the last resolution shell.
ASU, asymmetric unit.
Figure 2.
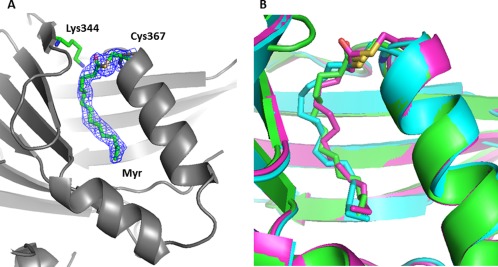
X‐ray structure of myristoylated human hTEAD4217–434. The coordinates have been deposited in the PDB databank (PDB access code = 5OAQ). For clarity, the bound YAP60–100 peptide is not displayed (see Supporting Information Fig. S1). (A) Acylation site. TEAD4 is represented by gray ribbons. hTEAD4 Cys367, Lys344, and the bound myristate moiety are indicated. The electron density (2Fo‐Fc map contoured at 1σ) for Cys367 and the bound myristate are shown (blue mesh). (B) The structure of hTEAD4217–434 (green), hTEAD2217–447 (pdb 5EMV, cyan) and hTEAD3216–435 (pdb 5EMW, purple) have been superimposed. The modified cysteines (hTEAD4 Cys367, hTEAD2 Cys380, hTEAD3 Cys371) and the myristate (TEAD4) or the palmitate (TEAD2/3) moieties are represented. The figures were generated with PyMol (Schrödinger LLC).
On the basis of these structural data, hTEAD4 Cys367 was mutated to serine. The corresponding protein (hereafter referred to as Cys367Ser) was purified, and LC‐MS analysis shows that it is not covalently modified [Fig. 1(D)], confirming the critical role of Cys367 in hTEAD4217–434 acylation. This also indicates that – under our experimental conditions ‐ in the absence of Cys367 there is little (if any) additional acylation on hTEAD4217–434. Cys367 is therefore the primary acylation site of hTEAD4217–434. The T m of Cys367Ser is 48.7 ± 0.2°C [Fig. 1(D)]. This value is significantly lower (5°C) than that for Acyl‐TEAD4, confirming that non‐acylated TEAD4 has reduced thermal stability. However, this T m is also higher (5°C) than that for Non‐Acyl‐TEAD4.
Interaction between the different TEAD4 proteins and hYAP51–99/hTAZ14–56
We used synthetic peptides mimicking the TEAD‐binding region of hYAP (region 51–99, hYAP51–99) and hTAZ (region 14–56, hTAZ14–56)13 to evaluate the ability of the different TEAD4 proteins to interact with YAP and TAZ.
The interaction between these peptides and the TEAD4 proteins was first evaluated in FTDA. The thermal stability of the different TEAD4 proteins is significantly changed when there is an excess of peptide and positive thermal shifts are measured (ΔT m = – > 0; Table 2). This shows that the different TEAD4 proteins bind to hYAP51–99 and hTAZ14–56.
Table 2.
Interaction Between the Different TEAD4 Proteins and the Synthetic Peptides hYAP51–99 and hTAZ14–56
| Peptides | T m (°C) | ΔT m (°C) | K d (nM) |
|---|---|---|---|
| Non‐Acyl‐TEAD4 | |||
| hYAP51–99 | 51.1 ± 0.2 | 7.4 ± 0.3 | 58 ± 3 |
| hTAZ14–56 | 51.8 ± 0.1 | 8.1 ± 0.3 | 16 ± 1 |
| Acyl‐TEAD4 | |||
| hYAP51–99 | 59.1 ± 0.2 | 5.0 ± 0.3 | 21 ± 1 |
| hTAZ14–56 | 59.3 ± 0.2 | 5.2 ± 0.3 | 5.3 ± 0.3 |
| Cys367Ser | |||
| hYAP51–99 | 54.5 ± 0.0 | 5.8 ± 0.2 | 58 ± 4 |
| hTAZ14–56 | 55.5 ± 0.0 | 6.8 ± 0.2 | 14 ± 1 |
| Ref‐Cys367Ser | |||
| hYAP51–99 | 52.7 ± 0.2 | 7.2 ± 0.2 | 78 ± 5 |
| hTAZ14–56 | 53.5 ±0.0 | 8.1 ± 0.2 | 18 ± 1 |
The melting temperature (T m) of the TEAD4 proteins (1–2 µM) in the presence of the peptides (20 µM) was measured in a FTDA. The effect of the peptides on the thermal stability of the proteins was calculated as follows: ΔT m = – . values are given in Table 1. Average and SEs of n ≥ 3 independent experiments are indicated. SEΔ T m = ( + )1/2. The dissociation constant (K d) was measured at equilibrium by SPR at 10°C with the different proteins immobilized on sensor chips. Average and SE of n ≥ 3 independent experiments are indicated.
To obtain more quantitative data, the interaction between these two peptides and the TEAD4 proteins was measured by surface plasmon resonance (SPR). The biotinylated N‐Avitagged TEAD4 proteins were immobilized on sensor chips coated with streptavidin, and the dissociation constant (K d) of the peptides was measured at equilibrium. When the SPR experiments were conducted at 25°C, we observed a clear binding of YAP/TAZ to Acyl‐TEAD4, Non‐Acyl‐TEAD4 and Cys367Ser. However, we also noticed a significant and progressive decrease of the measured signal over the course of the experiments with Non‐Acyl‐TEAD4 and Cys367Ser, while only a small signal loss was detected with Acyl‐TEAD4 (Supporting Information Fig. S2). In line with the thermal shift data, this shows that the non‐acylated TEAD4 forms are less stable than their acylated counterpart. This lower stability of the non‐acylated proteins prevented us from making accurate K d determinations at 25°C. The experiments were therefore repeated at 10°C. At this lower temperature, the proteins were more stable and the loss of signal over the course of an experiment was minimized (Supporting Information Fig. S2). Under such conditions, the peptides bind to the TEAD4 proteins in a dose‐dependent manner following a 1:1 binding mechanism, and the maximum signal (R max) measured was between 70 and 100% of the theoretical R max ( , see definition in “Material and Methods” section). The K d measured between hYAP51–99/hTAZ14–56 and Non‐Acyl‐TEAD4 and Cys367Ser were less than three times greater than the K d obtained with Acyl‐TEAD4 (Table 2). This indicates that, in SPR at 10°C, hYAP51–99/hTAZ14–56 have similar affinities for acylated and non‐acylated TEAD4217–434.
Chasing potential non‐covalent binders
Since low‐molecular weight compounds can bind in a non‐covalent manner to the “central pocket” of TEAD,10 the possibility cannot be ruled out that a fatty acid (or another E. coli metabolite) may occupy this pocket in Non‐Acyl‐TEAD4, having the same effect on the YAP/TAZ:TEAD interaction as a covalently bound fatty acid. Molecules non‐covalently bound to Non‐Acyl‐TEAD4 and Cys367Ser cannot be detected in our LC‐MS analyzes. We therefore decided to produce a TEAD4 protein that has a low likelihood of being bound non‐covalently to a ligand. The hTEAD4217–434 Cys367Ser mutant protein was denatured in the presence of urea (8 M), immobilized on a HiTrap Chelating HP column and extensively washed before being refolded (see “Material and Methods”, scheme D). The unfolding/refolding protocol was not optimized to obtain large quantities of refolded protein, but a sufficient amount of refolded hTEAD4217–434 Cys367Ser (hereafter referred to as Ref‐Cys367Ser) was obtained for it to be studied. LC‐MS shows that Ref‐Cys367Ser has the expected molecular weight and that it is not covalently modified [Fig. 1(E)]. This protein has a higher molecular weight than the other TEAD4 proteins, because it still harbors a His6‐tag at its N‐terminus. The T m of Ref‐Cys367Ser, 45.4 ± 0.2°C, is similar to that for one of its non‐refolded counterparts [Fig. 1(E), Supporting Information Fig. S3] suggesting that the overall structure of Cys367Ser is recovered after refolding. hYAP51–99 and hTAZ14–56 induce positive thermal shifts in the FTDA once incubated with Ref‐Cys367Ser, revealing that they bind to this protein (Fig. 3, Table 2). Similarly, SPR experiments (at 10°C) showed that hYAP51–99 and hTAZ14–56 bind in a dose‐dependent manner to Ref‐Cys367Ser following a 1:1 binding mechanism (Fig. 3). The Kd values measured with Ref‐Cys367Ser were close to those determined with Acyl‐TEAD4 (less than a fourfold difference, Table 2) showing that both proteins bind in a similar manner to hYAP51–99 and hTAZ14–56.
Figure 3.
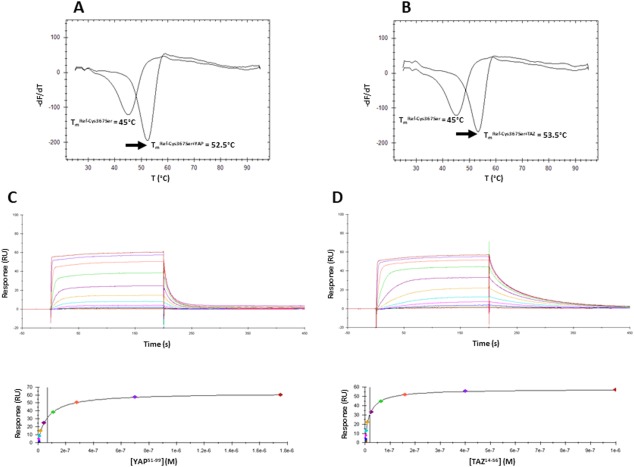
Interaction between Ref‐Cys367Ser and hYAP51–99/hTAZ14–56. A, B. Representative thermograms obtained in the FTDA with Ref‐Cys367Ser (1–2 µM) in the absence or presence of hYAP51–99 (A) or hTAZ14–56 (B) (20 µM). The melting temperatures (T m) were obtained by plotting the first derivative of the fluorescence emission (F) as a function of the temperature (–dF/dT). The curve minimum corresponds to T m. The arrow shows the thermal shift (ΔT m) observed upon peptide binding. The indicated T m have been measured for the experiments depicted on the figure. These T m are slightly different from the T m presented on Table II because these later are the average values obtained from several independent measurements. Upper panels: Representative sensorgrams obtained with Ref‐Cys367Ser immobilized on the chips and different concentrations of hYAP51–99 (C) hTAZ14–56 (D). Lower panels: Req vs [YAP60–100/TAZ17–56] plots. Req are the responses measured at equilibrium (plateau values in the above sensorgrams). These curves were fitted with 1 site binding equation model to determine K d.
Altogether our biochemical data show that—under our experimental conditions—hYAP51–99 and hTAZ14–56 bind in a similar fashion to recombinant acylated and non‐acylated hTEAD4217–434.
Study of the role of TEAD4 acylation in the cellular context
The previous biochemical data were generated with protein fragments under non‐physiological conditions. The effect of TEAD acylation on the YAP:TEAD interaction might be different in cells where full‐length proteins are present in their native context. We therefore studied the interaction between hTEAD4 Cys367Ser and YAP in cellular assays.
Wild‐type (wt) hYAP and N‐terminally V5‐tagged hTEAD4 were co‐transfected in HEK293FT cells, and YAP was immunoprecipitated. The western blot analysis shows that, when the relative amounts of wt and mutant TEAD4 expressed in cells are taken into account, similar quantities of wt and Cys367Ser hTEAD4 are co‐immunoprecipitated with YAP [Fig. 4(A)]. The hTEAD4 Cys367Ser mutation therefore has little effect on the YAP:TEAD interaction. To determine the impact of this mutation in a functional read out, we studied the ability of wt and Cys367Ser hTEAD4 to stimulate in the presence of YAP a luciferase reporter gene under the control of multiple MCAT sites that function as TEAD recognition sequences.18 The reporter activity measured in the presence of hTEAD4 Cys367Ser was about 35% lower than that detected in the presence of wt hTEAD4 [Fig. 4(B)]. However, this lower activity parallels a reduction in expression of hTEAD4 Cys367Ser, as estimated from western blot quantification [Fig. 4(B)]. Considering this difference in expression, there was no significant difference between wt and Cys367Ser hTEAD4 activity in the reporter gene assay. Overall our data show that the single hTEAD4 Cys367Ser mutation had little effect on the YAP:TEAD4 interaction. However, we noticed that hTEAD4 Cys367 appears to have a tendency to be expressed at lower levels than wt hTEAD4 [Fig. 4(A), 4(B)] suggesting that this mutant might be less stable than wt in cells.
Figure 4.
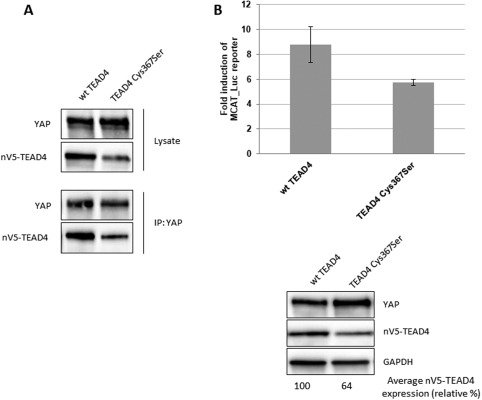
Effect of the hTEAD4 Cys367Ser mutation on the YAP:TEAD4 interaction in cells. (A) Co‐immunoprecipitation: N‐terminally V5‐tagged wt or Cys367Ser TEAD4 were co‐transfected with wt YAP into HEK293FT cells. YAP was immunoprecipitated, and co‐immunoprecipitated nV5‐TEAD4 was determined by anti‐V5 western Blot. (B) MCAT‐Luc reporter assay: N‐terminally V5‐tagged wt and Cys367Ser TEAD4 were co‐transfected with wt YAP into the HEK293::MCAT_Luc reporter model. Resazurin‐normalized luciferase activity was measured and is graphed as fold induction over baseline. YAP and nV5‐TEAD4 expression levels were determined in parallel by western blot. A representative example from one of three independent repeats is shown. The averaged expression levels of the TEAD4 Cys367Ser mutant versus wt TEAD4 were quantified by ImageJ software (https://imagej.nih.gov/ij/) and are indicated as relative percentage.
Discussion
Chan et al.11 have recently shown that TEAD palmitoylation is required for its interaction with YAP/TAZ. In the experiments reported by these authors, the single mutation hTEAD1 Cys359Ser has only a moderate effect on the YAP/TAZ:TEAD1 interaction. Similarly, we observed a reduction of the YAP:TEAD4 interaction in our cellular assays, but this reduction can be paralleled by a lower expression of hTEAD4 Cys367Ser. A more complete inhibition of YAP/TAZ:TEAD was obtained by Chan et al. when they mutated up to 2 additional cysteines, hTEAD1 Cys53Ser and Cys327Ser.11 Under our in vitro experimental conditions, when hTEAD4 Cys367 was mutated to serine, no significant acylation of hTEAD4217–434 was detected anymore, indicating that hTEAD4 is not acylated at Cys335 (equivalent to hTEAD1 Cys327). Furthermore, the structure of hTEAD436–139 did not reveal any acylation of Cys61 (equivalent to hTEAD1 Cys53).19 Altogether this indicates that Cys367 is likely the main acylation site on hTEAD4. In vitro, YAP/TAZ binds to Ref‐Cys367Ser, revealing that a non‐acylated TEAD4 protein which has a high likelihood to have an unoccupied “central pocket” can interact with these two co‐activators. This shows that, under our experimental conditions, hTEAD4217–434 acylation is not required for YAP/TAZ binding.
We also failed to find a significant difference between wt and Cys367Ser TEAD4 in our cellular assays. However, the interpretation of these data should be treated with caution. The hTEAD4 Cys367Ser mutation abolishes covalent binding of a fatty acid, but it may not prevent its non‐covalent binding to the “central pocket”. In cells, therefore, TEAD4 Cys367Ser may have a fatty acid molecule bound to its “central pocket”, and this may have an effect on the YAP/TAZ:TEAD interaction similar to that of a covalently bound fatty acid. This could explain why we failed to find a significant difference between wt and Cys367Ser TEAD4 in our cellular assays.
In agreement with Noland et al.,12 we observed in vitro that non‐acylated hTEAD4217–434 has a lower thermal stability than its acylated counterpart. Noland et al. have also pointed out that the hydroxylamine treatment they used to cleave the thioester bond between hTEAD2 Cys380 and the fatty acid does not preclude the possibility that it could still be bound in a non‐covalent manner to TEAD2.12 Accordingly, TEAD bound non‐covalently to a fatty acid might be less stable than its acylated counterpart. In cells, therefore, hTEAD4 Cys367Ser might be less stable than wt TEAD4 even when bound non‐covalently to a fatty acid molecule. TEAD acylation could thus be required for this protein to adopt and/or to maintain its fully active conformation. In the absence of a covalently bound fatty acid, TEAD might be more unstable and as a consequence less active, which would explain the different observations made in vitro on muscle differentiation and in Drosophila.11
Material and Methods
Cloning, protein expression, purification, and analytics of the TEAD4 proteins used for the biochemical studies
TEAD4 cloning
wt hTEAD4217–434 and Cys367Ser were cloned as previously described.13, 16 For cellular assays the hTEAD4 Cys367Ser mutation was introduced in a plasmid encoding for wt hTEAD416 by use of the QuikChange Lightning Site‐Directed Mutagenesis kit (Agilent, Santa Clara, CA) and the sequence was verified.
TEAD4 expression
E. coli BL21(DE3) competent cells (Novagen, Madison, WI), containing a plasmid encoding for biotin ligase BirA (acetyl‐CoA carboxylase biotin holoenzyme synthetase), were transformed with a pET‐28c expression vector encoding for Avi‐His6‐hTEAD4217–434. Transformed cells were added to LB medium containing 25 µg/mL kanamycin (Invitrogen, Carlsbad, CA) and 34 µg/mL chloramphenicol (Sigma‐Aldrich, Saint‐Louis, MO) and incubated overnight at 37°C under constant agitation (220 rpm). The pre‐cultures were diluted 1:10 into fresh medium containing antibiotics and grown under constant agitation (220 rpm) at 37°C until OD600 = 0.8. Protein expression was induced by adding 1 mM isopropyl β‐D‐1‐thiogalactopyranoside (IPTG, AppliChem GmbH, Darmstadt, Germany) and 135 µg/mL biotin (Sigma‐Aldrich, Saint‐Louis, MO) was added for in vivo biotinylation. The cells were grown overnight at 18°C under constant agitation (220 rpm), harvested by centrifugation at 6000 g, frozen in dry ice and stored at −80°C.
TEAD4 purification
Frozen cell pellets were thawed on ice and re‐suspended in Buffer A (50 mM TRIS·HCl pH 8.0, 1 M NaCl, 2 mM MgCl2, 1 mM Tris(2‐carboxyethyl)phosphine (TCEP), 20 mM imidazole, 0.1% (v:v) Tween 20, 5% (v:v) glycerol), freshly supplemented with cOmplete EDTA free protease inhibitor (1 tablet/50 mL buffer, Roche, Switzerland) and TurboNuclease (20 µL/50 mL buffer, Sigma‐Aldrich, Saint‐Louis, MO). The cells were disrupted by three passes through an EmulsiFlex C3 high pressure homogenizer (Avestin Inc., Ottawa, Canada) at 800–1000 bar and centrifuged at 43,000 × g for 30 min. All the purifications were carried out at 6–8°C. The cleared supernatant was loaded onto either 2 HisTrap HP 1 mL columns (GE Healthcare, Little Chalfont, UK) mounted in series or a single 5 mL column (GE Healthcare, Little Chalfont, UK) equilibrated with Buffer A. The columns were washed with Buffer A, followed by Buffer B (50 mM TRIS·HCl pH 8.5, 100 mM NaCl, 2 mM MgCl2, 1 mM TCEP, 20 mM imidazole, 5% (v:v) glycerol). Bound proteins were eluted with a linear gradient of Buffer B containing 250 mM imidazole. At this stage, 4 different purification schemes were used.
Scheme A ‐ purification of wt hTEAD4217–434 and Cys367Ser
The fractions containing TEAD4 were pooled and the His6‐tag was cleaved off by overnight incubation during dialysis at 4°C with of 1% (w:w) recombinant HRV 3C protease (produced in‐house) in 50 mM TRIS·HCl pH 8.0, 100 mM NaCl, 2 mM MgCl2, 1 mM TCEP, 5% (v:v) glycerol. The dialyzed protein was loaded onto a HiLoad 16/60 Superdex 75 prep grade column (GE Healthcare, Little Chalfont, UK) equilibrated with 20 mM TRIS·HCl pH 8.0, NaCl 100 mM, 2 MgCl2, 1 mM TCEP, 5% (v:v) glycerol. The fractions containing TEAD4 were concentrated by centrifugation in Amicon Ultra‐15 centrifugal units (Millipore, Billerica, MA), aliquoted, snap‐frozen in dry ice, and stored at −80°C.
Scheme B ‐ purification of Non‐Acyl‐TEAD4
The fractions containing TEAD4 were pooled and the His6‐tag was cleaved off by overnight incubation during dialysis at 4°C with 1% (w:w) recombinant HRV 3C protease (produced in‐house) in 50 mM TRIS·HCl pH 8.0, 100 mM NaCl, 2 mM MgCl2, 1 mM TCEP, 5% (v:v) glycerol. The dialyzed protein was loaded onto a HisTrap HP 5 mL column equilibrated with Buffer B. The TEAD4 protein was eluted with 5 column volumes Buffer B. Fractions containing TEAD4 were pooled and diluted 1:8 with IEX buffer A (20 mM TRIS.HCl pH 7.8, 2 mM MgCl2, 1 mM TCEP, 50 mM NaCl, 5% glycerol). The diluted protein was then loaded onto a Mono Q 10/100 GL anion‐exchange column (GE Healthcare, Little Chalfont, UK). Non‐Acyl‐TEAD4 was eluted by a gradient ranging from 27 to 40% IEX buffer B (IEX buffer A containing 500 mM NaCl) over 5 column volumes. Fractions containing Non‐Acyl‐TEAD4 were pooled and concentrated in an Amicon Ultra‐15 centrifugal unit. The protein was aliquoted, snap‐frozen on dry ice, and stored at −80°C.
Scheme C ‐ purification of acyl‐TEAD4
The fractions containing TEAD4 were pooled and the His6‐tag was cleaved off by overnight incubation during dialysis at 4°C with 1% (w:w) recombinant HRV 3C protease (produced in‐house) in Buffer B. The dialyzed protein was loaded onto two His‐Trap HP 1 mL columns mounted in series and equilibrated with Buffer B. The flow‐through fractions containing the cleaved protein were collected, pooled and concentrated by centrifugation in Amicon Ultra‐15 centrifugal units. After analysis of the protein content and determination of the concentration by Reversed‐Phase High‐Performance Liquid Chromatography (RP‐HPLC), a twofold molar equivalent of myristoyl Coenzyme A (Sigma‐Aldrich, Saint‐Louis, MO) was added, and the sample was incubated at room temperature for 30 min. The reaction products were loaded onto a HiLoad 16/60 Superdex 75 prep grade column equilibrated with 20 mM TRIS·HCl pH 8.0, 100 mM NaCl, 2 mM MgCl2, 1 mM TCEP, 5% (v:v) glycerol. Protein fractions containing Acyl‐TEAD4 were concentrated by centrifugation in Amicon Ultra‐15 cartridges. The protein was aliquoted, snap‐frozen in dry ice and stored at −80°C.
Scheme D ‐ purification of ref‐Cys367Ser
The fractions containing TEAD4 were diluted 1:8 in denaturation buffer (8 M urea, 50 mM NaH2PO4 pH 8.0, 100 mM NaCl, 0.25 mM TCEP) and incubated at room temperature for 20 min. The denatured protein was then loaded onto 2 HisTrap HP 1mL columns mounted in series and equilibrated with denaturation buffer. The columns were washed with 10 mL of denaturation buffer. The protein was then refolded while passing 20 mL of Buffer B over the column. Non‐aggregated protein was eluted from the column with 8 mL Buffer B containing 250 mM imidazole. Fractions containing TEAD4 were collected and further purified on a Superdex 75 10/300 size‐exclusion column (GE Healthcare, Little Chalfont, UK) equilibrated with 50 mM NaH2PO4 pH 7.8, 100 mM NaCl, 0.25 mM TCEP. Fractions containing Ref‐Cys367Ser were collected and pooled. The protein was aliquoted, snap‐frozen on dry ice and stored at −80°C.
Analytics of the TEAD4 proteins
All the TEAD4 proteins were cloned such that they possessed at their N‐terminus a His6‐tag and an Avi‐tag that was biotinylated during expression. The His6‐tag was removed from wt hTEAD4217–434, Acyl‐TEAD4, Non‐Acyl‐TEAD4, and Cys367Ser during purification while it was still present in Ref‐Cys367Ser. This explains the differences in mass measured between these different proteins. Samples were analyzed by SDS‐PAGE after each step of the purification process. Pre‐cast NuPAGE 4–12% Bis‐Tris protein gels (ThermoFisher Scientific, Waltham, MA) were run with NuPAGE MES SDS buffer (ThermoFisher Scientific, Waltham, MA) and stained with SimplyBlue SafeStain (ThermoFisher Scientific, Waltham, MA). The concentration and purity of the protein were assessed by analytical RP‐HPLC using a POROS R1/10 2.1 × 100 mm column (Dr Maisch GmbH, Ammerbuch, Germany) with a 20–80% gradient of water/acetonitrile at 80°C. Absorbance at 210 nm was monitored, and a BSA calibration curve was used to determine the protein concentration. The intact protein mass was determined by LC‐MS, using a Xevo‐G2‐S QTof mass‐spectrometer coupled to an ACQUITY Ultra‐Performance Liquid Chromatography system (Waters, Milford, MA).
Synthetic peptides
The synthetic N‐acetylated and C‐amidated hYAP51–99 (Ac‐GHQIVHVRGDSETDLEALFNAVxNPKTANVPQTVPxRLRKLPDSFFKPP‐NH2) and hTAZ14–56 (Ac‐GQQVIHVTQDLDTDLEALFNSVxNPKPSSWRKKILPESFFKEP‐NH2) peptides were purchased from Bachem (Bubendorf, Switzerland). To enhance the oxidative stability of the peptide, the methionine residues present in the wt sequences have been replaced with norleucines (x in the above sequences). The peptides were dissolved in 90% DMSO (10 mM) and stored at −20°C. The purity of hYAP51–99 and hTAZ14–56 was higher than 90% as determined by RP‐HPLC and LC‐MS.
Fluorescence‐based thermal denaturation assay
Proteins (1–2 µM) were diluted in 50 mM HEPES, 100 mM KCl, 0.25 mM TCEP, 1 mM EDTA, 2% (v/v) DMSO, pH 7.4, containing 2x SYPRO Orange dye (ThermoFisher Scientific, Waltham, MA) in the presence or absence of the peptides (20 µM). This mix was added to 384‐well, thin‐walled Hard‐Shell PCR microplates (BioRad, Hercules, CA) that were covered by optically clear adhesive seals to prevent evaporation. Measurements were carried out with a CFX384 Real‐Time PCR Detection System (BioRad, Hercules, CA). The temperature was increased from 25 to 95°C at 0.5°C/30 s, and the fluorescence intensity was measured with the excitation and emission filters set to 465 and 590 nm, respectively. The melting temperatures (T m) were determined by analyzing the thermal denaturation curves with CFX Manager (BioRa, Hercules, CA).
Surface plasmon resonance
All the experiments were carried out with a Biacore T200 optical biosensor and Series S sensor Chip SA (GE Healthcare, Little Chalfont, UK). The chips were washed 3 times with 1 M NaCl/50 mM NaOH. The N‐Avitagged hTEAD4217–434 proteins were injected at a flow rate of 5 μl/min in SPR immobilization buffer (50 mM HEPES, 100 mM KCl, 0.25 mM TCEP, 1 mM EDTA, 0.05% (v/v) Tween 20, 0.05% (w/v) bovine serum albumin (BSA), pH 7.4). The experiments were performed at 10 and 25°C in SPR running buffer (SPR immobilization buffer containing 2% (v/v) DMSO) at a flow rate of 50 mL/min. Peptides were diluted in SPR running buffer (2% (v/v) DMSO final). After baseline equilibration with a series of buffer blanks, a 1–3% DMSO correction series was performed. Each cycle consisted of an injection phase of 200 s followed by a dissociation phase of 400 s. All data were referenced for a blank streptavidin reference surface and blank injections of running buffer to minimize the influence of baseline drift upon binding. Each sensorgram was visually inspected, and low‐quality experiments (e.g., large bulk effect, equilibrium not reached, noisy baseline, etc.) were not further analyzed. The sensorgrams were globally fitted with a 1:1 interaction model using the Biacore T200 evaluation software. The dissociation constants (K d) were measured at equilibrium. Experiments with a standard error on fitted K d higher than 20% K d or a fitted maximum binding capacity ( ) <70% theoretical R max ( ) ( = (MWYAP/MWTEAD) xRTEAD × n with MWYAP and MWTEAD molecular weight of YAP and TEAD4, respectively, RTEAD level of immobilization of TEAD4 in response unit and n stoichiometry (here n = 1) were discarded.
Structure determination of the hYAP60–100:hTEAD4217–434 complex
Expression and purification of TEAD4 for crystallization
His6‐PreSc‐hTEAD4217–434 protein expression was outsourced to Novoprotein (China). Protein purification yielded after PreScission cleavage to TEAD4217–434 harboring at its N‐terminus additional glycine and proline residues (hereafter referred to as GP‐TEAD4). Partially acylated GP‐TEAD4 was myristoylated by incubation in vitro with myristoyl Coenzyme A and the buffer was exchanged on a NAP‐5 column (GE Healthcare, Little Chalfont, UK). The protein was complexed with a 1.2‐excess of hYAP60–100 (ThermoFisher Scientific, Waltham, MA). The complex was concentrated to 4 mg/mL. The final protein buffer was 25 mM TRIS.HCl pH 8.0, 100 mM NaCl, 1 mM TCEP.
Crystallization, data collection, structure solution, and refinement
Co‐crystals of GP‐TEAD4 in complex with YAP60–100 were obtained at 20°C by sitting drop vapor diffusion. The drops were made up of 1 µL of protein solution and 1 µL of well solution. The reservoir solution consisted of 15% (w/v) PEG3350 + 2% tacsimate, pH5.0 + 0.1 M Na citrate pH 6.0. The crystal was cryo‐protected in well solution supplemented with 20% ethylene glycol and flash‐frozen in liquid nitrogen. A data set was collected at the Swiss Light Source Facility (SLS, Villigen, Switzerland) on beamline X10SA. The data were processed with XDS.20 The structure of the YAP60–100:GP‐TEAD4 complex was determined by molecular replacement (PHASER21) using a previous in‐house TEAD4 X‐ray structure as the search model. Programs REFMAC22 and COOT23 were used for refinement and model (re)building. The final refined structure has an R (Rfree) value of 0.229 (0.252) and showed excellent geometry in the Ramachandran plot. All residues are in the permitted regions, except residue Lys277 (excellent electron density confirms the local structure). See Table 1 for details of the data collection and structure refinement. The crystallographic data have been deposited at the RSCB Protein Data Bank (PDB, www.pdb.org) with the access code 5OAQ.
Cellular assays
Cell culture, transfections, YAP/TEAD immunoprecipitations and luciferase assays were carried out as previously described in.16 Anti‐GAPDH antibody (Cell Signaling Technology, Danvers, MA) was used for western Blot analysis all the other antibodies have been described previously in.16
Supporting information
Supporting Information
Acknowledgments
We thank Bernd Gerhartz, who organized the fermentation outsourcing of TEAD4 for crystallization, and Bernard Mathis, who purified this protein.
Mesrouze Y and Meyerhofer M contributed equally to this work.
Statement: The TEAD transcription factors are the most distal elements of the Hippo pathway. These proteins are acylated at a conserved cysteine residue. Non‐acylated and acylated TEAD interact in a similar manner in vitro and in cells with the coactivators YAP/TAZ. However the acylation of TEAD enhances its stability exerting an important role in the function of this transcription factor.
References
- 1. Zhao B, Tumaneng K, Guan KL (2011) The Hippo pathway in organ size control, tissue regeneration and stem cell self‐renewal. Nat Cell Biol 13:877–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yu FX, Zhao B, Guan KL (2015) Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 163:811–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu J, Lin JD, Wang CY, Chinnaiyan AM, Lai ZC, Guan KL (2008) TEAD mediates YAP‐dependent gene induction and growth control. Genes Dev 22:1962–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Felley‐Bosco E, Stahel R (2014) Hippo/YAP pathway for targeted therapy. Transl Lung Cancer Res 3:75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gong R, Yu FX (2015) Targeting the Hippo pathway for anti‐cancer therapies. Curr Med Chem 22:4104–4117. [DOI] [PubMed] [Google Scholar]
- 6. Santucci M, Vignudelli T, Ferrari S, Mor M, Scalvini L, Bolognesi ML, Uliassi E, Costi MP (2015) The Hippo pathway and YAP/TAZ‐TEAD protein‐protein interaction as targets for regenerative medicine and cancer treatment. J Med Chem 58:4857–4873. [DOI] [PubMed] [Google Scholar]
- 7. Zhang K, Qi HX, Hu ZM, Chang YN, Shi ZM, Han XH, Han YW, Zhang RX, Zhang Z, Chen T, Hong W (2015) YAP and TAZ take center stage in cancer. Biochemistry 54:6555–6566. [DOI] [PubMed] [Google Scholar]
- 8. Chen L, Loh PG, Song H (2010) Structural and functional insights into the TEAD‐YAP complex in the Hippo signaling pathway. Protein Cell 1:1073–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li Z, Zhao B, Wang P, Chen F, Dong Z, Yang H, Guan KL, Xu Y (2010) Structural insights into the YAP and TEAD complex. Genes Dev 24:235–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pobatti AV, Han X, Hung AW, Weiguang S, Huda N, Chen GY, Kang C, Chia CSB, Luo X, Hong W, Poulsen A (2015) Targeting the central pocket in human transcription factor TEAD as a potential cancer theraputic strategy. Structure 23:2076–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chan PY, Han X, Zheng B, DeRan M, Yu J, Jarugumilli GK, Deng H, Pan D, Luo X, Wu X (2016) Autopalmitoylation of TEAD proteins regulates transcriptional output of the Hippo pathway. Nat Chem Biol 12:282–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Noland CL, Gierke S, Schnier PD, Murray J, Sandoval WN, Sagolla M, Dey A, Hannoush RN, Fairbrother WJ, Cunningham CN (2016) Palmitoylation of TEAD transcription factors is required for their stability and function in Hippo pathway signaling. Structure 24:179–186. [DOI] [PubMed] [Google Scholar]
- 13. Hau JC, Erdmann D, Mesrouze Y, Furet P, Fontana P, Zimmermann C, Schmelzle T, Hofmann F, Chène P (2013) The TEAD4‐YAP/TAZ protein‐protein interaction: expected similarities and unexpected differences. ChemBioChem 14:1218–1225. [DOI] [PubMed] [Google Scholar]
- 14. Mesrouze Y, Hau JC, Erdmann D, Zimmermann C, Fontana P, Schmelzle T, Chène P (2014) The surprising features of the TEAD4‐Vgll1 protein‐protein interactions. ChemBioChem 15:537–542. [DOI] [PubMed] [Google Scholar]
- 15. Mesrouze Y, Erdmann D, Zimmermann C, Fontana P, Schmelzle T, Chène P (2016) Different recognition of TEAD transcription factor by the conserved β‐strand:loop:α‐helix motif of the TEAD‐binding site of YAP and VGLL1. ChemistrySelect 1:2993–2997. [Google Scholar]
- 16. Mesrouze Y, Bokhovchuk F, Meyerhofer M, Fontana P, Zimmermann C, Martin T, Delaunay C, Erdmann D, Schmelzle T, Chene P (2017) Dissection of the interaction between the intrinsically disordered YAP protein and the transcription factor TEAD. Elife 6:e25068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen L, Chan SW, Zhang XQ, Walsh M, Lim CJ, Hong W, Song H (2010) Structural basis of YAP recognition by TEAD4 in the Hippo pathway. Genes Dev 24:290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Michaloglou C, Lehmann W, Martin T, Delaunay C, Hueber A, Barys L, Niu H, Billy E, Wartmann M, Ito M, Wilson CJ, Digan ME, Bauer A, Voshol H, Christofori G, Sellers WR, Hofmann F, Schmelzle T (2013) The tyrosine phosphatase PTPN14 is a negative regulator of YAP activity. PLoS One 8:e61916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shi Z, He F, Chen M, Hua L, Wang W, Jiao S, Zhou Z (2017) DNA‐binding mechanism of the Hippo pathway transcription factor TEAD4. Oncogene [VOL:PAGE #S]. [DOI] [PubMed] [Google Scholar]
- 20. Kabsch W (2010) XDS. Acta Cryst D66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Project CC (1994) The CCP4 suite: programs for protein crystallography. Acta Cryst D50:760–763. [DOI] [PubMed] [Google Scholar]
- 22. Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Cryst D60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 23. Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Cryst D66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information